
Unveiling the Role of Copper Valence States in Enhancing the Catalytic Performance of Copper-Modified ZSM-5 for Direct Methane Conversion

Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
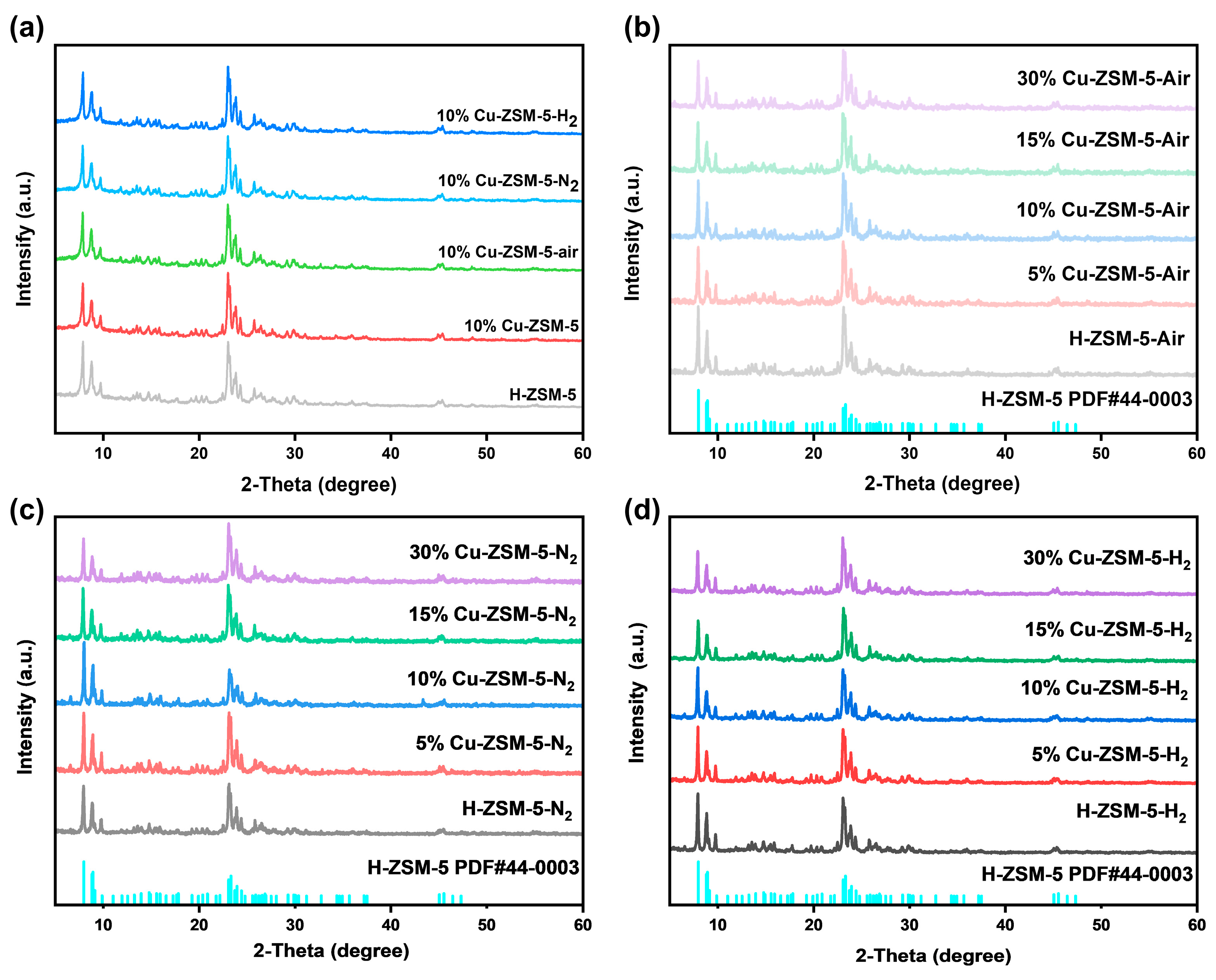
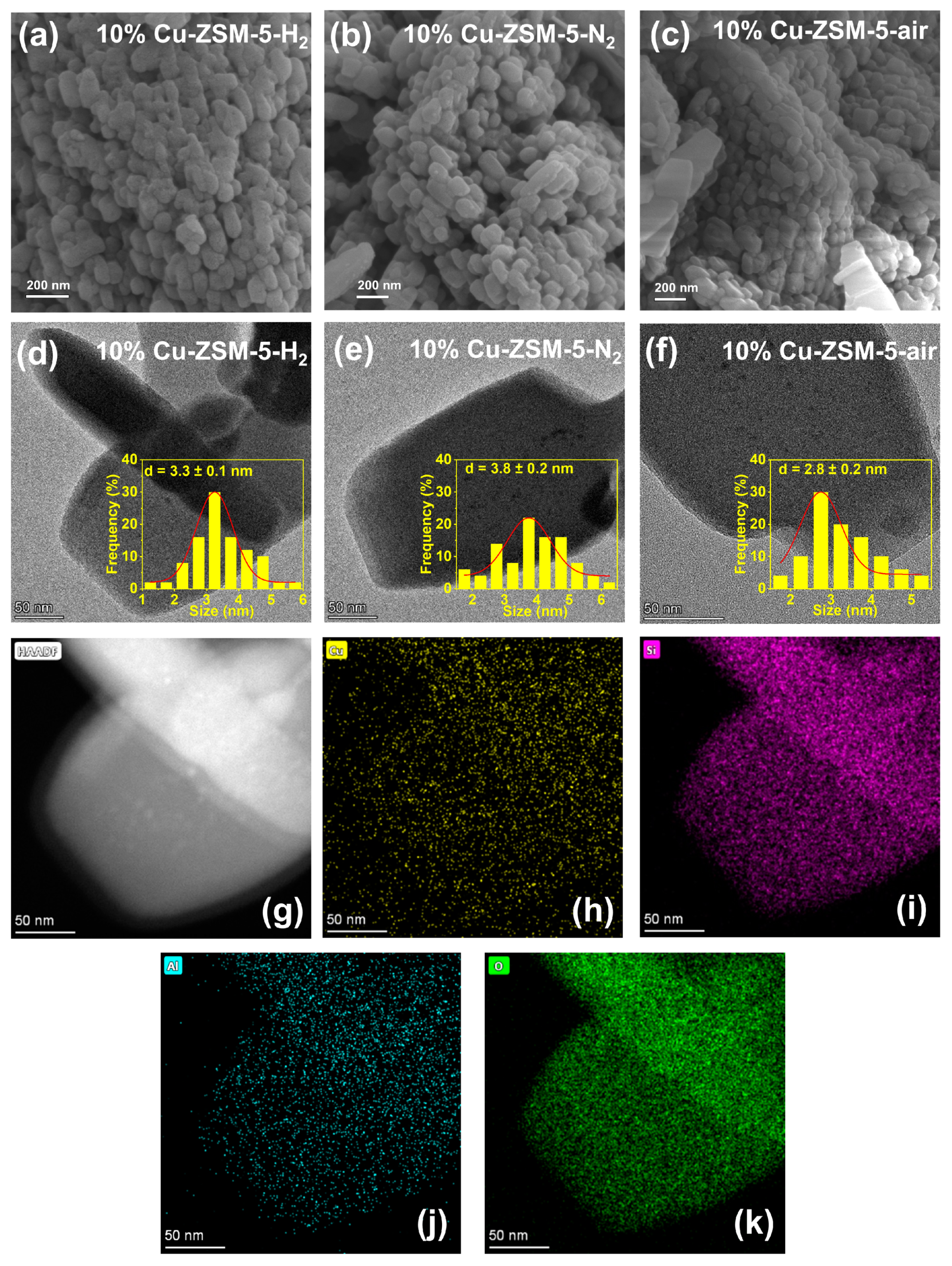
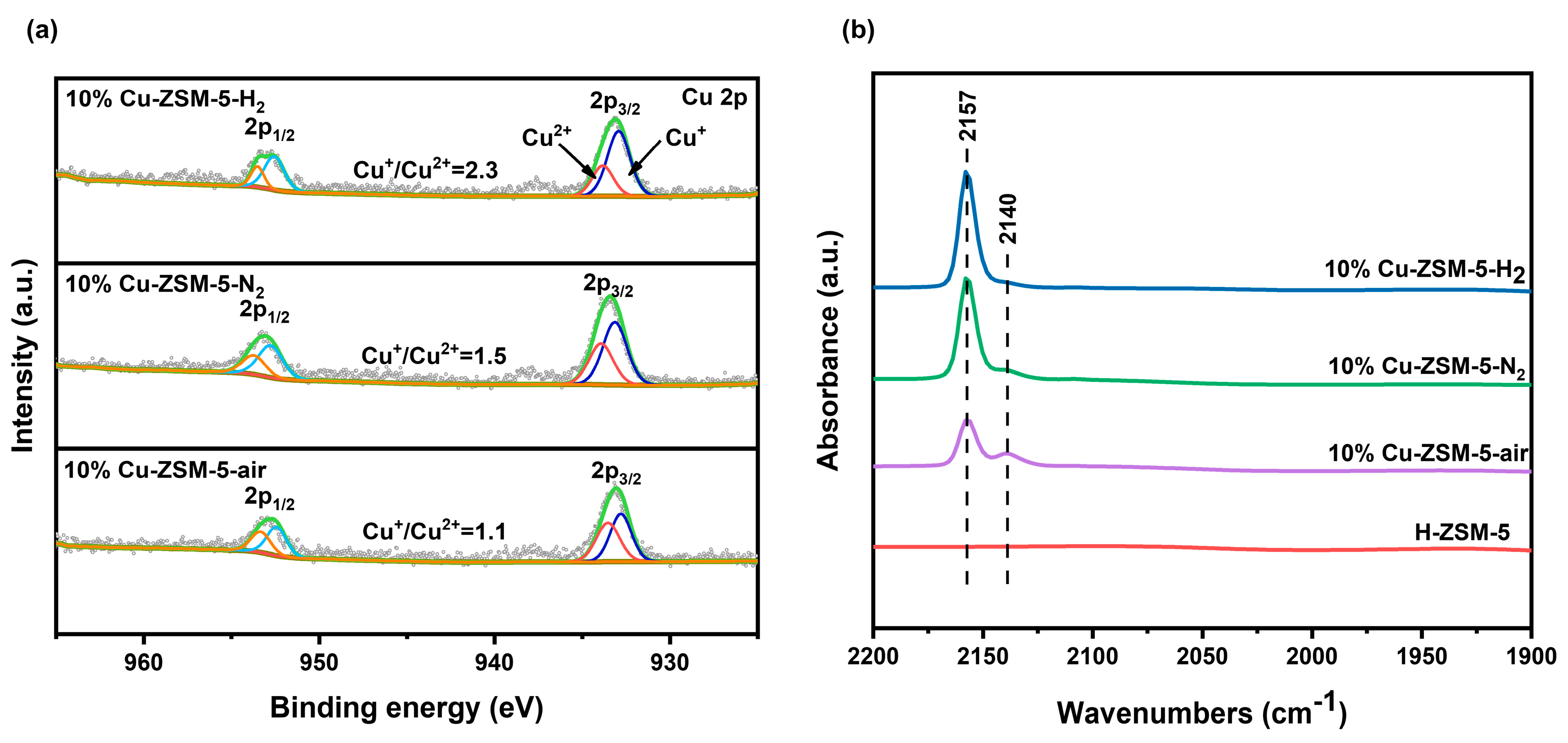
2.1. Structure and Chemical Properties of Catalysts
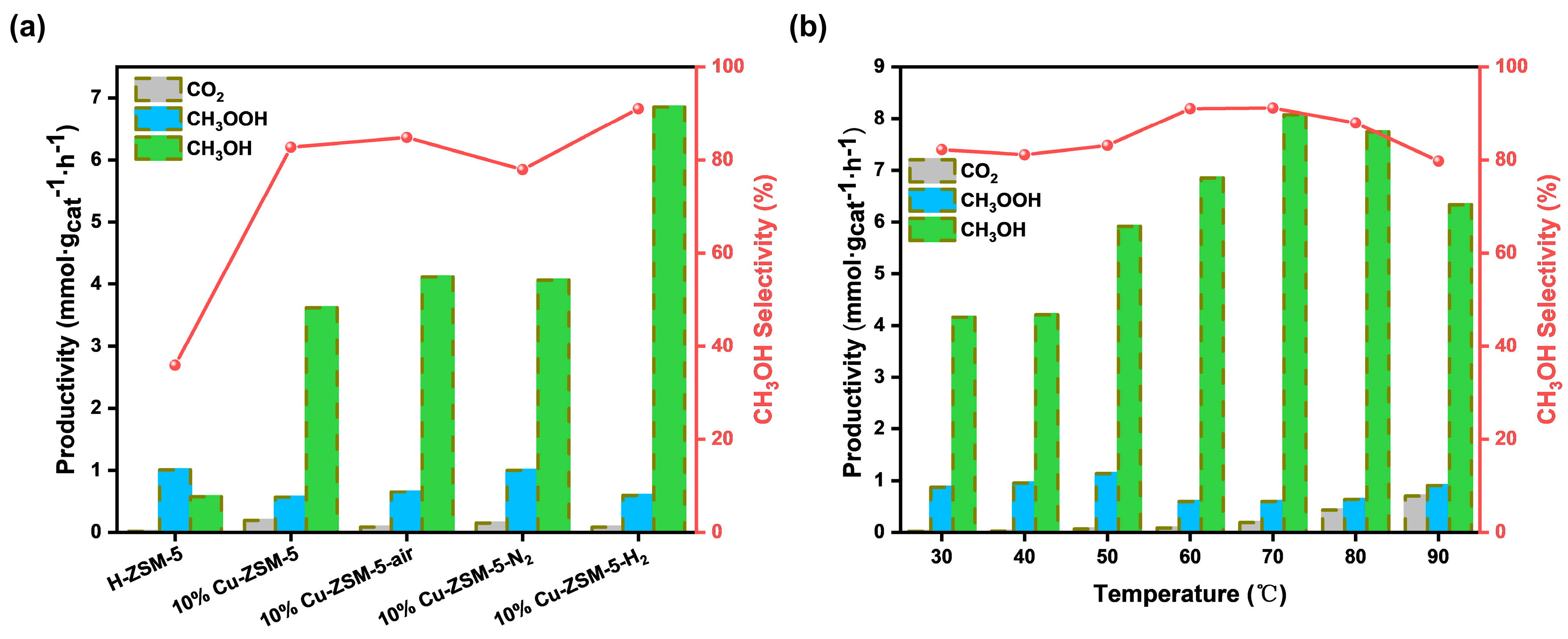
2.2. Catalytic Activity and Selectivity for CH4 Conversion
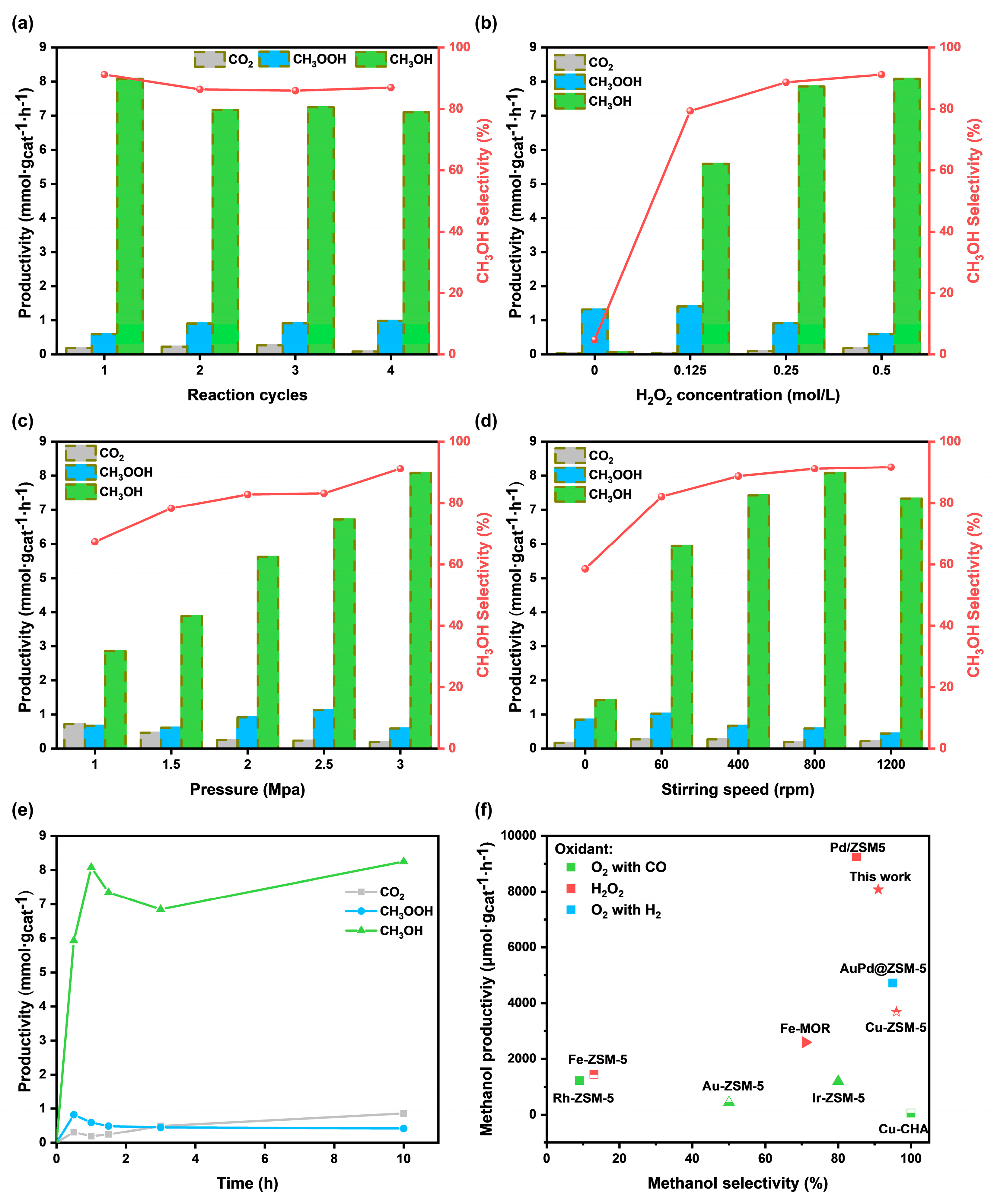
2.3. Mechanism Investigate
3. Materials and Methods
3.1. Materials and Reagents
3.2. Catalysts Preparation
3.3. Characterizations
3.4. Evaluation of Catalytic Performance
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Song, H.; Meng, X.; Wang, Z.; Liu, H.; Ye, J. Solar-Energy-Mediated Methane Conversion. Joule 2019, 3, 1606–1636. [Google Scholar] [CrossRef]
- Li, M.; Shan, J.; Giannakakis, G.; Ouyang, M.; Cao, S.; Lee, S.; Allard, L.F.; Flytzani-Stephanopoulos, M. Single-Step Selective Oxidation of Methane to Methanol in the Aqueous Phase on Iridium-Based Catalysts. Appl. Catal. B Environ. 2021, 292, 120124. [Google Scholar] [CrossRef]
- Mehmood, A.; Chae, S.Y.; Park, E.D. Low-Temperature Electrochemical Oxidation of Methane into Alcohols. Catalysts 2024, 14, 58. [Google Scholar] [CrossRef]
- Yvon-Durocher, G.; Allen, A.P.; Bastviken, D.; Conrad, R.; Gudasz, C.; St-Pierre, A.; Thanh-Duc, N.; Del Giorgio, P.A. Methane Fluxes Show Consistent Temperature Dependence across Microbial to Ecosystem Scales. Nature 2014, 507, 488–491. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Jiang, L.; Li, D.; Tian, S.; Zhu, X.; Wang, H.; He, C.; Li, K. Progress and Key Challenges in Catalytic Combustion of Lean Methane. J. Energy Chem. 2022, 75, 173–215. [Google Scholar] [CrossRef]
- Kang, J.; Park, E.D. Liquid-Phase Selective Oxidation of Methane to Methane Oxygenates. Catalysts 2024, 14, 167. [Google Scholar] [CrossRef]
- Meng, X.; Cui, X.; Rajan, N.P.; Yu, L.; Deng, D.; Bao, X. Direct Methane Conversion under Mild Condition by Thermo-, Electro-, or Photocatalysis. Chem 2019, 5, 2296–2325. [Google Scholar] [CrossRef]
- He, D.; Wu, S.; Cao, X.; Chen, D.; Zhang, L.; Zhang, Y.; Luo, Y. Dynamic Trap of Ni at Elevated Temperature for Yielding High-Efficiency Methane Dry Reforming Catalyst. Appl. Catal. B Environ. Energy 2024, 346, 123728. [Google Scholar] [CrossRef]
- Palmer, C.; Upham, D.C.; Smart, S.; Gordon, M.J.; Metiu, H.; McFarland, E.W. Dry Reforming of Methane Catalysed by Molten Metal Alloys. Nat. Catal. 2020, 3, 83–89. [Google Scholar] [CrossRef]
- Kang, J.; Park, E.D. Selective Oxidation of Methane over Fe-Zeolites by In Situ Generated H2O2. Catalysts 2020, 10, 299. [Google Scholar] [CrossRef]
- Colby, J.; Stirling, D.I.; Dalton, H. The Soluble Methane Mono-Oxygenase of Methylococcus Capsulatus (Bath). Its ability to oxygenate n-alkanes, n-alkenes, ethers, and alicyclic, aromatic and heterocyclic compounds. Biochem. J. 1977, 165, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Ross, M.O.; MacMillan, F.; Wang, J.; Nisthal, A.; Lawton, T.J.; Olafson, B.D.; Mayo, S.L.; Rosenzweig, A.C.; Hoffman, B.M. Particulate Methane Monooxygenase Contains Only Mononuclear Copper Centers. Science 2019, 364, 566–570. [Google Scholar] [CrossRef]
- Yu, X.; Wu, B.; Huang, M.; Lu, Z.; Li, J.; Zhong, L.; Sun, Y. IrFe/ZSM-5 Synergistic Catalyst for Selective Oxidation of Methane to Formic Acid. Energy Fuels 2021, 35, 4418–4427. [Google Scholar] [CrossRef]
- Knorpp, A.J.; Newton, M.A.; Sushkevich, V.L.; Zimmermann, P.P.; Pinar, A.B.; Van Bokhoven, J.A. The Influence of Zeolite Morphology on the Conversion of Methane to Methanol on Copper-Exchanged Omega Zeolite (MAZ). Catal. Sci. Technol. 2019, 9, 2806–2811. [Google Scholar] [CrossRef]
- Göltl, F.; Michel, C.; Andrikopoulos, P.C.; Love, A.M.; Hafner, J.; Hermans, I.; Sautet, P. Computationally Exploring Confinement Effects in the Methane-to-Methanol Conversion Over Iron-Oxo Centers in Zeolites. ACS Catal. 2016, 6, 8404–8409. [Google Scholar] [CrossRef]
- Kim, M.S.; Yang, G.S.; Park, E.D. Effects of Cu Species on Liquid-Phase Partial Oxidation of Methane with H2O2 over Cu-Fe/ZSM-5 Catalysts. Catalysts 2022, 12, 1224. [Google Scholar] [CrossRef]
- Smeets, P.J.; Groothaert, M.H.; Schoonheydt, R.A. Cu Based Zeolites: A UV–Vis Study of the Active Site in the Selective Methane Oxidation at Low Temperatures. Catal. Today 2005, 110, 303–309. [Google Scholar] [CrossRef]
- Cao, J.; Lewis, R.J.; Qi, G.; Bethell, D.; Howard, M.J.; Harrison, B.; Yao, B.; He, Q.; Morgan, D.J.; Ni, F.; et al. Methane Conversion to Methanol Using Au/ZSM-5 Is Promoted by Carbon. ACS Catal. 2023, 13, 7199–7209. [Google Scholar] [CrossRef]
- Qi, G.; Davies, T.E.; Nasrallah, A.; Sainna, M.A.; Howe, A.G.R.; Lewis, R.J.; Quesne, M.; Catlow, C.R.A.; Willock, D.J.; He, Q.; et al. Au-ZSM-5 Catalyses the Selective Oxidation of CH4 to CH3OH and CH3COOH Using O2. Nat. Catal. 2022, 5, 45–54. [Google Scholar] [CrossRef]
- Sheppard, T.; Daly, H.; Goguet, A.; Thompson, J.M. Improved Efficiency for Partial Oxidation of Methane by Controlled Copper Deposition on Surface-Modified ZSM-5. ChemCatChem 2016, 8, 562–570. [Google Scholar] [CrossRef]
- Pappas, D.K.; Borfecchia, E.; Dyballa, M.; Pankin, I.A.; Lomachenko, K.A.; Martini, A.; Signorile, M.; Teketel, S.; Arstad, B.; Berlier, G.; et al. Methane to Methanol: Structure–Activity Relationships for Cu-CHA. J. Am. Chem. Soc. 2017, 139, 14961–14975. [Google Scholar] [CrossRef] [PubMed]
- Pokhrel, J.; Shantz, D.F. Continuous Partial Oxidation of Methane to Methanol over Cu-SSZ-39 Catalysts. J. Catal. 2023, 421, 300–308. [Google Scholar] [CrossRef]
- Burnett, L.; Rysakova, M.; Wang, K.; González-Carballo, J.; Tooze, R.P.; García-García, F.R. Isothermal Cyclic Conversion of Methane to Methanol Using Copper-Exchanged ZSM-5 Zeolite Materials under Mild Conditions. Appl. Catal. Gen. 2019, 587, 117272. [Google Scholar] [CrossRef]
- Tang, X.; Wang, L.; Yang, B.; Fei, C.; Yao, T.; Liu, W.; Lou, Y.; Dai, Q.; Cai, Y.; Cao, X.-M.; et al. Direct Oxidation of Methane to Oxygenates on Supported Single Cu Atom Catalyst. Appl. Catal. B Environ. 2021, 285, 119827. [Google Scholar] [CrossRef]
- Hammond, C.; Forde, M.M.; Ab Rahim, M.H.; Thetford, A.; He, Q.; Jenkins, R.L.; Dimitratos, N.; Lopez-Sanchez, J.A.; Dummer, N.F.; Murphy, D.M.; et al. Direct Catalytic Conversion of Methane to Methanol in an Aqueous Medium by Using Copper-Promoted Fe-ZSM-5. Angew. Chem. Int. Ed. 2012, 51, 5129–5133. [Google Scholar] [CrossRef]
- Engedahl, U.; Boje, A.; Ström, H.; Grönbeck, H.; Hellman, A. Investigating the Composition of the Metal Dimer Site in Chabazite for Direct Methane-to-Methanol Conversion. J. Phys. Chem. C 2024, 128, 3641–3651. [Google Scholar] [CrossRef]
- Narsimhan, K.; Iyoki, K.; Dinh, K.; Román-Leshkov, Y. Catalytic Oxidation of Methane into Methanol over Copper-Exchanged Zeolites with Oxygen at Low Temperature. ACS Cent. Sci. 2016, 2, 424–429. [Google Scholar] [CrossRef]
- Yu, T.; Li, Z.; Lin, L.; Chu, S.; Su, Y.; Song, W.; Wang, A.; Weckhuysen, B.M.; Luo, W. Highly Selective Oxidation of Methane into Methanol over Cu-Promoted Monomeric Fe/ZSM-5. ACS Catal. 2021, 11, 6684–6691. [Google Scholar] [CrossRef]
- Dou, B.; Lv, G.; Wang, C.; Hao, Q.; Hui, K. Cerium Doped Copper/ZSM-5 Catalysts Used for the Selective Catalytic Reduction of Nitrogen Oxide with Ammonia. Chem. Eng. J. 2015, 270, 549–556. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, B.; Yang, J.; Rui, P.; Fan, N.; Liao, W.; Shu, X. Zeolite Fe-MFI as Catalysts in the Selective Liquid-Phase Dehydration of 1-Phenylethanol. Catal. Commun. 2018, 110, 97–101. [Google Scholar] [CrossRef]
- Li, Y.; Deng, J.; Song, W.; Liu, J.; Zhao, Z.; Gao, M.; Wei, Y.; Zhao, L. Nature of Cu Species in Cu–SAPO-18 Catalyst for NH3–SCR: Combination of Experiments and DFT Calculations. J. Phys. Chem. C 2016, 120, 14669–14680. [Google Scholar] [CrossRef]
- Pang, L.; Fan, C.; Shao, L.; Song, K.; Yi, J.; Cai, X.; Wang, J.; Kang, M.; Li, T. The Ce Doping Cu/ZSM-5 as a New Superior Catalyst to Remove NO from Diesel Engine Exhaust. Chem. Eng. J. 2014, 253, 394–401. [Google Scholar] [CrossRef]
- Romero-Sáez, M.; Divakar, D.; Aranzabal, A.; González-Velasco, J.R.; González-Marcos, J.A. Catalytic Oxidation of Trichloroethylene over Fe-ZSM-5: Influence of the Preparation Method on the Iron Species and the Catalytic Behavior. Appl. Catal. B Environ. 2016, 180, 210–218. [Google Scholar] [CrossRef]
- Tagliavini, M.; Weidler, P.G.; Njel, C.; Pohl, J.; Richter, D.; Böhringer, B.; Schäfer, A.I. Polymer-Based Spherical Activated Carbon—Ultrafiltration (UF-PBSAC) for the Adsorption of Steroid Hormones from Water: Material Characteristics and Process Configuration. Water Res. 2020, 185, 116249. [Google Scholar] [CrossRef]
- Li, H.; Cheng, R.; Liu, Z.; Du, C. Waste Control by Waste: Fenton–like Oxidation of Phenol over Cu Modified ZSM–5 from Coal Gangue. Sci. Total Environ. 2019, 683, 638–647. [Google Scholar] [CrossRef]
- Yu, B.; Cheng, L.; Dai, S.; Jiang, Y.; Yang, B.; Li, H.; Zhao, Y.; Xu, J.; Zhang, Y.; Pan, C.; et al. Silver and Copper Dual Single Atoms Boosting Direct Oxidation of Methane to Methanol via Synergistic Catalysis. Adv. Sci. 2023, 10, 2302143. [Google Scholar] [CrossRef]
- Punnoose, A.; Seehra, M.S.; Dunn, B.C.; Eyring, E.M. Characterization of CuCl2/PdCl2/Activated Carbon Catalysts for the Synthesis of Diethyl Carbonate. Energy Fuels 2002, 16, 182–188. [Google Scholar] [CrossRef]
- Wang, L.; Li, W.; Qi, G.; Weng, D. Location and Nature of Cu Species in Cu/SAPO-34 for Selective Catalytic Reduction of NO with NH3. J. Catal. 2012, 289, 21–29. [Google Scholar] [CrossRef]
- Agarwal, N.; Freakley, S.J.; McVicker, R.U.; Althahban, S.M.; Dimitratos, N.; He, Q.; Morgan, D.J.; Jenkins, R.L.; Willock, D.J.; Taylor, S.H.; et al. Aqueous Au-Pd Colloids Catalyze Selective CH4 Oxidation to CH3 OH with O2 under Mild Conditions. Science 2017, 358, 223–227. [Google Scholar] [CrossRef]
- Xie, P.; Ding, J.; Yao, Z.; Pu, T.; Zhang, P.; Huang, Z.; Wang, C.; Zhang, J.; Zecher-Freeman, N.; Zong, H.; et al. Oxo Dicopper Anchored on Carbon Nitride for Selective Oxidation of Methane. Nat. Commun. 2022, 13, 1375. [Google Scholar] [CrossRef]
- Armstrong, R.D.; Peneau, V.; Ritterskamp, N.; Kiely, C.J.; Taylor, S.H.; Hutchings, G.J. The Role of Copper Speciation in the Low Temperature Oxidative Upgrading of Short Chain Alkanes over Cu/ZSM-5 Catalysts. ChemPhysChem 2018, 19, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Li, Z.; Jones, W.; Liu, Y.; He, Q.; Song, W.; Du, P.; Yang, B.; An, H.; Farmer, D.M.; et al. Identifying Key Mononuclear Fe Species for Low-Temperature Methane Oxidation. Chem. Sci. 2021, 12, 3152–3160. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Wang, L.; Zuidema, E.; Mondal, K.; Zhang, M.; Zhang, J.; Wang, C.; Meng, X.; Yang, H.; Mesters, C.; et al. Hydrophobic Zeolite Modification for in Situ Peroxide Formation in Methane Oxidation to Methanol. Science 2020, 367, 193–197. [Google Scholar] [CrossRef]
- Shan, J.; Li, M.; Allard, L.F.; Lee, S.; Flytzani-Stephanopoulos, M. Mild Oxidation of Methane to Methanol or Acetic Acid on Supported Isolated Rhodium Catalysts. Nature 2017, 551, 605–608. [Google Scholar] [CrossRef]
- Fang, Z.; Murayama, H.; Zhao, Q.; Liu, B.; Jiang, F.; Xu, Y.; Tokunaga, M.; Liu, X. Selective Mild Oxidation of Methane to Methanol or Formic Acid on Fe–MOR Catalysts. Catal. Sci. Technol. 2019, 9, 6946–6956. [Google Scholar] [CrossRef]
- Huang, W.; Zhang, S.; Tang, Y.; Li, Y.; Nguyen, L.; Li, Y.; Shan, J.; Xiao, D.; Gagne, R.; Frenkel, A.I. Low-Temperature Transformation of Methane to Methanol on Pd1O4 Single Sites Anchored on the Internal Surface of Microporous Silicate. Angew. Chem. Int. Ed. 2016, 55, 13441–13445. [Google Scholar] [CrossRef]
- Sogukkanli, S.; Moteki, T.; Ogura, M. Selective Methanol Formation via CO-Assisted Direct Partial Oxidation of Methane over Copper-Containing CHA-Type Zeolites Prepared by One-Pot Synthesis. Green Chem. 2021, 23, 2148–2154. [Google Scholar] [CrossRef]
- Woertink, J.S.; Smeets, P.J.; Groothaert, M.H.; Vance, M.A.; Sels, B.F.; Schoonheydt, R.A.; Solomon, E.I. A [Cu2O]2+ Core in Cu-ZSM-5, the Active Site in the Oxidation of Methane to Methanol. Proc. Natl. Acad. Sci. USA 2009, 106, 18908–18913. [Google Scholar] [CrossRef]
- Vanelderen, P.; Snyder, B.E.R.; Tsai, M.-L.; Hadt, R.G.; Vancauwenbergh, J.; Coussens, O.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. Spectroscopic Definition of the Copper Active Sites in Mordenite: Selective Methane Oxidation. J. Am. Chem. Soc. 2015, 137, 6383–6392. [Google Scholar] [CrossRef]
- Grundner, S.; Markovits, M.A.C.; Li, G.; Tromp, M.; Pidko, E.A.; Hensen, E.J.M.; Jentys, A.; Sanchez-Sanchez, M.; Lercher, J.A. Single-Site Trinuclear Copper Oxygen Clusters in Mordenite for Selective Conversion of Methane to Methanol. Nat. Commun. 2015, 6, 7546. [Google Scholar] [CrossRef]
- Chen, L.; Duan, J.; Du, P.; Sun, W.; Lai, B.; Liu, W. Accurate Identification of Radicals by In-Situ Electron Paramagnetic Resonance in Ultraviolet-Based Homogenous Advanced Oxidation Processes. Water Res. 2022, 221, 118747. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, Z.; Zhang, H.; Liu, P.; Xie, Z.; Song, W.; Liu, B.; Zhao, Z. Efficient Catalysts of Surface Hydrophobic Cu-BTC with Coordinatively Unsaturated Cu(I) Sites for the Direct Oxidation of Methane. Proc. Natl. Acad. Sci. USA 2023, 120, e2206619120. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Yao, Q.; Xu, Y.; Cao, K.; Huang, X. Strong Synergy in a Lichen-like RuCu Nanosheet Boosts the Direct Methane Oxidation to Methanol. Nano Energy 2020, 71, 104566. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pu, Y.; Zhang, H.; Li, Y.; Yu, C.; Song, X.; Yang, C.; Fu, M. Unveiling the Role of Copper Valence States in Enhancing the Catalytic Performance of Copper-Modified ZSM-5 for Direct Methane Conversion. Catalysts 2025, 15, 277. https://doi.org/10.3390/catal15030277
Pu Y, Zhang H, Li Y, Yu C, Song X, Yang C, Fu M. Unveiling the Role of Copper Valence States in Enhancing the Catalytic Performance of Copper-Modified ZSM-5 for Direct Methane Conversion. Catalysts. 2025; 15(3):277. https://doi.org/10.3390/catal15030277
Chicago/Turabian StylePu, Yunhan, Huajie Zhang, Yanjun Li, Chuan Yu, Xiaofei Song, Chen Yang, and Mingli Fu. 2025. "Unveiling the Role of Copper Valence States in Enhancing the Catalytic Performance of Copper-Modified ZSM-5 for Direct Methane Conversion" Catalysts 15, no. 3: 277. https://doi.org/10.3390/catal15030277
APA StylePu, Y., Zhang, H., Li, Y., Yu, C., Song, X., Yang, C., & Fu, M. (2025). Unveiling the Role of Copper Valence States in Enhancing the Catalytic Performance of Copper-Modified ZSM-5 for Direct Methane Conversion. Catalysts, 15(3), 277. https://doi.org/10.3390/catal15030277