

Alkali-Boosted Catalytic Activity of Co-Based Catalysts Supported by Nanoporous Carbon in the Hydrodeoxygenation of Guaiacol

 ,
,  ,
,
Abstract
1. Introduction
2. Results and Discussion
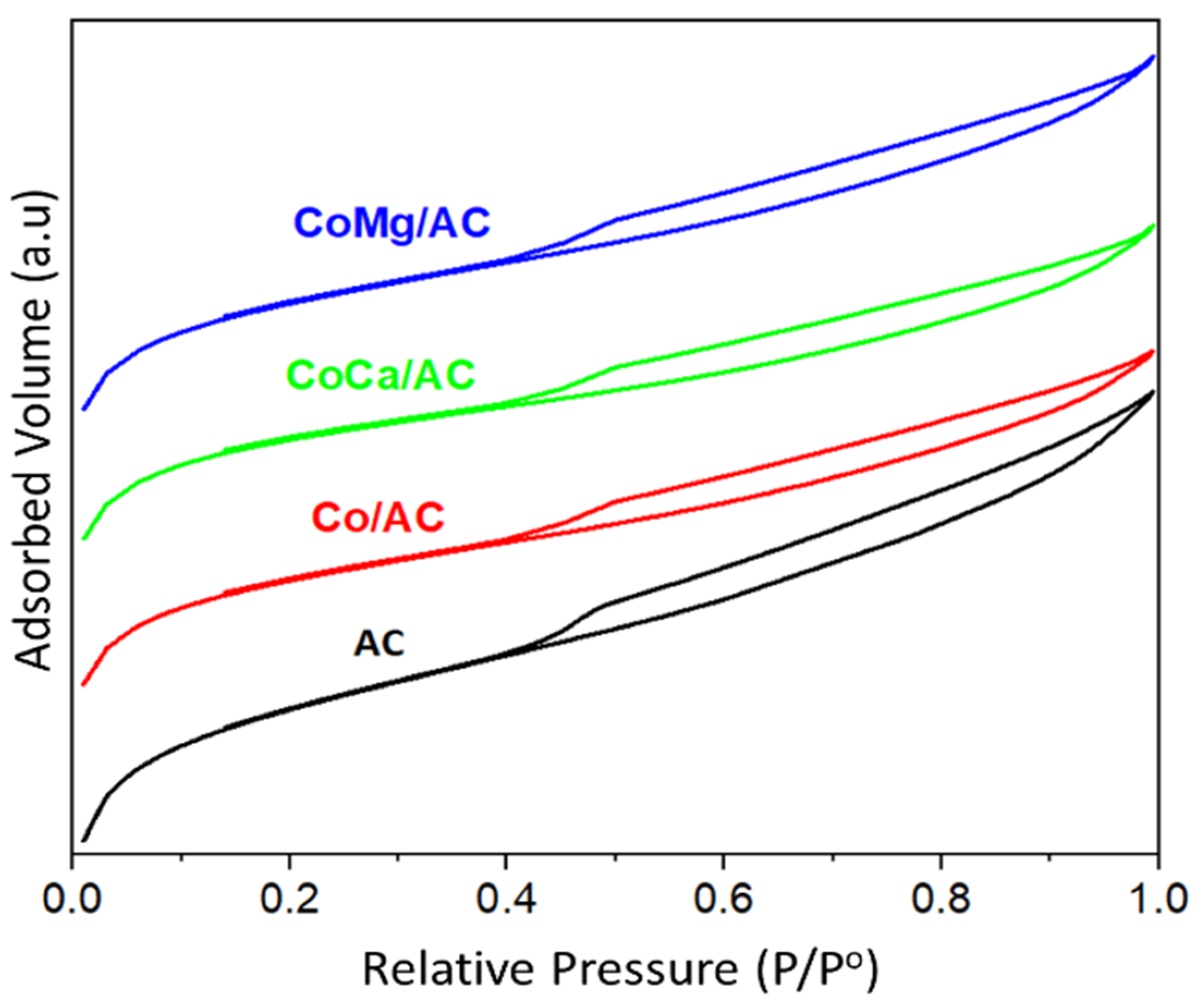
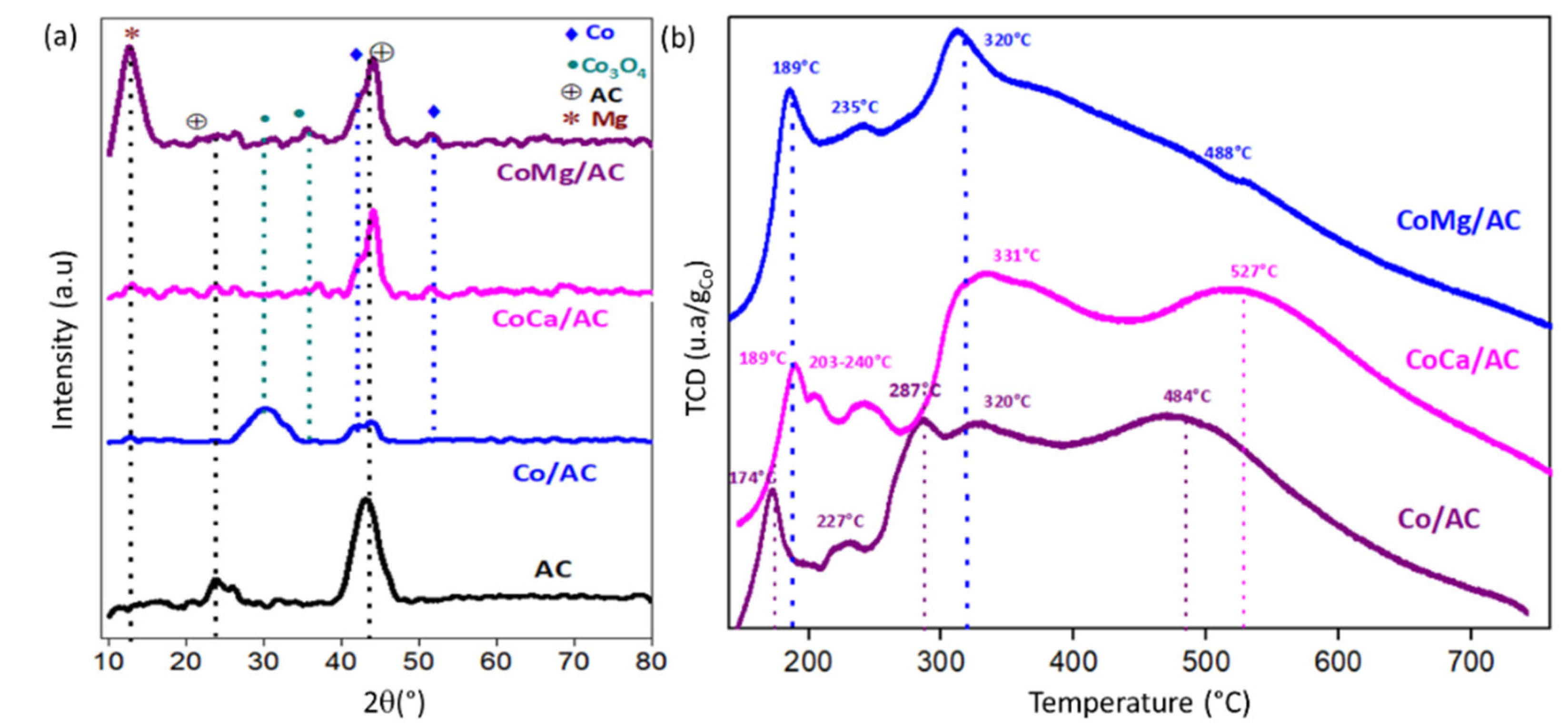
2.1. Characterization of Catalysts
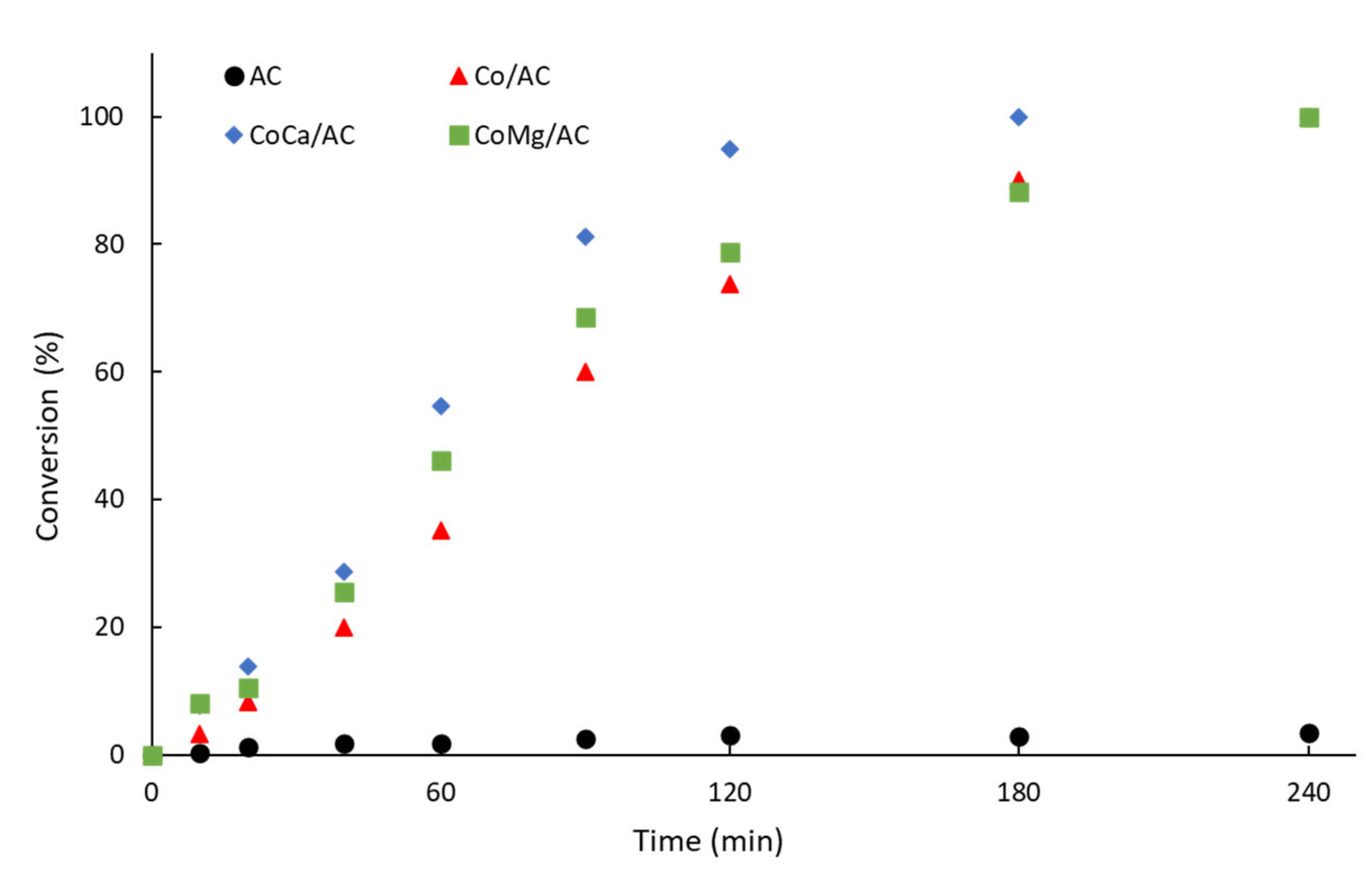
2.2. Catalytic Activity
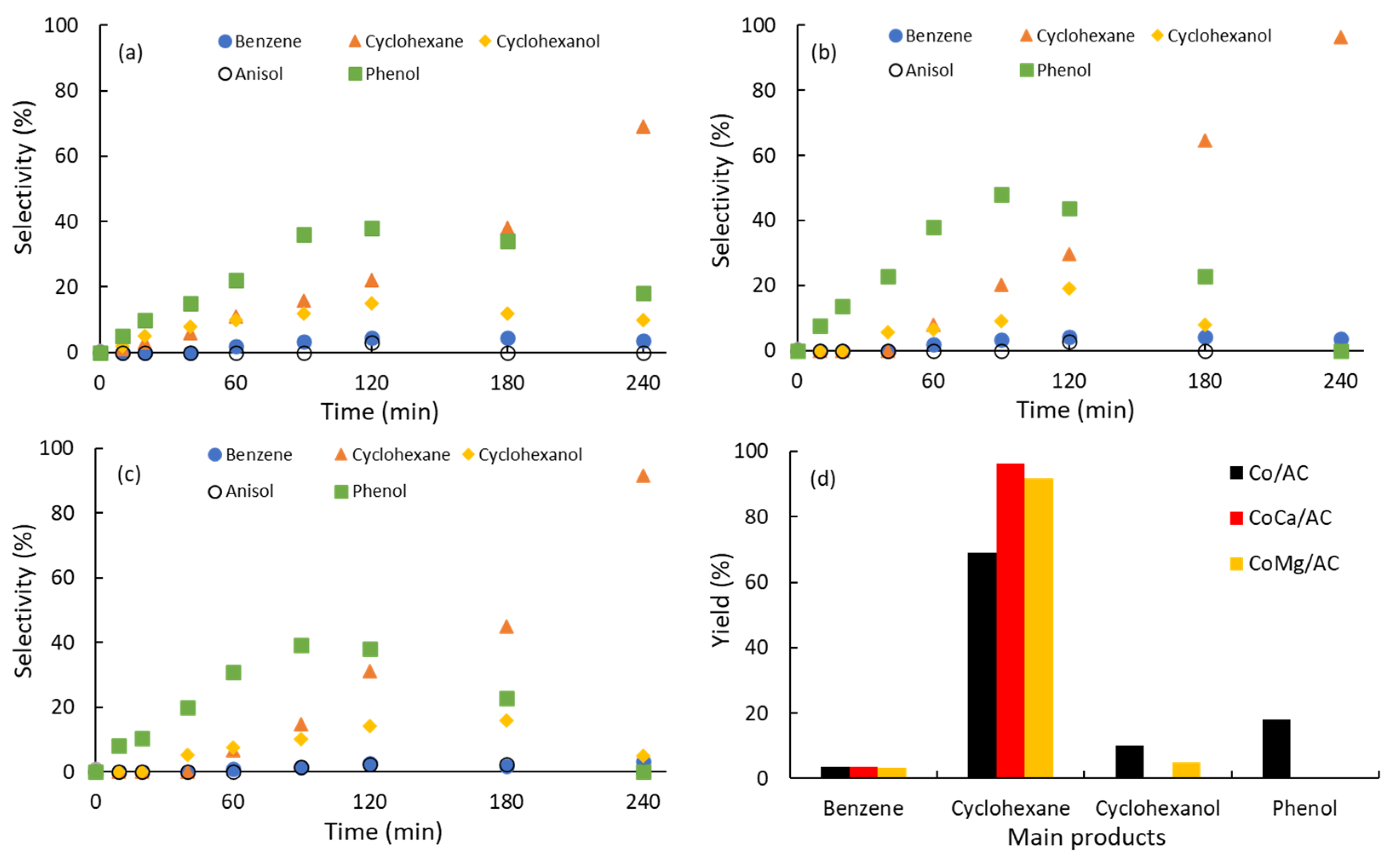
2.3. Selectivity of Products
2.4. General Discussion
3. Materials and Methods
3.1. Synthesis of Activated Carbon and Catalysts
3.2. Characterization
3.3. Catalytic Tests
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Cordero-Lanzac, T.; Palos, R.; Arandes, J.M.; Castaño, P.; Rodríguez-Mirasol, J.; Cordero, T.; Bilbao, J. Stability of an acid activated carbon based bifunctional catalyst for the raw bio-oil hydrodeoxygenation. Appl. Catal. B Environ. 2017, 203, 389–399. [Google Scholar] [CrossRef]
- Cordero-Lanzac, T.; Hita, I.; García-Mateos, F.J.; Castaño, P.; Rodríguez-Mirasol, J.; Cordero, T.; Bilbao, J. Adaptable kinetic model for the transient and pseudo-steady states in the hydrodeoxygenation of raw bio-oil. Chem. Engin. J. 2020, 400, 124679. [Google Scholar] [CrossRef]
- Fan, X.-D.; Wu, Y.-J.; Tu, R.; Sun, Y.; Jiang, E.-C.; Xu, X.-W. Hydrodeoxygenation of guaiacol via rice husk char supported Ni based catalysts: The influence of char supports. Renew. Energy 2020, 157, 1035–1045. [Google Scholar] [CrossRef]
- Wu, X.; Ge, Q.; Zhu, X. Vapor phase hydrodeoxygenation of phenolic compounds on group 10 metal-based catalysts: Reaction mechanism and product selectivity control. Catal. Today 2021, 365, 143–161. [Google Scholar] [CrossRef]
- Blanco, E.; Dongil, A.B.; García-Fierro, J.L.; Escalona, N. Insights in supported rhenium carbide catalysts for hydroconversion of lignin-derived compounds. Appl. Catal. A Gen. 2020, 599, 117600. [Google Scholar] [CrossRef]
- Blanco, E.; Cabeza, P.; Naharro Ovejero, V.; Contreras, C.; Dongil, A.B.; Ghampson, I.T.; Escalona, N. Effect of carbon support and functionalization on the synthesis of rhenium carbide and its use on HDO of guaiacol. Catal. Today 2023, 420, 114031. [Google Scholar] [CrossRef]
- Blanco, E.; Díaz de León, J.N.; García-Fierro, J.L.; Escalona, N. Study of supported bimetallic MoRe carbides catalysts for guaiacol conversion. Catal. Today 2021, 367, 290–296. [Google Scholar] [CrossRef]
- Matos, J.; Brito, J.; Laine, J. Activated carbon supported Ni-Mo: Effects of pretreatments and composition on catalyst reducibility and on ethylene conversion. Appl. Catal. A Gen. 1997, 152, 27–42. [Google Scholar] [CrossRef]
- Matos, J.; Laine, J. Ethylene conversion on activated carbon supported NiMo catalysts: Effect of the Support. Appl. Catal. A Gen. 2003, 241, 25–38. [Google Scholar] [CrossRef]
- Matos, J.; Díaz, K.; García, V.; Cordero, T.C.; Brito, J.L. Methane transformation in presence of carbon dioxide on activated carbon supported nickel-calcium catalysts. Catal. Lett. 2006, 109, 163–169. [Google Scholar] [CrossRef]
- Díaz, K.; García, V.; Matos, J. Activated carbon supported Ni-Ca: Influence of reaction parameters on activity and stability of catalyst on methane reformation. Fuel 2007, 86, 1337–1344. [Google Scholar] [CrossRef]
- Goscianska, J.; Pietzrak, R.; Matos, J. Catalytic performance of ordered mesoporous carbons modified with lanthanides in dry methane reforming. Catal. Today 2018, 301, 204–216. [Google Scholar] [CrossRef]
- Tian, F.; Wang, W.; Liu, B.; Pan, Y.; Dong, B.; Li, Y.; Guo, H.; Chai, Y.; Liu, C. Synergistic effect between CoSx and MoS2 at the micrometer scale: Considerable promotion of the hydrodesulfurization of DBT. Chem. Engin. J. 2024, 484, 149579. [Google Scholar] [CrossRef]
- Medina Cervantes, J.A.; Díaz de León, J.N.; Fuentes Moyado, S.; Alonso-Núñez, G. Influence of precursor compounds on the structural and catalytic properties of CoNiMo/SBA-15 catalysts used in the hydrodesulfurization of dibenzothiophene. Mol. Catal. 2023, 547, 113399. [Google Scholar] [CrossRef]
- Wang, E.; Yang, F.; Song, M.; Chen, G.; Zhang, Q.; Wang, F.; Bing, L.; Wang, G.; Han, D. Recent advances in the unsupported catalysts for the hydrodesulfurization of fuel. Fuel Proc. Technol. 2022, 235, 107386. [Google Scholar] [CrossRef]
- Zhao, L.; Huang, W.; Xu, Z.; Xu, Z.; Wang, X.; Wei, Q.; Zhou, Y. In situ synthesis of Co modified SAPO-5 molecular sieves and the application in quinoline hydrodenitrogenation of their NiWS supported catalysts. Chem. Engin. Sci. 2023, 284, 119428. [Google Scholar] [CrossRef]
- Paz Carmona, H.; Tišler, Z.; Svobodová, E.; Akhmetzyanova, U. Co-processing of atmospheric gas oil with rapeseed oil over sulfur-free supported and phosphorus-modified Co-Mo and Ni-Mo carbide catalysts. Catal. Lett. 2022, 152, 3814–3824. [Google Scholar] [CrossRef]
- Klimov, O.V.; Vatutina, Y.V.; Nadeina, K.A.; Kazakov, M.O.; Gerasimov, E.Y.; Prosvirin, I.P.; Larina, T.V.; Noskov, A.S. CoMoB/Al2O3 catalysts for hydrotreating of diesel fuel. The effect of the way of the boron addition to a support or an impregnating solution. Catal. Today 2018, 305, 192–202. [Google Scholar] [CrossRef]
- Matos, J.; Samudio-González, D.; Blanco, E.; Poon, P.S.; Escalona, N. Alkali-driven selectivity of products on carbon-supported Ni-based catalysts during the HDO of guaiacol. Fuel 2024, 374, 132442. [Google Scholar] [CrossRef]
- Jiang, D.; Lin, M.; Yan, Y.; Zhan, L.; Li, R.; Wu, Y. Highly selective hydrogenation of guaiacol to cyclohexanol over carbon-encapsulated highly dispersed cobalt catalyst. Chem. Eng. Sci. 2024, 290, 119779. [Google Scholar] [CrossRef]
- Kim, H.; Lim, Y.H.; Park, J.H.; Ha, J.-M.; Kim, D.H. Hydrodeoxygenation of guaiacol over physically mixed Co/TiO2 and WO3/TiO2 catalysts. Green Chem. 2024, 26, 2692–2704. [Google Scholar] [CrossRef]
- Chen, J.; Ma, Z.; Qin, J.; Chen, M.; Dong, L.; Mao, W.; Zhou, X.; Long, Y.; Ma, J. Highly efficient and selective hydrodeoxygenation of guaiacol to cyclohexanol over a rod-like CoNi-C catalyst. Fuel 2023, 353, 129216. [Google Scholar] [CrossRef]
- Hongkailers, S.; Pattiya, A.; Hinchiranan, N. Hydrodeoxygenation of oxygenates derived from biomass pyrolysis using titanium dioxide-supported cobalt catalysts. Molecules 2023, 28, 7468. [Google Scholar] [CrossRef]
- Wu, L.; Wei, J.; Zhang, Y.; He, Y.; Wang, X.; Guo, H.; Tang, Y.; Tan, L. The selective hydrodeoxygenation of guaiacol to cyclohexanol over cobalt-modified TS-1 catalysts. Micro. Mesoporous Mater. 2023, 348, 112347. [Google Scholar] [CrossRef]
- Li, X.; Wang, Y.; Zhang, G.; Sun, W.; Bai, Y.; Zheng, L.; Han, X.; Wu, L. Influence of Mg-promoted Ni-based catalyst supported on coconut shell carbon for CO2 methanation. Chem. Sel. 2019, 4, 838–845. [Google Scholar] [CrossRef]
- Zhang, J.; Sudduth, B.; Sun, J.; Kovarik, L.; Engelhard, M.H.; Wang, Y. Elucidating the active site and the role of alkali metals in selective hydrodeoxygenation of phenols over iron-carbide-based catalyst. ChemSusChem 2021, 14, 4546–4555. [Google Scholar]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Helmich, M.; Luckas, M.; Pasel, C.; Bathen, D. Characterization of microporous activated carbons using molecular probe method. Carbon 2014, 74, 22–31. [Google Scholar]
- Shao, M.; Wang, D.; Yu, G.; Hu, B.; Yu, W.; Qian, Y. The synthesis of carbon nanotubes at low temperature via carbon suboxide disproportionation. Carbon 2004, 42, 183–185. [Google Scholar] [CrossRef]
- Ghods, B.; Meshkani, F.; Rezaei, M. Effects of alkaline earth promoters on the catalytic performance of the nickel catalysts supported on high surface area mesoporous magnesium silicate in dry reforming reaction. Inter. J. Hydrog. Energy 2016, 41, 22913–22921. [Google Scholar] [CrossRef]
- Chiou, J.Y.Z.; Liu, S.W.; Ho, K.F.; Huang, H.H.; Tang, C.W.; Wang, C.B. Ca-modified Co/SBA-15 catalysts for hydrogen production through ethanol steam reforming. Inter. Lett. Chem. Phys. Astron. 2013, 24, 1–16. [Google Scholar]
- He, K.; Dong, Y.M.; Li, Z.; Yin, L.; Zhang, A.M.; Zheng, Y.C. Catalytic ozonation of phenol in water with natural brucite and magnesia. J. Hazardous Mater. 2008, 159, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Vizcaíno, A.J.; Carrero, A.; Calles, J.A. Comparison of ethanol steam reforming using Co and Ni catalysts supported on SBA-15 modified by Ca and Mg. Fuel Process. Technol. 2016, 146, 99–109. [Google Scholar] [CrossRef]
- Du, H.; Zhu, H.; Chen, X.; Dong, W.; Lu, W.; Luo, W.; Ding, Y. Study on CaO-promoted Co/AC catalysts for synthesis of higher alcohols from syngas. Fuel 2016, 182, 42–49. [Google Scholar] [CrossRef]
- Dongil, A.B.; Ghampson, I.T.; García, R.; Fierro, J.L.G.; Escalona, N. Hydrodeoxygenation of guaiacol over Ni/carbon catalysts: Effect of the support and Ni loading. RSC Adv. 2016, 6, 2611. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SBET (m2 g−1) a | Vtotal (cm3 g−1) b | Vmicro (cm3 g−1) c | Vmeso (cm3 g−1) d | Wp (nm) e | DCo (nm) f |
|---|---|---|---|---|---|---|
| AC | 966 | 0.60 | 0.43 | 0.17 | 2.48 | - |
| Co/AC | 792 | 0.48 | 0.33 | 0.15 | 2.42 | 23 |
| CoCa/AC | 795 | 0.46 | 0.34 | 0.12 | 2.31 | 11 |
| CoMg/AC | 794 | 0.48 | 0.35 | 0.13 | 2.42 | 12 |
| Reduction Peaks (°C) | |||||
|---|---|---|---|---|---|
| Catalysts | 1 | 2 | 3 | 4 | 5 |
| Co/AC | 174 | 227 | 287 | 320 | 484 |
| CoCa/AC | 189 | 203 | 240 | 331 | 527 |
| CoMg/AC | 189 | 235 | - | 320 | 488 |
| Catalysts | CO Consumption (mmol·g−1) | Metallic Dispersion (%) | Particle Size (nm) | |
|---|---|---|---|---|
| Chemisorption | XRD | |||
| Co/AC | 22.2 | 4 | 20 | 23 |
| CoCa/AC | 38.5 | 8 | 11 | 11 |
| CoMg/AC | 23.0 | 4 | 20 | 12 |
| Catalysts | ro (mmol·g−1·s−1) a | (ro-i/ro-Co) (a.u) b | TOF (s−1) c |
|---|---|---|---|
| AC | 0.41 | 0.06 | -- |
| Co/AC | 6.78 | 1.00 | 0.31 |
| CoCa/AC | 15.8 | 2.33 | 0.41 |
| CoMg/AC | 16.7 | 2.46 | 0.72 |
| Catalysts | ro (mmol·g−1·s−1) a | TOF (s−1) b | SCyhex240 (%) c | SPhOH240 (%) d | Ref |
|---|---|---|---|---|---|
| Co/AC | 6.78 | 0.31 | 70 | 18 | This work |
| CoCa/AC | 15.8 | 0.41 | 96 | 0 | This work |
| CoMg/AC | 16.7 | 0.72 | 92 | 0 | This work |
| Ni/AC | 10.7 | 0.08 | 61 | 1 | [19] |
| NiCa/AC | 14.4 | 0.18 | 69 | 12 | [19] |
| NiMg/AC | 13.1 | 0.16 | 69 | 13 | [19] |
| Ni(10 wt.%.)/CNT | 14.0 | 0.36 | 10 | 0 | [35] |
| Ni(15 wt.%)/CNT | 43.0 | 0.77 | 48 | 0 | [35] |
| ReC | 15.3 | 0.15 | 5 | 30 | [5] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matos, J.; Samudio-González, D.; Blanco, E.; Poon, P.S.; Escalona, N. Alkali-Boosted Catalytic Activity of Co-Based Catalysts Supported by Nanoporous Carbon in the Hydrodeoxygenation of Guaiacol. Catalysts 2025, 15, 17. https://doi.org/10.3390/catal15010017
Matos J, Samudio-González D, Blanco E, Poon PS, Escalona N. Alkali-Boosted Catalytic Activity of Co-Based Catalysts Supported by Nanoporous Carbon in the Hydrodeoxygenation of Guaiacol. Catalysts. 2025; 15(1):17. https://doi.org/10.3390/catal15010017
Chicago/Turabian StyleMatos, Juan, Diana Samudio-González, Elodie Blanco, Po S. Poon, and Néstor Escalona. 2025. "Alkali-Boosted Catalytic Activity of Co-Based Catalysts Supported by Nanoporous Carbon in the Hydrodeoxygenation of Guaiacol" Catalysts 15, no. 1: 17. https://doi.org/10.3390/catal15010017
APA StyleMatos, J., Samudio-González, D., Blanco, E., Poon, P. S., & Escalona, N. (2025). Alkali-Boosted Catalytic Activity of Co-Based Catalysts Supported by Nanoporous Carbon in the Hydrodeoxygenation of Guaiacol. Catalysts, 15(1), 17. https://doi.org/10.3390/catal15010017