
Structured Mesh-Type Pt/Mn/γ-Al2O3/Al Catalyst Enhanced the CO Oxidation at Room Temperature by In Situ Generation of Hydroxyl: Behavior and Mechanism


Abstract

1. Introduction
2. Results and Discussions
2.1. DFT Investigation of Water Dissociation and CO Oxidation Behaviors on Mn-Modified Pt-Based Catalysts
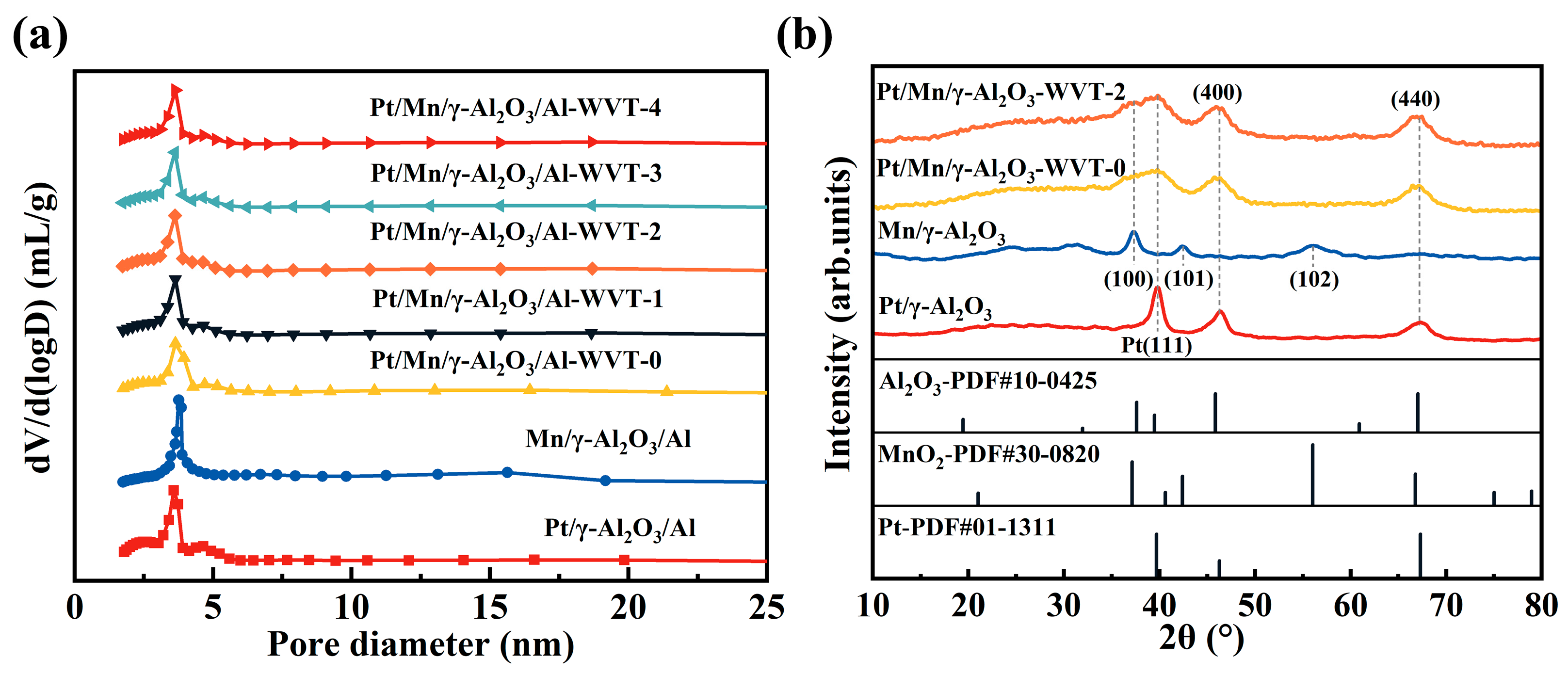
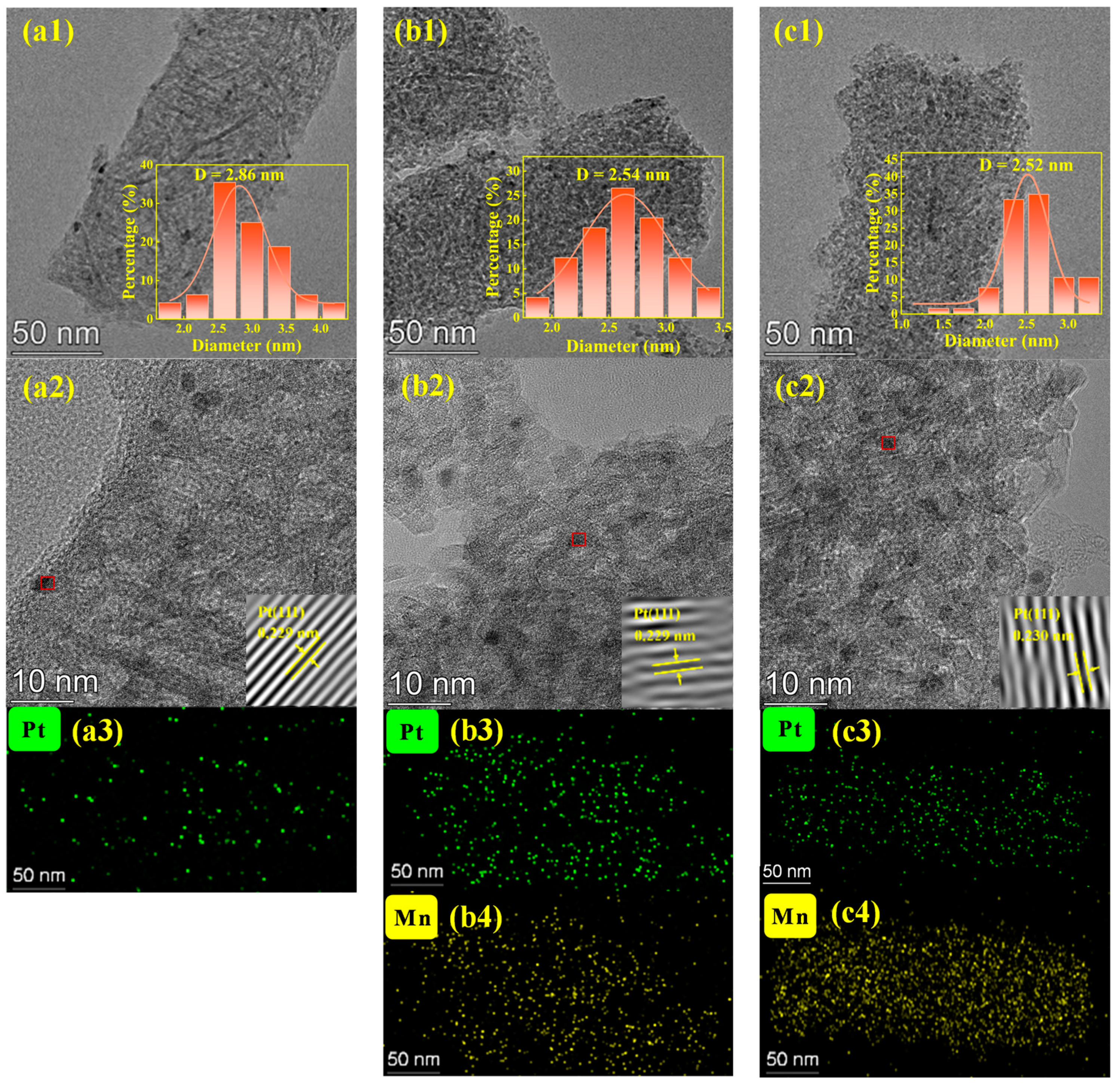
2.2. Surface Properties of the Mesh-Type γ-Al2O3 Carrier and the Effect of Mn-Loading on Pt-Based Catalyst
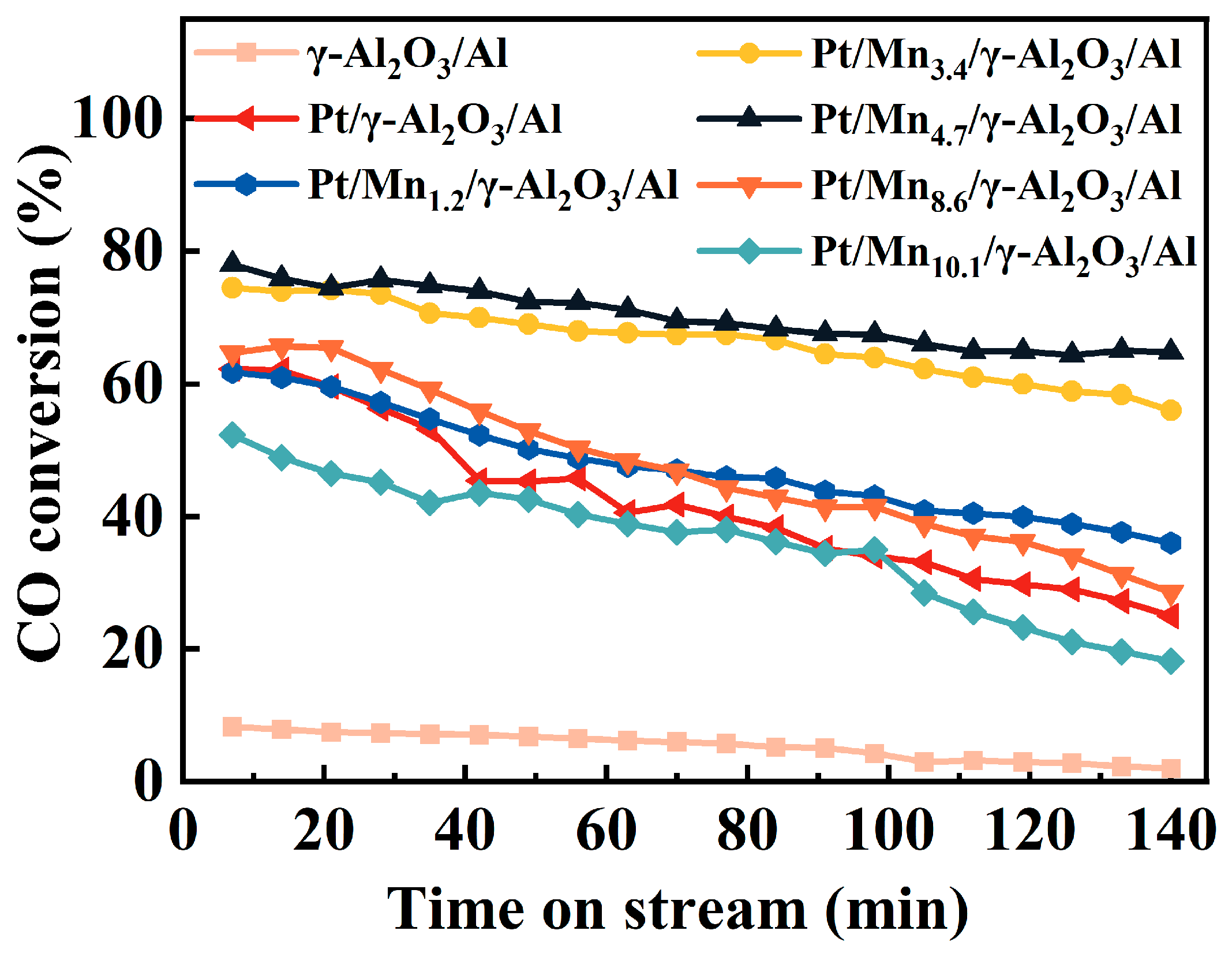
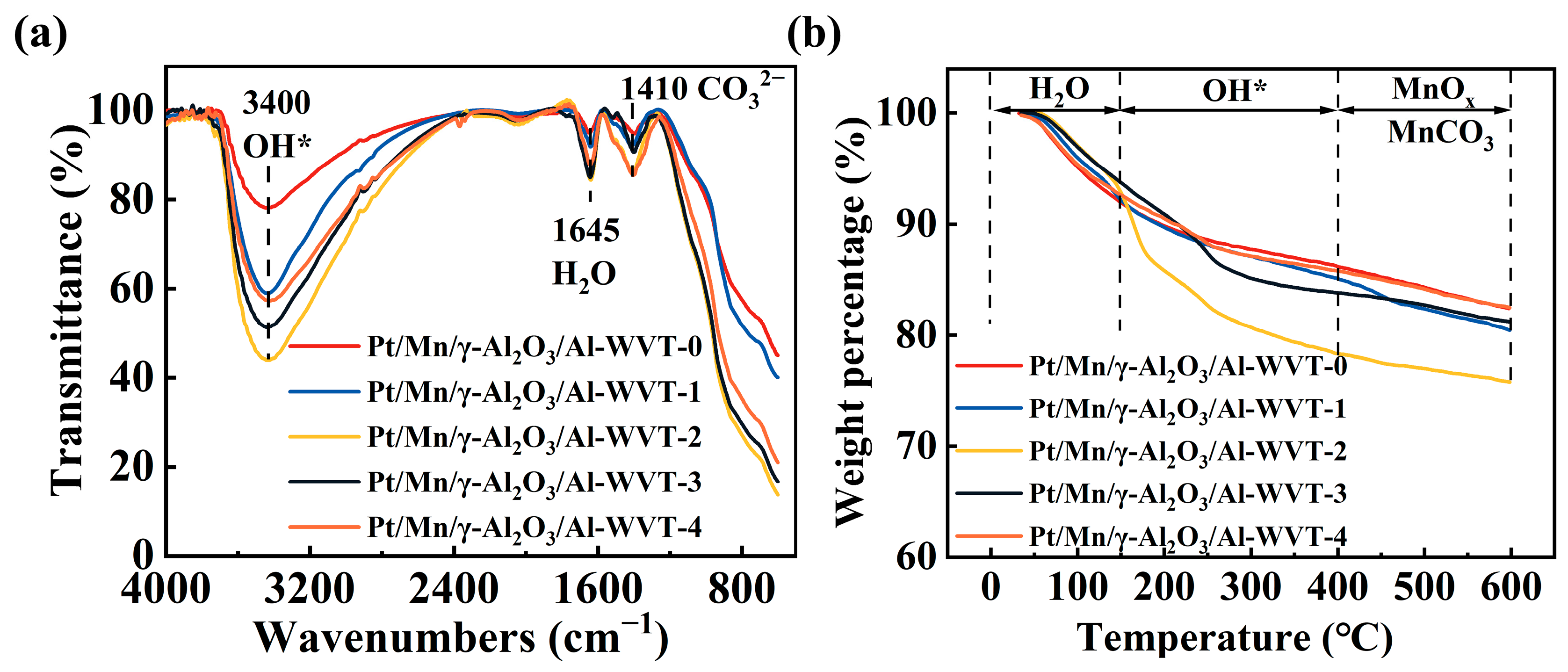
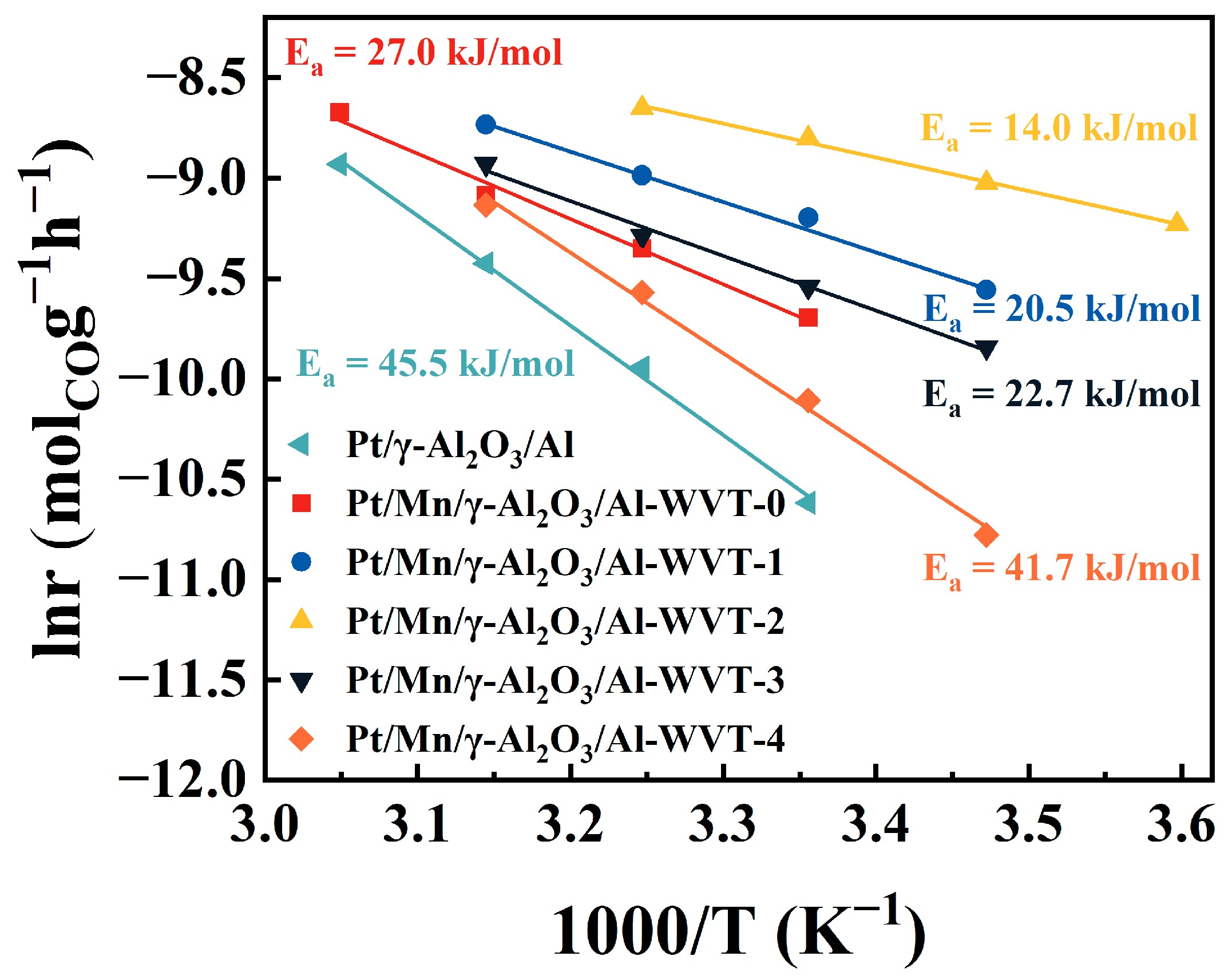
2.3. The Effects of Mn-Modification on Enhancing the CO Removal Efficiency by Water Vapor Treatment
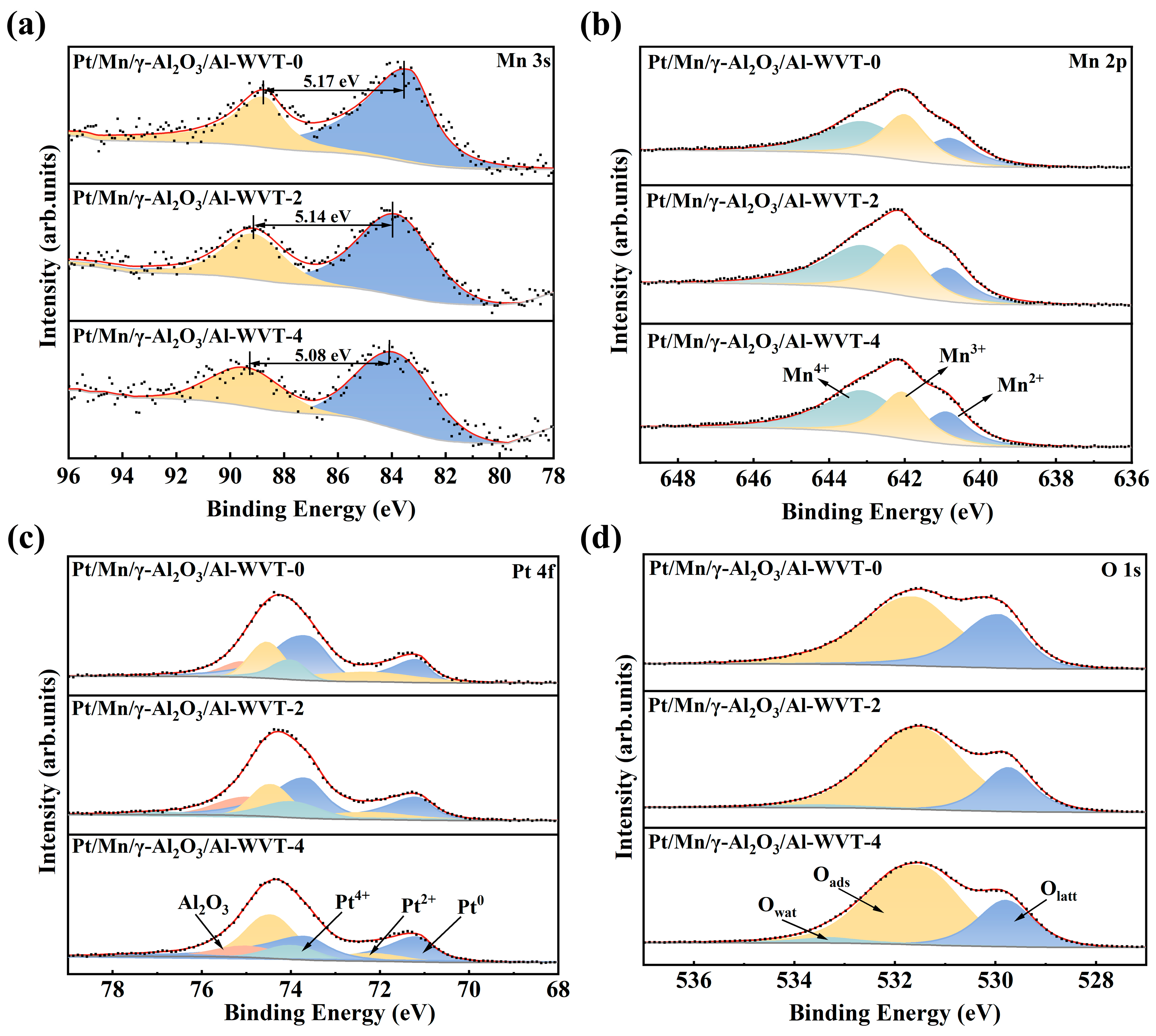
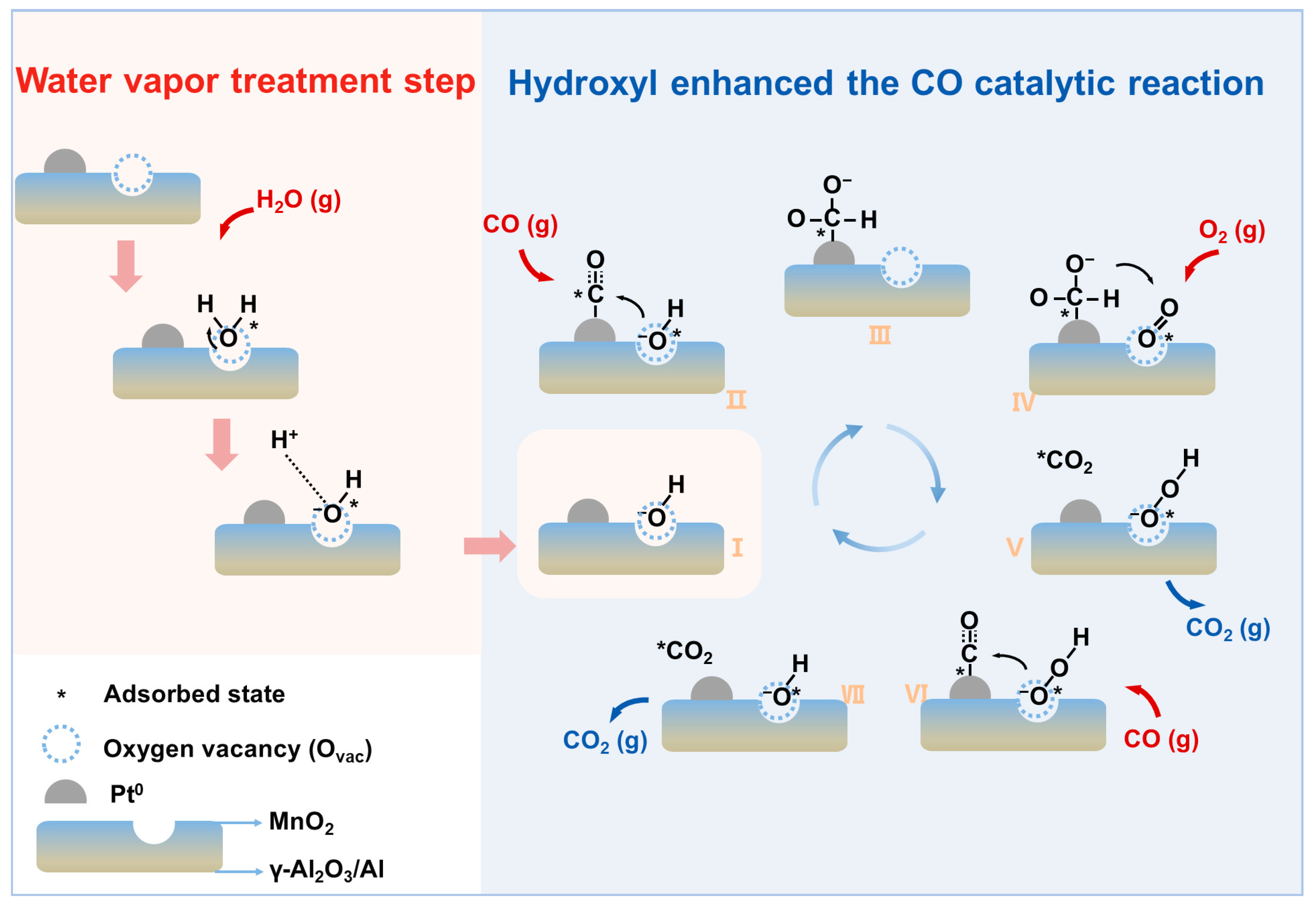
2.4. Mechanism of Water Vapor Treatment and CO Oxidation Reaction
3. Materials and Methods
3.1. Catalyst Preparation
3.2. Catalyst Characterization
3.3. Catalytic Activity Evaluation
3.4. DFT Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Miller, S.M.; Matross, D.M.; Andrews, A.E.; Millet, D.B.; Longo, M.; Gottlieb, E.W.; Hirsch, A.I.; Gerbig, C.; Lin, J.C.; Daube, B.C.; et al. Sources of Carbon Monoxide and Formaldehyde in North America Determined from High-Resolution Atmospheric Data. Atmos. Chem. Phys. 2008, 8, 7673–7696. [Google Scholar] [CrossRef]
- Wang, Y.; Yuan, Q.; Li, T.; Zhu, L. Global Spatiotemporal Estimation of Daily High-Resolution Surface Carbon Monoxide Concentrations Using Deep Forest. J. Clean. Prod. 2022, 350, 131500. [Google Scholar] [CrossRef]
- Dent, M.R.; Rose, J.J.; Tejero, J.; Gladwin, M.T. Carbon Monoxide Poisoning: From Microbes to Therapeutics. Annu. Rev. Med. 2024, 75, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Jing, P.; Gong, X.; Liu, B.; Zhang, J. Recent Advances in Synergistic Effect Promoted Catalysts for Preferential Oxidation of Carbon Monoxide. Catal. Sci. Technol. 2020, 10, 919–934. [Google Scholar] [CrossRef]
- Rao, R.; Sun, H.; Dong, X.; Dong, H.; Fang, W.; Tang, Y.; Fang, S.; Hu, C. A Facile and Large-Scale Synthesis of Co3O4/N-Doped Graphene for CO Oxidation: Low-Temperature Catalytic Activity and the Role of Nitrogen States. Appl. Surf. Sci. 2020, 513, 145800. [Google Scholar] [CrossRef]
- Boronin, A.I.; Slavinskaya, E.M.; Figueroba, A.; Stadnichenko, A.I.; Kardash, T.Y.; Stonkus, O.A.; Fedorova, E.A.; Muravev, V.V.; Svetlichnyi, V.A.; Bruix, A.; et al. CO Oxidation Activity of Pt/CeO2 Catalysts below 0 Degrees C: Platinum Loading Effects. Appl. Catal. B-Environ. 2021, 286, 119931. [Google Scholar] [CrossRef]
- Luo, Y.; He, T.; Li, G.; Wu, D.; Liu, W.; Zhang, S.; Peng, H. Synthetic Pt-Fe(OH)x Catalysts by One-Pot Method for CO Catalytic Oxidation. Front. Chem. 2024, 12, 1413489. [Google Scholar] [CrossRef]
- Zhang, T.; Zheng, P.; Gao, J.; Liu, X.; Ji, Y.; Tian, J.; Zou, Y.; Sun, Z.; Hu, Q.; Chen, G.; et al. Simultaneously Activating Molecular Oxygen and Surface Lattice Oxygen on Pt/TiO2 for Low-Temperature CO Oxidation. Nat. Commun. 2024, 15, 6827. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Xing, J.-Y.; Jia, A.-P.; Tang, C.; Wang, Y.-J.; Luo, M.-F.; Lu, J.-Q. CO Oxidation over Pt/Cr1.3Fe0.7O3 Catalysts: Enhanced Activity on Single Pt Atom by H2O Promotion. J. Catal. 2020, 382, 192–203. [Google Scholar] [CrossRef]
- Li, J.-J.; Zhu, B.-L.; Wang, G.-C.; Liu, Z.-F.; Huang, W.-P.; Zhang, S.-M. Enhanced CO Catalytic Oxidation over an Au–Pt Alloy Supported on TiO2 Nanotubes: Investigation of the Hydroxyl and Au/Pt Ratio Influences. Catal. Sci. Technol. 2018, 8, 6109–6122. [Google Scholar] [CrossRef]
- Zhang, N.; Li, L.; Wu, R.; Song, L.; Zheng, L.; Zhang, G.; He, H. Activity Enhancement of Pt/MnOx Catalyst by Novel β-MnO2 for Low-Temperature CO Oxidation: Study of the CO–O2 Competitive Adsorption and Active Oxygen Species. Catal. Sci. Technol. 2019, 9, 347–354. [Google Scholar] [CrossRef]
- Deng, Y.; Liu, S.; Fu, L.; Yuan, Y.; Zhao, A.; Wang, D.; Zheng, H.; Ouyang, L.; Yuan, S. Crystal Plane Induced Metal-Support Interaction in Pd/Pr-CeO2 Catalyst Boosts H2O-Assisted CO Oxidation. J. Catal. 2023, 417, 60–73. [Google Scholar] [CrossRef]
- Lin, J.; Zhao, S.; Yang, J.; Huang, W.-H.; Chen, C.-L.; Chen, T.; Zhao, Y.; Chen, G.; Qiu, Y.; Gu, L. Hydrogen Spillover Induced PtCo/CoOx Interfaces with Enhanced Catalytic Activity for CO Oxidation at Low Temperatures in Humid Conditions. Small 2023, 20, 2309181. [Google Scholar] [CrossRef]
- Liu, J.; Liang, Z.; Xie, Z.; Shu, Q.; Zhu, Y.; Zhang, Q. Fe-Modified Hydroxyl-Rich Structured Pt/Fex/γ-Al2O3/Al Catalyst for CO Oxidationat Room Temperature: Behavior and Mechanism. React. Kinet. Mech. Catal. 2023, 136, 1283–1299. [Google Scholar] [CrossRef]
- Zhao, S.; Wen, Y.; Liu, X.; Pen, X.; Lu, F.; Gao, F.; Xie, X.; Du, C.; Yi, H.; Kang, D.; et al. Formation of Active Oxygen Species on Single-Atom Pt Catalyst and Promoted Catalytic Oxidation of Toluene. Nano Res. 2020, 13, 1544–1551. [Google Scholar] [CrossRef]
- Zhang, H.; Sui, S.; Zheng, X.; Cao, R.; Zhang, P. One-Pot Synthesis of Atomically Dispersed Pt on MnO2 for Efficient Catalytic Decomposition of Toluene at Low Temperatures. Appl. Catal. B-Environ. 2019, 257, 117878. [Google Scholar] [CrossRef]
- Li, X.; Ren, S.; Chen, Z.; Jiang, Y.; Wang, M.; Wang, L.; Liu, M. Unraveling the Morphology and Crystal Plane Dependence of Bifunctional MnO2 Catalyst for Simultaneous Removal of NO and CO at Low Temperature. Sep. Purif. Technol. 2023, 325, 124760. [Google Scholar] [CrossRef]
- Li, X.; Jiang, Y.; Ren, S.; Chen, Z.; Wang, M.; Li, J.; Yang, J.; Chen, H. A Comparison of Bifunctional MnOx Catalysts Prepared via Different Precipitants for Simultaneous Removal NO and CO. Mol. Catal. 2023, 549, 113524. [Google Scholar] [CrossRef]
- Shan, R.; Sheng, Z.; Hu, S.; Xiao, H.; Zhang, Y.; Zhang, J.; Wang, L.; Zhang, C.; Li, J. Enhancing Oxidation Reaction over Pt-MnO2 Catalyst by Activation of Surface Oxygen. J. Environ. Sci. 2023, 134, 117–125. [Google Scholar] [CrossRef]
- Yan, X.; Gan, T.; Shi, S.; Du, J.; Xu, G.; Zhang, W.; Yan, W.; Zou, Y.; Liu, G. Potassium-Incorporated Manganese Oxide Enhances the Activity and Durability of Platinum Catalysts for Low-Temperature CO Oxidation. Catal. Sci. Technol. 2021, 11, 6369–6373. [Google Scholar] [CrossRef]
- Wu, D.; Song, L.; Zhang, B.; Li, Y. Effect of the Mechanical Failure of Catalyst Pellets on the Pressure Drop of a Reactor. Chem. Eng. Sci. 2003, 58, 3995–4004. [Google Scholar] [CrossRef]
- Zhang, W.; Xie, W.; Ma, T.; Zhang, Q. Development of Zn Doping Fe-Pd Bifunctional Mesh-Type Catalyst for Heterogeneous Electro-Fenton System. AIChE J. 2025, 71, e18604. [Google Scholar] [CrossRef]
- Zhang, Q.; Luan, H.; Li, T.; Wu, Y.; Ni, Y. Study on Pt-Structured Anodic Alumina Catalysts for Catalytic Combustion of Toluene: Effects of Competitive Adsorbents and Competitive Impregnation Methods. Appl. Surf. Sci. 2016, 360, 1066–1074. [Google Scholar] [CrossRef]
- Li, R.; Zheng, X.; Yan, H.; Tuo, Y.; Liu, Y.; Feng, X.; Chen, X.; Chen, D.; Yang, C. Understanding the Promotion Effect of Pt in the Reaction System of Water Gas Shift Reaction Catalyzed by Pt/α-MoC from Theoretical Perspectives. Appl. Surf. Sci. 2024, 644, 158800. [Google Scholar] [CrossRef]
- Wang, A.; Wu, Y.; Shen, X.; Zhang, Q.; Jian, H.; Han, C. Efficient Ozone Decomposition by Amorphous Mn-Ni Bimetallic Catalysts under an Entire Humidity Environment. J. Environ. Chem. Eng. 2024, 12, 113848. [Google Scholar] [CrossRef]
- Zhang, Q.; Fan, F.; Xu, G.; Ye, D.; Wang, W.; Zhu, Z. Steam Reforming of Dimethyl Ether over a Novel Anodic γ-Al2O3 Supported Copper Bi-Functional Catalyst. Int. J. Hydrogen Energy 2013, 38, 10305–10314. [Google Scholar] [CrossRef]
- Zhang, Q.; Sun, D.; Fan, F.; Zhang, Q.; Zhu, Z. A Novel Monolith Catalyst of Plate-Type Anodic Alumina for the Hydrolysis of Dimethyl Ether. Catal. Commun. 2013, 34, 64–68. [Google Scholar] [CrossRef]
- Lü, H.; Gao, J.; Jiang, Z.; Jing, F.; Yang, Y.; Wang, G.; Li, C. Ultra-Deep Desulfurization of Diesel by Selective Oxidation with [C18H37N(CH3)3]4[H2NaPW10O36] Catalyst Assembled in Emulsion Droplets. J. Catal. 2006, 239, 369–375. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, J.; Rong, S.; Wang, H.; Zhang, P. Cerium Modified Birnessite-Type MnO2 for Gaseous Formaldehyde Oxidation at Low Temperature. Appl. Catal. B Environ. 2017, 211, 212–221. [Google Scholar] [CrossRef]
- Liu, X.; Wu, J.; Zhang, S.; Li, Q.; Wu, Z.; Zhang, J. Evaluation of an ε-Manganese (IV) Oxide/Manganese Vanadium Oxide Composite Catalyst Enriched with Oxygen Vacancies for Enhanced Formaldehyde Removal. Appl. Catal. B Environ. 2023, 320, 121994. [Google Scholar] [CrossRef]
- Shu, Q.; Xiang, Y.; Zhang, Q. Planar Growth, Facet-Oriented La2O3 (003) in CuLa Catalysts: Enhancement in Charge Transport and Water Adsorption for Methanol Steam Reforming. Fuel 2025, 381, 133612. [Google Scholar] [CrossRef]
- Muravev, V.; Parastaev, A.; van den Bosch, Y.; Ligt, B.; Claes, N.; Bals, S.; Kosinov, N.; Hensen, E.J.M. Size of Cerium Dioxide Support Nanocrystals Dictates Reactivity of Highly Dispersed Palladium Catalysts. Science 2023, 380, 1174–1179. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Jin, H.; Zhou, H.; Liu, B.; Hu, C.; Chen, D.; Wang, Z.; Hu, Z.; Zhao, Y.; Li, H.-W.; et al. Cobalt Single Atom Site Isolated Pt Nanoparticles for Efficient ORR and HER in Acid Media. Nano Energy 2021, 88, 106221. [Google Scholar] [CrossRef]
- Ma, C.; Pan, J.; Chen, C.; Dong, Y.; Yao, F.; Wang, F.; Song, M. Investigation into the Roles of Interfacial H2O Structure in Catalytic Oxidation of HCHO and CO over CuMnO2 Catalysts. J. Environ. Sci. 2024, 137, 310–320. [Google Scholar] [CrossRef]
- Lizarbe, A.J.; Major, G.H.; Fernandez, V.; Fairley, N.; Linford, M.R. Insight Note: X-Ray Photoelectron Spectroscopy (XPS) Peak Fitting of the Al 2p Peak from Electrically Isolated Aluminum Foil with an Oxide Layer. Surf. Interface Anal. 2023, 55, 651–657. [Google Scholar] [CrossRef]
- Ma, L.; Ding, C.; Ma, Z.; Wang, J.; Xu, H.; Zhang, K.; Li, G. Defect Engineering by Steam Treatment over Pt/CeO2 Catalyst Promoting C–H Activation in Partial Oxidation of Methane. Int. J. Hydrogen Energy 2023, 48, 17055–17064. [Google Scholar] [CrossRef]
- Chen, Y.; Feng, Y.; Li, L. Identification of Active Sites on High-Performance Pt/Al2O3 Catalyst for Cryogenic CO Oxidation. Available online: https://pubs.acs.org/doi/epdf/10.1021/acscatal.0c02253 (accessed on 5 September 2023).
- Xie, Z.; Ji, L.; Bian, H.; Zhang, Q. Generation of Abundant Oxygen Vacancies in Sandwich Structured ε-MnO2/γ-Al2O3/Al by a Facile Modification Strategy for Enhanced Catalytic Decomposition of Ozone in Humid Conditions. Chem. Eng. J. 2025, 509, 161202. [Google Scholar] [CrossRef]
- Li, H.; Xu, S.; Du, J.; Tang, J.; Zhou, Q. Cu@Co-MOFs as a Novel Catalyst of Peroxymonosulfate for the Efficient Removal of Methylene Blue. RSC Adv. 2019, 9, 9410–9420. [Google Scholar] [CrossRef]
- Ma, C.; Yang, C.; Wang, B.; Chen, C.; Wang, F.; Yao, X.; Song, M. Effects of H2O on HCHO and CO Oxidation at Room-Temperature Catalyzed by MCo2O4 (M=Mn, Ce and Cu) Materials. Appl. Catal. B Environ. 2019, 254, 76–85. [Google Scholar] [CrossRef]
- Merin Saju, S.; Vargeese, A.A. Synthesis of Perforated Single-Crystalline Mn2O3 Microcubes for Thermocatalytic Applications. New J. Chem. 2024, 48, 17150–17158. [Google Scholar] [CrossRef]
- Meunier, F.C.; Reid, D.; Goguet, A.; Shekhtman, S.; Hardacre, C.; Burch, R.; Deng, W.; Flytzani-Stephanopoulos, M. Quantitative Analysis of the Reactivity of Formate Species Seen by DRIFTS over a Au/Ce(La)O2 Water–Gas Shift Catalyst: First Unambiguous Evidence of the Minority Role of Formates as Reaction Intermediates. J. Catal. 2007, 247, 277–287. [Google Scholar] [CrossRef]
- Chen, G.; Zhao, Y.; Fu, G.; Duchesne, P.N.; Gu, L.; Zheng, Y.; Weng, X.; Chen, M.; Zhang, P.; Pao, C.-W.; et al. Interfacial Effects in Iron-Nickel Hydroxide-Platinum Nanoparticles Enhance Catalytic Oxidation. Science 2014, 344, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Pereira-Hernández, X.I.; DeLaRiva, A.; Muravev, V.; Kunwar, D.; Xiong, H.; Sudduth, B.; Engelhard, M.; Kovarik, L.; Hensen, E.J.M.; Wang, Y.; et al. Tuning Pt-CeO2 Interactions by High-Temperature Vapor-Phase Synthesis for Improved Reducibility of Lattice Oxygen. Nat. Commun. 2019, 10, 1358. [Google Scholar] [CrossRef]
- Ogihara, Y.; Yano, H.; Matsumoto, T.; Tryk, D.A.; Iiyama, A.; Uchida, H. In Situ FTIR Analysis of CO-Tolerance of a Pt-Fe Alloy with Stabilized Pt Skin Layers as a Hydrogen Anode Catalyst for Polymer Electrolyte Fuel Cells. Catalysts 2017, 7, 8. [Google Scholar] [CrossRef]
- Zhang, Z.; Xu, J.; Kang, J.; Chen, Y. Role of Bridge-bonded Formate in Formic Acid Dehydration to CO at Pt Electrode: Electrochemial In-situ Infrared Spectroscopic Study. Chin. J. Chem. Phys. 2013, 26, 471–476. [Google Scholar] [CrossRef]
- Zhang, C.; He, H.; Tanaka, K. Catalytic Performance and Mechanism of a Pt/TiO2 Catalyst for the Oxidation of Formaldehyde at Room Temperature. Appl. Catal. B-Environ. 2006, 65, 37–43. [Google Scholar] [CrossRef]
- Yajima, T.; Uchida, H.; Watanabe, M. In-Situ ATR-FTIR Spectroscopic Study of Electro-Oxidation of Methanol and Adsorbed CO at Pt−Ru Alloy. J. Phys. Chem. B 2004, 108, 2654–2659. [Google Scholar] [CrossRef]
- Tou, A.; Kim, H.-H.; Einaga, H.; Teramoto, Y.; Ogata, A. Ozone-Assisted Catalysis of CO: In Situ Fourier Transform IR Evidence of the Cooperative Effect of a Bimetallic Ag-Pd Catalyst. Chem. Eng. J. 2019, 355, 380–389. [Google Scholar] [CrossRef]
- Belletti, G.D.; Colombo, E.; Cabana, N.; Quaino, P.; Collins, S. Mechanistic Investigation of Methanol Oxidation on Au/TiO2: A Combined DRIFT and DFT Study. Top. Catal. 2022, 65, 915–925. [Google Scholar] [CrossRef]
- Hao, Y.; Hung, S.-F.; Zeng, W.-J.; Wang, Y.; Zhang, C.; Kuo, C.-H.; Wang, L.; Zhao, S.; Zhang, Y.; Chen, H.-Y.; et al. Switching the Oxygen Evolution Mechanism on Atomically Dispersed Ru for Enhanced Acidic Reaction Kinetics. J. Am. Chem. Soc. 2023, 145, 23659–23669. [Google Scholar] [CrossRef]
- Zhu, Y.; Yang, L.; Ma, J.; Fang, Y.; Yang, J.; Chen, X.; Zheng, J.; Zhang, S.; Chen, W.; Pan, C.; et al. Rapid Ozone Decomposition over Water-Activated Monolithic MoO3/Graphdiyne Nanowalls under High Humidity. Angew. Chem. 2023, 135, e202309158. [Google Scholar] [CrossRef]
- Vayssilov, G.N.; Mihaylov, M.; Petkov, P.S.; Hadjiivanov, K.I.; Neyman, K.M. Reassignment of the Vibrational Spectra of Carbonates, Formates, and Related Surface Species on Ceria: A Combined Density Functional and Infrared Spectroscopy Investigation. J. Phys. Chem. C 2011, 115, 23435–23454. [Google Scholar] [CrossRef]
- Lou, Y.; Liu, J. CO Oxidation on Metal Oxide Supported Single Pt Atoms: The Role of the Support. Ind. Eng. Chem. Res. 2017, 56, 6916–6925. [Google Scholar] [CrossRef]
- Ke, Q.; Zhang, Y.; Wan, C.; Tang, J.; Li, S.; Guo, X.; Han, M.; Hamada, T.; Osman, S.M.; Kang, Y.; et al. Sunlight-Driven and Gram-Scale Vanillin Production via Mn-Defected γ-MnO2 Catalyst in Aqueous Environment. Chem. Sci. 2024, 15, 5368–5375. [Google Scholar] [CrossRef]
- Xu, B.; Qin, H.; Wu, X.; Sun, Y. Ozone Decomposition Mechanism at Different Structural Oxygen Vacancies on Manganese Dioxide. J. Phys. Chem. C 2022, 126, 17076–17083. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, K.; He, F.; Gao, X.; Fan, S.; Ma, Q.; Zhao, T.; Zhang, J. H2O Derivatives Mediate CO Activation in Fischer-Tropsch Synthesis: A Review. Molecules 2023, 28, 5521. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- John, P.P.; Kieron, B. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Pt Loading | Mn Loading | WVT a | SBET b | Vpore c | Dpore c |
|---|---|---|---|---|---|---|
| (wt.%) | (wt.%) | (h) | (m2·g−1) | (mL·g−1) | (nm) | |
| Pt/γ-Al2O3/Al | 0.91 | / | 0 | 112 | 0.08 | 4.1 |
| Mn/γ-Al2O3/Al | / | 4.7 | 0 | 109 | 0.15 | 4.7 |
| Pt/Mn/γ-Al2O3/Al-WVT-0 | 0.90 | 4.7 | 0 | 164 | 0.17 | 3.9 |
| Pt/Mn/γ-Al2O3/Al-WVT-1 | 0.90 | 4.7 | 1 | 160 | 0.21 | 5.0 |
| Pt/Mn/γ-Al2O3/Al-WVT-2 | 0.90 | 4.7 | 2 | 161 | 0.20 | 4.8 |
| Pt/Mn/γ-Al2O3/Al-WVT-3 | 0.90 | 4.7 | 3 | 162 | 0.21 | 4.9 |
| Pt/Mn/γ-Al2O3/Al-WVT-4 | 0.90 | 4.7 | 4 | 157 | 0.21 | 5.0 |
| Catalyst | Mn 3s | Mn 2p | Pt 4f | O 1s | |
|---|---|---|---|---|---|
| AOS | Mn3+/ Mntotal | Mn4+/ Mntotal | Pt0/ (Pt0 + Pt2+ + Pt4+) | Oads/ (Olatt + Oads + Owat) | |
| Pt/Mn/γ-Al2O3/Al-WVT-0 | 3.13 | 0.34 | 0.42 | 0.55 | 0.65 |
| Pt/Mn/γ-Al2O3/Al-WVT-2 | 3.17 | 0.31 | 0.45 | 0.50 | 0.74 |
| Pt/Mn/γ-Al2O3/Al-WVT-4 | 3.23 | 0.29 | 0.48 | 0.47 | 0.69 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, M.; Shu, Q.; Zhang, R.; Zhang, Q. Structured Mesh-Type Pt/Mn/γ-Al2O3/Al Catalyst Enhanced the CO Oxidation at Room Temperature by In Situ Generation of Hydroxyl: Behavior and Mechanism. Catalysts 2025, 15, 430. https://doi.org/10.3390/catal15050430
Cao M, Shu Q, Zhang R, Zhang Q. Structured Mesh-Type Pt/Mn/γ-Al2O3/Al Catalyst Enhanced the CO Oxidation at Room Temperature by In Situ Generation of Hydroxyl: Behavior and Mechanism. Catalysts. 2025; 15(5):430. https://doi.org/10.3390/catal15050430
Chicago/Turabian StyleCao, Meijia, Qingli Shu, Ran Zhang, and Qi Zhang. 2025. "Structured Mesh-Type Pt/Mn/γ-Al2O3/Al Catalyst Enhanced the CO Oxidation at Room Temperature by In Situ Generation of Hydroxyl: Behavior and Mechanism" Catalysts 15, no. 5: 430. https://doi.org/10.3390/catal15050430
APA StyleCao, M., Shu, Q., Zhang, R., & Zhang, Q. (2025). Structured Mesh-Type Pt/Mn/γ-Al2O3/Al Catalyst Enhanced the CO Oxidation at Room Temperature by In Situ Generation of Hydroxyl: Behavior and Mechanism. Catalysts, 15(5), 430. https://doi.org/10.3390/catal15050430