
Liquid-Phase Selective Oxidation of Methane to Methane Oxygenates



Abstract
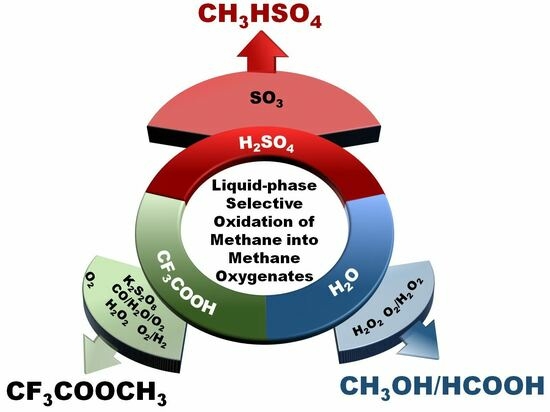
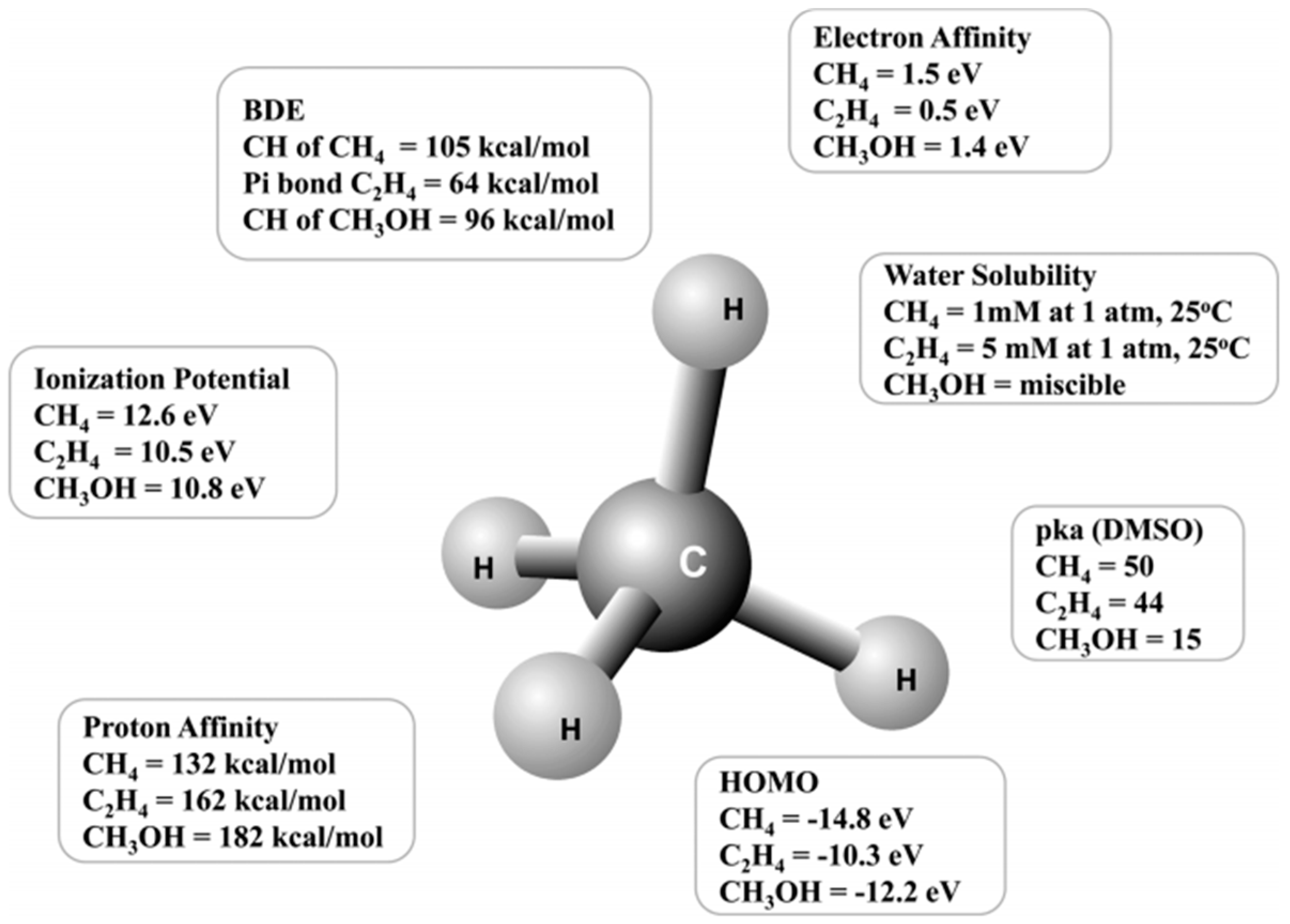
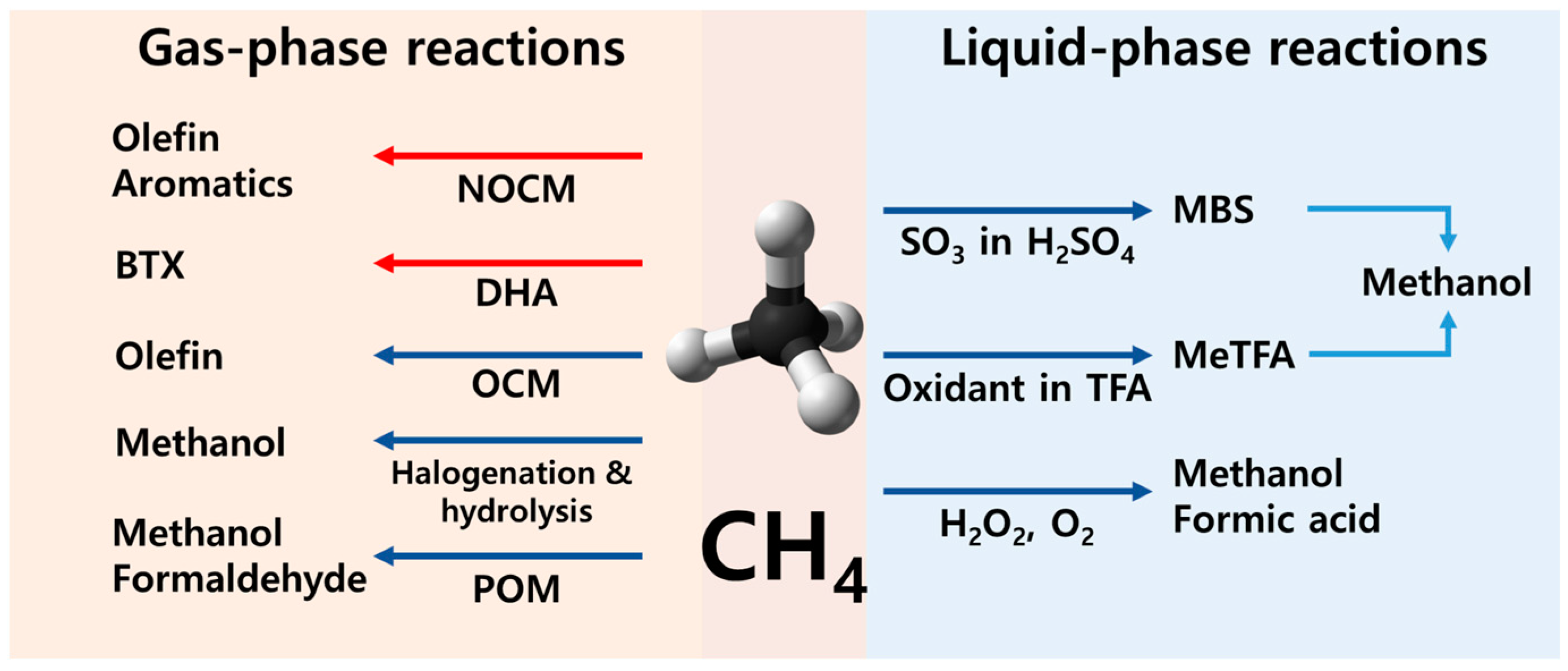
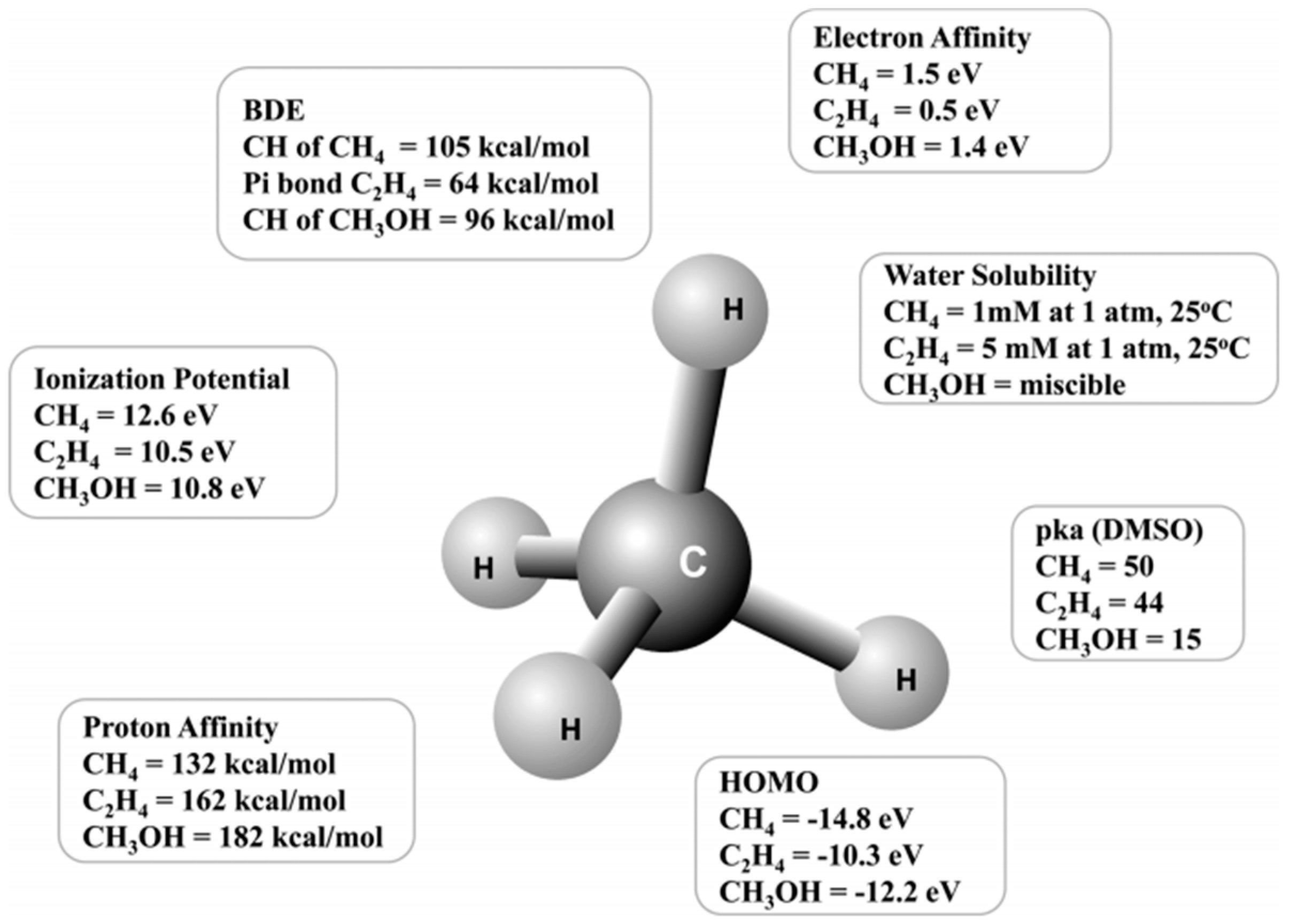
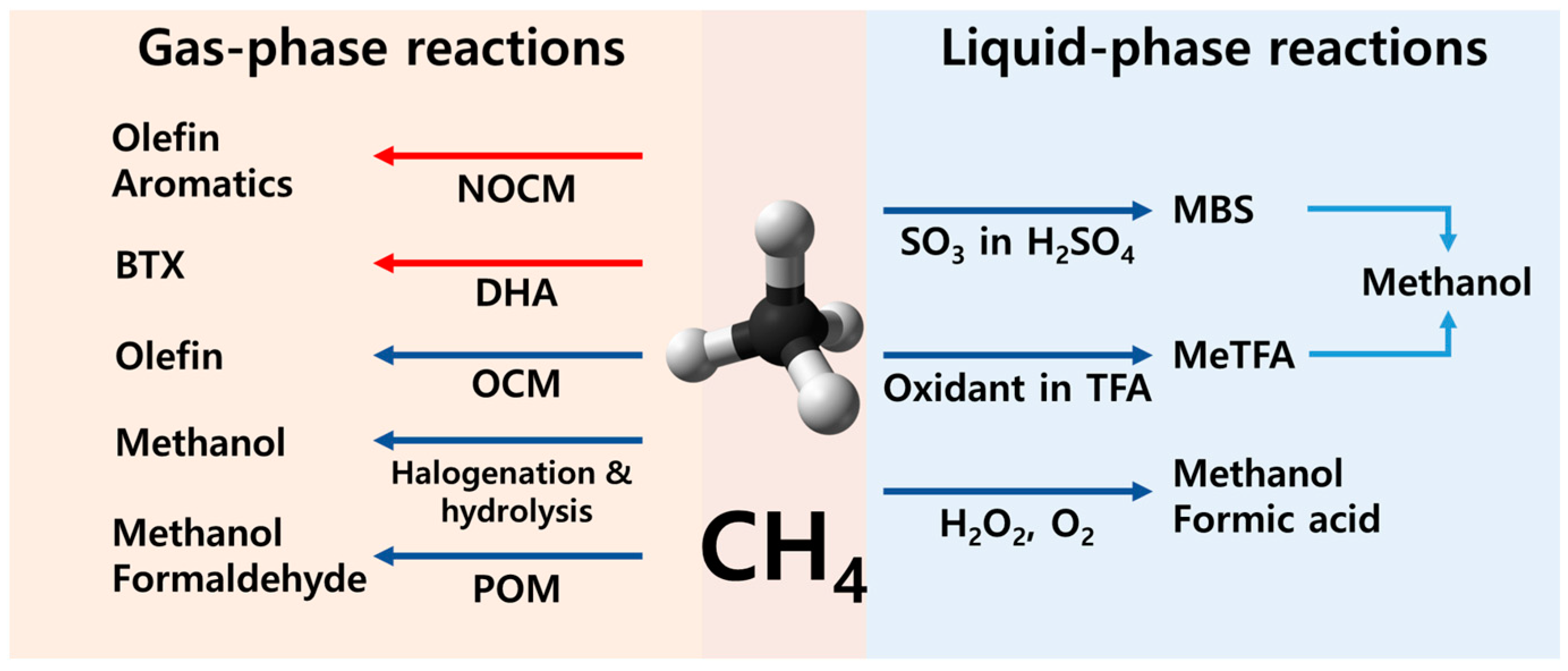
1. Introduction
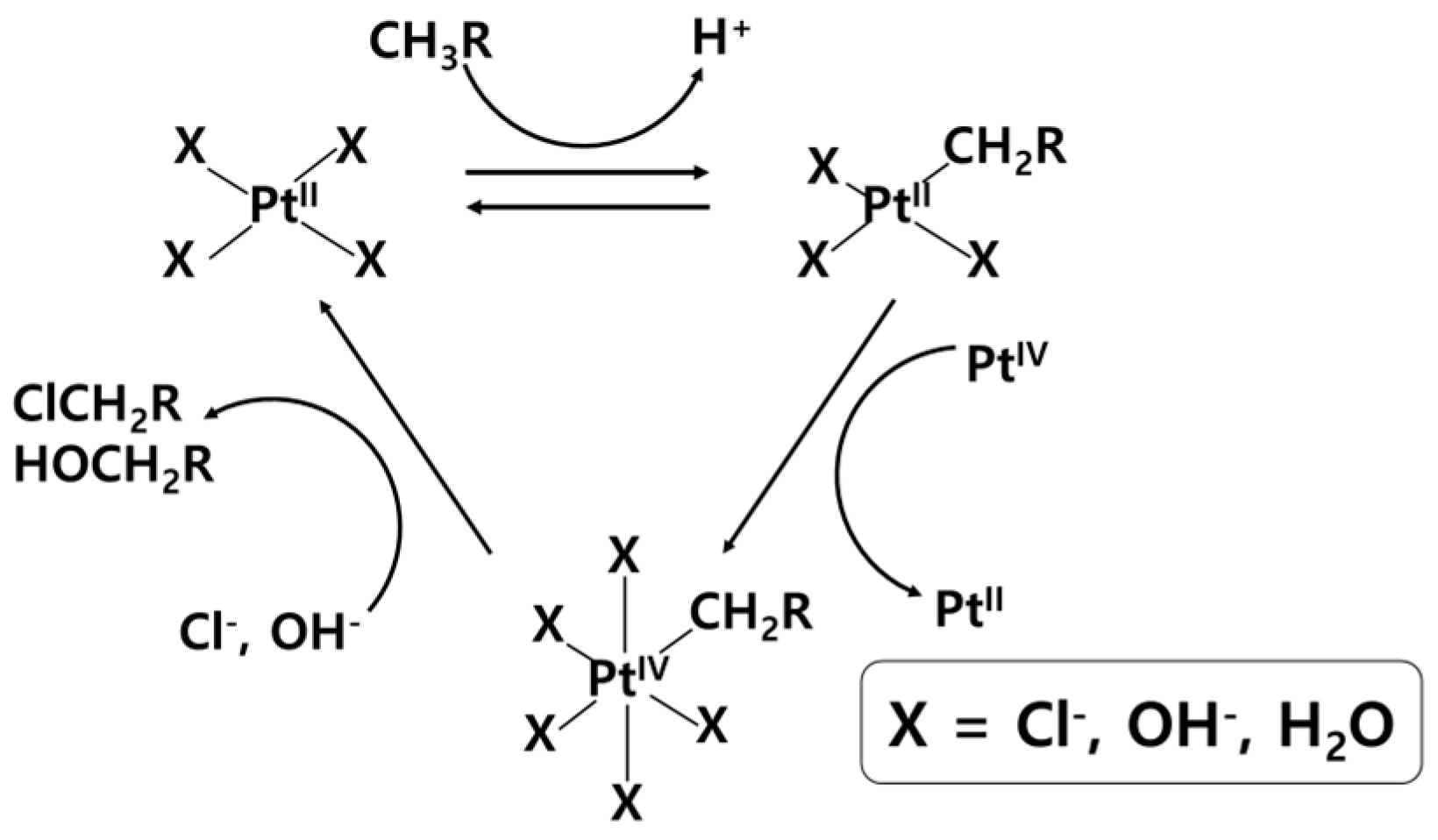
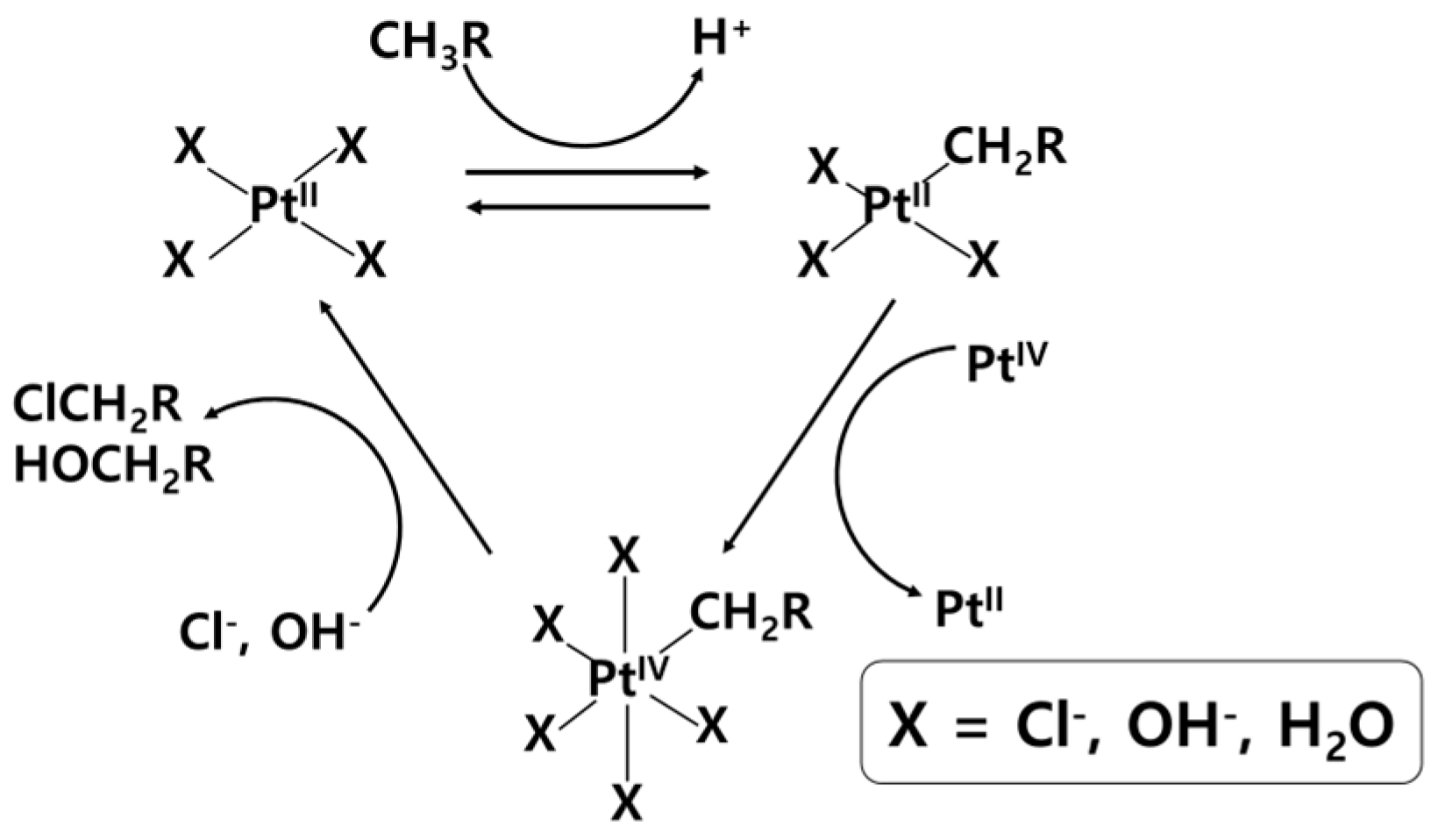
2. Liquid-Phase Partial Oxidation of Methane in Strong Acids
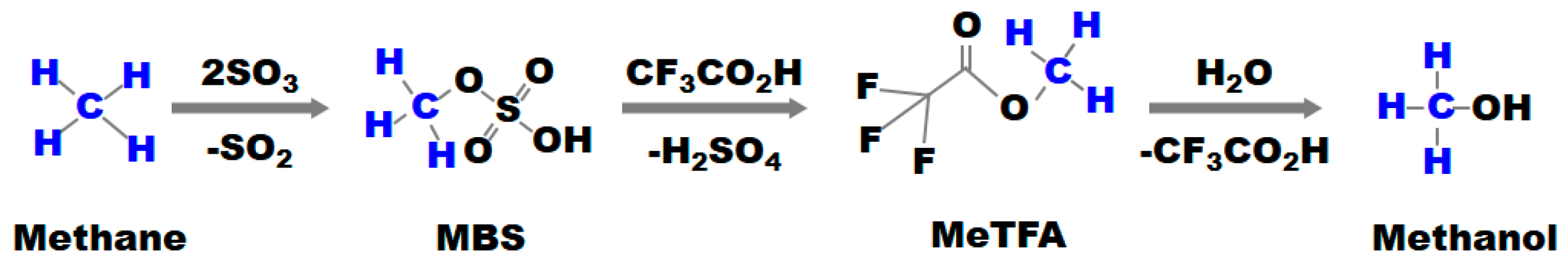
2.1. HTFA
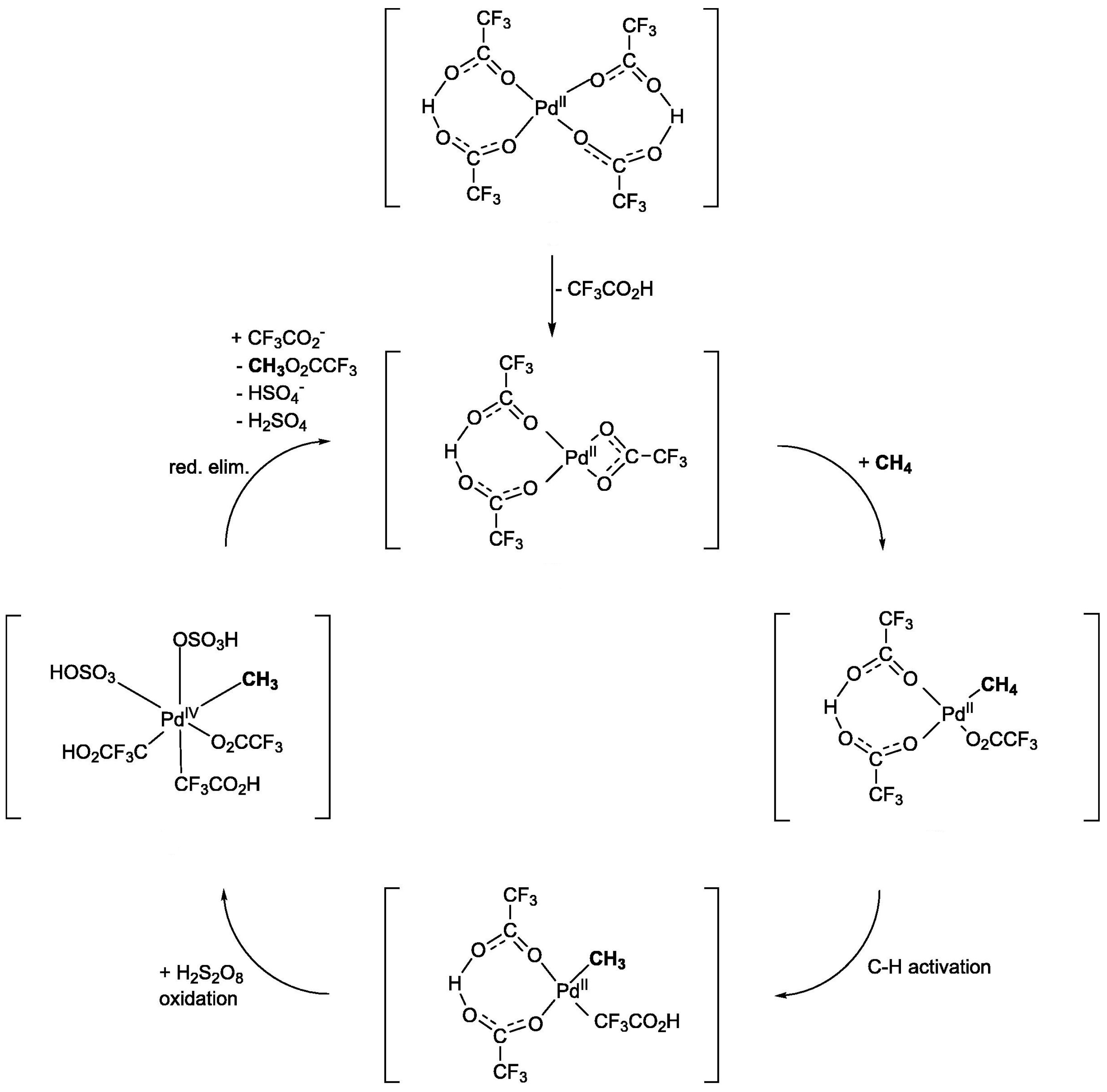
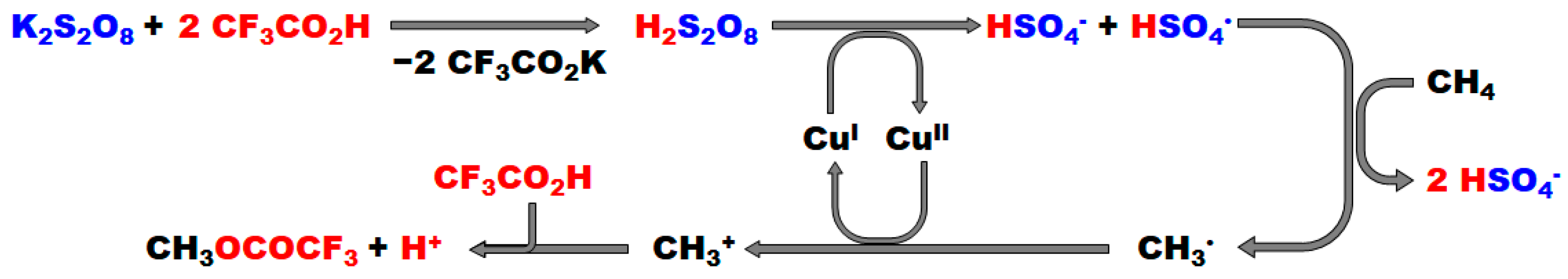
2.1.1. Potassium Persulfate (K2S2O8)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | Temp. (°C) | K2S2O8 (mmol) | Gas Composition (bar) | TON | TOF (h−1) | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | Pd(CH3COO)2 | 80 | 21 | CH4 = 20 | 3.8 | 0.2 | [13] |
| 2 | Pd-NHC * | 90 | 21 | CH4 = 30 | 30 | 2.1 | [13] |
| 3 | [Me4N]2[PdCl4] | 80 | 10 | CH4 = 20 | 330 | 22.0 | [16] |
| 4 | Pyr-POPs-Pd * | 80 | 20 | CH4 = 1 | 664 | 33.2 | [17] |
| 5 | Cu(CH3COO)2 | 100 | 5 | CH4:N2 = 5:25 | 30.4 | 1.5 | [18] |
| 6 | CuO | 90 | 2.8 | CH4 = 5.2 | 33 | 1.9 | [19] |
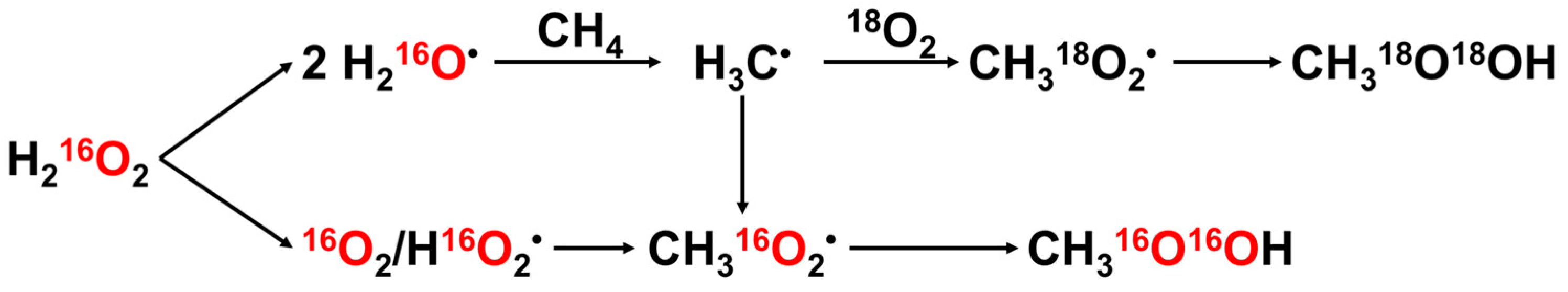
2.1.2. Hydrogen Peroxide (H2O2)
2.1.3. O2
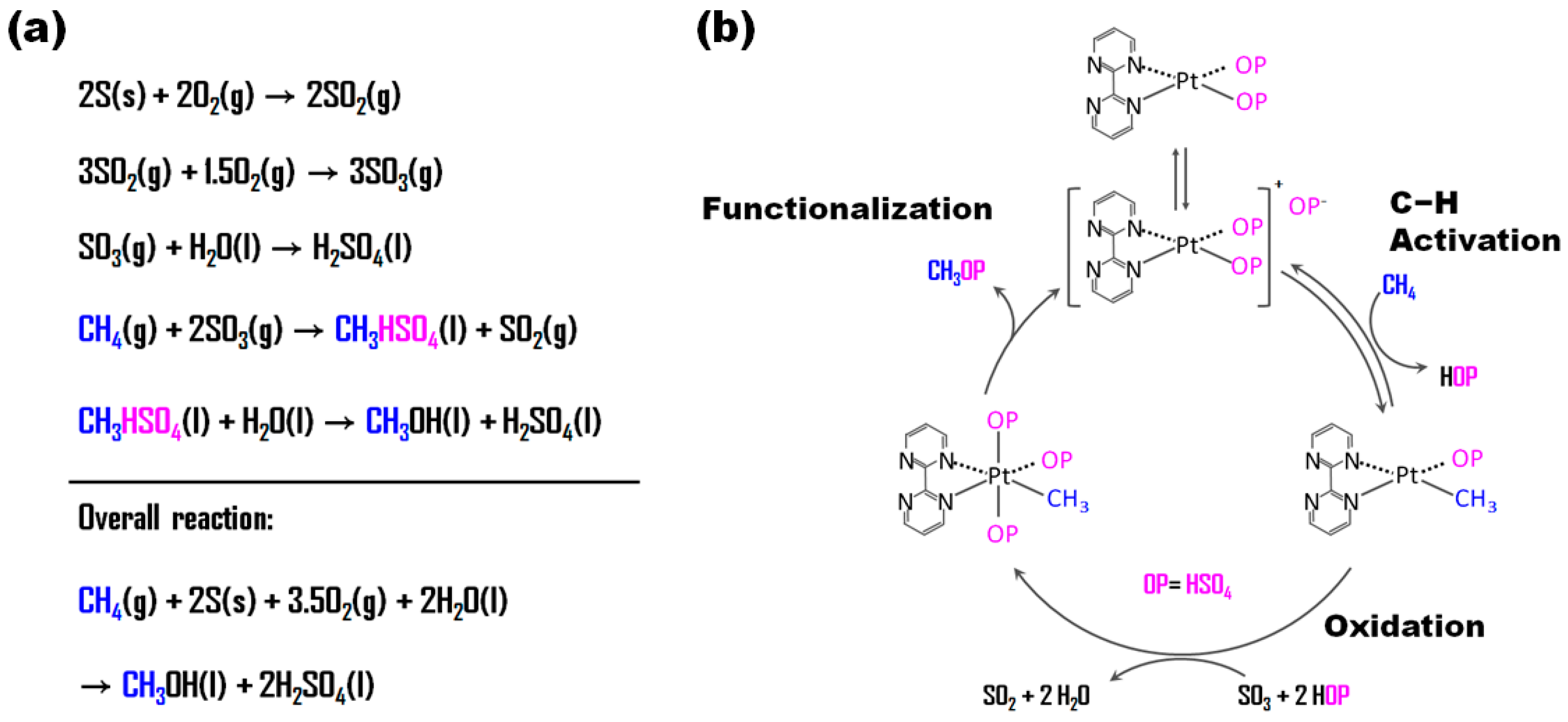
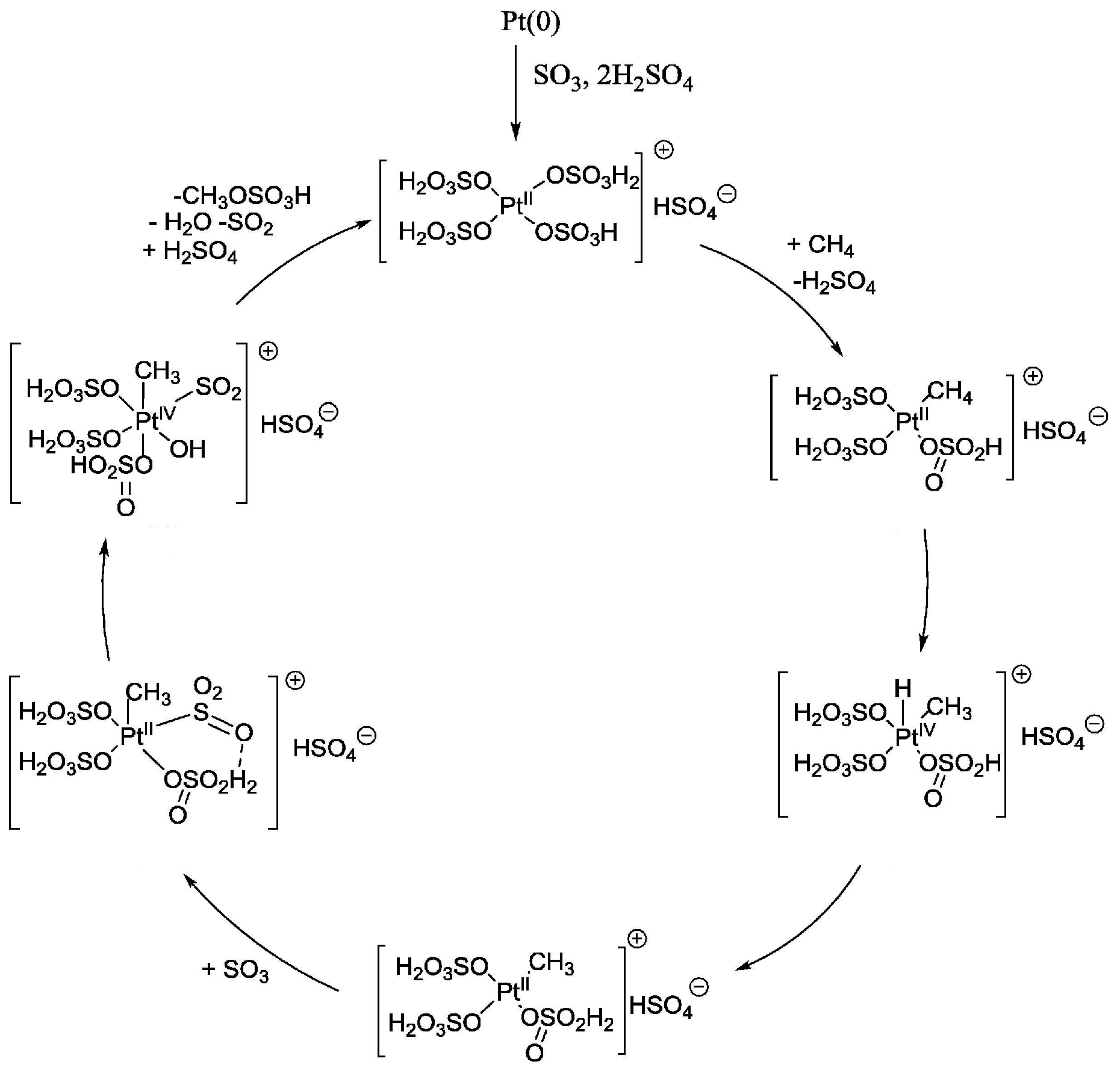
2.2. H2SO4
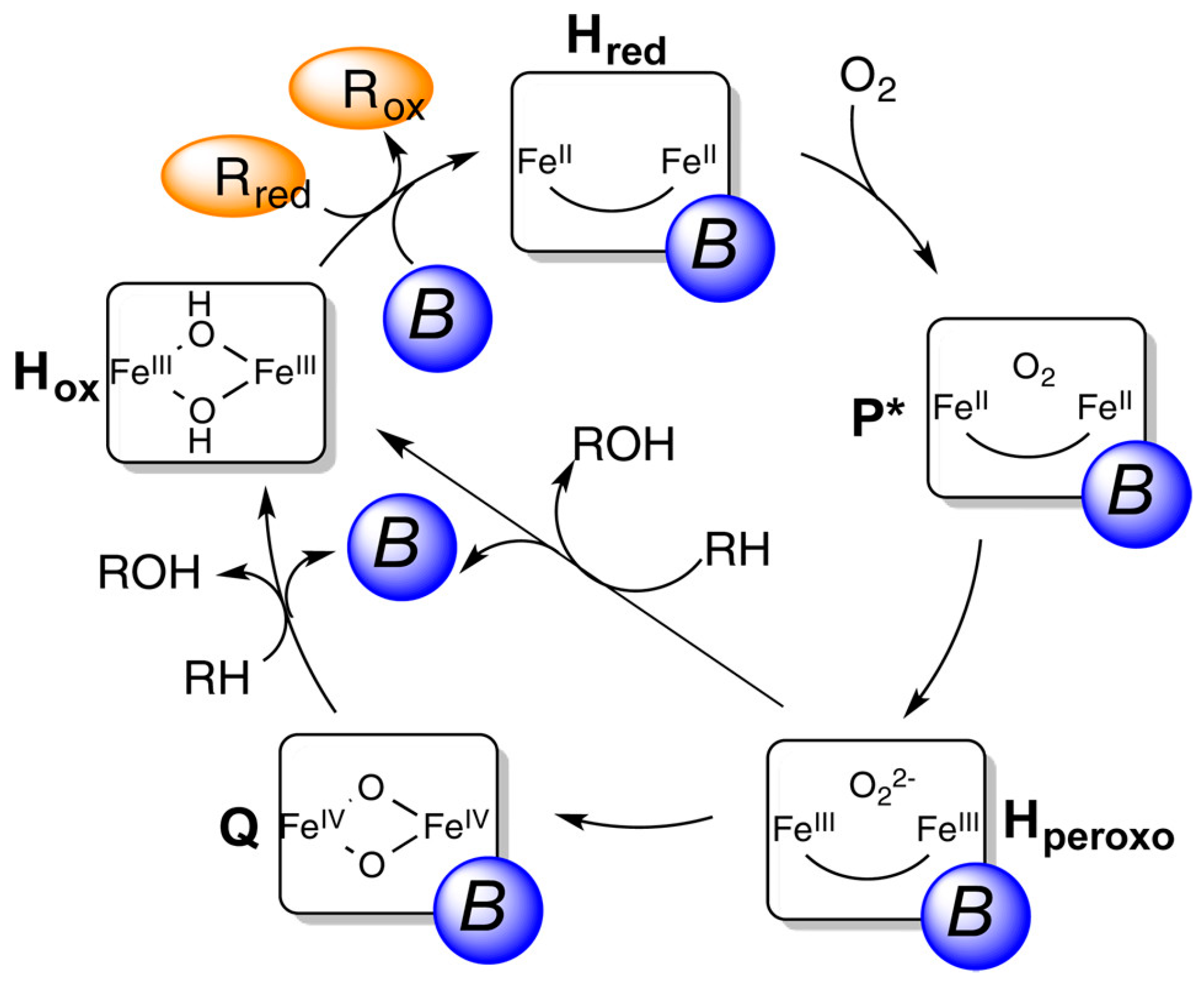
3. Liquid-Phase Partial Oxidation of Methane in Water
3.1. H2O2
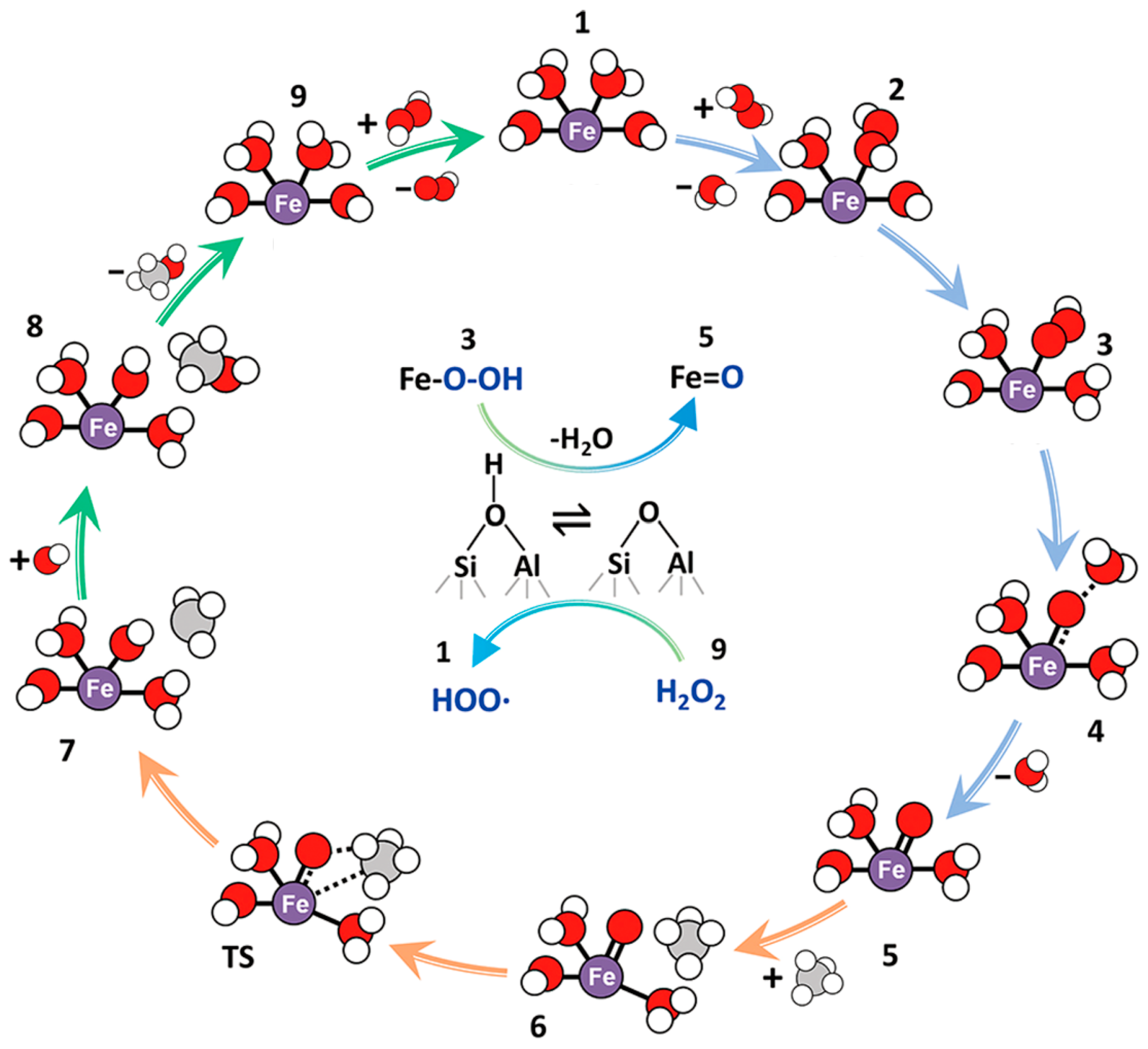
3.1.1. Fe-Zeolite
| Entry | Catalyst | Temp. (°C) | H2O2 (mmol) | CH4 (bar) | Total Productivity (mmol/gcat./h) | Product Selectivity (%) | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | H-ZSM-5 | 100 | 122 | 26 | 2.3 | CH3OH: 0.1 HCOOH: 55 CO2: 45 | [56] |
| 2 | 2.5%Fe/ZSM-5 | 50 | 5 | 30.5 | 16.8 | CH3OH: 10 HCOOH: 72 CO2: 17 | [57] |
| 3 | Fe-silicalite-1 | 50 | 5 | 30.5 | 9.5 | CH3OH: 19 HCOOH: 67 CO2: 9 | [62] |
| 4 | 0.03%Fe/ZSM-5(66) | 80 | 5 | 30 | 54.1 | CH3OH: 1 HCOOH: 84 CO2: 5 | [68] |
| 5 | 0.45%Fe-ZSM-5 | 50 | 5 | 30 | 45.2 | CH3OH: 2 HCOOH: 92 CO2: 0 | [69] |
| 6 | ZSM-5(30) | 50 | 5 | 10 | 26.7 | CH3OH: 11 HCOOH: 54 CO2: 1 | [71] |
| 7 | Ga,Fe-MFI(50) | 55 | 5 | 30 | 51.2 | CH3OH: 5 HCOOH: 90 CO2: 3 | [74] |
| 8 | Al,Fe-MFI(50) | 55 | 5 | 30 | 44.0 | CH3OH: 5 HCOOH: 87 CO2: 7 | [74] |
| 9 | Fe/ZSM-5 | 50 | 5 | 30.5 | 3.5 | - | [75] |
| 10 b | Fe-MOR | 80 | 10 | 28.5 | 8.9 | CH3OH: 17 HCOOH: 37 CO2: 9 | [76] |
| 11 a,b | Fe-MFI | 50 | 27 | 30 | 11.3 | CH3OH: 84 HCOOH: 11 CO2: 0 | [77] |
| 12 b | Fe-MFI | 50 | 27 | 30 | 13.1 | CH3OH: 1 HCOOH: 35 CO2: 63 | [77] |
3.1.2. Promoted Fe-Zeolites
3.1.3. Metal–Organic Framework (MOF)-Based Catalysts
3.1.4. Other Catalysts
| Entry | Catalyst | Temp. (°C) | H2O2 (mmol) | Feed Composition (bar) | Total Productivity (mmol/gcat./h) | Product Selectivity (%) a | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | Au-Pd/TiO2 | 90 | 5 | CH4 = 30.5 | 1.9 | CH3OH: 88 HCOOH: 0 CO2: 12 | [88] |
| 2 | Au-Pd colloid | 50 | 1 | CH4:O2 = 30:5 | 53.6 | CH3OH: 88 HCOOH: 6 CO2: 5 | [89] |
| 3 | AuPd@ZIF-8 | 50 | 0.5 | CH4:O2 = 30:5 | 4.5 | CH3OH: 59 HCOOH: 26 CO2: 14 | [90] |
| 4 | 2.7%FeN4/GN | 25 | 49 | CH4:N2 = 18:2 | 0.2 | CH3OH: 39 HCOOH: 29 CO2: 6 | [94] |
| 5 | 2.5%Fe/NC-HH | 25 | 5 | CH4 = 40 | 1.6 | CH3OH: 29 HCOOH: 51 CO2: 20 | [95] |
| 6 | Cr/TiO2 | 50 | 5 | CH4 = 30 | 4.4 | CH3OH: 48 HCOOH: 5 CO2: 0 | [96] |
3.2. In Situ Generated H2O2
3.2.1. Pd-Based Catalyst and Transition Metal-Based Catalyst
3.2.2. Pd-Au-Based Catalyst
| Entry | Catalyst | Temp. (°C) | Feed Composition (bar) | Total Productivity (mmol/gcat./h) | Product Selectivity (%) a | Ref. |
|---|---|---|---|---|---|---|
| 1 | FeSO4 + Pd/C | 20 | CH4:H2:Air = 15:3:10 | 64.2 b | CH3OH: 5 HCOOH: 61 CO2: 34 | [111] |
| 2 | Fe/ZSM-5 + Pd/c-s-HCPP c | 50 | CH4:H2:Air = 15:3:10 | 3.4 | CH3OH: 28 HCOOH: 61 CO2: 11 | [112] |
| 3 | Pd-Fe/ZSM-5 | 50 | CH4:H2:Air = 15:3:10 | 0.5 | CH3OH: 52 HCOOH: 37 CO2: 11 | [114] |
| 4 | Fe/ZSM-5 + Pd/AC | 50 | CH4:H2:Air = 15:3:10 | 3.5 | CH3OH: 34 HCOOH: 45 CO2: 20 | [115] |
| 5 | Pd-Cu/ZSM-5 d | 120 | CH4:H2:O2 = 73:24:9 | 2.2 | CH3OH: 55 HCOOH: 40 CO2: 5 | [116] |
| 6 | Pd/CsPMA-H e | 25 | CH4:O2 = 20:0.3 | 0.067 | CH3OH: 100 | [117] |
| 7 | AuPd/TiO2 | 50 | CH4:H2:O2:N2 = 30.5:0.3:0.7:8.7 | 0.14 | CH3OH: 83 HCOOH: 0 CO2: 17 | [88] |
| 8 | Pd-Au/CNTs | 50 | CH4:H2:O2:Ar = 15.5:1.3:2.6:13.5 | 0.4 | CH3OH: 78 HCOOH: 22 CO2: 0 | [118] |
| 9 | AuPd@ZSM-5-C16 | 70 | CH4:H2:O2:Ar = 0.5:0.9:1.8:27 | 5.0 | CH3OH: 95 HCOOH: 5 CO2: 0 | [120] |
4. Summary and Outlook
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schwach, P.; Pan, X.; Bao, X. Direct Conversion of Methane to Value-Added Chemicals over Heterogeneous Catalysts: Challenges and Prospects. Chem. Rev. 2017, 117, 8497–8520. [Google Scholar] [CrossRef] [PubMed]
- Alola, A.A.; Onifade, S.T.; Magazzino, C.; Obekpa, H.O. The Effects of Gas Flaring as Moderated by Government Quality in Leading Natural Gas Flaring Economies. Sci. Rep. 2023, 13, 14394. [Google Scholar] [CrossRef] [PubMed]
- Ahlquist, M.; Nielsen, R.J.; Periana, R.A.; Goddard, W.A. Product Protection, the Key to Developing High Performance Methane Selective Oxidation Catalysts. J. Am. Chem. Soc. 2009, 131, 17110–17115. [Google Scholar] [CrossRef] [PubMed]
- Gunsalus, N.J.; Koppaka, A.; Park, S.H.; Bischof, S.M.; Hashiguchi, B.G.; Periana, R.A. Homogeneous Functionalization of Methane. Chem. Rev. 2017, 117, 8521–8573. [Google Scholar] [CrossRef]
- Biswal, T.; Shadangi, K.P.; Sarangi, P.K.; Srivastava, R.K. Conversion of Carbon Dioxide to Methanol: A Comprehensive Review. Chemosphere 2022, 298, 134299. [Google Scholar] [CrossRef]
- Dieterich, V.; Buttler, A.; Hanel, A.; Spliethoff, H.; Fendt, S. Power-to-Liquid via Synthesis of Methanol, DME or Fischer–Tropsch-Fuels: A Review. Energy Environ. Sci. 2020, 13, 3207–3252. [Google Scholar] [CrossRef]
- Bao, J.; Yang, G.; Yoneyama, Y.; Tsubaki, N. Significant Advances in C1 Catalysis: Highly Efficient Catalysts and Catalytic Reactions. ACS Catal. 2019, 9, 3026–3053. [Google Scholar] [CrossRef]
- Xu, Z.C.; Park, E.D. Gas-Phase Selective Oxidation of Methane into Methane Oxygenates. Catalysts 2022, 12, 314. [Google Scholar] [CrossRef]
- Ravi, M.; Ranocchiari, M.; van Bokhoven, J.A. The Direct Catalytic Oxidation of Methane to Methanol—A Critical Assessment. Angew. Chem.—Int. Ed. 2017, 56, 16464–16483. [Google Scholar] [CrossRef] [PubMed]
- Dummer, N.F.; Willock, D.J.; He, Q.; Howard, M.J.; Lewis, R.J.; Qi, G.; Taylor, S.H.; Xu, J.; Bethell, D.; Kiely, C.J.; et al. Methane Oxidation to Methanol. Chem. Rev. 2023, 123, 6359–6411. [Google Scholar] [CrossRef] [PubMed]
- Shilov, A.E.; Shul’pin, G.B. Activation and Catalytic Reactions of Saturated Hydrocarbons in the Presence of Metal Complexes; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2001; Volume 21, ISBN 1402004206. [Google Scholar]
- Gretz, E.; Oliver, T.F.; Sen, A. Carbon-Hydrogen Bond Activation by Electrophilic Transition-Metal Compounds. Palladium (II)-Mediated Oxidation of Arenes and Alkanes Including Methane. J. Am. Chem. Soc. 1987, 109, 8109–8111. [Google Scholar] [CrossRef]
- Muehlhofer, M.; Strassner, T.; Herrmann, W.A. New Catalyst Systems for the Catalytic Conversion of Methane into Methanol. Angew. Chem.—Int. Ed. 2002, 41, 1745–1747. [Google Scholar] [CrossRef]
- Strassner, T.; Muehlhofer, M.; Zeller, A.; Herdtweck, E.; Herrmann, W.A. The Counterion Influence on the CH-Activation of Methane by Palladium(II) Biscarbene Complexes—Structures, Reactivity and DFT Calculations. J. Organomet. Chem. 2004, 689, 1418–1424. [Google Scholar] [CrossRef]
- Ahrens, S.; Strassner, T. Detour-Free Synthesis of Platinum-Bis-NHC Chloride Complexes, Their Structure and Catalytic Activity in the CH Activation of Methane. Inorganica Chim. Acta 2006, 359, 4789–4796. [Google Scholar] [CrossRef]
- Cheong, S.H.; Kim, D.; Dang, H.T.; Kim, D.; Seo, B.; Cheong, M.; Hong, S.H.; Lee, H. Methane Oxidation to Methyl Trifluoroacetate by Simple Anionic Palladium Catalyst: Comprehensive Understanding of K2S2O8-Based Methane Oxidation in CF3CO2H. J. Catal. 2022, 413, 803–811. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, M.; Han, Z.; Huang, S.; Yuan, D.; Su, W. Atmosphere-Pressure Methane Oxidation to Methyl Trifluoroacetate Enabled by a Porous Organic Polymer-Supported Single-Site Palladium Catalyst. ACS Catal. 2021, 11, 1008–1013. [Google Scholar] [CrossRef]
- Yin, G.; Piao, D.-G.; Kitamura, T.; Fujiwara, Y. Cu(OAc)2-Catalyzed Partial Oxidation of Methane to Methyl Trifuoroacetate in the Liquid Phase. Appl. Organomet. Chem. 2000, 14, 438–442. [Google Scholar] [CrossRef]
- Ravi, M.; van Bokhoven, J.A. Homogeneous Copper-Catalyzed Conversion of Methane to Methyl Trifluoroacetate in High Yield at Low Pressure. ChemCatChem 2018, 10, 2383–2386. [Google Scholar] [CrossRef]
- Goyal, R.; Singh, O.; Agrawal, A.; Samanta, C.; Sarkar, B. Advantages and Limitations of Catalytic Oxidation with Hydrogen Peroxide: From Bulk Chemicals to Lab Scale Process. Catal. Rev. 2022, 64, 229–285. [Google Scholar] [CrossRef]
- Puértolas, B.; Hill, A.K.; García, T.; Solsona, B.; Torrente-Murciano, L. In-Situ Synthesis of Hydrogen Peroxide in Tandem with Selective Oxidation Reactions: A Mini-Review. Catal. Today 2015, 248, 115–127. [Google Scholar] [CrossRef]
- Samanta, C. Direct Synthesis of Hydrogen Peroxide from Hydrogen and Oxygen: An Overview of Recent Developments in the Process. Appl. Catal. A Gen. 2008, 350, 133–149. [Google Scholar] [CrossRef]
- Dittmeyer, R.; Grunwaldt, J.D.; Pashkova, A. A Review of Catalyst Performance and Novel Reaction Engineering Concepts in Direct Synthesis of Hydrogen Peroxide. Catal. Today 2015, 248, 149–159. [Google Scholar] [CrossRef]
- Ranganathan, S.; Sieber, V. Recent Advances in the Direct Synthesis of Hydrogen Peroxide Using Chemical Catalysis—A Review. Catalysts 2018, 8, 379. [Google Scholar] [CrossRef]
- Piao, D.-G.; Inoue, K.; Shibasaki, H.; Taniguchi, Y.; Kitamura, T.; Fujiwara, Y. An Efficient Partial Oxidation of Methane in Trifluoroacetic Acid Using Vanadium-Containing Heteropolyacid Catalysts. J. Organomet. Chem. 1999, 574, 116–120. [Google Scholar] [CrossRef]
- Ingrosso, G.; Midollini, N. Palladium(II)- or Copper(II)-Catalysed Solution-Phase Oxyfunctionalisation of Methane and Other Light Alkanes by Hydrogen Peroxide in Trifluoroacetic Anhydride. J. Mol. Catal. A Chem. 2003, 204–205, 425–431. [Google Scholar] [CrossRef]
- Park, E.D.; Hwang, Y.-S.; Lee, J.S. Direct Conversion of Methane into Oxygenates by H2O2 Generated in Situ from Dihydrogen and Dioxygen. Catal. Commun. 2001, 2, 187–190. [Google Scholar] [CrossRef]
- Park, E.D.; Hwang, Y.S.; Lee, C.W.; Lee, J.S. Copper- and Vanadium-Catalyzed Methane Oxidation into Oxygenates with in Situ Generated H2O2 over Pd/C. Appl. Catal. A Gen. 2003, 247, 269–281. [Google Scholar] [CrossRef]
- Lin, M.; Hogan, T.; Sen, A. A Highly Catalytic Bimetallic System for the Low-TemperatureSelective Oxidation of Methane and Lower Alkanes with Dioxygen as the Oxidant. J. Chem. Soc. Chem. Commun. 1997, 119, 6048–6053. [Google Scholar] [CrossRef]
- Park, E.D.; Choi, S.H.; Lee, J.S. Characterization of Pd/C and Cu Catalysts for the Oxidation of Methane to a Methanol Derivative. J. Catal. 2000, 194, 33–44. [Google Scholar] [CrossRef]
- Seki, Y.; Mizuno, N.; Misono, M. High-Yield Liquid-Phase Oxygenation of Methane with Hydrogen Peroxide Catalyzed by 12-Molybdovanadophosphoric Acid Catalyst Precursor. Appl. Catal. A Gen. 1997, 158, 47–51. [Google Scholar] [CrossRef]
- Vargaftik, M.N.; Stolarov, I.P.; Moiseev, I.I. Highly Selective Partial Oxidation of Methane to Methyl Trifluoroacetate. J. Chem. Soc. Chem. Commun. 1990, 1049–1050. [Google Scholar] [CrossRef]
- Strassner, T.; Ahrens, S.; Muehlhofer, M.; Munz, D.; Zeller, A. Cobalt-Catalyzed Oxidation of Methane to Methyl Trifluoroacetate by Dioxygen. Eur. J. Inorg. Chem. 2013, 2013, 3659–3663. [Google Scholar] [CrossRef]
- Blankenship, A.N.; Ravi, M.; Newton, M.A.; van Bokhoven, J.A. Heterogeneously Catalyzed Aerobic Oxidation of Methane to a Methyl Derivative. Angew. Chem.—Int. Ed. 2021, 60, 18138–18143. [Google Scholar] [CrossRef]
- Ji, Y.; Blankenship, A.N.; van Bokhoven, J.A. Heterogeneous Mn-Based Catalysts for the Aerobic Conversion of Methane-to-Methyl Trifluoroacetate. ACS Catal. 2023, 13, 3896–3901. [Google Scholar] [CrossRef]
- Periana, R.A.; Taube, D.J.; Gamble, S.; Taube, H.; Satoh, T.; Fujii, H. Platinum Catalysts for the High-Yield Oxidationof Methane to a Methanol Derivative. Science 1998, 280, 560–564. [Google Scholar] [CrossRef]
- Periana, R.A.; Taube, D.J.; Evitt, E.R.; Loffler, D.G.; Wentrcek, P.R.; Voss, G.; Masuda, T. A Mercury-Catalyzed, High-Yield System for the Oxidation of Methane to Methanol. Science 1993, 259, 340–343. [Google Scholar] [CrossRef]
- Zimmermann, T.; Bilke, M.; Soorholtz, M.; Schüth, F. Influence of Catalyst Concentration on Activity and Selectivity in Selective Methane Oxidation with Platinum Compounds in Sulfuric Acid and Oleum. ACS Catal. 2018, 8, 9262–9268. [Google Scholar] [CrossRef]
- Dang, H.T.; Lee, H.W.; Lee, J.; Choo, H.; Hong, S.H.; Cheong, M.; Lee, H. Enhanced Catalytic Activity of (DMSO)2PtCl2 for the Methane Oxidation in the SO3-H2SO4 System. ACS Catal. 2018, 8, 11854–11862. [Google Scholar] [CrossRef]
- Lee, H.W.; Dang, H.T.; Kim, H.; Lee, U.; Ha, J.M.; Jae, J.; Cheong, M.; Lee, H. Pt Black Catalyzed Methane Oxidation to Methyl Bisulfate in H2SO4-SO3. J. Catal. 2019, 374, 230–236. [Google Scholar] [CrossRef]
- Palkovits, R.; Antonietti, M.; Kuhn, P.; Thomas, A.; Schüth, F. Solid Catalysts for the Selective Low-Temperature Oxidation of Methane to Methanol. Angew. Chem.—Int. Ed. 2009, 48, 6909–6912. [Google Scholar] [CrossRef]
- Zimmermann, T.; Soorholtz, M.; Bilke, M.; Schüth, F. Selective Methane Oxidation Catalyzed by Platinum Salts in Oleum at Turnover Frequencies of Large-Scale Industrial Processes. J. Am. Chem. Soc. 2016, 138, 12395–12400. [Google Scholar] [CrossRef]
- Park, E.D.; Lee, K.H.; Lee, J.S. Easily Separable Molecular Catalysis. Catal. Today 2000, 63, 147–157. [Google Scholar] [CrossRef]
- Soorholtz, M.; White, R.J.; Zimmermann, T.; Titirici, M.M.; Antonietti, M.; Palkovits, R.; Schüth, F. Direct Methane Oxidation over Pt-Modified Nitrogen-Doped Carbons. Chem. Commun. 2013, 49, 240–242. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Zerella, M.; Bell, A.T. A High-Yield, Liquid-Phase Approach for the Partial Oxidation of Methane to Methanol Using SO3 as the Oxidant. Adv. Synth. Catal. 2005, 347, 1203–1206. [Google Scholar] [CrossRef]
- Im, J.; Cheong, S.H.; Dang, H.T.; Kim, N.K.; Hwang, S.; Lee, K.B.; Kim, K.; Lee, H.; Lee, U. Economically Viable Co-Production of Methanol and Sulfuric Acid via Direct Methane Oxidation. Commun. Chem. 2023, 6, 282. [Google Scholar] [CrossRef]
- Koo, C.W.; Rosenzweig, A.C. Biochemistry of Aerobic Biological Methane Oxidation. Chem. Soc. Rev. 2021, 50, 3424–3436. [Google Scholar] [CrossRef]
- Hanson, R.S.; Hanson, T.E. Methanotrophic Bacteria. Microbiol. Rev. 1996, 60, 439–471. [Google Scholar] [CrossRef]
- Banerjee, R.; Proshlyakov, Y.; Lipscomb, J.D.; Proshlyakov, D.A. Structure of the Key Species in the Enzymatic Oxidation of Methane to Methanol. Nature 2015, 518, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liang, A.D.; Lippard, S.J. Coupling Oxygen Consumption with Hydrocarbon Oxidation in Bacterial Multicomponent Monooxygenases. Acc. Chem. Res. 2015, 48, 2632–2639. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, R.L.; Rosenzweig, A.C. Crystal Structure of a Membrane-Bound Metalloenzyme That Catalyses the Biological Oxidation of Methane. Nature 2005, 434, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Lawton, T.J.; Ham, J.; Sun, T.; Rosenzweig, A.C. Structural conservation of the B subunit in the ammonia monooxygenase/particulate methane monooxygenase superfamily. Proteins 2014, 82, 2263–2267. [Google Scholar] [CrossRef]
- Cao, L.; Caldararu, O.; Rosenzweig, A.C.; Ryde, U. Quantum Refinement Does Not Support Dinuclear Copper Sites in Crystal Structures of Particulate Methane Monooxygenase. Angew. Chem.—Int. Ed. 2018, 57, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Shiota, Y.; Yoshizawa, K. Comparison of the Reactivity of Bis(μ-Oxo)CuIICuIII and CuIIICuIII Species to Methane. Inorg. Chem. 2009, 48, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, K.; Shiota, Y. Conversion of Methane to Methanol at the Mononuclear and Dinuclear Copper Sites of Particulate Methane Monooxygenase (PMMO): A DFT and QM/MM Study. J. Am. Chem. Soc. 2006, 128, 9873–9881. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.K.M.L.; Kumashiro, M.; Ishihara, T. Direct Synthesis of Formic Acid by Partial Oxidation of Methane on H-ZSM-5 Solid Acid Catalyst. Catal. Commun. 2011, 12, 1198–1200. [Google Scholar] [CrossRef]
- Hammond, C.; Forde, M.M.; Ab Rahim, M.H.; Thetford, A.; He, Q.; Jenkins, R.L.; Dimitratos, N.; Lopez-Sanchez, J.A.; Dummer, N.F.; Murphy, D.M.; et al. Direct Catalytic Conversion of Methane to Methanol in an Aqueous Medium by Using Copper-Promoted Fe-ZSM-5. Angew. Chem.—Int. Ed. 2012, 51, 5129–5133. [Google Scholar] [CrossRef]
- Bordiga, S.; Buzzoni, R.; Geobaldo, F.; Lamberti, C.; Giamello, E.; Zecchina, A.; Leofanti, G.; Petrini, G.; Tozzola, G.; Vlaic, G. Structure and Reactivity of Framework and Extraframework Iron in Fe-Silicalite as Investigated by Spectroscopic and Physicochemical Methods. J. Catal. 1996, 158, 486–501. [Google Scholar] [CrossRef]
- Voskoboinikov, T.V.; Chen, H.-Y.; Sachtler, W.M.H. On the Nature of Active Sites in Fe/ZSM-5 Catalysts for NOx Abatement. Appl. Catal. B 1998, 19, 279–287. [Google Scholar] [CrossRef]
- Chen, H.-Y.; Sachtler, W.M.H. Activity and Durability of Fe/ZSM-5 Catalysts for Lean Burn NOx Reduction in the Presence of Water Vapor. Catal. Today 1998, 42, 73–83. [Google Scholar] [CrossRef]
- Hammond, C.; Jenkins, R.L.; Dimitratos, N.; Lopez-Sanchez, J.A.; Ab Rahim, M.H.; Forde, M.M.; Thetford, A.; Murphy, D.M.; Hagen, H.; Stangland, E.E.; et al. Catalytic and Mechanistic Insights of the Low-Temperature Selective Oxidation of Methane over Cu-Promoted Fe-ZSM-5. Chem.—A Eur. J. 2012, 18, 15735–15745. [Google Scholar] [CrossRef]
- Hammond, C.; Dimitratos, N.; Jenkins, R.L.; Lopez-Sanchez, J.A.; Kondrat, S.A.; Hasbi Ab Rahim, M.; Forde, M.M.; Thetford, A.; Taylor, S.H.; Hagen, H.; et al. Elucidation and Evolution of the Active Component within Cu/Fe/ZSM-5 for Catalytic Methane Oxidation: From Synthesis to Catalysis. ACS Catal. 2013, 3, 689–699. [Google Scholar] [CrossRef]
- Forde, M.M.; Armstrong, R.D.; McVicker, R.; Wells, P.P.; Dimitratos, N.; He, Q.; Lu, L.; Jenkins, R.L.; Hammond, C.; Lopez-Sanchez, J.A.; et al. Light Alkane Oxidation Using Catalysts Prepared by Chemical Vapour Impregnation: Tuning Alcohol Selectivity through Catalyst Pre-Treatment. Chem. Sci. 2014, 5, 3603–3616. [Google Scholar] [CrossRef]
- Hammond, C.; Hermans, I.; Dimitratos, N. Biomimetic Oxidation with Fe-ZSM-5 and H2O2-Identification of an Active, Extra-Framework Binuclear Core and an FeIII-OOH Intermediate with Resonance-Enhanced Raman Spectroscopy. ChemCatChem 2015, 7, 434–440. [Google Scholar] [CrossRef]
- Xu, J.; Armstrong, R.D.; Shaw, G.; Dummer, N.F.; Freakley, S.J.; Taylor, S.H.; Hutchings, G.J. Continuous Selective Oxidation of Methane to Methanol over Cu- and Fe-Modified ZSM-5 Catalysts in a Flow Reactor. Catal. Today 2016, 270, 93–100. [Google Scholar] [CrossRef]
- Kim, M.S.; Park, E.D. Aqueous-Phase Partial Oxidation of Methane with H2O2 over Fe-ZSM-5 Catalysts Prepared from Different Iron Precursors. Microporous Mesoporous Mater. 2021, 324, 111278. [Google Scholar] [CrossRef]
- Kim, M.S.; Park, K.H.; Cho, S.J.; Park, E.D. Partial Oxidation of Methane with Hydrogen Peroxide over Fe-ZSM-5 Catalyst. Catal. Today 2021, 376, 113–118. [Google Scholar] [CrossRef]
- Zhu, K.; Liang, S.; Cui, X.; Huang, R.; Wan, N.; Hua, L.; Li, H.; Chen, H.; Zhao, Z.; Hou, G.; et al. Highly Efficient Conversion of Methane to Formic Acid under Mild Conditions at ZSM-5-Confined Fe-Sites. Nano Energy 2021, 82, 105718. [Google Scholar] [CrossRef]
- Oda, A.; Aono, K.; Murata, N.; Murata, K.; Yasumoto, M.; Tsunoji, N.; Sawabe, K.; Satsuma, A. Rational Design of ZSM-5 Zeolite Containing a High Concentration of Single Fe Sites Capable of Catalyzing the Partial Oxidation of Methane with High Turnover Frequency. Catal. Sci. Technol. 2022, 12, 542–550. [Google Scholar] [CrossRef]
- Yu, T.; Li, Z.; Lin, L.; Chu, S.; Su, Y.; Song, W.; Wang, A.; Weckhuysen, B.M.; Luo, W. Highly Selective Oxidation of Methane into Methanol over Cu-Promoted Monomeric Fe/ZSM-5. ACS Catal. 2021, 11, 6684–6691. [Google Scholar] [CrossRef]
- Al-Shihri, S.; Richard, C.J.; Chadwick, D. Selective Oxidation of Methane to Methanol over ZSM-5 Catalysts in Aqueous Hydrogen Peroxide: Role of Formaldehyde. ChemCatChem 2017, 9, 1276–1283. [Google Scholar] [CrossRef]
- Al-Shihri, S.; Richard, C.J.; Al-Megren, H.; Chadwick, D. Insights into the Direct Selective Oxidation of Methane to Methanol over ZSM-5 Zeolytes in Aqueous Hydrogen Peroxide. Catal. Today 2020, 353, 269–278. [Google Scholar] [CrossRef]
- Hammond, C.; Dimitratos, N.; Lopez-Sanchez, J.A.; Jenkins, R.L.; Whiting, G.; Kondrat, S.A.; Ab Rahim, M.H.; Forde, M.M.; Thetford, A.; Hagen, H.; et al. Aqueous-Phase Methane Oxidation over Fe-MFI Zeolites; Promotion through Isomorphous Framework Substitution. ACS Catal. 2013, 3, 1835–1844. [Google Scholar] [CrossRef]
- Shahami, M.; Shantz, D.F. Zeolite Acidity Strongly Influences Hydrogen Peroxide Activation and Oxygenate Selectivity in the Partial Oxidation of Methane over M,Fe-MFI (M: Ga, Al, B) Zeolites. Catal. Sci. Technol. 2019, 9, 2945–2951. [Google Scholar] [CrossRef]
- Kalamaras, C.; Palomas, D.; Bos, R.; Horton, A.; Crimmin, M.; Hellgardt, K. Selective Oxidation of Methane to Methanol over Cu- And Fe-Exchanged Zeolites: The Effect of Si/Al Molar Ratio. Catal. Lett. 2016, 146, 483–492. [Google Scholar] [CrossRef]
- Fang, Z.; Murayama, H.; Zhao, Q.; Liu, B.; Jiang, F.; Xu, Y.; Tokunaga, M.; Liu, X. Selective Mild Oxidation of Methane to Methanol or Formic Acid on Fe-MOR Catalysts. Catal. Sci. Technol. 2019, 9, 6946–6956. [Google Scholar] [CrossRef]
- Xiao, P.; Wang, Y.; Nishitoba, T.; Kondo, J.N.; Yokoi, T. Selective Oxidation of Methane to Methanol with H2O2 over an Fe-MFI Zeolite Catalyst Using Sulfolane Solvent. Chem. Commun. 2019, 55, 2896–2899. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Yang, G.S.; Park, E.D. Effects of Cu Species on Liquid-Phase Partial Oxidation of Methane with H2O2 over Cu-Fe/ZSM-5 Catalysts. Catalysts 2022, 12, 1224. [Google Scholar] [CrossRef]
- Sun, S.; Barnes, A.J.; Gong, X.; Lewis, R.J.; Dummer, N.F.; Bere, T.; Shaw, G.; Richards, N.; Morgan, D.J.; Hutchings, G.J. Lanthanum Modified Fe-ZSM-5 Zeolites for Selective Methane Oxidation with H2O2. Catal. Sci. Technol. 2021, 11, 8052–8064. [Google Scholar] [CrossRef]
- Yu, X.; Wu, B.; Huang, M.; Lu, Z.; Li, J.; Zhong, L.; Sun, Y. IrFe/ZSM-5 Synergistic Catalyst for Selective Oxidation of Methane to Formic Acid. Energy Fuels 2021, 35, 4418–4427. [Google Scholar] [CrossRef]
- Huang, W.; Zhang, S.; Tang, Y.; Li, Y.; Nguyen, L.; Li, Y.; Shan, J.; Xiao, D.; Gagne, R.; Frenkel, A.I.; et al. Low-Temperature Transformation of Methane to Methanol on Pd1O4 Single Sites Anchored on the Internal Surface of Microporous Silicate. Angew. Chem.—Int. Ed. 2016, 55, 13441–13445. [Google Scholar] [CrossRef]
- Osadchii, D.Y.; Olivos-Suarez, A.I.; Szécsényi, Á.; Li, G.; Nasalevich, M.A.; Dugulan, I.A.; Crespo, P.S.; Hensen, E.J.M.; Veber, S.L.; Fedin, M.V.; et al. Isolated Fe Sites in Metal Organic Frameworks Catalyze the Direct Conversion of Methane to Methanol. ACS Catal. 2018, 8, 5542–5548. [Google Scholar] [CrossRef]
- Szécsényi, A.; Li, G.; Gascon, J.; Pidko, E.A. Unraveling Reaction Networks behind the Catalytic Oxidation of Methane with H2O2 over a Mixed-Metal MIL-53(Al,Fe) MOF Catalyst. Chem. Sci. 2018, 9, 6765–6773. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Shi, Y.; Jiang, Y.; Zhang, X.; Long, C.; An, P.; Zhu, Y.; Shao, S.; Yan, Z.; Li, G.; et al. Fe-O Clusters Anchored on Nodes of Metal–Organic Frameworks for Direct Methane Oxidation. Angew. Chem.—Int. Ed. 2021, 60, 5811–5815. [Google Scholar] [CrossRef]
- Lee, H.; Kwon, C.; Keum, C.; Kim, H.E.; Lee, H.; Han, B.; Lee, S.Y. Methane Partial Oxidation by Monomeric Cu Active Center Confined on ZIF-7. Chem. Eng. J. 2022, 450, 138472. [Google Scholar] [CrossRef]
- Lee, H.; Kwon, C.; Vikneshvaran, S.; Lee, S.; Lee, S.Y. Partial Oxidation of Methane to Methyl Oxygenates with Enhanced Selectivity Using a Single-Atom Copper Catalyst on Amorphous Carbon Support. Appl. Surf. Sci. 2023, 639, 158289. [Google Scholar] [CrossRef]
- Lee, H.; Lee, S.Y. High Metal Loaded Cu(i)N3 Single-Atom Catalysts: Superior Methane Conversion Activity and Selectivity under Mild Conditions. J. Mater. Chem. A Mater. 2023, 11, 15691–15701. [Google Scholar] [CrossRef]
- Ab Rahim, M.H.; Forde, M.M.; Jenkins, R.L.; Hammond, C.; He, Q.; Dimitratos, N.; Lopez-Sanchez, J.A.; Carley, A.F.; Taylor, S.H.; Willock, D.J.; et al. Oxidation of Methane to Methanol with Hydrogen Peroxide Using Supported Gold-Palladium Alloy Nanoparticles. Angew. Chem.—Int. Ed. 2013, 52, 1280–1284. [Google Scholar] [CrossRef]
- Agarwal, N.; Freakley, S.J.; McVicker, R.U.; Althahban, S.M.; Dimitratos, N.; He, Q.; Morgan, D.J.; Jenkins, R.L.; Willock, D.J.; Taylor, S.H.; et al. Aqueous Au-Pd Colloids Catalyze Selective CH4 Oxidation to CH3OH with O2 under Mild Conditions. Science 2017, 358, 223–227. [Google Scholar] [CrossRef]
- Xu, G.; Yu, A.; Xu, Y.; Sun, C. Selective Oxidation of Methane to Methanol Using AuPd@ZIF-8. Catal. Commun. 2021, 158, 106338. [Google Scholar] [CrossRef]
- McVicker, R.; Agarwal, N.; Freakley, S.J.; He, Q.; Althahban, S.; Taylor, S.H.; Kiely, C.J.; Hutchings, G.J. Low Temperature Selective Oxidation of Methane Using Gold-Palladium Colloids. Catal. Today 2020, 342, 32–38. [Google Scholar] [CrossRef]
- Colby, J.; Stirling, D.I.; Dalton, H. The Soluble Methane Mono-Oxygenase of Methylococcus Capsulatus (Bath). Its Ability to Oxygenate n-Alkanes, n-Alkenes, Ethers, and Alicyclic, Aromatic and Heterocyclic Compounds. Biochem. J. 1977, 165, 395–402. [Google Scholar] [CrossRef]
- Yan, Y.; Chen, C.; Zou, S.; Liu, J.; Xiao, L.; Fan, J. High H2O2 Utilization Promotes Selective Oxidation of Methane to Methanol at Low Temperature. Front. Chem. 2020, 8, 252. [Google Scholar] [CrossRef]
- Cui, X.; Li, H.; Wang, Y.; Hu, Y.; Hua, L.; Li, H.; Han, X.; Liu, Q.; Yang, F.; He, L.; et al. Room-Temperature Methane Conversion by Graphene-Confined Single Iron Atoms. Chem 2018, 4, 1902–1910. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, Y. Facile Synthesis of N-Doped Carbon Supported Iron Species for Highly Efficient Methane Conversion with H2O2 at Ambient Temperature. Appl. Catal. A Gen. 2021, 615, 118052. [Google Scholar] [CrossRef]
- Shen, Q.; Cao, C.; Huang, R.; Zhu, L.; Zhou, X.; Zhang, Q.; Gu, L.; Song, W. Single Chromium Atoms Supported on Titanium Dioxide Nanoparticles for Synergic Catalytic Methane Conversion under Mild Conditions. Angew. Chem.—Int. Ed. 2020, 59, 1216–1219. [Google Scholar] [CrossRef]
- Choudhary, V.R.; Sansare, S.D.; Gaikwad, A.G. Direct Oxidation of H2 to H2O2 and Decomposition of H2O2 over Oxidized and Reduced Pd-Containing Zeolite Catalysts in Acidic Medium. Catal. Lett. 2002, 84, 81–87. [Google Scholar] [CrossRef]
- Gaikwad, A.; Sansare, S.; Choudhary, V. Direct Oxidation of Hydrogen to Hydrogen Peroxide over Pd-Containing Fluorinated or Sulfated Al2O3, ZrO2, CeO2, ThO2, Y2O3 and Ga2O3 Catalysts in Stirred Slurry Reactor at Ambient Conditions. J. Mol. Catal. A Chem. 2002, 181, 143–149. [Google Scholar] [CrossRef]
- Park, S.; Kim, T.J.; Chung, Y.M.; Oh, S.H.; Song, I.K. Direct Synthesis of Hydrogen Peroxide from Hydrogen and Oxygen over Palladium Catalyst Supported on SO3H-Functionalized SBA-15. Catal. Lett. 2009, 130, 296–300. [Google Scholar] [CrossRef]
- Park, S.; Baeck, S.H.; Kim, T.J.; Chung, Y.M.; Oh, S.H.; Song, I.K. Direct Synthesis of Hydrogen Peroxide from Hydrogen and Oxygen over Palladium Catalyst Supported on SO3H-Functionalized Mesoporous Silica. J. Mol. Catal. A Chem. 2010, 319, 98–107. [Google Scholar] [CrossRef]
- Blanco-Brieva, G.; de Frutos Escrig, M.P.; Campos-Martin, J.M.; Fierro, J.L.G. Direct Synthesis of Hydrogen Peroxide on Palladium Catalyst Supported on Sulfonic Acid-Functionalized Silica. Green. Chem. 2010, 12, 1163–1166. [Google Scholar] [CrossRef]
- Park, S.-E.; Huang, L.; Lee, C.W.; Chang, J.-S. Generation of H2O2 from H2 and O2 over Zeolite Beta Containing Pd and Heterogenized Organic Compounds. Catal. Today 2000, 61, 117–122. [Google Scholar] [CrossRef]
- Li, G.; Edwards, J.; Carley, A.F.; Hutchings, G.J. Direct Synthesis of Hydrogen Peroxide from H2 and O2 Using Zeolite-Supported Au-Pd Catalysts. Catal. Today 2007, 122, 361–364. [Google Scholar] [CrossRef]
- Li, G.; Edwards, J.; Carley, A.F.; Hutchings, G.J. Direct Synthesis of Hydrogen Peroxide from H2 and O2 and in Situ Oxidation Using Zeolite-Supported Catalysts. Catal. Commun. 2007, 8, 247–250. [Google Scholar] [CrossRef]
- Park, S.; Lee, S.H.; Song, S.H.; Park, D.R.; Baeck, S.H.; Kim, T.J.; Chung, Y.M.; Oh, S.H.; Song, I.K. Direct Synthesis of Hydrogen Peroxide from Hydrogen and Oxygen over Palladium-Exchanged Insoluble Heteropolyacid Catalysts. Catal. Commun. 2009, 10, 391–394. [Google Scholar] [CrossRef]
- Alotaibi, F.; Al-Mayman, S.; Alotaibi, M.; Edwards, J.K.; Lewis, R.J.; Alotaibi, R.; Hutchings, G.J. Direct Synthesis of Hydrogen Peroxide Using Cs-Containing Heteropolyacid-Supported Palladium–Copper Catalysts. Catal. Lett. 2019, 149, 998–1006. [Google Scholar] [CrossRef]
- Puthiaraj, P.; Yu, K.; Ahn, W.S.; Chung, Y.M. Pd Nanoparticles on a Dual Acid-Functionalized Porous Polymer for Direct Synthesis of H2O2: Contribution by Enhanced H2 Storage Capacity. J. Ind. Eng. Chem. 2020, 81, 375–384. [Google Scholar] [CrossRef]
- Choudhary, V.R.; Samanta, C. Role of Chloride or Bromide Anions and Protons for Promoting the Selective Oxidation of H2 by O2 to H2O2 over Supported Pd Catalysts in an Aqueous Medium. J. Catal. 2006, 238, 28–38. [Google Scholar] [CrossRef]
- Lee, M.W.; Jo, D.Y.; Han, G.H.; Lee, K.Y. DFT Calculations on Selectivity Enhancement by Br Addition on Pd Catalysts in the Direct Synthesis of Hydrogen Peroxide. Catal. Today 2022, 397–399, 232–239. [Google Scholar] [CrossRef]
- Melada, S.; Rioda, R.; Menegazzo, F.; Pinna, F.; Strukul, G. Direct Synthesis of Hydrogen Peroxide on Zirconia-Supported Catalysts under Mild Conditions. J. Catal. 2006, 239, 422–430. [Google Scholar] [CrossRef]
- Kang, J.; Park, E.D. Aqueous-Phase Selective Oxidation of Methane with Oxygen over Iron Salts and Pd/C in the Presence of Hydrogen. ChemCatChem 2019, 11, 4247–4251. [Google Scholar] [CrossRef]
- Kang, J.; Park, E.D. Selective Oxidation of Methane over Fe-Zeolites by In Situ Generated H2O2. Catalysts 2020, 10, 299. [Google Scholar] [CrossRef]
- Kang, J.; Puthiaraj, P.; Ahn, W.S.; Park, E.D. Direct Synthesis of Oxygenates via Partial Oxidation of Methane in the Presence of O2 and H2 over a Combination of Fe-ZSM-5 and Pd Supported on an Acid-Functionalized Porous Polymer. Appl. Catal. A Gen. 2020, 602, 117711. [Google Scholar] [CrossRef]
- Yang, G.S.; Kang, J.; Park, E.D. Aqueous-Phase Partial Oxidation of Methane over Pd−Fe/ZSM-5 with O2 in the Presence of H2. ChemCatChem 2023, 15, e202201630. [Google Scholar] [CrossRef]
- Kang, J.; Park, E.D. Partial Oxidation of Methane over Fe/ZSM-5 with Hydrogen Peroxide Generated in Situ over Pd/C in the Presence of Halide Ions. Catal. Today 2024, 426, 114367. [Google Scholar] [CrossRef]
- Wu, B.; Lin, T.; Huang, M.; Li, S.; Li, J.; Yu, X.; Yang, R.; Sun, F.; Jiang, Z.; Sun, Y.; et al. Tandem Catalysis for Selective Oxidation of Methane to Oxygenates Using Oxygen over PdCu/Zeolite. Angew. Chem.—Int. Ed. 2022, 61, e2022041. [Google Scholar] [CrossRef]
- Wang, S.; Fung, V.; Hülsey, M.J.; Liang, X.; Yu, Z.; Chang, J.; Folli, A.; Lewis, R.J.; Hutchings, G.J.; He, Q.; et al. H2-Reduced Phosphomolybdate Promotes Room-Temperature Aerobic Oxidation of Methane to Methanol. Nat. Catal. 2023, 6, 895–905. [Google Scholar] [CrossRef]
- He, Y.; Luan, C.; Fang, Y.; Feng, X.; Peng, X.; Yang, G.; Tsubaki, N. Low-Temperature Direct Conversion of Methane to Methanol over Carbon Materials Supported Pd-Au Nanoparticles. Catal. Today 2020, 339, 48–53. [Google Scholar] [CrossRef]
- He, Y.; Liang, J.; Imai, Y.; Ueda, K.; Li, H.; Guo, X.; Yang, G.; Yoneyama, Y.; Tsubaki, N. Highly Selective Synthesis of Methanol from Methane over Carbon Materials Supported Pd-Au Nanoparticles under Mild Conditions. Catal. Today 2020, 352, 104–110. [Google Scholar] [CrossRef]
- Jin, Z.; Wang, L.; Zuidema, E.; Mondal, K.; Zhang, M.; Zhang, J.; Wang, C.; Meng, X.; Yang, H.; Mesters, C.; et al. Hydrophobic Zeolite Modification for in Situ Peroxide Formation in Methane Oxidation to Methanol. Science 2020, 367, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Qi, G.; Davies, T.E.; Nasrallah, A.; Sainna, M.A.; Howe, A.G.R.; Lewis, R.J.; Quesne, M.; Catlow, C.R.A.; Willock, D.J.; He, Q.; et al. Au-ZSM-5 Catalyses the Selective Oxidation of CH4 to CH3OH and CH3COOH Using O2. Nat. Catal. 2022, 5, 45–54. [Google Scholar] [CrossRef]
- Mehmood, A.; Chae, S.Y.; Park, E.D. Photoelectrochemical Conversion of Methane into Value-Added Products. Catalysts 2021, 11, 1387. [Google Scholar] [CrossRef]
- Mehmood, A.; Chae, S.Y.; Park, E.D. Low-Temperature Electrochemical Oxidation of Methane into Alcohols. Catalysts 2024, 14, 58. [Google Scholar] [CrossRef]
- Yuniar, G.; Saputera, W.H.; Sasongko, D.; Mukti, R.R.; Rizkiana, J.; Devianto, H. Recent Advances in Photocatalytic Oxidation of Methane to Methanol. Molecules 2022, 27, 5496. [Google Scholar] [CrossRef] [PubMed]
- Belousov, A.S.; Shafiq, I. Heterogeneous Photocatalysis for C–H Bond Activation. J. Environ. Chem. Eng. 2023, 11, 110970. [Google Scholar] [CrossRef]
- Jia, T.; Wang, W. Research Progress and Outlook on Photocatalytic Conversion of Methane to Methanol. ChemCatChem 2024, e202301279. [Google Scholar] [CrossRef]
- Liang, C.; He, B. A Titration Method for Determining Individual Oxidant Concentration in the Dual Sodium Persulfate and Hydrogen Peroxide Oxidation System. Chemosphere 2018, 198, 297–302. [Google Scholar] [CrossRef]
- Forde, M.M.; Armstrong, R.D.; Hammond, C.; He, Q.; Jenkins, R.L.; Kondrat, S.A.; Dimitratos, N.; Lopez-Sanchez, J.A.; Taylor, S.H.; Willock, D.; et al. Partial Oxidation of Ethane to Oxygenates Using Fe- and Cu-Containing ZSM-5. J. Am. Chem. Soc. 2013, 135, 11087–11099. [Google Scholar] [CrossRef]
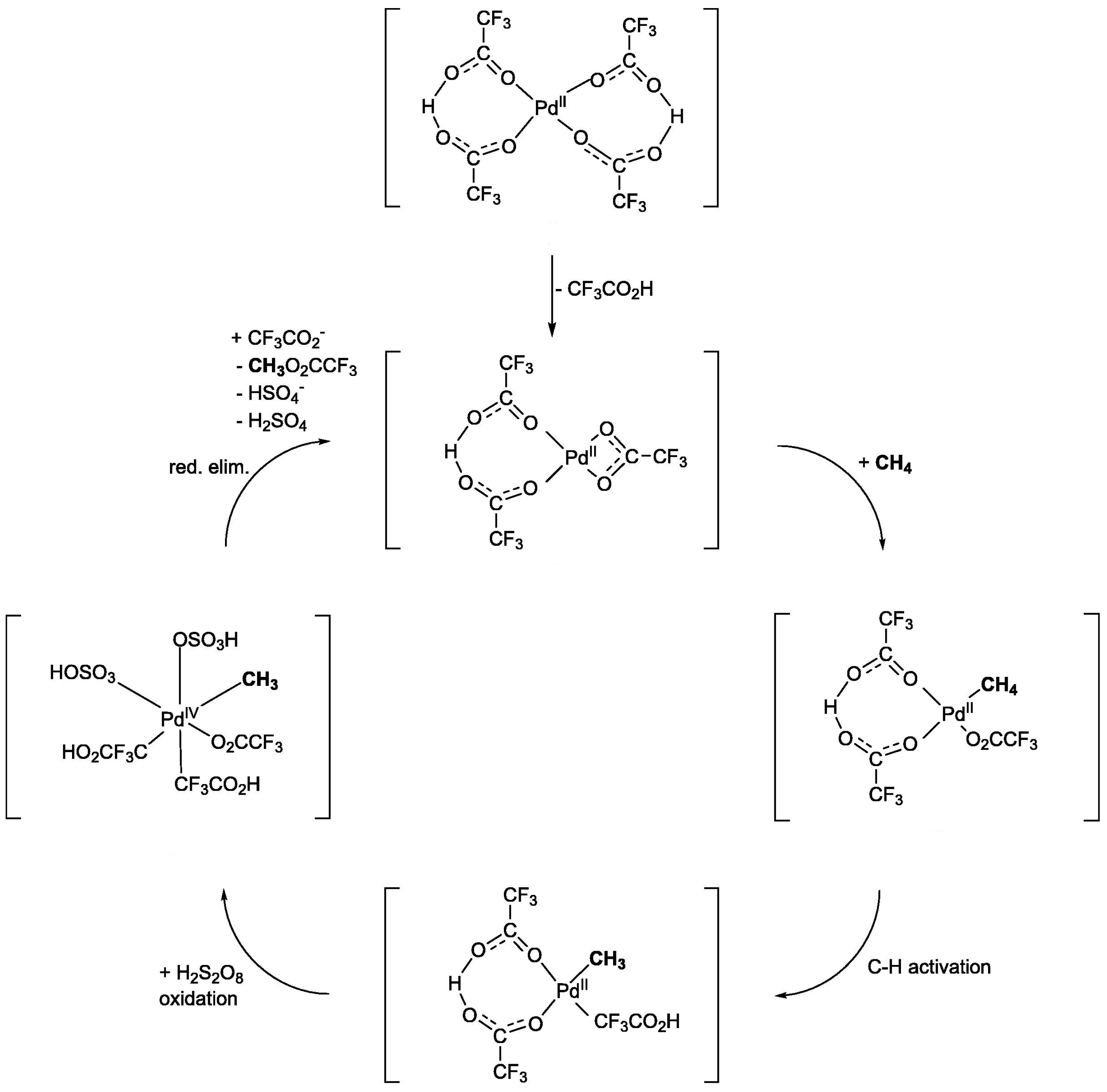

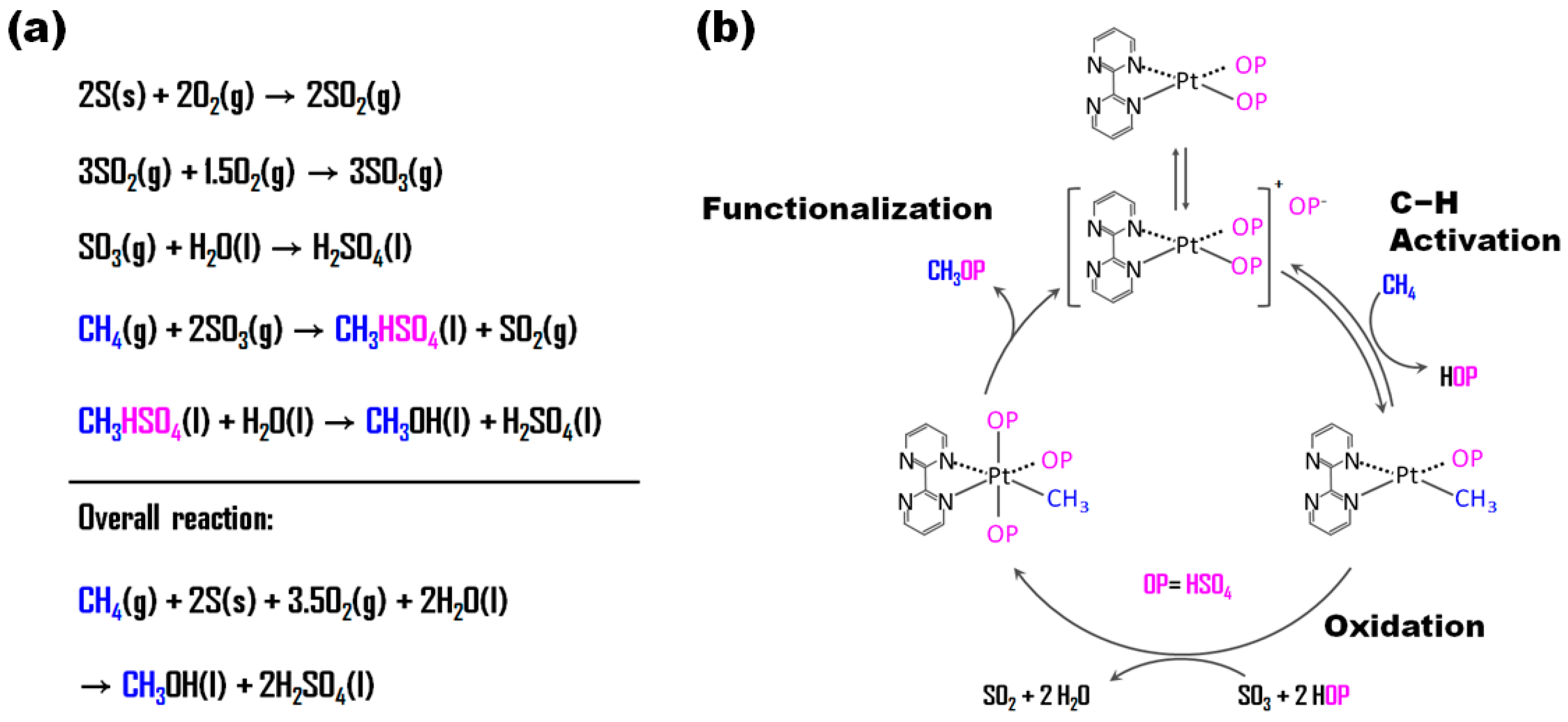

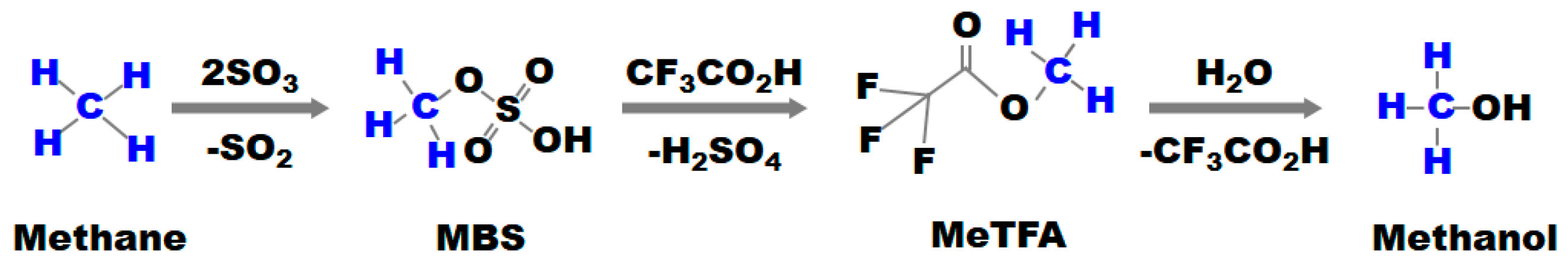
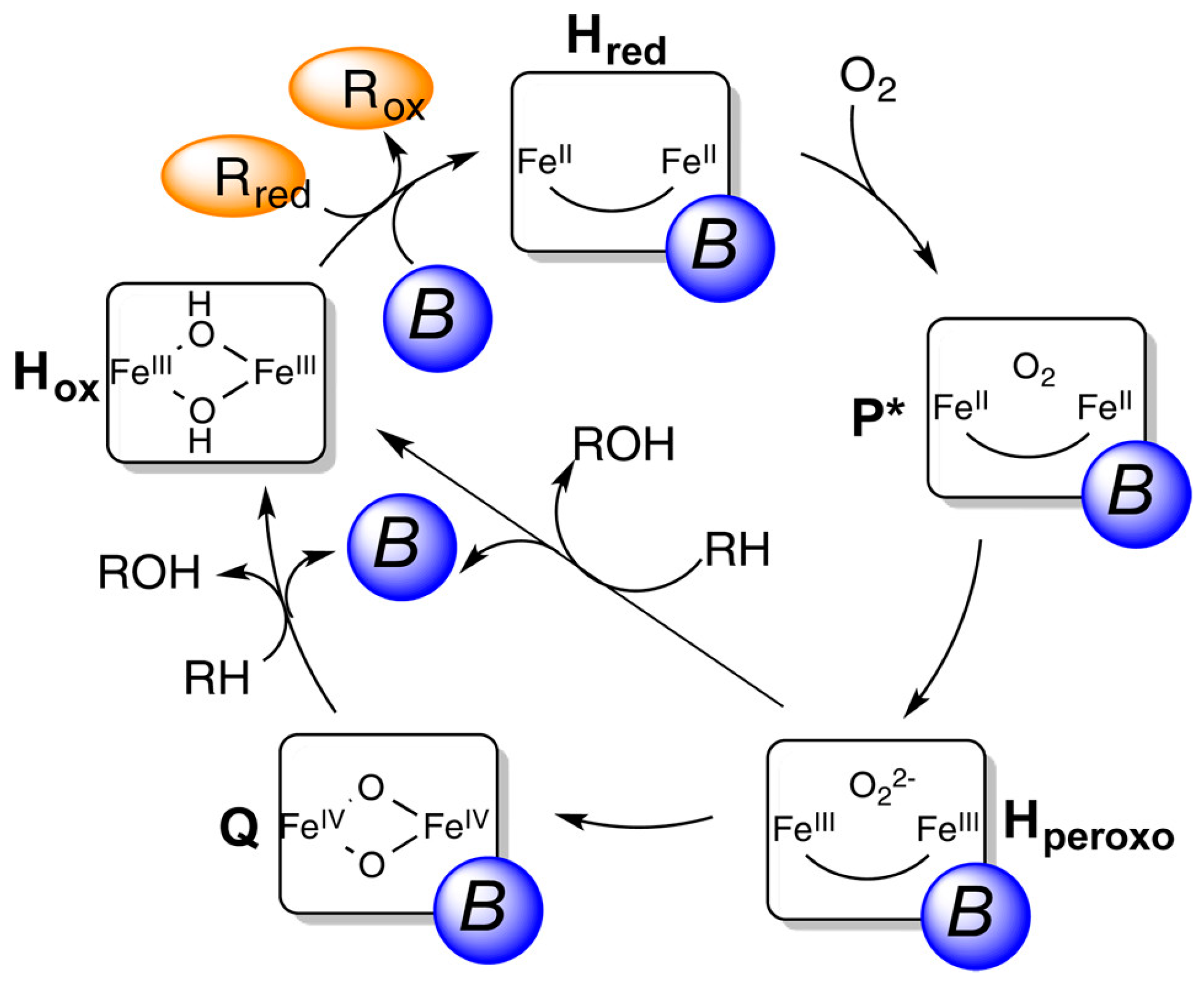

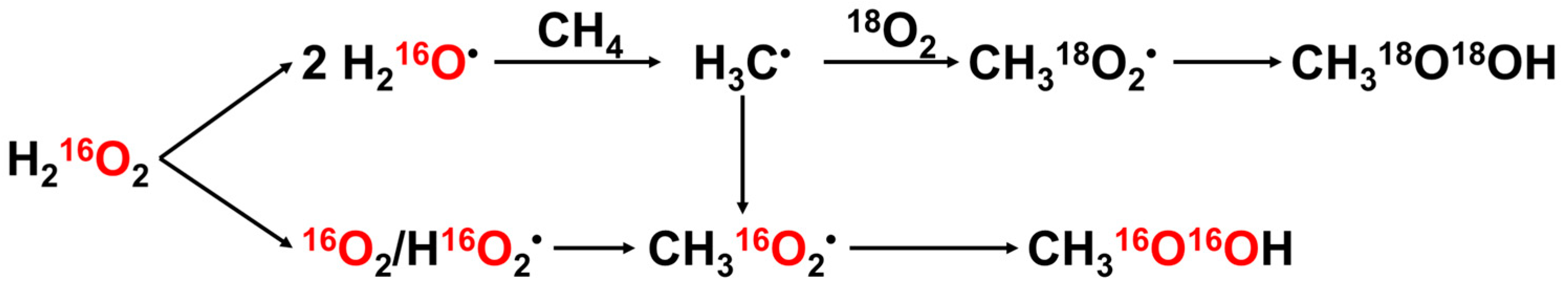
| Entry | Catalyst | Temp. (°C) | H2O2 (mmol) | Gas Composition (bar) | Solvent Composition (vol.%) | TON | TOF (h−1) | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | CuCl2 + Pd/C | 90 | - | CH4:O2:CO = 61:6.8:13.6 | HTFA:H2O = 75:25 | 30 | 0.33 | [29] |
| 2 | H4PVMo11O40 | 80 | 10 | CH4 = 50 | TFAA = 100 | 260 | 10.8 | [31] |
| 3 | 5%Pd/C + Cu(OAc)2 | 80 | - | CH4:O2:H2 = 34:6.8:6.8 | HTFA/TFAA = 80:20 | 20 | 4 | [28] |
| 4 | 5%Pd/C + NH4VO3 | 80 | - | CH4:O2:H2 = 34:6.8:6.8 | HTFA/TFAA = 80:20 | 3.7 | 0.7 | [28] |
| 5 | Cu(OAc)2 | 80 | 10 | CH4:N2 = 34:13.6 | TFAA = 100 | 1.0 | 0.5 | [28] |
| Entry | Catalyst | Temp. (°C) | SO3 (mmol) | PCH4 (bar) | TON c | TOF (h−1) | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | HgSO4 | 180 | - | 34.5 | 10.8 | 3.6 | [37] |
| 2 | K2PtCl4 | 215 | 70 | 72 | n.d. | 22,998 | [38] |
| 3 | (DMSO)2PtCl2 a | 180 | 75 | 35 | 19,125 | 6375 | [39] |
| 4 | Pt black | 180 | 75 | 35 | 1982 | 661 | [40] |
| 5 | K2[PtCl4]-CTF b | 215 | 103 | 40 | 201 | 80.4 | [41] |
| 6 | Pt-CTF b | 215 | 103 | 40 | 246 | 98.4 | [41] |
| Entry | Catalyst | Temp. (°C) | H2O2 (mmol) | CH4 (bar) | Total Productivity (mmol/gcat./h) | Product Selectivity (%) | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | 2.5%Cu-2.5%Fe/ZSM-5 | 50 | 5 | 30.5 | 16.5 | CH3OH: 85 HCOOH: 0 CO2: 15 | [57] |
| 2 | 0.5%LaFe-ZSM-5(H2) | 50 | 5 | 30.5 | 59.5 | CH3OH: 6 HCOOH: 90 CO2: 4 | [79] |
| 3 | 0.5%LaFeCu-ZSM-5(H2) | 50 | 5 | 30.5 | 12.6 | CH3OH: 85 HCOOH: 0 CO2: 15 | [79] |
| 4 | 0.5%LaFeCu-ZSM-5(Air) | 50 | 5 | 30.5 | 4.6 | CH3OH: 51 HCOOH: 43 CO2: 2 | [79] |
| 5 | 0.1%Ir0.6%Fe/ZSM-5 | 50 | 5 | 28.5 | 3.5 | CH3OH: 16 HCOOH: 71 CO2: 8 | [80] |
| 6 | 0.7%Fe/ZSM-5 | 50 | 5 | 28.5 | 1.0 | CH3OH: 17 HCOOH: 54 CO2: 4 | [80] |
| 7 | 0.01%Pd/ZSM-5 | 50 | 5 | 30 | 8.0 | CH3OH: 7 HCOOH: 54 CO2: 14 | [81] |
| Entry | Catalyst | Temp. (°C) | H2O2 (mmol) | Feed Composition (bar) | Total Productivity (mmol/gcat./h) | Product Selectivity (%) a | Ref. |
|---|---|---|---|---|---|---|---|
| 1 b | MIL-53(Al,Fe) | 40–60 | 5 | CH4 = 30.5 | 7.8 | CH3OH: 44 HCOOH: 21 CO2:36 | [82] |
| 2 | UiO-66(2.5TFA)-Fe | 50 | 3 | CH4 = 30 | 4.9 | CH3OH: 13 HCOOH: 63 CO2: 2 | [84] |
| 3 | Cu-ZIF-7 | 50 | 5 | CH4 = 28.5 | 1.1 | CH3OH: 22 HCOOH: 0 CO2: 71 | [85] |
| 4 | CuCZ8-20 | 40 | 10 | CH4 = 30 | 0.5 | CH3OH: 41 HCOOH: 0 CO2: 49 | [86] |
| 5 | CuNC-600 | 50 | 5 | CH4 = 30 | 4.0 | CH3OH: 80 HCOOH: 0 CO2: 11 | [87] |
| Entry | Solvent | Oxidant | Advantages | Disadvantages |
|---|---|---|---|---|
| 1 | CF3COOH | K2S2O8 | Relatively high yields of MeTFA | Corrosive solvent Solvent decomposition Waste (KHSO4) from an oxidant |
| 2 | CF3COOH | H2O2 | Relatively high yields of MeTFA | Corrosive solvent TFAA is required H2O2 is expensive |
| 3 | CF3COOH | CO/H2O/O2 | Relatively high yields of MeTFA O2 can be indirectly used | Corrosive solvent CO is required Waste (CO2) from an oxidant |
| 4 | CF3COOH | H2/O2 | Relatively high yields of MeTFA O2 can be indirectly used | Corrosive solvent H2 and TFAA are required |
| 5 | CF3COOH | O2 | O2 can be directly used | Corrosive solvent Relatively high reaction temperatures Very low yields of MeTFA Solvent decomposition |
| 6 | H2SO4 | SO3 | High yields of MBS O2 can be indirectly used | Corrosive solvent Inevitable H2SO4 co-production |
| 7 | H2O | H2O2 | Relatively low product yields with an exception (Table S1) No waste from an oxidant | H2O2 is expensive |
| 8 | H2O | H2/O2 | Relatively low product yields with an exception [119] O2 can be indirectly used No waste from an oxidant | H2 is required |
| 9 | H2O | CO/H2O/O2 | Relatively low product yields O2 can be indirectly used | CO is required Waste (CO2) from an oxidant |
| 10 | H2O | O2 | O2 can be directly used No waste from an oxidant | Very low product yields |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, J.; Park, E.D. Liquid-Phase Selective Oxidation of Methane to Methane Oxygenates. Catalysts 2024, 14, 167. https://doi.org/10.3390/catal14030167
Kang J, Park ED. Liquid-Phase Selective Oxidation of Methane to Methane Oxygenates. Catalysts. 2024; 14(3):167. https://doi.org/10.3390/catal14030167
Chicago/Turabian StyleKang, Jongkyu, and Eun Duck Park. 2024. "Liquid-Phase Selective Oxidation of Methane to Methane Oxygenates" Catalysts 14, no. 3: 167. https://doi.org/10.3390/catal14030167
APA StyleKang, J., & Park, E. D. (2024). Liquid-Phase Selective Oxidation of Methane to Methane Oxygenates. Catalysts, 14(3), 167. https://doi.org/10.3390/catal14030167