1. Introduction
Fuel cells are a specialized electrochemical device that convert the stored chemical energy of a fuel into electrical energy. During the process, the anode receives a continuous fuel supply, and the cathode is fed with an oxidant [
1]. These fuel cells can produce electrical energy continuously if supplied constantly. Due to their absence of intermediate heat-producing steps or thermodynamic limitations for mechanical work [
1], in addition to their low contamination due to a lack of a combustion process [
2,
3], the fuel cells are advantaged over other electrochemical cells like batteries and even engines and devices that employ fossil fuels.
Reducing the size of the electrocatalysts to the nanoparticle scale significantly alters their thermodynamic and transport properties. The large relative fraction of surface atoms in nanoparticles determines the size dependencies of the melting temperature and pressure and, therefore, it has an impact on heat capacity, thermal and electrical conductivity, and, most importantly for this work, chemical potential [
4,
5,
6,
7,
8,
9,
10]. For instance, quantum confinement modifies the band structure of metallic nanoparticles and nanocrystals, affecting their electronic, conductive, and optical properties. This modification induces anomalous behaviors of the band gap energy at different temperatures [
7,
8]. In addition, the chemical potential of a nanoparticle is influenced by the surface energy and the surface structure [
11]. As nanoparticles have a much larger surface area to volume ratio compared to bulk materials, their chemical affinity increases and, therefore, the nanoparticles are more reactive [
4,
5,
7,
12,
13,
14,
15,
16,
17,
18]. Notably, the classical equations of chemical kinetics can be used to describe the processes on the surface of nanoparticles [
7].
In recent years, considerable progress has been made in developing nanoelectrocatalysts and their synthesis techniques over the past few decades [
19,
20,
21]. Many efforts have been focused on optimizing their performance for alcohol oxidation (methanol, ethanol) and oxygen reduction; see, for instance, [
21,
22,
23,
24,
25,
26,
27,
28].
In the present study, we are interested in the methanol oxidation reaction (MOR), which involves the oxidation of methanol (CH
3OH) into carbon dioxide (CO
2) as a final product [
28,
29,
30]. However, in the case of Pd-based nanocatalysts, their sensitivity to CO or palladium oxide poisoning can significantly degrade their catalytic performance [
19,
25]. Hence, it is of primary importance to develop and understand the performance of Pd-based catalysts combined with other metals like Ni or Cu, since the addition of Ni to Pd can significantly enhance the catalytic activity towards MOR and reduces significantly the costs of practical implementation [
19,
29,
30,
31,
32].
In the case of bimetallic particles like the NiPd nanoalloys that we will study here, the evolution of the reactions takes place, apparently, along a bifunctional mechanism [
19,
29,
31,
33]. This mechanism is characterized by the synergistic effect of the NiPd composition, which leads to the enhanced catalytic performance of NiPd nanocatalysts compared to the monometallic Pd case [
29,
31].
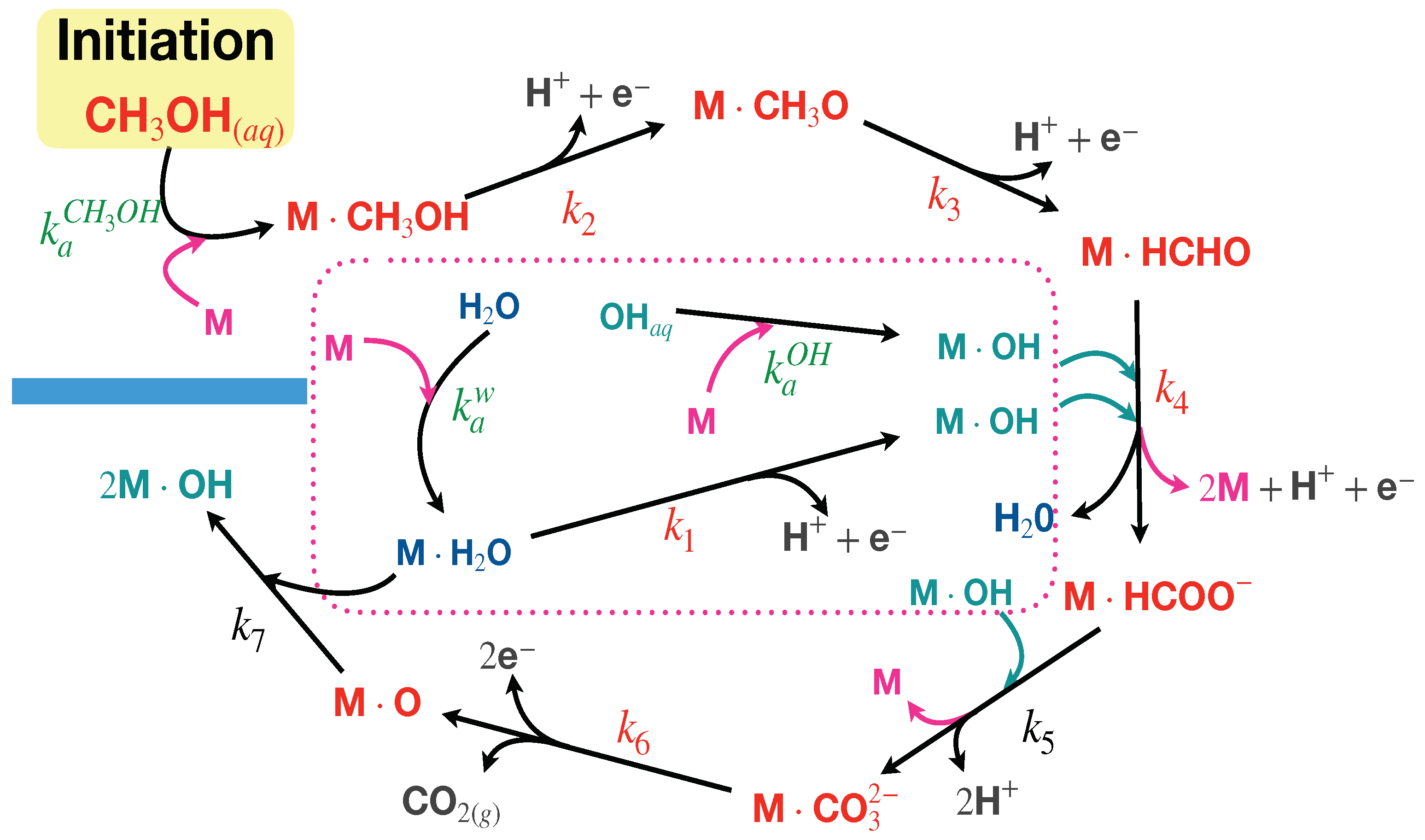
According to this mechanism, methanol is adsorbed at Pd sites and first dehydrogenated to methoxy. Then, formaldehyde and subsequently formate are then oxidized with the help of adsorbed OH
− ions. OH
− ions have, in general, two sources: one by direct adsorption from the solution, and, complementary, as a product of the hydrolysis of previously adsorbed water molecules [
19,
21,
25,
29,
31,
32]. Therefore, the oxidation steps run simultaneously with the hydrolysis of water molecules and the adsorption of OH
− radicals at the Ni sites. Notice that these processes compete for the active sites over the nanoparticle’s surface. The last step suggested is the oxidation of adsorbed carbonate, which decomposes in carbon dioxide (instantaneously released to the solution), producing two electrons and protons. This step also yields an oxidation of an active site, therefore having a poisoning effect over the surface [
29,
31,
34].
In this work, we conduct a combined theoretical and experimental investigation of MOR on nickel–palladium electrocatalysts. Using linear voltammetry in an alkaline medium, we examine the performance of catalysts with varying Ni:Pd ratios (3:1, 1:1, 1:3). Our approach aims to elucidate the synergistic effects of nickel and palladium on MOR activity by systematically probing different nanoalloy compositions and analyzing the results obtained through the comparison with a kinetic model of the mechanism. Hence, the synergistic effects are then assessed by analyzing the behavior and tendencies of the reaction rate constants for different bulk methanol concentrations.
Concerning experiments, our work builds upon the findings of Araujo et al. (2020) [
33], highlighting the crucial role of Pd-Ni interactions in enhancing MOR performance on Pd-based electrocatalysts. By systematically varying the Ni:Pd ratio, we aim to gain deeper insights into the underlying mechanisms and the optimal nanoparticle composition for maximizing methanol oxidation activity. This mesoscopic study of the chemical mechanisms could be complemented by molecular-level studies such as SEM, XRD, and XPS, as well as heuristic DFT and molecular dynamics studies that would deepen our understanding of the relationship between the electronic structure of nanoparticles and their efficiency [
35]. However, the present study, based primarily on theoretical modeling of electrochemical measurements, seems to be sufficient to explain the differentiated performance of the cell as a function of the nickel-to-palladium ratio.
Our theoretical analysis of the oxidation curves is based on the proposal of a reaction mechanism of the MOR that approximately follows the mechanisms reported in the literature [
19,
25,
29,
31,
32], as already discussed in previous paragraphs. Specifically, in the model mechanism we presented in Ref. [
25], the process begins with water and methanol molecules diffusing into the electrode’s pores with a limited number of active sites. Methanol adsorbs onto active metal sites independently of voltage, forming neutral layers. The chemical potentials determine the adsorption affinity and Gibbs energy. Using irreversible thermodynamics, the dynamics are proposed to follow a Langmuir–Hinshelwood-like equation. At equilibrium, the Nernst relation adjusts the affinity based on the external potential. This affinity, linked to reaction rates via flow–force relationships, leads to the Butler–Volmer equation, describing current as a function of concentrations and the applied voltage. In this work, we will follow a similar thermodynamic strategy, but instead applying it to the chemical mechanism of the new catalyst. In the present case, direct adsorption of OH ions is also included, as suggested in the literature, and the path followed differs from others since it is assumed that bimetallic nanoparticles promote the formation of formate and carbonate as intermediates and, therefore, the poisoning of the active sites occurs by direct oxidation of palladium atoms and not by the formation of carbon monoxide molecules, as assumed in the case of pure Pd-based catalysts [
25].
The article is organized as follows:
Section 3 describes the experimental procedure from synthesis to linear voltammetry followed to obtain the oxidation curves. Then,
Section 4 presents the theoretical model starting from the postulation of the mechanism to the formulation of the corresponding set of kinetic equations.
Section 5 is then devoted to presenting our results and their discussion in terms of the comparison with the theoretical model. Finally, the conclusions
Section 2 summarizes our results and contextualizes this study.
2. Results and Discussion
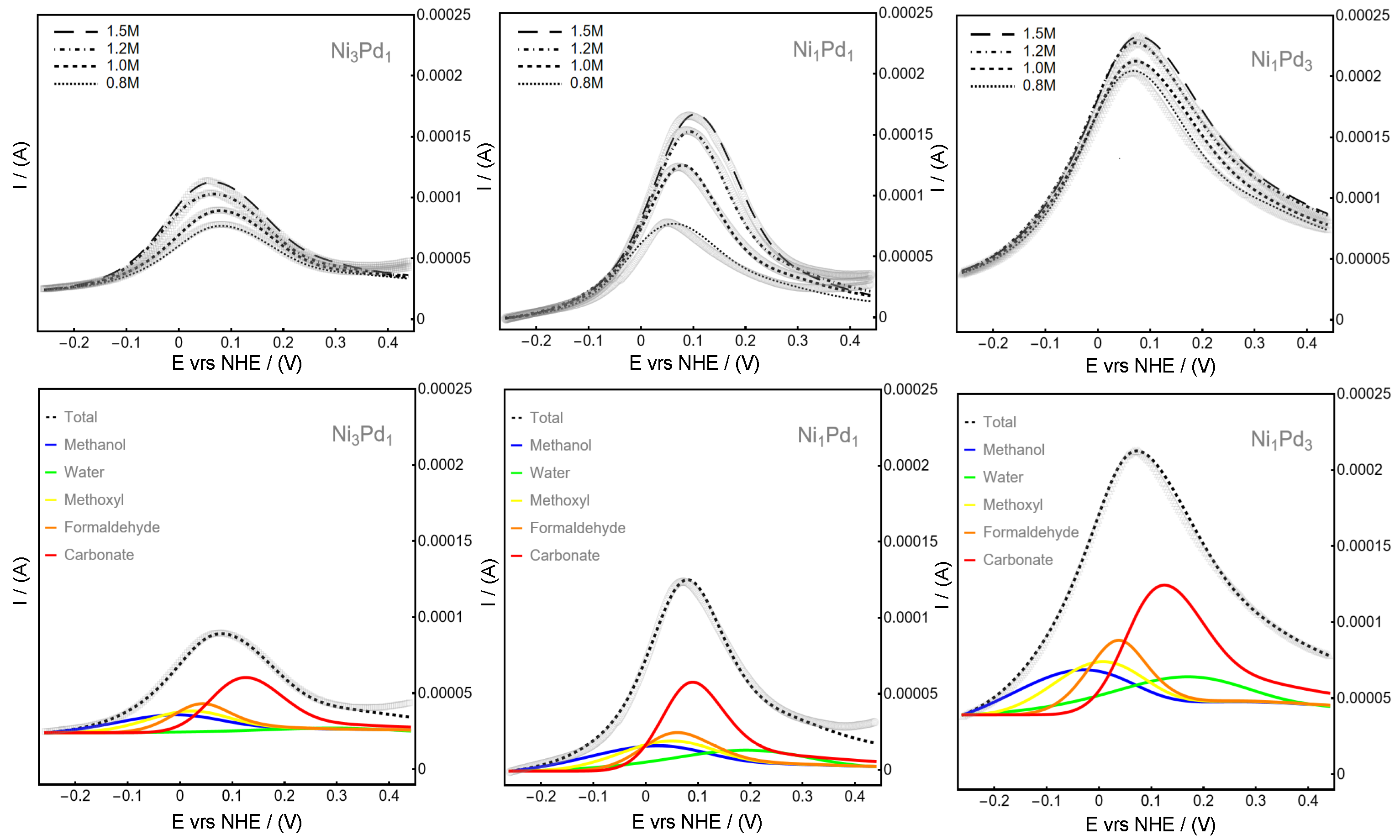
Figure 1 presents the fit (lines) of the experimental data (symbols) obtained using our synthesized Ni
xPd
y catalysts (see
Section 3). Subplots illustrate results for specific compositions: (a) Ni
3Pd
1, (b) Ni
1Pd
1, and (c) Ni
1Pd
3. The data indicate that nanoparticles with a higher relative proportion of Pd (Ni
1Pd
3) exhibit superior oxidation performance compared to those with higher Ni content.
The numerical procedure for constructing
Figure 1 involves solving the differential Equations (
11)–(
17) and (
23) with a fourth-order Runge–Kutta method, followed by the calculation of the total oxidation current using Equation (
22). The dependent variables include the concentrations
,
,
,
,
,
,
,
,
, and
. These equations are solved parametrically over a wide range of fitting parameters:
,
,
,
,
,
, and
–
. The total simulation time is 35 s, with a maximum time step of
s. Initial conditions are set to zero for all variables except
.
In the top row of
Figure 1, we show the fits for different methanol concentrations, from 0.8 to 1.5, ranging from lowest to highest. The peak current for the Ni
3Pd
1 case shifts towards lower overpotential values, contrasting with the behavior observed in the other two cases. However, this shift is relatively minor across all cases studied. Notably, for potentials exceeding 0.3 V, the Ni
1Pd
3 material does not exhibit a significant increase in oxidation, facilitating the alignment of experimental results with the proposed model. This absence of higher-potential oxidation appears linked to a delay in water hydrolysis for materials with higher Ni content.
To confirm this observation, the bottom row of
Figure 1 details the contributions of each reactant to the total oxidation current. The reaction involving water, depicted by the green line, demonstrates the delayed production and depletion of water relative to other intermediates contributing to the overall current, as outlined in Equation (
22). The green line for water hydrolysis peaks at different potentials depending on the material composition: 0.31 V for Ni
3Pd
1, 0.22 V for Ni
1Pd
1, and 0.185 V for Ni
1Pd
3. This trend suggests that the delay in water hydrolysis decreases as the oxidation activity increases.
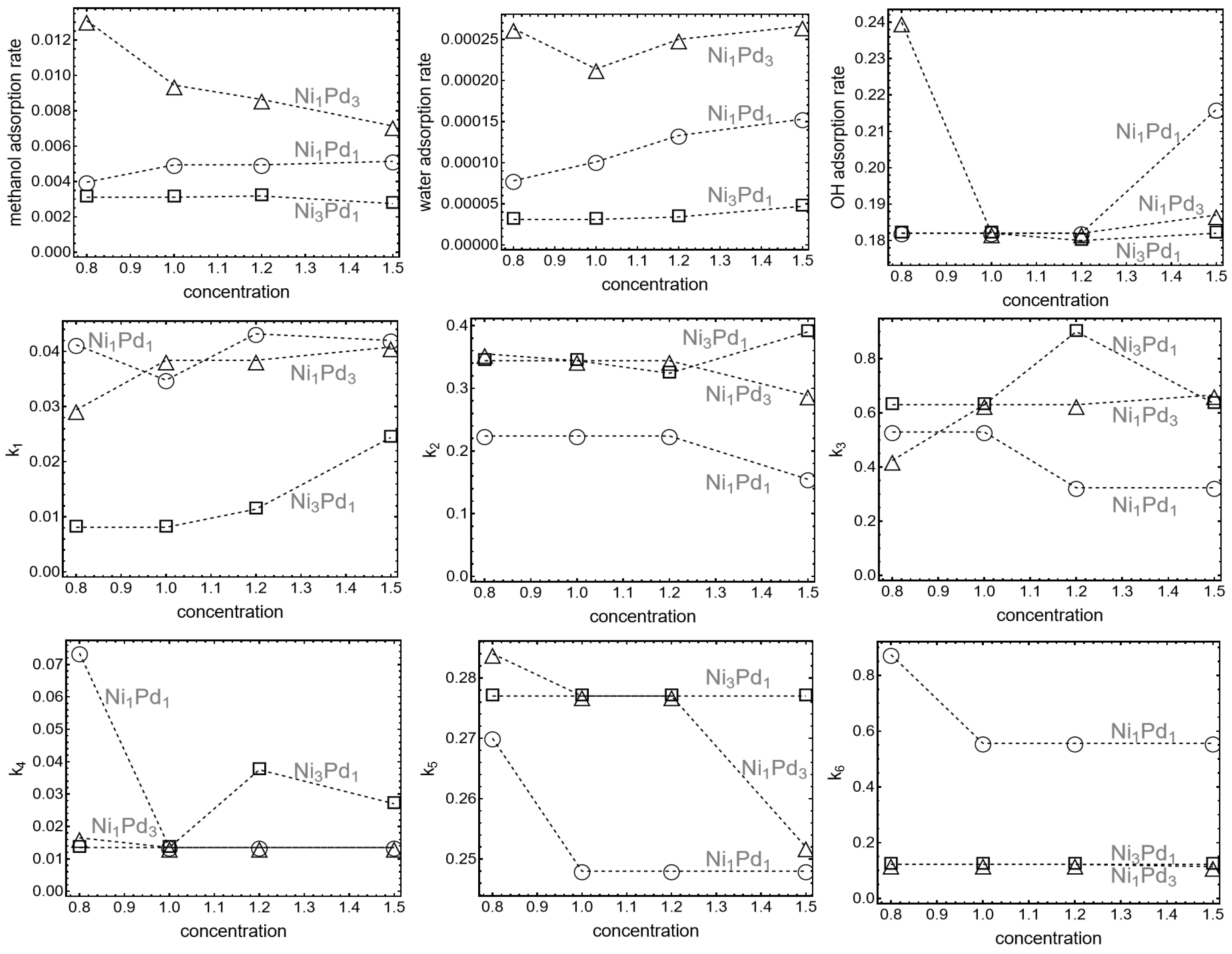
To better understand the progression of key reaction steps in the oxidation process including the role played by hydrolysis it is important to compare the reaction rates in the different processes.
Figure 2 displays the adsorption and reaction rate constants used for the fit: in the first row, the adsorption rate constants for surface reactants: methanol, water and OH in
,
, and
, respectively; in the second row the reaction constants for water hydrolysis
, methanol oxidation
, and methoxy oxidation
and finally, in the third row, constants for formaldehyde oxidation
, carbonate formation
, and carbonate oxidation
. The value of the fitting parameters is plotted against methanol concentration to show possible trends.
The second row of
Figure 2 shows that the water hydrolysis rate constant
for Ni
3Pd
1 is four times smaller than the corresponding constants for Ni
1Pd
1 and Ni
1Pd
3, which exhibit similar maxima positions. The green curve heights also align with the first-row results in
Figure 2, where the methanol and water adsorption rates for Ni
1Pd
3 are higher than for the other two cases. Besides, regarding adsorption rate constants,
for Ni
3Pd
1 is nearly double that of Ni
1Pd
1 but approximately one-third of the value for Ni
1Pd
3. This allows to conclude that the influence of water depends crucially on the characteristic time dictated fundamentally by hydrolysis on the constant
rather than by adsorption, and that this time is fundamentally affected by the nickel/palladium ratio.
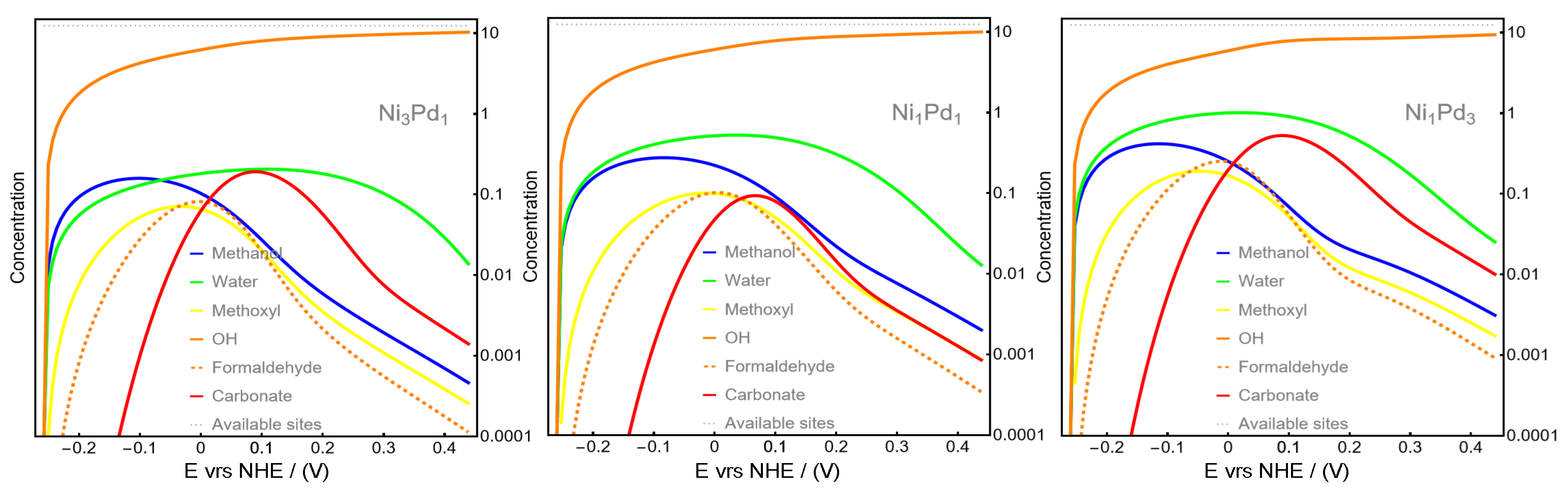
Figure 3 illustrates the surface concentration evolution of all species involved in the methanol oxidation reaction (MOR) as a function of applied voltage. The green line, representing water surface concentration, confirms that Ni
3Pd
1 nanoparticles adsorb considerably less water than the other two compositions. This observation supports the conclusion that a higher proportion of Pd atoms on the nanoparticle surface enhances water adsorption and accelerates its oxidation rate.
Additionally, this finding suggests that, at lower Ni content, the dominant mechanism, characterized by direct OH adsorption (depicted by the orange line in
Figure 3, representing OH surface concentration), may compete with the mechanism described in Ref. [
25]. In that study, direct OH adsorption was not considered, and surface poisoning was attributed to adsorbed CO formation rather than the formation of palladium oxides or the overpopulation of adsorbed OH. Here, it could be useful to delve deeper into the comparison between both mechanisms. The mechanism proposed in [
25] was based on the assumption that, in an aqueous solution of 0.3 M KOH, water molecules generally adsorb more strongly onto Pd than OH ions. This assumption arises from the fact that water molecules, despite being present at a much higher concentration, are less charged and experience weaker repulsive forces from the negatively charged Pd surface. This allows for water molecules to compete more effectively for adsorption sites on the Pd surface compared to OH ions. Additionally, the high concentration of OH ions in the solution can lead to strong electrostatic repulsion between the negatively charged Pd surface (due to adsorbed OH species) and incoming OH ions, further hindering their adsorption. According to this mechanism, water hydrolysis becomes a relevant factor during the MOR on pure Pd catalysts, as it becomes noticeable even at low applied potentials and reaches a maximum of around 0.06 V (see
Figure 3 in Ref. [
25]). This behavior contrasts with the results obtained in the mechanism proposed here, where the water hydrolysis maxima occur at potentials higher than 1.5 V (see
Figure 1). Therefore, water hydrolysis seems to be significantly delayed in NiPd catalysts compared to pure Pd. Water adsorption on Pd catalysts has been related to
d-orbitals through first-principles calculations [
36,
37]. Furthermore, in this pathway in the former mechanism, the oxidation of methanol and its intermediates over Pd proceeds directly toward the formation of adsorbed CO without the necessity of OH ions, which only participate in the slow formation of COOH before the final production of CO
2. This highlights CO as the primary poisoning species.
In contrast, the presence of Ni on the surface of NiPd alloys modifies the adsorption properties of the nanoparticles, as Ni exhibits stronger adsorption of OH ions compared to Pd, which has a higher affinity for water, but is now less abundant on the surface. Since Ni is more electropositive than Pd, it tends to have a higher electron density at the surface due to its electronic structure and higher d-band center. This increased electron density can stabilize and interact more effectively with the negatively charged OH species, leading to stronger adsorption. Therefore, Ni generally exhibits a stronger affinity for OH compared to Pd. Hence, due to its higher affinity for OH ions and the competitive adsorption effects in the presence of high OH concentrations, OH ions are likely to adsorb more strongly onto Ni than onto Pd. Consequently, in the present study, both OH and water adsorption have been considered to account for these two sources of surface OH. This, along with the high binding energy of OH on Ni and Pd [
34,
38], results in OH ions contributing earlier and more significantly than water to the oxidation of intermediates in the MOR (see
Figure 3), thereby favoring the alternative pathway described in this work. Increasing the proportion of Pd reduces the surface concentration of OH, as observed in
Figure 3. In this pathway, OH ions are required for the formation of formate from formaldehyde (the rate-determining step, see
Figure 1) and for the conversion of formate to carbonate (steps 4 and 5 of the mechanism). The combination of OH adsorption and production via water hydrolysis significantly increases the surface coverage by OH ions, making them the primary poisoning species, in contrast to pure Pd catalysts. Further research is necessary to determine the relative contribution of each mechanism to MOR activity and how it relates to the detailed structure of the catalyst surfaces. To conclude this discussion, it is worth mentioning that the presence of OH can be confirmed, for example, through Fourier-transform infrared spectroscopy (FTIR) [
35,
39].
Consistent with previous observations, the rate constants in
Figure 2 further clarify the influence of Pd content on methanol adsorption and oxidation. For example, it is clear that, for nanoparticles with higher Pd content (Ni
1Pd
3), methanol adsorption rates are strongly dependent on methanol bulk concentration. Below 1 M, the adsorption rate is approximately three times larger compared to the other two compositions, but it decreases by nearly half when the concentration reaches 1.5 M. In contrast, Ni
3Pd
1 and Ni
1Pd
1 nanoparticles exhibit only a minor increase in adsorption rates with methanol concentration, suggesting a less dynamic response. In the case of OH adsorption rates, Ni
3Pd
1 nanoparticles remain constant at
∼0.182, independent of methanol concentration. However, for Ni
1Pd
3 nanoparticles, the rate starts higher at 0.24 and decreases rapidly to converge with the rate of Ni
3Pd
1.
The methanol oxidation rate (
) is comparable for Ni
3Pd
1 and Ni
1Pd
3 nanoparticles, both exceeding the rate observed for Ni
1Pd
1. This highlights the efficiency of these two compositions in oxidizing methanol. For carbonate oxidation, remarkably, Ni
1Pd
1 nanoparticles exhibit a significantly faster oxidation rate of adsorbed carbonate (
), approximately three times higher than in the other two cases (as can be seen in
Figure 1, second row, red lines). The maximum of the red line for Ni
1Pd
1 appears at lower voltages, contributing to a more symmetric oxidation peak compared to the other compositions. The oxidation steps for methanol (
), methoxy (
), and formaldehyde (
) are similar across all compositions. However, nickel carbonate is more readily oxidized than palladium carbonate, as evidenced by the general trends in the data.
To further clarify the role of OH in relation to other intermediates, including metal oxides,
Figure 4 presents the surface concentrations of formate (magenta), metal oxide (brown), and the combined concentration of all other species (orange), and, as observed in
Figure 3, these other species are predominantly constituted by OH.
Figure 4 reveals a continuous increase in OH surface concentration, underscoring its significant role in surface poisoning during the anodic reaction. This finding highlights the critical influence of OH adsorption on the reaction’s efficiency and mechanism. Additionally, the presence of palladium oxides shows a slight increase from left to right, suggesting that their contribution to surface poisoning does not significantly impair cell efficiency. Interestingly, a higher Ni content inhibits palladium oxide formation while promoting an increase in OH concentration, which emerges as the primary blocker of active sites due to its high binding energy. This strong binding leads to an excessive amount of adsorbed OH on the surface, blocking active sites and preventing the adsorption of other reactants (such as methanol, in this case) necessary for the reaction to proceed. This conclusion aligns with the finding of Ref. [
34]. From a microscopic perspective, several mechanisms can influence the adsorption and binding process. In bimetallic systems such as Ni-Pd catalysts, the d-band center can be modulated through alloying, strain engineering, or surface modifications. For instance, modifying the d-band center of Pd using ethylene glycol has been shown to enhance catalytic performance by altering hydrogen adsorption characteristics [
40]. Furthermore, the increased binding energy can be attributed to a ligand effect, as the presence of Ni in the alloy can alter the electronic structure of Pd. Being more electropositive than Pd, Ni can donate electrons to Pd, weaken the Pd-O bond, and facilitate OH adsorption while simultaneously increasing site poisoning due to the relatively high OH binding energy [
38,
40]. Another possible factor that can be considered involves strain effects. Incorporating Ni into the Pd lattice can induce strain, altering the adsorption energies of reaction intermediates. Strain can modify bond lengths and bond angles on the surface, impacting the interaction between the catalyst and adsorbates [
41]. Further analysis, such as ab initio computational studies to determine OH binding energies on Ni and Pd sites, could help to elucidate the optimal OH binding strength and the spatial distribution of species during adsorption and OH production in this NiPd catalyst system. However, these studies fall beyond the scope of the present work.
This equilibrium differs from the mechanism proposed in Ref. [
25], where surface poisoning was attributed to CO formation rather than OH adsorption or palladium oxide formation. This difference arises from the fact that, the in direct pathway for the CO-CO
2 formation mechanism [
25], it seems that water adsorption is predominant compared to OH adsorption. Dominant CO formation in MOR against OH adsorption and Pd·O formation, as suggested by the mechanism analyzed in this study, has been recently reported in a study analyzing the role of Sb in mitigating CO poisoning in Pd-based MOR catalysts [
42]. The authors concluded that, by modifying the electronic properties of Pd and potentially facilitating the removal of adsorbed CO through interactions with adsorbed OH and alkali metal cations, like in the mechanism proposed in [
25], the Sb-Pd catalyst demonstrates enhanced activity and durability [
42]. For the present study, we may conclude that the balance between Ni and Pd plays a pivotal role in modulating the reaction pathway and efficiency.
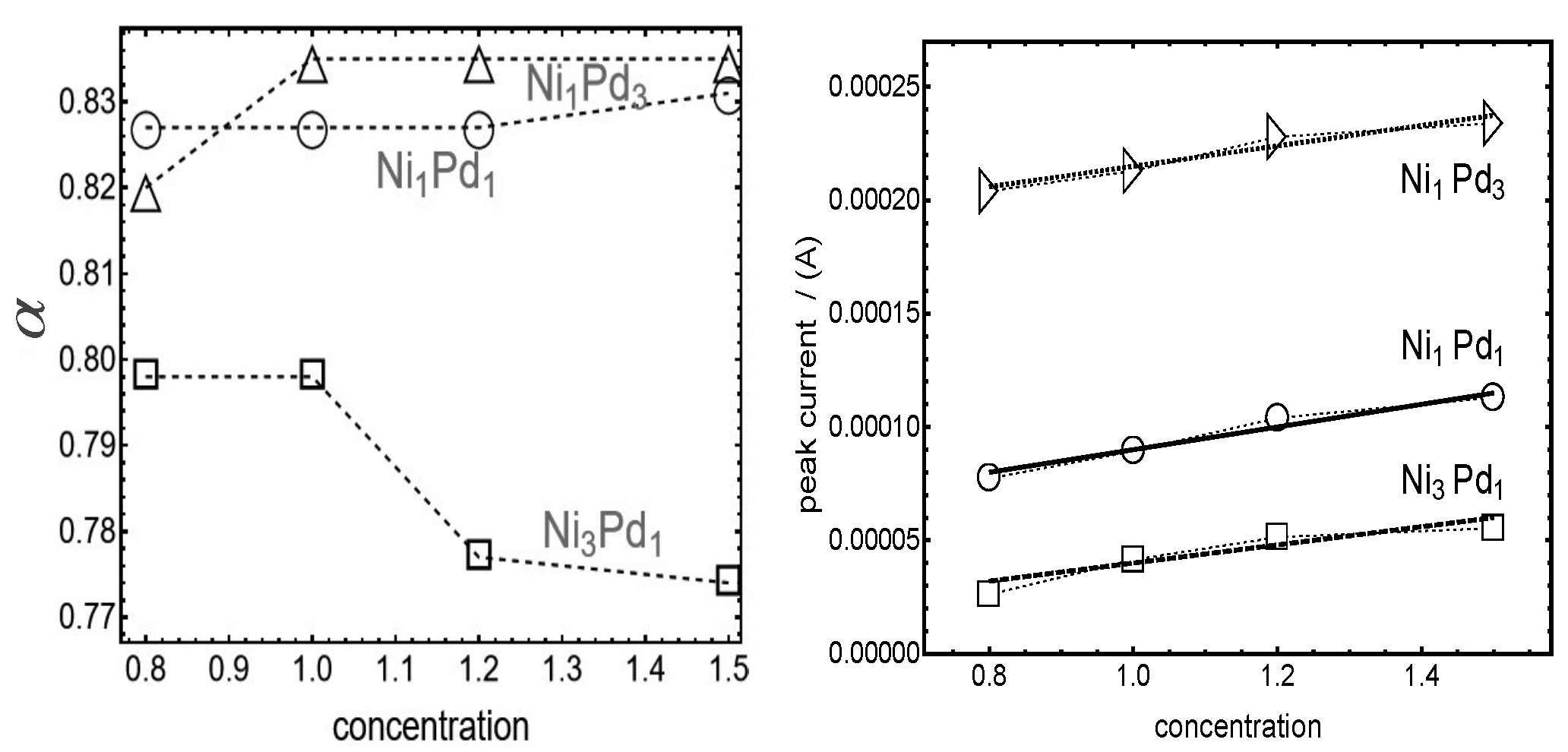
The most significant differences arising from varying proportions of Nickel (Ni) and Palladium (Pd) are highlighted in
Figure 5, which displays the transfer parameter (
) and the peak current for the three types of nanoparticles. Two notable results are evident from the figure.
As first result at the left of
Figure 5 we can see that the different Ni/Pd ratios during nanoparticle synthesis lead to distinct energy barrier asymmetries. For Ni
3Pd
1 nanoparticles, the transfer parameter
is notably larger than 0.5, indicating that the energy barrier for the oxidation reaction surpasses that of the reduction reaction. However, the value of
for Ni
3Pd
1 (
∼0.77–0.8) is lower than the nearly constant
∼0.83 observed for both Ni
1Pd
1 and Ni
1Pd
3. This suggests that higher Ni proportions on the nanoparticle surface enhance the asymmetry of the oxidation energy barrier compared to nanoparticles with higher Pd proportions. Moreover, the transfer factor of the Ni
3Pd
1 alloy exhibits greater sensitivity to the surrounding methanol concentration, decreasing as methanol bulk concentration increases.
The second result concerns the linear relationship between the peak current and methanol concentration, confirming that the adsorption mechanism we adopted is valid in the range of methanol and OH concentrations used (see [
43]). Furthermore, the slope of this linear trend correlates with the surface energy of the nanoparticles, as detailed in Ref. [
43]. This finding supports the notion that variations in surface composition significantly influence the nanoparticles’ catalytic activity and adsorption behavior.
More quantitatively, from
Figure 5, it is possible to observe that the peak current for Ni
1Pd
3 is higher, with fluctuations among the four maxima remaining within ±20% of the average peak current,
∼2.375
A. In contrast, Ni
1Pd
1 shows greater variability in peak current, reaching up to ±33% around the average value
∼2.375
A. For Ni
3Pd
1, the average peak current is significantly lower, at
∼9.55
A, but the variations are similar to Ni
1Pd
3, around ±20%. These findings suggest that the current variability increases when the relative proportions of Ni and Pd approach equimolar ratios.
5. Kinetics for Linear Voltammetry and Oxidation Current
From the previous mechanism, it is possible to obtain a set of differential equations for the surface concentration by following the laws of non-equilibrium thermodynamics [
25]. Surface concentrations will be denoted by
in Mol/m
2, and the set of reactions in (
1)–(
10) leads to the following set of equations. From the reactions accounting for adsorption processes, we have that the change in time of the surface concentration of water, with notation
for shortness, is
whereas for the surface concentration the methanol,
, is given by
The time evolution of the surface OH
− is given by
In these equations, the first two terms at the right-hand side (RHS) represent the adsorption and desorption of bulk methanol, water, and hydroxyl, having bulk concentrations of
,
and
, respectively.
counts the number of active sites not occupied by the reacting species, and its explicit dependence will be given later. The respective adsorption velocity constants were discussed above. The third terms at the RHS correspond to the first oxidation step of the mechanism, being
and
, or the oxidation constants of surface-adsorbed methanol and water. The hydrolysis reaction leads to the formation of surface
with the rate
. Additionally, (
13) contains two terms corresponding to the oxidation of formaldehyde
and formate
. The surface concentration of palladium oxide is denoted by
. The oxidation processes are induced by an over-potential
and are modulated by the transfer coefficient
. Finally,
F is the Faraday constant and
is the molar thermal energy of the system.
Denoting by
the surface concentration of CH
3O, we have
where
is the oxidation velocity constant of CH
3O. The following steps are as follows:
with
being the surface concentration of formaldehyde (HCHO) and
being its oxidation velocity constant. Then, we have
where
is the surface concentration of HCOO
− concentration and
the corresponding reaction constant. Carbonate (
) surface concentration is
, and therefore is as follows:
The subsequent reaction is the regeneration of active sites from palladium oxide (M·O)
:
Carbon dioxide gas production is, therefore,
In the previous reaction mechanism, the oxidation contributions finish at Equation (
17), in which the six electrons produced by methanol oxidation are completed [
25]. The subsequent reactions do not influence the oxidation peak and will not be taken into account in the following analysis, since they took place in the cathodic part of the cyclic voltammetry.
In the adsorption steps of methanol, water, and OH
− ions, the concentration of free surface active sites,
, is given by a Langmuir–Hinshelwood-like term:
The last term in the previous equation is likely responsible for surface poisoning because of the existence of adsorbed OH ions and the specific formation of palladium oxides (MO). These poisoning effects differ from the one previously considered during MOR on pure Pd nanoparticles, in which the formation of adsorbed CO was responsible for the blocking of active sites [
25].
Although we have called it a Langmuir–Hinshelwood-like model for the reader’s reference, it is convenient to clarify here that our adsorption model, represented by the set of Equations (
11)–(
13) and (
20), in fact goes beyond the classical LH model since the standard LH model assumes that the availability of free sites is determined solely by the total number of sites and the fractional coverage of a single adsorbate (methanol, water, or OH). It does not explicitly consider the influence of multiple adsorbed species on the availability of free sites (in our case, all of the intermediates of the mechanism). Therefore, although competitive adsorption among three bulk species can still be considered a classical LH model, our description cannot be considered to be the same rigorous type, since it incorporates surface interactions and competitive adsorption through Equation (
20). Our kinetic model, by incorporating Equation (
20) (implying that several species compete for adsorption sites on the surface) into three different adsorption kinetic equations with different adsorption rates (and, implicitly, different adsorption energies), couples the whole mechanism into the adsorption kinetics, giving rise to a highly non-linear feedback that is not accounted for in the classical LH case. Thus, despite the apparent simple form of the equations, a highly competitive adsorption and intermediate occupancy of active sites implicitly incorporates lateral interactions and surface heterogeneity in an averaged form. What is not directly incorporated in our model is the lateral diffusion of the species over the surface of the catalyst. However, we believe that, in the case of nanoscale catalysts, this effect is not too relevant due to the small characteristic size of the system. Finally, we want to add that, despite the possible limitations of the LH model and our more general approach, some reports suggest that the use of the LH model for methanol adsorption can still be quantitatively acceptable [
44,
45].
The total current observed in a methanol oxidation reaction (MOR) is the result of multiple individual oxidation reactions occurring simultaneously or sequentially at the electrode surface. Each of these steps involves an oxidation reaction, and each oxidation reaction contributes to the overall current. This electrochemical current
can be written as the sum of the contributions of the oxidation reactions in the mechanism:
where
is the electrode area. In the present case, the current is given by
Using the previous relations and the evolution equation for the applied voltage:
with
being the sweep rate satisfying
, one can calculate how the current changes with time and applied voltage during a linear voltammetry experiment.
6. Conclusions
This study analyzes the impact of the relative proportion of nickel (Ni) and palladium (Pd) in catalytic nanoparticles on the methanol oxidation reaction (MOR). The results are presented in terms of kinetic parameters, surface concentrations, and peak currents, showing significant differences between three main compositions: Ni3Pd1, Ni1Pd1, and Ni1Pd3.
It is observed that a higher Pd content on the surface promotes water adsorption and increases its oxidation rate, thereby reducing the need for direct OH adsorption. This trend is evident in the lower concentration of water adsorbed on Ni3Pd1 nanoparticles compared to the other two compositions. Furthermore, the increase in Pd favors mechanisms in which the formation of Pd oxides plays a minor role in surface poisoning, unlike Ni, which increases the concentrations of adsorbed OH, contributing significantly to the blocking of active sites.
The analysis of the rate constants shows that, for Ni1Pd3, the adsorption of methanol depends markedly on its concentration in the medium, being approximately three times higher for concentrations lower than 1 M, compared to the other two compositions. In contrast, the adsorption rate of OH on Ni3Pd1 remains constant. These results suggest that the predominant presence of Pd favors the oxidation of water and reduces the dependence on OH-based mechanisms. On the other hand, nanoparticles with equimolar proportions (Ni1Pd1) stand out for oxidizing adsorbed carbonates approximately three times faster than the other two compositions, which influences the symmetry of the oxidation peaks. Regarding the transfer parameter, Ni3Pd1 shows a higher degree of asymmetry in the oxidation energy barrier, while Ni1Pd1 and Ni1Pd3 have more constant values and closer to 0.83.
The results suggest that a higher Pd content favors more efficient oxidation mechanisms by reducing the formation of intermediate products that cause surface poisoning, such as CO, carbonates, or palladium oxide. However, as the proportion of Ni increases, an increase in the concentration of adsorbed OH is observed, which dominates the blocking of active sites even above from the palladium oxide blocking. This behavior clearly differs from the that observed in the direct methanol–CO mechanism in which carbonate formation is avoided in Pd-dominant alloys. This relationship suggests that the surface composition can be tuned to optimize catalytic performance, depending on the desired dominant mechanism.
Finally, this study highlights how the relative proportion of Ni and Pd affects both the kinetic characteristics and the intermediate products, providing a basis for the rational design of catalysts in methanol oxidation applications.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}