
Diphenyl Carbonate: Recent Progress on Its Catalytic Synthesis by Transesterification

Abstract
1. Introduction to Diphenyl Carbonate
2. Reaction Pathways and Reaction Thermodynamics of Transesterification of Phenol with Dimethyl Carbonate
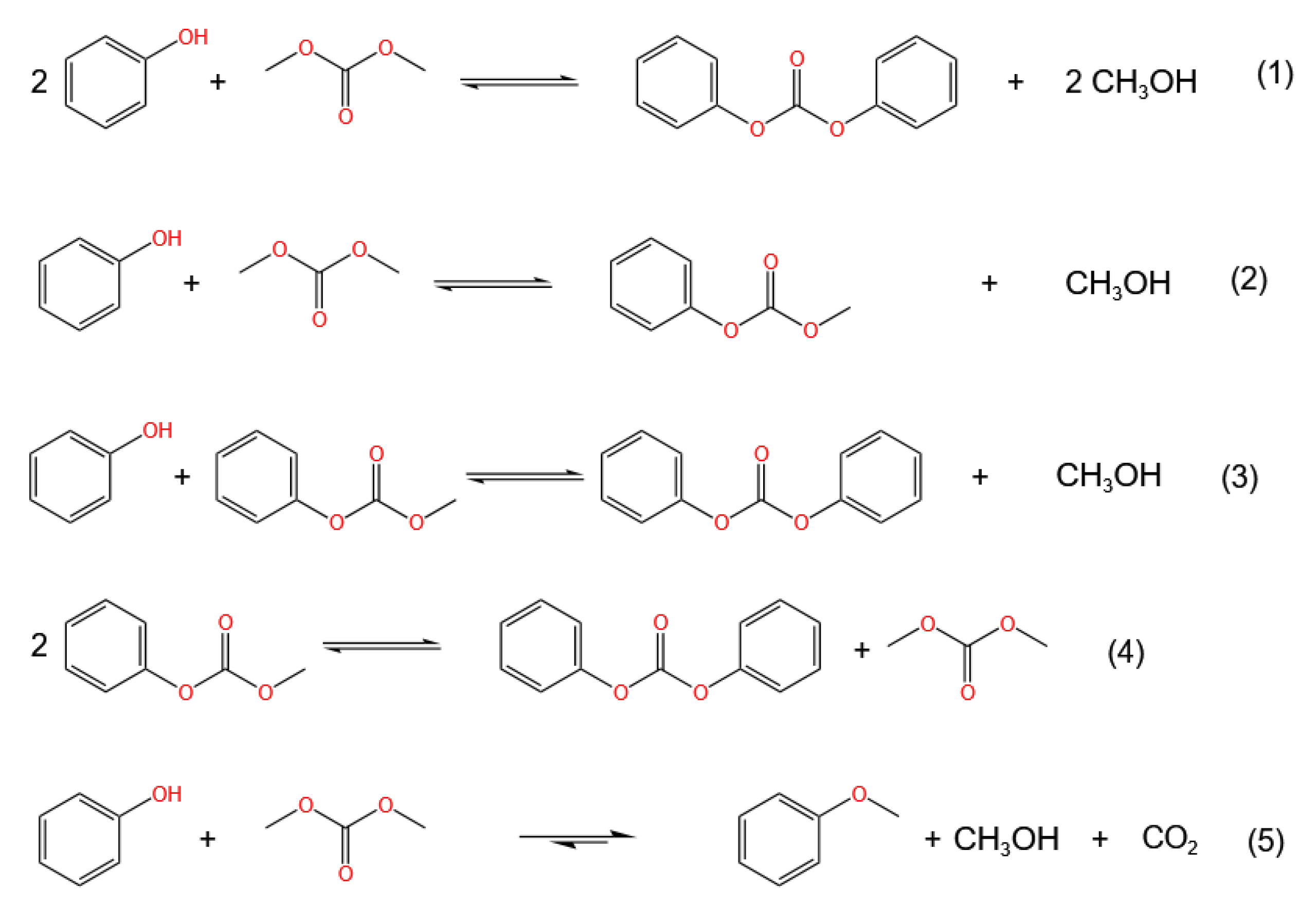
2.1. Reaction Pathways for Transesterification
2.2. Thermodynamic Analysis of Transesterification
2.2.1. Methodology of Thermodynamic Analysis
2.2.2. Thermodynamic Constraint in Transesterification by Calculation
3. Homogeneous Catalysts for Transesterification
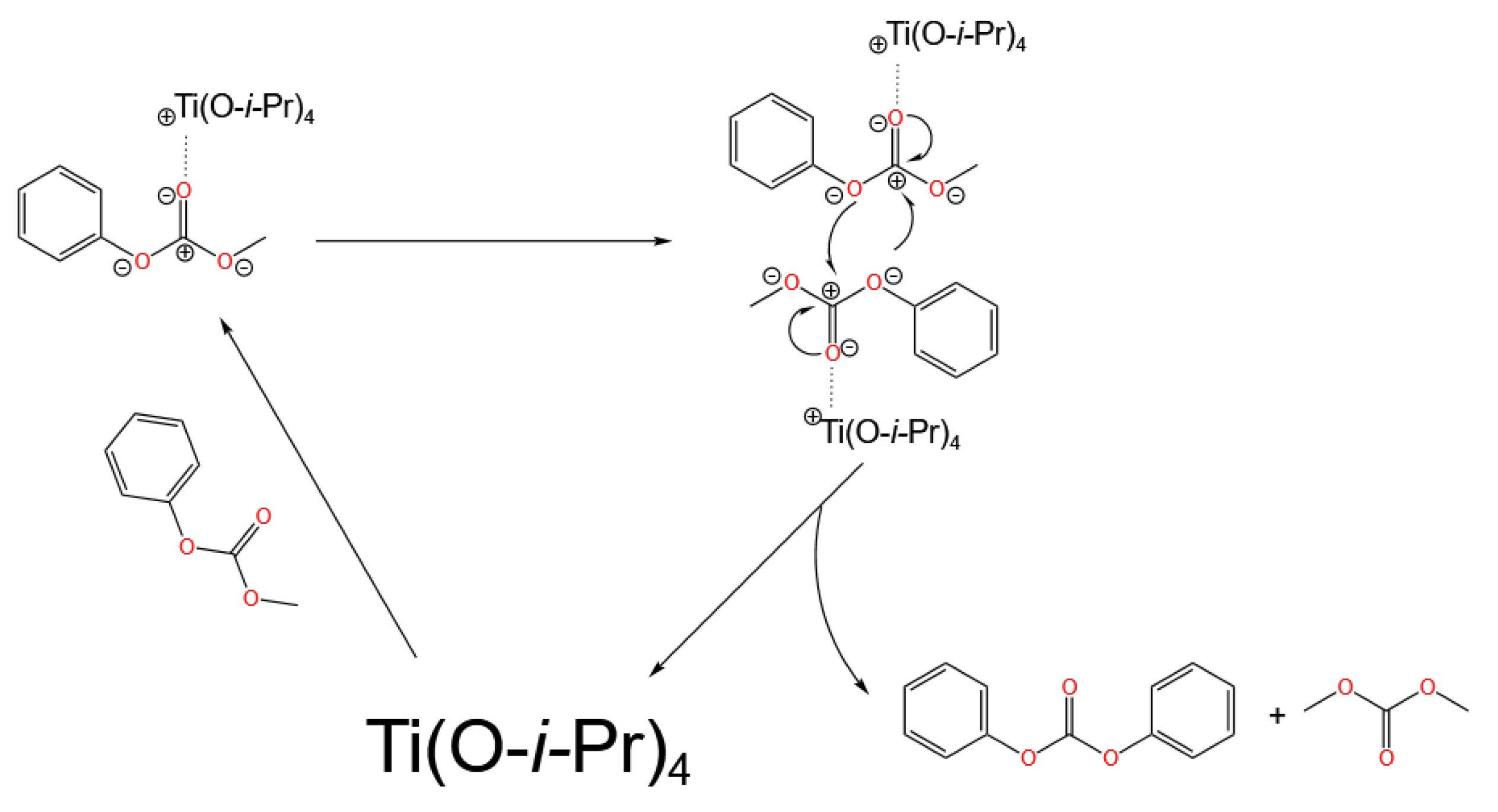
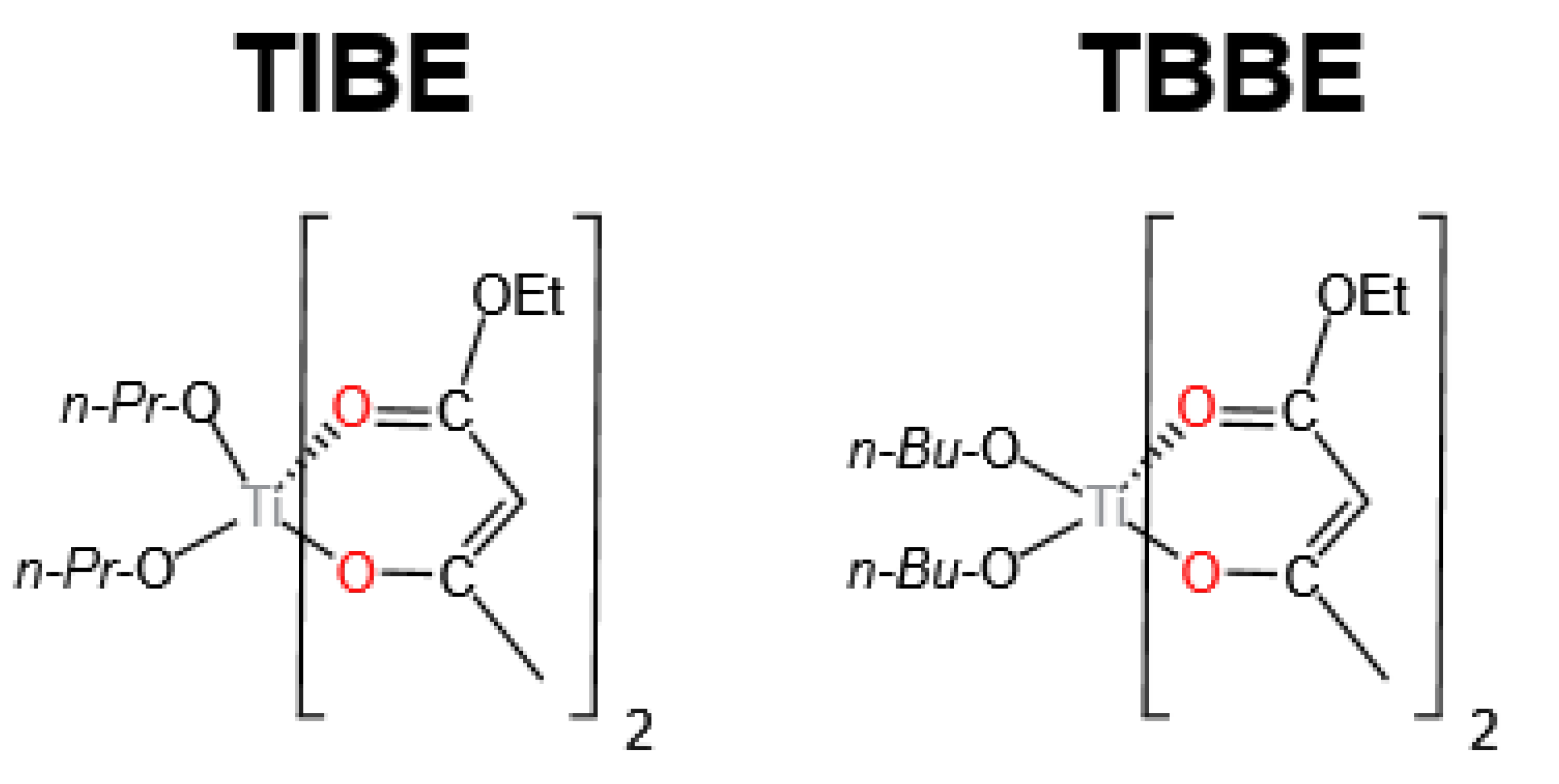
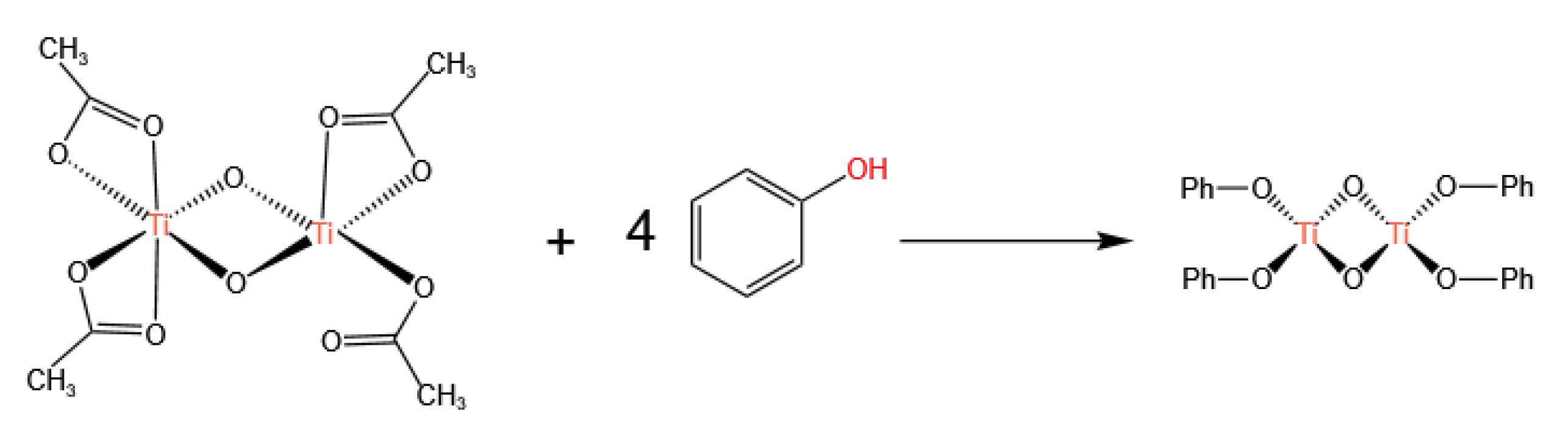
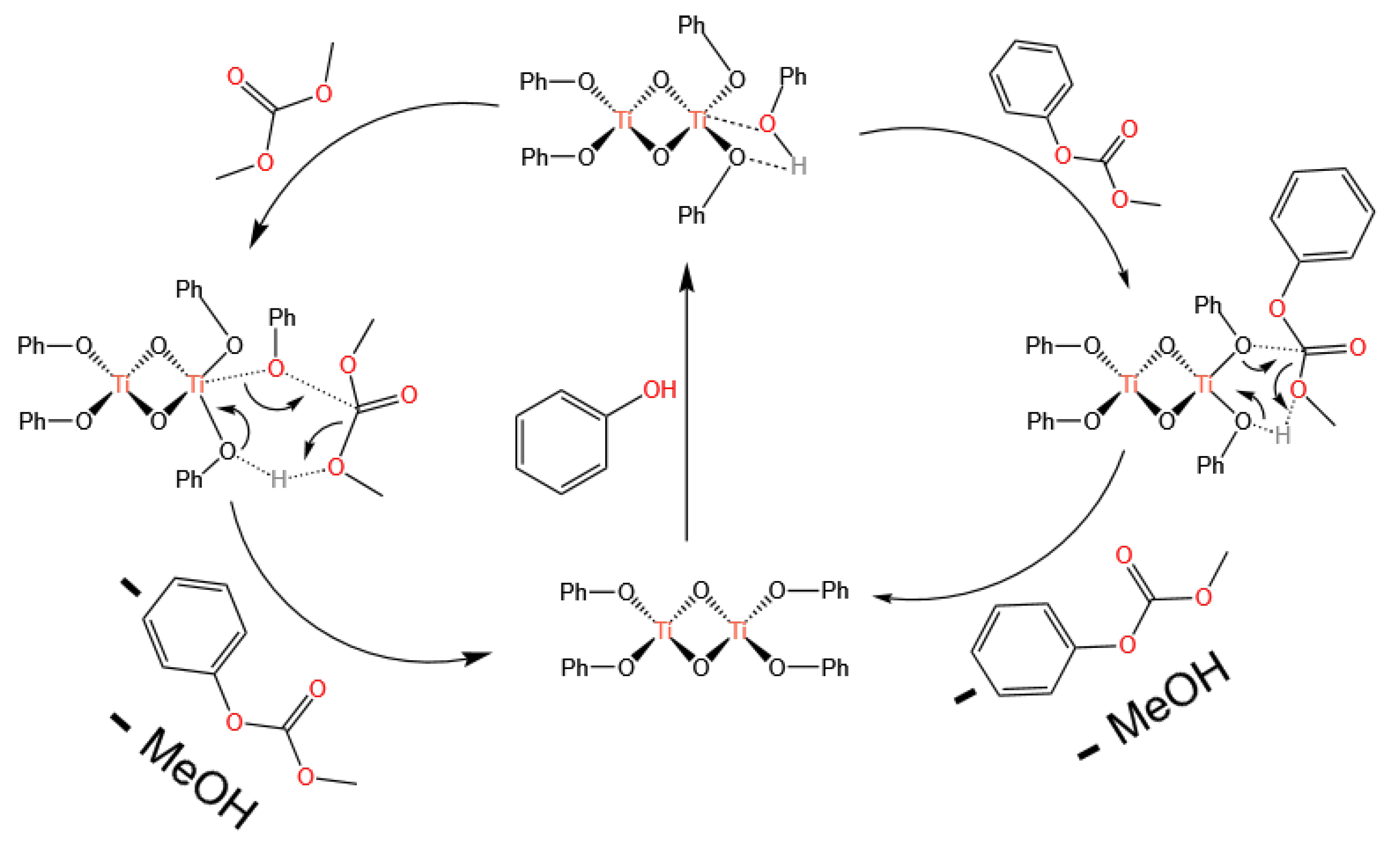
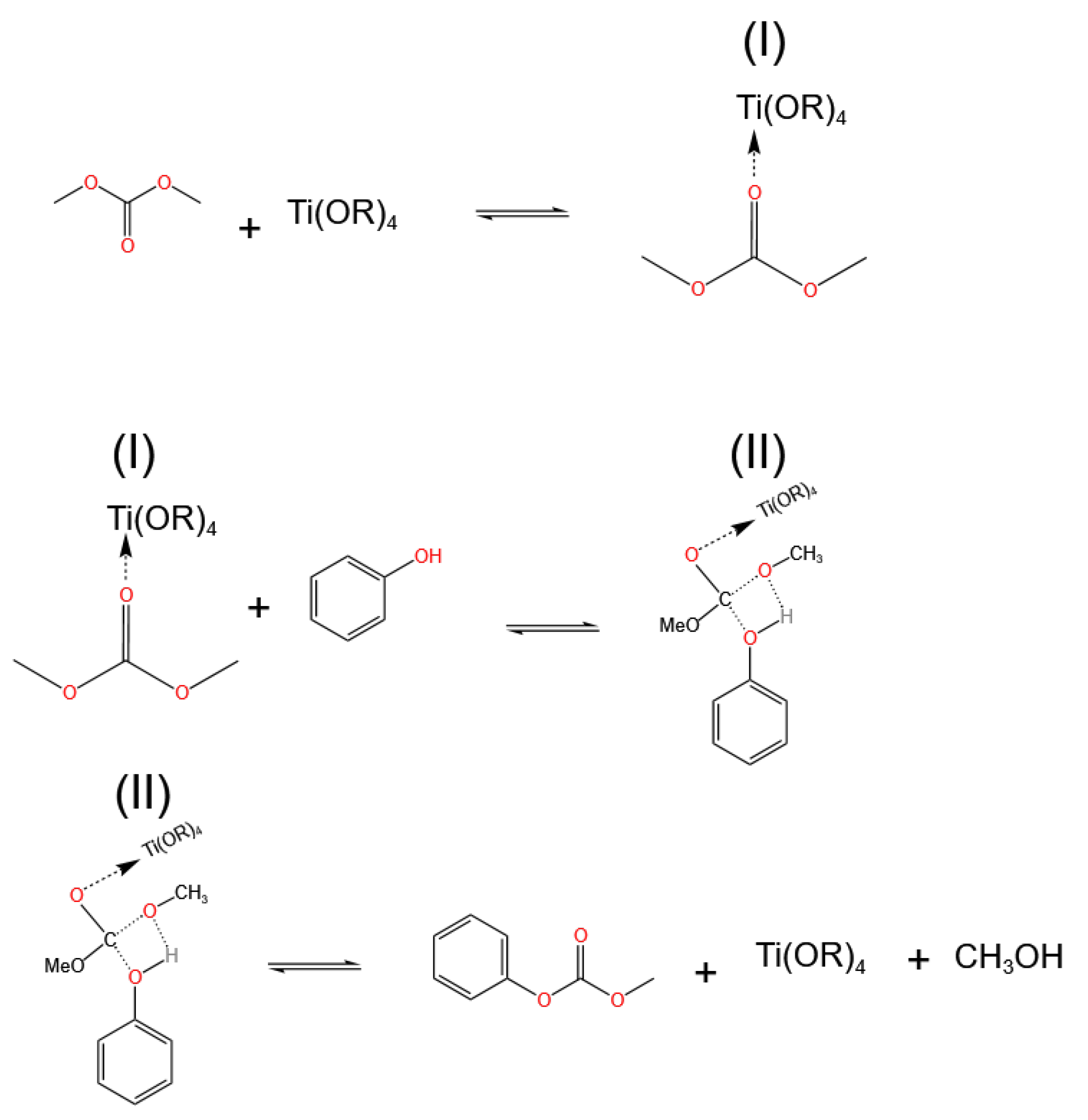
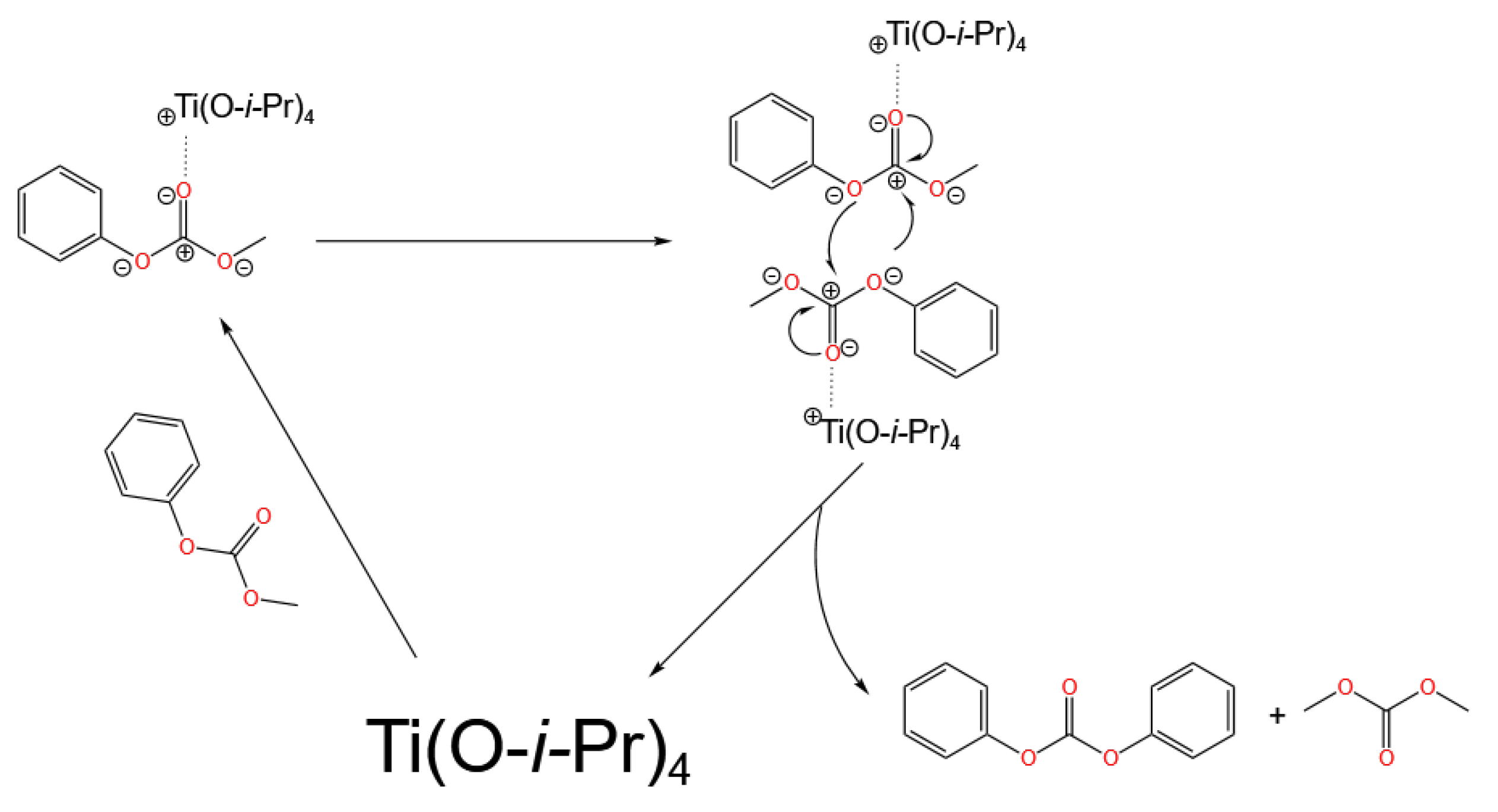
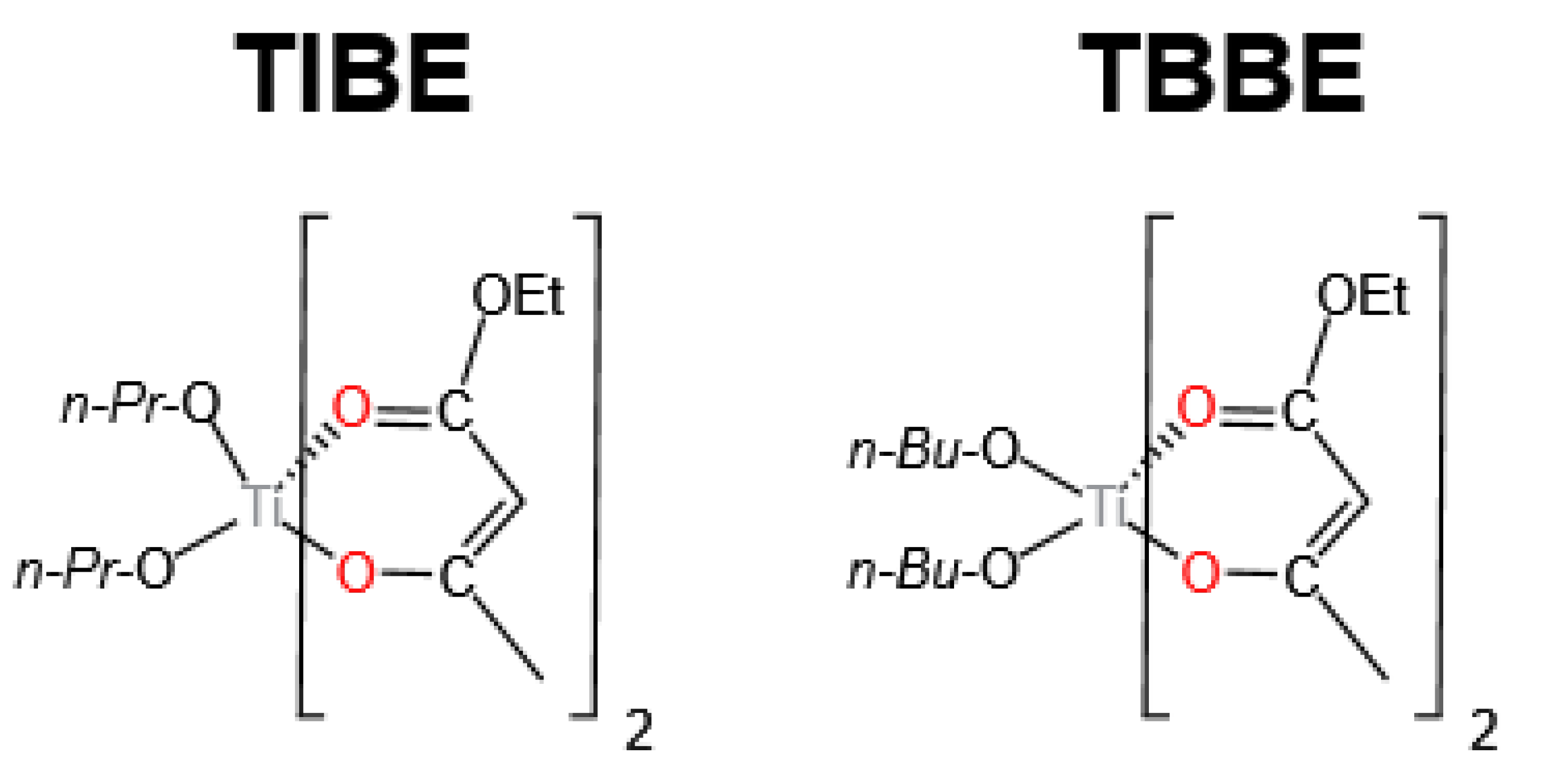
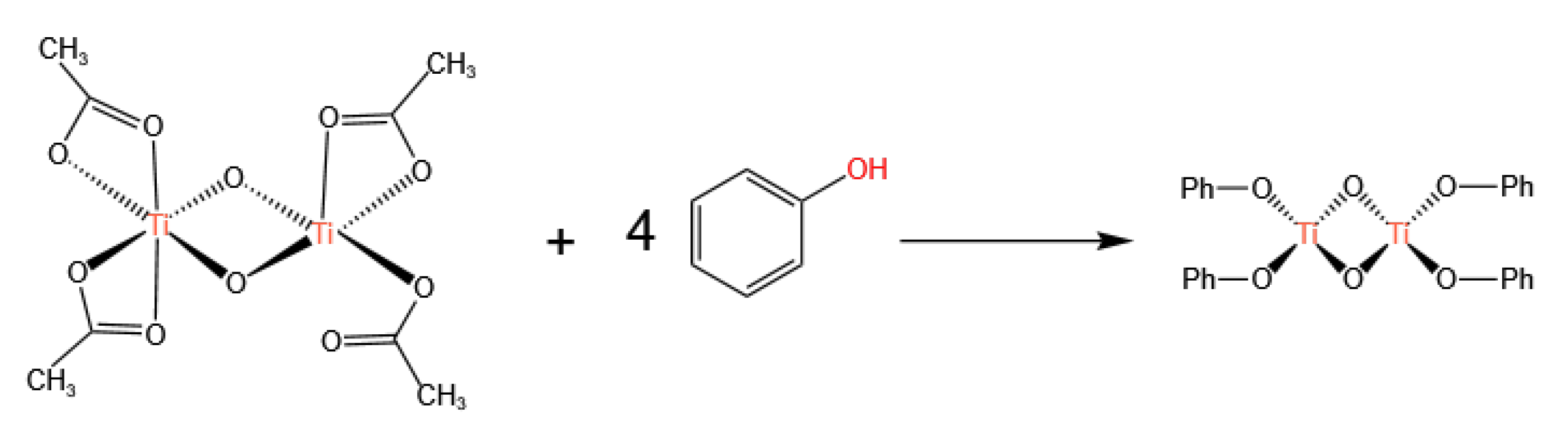
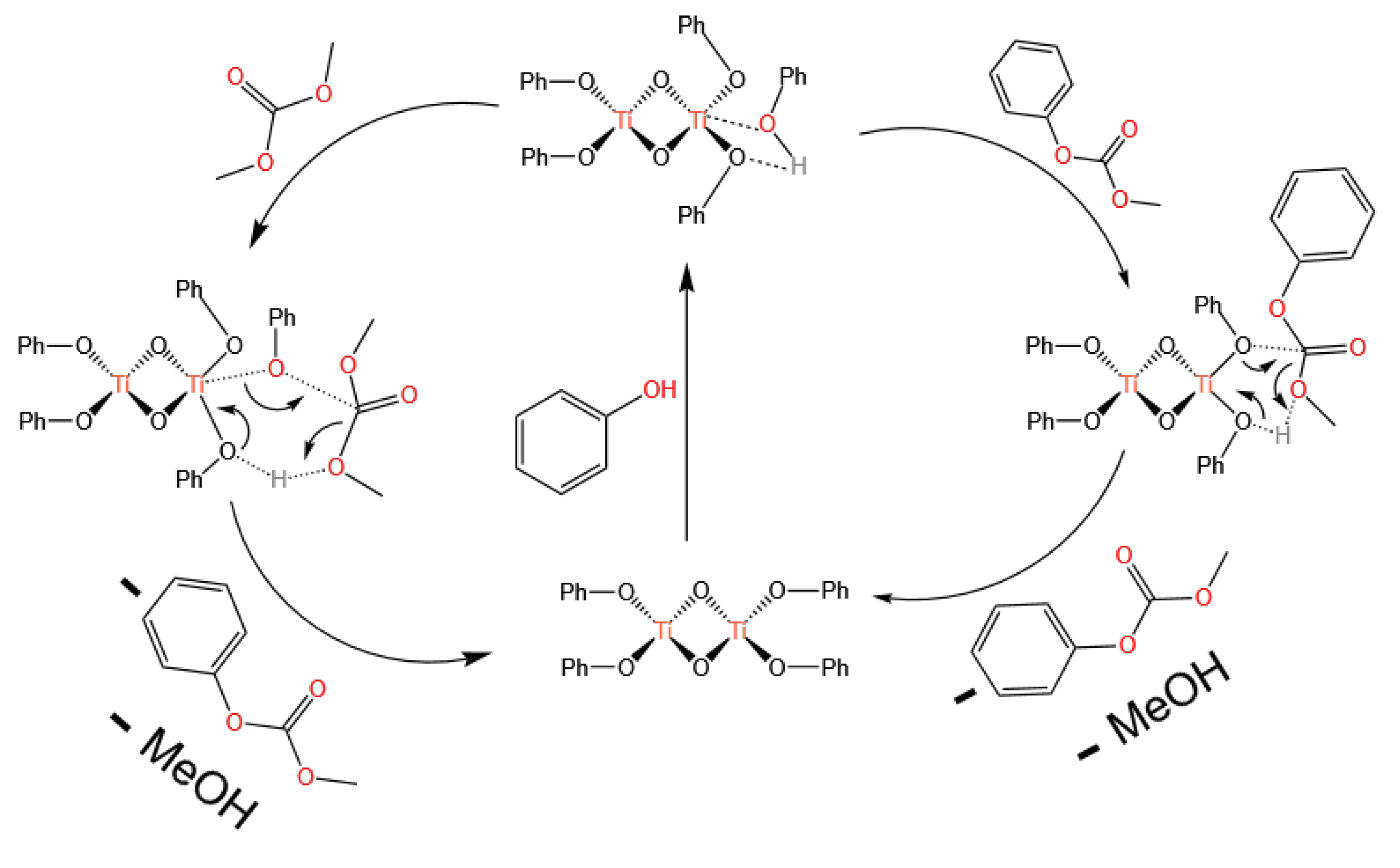
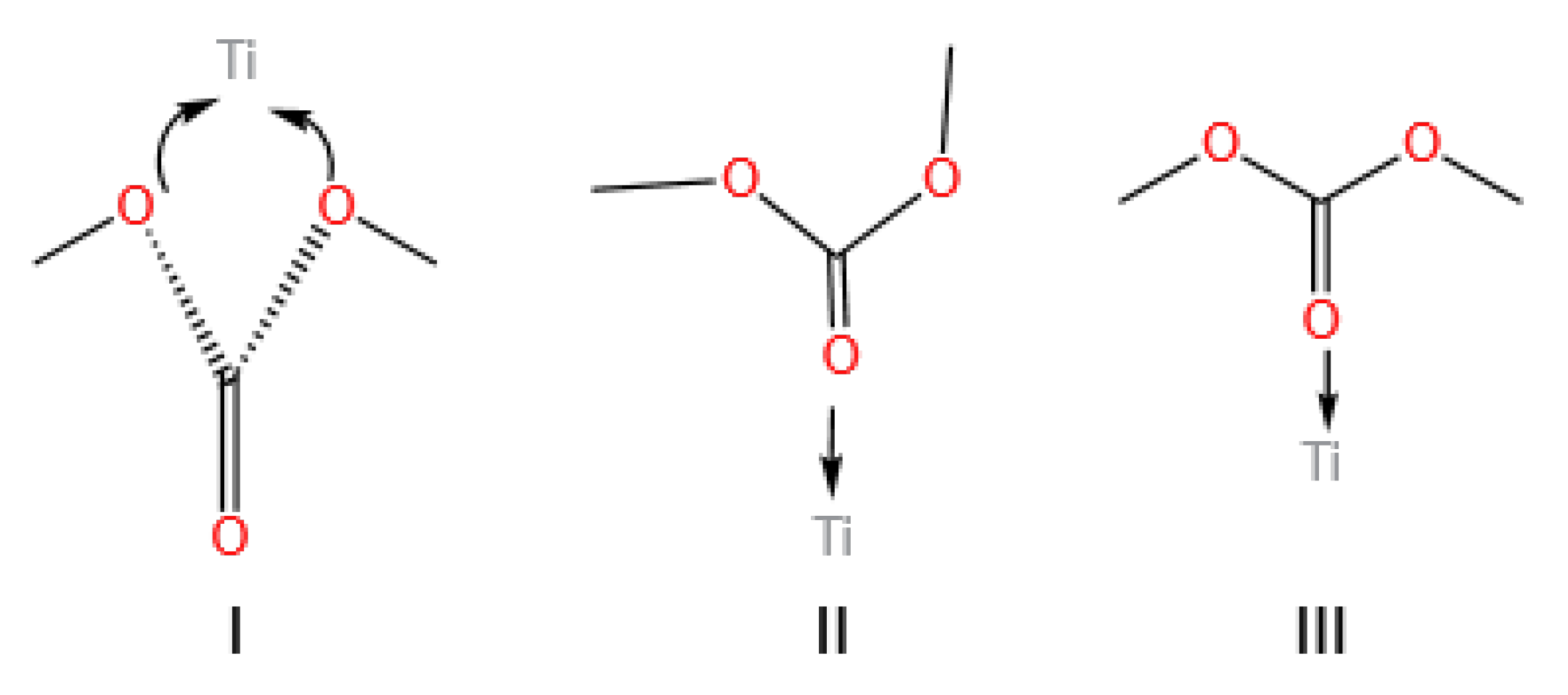
3.1. Ti-Based Catalysts
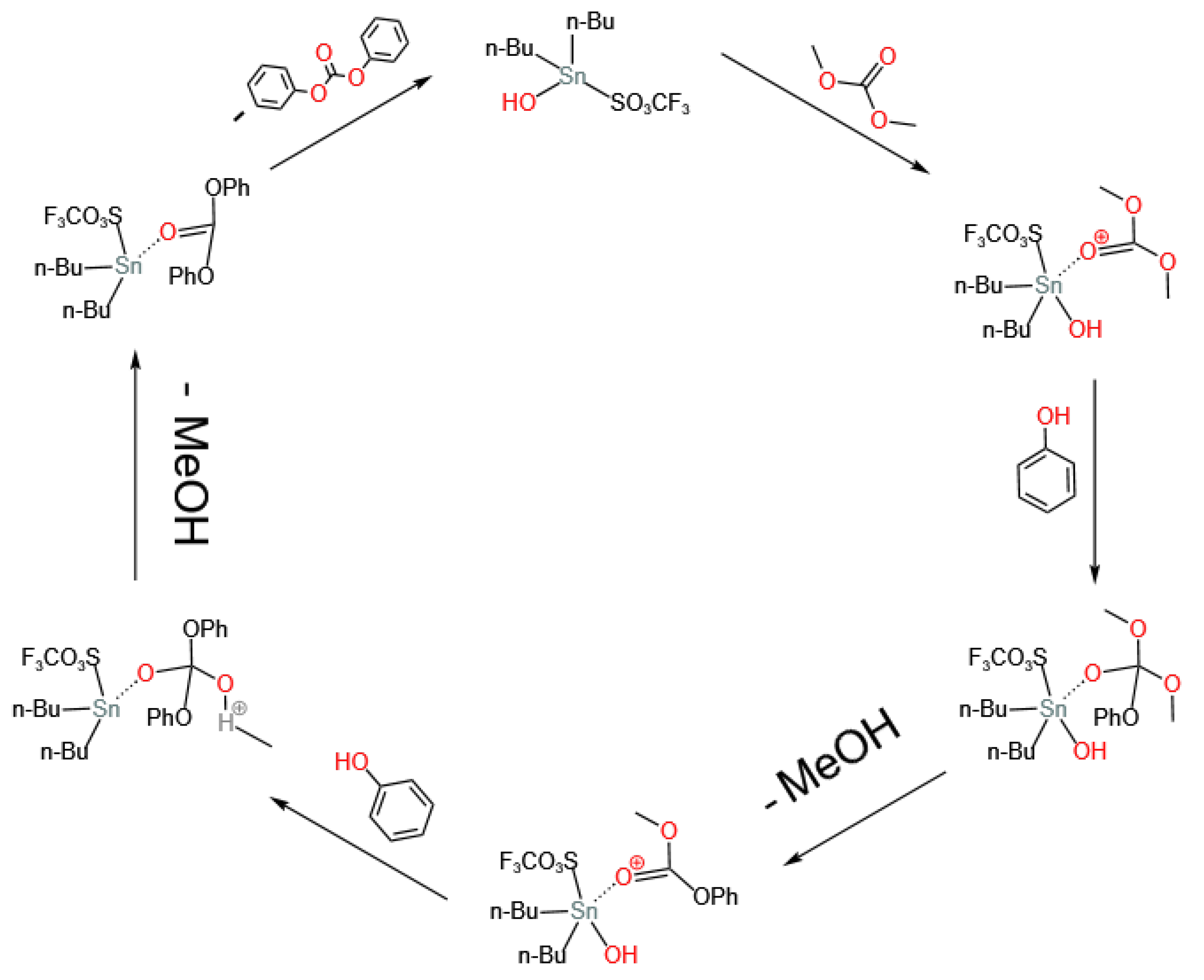
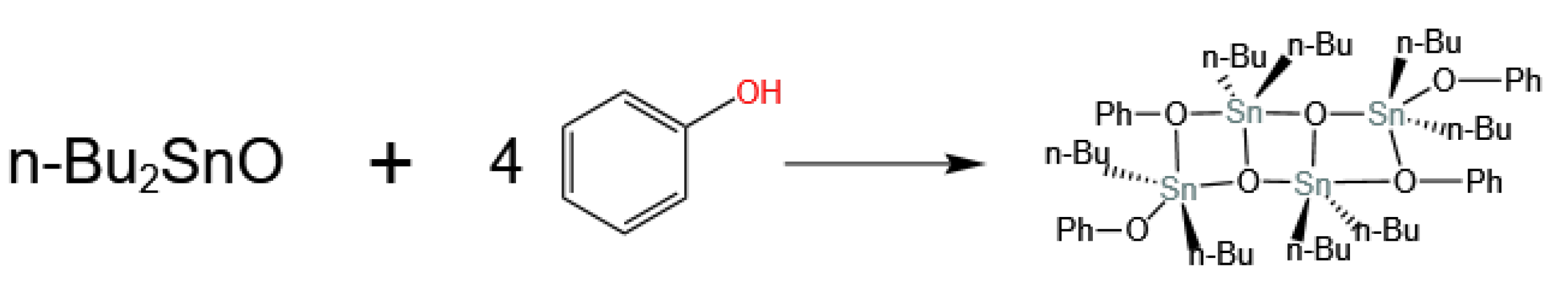
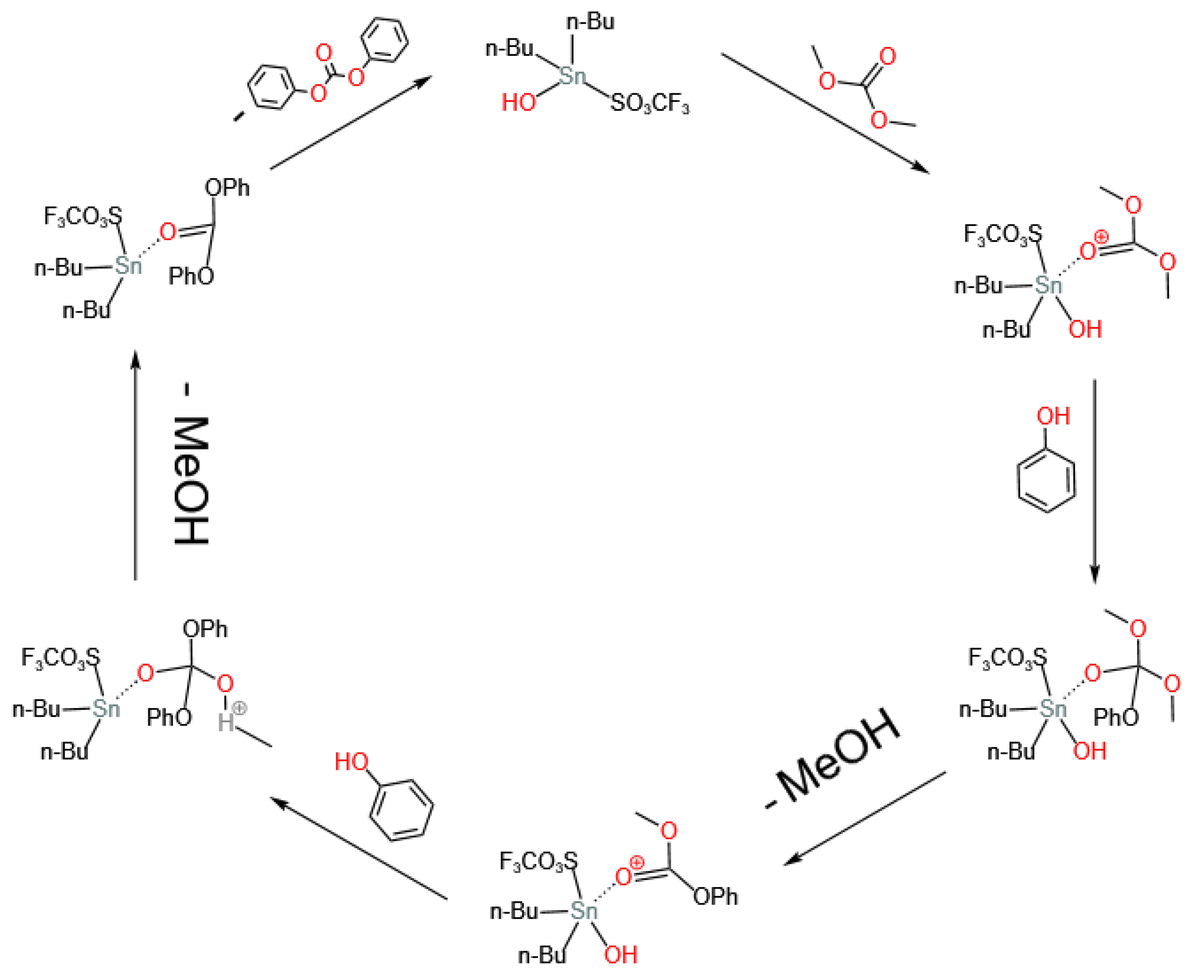
3.2. Sn-Based Catalysts
3.3. Lead-Based Catalysts
3.4. Rare-Earth-Metal-Based Catalysts
4. Heterogeneous Catalysts for Transesterification
4.1. Oxides
4.1.1. TiO2
4.1.2. Lead Oxides
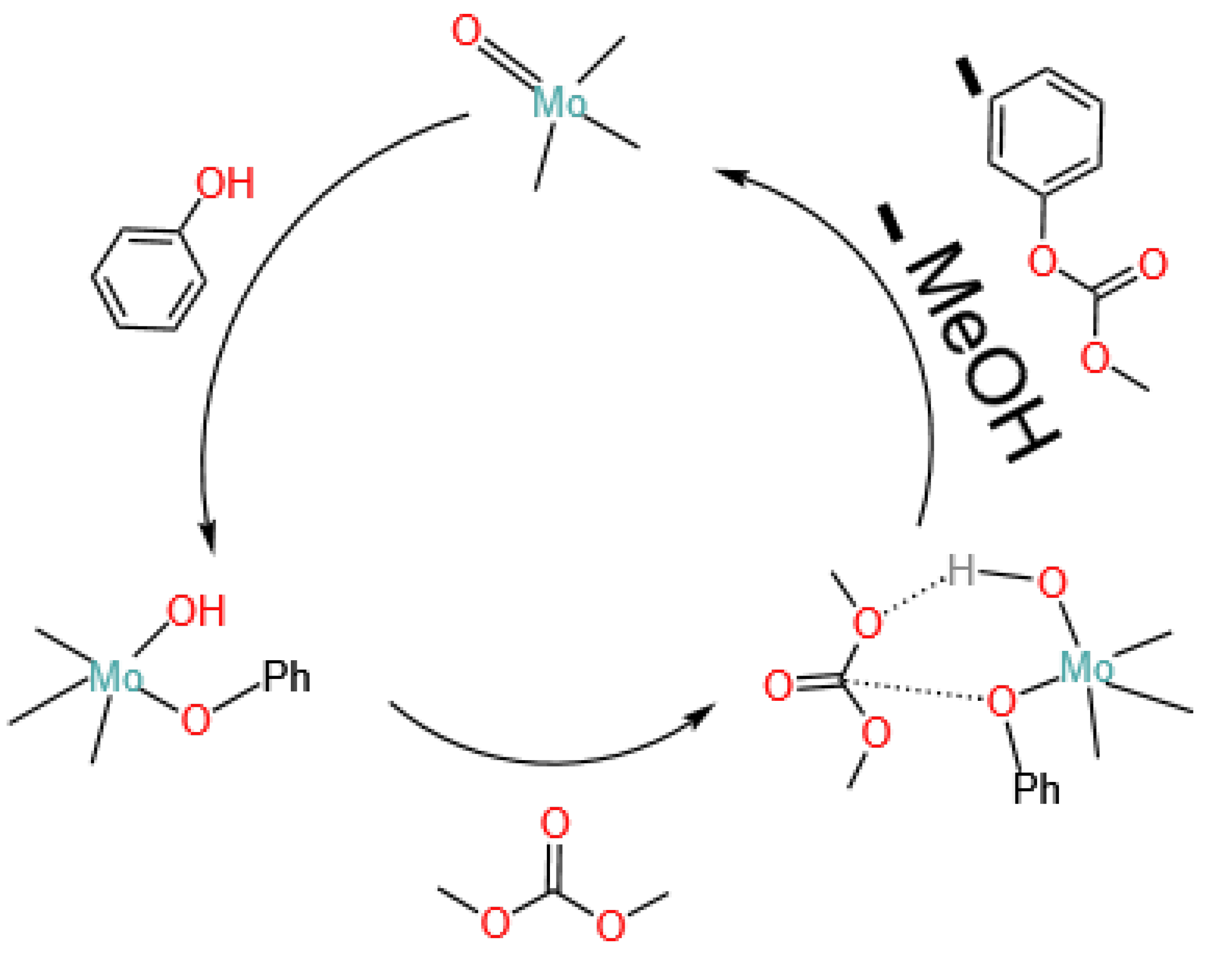
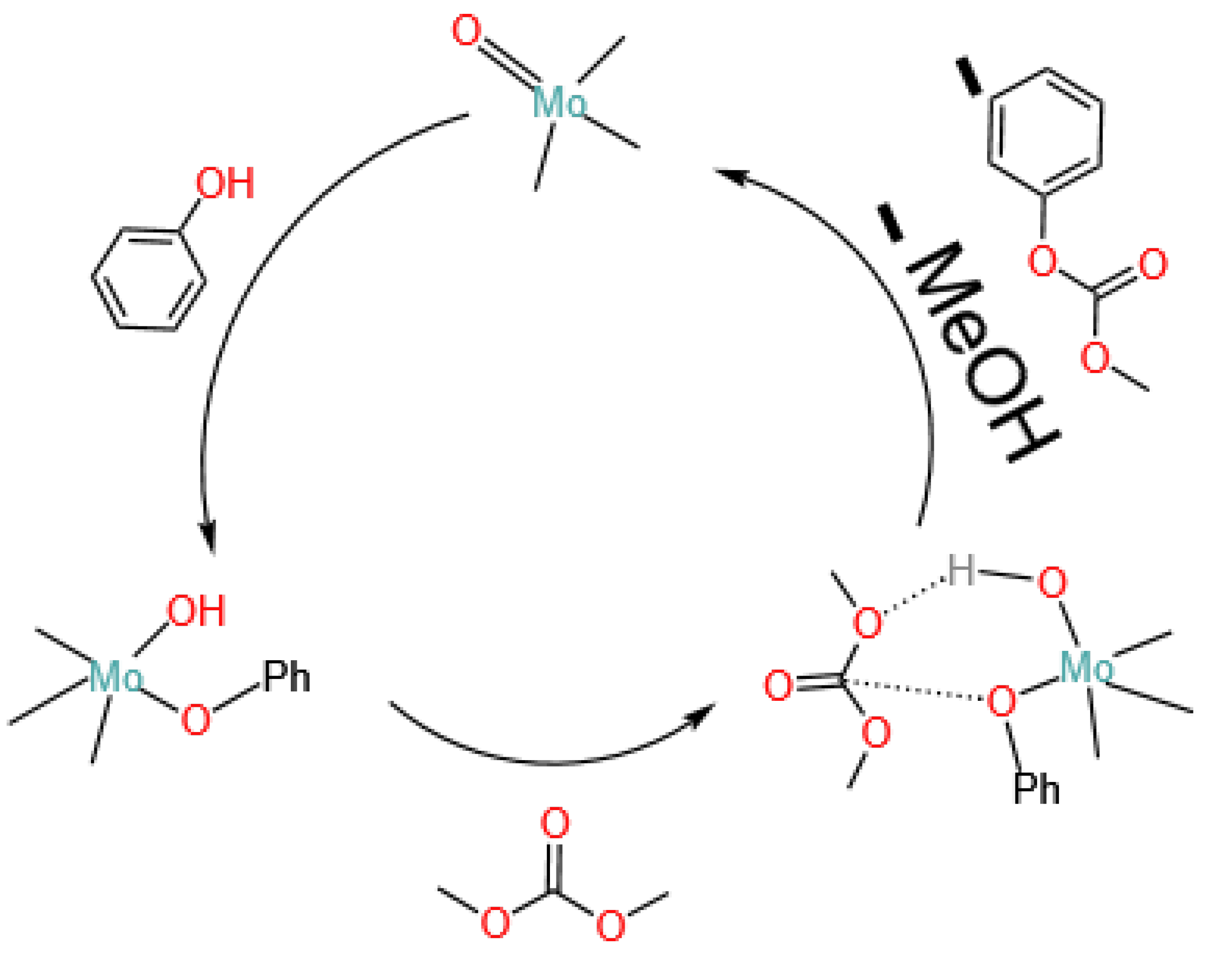
4.1.3. Molybdenum Oxides
4.1.4. Other Oxides or Hydroxides
4.2. Molecular Sieves
4.3. Hydrotalcite
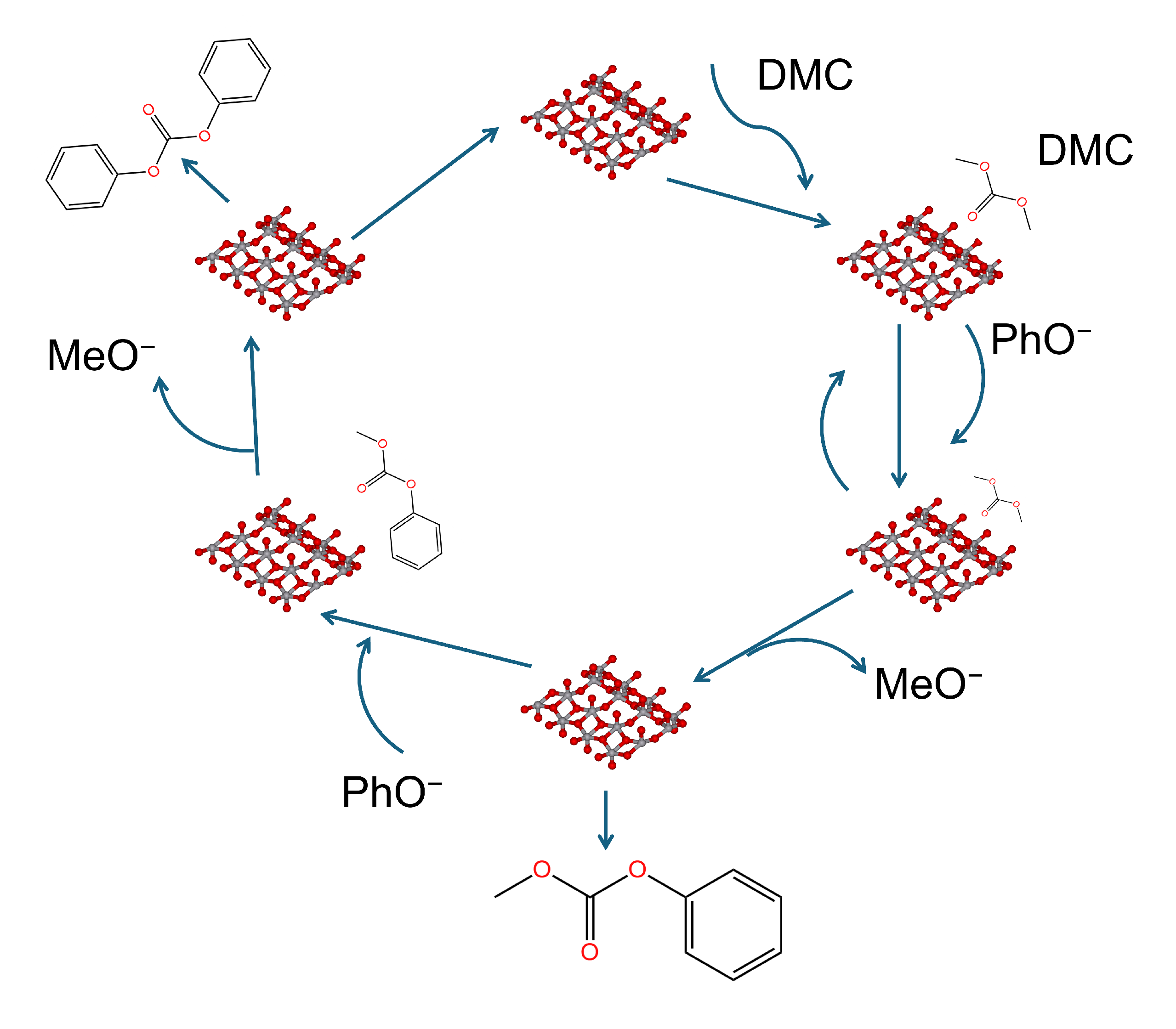
4.4. Heteropolyacids and Heteropolyacid Salts
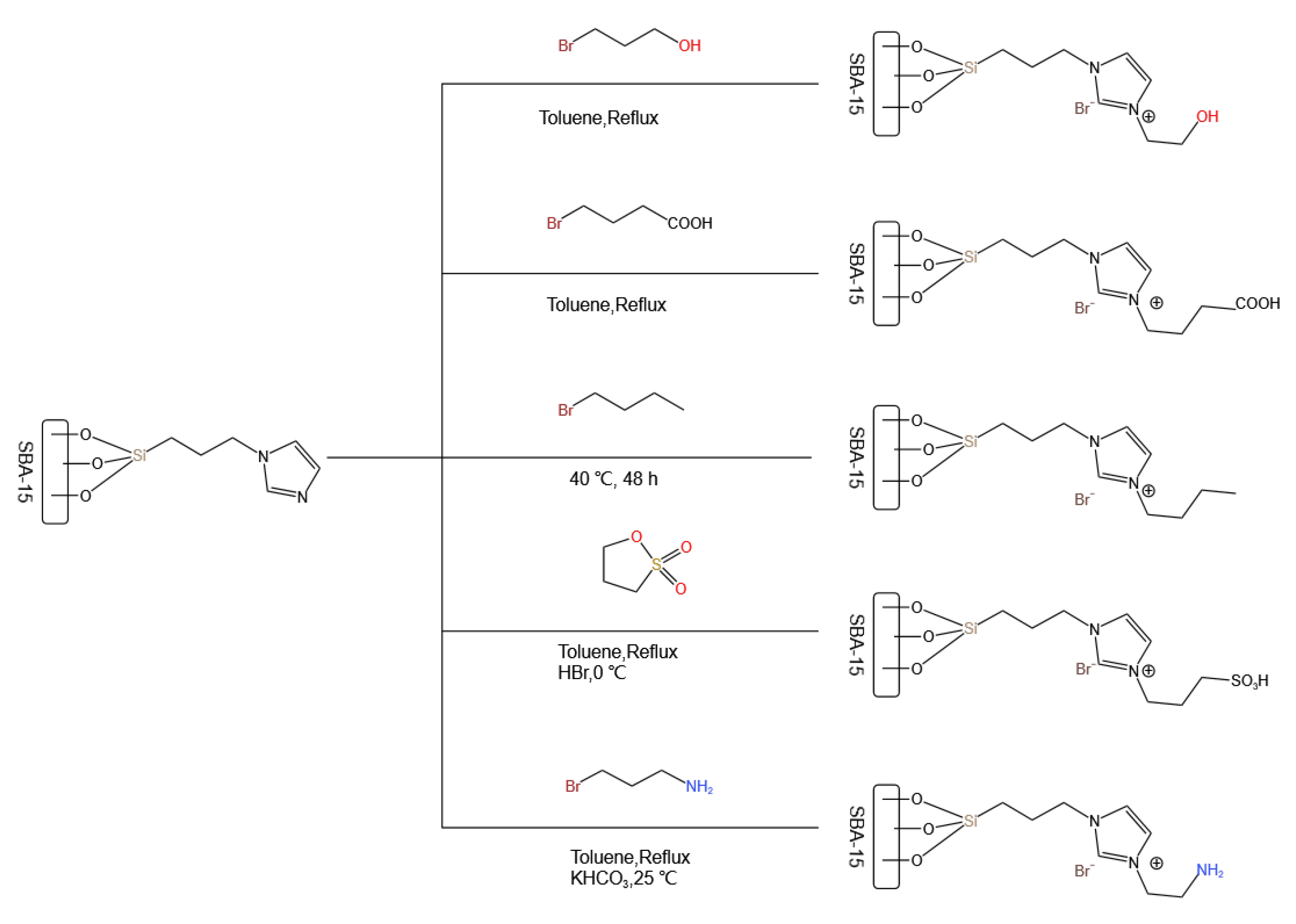
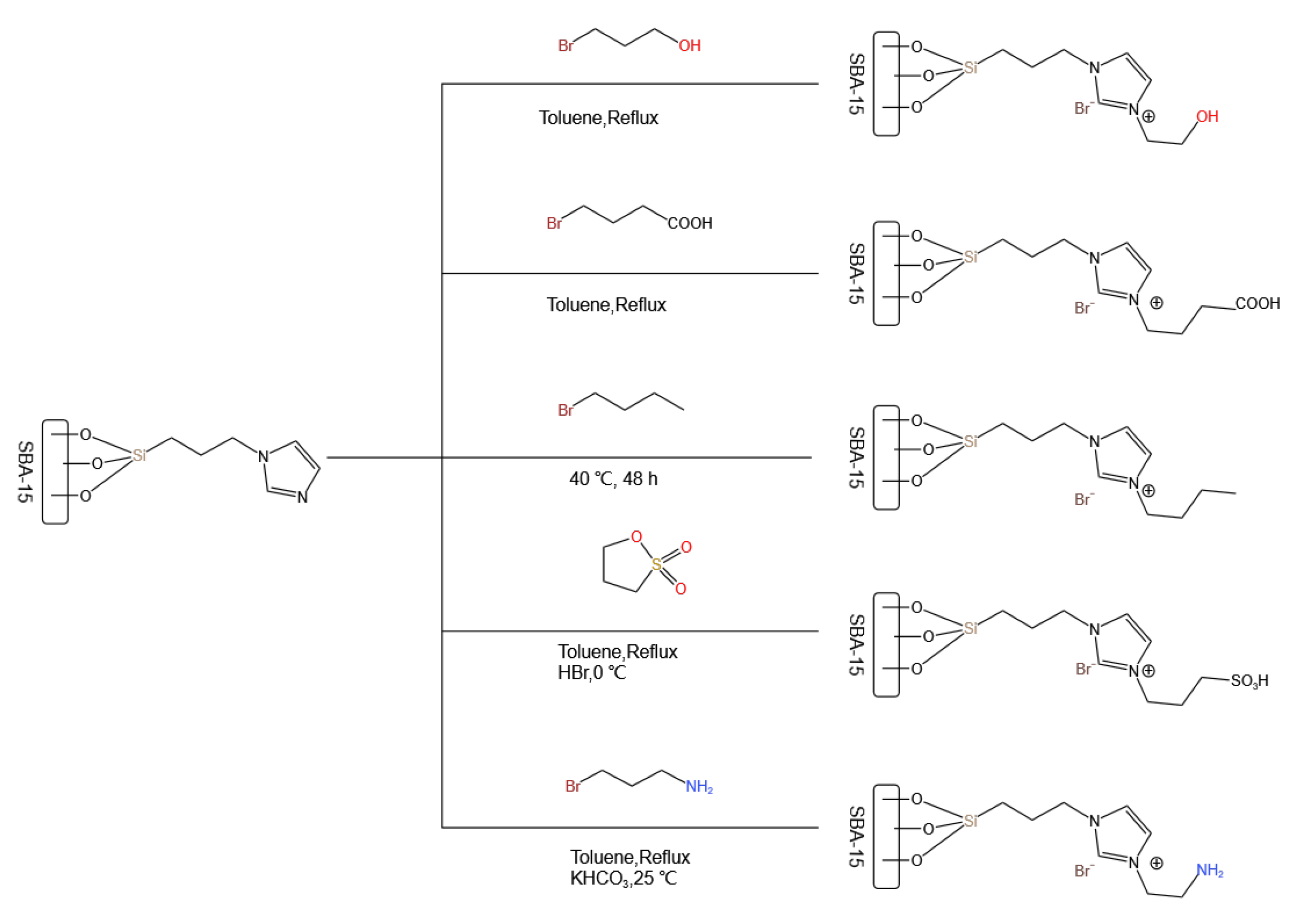
5. Bridge between Homogeneous and Heterogeneous Catalysis: Ionic Liquids
6. Potential Alternative Routes: Dimethyl Oxalate and Phenyl Acetate as Raw Materials
7. Conclusions and Prospects
- Homogeneous catalysts, represented by Ti-based, Sn-based, and Pb-based catalysts, follow the Lewis acid catalysis mechanism. The catalytic centers rely on the Lewis acidity of the metal center: Lewis acid sites first activate the carbonyl group of DMC, followed by the nucleophilic addition of phenol, ultimately generating DPC. Currently, Pb-based catalysts are no longer used due to environmental issues, and Ti-based catalysts are the mainstream. However, Ti-based catalysts, mainly titanium alkoxides, are still limited by their poor stability.
- Heterogeneous catalysts include oxides, molecular sieves, hydrotalcites, and polyoxometalates/polyoxometalate salts, among others. Their design concept borrows from the catalysis of metal Lewis acid centers, with the basic idea being the heterogenization of homogeneous catalysts. Among them, TiO2-based catalysts are more frequently reported, but their activity and stability still need to be improved.
- New types of catalysts, represented by ionic liquids, combining the characteristics of homogeneous and heterogeneous catalysts, can act as homogeneous catalysts during the reaction and be separated from the catalytic system after the reaction, which is the direction of future basic research on catalysts. However, they are still limited by high preparation costs, complex preparation processes, and issues related to the loss of catalytically active components. Additionally, their catalytic reaction mechanism still needs to be further clarified.
- Currently, the transesterification reaction between dimethyl carbonate and phenol is still limited by thermodynamics, with a relatively small equilibrium constant. However, using dimethyl oxalate or phenyl acetate for transesterification can, to some extent, increase the equilibrium constant of the transesterification and alleviate the thermodynamic constraints.
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| DMC | Dimethyl carbonate |
| DPC | Diphenyl carbonate |
| MPC | Methyl phenyl carbonate |
| PhOH | Phenol |
| DMO | Dimethyl oxalate |
| PA | Phenyl acetate |
| DPO | Diphenyl oxalate |
References
- Freitag, D.; Fengler, G.; Morbitzer, L. Routes to new aromatic polycarbonates with special material properties. Angew. Chem. Int. Ed. Engl. 1991, 30, 1598–1610. [Google Scholar] [CrossRef]
- Domingo-Espin, M.; Puigoriol-Forcada, J.M.; Garcia-Granada, A.A.; Llumà, J.; Borros, S.; Reyes, G. Mechanical property characterization and simulation of fused deposition modeling Polycarbonate parts. Mater. Des. 2015, 83, 670–677. [Google Scholar] [CrossRef]
- Park, S.J.; Lee, J.E.; Lee, H.B.; Park, J.; Lee, N.K.; Son, Y.; Park, S.H. 3D printing of bio-based polycarbonate and its potential applications in ecofriendly indoor manufacturing. Addit. Manuf. 2020, 31, 100974. [Google Scholar] [CrossRef]
- Sai, T.; Ran, S.; Guo, Z.; Yan, H.; Zhang, Y.; Wang, H.; Song, P.; Fang, Z. Transparent, highly thermostable and flame retardant polycarbonate enabled by rod-like phosphorous-containing metal complex aggregates. Chem. Eng. J. 2021, 409, 128223. [Google Scholar] [CrossRef]
- Yu, W.; Maynard, E.; Chiaradia, V.; Arno, M.C.; Dove, A.P. Aliphatic polycarbonates from cyclic carbonate monomers and their application as biomaterials. Chem. Rev. 2021, 121, 10865–10907. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Jin, Z.; Chu, F.; Cai, W.; Zhu, Y.; Yu, B.; Song, L.; Hu, Y. High-performance flame-retardant polycarbonate composites: Mechanisms investigation and fire-safety evaluation systems establishment. Compos. Part Eng. 2022, 238, 109873. [Google Scholar] [CrossRef]
- Bian, Y.; Liu, Q.; Feng, Z.; Hua, J.; Xie, H.; Chen, S.; Cai, Y.; Yao, X.; Luo, S. High-speed penetration dynamics of polycarbonate. Int. J. Mech. Sci. 2022, 223, 107250. [Google Scholar] [CrossRef]
- Fromel, M.; Pester, C.W. Polycarbonate Surface Modification via Aqueous SI-PET-RAFT. Macromolecules 2022, 55, 4907–4915. [Google Scholar] [CrossRef]
- Mehrara, R.; Malekie, S.; Kotahi, S.M.S.; Kashian, S. Introducing a novel low energy gamma ray shield utilizing polycarbonate bismuth oxide composite. Sci. Rep. 2021, 11, 10614. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.B.; Joshi, U.A.; Lee, J.S. Making polycarbonates without employing phosgene: An overview on catalytic chemistry of intermediate and precursor syntheses for polycarbonate. Ind. Eng. Chem. Res. 2004, 43, 1897–1914. [Google Scholar] [CrossRef]
- Darensbourg, D.J. Making plastics from carbon dioxide: Salen metal complexes as catalysts for the production of polycarbonates from epoxides and CO2. Chem. Rev. 2007, 107, 2388–2410. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Worch, J.C.; Dove, A.P.; Coulembier, O. Update and challenges in carbon dioxide-based polycarbonate synthesis. ChemSusChem 2020, 13, 469–487. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Ma, X.; Wang, S. Phosgene-free approaches to catalytic synthesis of diphenyl carbonate and its intermediates. Appl. Catal. Gen. 2007, 316, 1–21. [Google Scholar] [CrossRef]
- Song, Z.; Jin, W.; Gao, F.; Jin, X. Recent advances in catalyst development for transesterification of dialkyl carbonates with phenol. Ind. Eng. Chem. Res. 2020, 59, 20630–20645. [Google Scholar] [CrossRef]
- Lee, H.Y.; Novita, F.J.; Weng, K.C. Hybrid heat-integrated design and control for a diphenyl carbonate reactive distillation process. Chem. Eng. Process.-Process. Intensif. 2021, 162, 108344. [Google Scholar] [CrossRef]
- Otera, J. Transesterification. Chem. Rev. 1993, 93, 1449–1470. [Google Scholar] [CrossRef]
- Shen, W.; Ge, Q.; Gu, K.; Nie, Y.; Jiao, L.; Zhu, Z.; Fang, Y. Stable organic titanium catalysts and reactive distillation used for the transesterification of dimethyl carbonate with phenol. Chem. Eng. Technol. 2020, 43, 2359–2364. [Google Scholar] [CrossRef]
- Yin, X.; Zeng, Y.; Yao, J.; Zhang, H.; Deng, Z.; Wang, G. Kinetic modeling of the transesterification reaction of dimethyl carbonate and phenol in the reactive distillation reactor. Ind. Eng. Chem. Res. 2014, 53, 19087–19093. [Google Scholar] [CrossRef]
- Aihua, X.; Minqing, Z.; Zhimin, H.; Jianping, Z. Thermodynamic analysis of synthesis of diphenyl carbonate by transesterification of dm ethyl carbonate and phenol. Chem. Eng. 2006, 34, 40–43. [Google Scholar]
- Mei, F.; Li, G. The thermodynamic properties and homogeneous catalysts for the synthesis of diphenyl carbonate by transesterification of dimethyl carbonate with phenol. Chin. J. Synth. Chem. 2003, 11, 320–326. [Google Scholar]
- Guo, X.; Shi, Y.; Zheng, H.; Cheng, J.; Zhao, X.; Yang, B. Thermodynamic study on diphenyl carbonate synthesis with different feedstocks. J. Chem. Eng. Chin. Univ. 2016, 30, 754–760. [Google Scholar]
- Haubrock, J.; Raspe, M.; Versteeg, G.F.; Kooijman, H.A.; Taylor, R.; Hogendoorn, J.A. Reaction from dimethyl carbonate to diphenyl carbonate. 1. experimental determination of the chemical equilibria. Ind. Eng. Chem. Res. 2008, 47, 9854–9861. [Google Scholar] [CrossRef]
- Sun, W.; Shao, J.; Xi, Z.; Zhao, L. Thermodynamics and kinetics of transesterification reactions to produce diphenyl carbonate from dimethyl carbonate catalyzed by tetrabutyl titanate and dibutyltin oxide. Can. J. Chem. Eng. 2017, 95, 353–358. [Google Scholar] [CrossRef]
- Bandsode, S.P.; Besta, C.S. Dynamic analysis and decentralised control system design for diphenyl carbonate reactive distillation process. Indian Chem. Eng. 2020, 64, 151–161. [Google Scholar] [CrossRef]
- Mofijur, M.; Siddiki, S.Y.A.; Shuvho, M.B.A.; Djavanroodi, F.; Fattah, I.R.; Ong, H.C.; Chowdhury, M.; Mahlia, T. Effect of nanocatalysts on the transesterification reaction of first, second and third generation biodiesel sources—A mini-review. Chemosphere 2021, 270, 128642. [Google Scholar] [CrossRef] [PubMed]
- Orege, J.I.; Oderinde, O.; Kifle, G.A.; Ibikunle, A.A.; Raheem, S.A.; Ejeromedoghene, O.; Okeke, E.S.; Olukowi, O.M.; Orege, O.B.; Fagbohun, E.O.; et al. Recent advances in heterogeneous catalysis for green biodiesel production by transesterification. Energy Convers. Manag. 2022, 258, 115406. [Google Scholar] [CrossRef]
- Nayab, R.; Imran, M.; Ramzan, M.; Tariq, M.; Taj, M.B.; Akhtar, M.N.; Iqbal, H.M. Sustainable biodiesel production via catalytic and non-catalytic transesterification of feedstock materials—A review. Fuel 2022, 328, 125254. [Google Scholar] [CrossRef]
- Maleki, B.; Ashraf Talesh, S.; Mansouri, M. Comparison of catalysts types performance in the generation of sustainable biodiesel via transesterification of various oil sources: A review study. Mater. Today Sustain. 2022, 18, 100157. [Google Scholar] [CrossRef]
- Gao, J.; Yao, J.; Mei, H.; Wang, G. Transesterification of dimethyl carbonate and phenol with titanate catalysts. Chin. J. Catal. 2001, 22, 406–407. [Google Scholar]
- Niu, H.; Yao, J.; Wang, Y.; Wang, G. Cp2TiCl2 used as a catalyst for the transesterification between dimethyl carbonate and phenol to diphenyl carbonate. J. Mol. Catal. Chem. 2005, 235, 240–243. [Google Scholar] [CrossRef]
- Hongying, N.; Haiming, G.; Jie, Y.; Yue, W.; Gongying, W. Transesterification of dimethyl carbonate and phenol to diphenyl carbonate catalyzed by titanocene complexes. Acta Chim. Sin. 2006, 64, 1269. [Google Scholar]
- Rongzhi, T.; Songlin, W.; Yuanzhuo, Z.; Tong, C.; Gongying, W. Catalytic property of titanyl acetate in the transesterification reaction of dimethyl carbonate and phenol. Chem. J. Chin. Univ. 2014, 35, 2418–2424. [Google Scholar]
- Wang, S.; Chen, T.; Wang, G.; Cui, C.; Niu, H.; Li, C. Influence of coordination groups on the catalytic performances of organo-titanium compounds for disproportionation of methyl phenyl carbonate to synthesize diphenyl carbonate. Appl. Catal. Gen. 2017, 540, 1–6. [Google Scholar] [CrossRef]
- Lee, H.; Joon Kim, S.; Sung Ahn, B.; Koo Lee, W.; Sik Kim, H. Role of sulfonic acids in the Sn-catalyzed transesterification of dimethyl carbonate with phenol. Catal. Today 2003, 87, 139–144. [Google Scholar] [CrossRef]
- Du, Z.; Chen, S.; Shen, C.; Zhou, B.; Huang, L.; Wang, G.; Wu, Y. Effect of copper compounds on the synthesis of diphenyl carbonate from transesterification catalyzed by n-Bu2SnO. Adv. Mater. Res. 2012, 396–398, 759–763. [Google Scholar]
- Du, Z.; Kang, W.; Cheng, T.; Yao, J.; Wang, G. Novel catalytic systems containing n-BuSn(O)OH for the transesterification of dimethyl carbonate and phenol. J. Mol. Catal. Chem. 2006, 246, 200–205. [Google Scholar] [CrossRef]
- Shaikh, A.A.G.; Sivaram, S. Dialkyl and diaryl carbonates by carbonate interchange reaction with dimethyl carbonate. Ind. Eng. Chem. Res. 1992, 31, 1167–1170. [Google Scholar] [CrossRef]
- Jie, Y.; Junjie, G.; Yi, Z.; Yue, W.; Gongying, W. Influences of catalysts on the transesterification of dimethyl carbonate and phenol. Chin. J. Appl. Chem. 2003, 20, 898–902. [Google Scholar]
- Yue, W.; Jie, Y.; Yi, Z.; Gongying, W. Mechanism study of di-n-butyltin oxide catalyzed transesterificationof dimethyl carbonate with phenol. Acta Chim. Sin. 2005, 63, 603. [Google Scholar]
- Lee, H.; Yong Bae, J.; Kwon, O.S.; Joon Kim, S.; Deuk Lee, S.; Sik Kim, H. Sulfonate-bonded tin complexes for the production of diphenyl carbonate. J. Organomet. Chem. 2004, 689, 1816–1820. [Google Scholar] [CrossRef]
- Fukuoka, S.; Fukawa, I.; Tojo, M.; Oonishi, K.; Hachiya, H.; Aminaka, M.; Hasegawa, K.; Komiya, K. A novel non-phosgene process for polycarbonate production from CO2: Green and sustainable chemistry in practice. Catal. Surv. Asia 2010, 14, 146–163. [Google Scholar] [CrossRef]
- Fuming, M.; Guangxing, L.; Jin, N.; Huibi, X. A novel catalyst for transesterification of dimethyl carbonate with phenol to diphenyl carbonate: Samarium trifluoromethanesulfonate. J. Mol. Catal. Chem. 2002, 184, 465–468. [Google Scholar] [CrossRef]
- Niu, H.; Guo, H.; Yao, J.; Wang, Y.; Wang, G. Transesterification of dimethyl carbonate and phenol to diphenyl carbonate catalyzed by samarium diiodide. J. Mol. Catal. Chem. 2006, 259, 292–295. [Google Scholar] [CrossRef]
- Gao, Y.; Li, Z.; Su, K.; Cheng, B. Excellent performance of TiO2 (B) nanotubes in selective transesterification of DMC with phenol derivatives. Chem. Eng. J. 2016, 301, 12–18. [Google Scholar] [CrossRef]
- Kim, W.B.; Lee, J.S. Gas phase transesterification of dimethyl carbonate and phenol over supported titanium dioxide. J. Catal. 1999, 185, 307–313. [Google Scholar] [CrossRef]
- Qu, Y.; Wang, S.; Chen, T.; Wang, G. Zn-promoted synthesis of diphenyl carbonate via transesterification over Ti–Zn double oxide catalyst. Res. Chem. Intermed. 2017, 43, 2725–2735. [Google Scholar] [CrossRef]
- Xin, G.; Bijing, L.; Jing, H.; Tong, C.; Gongying, W.; Xuteng, H. Effect of the surfactant on the catalytic activity of TiO2/CNT for transesterification between dimethyl carbonate and phenol. Acta Chim. Sin. 2011, 69, 2328. [Google Scholar]
- Qu, Y.; Yang, H.; Wang, S.; Chen, T.; Wang, G. High selectivity to diphenyl carbonate synthesized via transesterification between dimethyl carbonate and phenol with C60-doped TiO2. Chem. Res. Chin. Univ. 2017, 33, 804–810. [Google Scholar] [CrossRef]
- Yang, H.; Xiao, Z.; Qu, Y.; Chen, T.; Chen, Y.; Wang, G. The role of RGO in TiO2–RGO composites for the transesterification of dimethyl carbonate with phenol to diphenyl carbonate. Res. Chem. Intermed. 2018, 44, 799–812. [Google Scholar] [CrossRef]
- Tang, R.; Chen, T.; Chen, Y.; Zhang, Y.; Wang, G. Core-shell TiO2@SiO2 catalyst for transesterification of dimethyl carbonate and phenol to diphenyl carbonate. Chin. J. Catal. 2014, 35, 457–461. [Google Scholar] [CrossRef]
- Kim, W.B.; Lee, J.S. A new process for the synthesis of diphenyl carbonate from dimethyl carbonate and phenol over heterogeneous catalysts. Catal. Lett. 1999, 59, 83–88. [Google Scholar] [CrossRef]
- Weiqing, Z.; Xinqiang, Z.; Wang, Y.; Zhang, J. Synthesis of diphenyl carbonate by transesterification over lead and zinc double oxide catalyst. Appl. Catal. Gen. 2004, 260, 19–24. [Google Scholar] [CrossRef]
- Cao, M.; Meng, Y.; Lu, Y. Synthesis of diphenyl carbonate from dimethyl carbonate and phenol using O2-promoted PbO/MgO catalysts. Catal. Commun. 2005, 6, 802–807. [Google Scholar] [CrossRef]
- Wang, S.; Li, C.; Xiao, Z.; Chen, T.; Wang, G. Highly efficient and stable PbO–ZrO2 catalyst for the disproportionation of methyl phenyl carbonate to synthesize diphenyl carbonate. J. Mol. Catal. Chem. 2016, 420, 26–33. [Google Scholar] [CrossRef]
- Wang, S.; Jiang, N.; Liang, L.; Niu, H.; Chen, T.; Wang, G. A facile route to prepare PbZr nanocomposite catalysts for the efficient synthesis of diphenyl carbonate. Catal. Lett. 2021, 151, 3250–3260. [Google Scholar] [CrossRef]
- Hao, Y.; Wangming, H.; Lvming, S. Transesterification of dimethyl carbonate and phenol over PbO-Yb2O3 catalyst. J. Chem. Eng. Chin. Univ. 2007, 21, 146–149. [Google Scholar]
- Wang, S.; Niu, H.; Wang, J.; Chen, T.; Wang, G.; Zhang, J. Highly effective transformation of methyl phenyl carbonate to diphenyl carbonate with recyclable Pb nanocatalyst. RSC Adv. 2019, 9, 20415–20423. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yang, G.; Li, Y.; Zheng, H.; Cui, L. PbTiO3 catalyst for transesterification of dimethyl carbonate with phenol. J. Taiyuan Univ. Technol. 2010, 41, 723–727. [Google Scholar]
- Li, Z.; Wang, Y.; Ding, X.; Zhao, X. Investigation on the deactivation cause of lead-zinc double oxide for the synthesis of diphenyl carbonate by transesterification. J. Nat. Gas Chem. 2009, 18, 104–109. [Google Scholar] [CrossRef]
- Wang, S.; Niu, H.; Guo, M.; Wang, J.; Chen, T.; Wang, G. Effect of zirconia polymorph on the synthesis of diphenyl carbonate over supported lead catalysts. Mol. Catal. 2019, 468, 117–124. [Google Scholar] [CrossRef]
- Fu, Z.H.; Ono, Y. Two-step synthesis of diphenyl carbonate from dimethyl carbonate and phenol using MoO3/SiO2 catalysts. J. Mol. Catal. Chem. 1997, 118, 293–299. [Google Scholar] [CrossRef]
- Tong, D.; Chen, T.; Ma, F.; Kang, T.; Lei, Y.; Hu, J.; Wang, Y.; Wang, G. Transesterification of dimethyl carbonate with phenol over a bimetallic molybdenum and copper catalyst. React. Kinet. Catal. Lett. 2008, 94, 121–129. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Y.; Chen, T.; Wang, G. Preparation and catalytic property of MoO3/SiO2 for disproportionation of methyl phenyl carbonate to diphenyl carbonate. J. Mol. Catal. Chem. 2015, 398, 248–254. [Google Scholar] [CrossRef]
- Li, Y.; He, S. Enhanced catalytic behavior for synthesis of diphenyl carbonate over SBA-16 composited with α-MoO3. Mol. Cryst. Liq. Cryst. 2022, 757, 125–137. [Google Scholar] [CrossRef]
- Wang, C.; Guo, T.; Sun, M.; Li, C.; Guo, M.; Li, C.; Wang, Q. Transesterification of dimethyl carbonate with phenol to diphenyl carbonate over magnesium oxide nanosheets. Sci. Adv. Mater. 2018, 10, 779–784. [Google Scholar] [CrossRef]
- Wang, Q.; Li, C.; Guo, M.; Luo, S.; Hu, C. Transesterification of dimethyl carbonate with phenol to diphenyl carbonate over hexagonal Mg(OH)2 nanoflakes. Inorg. Chem. Front. 2015, 2, 47–54. [Google Scholar] [CrossRef]
- Tong, D.S.; Yao, J.; Wang, Y.; Niu, H.Y.; Wang, G.Y. Transesterification of dimethyl carbonate with phenol to diphenyl carbonate over V2O5 catalyst. J. Mol. Catal. Chem. 2007, 268, 120–126. [Google Scholar] [CrossRef]
- Tong, D.; Chen, T.; Yao, J.; Wang, Y.; Wang, G.; Shi, D.; Li, Z.; Chen, Z. V-Cu composite oxide catalyst for transesterification of dimethyl carbonate with phenol to diphenyl carbonate. Chin. J. Catal. 2007, 28, 190–192. [Google Scholar] [CrossRef]
- Wang, Q.; Sun, M.M.; Guo, M.; Luo, S.J.; Li, C.H.; Hu, C.W. Heterogeneous transesterification of dimethyl carbonate and phenol to diphenyl carbonate over vanadium pentoxide microstructures. Integr. Ferroelectr. 2015, 164, 154–164. [Google Scholar] [CrossRef]
- Li, Z.; Qin, Z.; Wang, J. Catalytic synthesis of diphenyl carbonate by transesterification of methyl-carbonate with phenol. Petrochem. Technol. 2006, 35, 528–532. [Google Scholar]
- Wu, Q.; Xu, C.; Zhu, L.; Meng, X.; Xiao, F.S. Recent strategies for synthesis of metallosilicate zeolites. Catal. Today 2022, 390–391, 2–11. [Google Scholar] [CrossRef]
- Deng, X.; Yang, D.; Li, W.; Chai, Y.; Wu, G.; Li, L. Chemistry of coordinatively unsaturated centers in zeolites. Trends Chem. 2023, 5, 892–905. [Google Scholar] [CrossRef]
- Zhao, P.; Li, Z.; Zhang, Y.; Cui, D.; Guo, Q.; Dong, Z.; Qi, G.; Xu, J.; Deng, F. Tuning Lewis acid sites in TS-1 zeolites for hydroxylation of anisole with hydrogen peroxide. Microporous Mesoporous Mater. 2022, 335, 111840. [Google Scholar] [CrossRef]
- Ravi, M.; Sushkevich, V.L.; van Bokhoven, J.A. Towards a better understanding of Lewis acidic aluminium in zeolites. Nat. Mater. 2020, 19, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Shudong, Z.; Chenghua, X.; Liangrong, F.; Fali, Q. Synthesis of diphenyl carbonate over Ti-β molecular sieves. Fine Chem. 2005, 22, 115–117. [Google Scholar]
- Luo, S.; Chen, T.; Tong, D.; Zeng, Y.; Lei, Y.; Wang, G. Synthesis of diphenyl carbonate via transesterification catalyzed by HMS mesoporous molecular sieves containing heteroelements. Chin. J. Catal. 2007, 28, 937–939. [Google Scholar] [CrossRef]
- Zhang, J.; Gao, Y.; Zhang, J.; Zhao, J.; Shen, H. Synthesis and characterization of TiO2-V2O5-MCM-41 for catalyzing transesterification of dimethyl carbonate with phenol. Chem. Cent. J. 2018, 12. [Google Scholar] [CrossRef]
- Yuanzhuo, Z.; Songlin, W.; Zhongliang, X.; Tong, C.; Gongying, W. Effect of silanol content in silica support on catalytic performance ofsilica-anchored organotin catalyst for transesterification. CIESC J. 2017, 68, 1892–1898. [Google Scholar]
- Fuming, M.; Zhi, P.; Guangxing, L. The transesterification of dimethyl carbonate with phenol over Mg-Al-hydrotalcite catalyst. Org. Process. Res. Dev. 2004, 8, 372–375. [Google Scholar] [CrossRef]
- Qinqin, Y.; Shu, W.; Rongxian, B.; Fuming, M.; Guangxing, L. Transesterification of dimethyl carbonate and phenol catalyzed by Zn-Al hydrotalcite. Chem. J. Chin. Univ. 2005, 26, 1502–1506. [Google Scholar]
- Shu, W.; Qinqin, Y.; Jinming, H.; Rongxian, B.; Guangxing, L. Study on the catalytic activity, life time and deactivation of hydrotalcite-like catalysts in transesterification. Acta Chim. Sin. 2005, 63, 1575. [Google Scholar]
- Chen, T.; Han, H.; Yao, J.; Wang, G. The transesterification of dimethyl carbonate and phenol catalyzed by 12-molybdophosphoric salts. Catal. Commun. 2007, 8, 1361–1365. [Google Scholar] [CrossRef]
- Huajun, H.; Tong, C.; Jie, Y.; Gongying, W.; Desheng, Z.; Dachuan, S.; Zhiming, C. Effect of caclining temperature on structure and catalytic performance of SiO2 supported Cu3/2PMo12O40. J. Mol. Catal. 2006, 20, 496–499. [Google Scholar]
- Han, H.; Chen, T.; Yao, J.; Wang, G. A heterogeneous catalyst for the transesterification of dimethyl carbonate and phenol to form diphenyl carbonate. Chin. J. Catal. 2006, 27, 7–8. [Google Scholar] [CrossRef]
- Wang, S.; Tang, R.; Zhang, Y.; Chen, T.; Wang, G. 12-Molybdophosphoric acid supported on titania: A highly active and selective heterogeneous catalyst for the transesterification of dimethyl carbonate and phenol. Chem. Eng. Sci. 2015, 138, 93–98. [Google Scholar] [CrossRef]
- Luo, S.; Chi, Y.; Sun, L.; Wang, Q.; Hu, C. Single-step catalytic synthesis of diphenyl carbonate over transition-metal-substituted Keggin-type tungstophosphoric acid. Catal. Commun. 2008, 9, 2560–2564. [Google Scholar] [CrossRef]
- Tae Kim, Y.; Duck Park, E. Transesterification between dimethyl carbonate and phenol in the presence of (NH4)8Mo10O34 as a catalyst precursor. Appl. Catal. Gen. 2009, 361, 26–31. [Google Scholar] [CrossRef]
- Deshmukh, K.M.; Qureshi, Z.S.; Dhake, K.P.; Bhanage, B.M. Transesterification of dimethyl carbonate with phenol using Brønsted and Lewis acidic ionic liquids. Catal. Commun. 2010, 12, 207–211. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Q.; Cui, C.; Niu, H.; Wu, C.; Wang, J. Ionic liquids-SBA-15 hybrid catalysts for highly efficient and solvent-free synthesis of diphenyl carbonate. Green Energy Environ. 2023, 8, 183–193. [Google Scholar] [CrossRef]
- Jiang, Z.; Wang, H.; Shan, L.; Zheng, R.; Zhao, X.; Liao, Z.; Guo, L. Preparation of temperature-controlled heteropolyacid ionic liquids and their application for synthesis of diphenyl carbonate. Catal. Lett. 2022, 153, 1308–1318. [Google Scholar] [CrossRef]
- Ma, X.; Guo, H.; Wang, S.; Sun, Y. Transesterification of dimethyl oxalate with phenol over TS-1 catalyst. Fuel Process. Technol. 2003, 83, 275–286. [Google Scholar] [CrossRef]
- Wang, S.; Ma, X.; Gong, J.; Gao, N.; Guo, H.; Yang, X.; Xu, G. Characterization and activity of stannum modified Hβ catalysts for transesterification of dimethyl oxalate with phenol. Catal. Today 2004, 93–95, 377–381. [Google Scholar] [CrossRef]
- Ma, X.; Gong, J.; Wang, S.; He, F.; Guo, H.; Yang, X.; Xu, G. Characterization and reactivity of stannum modified titanium silicalite TS-1 catalysts for transesterification of dimethyl oxalate with phenol. J. Mol. Catal. Chem. 2005, 237, 1–8. [Google Scholar] [CrossRef]
- Wang, S.; Ma, X.; Guo, H.; Gong, J.; Yang, X.; Xu, G. Characterization and catalytic activity of TiO2/SiO2 for transesterification of dimethyl oxalate with phenol. J. Mol. Catal. Chem. 2004, 214, 273–279. [Google Scholar] [CrossRef]
- GONG, J.; MA, X.; WANG, S.; LIU, M.; YANG, X.; XU, G. Transesterification of dimethyl oxalate with phenol over MoO3/SiO2 catalysts. J. Mol. Catal. Chem. 2004, 207, 215–220. [Google Scholar] [CrossRef]
- Wang, S.; Ma, X.; Gong, J.; Yang, X.; Guo, H.; Xu, G. Transesterification of dimethyl oxalate with phenol under SnO2/SiO2 catalysts. Ind. Eng. Chem. Res. 2004, 43, 4027–4030. [Google Scholar] [CrossRef]
- Biradar, A.; Umbarkar, S.; Dongare, M. Transesterification of diethyl oxalate with phenol using MoO3/SiO2 catalyst. Appl. Catal. Gen. 2005, 285, 190–195. [Google Scholar] [CrossRef]
- Liu, Y.; Ma, X.; Wang, S.; Gong, J. The nature of surface acidity and reactivity of MoO3/SiO2 and MoO3/TiO2–SiO2 for transesterification of dimethyl oxalate with phenol: A comparative investigation. Appl. Catal. Environ. 2007, 77, 125–134. [Google Scholar] [CrossRef]
- Yang, X.; Ma, X.; Wang, S.; Gong, J. Transesterification of dimethyl oxalate with phenol over TiO2/SiO2: Catalyst screening and reaction optimization. AIChE J. 2008, 54, 3260–3272. [Google Scholar] [CrossRef]
- Cao, P.; Yang, J.; Yang, X.; Yao, J.; Wang, Y.; Wang, G. Organotitanium compound catalysts for transesterification of dimethyl carbonate and phenyl acetate to diphenyl carbonate. Chin. J. Catal. 2009, 30, 65–68. [Google Scholar] [CrossRef]
- Cao, P.; Yang, X.; Tang, C.; Yang, J.; Yao, J.; Wang, Y.; Wang, G. Molybdenum trioxide catalyst for transesterification of dimethyl carbonate and phenyl acetate to diphenyl carbonate. Chin. J. Catal. 2009, 30, 853–855. [Google Scholar] [CrossRef]
- Cao, P.; Yang, X.; Ma, F.; Tang, C.; Kang, T.; Wang, G. MoO3 catalyst for transesterification of dimethyl carbonate and phenyl acetate to diphenyl carbonate. Petrochem. Technol. 2011, 40, 1037–1041. [Google Scholar]
- Wang, L.; Yang, X.; Liu, L.; Wang, G. Transesterification of dimethyl carbonate and phenyl acetate to diphenyl carbonate over TiO2/SiO2 catalysts. Petrochem. Technol. 2012, 41, 770–777. [Google Scholar]
- Tang, R.; Wang, S.; Zhang, Y.; Chen, T.; Wang, G. Catalytic property of titanyl acetate in the transesterification reaction of dimethyl carbonate and phenol. Chem. J. Chin. Univ. 2014, 35, 2418–2424. [Google Scholar]
- Cao, P.; Wang, G. Supported MoO3/SiO2 catalyst for transesterification of dimethyl carbonate and phenyl acetate to diphenyl carbonate. Acta Pet. Sin. (Petroleum Process. Sect.) 2014, 30, 823–828. [Google Scholar]
- Jia, B.; Cao, P.; Zhang, H.; Wang, G. Mesoporous amorphous TiO2 shell-coated ZIF-8 as an efficient and recyclable catalyst for transesterification to synthesize diphenyl carbonate. J. Mater. Sci. 2019, 54, 9466–9477. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature/°C | /kJmol−1 | /kJmol−1 | /Jmol−1K−1 | K |
|---|---|---|---|---|
| 25 | 65.4 | 64.4 | −3.6 | 3.5 × 10−12 |
| 100 | 25.2 | 49.6 | 65.3 | 2.9 × 10−4 |
| 150 | 22.2 | 45.9 | 56 | 1.8 × 10−3 |
| 180 | 20.8 | 43 | 49.1 | 4.0 × 10−3 |
| 220 | 26.8 | 45.2 | −146.1 | 1.4 × 10−3 |
| 250 | 28.8 | 41.2 | −133.9 | 1.3 × 10−3 |
| Temperature/°C | /kJmol−1 | /kJmol−1 | /Jmol−1K−1 | K |
|---|---|---|---|---|
| 25 | −32.6 | 25.9 | 192.9 | 5.1 × 105 |
| 100 | −45.7 | 10.7 | 151.1 | 2.50 × 106 |
| 150 | −53.5 | 13.4 | 158.2 | 4.10 × 106 |
| 180 | −59.7 | 52.5 | 247.6 | 7.60 × 106 |
| 220 | −65.7 | 10.7 | 154.9 | 9.10 × 106 |
| 250 | −71.8 | 14.8 | 165.6 | 15.00 × 106 |
| Catalyst | Temperature (°C) | Time (h) | PhOH/DMC | Cat/PhOH (in Mole) | Conversion ** (%) | MPC Yield (%) | DPC Yield (%) | Transesterification Selectivity * (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Ti(OBu)4 | 175 | 8 | 1 | 0.1 | 28.7 | 26.1 | 2.6 | 99.99 | [29] |
| Ti(OPh)4 | 175 | 8 | 1 | 0.1 | 25 | 23.1 | 1.9 | 100 | [29] |
| TiCp2Cl2 | 150–180 | 10 | 1 | 0.005 | 46.8 | 20.3 | 25.7 | 98.3 | [30] |
| Ti(OEt)4 | 150–180 | 10 | 1 | 0.005 | 36.8 | 13.2 | 22.9 | 98.1 | [30] |
| Ti(OiPr)4 | 150–180 | 10 | 1 | 0.005 | 30 | 17.2 | 12.4 | 97.7 | [31] |
| CpTiCl3 | 150–180 | 10 | 1 | 0.005 | 40.2 | 20.3 | 25.7 | 98.3 | [31] |
| [(Me)5Cp]2TiCl2 | 150–180 | 10 | 1 | 0.005 | 30.7 | 16.2 | 22.6 | 96.5 | [31] |
| [(Me)5Cp]TiCl3 | 150–180 | 10 | 1 | 0.005 | 6.8 | 17.4 | 12.9 | 100 | [31] |
| [(Et−Cp)2]2TiCl2 | 150–180 | 10 | 1 | 0.005 | 40.2 | 6.8 | N.D.*** | 100 | [31] |
| [(Me)5Cp]Ti(OMe)3 | 150–180 | 10 | 1 | 0.005 | 6.6 | 22.2 | 16.6 | 96.5 | [31] |
| Cp2Ti(SO3CF3)2 | 150–180 | 10 | 1 | 0.005 | 36.8 | 6.1 | 0.5 | 100 | [31] |
| [i−PrO]4TiCl | 150–180 | 10 | 1 | 0.005 | 40.1 | 6.1 | 4.7 | 29.3 | [31] |
| Cp2TiPh2 | 150–180 | 10 | 1 | 0.005 | 47.6 | 17.2 | 12.4 | 97.7 | [31] |
| Cp2Ti(OPh)2 | 150–180 | 10 | 1 | 0.005 | 46 | 23.9 | 23.2 | 98.9 | [31] |
| TIBE | 185 | 3 | 0.125 | 0.0047 | 48.6 | 48.4 | N.D.*** | 99.5 | [17] |
| TBBE | 185 | 3 | 0.125 | 0.0047 | 50.7 | 50.4 | N.D.*** | 99.4 | [17] |
| TiO(OOCMe)2 | 150–180 | 9 | 1 | 0.0033 | 47.8 | 25.6 | 22.2 | 99.9 | [32] |
| TiCp2Cl2 ***** | 180 | 3 | N.A.**** | 0.04 | 31.4 | N.A.**** | 30.3 | 96.5 | [33] |
| TiO(acac)2 ***** | 180 | 3 | N.A.**** | 0.04 | 69.2 | N.A.**** | 68.3 | 98.7 | [33] |
| TiO(OOCMe)2 ***** | 180 | 3 | N.A.**** | 0.04 | 71.1 | N.A.**** | 69.5 | 97.7 | [33] |
| Ti(OBu)4 ***** | 180 | 3 | N.A.**** | 0.04 | 86.2 | N.A. **** | 85.6 | 99.3 | [33] |
| Ti(OPh)4 ***** | 180 | 3 | N.A. **** | 0.04 | 89.1 | N.A. **** | 87.8 | 98.5 | [33] |
| Ti(OiPr)4 ***** | 180 | 3 | N.A. **** | 0.04 | 90.4 | N.A. **** | 90 | 99.6 | [33] |
| Catalyst | Temperature (°C) | Time (h) | PhOH/DMC | Cat/PhOH (in Mole) | Conversion ** (%) | MPC Yield (%) | DPC Yield (%) | Transesterification Selectivity * (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Bu2SnO | 175 | 8 | 1 | 0.01 | 20.2 | 19.8 | 0.4 | 100 | [20] |
| Bu4Sn | 175 | 8 | 1 | 0.01 | 0.1 | 0.1 | 0 | 100 | [20] |
| Bu2SnO−CF3SO3H | 180 | 3 | 5 | 0.002 | 58.5 | 39.8 | 18.3 | 99.3 | [34] |
| Bu2SnO−CH3SO3H | 180 | 3 | 5 | 0.002 | 56.5 | 36.5 | 19.7 | 99.5 | [34] |
| Bu2SnO−CH3C6H4SO3H | 180 | 3 | 5 | 0.002 | 60.4 | 39.4 | 20.8 | 99.7 | [34] |
| Bu2SnO−C6H5SO3H | 180 | 3 | 5 | 0.002 | 57.9 | 38.2 | 19.4 | 99.5 | [34] |
| Bu2SnO−Cu2O | 150–180 | 8 | 2 | 0.008 | 50.8 *** | 35.4 | 15.4 | 99.9 | [35] |
| BuSn(O)OH−CuI | 150–180 | 8 | 2 | 0.0113 | 57.8 *** | 39.2 | 18.6 | 99.9 | [36] |
| Catalyst | MPC | DPC | ||
|---|---|---|---|---|
| Yield (%) | Selectivity (%) | Yield (%) | Selectivity (%) | |
| Ti(OPh)4 | 6.2 | 98.0 | 0.1 | 1.0 |
| Bu2SnO | 7.9 | 98.0 | 0.1 | 1.0 |
| Pb(OPh)2 | 11.3 | 97.0 | 0.2 | 2.0 |
| Catalyst | Temperature (°C) | Time (h) | PhOH/DMC | Cat/PhOH (in Mass) | Conversion ** (%) | MPC Yield (%) | DPC Yield (%) | Transesterification Selectivity (%) * | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| SmI2 | 150–180 | 8 | 1 | 0.002 | 47.7 | 26 | 18.7 | 93.7 | [43] |
| Sm(OTf)3 | 190 | 12 | 4 | 0.0036 | 34.4 *** | 2.1 | 31.1 | 96.5 | [42] |
| Catalyst | Temperature (°C) | Time (h) | PhOH/DMC | Cat/PhOH (in Mass) | Conversion ** (%) | MPC Yield (%) | DPC Yield (%) | Transesterification Selectivity (%) * | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| TiO2 | 150–180 | 9 | 1 | 0.013 | 29.5 | 8.9 | 20.4 | 99.7 | [44] |
| TiO2/SiO2 | 430 | N.A.*** | 0.2 | N.A. *** | 37 | 31 | 0 | 87 | [45] |
| TiO2−ZnO | 160–180 | 8 | 1 | 0.015 | 41.2 | 22.9 | 17.6 | 98.2 | [46] |
| TiO2/CNT | 150–180 | 9 | 1 | 0.053 | 25.3 | 14.1 | 11.2 | 99.9 | [47] |
| TiO2/CNT-S | 150–180 | 9 | 1 | 0.053 | 41.6 | 18.3 | 23.3 | 99.9 | [47] |
| TiO2/CNT-C | 150–180 | 9 | 1 | 0.053 | 43.9 | 23.9 | 20 | 99.9 | [47] |
| TiO2-C60-8 | 160–180 | 8 | 1 | 0.015 | 34.4 | 17.1 | 17.3 | 99.9 | [48] |
| TiO2-RGO-50 | 150–180 | 9 | 1 | 0.013 | 53.5 | 17.6 | 35.8 | 99.9 | [49] |
| TiO2@SiO2 | 150–180 | 9 | 1 | 0.013 | 41.8 | 17.7 | 24.1 | 100 | [50] |
| TiO2-Nanotube | 170 | 10 | 0.056 | 0.106 | 54.6 | 10.1 | 44.1 | 99.2 | [44] |
| Catalyst | Temperature (°C) | Time (h) | PhOH/DMC | Cat/PhOH (in Mass) | Conversion ** (%) | MPC Yield (%) | DPC Yield (%) | Transesterification Selectivity * (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| PbO | 180 | 8–10 | 0.82 | 0.06 | 22.0 | 7.6 | 14.4 | 100 | [52] |
| O2-PbO/MgO | 180 | 8–10 | 0.82 | 0.06 | 36.8 | 10.0 | 26.6 | 99.5 | [53] |
| PbO-ZnO | 180 | N.A.*** | 0.8 | 0.02 | 64.6 | N.D. | 45.6 | 72 | [52] |
| 15.2PbZr *** | 200 | 2.5 | N.A.*** | 0.053 | 76.6 | N.A. | 76.1 | 99.3 | [54,55] |
| 15.2PbTi *** | 200 | 2.5 | N.A. *** | 0.053 | 34.2 | N.A. | 32.9 | 96.2 | [54] |
| 15.2PbAl *** | 200 | 2.5 | N.A.*** | 0.053 | 47.3 | N.A. | 43.1 | 91.1 | [54] |
| 15.2PbSi *** | 200 | 2.5 | N.A.*** | 0.053 | 56.6 | N.A. | 55.1 | 97.3 | [54] |
| 15.2PbMg *** | 200 | 2.5 | N.A.*** | 0.053 | 67.5 | N.A. | 62.1 | 92 | [54] |
| PbO-ZnO | 160 | 10 | 2 | 0.04 | 32.0 **** | 28.0 | 2.5 | 95.3 | [56] |
| PbO-Fe2O3 | 160 | 10 | 2 | 0.04 | 33.7 **** | 31.7 | 2.0 | 100.0 | [56] |
| PbO-La2O3 | 160 | 10 | 2 | 0.04 | 50.0 **** | 39.6 | 4.7 | 88.6 | [56] |
| PbO-NiO | 160 | 10 | 2 | 0.04 | 61.1 **** | 37.4 | 11.8 | 80.5 | [56] |
| PbO-ZrO2 | 160 | 10 | 2 | 0.04 | 22.7 **** | 21.7 | 0 | 95.6 | [56] |
| PbO-Ce2O3 | 160 | 10 | 2 | 0.04 | 62.2 **** | 51.7 | 7.2 | 94.7 | [56] |
| PbO-Sm2O3 | 160 | 10 | 2 | 0.04 | 62.0 **** | 36.2 | 13.8 | 80.6 | [56] |
| PbO-Y2O3 | 160 | 10 | 2 | 0.04 | 47.2 **** | 36.9 | 5.2 | 89.2 | [56] |
| PbO-Yb2O3 | 160 | 10 | 2 | 0.04 | 57.5 **** | 39.7 | 11.0 | 88.2 | [56] |
| PbO/MgO***** | 200 | 2.5 | N.A. | 0.053 | 60.1 | N.A | 56.0 | 93.2 | [57] |
| PbO/ZrO2***** | 200 | 2.5 | N.A. | 0.053 | 56.3 | N.A. | 55.2 | 98.0 | [57] |
| PbO/SiO2 ***** | 200 | 2.5 | N.A. | 0.053 | 47.7 | N.A. | 46.2 | 96.9 | [57] |
| PbO/Al2O3***** | 200 | 2.5 | N.A. | 0.053 | 30.2 | N.A. | 26.5 | 87.7 | [57] |
| PbO/TiO2***** | 200 | 2.5 | N.A. | 0.053 | 24.3 | N.A. | 22.9 | 94.2 | [57] |
| PbTiO3***** | 180 | 9 | 2 | N.A. | 27.8 | 16.3 | 9.2 | 91.7 | [58] |
| Catalyst | Temperature (°C) | Time (h) | PhOH/DMC | Cat/PhOH (in Mass) | Conversion ** (%) | MPC Yield (%) | DPC Yield (%) | Transesterification Selectivity * (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| MoO3 | 160 | 4 | 0.2 | 0.09 | 3.8 | 3.8 | N.D.*** | 100 | [61] |
| MoO3/SiO2 | 160 | 4 | 0.2 | 0.09 | 17.3 | 17.1 | 0.2 | 100 | [61] |
| MoO3/FSM-36 | 160 | 4 | 0.2 | 0.09 | 14.7 | 14.3 | 0.1 | 97.3 | [61] |
| MoO3/ZrO2 | 160 | 4 | 0.2 | 0.09 | 13.9 | 13.5 | 0.1 | 97.1 | [61] |
| MoO3/TiO2 | 160 | 4 | 0.2 | 0.09 | 9.3 | 8.9 | 0.0 | 95.7 | [61] |
| MoO3/SiO2−Al2O3 | 160 | 4 | 0.2 | 0.09 | 18.3 | 4.3 | 0.0 | 23.5 | [61] |
| MoO3/Al2O3 | 160 | 4 | 0.2 | 0.09 | 20.0 | 3.6 | 0.0 | 18 | [61] |
| MoO3/CaO | 160 | 4 | 0.2 | 0.09 | 23.2 | 2.0 | 0.0 | 8.6 | [61] |
| Mo-K | 150–180 | 9 | 1 | 0.048 | 15.6 | 15.7 | 15.3 | 92.9 | [62] |
| Mo-Mg | 150–180 | 9 | 1 | 0.048 | 17.9 | 6.7 | 7.8 | 90.5 | [62] |
| Mo-Pb | 150–180 | 9 | 1 | 0.048 | 17.1 | 9.1 | 7.1 | 93.0 | [62] |
| Mo-Bi | 150–180 | 9 | 1 | 0.048 | 14.2 | 9.3 | 6.6 | 97.9 | [62] |
| Mo-Co | 150–180 | 9 | 1 | 0.048 | 21.4 | 5.4 | 8.5 | 96.3 | [62] |
| Mo-Zn | 150–180 | 9 | 1 | 0.048 | 24.2 | 9.7 | 10.9 | 96.3 | [62] |
| Mo-Cr | 150–180 | 9 | 1 | 0.048 | 25.3 | 11.4 | 11.9 | 97.6 | [62] |
| Mo-Sb | 150–180 | 9 | 1 | 0.048 | 32.7 | 10.2 | 14.5 | 97.2 | [62] |
| Mo-Cu | 150–180 | 9 | 1 | 0.048 | 41.6 | 14.0 | 17.8 | 93.3 | [62] |
| Catalyst | Temperature (°C) | Time (h) | PhOH/DMC | Cat/PhOH (in Mass) | Conversion ** (%) | MPC Yield (%) | DPC Yield (%) | Transesterification Selectivity * (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Nanosheet-MgO | 180 | 13 | 2 | 0.002 **** | N.A.*** | N.A.*** | N.A.*** | 95.7 | [65] |
| Flake-Mg(OH)2 | 180 | 13 | 2 | 0.002 **** | 25.9 | 6.1 | 17.8 | 92.3 | [66] |
| V2O5 | 150–180 | 9 | 1.5 | 0.016 **** | 42.0 | 17.5 | 22.6 | 95.5 | [67] |
| V-Cu | 150–180 | 9 | 1 | 0.013 **** | 37.0 | 20.2 | 15.6 | 96.8 | [68] |
| V2O5-HCl | 180 | 13 | 2 | 0.0025 **** | 23.23 ***** | 0.93 | 12.94 | 100.0 | [69] |
| V2O5-NHO3 | 180 | 13 | 2 | 0.0025 **** | 6.81 | 2.49 | 20.95 | 100.0 | [69] |
| V2O5-H2SO4 | 180 | 13 | 2 | 0.0025 **** | 15.4 | 5.58 | 1.15 | 98.7 | [69] |
| V2O5-H3PW12O40 | 180 | 13 | 2 | 0.0025 **** | 50.64 | 2.64 | 12.73 | 99.5 | [69] |
| V2O5-H4SiW12O40 | 180 | 13 | 2 | 0.0025 **** | 1.71 | 0.34 | 0.29 | 97.5 | [69] |
| V2O5/SiO2 | 180 | 12 | 10 | 0.053 | 42.4 | 16.7 | 25.7 | 100.0 | [70] |
| Catalyst | Temperature (°C) | Time (h) | PhOH/DMC | Cat/PhOH (in Mass) | Conversion ** (%) | MPC Yield (%) | DPC Yield (%) | Transesterification Selectivity * (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Ti-Beta | 150–180 | 10 | 1 | 0.106 | 20.22 | 8.24 | 2.53 | 53.2 | [75] |
| Mo-HMS | 150–180 | 9 | 1 | 0.066 | 5.6 | 4.4 | 0.8 | 92.5 | [76] |
| Sn-HMS | 150–180 | 9 | 1 | 0.066 | 5.3 | 4.3 | 0.6 | 91.9 | [76] |
| Al-HMS | 150–180 | 9 | 1 | 0.066 | 14.6 | 2.1 | 1.4 | 23.4 | [76] |
| Ti-HMS | 150–180 | 9 | 1 | 0.066 | 31.4 | 14.6 | 16.8 | 99.9 | [76] |
| 10%V-20%Ti-MCM-41 | 180 | 8 | 1 | 0.017 | 33.88 *** | 21.47 | 12.41 | 100 | [77] |
| Sn-SiO2 | 160–180 | 9 | 1 | 0.033 | 29.4 | 14.7 | 14.7 | 99.9 | [78] |
| Sn-MSiO2 | 160–180 | 9 | 1 | 0.033 | 41.2 | 24.3 | 16.9 | 99.9 | [78] |
| Sn-MSiO2-Cal | 160–180 | 9 | 1 | 0.033 | 34.7 | 19.4 | 15.3 | 99.9 | [78] |
| Catalyst | Temperature (°C) | Time (h) | PhOH/DMC | Cat/PhOH (in Mass) | Conversion** (%) | MPC Yield (%) | DPC Yield (%) | Transesterification Selectivity * (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Mg-Al-HT | 160–180 | 10 | 2 | 0.015 | 31.9 | 14.7 | 11.6 | 82.4 | [79] |
| Mg-Al-HT-Cal | 160–180 | 10 | 2 | 0.015 | 15.0 | 7.5 | 1.3 | 58.7 | [79] |
| Zn-Al-HT-3 | 150–180 | 8 | 2 | 0.015 | 52.6 | 12.6 | 37.0 | 94.3 | [80] |
| MgAl | 150–180 | 8 | 2 | 0.01 | 51.8 | 48.8 *** | 94.2 | [81] | |
| ZnAl | 150–180 | 8 | 2 | 0.01 | 57.7 | 53.1 *** | 92.0 | [81] | |
| ZnFe | 150–180 | 8 | 2 | 0.01 | 35.4 | 31.9 *** | 90.1 | [81] | |
| NiFe | 150–180 | 8 | 2 | 0.01 | 37.6 | 31.3 *** | 83.2 | [81] | |
| ZnCr | 150–180 | 8 | 2 | 0.01 | 11.8 | 11.8 *** | 100 | [81] | |
| Catalyst | Temperature (°C) | Time (h) | PhOH/DMC | Cat/PhOH (in Mass) | Conversion ** (%) | MPC Yield (%) | DPC Yield (%) | Transesterification Selectivity * (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| KPMo | 150–180 | 8 | 1 | 0.004 ***** | 1.0 | 1.0 | N.D.**** | 99.9 | [82] |
| H0.5CsPMo | 150–180 | 8 | 1 | 0.004 ***** | 12.5 | 10.8 | 1.7 | 99.9 | [82] |
| LiPMo | 150–180 | 8 | 1 | 0.004 ***** | 20.4 | 10.6 | 9.8 | 99.9 | [82] |
| NaPMo | 150–180 | 8 | 1 | 0.004 ***** | 23.0 | 13.2 | 9.8 | 99.9 | [82] |
| MgPMo | 150–180 | 8 | 1 | 0.004 ***** | 12.5 | 5.4 | 6.3 | 93.6 | [82] |
| CaPMo | 150–180 | 8 | 1 | 0.004 ***** | 11.0 | 2.5 | 8.2 | 97.3 | [82] |
| BaPMo | 150–180 | 8 | 1 | 0.004 ***** | 7.1 | 5.4 | 1.7 | 99.9 | [82] |
| PbPMo | 150–180 | 8 | 1 | 0.004 ***** | 17.9 | 10.7 | 6.4 | 95.5 | [82] |
| AIPMo | 150–180 | 8 | 1 | 0.004 ***** | 30.6 | 7.0 | 6.5 | 44.1 | [82] |
| CuPMo | 150–180 | 8 | 1 | 0.004 ***** | 26.2 | 13.2 | 13.0 | 99.9 | [82] |
| ZnPMo | 150–180 | 8 | 1 | 0.004 ***** | 28.2 | 13.8 | 13.0 | 95.0 | [82] |
| MnPMo | 150–180 | 8 | 1 | 0.004 ***** | 10.3 | 3.7 | 6.3 | 97.1 | [82] |
| AgPMo | 150–180 | 8 | 1 | 0.004 ***** | 17.6 | 8.0 | 8.4 | 93.2 | [82] |
| CePMo | 150–180 | 8 | 1 | 0.004 ***** | 8.2 | 4.8 | 3.4 | 99.9 | [82] |
| FePMo | 150–180 | 8 | 1 | 0.004 ***** | 22.3 | 9.9 | 11.6 | 96.4 | [82] |
| NiPMo | 150–180 | 8 | 1 | 0.004 ***** | 19.5 | 9.5 | 9.0 | 94.9 | [82] |
| Cu1.5PMo12O40/SiO2 | 150–180 | 8 | 1 | 0.2 | 25.2 | 13.5 | 11.5 | 99.2 | [83,84] |
| HPMo/TiO2 | 160–180 | 8 | 1 | 0.04 | 44.7 | 22.8 | 21.7 | 99.6 | [85] |
| ZnPWO/TiO2 | N.A. *** | N.A.*** | N.A.*** | N.A.*** | 12.8 | N.A.*** | 10.7 | 83.6 | [85] |
| Catalyst | Temperature (°C) | Time (h) | PhOH/DMC | Cat/PhOH (in Mass) | Conversion ** (%) | MPC Yield (%) | DPC Yield (%) | Transesterification Selectivity * (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Bu2SnO−[NMP][HSO4] | 180–200 | 12 | 1 | 0.021 | 30.7 | 2.4 | 12.8 | 99.9 | [88] |
| Bu2SnO−[HMim][pTSA] | 180–200 | 12 | 1 | 0.021 | 29.3 | 4.9 | 12.2 | 99.9 | [88] |
| Bu2SnO−[C4Mim][pTSA] | 180–200 | 12 | 1 | 0.021 | 35 | 9.4 | 12.8 | 99.9 | [88] |
| Bu2SnO−[BMim]CI[ZnCl2] | 180–200 | 12 | 1 | 0.021 | 38.5 | 2.8 | 17.8 | 99.5 | [88] |
| Bu2SnO−[BMim]CI[ZnCl2]2 | 180–200 | 12 | 1 | 0.021 | 31.4 | 12.1 | 9.4 | 97.7 | [88] |
| Bu2SnO−[ChCl][ZnCl2] | 180–200 | 12 | 1 | 0.021 | 39 | 1.8 | 18.6 | 99.9 | [88] |
| Bu2SnO−[ChCl][ZnCl2]2 | 180–200 | 12 | 1 | 0.021 | 29.3 | 7.5 | 10.7 | 99.9 | [88] |
| Bu2SnO−[ChCl][SnCl2] | 180–200 | 12 | 1 | 0.021 | 18.1 | 3.1 | 7.5 | 99.9 | [88] |
| Bu2SnO−[ChCl][SnCl2]2 | 180–200 | 12 | 1 | 0.021 | 14.5 | 1.5 | 6.4 | 97.5 | [88] |
| Bu2SnO−[ChCl][FeCl3]2 | 180–200 | 12 | 1 | 0.021 | 14.9 | 4.3 | 5.3 | 99.9 | [88] |
| [SBA−15−IL−CH3]Br *** | 170 | 2 | N.A. | 0.035 | 51.0 | N.A. | 50.8 | 99.6 | [89] |
| [SBA−15−IL−OH]Br *** | 170 | 2 | N.A. | 0.035 | 80.5 | N.A. | 80.2 | 99.6 | [89] |
| [SBA−15−IL−COOH]Br *** | 170 | 2 | N.A. | 0.035 | 81.2 | N.A. | 73.4 | 90.4 | [89] |
| [SBA−15−IL−NH2]Br *** | 170 | 2 | N.A.**** | 0.035 | 82.6 | N.A. | 74.0 | 89.6 | [89] |
| [SBA−15−IL−SO4H]Br *** | 170 | 2 | N.A.**** | 0.035 | 84.6 | N.A. | 74.4 | 87.9 | [89] |
| [SBA−15−IL−2C−OH]Br *** | 170 | 2 | N.A.**** | 0.035 | 80.1 | N.A. | 79.6 | 99.4 | [89] |
| [SBA−15−IL−4C−OH]Br *** | 170 | 2 | N.A.**** | 0.035 | 77.4 | N.A. | 76.8 | 99.2 | [89] |
| [SBA−15−IL−6C−OH]Br *** | 170 | 2 | N.A.**** | 0.035 | 75.7 | N.A. | 75.2 | 99.3 | [89] |
| [SBA−15−IL−8C−OH]Br *** | 170 | 2 | N.A.**** | 0.035 | 74.7 | N.A. | 74.3 | 99.5 | [89] |
| [SBA−15−IL−OH]BF4 *** | 170 | 2 | N.A.**** | 0.035 | 79.6 | N.A. | 71.8 | 90.2 | [89] |
| [SBA−15−IL−OH]PF6 *** | 170 | 2 | N.A.**** | 0.035 | 77.2 | N.A. | 70.5 | 91.3 | [89] |
| [SBA−15−IL−OH]HSO4 *** | 170 | 2 | N.A.**** | 0.035 | 83.1 | N.A. | 70.8 | 85.2 | [89] |
| [SBA−15−IL−OH]OH *** | 170 | 2 | N.A.**** | 0.035 | 81.7 | N.A. | 70.2 | 85.9 | [89] |
| Na4[Ti(H2O)TiMo11O39] | 160 | 6 | 0.5 | 0.015 | 37.32 | 20.43 | 14.93 | 94.74 | [90] |
| Bmim6[Sn(H2O)TiMo11O39] | 160 | 6 | 0.5 | 0.015 | 31.92 | 15.92 | 15.56 | 98.62 | [90] |
| Bmim6[Zn(H2O)TiMo11O39] | 160 | 6 | 0.5 | 0.015 | 23.45 | 11.25 | 11.46 | 96.84 | [90] |
| Bmim6[Fe(H2O)TiMo11O39] | 160 | 6 | 0.5 | 0.015 | 29.10 | 9.45 | 18.82 | 97.15 | [90] |
| Bmim6[Cu(H2O)TiMo11O39] | 160 | 6 | 0.5 | 0.015 | 21.47 | 10.64 | 9.51 | 93.85 | [90] |
| Bmim4[Ti(H2O)TiMo11O39] | 160 | 6 | 0.5 | 0.015 | 42.45 | 18.38 | 23.50 | 98.65 | [90] |
| Bmim4[Pb(H2O)TiMo11O39] | 160 | 6 | 0.5 | 0.015 | 27.08 | 13.12 | 12.78 | 95.64 | [90] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; Shi, F.; Yang, G. Diphenyl Carbonate: Recent Progress on Its Catalytic Synthesis by Transesterification. Catalysts 2024, 14, 250. https://doi.org/10.3390/catal14040250
Wang D, Shi F, Yang G. Diphenyl Carbonate: Recent Progress on Its Catalytic Synthesis by Transesterification. Catalysts. 2024; 14(4):250. https://doi.org/10.3390/catal14040250
Chicago/Turabian StyleWang, Dong, Feng Shi, and Guochao Yang. 2024. "Diphenyl Carbonate: Recent Progress on Its Catalytic Synthesis by Transesterification" Catalysts 14, no. 4: 250. https://doi.org/10.3390/catal14040250
APA StyleWang, D., Shi, F., & Yang, G. (2024). Diphenyl Carbonate: Recent Progress on Its Catalytic Synthesis by Transesterification. Catalysts, 14(4), 250. https://doi.org/10.3390/catal14040250