
Electro-Refinery in Organics to Produce Energy Carriers: Co-Generation of Green Hydrogen and Carboxylic Acids by Glycerol Electrooxidation Using Dimensionally Stable Anode


 , , , , , , and
, , , , , , and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Polarization Curves
2.2. Reactor Characterization
2.3. EO of Glycerol
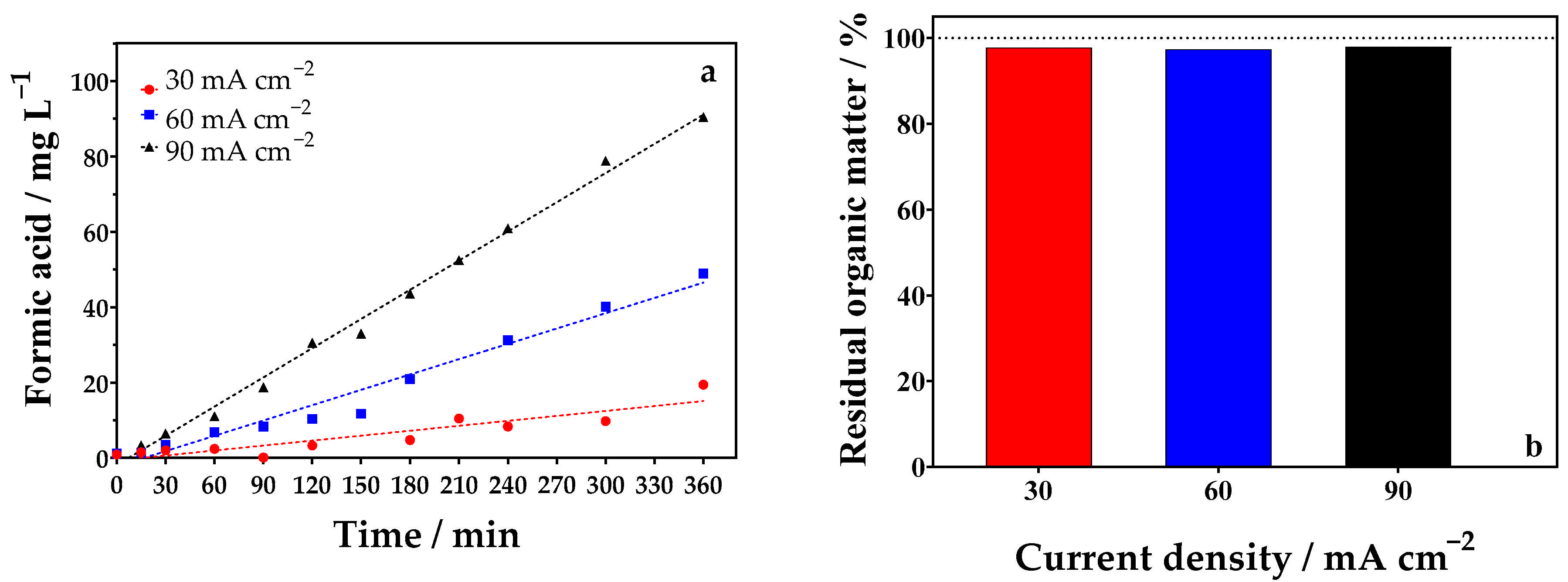
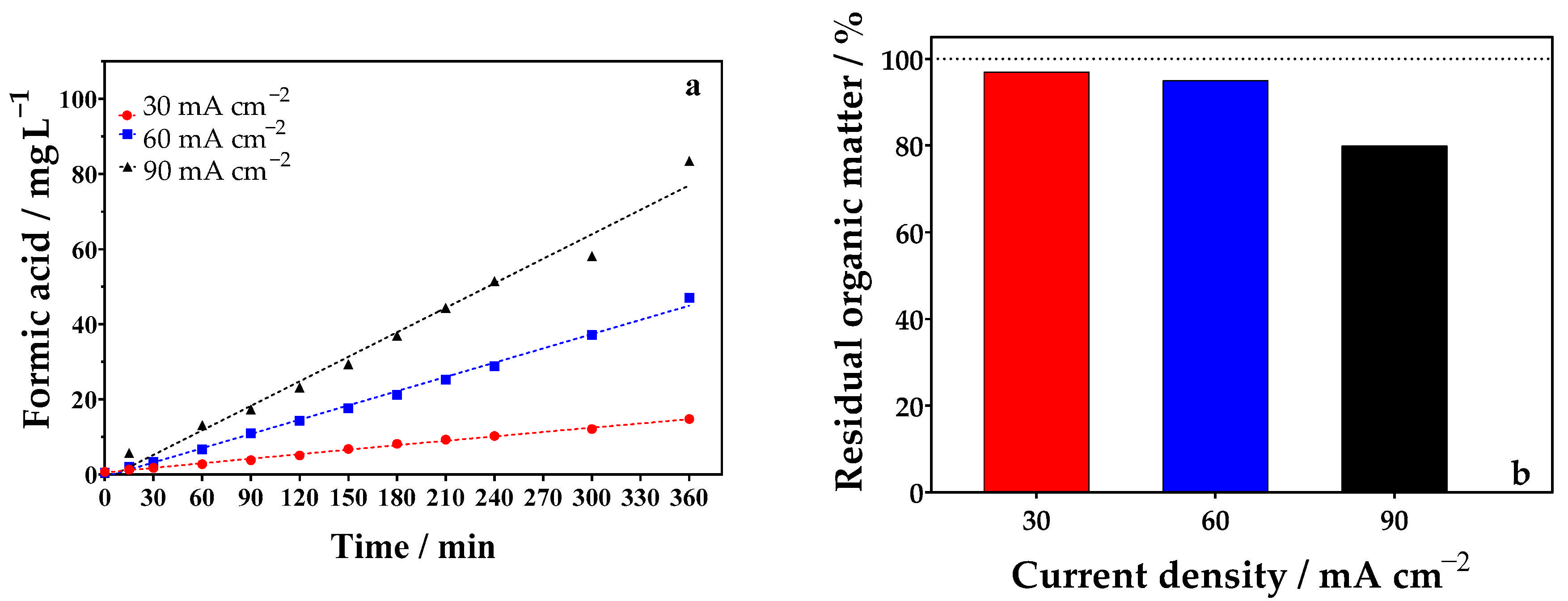
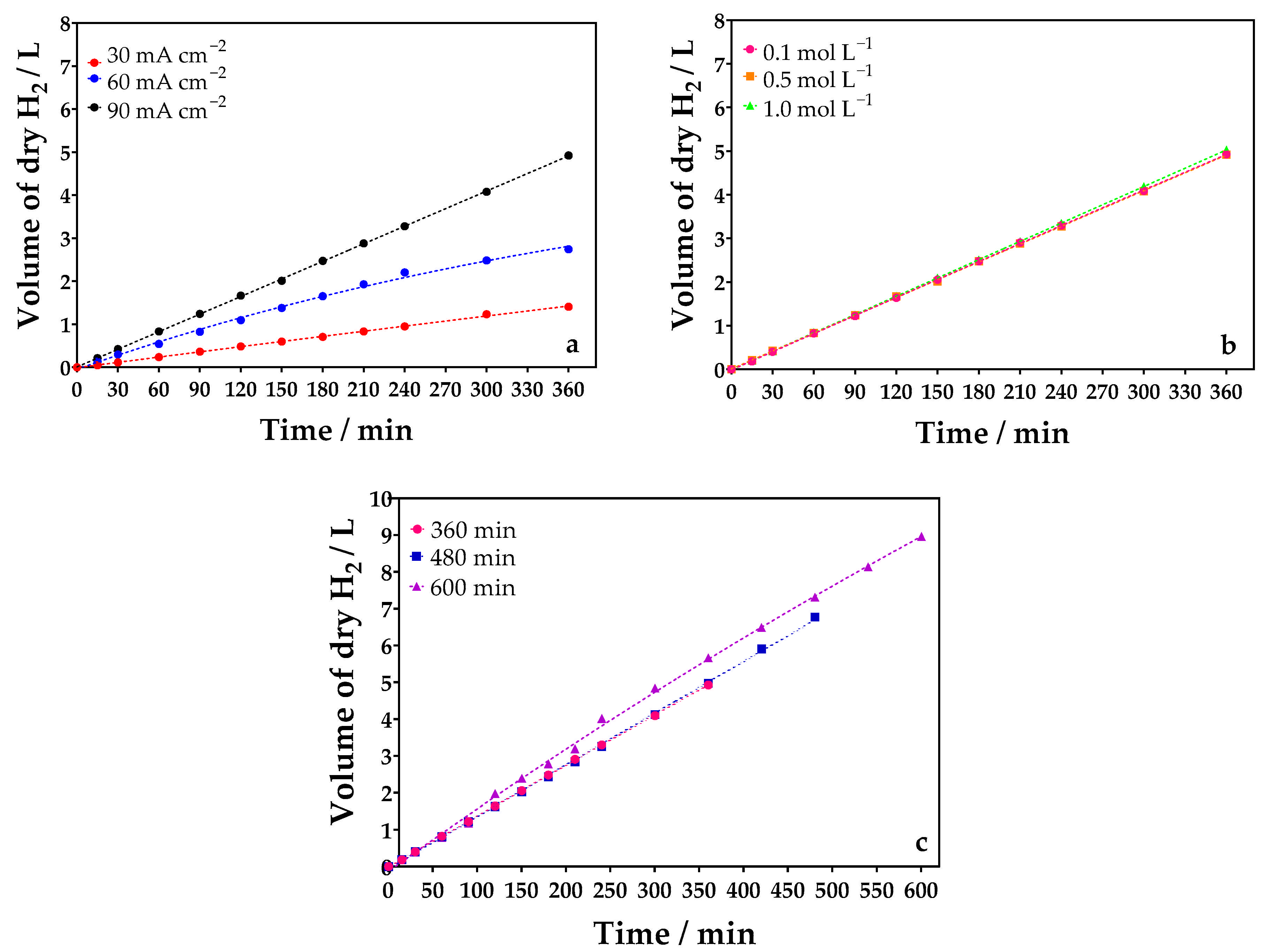
2.3.1. Influence of Current Density
2.3.2. Influence of the Concentration of the Supporting Electrolyte
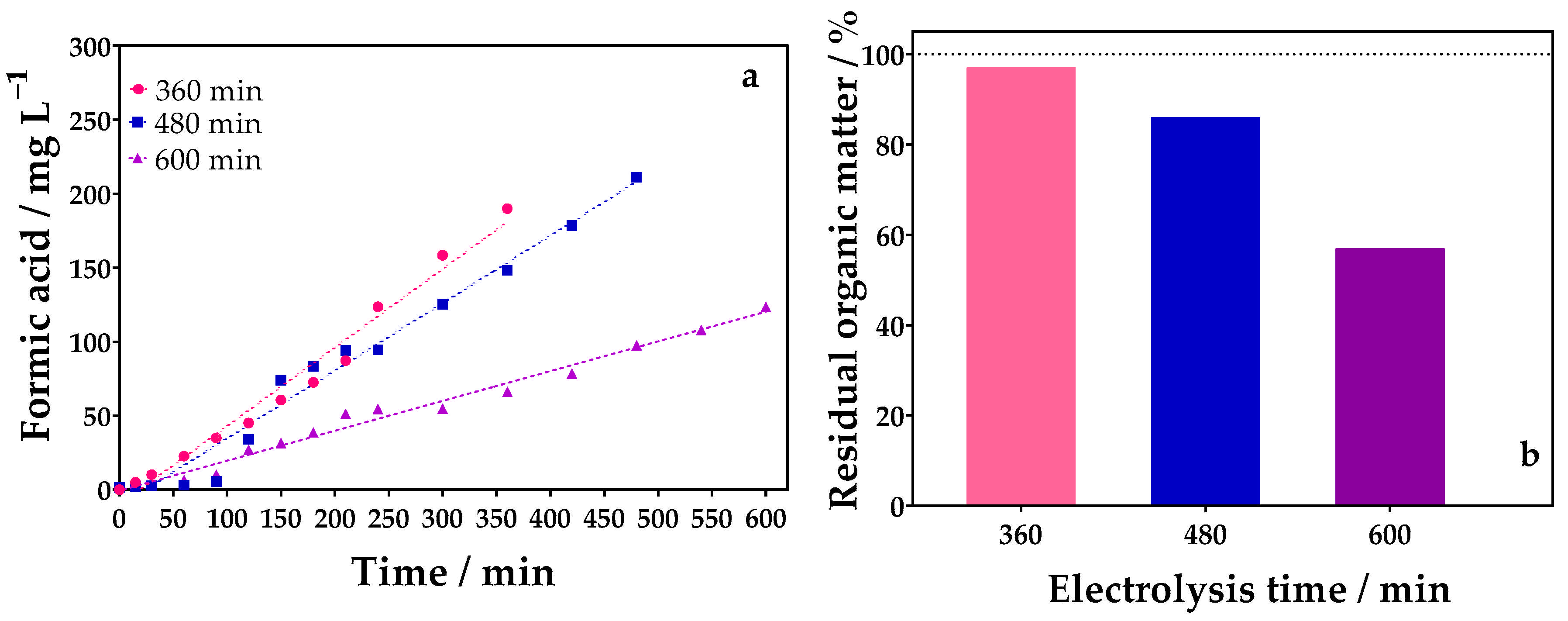
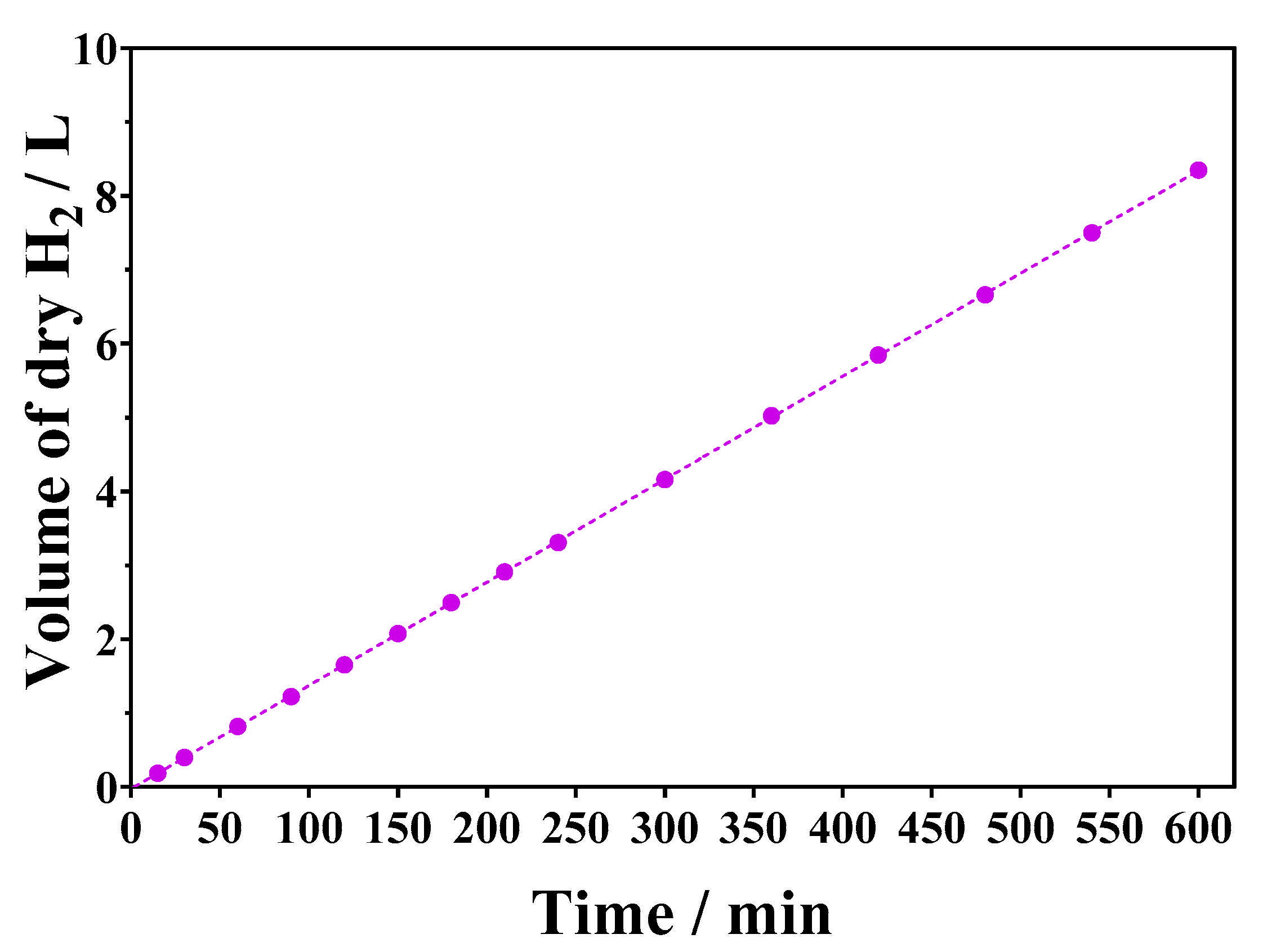
2.3.3. Influence of Treatment Time
2.4. Production of Hydrogen
2.5. EO of a Real Sample
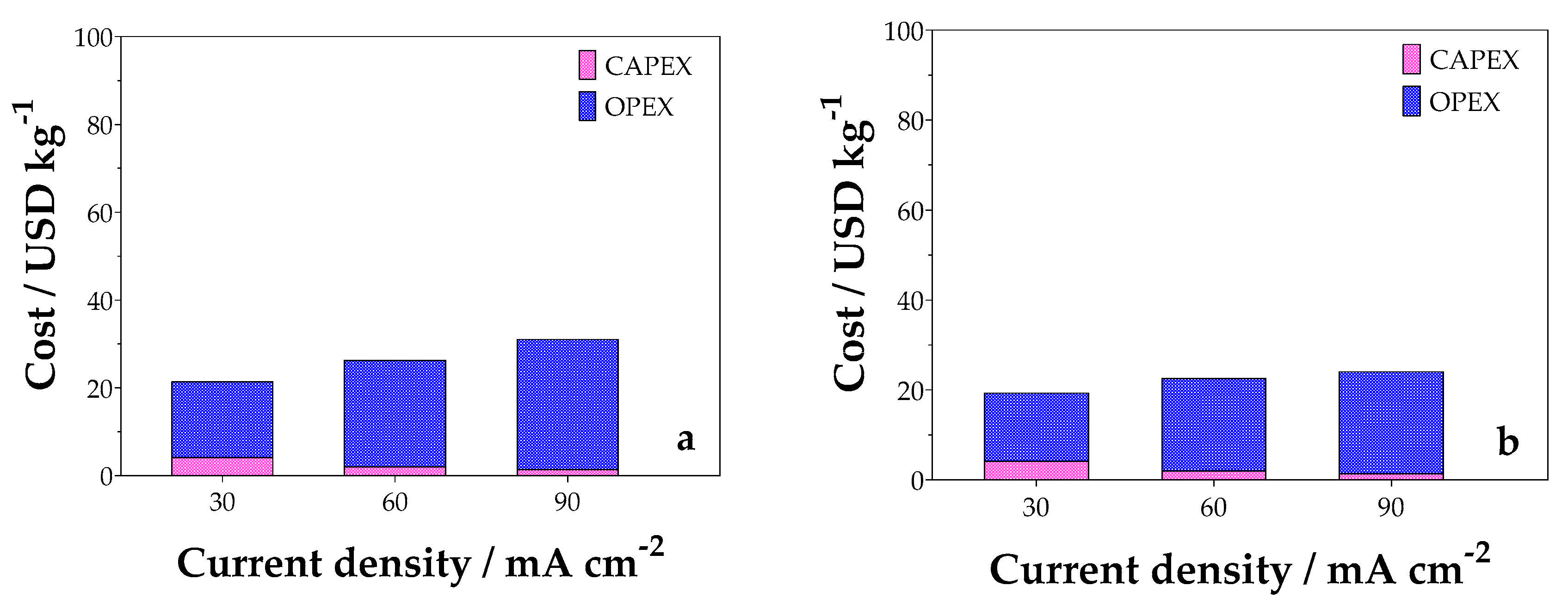
2.6. Preliminary Economic Analysis
3. Experimental Section
3.1. Chemicals
3.2. Electrochemical System
3.3. Polarization Curves
3.4. Electrochemical Cells Characterization
3.5. Analytical Techniques
3.6. Application to Real Sample
3.7. Economic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ji, J.; Lou, W.; Shen, P. Modular Design in Metal-Organic Frameworks for Oxygen Evolution Reaction. Int. J. Hydrogen Energy 2022, 47, 39443–39469. [Google Scholar] [CrossRef]
- Cano, Z.P.; Banham, D.; Ye, S.; Hintennach, A.; Lu, J.; Fowler, M.; Chen, Z. Batteries and Fuel Cells for Emerging Electric Vehicle Markets. Nat. Energy 2018, 3, 279–289. [Google Scholar] [CrossRef]
- Ma, N.; Gong, C.; Xie, H.; Shi, C.; Sha, J.; He, C.; He, F.; Zhao, N.; Liu, E. Metal-Oxygen Bonding Nanoarchitectonics for Regulation of Oxygen Evolution Reaction Performance in FeNi-Codoped CoOOH. Int. J. Hydrogen Energy 2022, 47, 29762–29770. [Google Scholar] [CrossRef]
- Suen, N.T.; Hung, S.F.; Quan, Q.; Zhang, N.; Xu, Y.J.; Chen, H.M. Electrocatalysis for the Oxygen Evolution Reaction: Recent Development and Future Perspectives. Chem. Soc. Rev. 2017, 46, 337–365. [Google Scholar] [CrossRef]
- Grigoriev, S.A.; Fateev, V.N.; Bessarabov, D.G.; Millet, P. Current Status, Research Trends, and Challenges in Water Electrolysis Science and Technology. Int. J. Hydrogen Energy 2020, 45, 26036–26058. [Google Scholar] [CrossRef]
- Du, L.; Sun, Y.; You, B. Hybrid Water Electrolysis: Replacing Oxygen Evolution Reaction for Energy-Efficient Hydrogen Production and Beyond. Mater. Rep. Energy 2021, 1, 100004. [Google Scholar] [CrossRef]
- Guenot, B.; Cretin, M.; Lamy, C. Clean Hydrogen Generation from the Electrocatalytic Oxidation of Methanol inside a Proton Exchange Membrane Electrolysis Cell (PEMEC): Effect of Methanol Concentration and Working Temperature. J. Appl. Electrochem. 2015, 45, 973–981. [Google Scholar] [CrossRef]
- Wang, T.; Cao, X.; Jiao, L. Progress in Hydrogen Production Coupled with Electrochemical Oxidation of Small Molecules. Angew. Chem. Int. Ed. 2022, 61, e202213328. [Google Scholar] [CrossRef]
- de Araujo, D.M.; Barbosa Segundo, I.D.; Cardozo, J.C.; Santos, J.E.L.; Nascimento, J.H.O.; Gondim, A.D.; dos Santos, E.V.; Martínez-Huitle, C.A. Produced Water Electrolysis with Simultaneous Green H2 Generation: From Wastewater to the Future of the Energetic Industry. Fuel 2024, 373, 132369. [Google Scholar] [CrossRef]
- Xavier, F.F.S.; Cunha, A.C.; Napporn, T.W.; Olivi, P. Replacing Oxygen Evolution Reaction by Glycerol Electrooxidation on Rh Modified Ni(OH)2/C for Energy-Efficient Hydrogen Production. Int. J. Hydrogen Energy 2023, 48, 31091–31100. [Google Scholar] [CrossRef]
- Campos da Paixão, I.; Cardozo, J.C.; Sales Monteiro, M.K.; Gondim, A.D.; Cavalcanti, L.N.; Fabiano de Santana Souza, D.; Martínez-Huitle, C.A.; Vieira dos Santos, E. A Sustainable Solar-Driven Electrochemical Process for Reforming Lignocellulosic Biomass Effluent into High Value-Added Products: Green Hydrogen, Carboxylic and Vanillic Acids. RSC Adv. 2023, 13, 35755–35765. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, E.V.; Martínez-Huitle, C.A.; Rodrigo, M.A. The Electro-Refinery in Organics: A New Arising Concept for Valorization of Wastes. Curr. Opin. Electrochem. 2023, 39, 101267. [Google Scholar] [CrossRef]
- Tang, C.; Zheng, Y.; Jaroniec, M.; Qiao, S.Z. Electrocatalytic Refinery for Sustainable Production of Fuels and Chemicals. Angew. Chem. Int. Ed. 2021, 60, 19572–19590. [Google Scholar] [CrossRef]
- Pirzadi, Z.; Meshkani, F. From Glycerol Production to Its Value-Added Uses: A Critical Review. Fuel 2022, 329, 125044. [Google Scholar] [CrossRef]
- Yang, F.; Hanna, M.A.; Sun, R. Value-Added Uses for Crude Glycerol—A Byproduct of Biodiesel Production. Biotechnol. Biofuels 2012, 5, 13. [Google Scholar] [CrossRef]
- Zhang, M.; Wu, H. Effect of Major Impurities in Crude Glycerol on Solubility and Properties of Glycerol/Methanol/Bio-Oil Blends. Fuel 2015, 159, 118–127. [Google Scholar] [CrossRef]
- Marshall, A.T.; Haverkamp, R.G. Production of Hydrogen by the Electrochemical Reforming of Glycerol–Water Solutions in a PEM Electrolysis Cell. Int. J. Hydrogen Energy 2008, 33, 4649–4654. [Google Scholar] [CrossRef]
- Brix, A.C.; Morales, D.M.; Braun, M.; Jambrec, D.; Junqueira, J.R.C.; Cychy, S.; Seisel, S.; Masa, J.; Muhler, M.; Andronescu, C.; et al. Electrocatalytic Oxidation of Glycerol Using Solid-State Synthesised Nickel Boride: Impact of Key Electrolysis Parameters on Product Selectivity. ChemElectroChem 2021, 8, 2336–2342. [Google Scholar] [CrossRef]
- Ahmad, M.S.; Cheng, C.K.; Kumar, R.; Singh, S.; Saeed, K.A.; Ong, H.R.; Abdullah, H.; Khan, M.R. Pd/CNT Catalysts for Glycerol Electro-Oxidation: Effect of Pd Loading on Production of Valuable Chemical Products. Electroanalysis 2020, 32, 1139–1147. [Google Scholar] [CrossRef]
- Sankar, J.; Onyeozili, E.N.; Kalu, E.E. Oxidation of Glycerol with Unactivated Electroless CuNiMoP Catalyst. ChemEngineering 2017, 1, 11. [Google Scholar] [CrossRef]
- Yu, X.; Campos, E.; Santos, D.; White, J.; Salazar-Alvarez, G.; Pettersson, L.G.M.; Cornell, A.; Johnsson, M.; Yu, X.; Johnsson, M.; et al. Electrocatalytic Glycerol Oxidation with Concurrent Hydrogen Evolution Utilizing an Efficient MoOx/Pt Catalyst. Small 2021, 17, 2104288. [Google Scholar] [CrossRef] [PubMed]
- Schwengber, C.A.; Alves, H.J.; Schaffner, R.A.; Da Silva, F.A.; Sequinel, R.; Bach, V.R.; Ferracin, R.J. Overview of Glycerol Reforming for Hydrogen Production. Renew. Sustain. Energy Rev. 2016, 58, 259–266. [Google Scholar] [CrossRef]
- Bastan, F.; Kazemeini, M.; Larimi, A.; Maleki, H. Production of Renewable Hydrogen through Aqueous-Phase Reforming of Glycerol over Ni/Al2O3MgO Nano-Catalyst. Int. J. Hydrogen Energy 2018, 43, 614–621. [Google Scholar] [CrossRef]
- Wu, J.; Yang, X.; Gong, M. Recent Advances in Glycerol Valorization via Electrooxidation: Catalyst, Mechanism and Device. Chin. J. Catal. 2022, 43, 2966–2986. [Google Scholar] [CrossRef]
- Rahim, S.A.N.M.; Lee, C.S.; Abnisa, F.; Aroua, M.K.; Daud, W.A.W.; Cognet, P.; Pérès, Y. A Review of Recent Developments on Kinetics Parameters for Glycerol Electrochemical Conversion—A by-Product of Biodiesel. Sci. Total Environ. 2020, 705, 135137. [Google Scholar] [CrossRef]
- Braun, M.; Santana, C.S.; Garcia, A.C.; Andronescu, C. From Waste to Value—Glycerol Electrooxidation for Energy Conversion and Chemical Production. Curr. Opin. Green Sustain. Chem. 2023, 41, 100829. [Google Scholar] [CrossRef]
- Jeon, H.; Jeong, B.; Choun, M.; Lee, J. In-Situ Electrochemical Extended X-Ray Absorption Fine Structure Spectroscopy Study on the Reactivation of Pd Electrocatalyst in Formic Acid Oxidation. Electrochim. Acta 2014, 140, 525–528. [Google Scholar] [CrossRef]
- Fan, L.; Liu, B.; Liu, X.; Senthilkumar, N.; Wang, G.; Wen, Z. Recent Progress in Electrocatalytic Glycerol Oxidation. Energy Technol. 2021, 9, 2000804. [Google Scholar] [CrossRef]
- Martínez-Huitle, C.A.; Rodrigo, M.A.; Sirés, I.; Scialdone, O. A Critical Review on Latest Innovations and Future Challenges of Electrochemical Technology for the Abatement of Organics in Water. Appl. Catal. B Environ. 2023, 328, 122430. [Google Scholar] [CrossRef]
- Espinoza, L.C.; Sepúlveda, P.; García, A.; Martins de Godoi, D.; Salazar, R. Degradation of Oxamic Acid Using Dimensionally Stable Anodes (DSA) Based on a Mixture of RuO2 and IrO2 Nanoparticles. Chemosphere 2020, 251, 126674. [Google Scholar] [CrossRef]
- Aquino Neto, S.; de Andrade, A.R. Electrooxidation of Glyphosate Herbicide at Different DSA® Compositions: PH, Concentration and Supporting Electrolyte Effect. Electrochim. Acta 2009, 54, 2039–2045. [Google Scholar] [CrossRef]
- Salazar-Banda, G.R.; Santos, G.d.O.S.; Duarte Gonzaga, I.M.; Dória, A.R.; Barrios Eguiluz, K.I. Developments in Electrode Materials for Wastewater Treatment. Curr. Opin. Electrochem. 2021, 26, 100663. [Google Scholar] [CrossRef]
- Comninellis, C.; Chen, G. Electrochemistry for the Environment; Springer: Berlin/Heidelberg, Germany, 2010; pp. 1–563. [Google Scholar] [CrossRef]
- Kaur, R.; Kushwaha, J.P.; Singh, N. Electro-Oxidation of Amoxicillin Trihydrate in Continuous Reactor by Ti/RuO2 Anode. Sci. Total Environ. 2019, 677, 84–97. [Google Scholar] [CrossRef]
- Kim, S.; Lee, T.; Han, S.; Lee, C.; Kim, C.; Yoon, J. Ir0.11Fe0.25O0.64 as a Highly Efficient Electrode for Electrochlorination in Dilute Chloride Solutions. J. Ind. Eng. Chem. 2021, 102, 155–162. [Google Scholar] [CrossRef]
- Li, C.; Tian, L.; Yuan, X.; Jiang, H.; Hu, Z.; Yin, Y. Review of Acidic Titanium-Based Oxygen Evolution Anode Catalyst Design: Mechanistic, Compositional Design, and Research Status. J. Alloys Compd. 2024, 979, 173576. [Google Scholar] [CrossRef]
- Santos, G.O.S.; Eguiluz, K.I.B.; Salazar-Banda, G.R.; Sáez, C.; Rodrigo, M.A. Understanding the Electrolytic Generation of Sulfate and Chlorine Oxidative Species with Different Boron-Doped Diamond Anodes. J. Electroanal. Chem. 2020, 857, 113756. [Google Scholar] [CrossRef]
- Duarte, J.L.S.; Meili, L.; Gomes, L.M.; Melo, J.M.O.; Ferro, A.B.; Tavares, M.G.; Tonholo, J.; Zanta, C.L.P.S. Electrochemical Degradation of 17-α-Methyltestosterone over DSA® Electrodes. Chem. Eng. Process. Process Intensif. 2019, 142, 107548. [Google Scholar] [CrossRef]
- Medeiros, M.C.; dos Santos, E.V.; Martínez-Huitle, C.A.; Fajardo, A.S.; Castro, S.S.L. Obtaining High-Added Value Products from the Technical Cashew-Nut Shell Liquid Using Electrochemical Oxidation with BDD Anodes. Sep. Purif. Technol. 2020, 250, 117099. [Google Scholar] [CrossRef]
- Castro, R.S.S.; Santos, G.O.S.; Lanza, M.R.V.; Salaza-Banda, G.R.; Eguiluz, K.I.B.; Rodrigo, M.A.; Sáez, C. New MMO Coatings for Electro-Refinery Applications: Promoting the Production of Carboxylates. Chemosphere 2024, 363, 142941. [Google Scholar] [CrossRef]
- Medeiros, M.C.; Castro, S.S.L.; dos Santos, E.V.; Rodrigo, M.A.; Martínez-Huitle, C.A. A Proof of Concept for the Electro-Refinery: Selective Electroproduction of Acetic Acid from t-CNSL Waste by Using DSA Electrode. Electrochem. Commun. 2022, 141, 107356. [Google Scholar] [CrossRef]
- Quiroz, M.A.; Martínez-Huitle, U.A.; Martínez-Huitle, C.A. Mass Transfer Measurements in a Parallel Disk Cell Using the Limiting Current Technique. J. Mex. Chem. Soc. 2005, 49, 279–283. [Google Scholar]
- Quiroz, M.A.; Córdova, F.; Lamy-Pitara, E.; Barbier, J. Electrocatalytic Hydrogenation of M-Xylene on Platinized-Platinum Electrodes. Electrochim. Acta 2000, 45, 4291–4298. [Google Scholar] [CrossRef]
- Ciobanu, M.; Wilburn, J.P.; Krim, M.L.; Cliffel, D.E. Fundamentals. In Handbook of Electrochemistry; Elsevier: Amsterdam, The Netherlands, 2007; pp. 3–29. [Google Scholar] [CrossRef]
- dos Santos, E.V.; Cardozo, J.C.; Feijoó Zambrano, T.N.; Quiroz Alfaro, M.A.; Martínez-Huitle, C.A. Towards Win-Win Electrochemical Alternative for Depolluting Water and Producing Green H2: Understanding and Unrevealing the Participation of Sacrificial Organic Compound on H2 Production. Int. J. Hydrogen Energy 2024, 110, 598–608. [Google Scholar] [CrossRef]
- Harris, D.C. Quantitative Chemical Analysis, 8th ed.; W.H. Freeman and Co.: New York, NY, USA, 2010; ISBN 1429239891. [Google Scholar]
- Yan, Y.; Hao, P.; Fu, Y.; Chen, W.; Shi, Q.; Zhou, H.; Kong, X.; Li, Z.; Shao, M.; Duan, X. Selective Electrooxidation Glycerol to Lactic Acid Coupled with Hydrogen Production over a Cooperative BiOx/Au Catalyst. AIChE J. 2024, 70, e18370. [Google Scholar] [CrossRef]
- Song, Y.; Wan, X.; Miao, Y.; Li, J.; Ren, Z.; Jin, B.; Zhou, H.; Li, Z.; Shao, M. Blocking Oxygen Evolution Reaction for Efficient Organic Electrooxidation Coupling Hydrogen Production by Using Layered Double Hydroxide Rich in Active Oxygen. Appl. Catal. B Environ. Energy 2023, 333, 122808. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, M.; Wang, S.; Ren, K.; Wang, M.; Wang, Z.; Li, X.; Wang, L.; Wang, H. Integrating Electrocatalytic Hydrogen Generation with Selective Oxidation of Glycerol to Formate over Bifunctional Nitrogen-Doped Carbon Coated Nickel-Molybdenum-Nitrogen Nanowire Arrays. Appl. Catal. B Environ. 2021, 298, 120493. [Google Scholar] [CrossRef]
- Wenderich, K.; Nieuweweme, B.A.M.; Mul, G.; Mei, B.T. Selective Electrochemical Oxidation of H2O to H2O2Using Boron-Doped Diamond: An Experimental and Techno-Economic Evaluation. ACS Sustain. Chem. Eng. 2021, 9, 7803–7812. [Google Scholar] [CrossRef]
- Khan, M.A.; Al-Attas, T.A.; Yasri, N.G.; Zhao, H.; Larter, S.; Hu, J.; Kibria, M.G. Techno-Economic Analysis of a Solar-Powered Biomass Electrolysis Pathway for Coproduction of Hydrogen and Value-Added Chemicals. Sustain. Energy Fuels 2020, 4, 5568–5577. [Google Scholar] [CrossRef]
- Le Bideau, D.; Chocron, O.; Mandin, P.; Kiener, P.; Benbouzid, M.; Sellier, M.; Kim, M.; Ganci, F.; Inguanta, R. Evolutionary Design Optimization of an Alkaline Water Electrolysis Cell for Hydrogen Production. Appl. Sci. 2020, 10, 8425. [Google Scholar] [CrossRef]
- Bansod, Y.; Crabbe, B.; Forster, L.; Ghasemzadeh, K.; D’Agostino, C. Evaluating the Environmental Impact of Crude Glycerol Purification Derived from Biodiesel Production: A Comparative Life Cycle Assessment Study. J. Clean. Prod. 2024, 437, 140485. [Google Scholar] [CrossRef]
- Xie, Y.; Sun, L.; Pan, X.; Zhou, Z.; Zheng, Y.; Yang, X.; Zhao, G. Carbon Paper Supported Gold Nanoflowers for Tunable Glycerol Electrooxidation Boosting Efficient Hydrogen Evolution. Carbon 2023, 203, 88–96. [Google Scholar] [CrossRef]
- dos Santos, E.C.; Araujo, R.B.; Valter, M.; Salazar-Alvarez, G.; Johnsson, M.; Bajdich, M.; Abild-Pedersen, F.; Pettersson, L.G.M. Efficient Screening of Bi–Metallic Electrocatalysts for Glycerol Valorization. Electrochim. Acta 2021, 398, 139283. [Google Scholar] [CrossRef]
- Barreto, J.P.d.P.; Santos, J.E.L.; Cardozo, J.C.; de Santana Souza, D.F.; Cavalcanti, L.N.; Gondim, A.D.; Martínez-Huitle, C.A.; dos Santos, E.V. Energy-Saving Electrochemical Green Hydrogen Production Coupled with Persulfate or Hydrogen Peroxide Valorization at Boron-Doped Diamond Electrodes. J. Environ. Chem. Eng 2024, 12, 114837. [Google Scholar] [CrossRef]
- Jagannadha Raju, G.M.; Sarma, G.V.S.; Ramesh, K.V.; Bhaskara Sarma, C. Mass Transfer at the Confining Wall of an Electrochemical Cell in Annular Flow with a Rotating Central Rod. Indian Chem. Eng. 2021, 63, 512–521. [Google Scholar] [CrossRef]
- Scott, K.; Lobato, J. Determination of a Mass-Transfer Coefficient Using the Limiting-Current Technique. Chem. Educ. 2002, 7, 214–219. [Google Scholar] [CrossRef]
- González, J.; Laborda, E.; Molina, Á. Voltammetric Kinetic Studies of Electrode Reactions: Guidelines for Detailed Understanding of Their Fundamentals. J. Chem. Educ. 2023, 100, 697–706. [Google Scholar] [CrossRef]
- De França Neta, L.S.; Borges, C.P.; Habert, A.C. Evaluation of Mass Transfer in a Novel Hollow Fiber Module Design Using an Electrochemical Technique. Braz. J. Chem. Eng. 2017, 34, 789–798. [Google Scholar] [CrossRef]
- de Castro, C.M.; Olivi, P.; de Freitas Araújo, K.C.; Barbosa Segundo, I.D.; dos Santos, E.V.; Martínez-Huitle, C.A. Environmental Application of a Cost-Effective Smartphone-Based Method for COD Analysis: Applicability in the Electrochemical Treatment of Real Wastewater. Sci. Total Environ. 2023, 855, 158816. [Google Scholar] [CrossRef]
- Cardozo, J.C.; Barbosa Segundo, I.D.; Galvão, E.R.V.P.; da Silva, D.R.; dos Santos, E.V.; Martínez-Huitle, C.A. Decentralized Environmental Applications of a Smartphone-Based Method for Chemical Oxygen Demand and Color Analysis. Sci. Rep. 2023, 13, 11082. [Google Scholar] [CrossRef]
- Ren, H.; Huang, Z.; Cai, G.; Guo, J.; Sun, Y.; Yao, W.; Ye, D.; Qian, H.; Zhang, J.; Zhao, H. Cobalt (II) Oxide for Efficient Glycerol Electrooxidation for Formic Acid Coupled with Hydrogen Production. J. Alloys Compd. 2024, 996, 174781. [Google Scholar] [CrossRef]
- Feng, X.; Liu, B.W.; Guo, K.X.; Fan, L.F.; Wang, G.X.; Ci, S.Q.; Wen, Z.H. Anodic Electrocatalysis of Glycerol Oxidation for Hybrid Alkali/Acid Electrolytic Hydrogen Generation. J. Electrochem. 2023, 29, 2215005. [Google Scholar] [CrossRef]
- Wu, G.; Dong, X.; Mao, J.; Li, G.; Zhu, C.; Li, S.; Chen, A.; Feng, G.; Song, Y.; Chen, W.; et al. Anodic Glycerol Oxidation to Formate Facilitating Cathodic Hydrogen Evolution with Earth-Abundant Metal Oxide Catalysts. Chem. Eng. J. 2023, 468, 143640. [Google Scholar] [CrossRef]
- Fan, L.; Ji, Y.; Wang, G.; Chen, J.; Chen, K.; Liu, X.; Wen, Z. High Entropy Alloy Electrocatalytic Electrode toward Alkaline Glycerol Valorization Coupling with Acidic Hydrogen Production. J. Am. Chem. Soc. 2022, 144, 7224–7235. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Song, F.; Hou, Y.; Wu, D.; Xu, F.; Jiang, K.; Gao, Z. Molybdenum, Tungsten Doped Cobalt Phosphides as Efficient Catalysts for Coproduction of Hydrogen and Formate by Glycerol Electrolysis. J. Colloid Interface Sci. 2024, 665, 152–162. [Google Scholar] [CrossRef]
- Li, Y.; Wei, X.; Chen, L.; Shi, J.; He, M. Nickel-Molybdenum Nitride Nanoplate Electrocatalysts for Concurrent Electrolytic Hydrogen and Formate Productions. Nat. Commun. 2019, 10, 5335. [Google Scholar] [CrossRef]
- Fang, Y.; Dai, C.; Liu, X.; Wang, Y.; Ju, C.; He, S.; Shi, R.; Liu, Y.; Zhang, J.; Zhu, Y.; et al. Sulfur-Doped Manganese-Cobalt Hydroxide with Promoted Surface Reconstruction for Glycerol Electrooxidation Assisted Hydrogen Production. Nano Energy 2024, 127, 109754. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | j/mA cm−2 | Oxidation Time/min | Undivided Reactor | Divided Reactor | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compartment | [FA]/mg L−1 | FE/% | Compartment | [FA]/mg L−1 | FE/% | Volume of Dry Green H2/L | ||||
| Anodic | Cathodic | |||||||||
| 1 | 30 | 360 | 0.1 mol L−1 glycerol + 0.5 mol L−1 Na2SO4 | 19.47 | 0.81 | 0.1 mol L−1 glycerol + 0.5 mol L−1 Na2SO4 | 0.5 mol L−1 Na2SO4 | 14.79 | 0.61 | 1.406 |
| 2 | 60 | 360 | 0.1 mol L−1 glycerol + 0.5 mol L−1 Na2SO4 | 48.96 | 1.02 | 0.1 mol L−1 glycerol + 0.5 mol L−1 Na2SO4 | 0.5 mol L−1 Na2SO4 | 47.08 | 0.98 | 2.744 |
| 3 | 90 | 360 | 0.1 mol L−1 glycerol + 0.5 mol L−1 Na2SO4 | 90.45 | 1.25 | 0.1 mol L−1 glycerol + 0.5 mol L−1 Na2SO4 | 0.5 mol L−1 Na2SO4 | 83.45 | 1.15 | 4.922 |
| 4 | 90 | 360 | 0.1 mol L−1 glycerol + 0.1 mol L−1 Na2SO4 | 188.93 | 2.61 | 0.1 mol L−1 glycerol + 0.1 mol L−1 Na2SO4 | 0.1 mol L−1 Na2SO4 | 189.93 | 2.63 | 4.927 |
| 5 | 90 | 360 | 0.1 mol L−1 glycerol + 0.5 mol L−1 Na2SO4 | 87.98 | 1.22 | 0.1 mol L−1 glycerol + 0.5 mol L−1 Na2SO4 | 0.5 mol L−1 Na2SO4 | 83.45 | 1.15 | 4.922 |
| 6 | 90 | 360 | 0.1 mol L−1 glycerol + 1.0 mol L−1 Na2SO4 | 88.49 | 1.22 | 0.1 mol L−1 glycerol + 1.0 mol L−1 Na2SO4 | 1.0 mol L−1 Na2SO4 | 44.41 | 0.61 | 5.033 |
| 7 | 90 | 360 | 0.1 mol L−1 glycerol + 0.1 mol L−1 Na2SO4 | 188.93 | 2.61 | 0.1 mol L−1 glycerol + 0.1 mol L−1 Na2SO4 | 0.1 mol L−1 Na2SO4 | 189.93 | 2.63 | 4.927 |
| 8 | 90 | 480 | 0.1 mol L−1 glycerol + 0.1 mol L−1 Na2SO4 | 256.21 | 2.66 | 0.1 mol L−1 glycerol + 0.1 mol L−1 Na2SO4 | 0.1 mol L−1 Na2SO4 | 211.17 | 2.19 | 6.769 |
| 9 | 90 | 600 | 0.1 mol L−1 glycerol + 0.1 mol L−1 Na2SO4 | 245.88 | 2.04 | 0.1 mol L−1 glycerol + 0.1 mol L−1 Na2SO4 | 0.1 mol L−1 Na2SO4 | 123.67 | 1.03 | 7.603 |
| Real Sample Parameters | Before Treatment | After Treatment | |
|---|---|---|---|
| Undivided Reactor | Divided Reactor | ||
| pH | 4.13 | 2.66 | 1.37 |
| Electrical Conductivity (μS cm−1) | 2755.0 | 1793.0 | 3828.0 |
| Apparent color (units PtCo) | 1640 | 37 | 103 |
| True color (units PtCo) | 845 | 5 | 53 |
| Turbidity (NTU) | 188.0 | 5.02 | 8.37 |
| Suspended solids (mg L−1) | 283.0 | 33 | 36 |
| Total dissolved solids (mg L−1) | 1377.0 | 8690.0 | 1964.0 |
| Free Chlorine (mg L−1) | 0.06 | <LD | <LD |
| Residual Chlorine (mg L−1) | 0.13 | <LD | <LD |
| Total Nitrogen (mg L−1) | 32.8 | <LD | <LD |
| COD (mg L−1) | 2238.0 | 1460.0 | 1942.0 |
| TOC (mg L−1) | 3268.0 | 2467.0 | 2063.0 |
| Phenol (mg L−1) | 0.072 | 1.02 | 1.38 |
| Anionic Surfactant (mg L−1) | 5.17 | 3.63 | 3.52 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
da Silva, L.M.G.; Costa, L.G.A.; Santos, J.E.L.; de A. Costa, E.C.T.; de Morais Araújo, A.M.; Gondim, A.D.; Cavalcanti, L.N.; Quiroz, M.A.; dos Santos, E.V.; Martínez-Huitle, C.A. Electro-Refinery in Organics to Produce Energy Carriers: Co-Generation of Green Hydrogen and Carboxylic Acids by Glycerol Electrooxidation Using Dimensionally Stable Anode. Catalysts 2025, 15, 333. https://doi.org/10.3390/catal15040333
da Silva LMG, Costa LGA, Santos JEL, de A. Costa ECT, de Morais Araújo AM, Gondim AD, Cavalcanti LN, Quiroz MA, dos Santos EV, Martínez-Huitle CA. Electro-Refinery in Organics to Produce Energy Carriers: Co-Generation of Green Hydrogen and Carboxylic Acids by Glycerol Electrooxidation Using Dimensionally Stable Anode. Catalysts. 2025; 15(4):333. https://doi.org/10.3390/catal15040333
Chicago/Turabian Styleda Silva, Letícia M. G., Letícia G. A. Costa, José E. L. Santos, Emily C. T. de A. Costa, Aruzza M. de Morais Araújo, Amanda D. Gondim, Lívia N. Cavalcanti, Marco A. Quiroz, Elisama V. dos Santos, and Carlos A. Martínez-Huitle. 2025. "Electro-Refinery in Organics to Produce Energy Carriers: Co-Generation of Green Hydrogen and Carboxylic Acids by Glycerol Electrooxidation Using Dimensionally Stable Anode" Catalysts 15, no. 4: 333. https://doi.org/10.3390/catal15040333
APA Styleda Silva, L. M. G., Costa, L. G. A., Santos, J. E. L., de A. Costa, E. C. T., de Morais Araújo, A. M., Gondim, A. D., Cavalcanti, L. N., Quiroz, M. A., dos Santos, E. V., & Martínez-Huitle, C. A. (2025). Electro-Refinery in Organics to Produce Energy Carriers: Co-Generation of Green Hydrogen and Carboxylic Acids by Glycerol Electrooxidation Using Dimensionally Stable Anode. Catalysts, 15(4), 333. https://doi.org/10.3390/catal15040333