

Gas-Phase Oxidative Dehydrogenation of n-Octane over Metal Oxide Catalysts: A Review



Abstract
1. Introduction
1.1. Importance of Alkane Activation and Associated Products
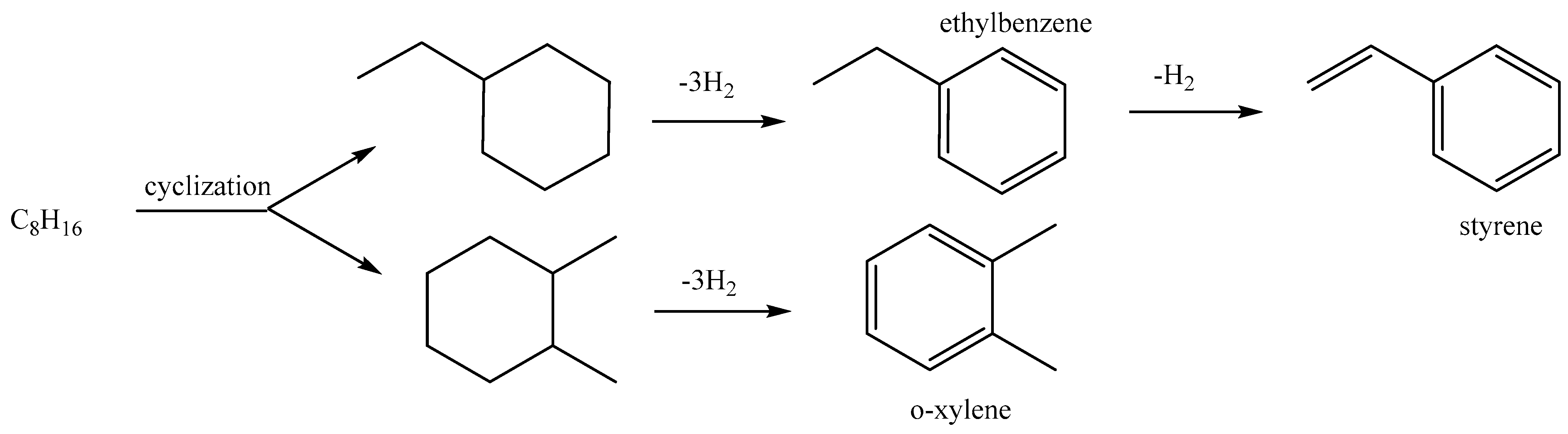
1.2. Conversion of Intermediate Chain Alkanes
1.3. Challenges Associated with Converting n-Octane to Value-Added Products
2. Oxidants
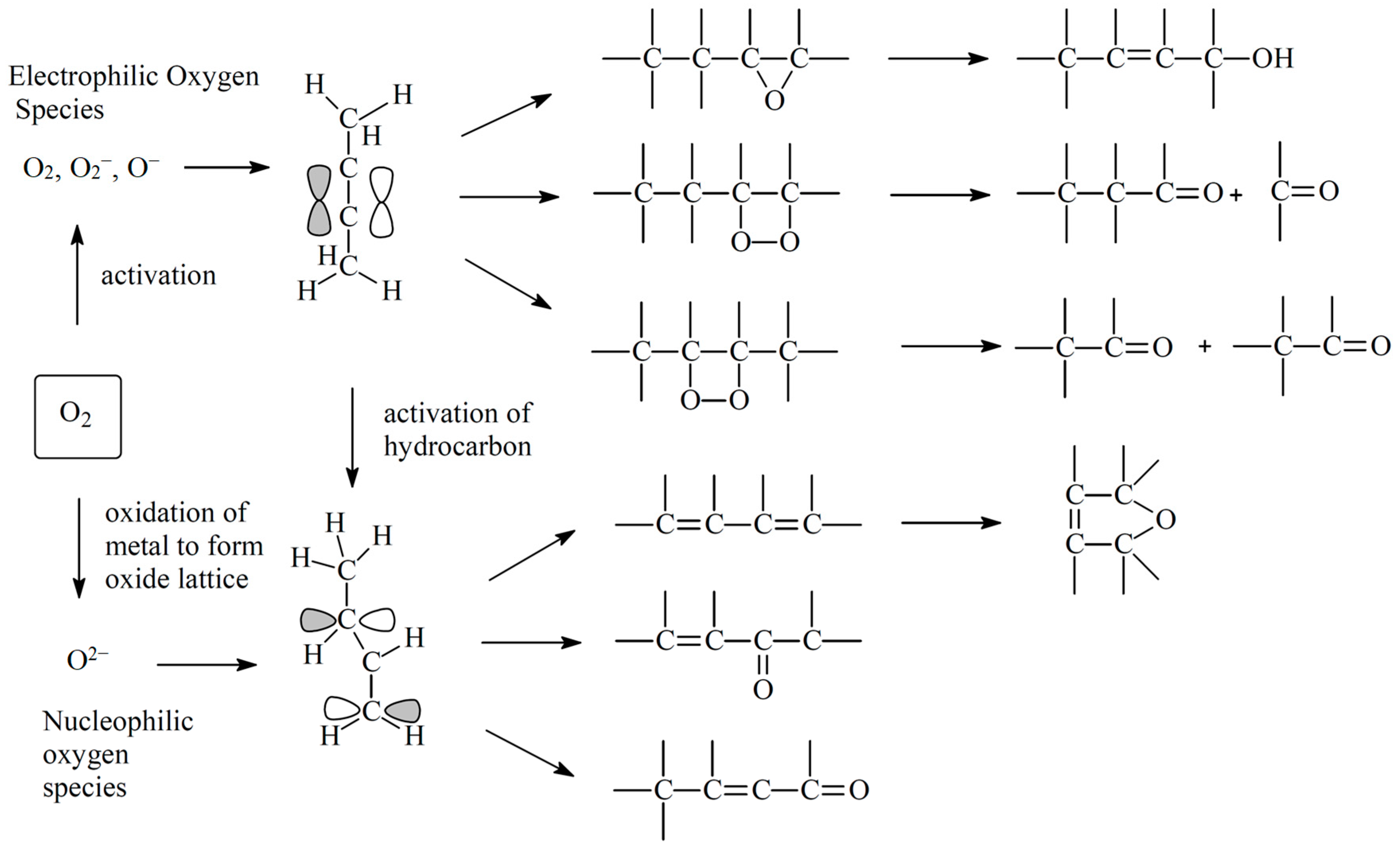
2.1. Molecular Oxygen
2.2. Carbon Dioxide
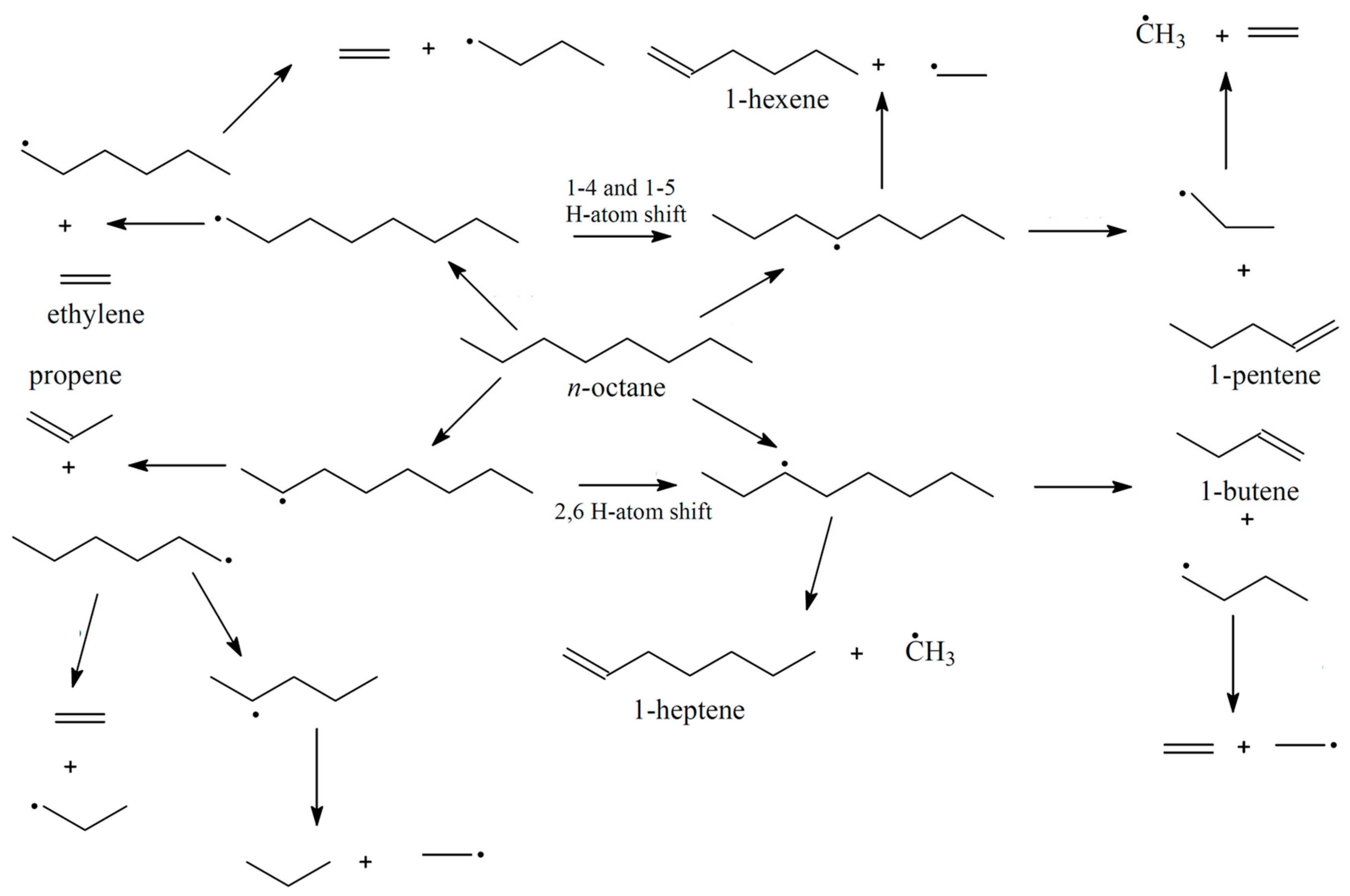
3. Mechanisms of Alkane Oxidation
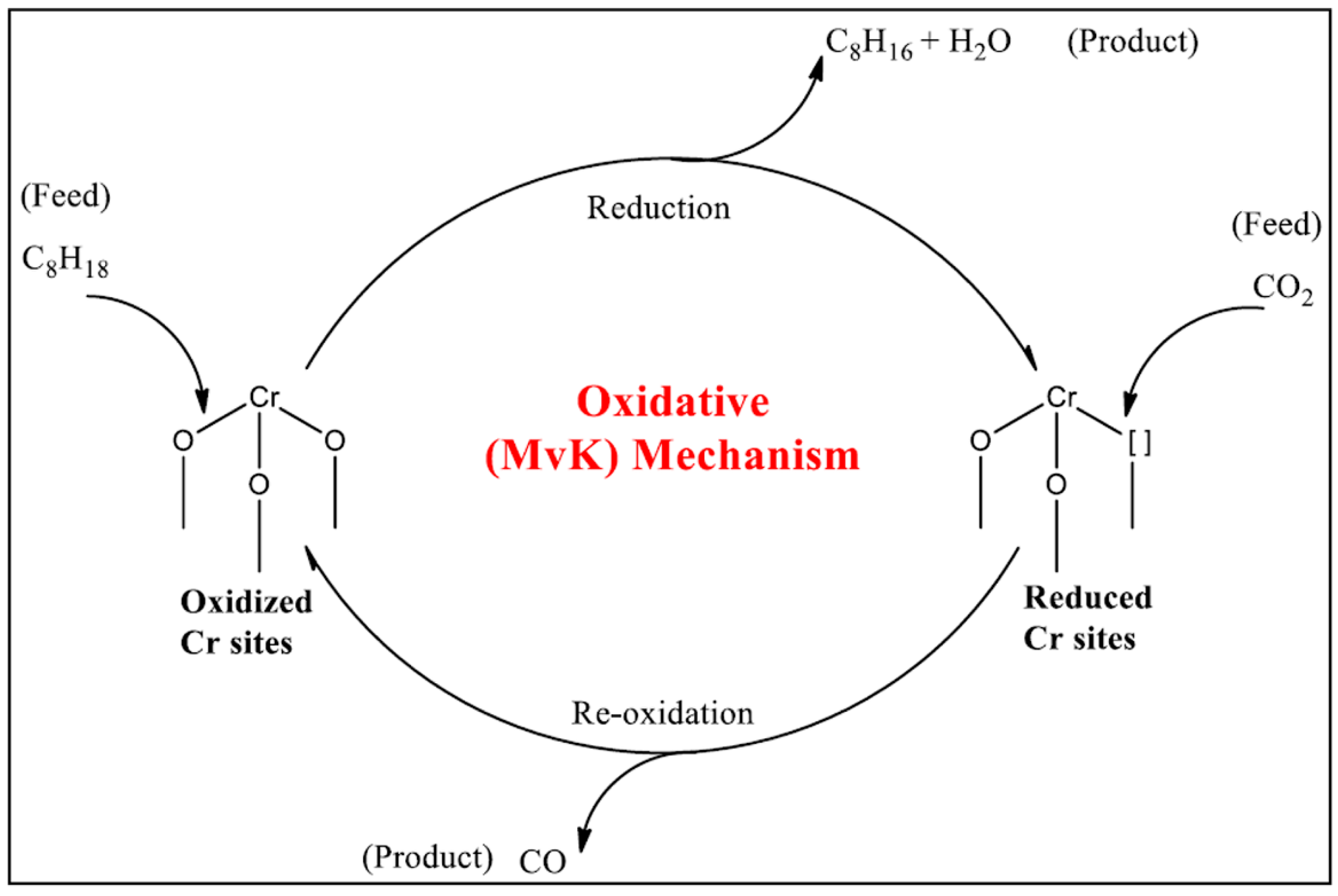
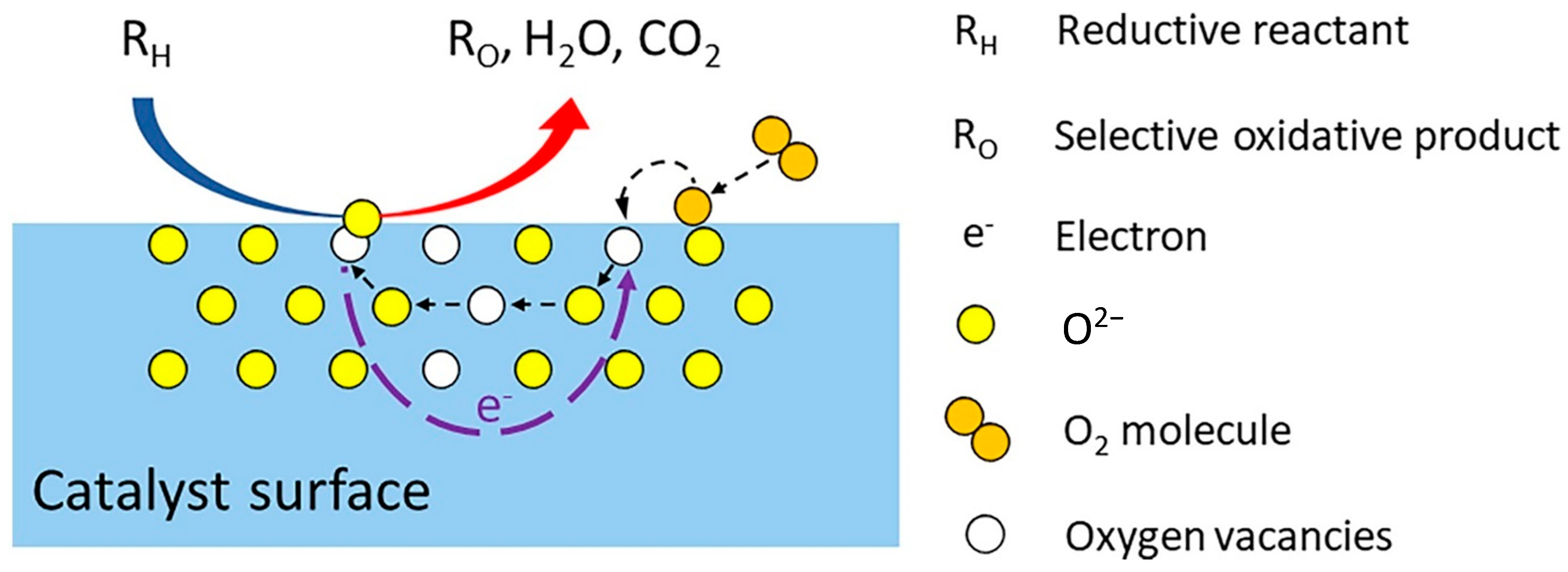
3.1. Mars and Van Krevelen
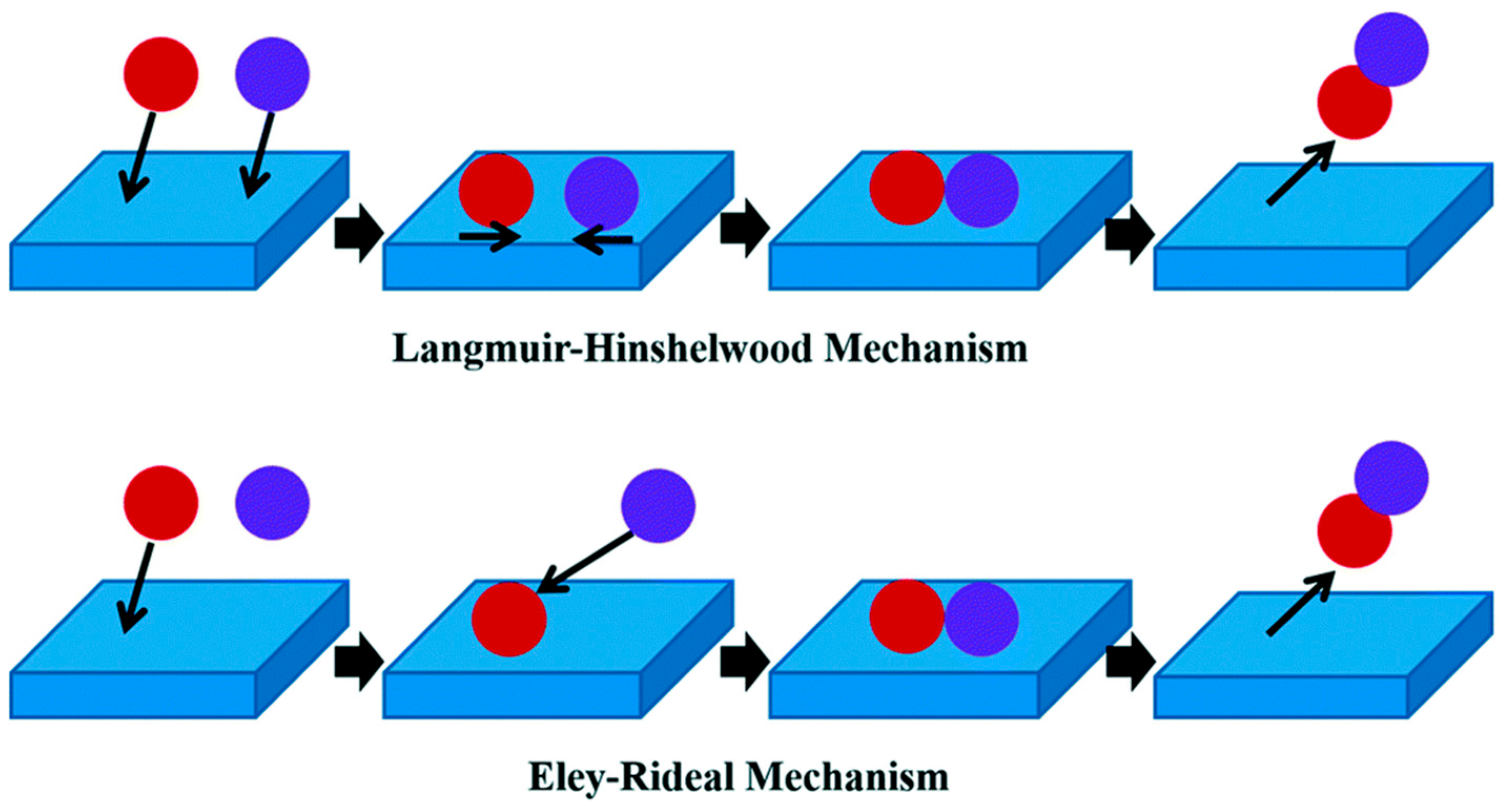
3.2. Langmuir–Hinshelwood and Eley-Rideal
4. Catalysts Used
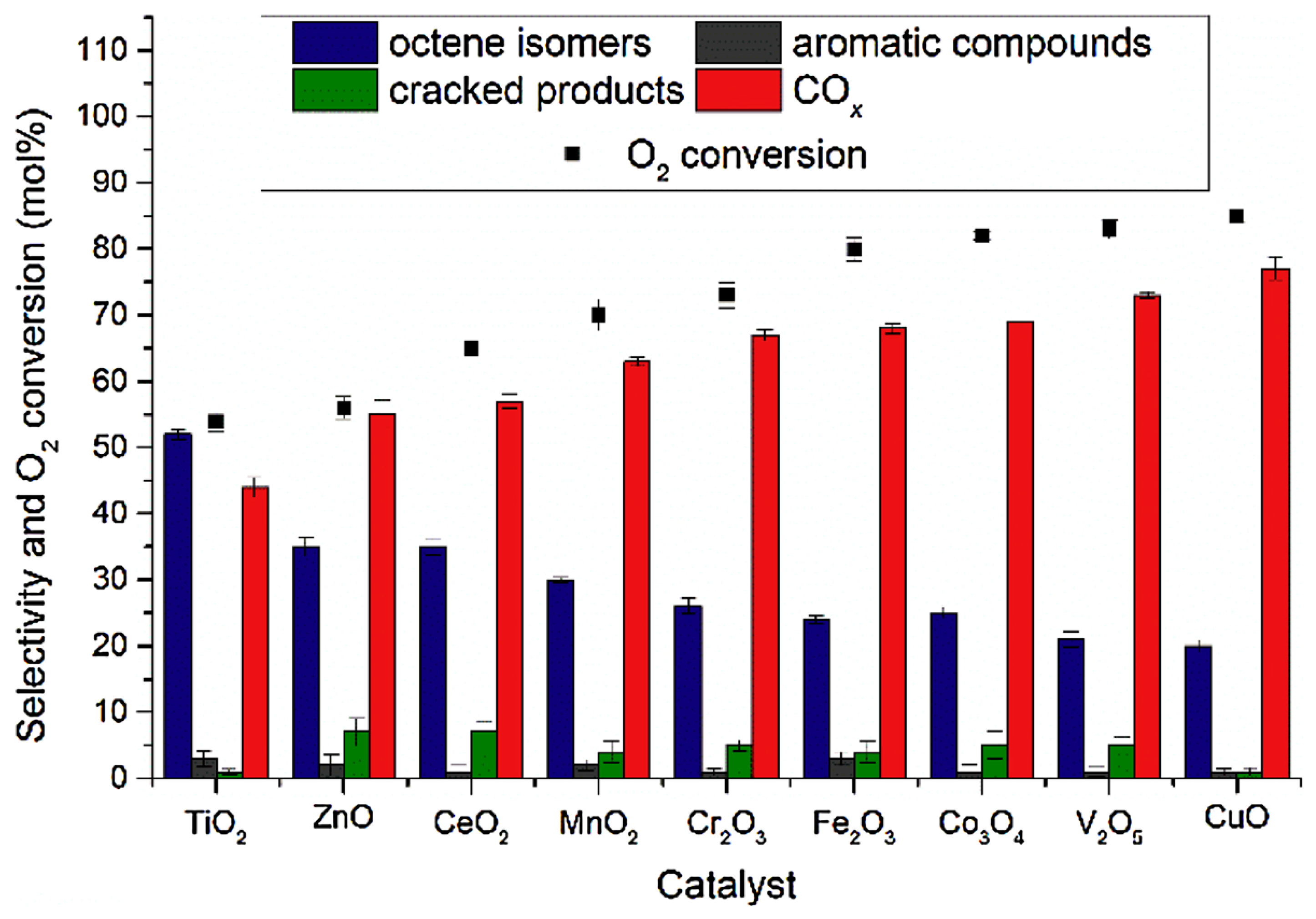
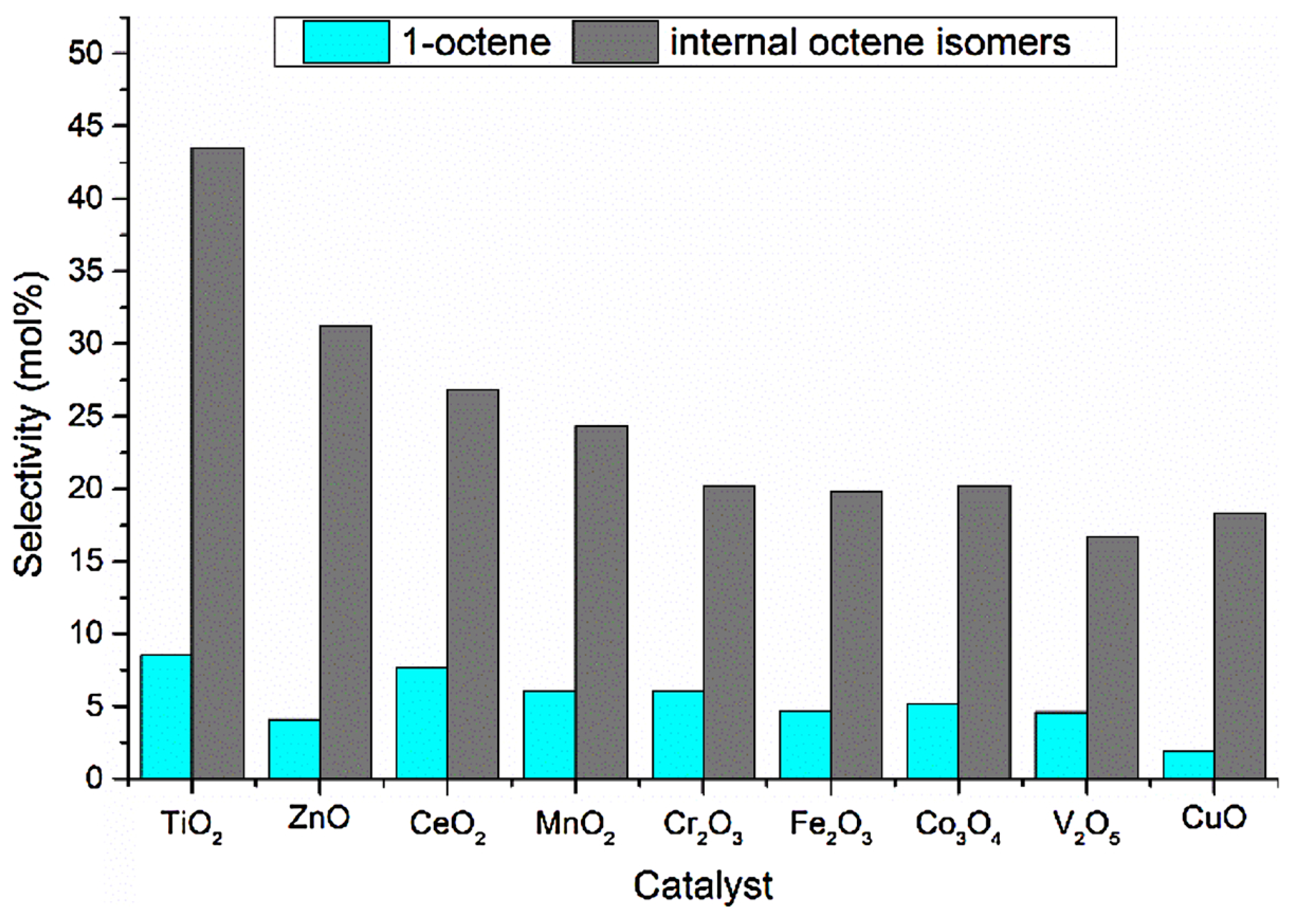
4.1. Unsupported Metal Oxides
4.2. Supported Metal Oxides
4.2.1. Acid–Base Properties
4.2.2. Redox Properties
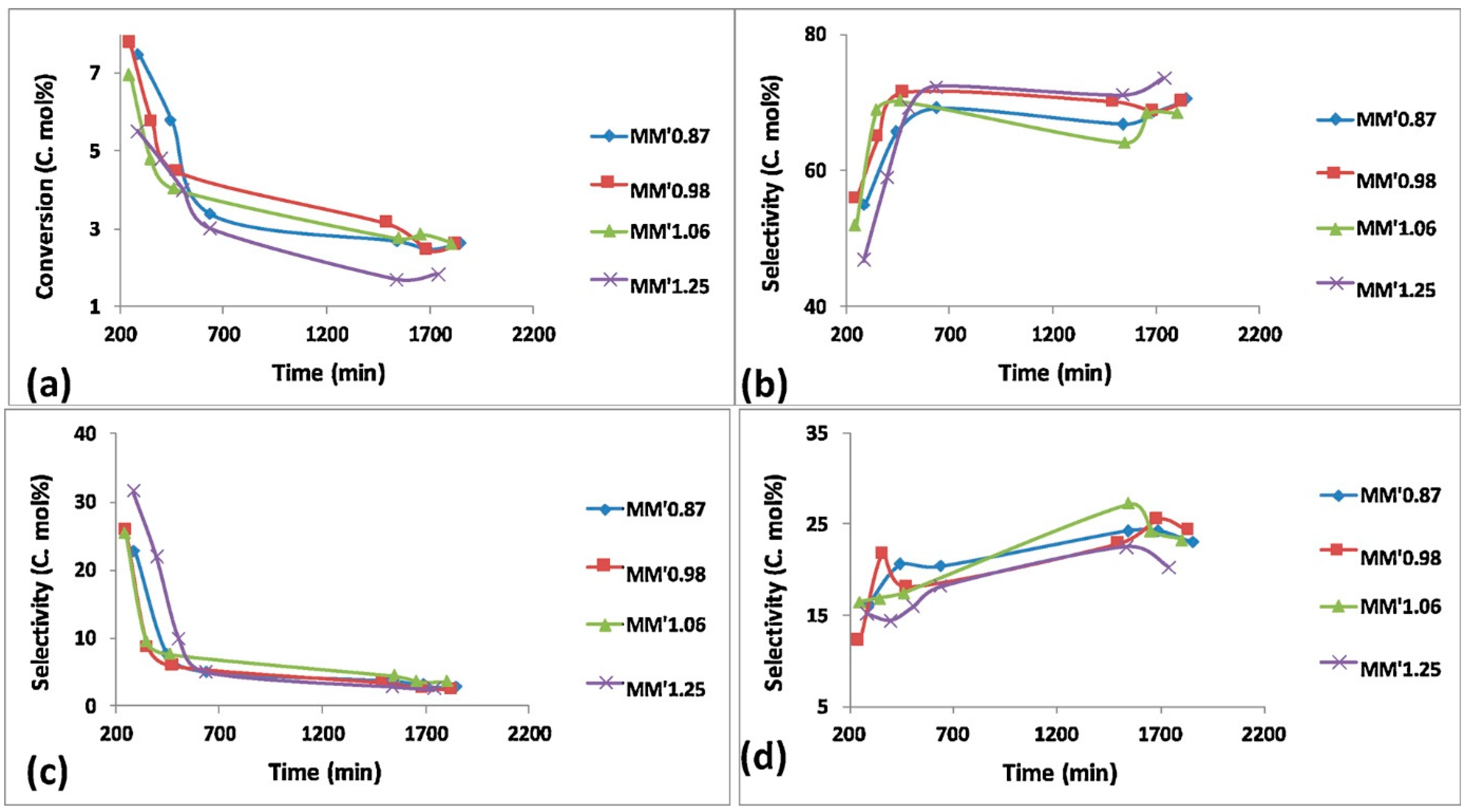
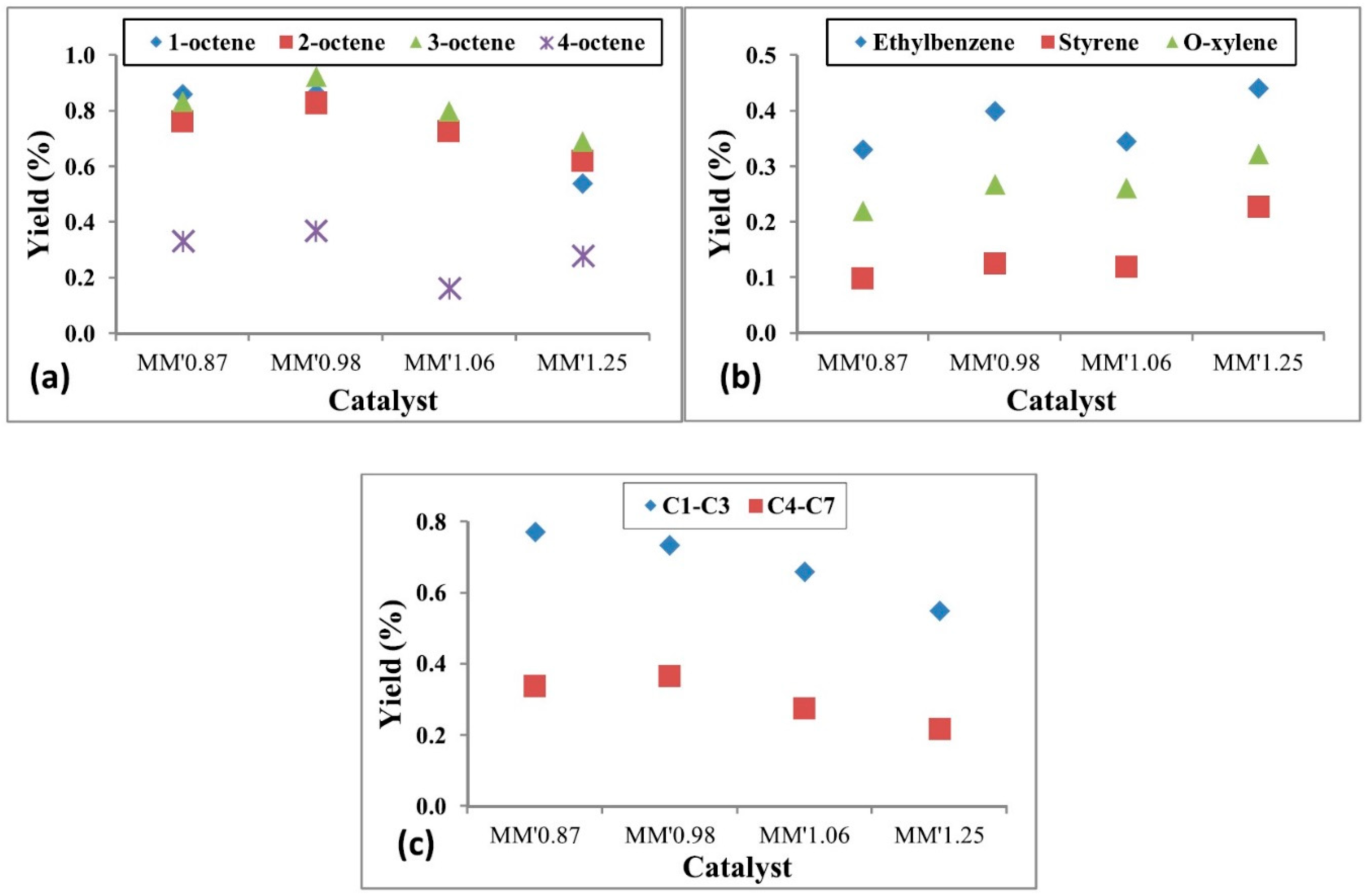
4.2.3. The Effect of Catalyst Synthesis Methods
Wet Impregnation
Solution Combustion Synthesis
Precipitation
5. Perspectives
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Dhar, A.; Vekariya, R.L.; Bhadja, P. n-Alkane isomerization by catalysis—A method of industrial importance: An overview. Cogent Chem. 2018, 4, 1514686. [Google Scholar] [CrossRef]
- Anderson, J.; Wells, R.; Galadima, A.; Ibrahim, B. Solid acid catalysts in heterogeneous n-alkanes hydroisomerisation for increasing octane number of gasoline. Afr. Sci. 2021, 11, 53–61. [Google Scholar]
- Budweg, S.; Junge, K.; Beller, M. Catalytic oxidations by dehydrogenation of alkanes, alcohols and amines with defined (non)-noble metal pincer complexes. Catal. Sci. Technol. 2020, 10, 3825–3842. [Google Scholar] [CrossRef]
- Wu, L.; Fleischer, I.; Jackstell, R.; Profir, I.; Franke, R.; Beller, M. Ruthenium-catalyzed hydroformylation/reduction of olefins to alcohols: Extending the scope to internal alkenes. J. Am. Chem. Soc. 2013, 135, 14306–14312. [Google Scholar] [CrossRef] [PubMed]
- Jeske, K.; Rösler, T.; Belleflamme, M.; Rodenas, T.; Fischer, N.; Claeys, M.; Leitner, W.; Vorholt, A.J.; Prieto, G. Direct conversion of syngas to higher alcohols via tandem integration of Fischer–Tropsch synthesis and reductive hydroformylation. Angew. Chem. 2022, 134, e202201004. [Google Scholar] [CrossRef]
- Yin, M.; Natelson, R.H.; Campos, A.A.; Kolar, P.; Roberts, W.L. Aromatization of n-octane over Pd/C catalysts. Fuel 2013, 103, 408–413. [Google Scholar] [CrossRef]
- Lappin, G. Alpha Olefins Applications Handbook; CRC Press: Boca Raton, FL, USA, 2014. [Google Scholar]
- Hájeková, E.; Špodová, L.; Bajus, M.; Mlynková, B. Separation and characterization of products from thermal cracking of individual and mixed polyalkenes. Chem. Pap. 2007, 61, 262–270. [Google Scholar] [CrossRef]
- Derouane, E.G.; Haber, J.; Lemos, F.; Ribeiro, F.R.; Guisnet, M. Catalytic Activation and Functionalisation of Light Alkanes: Advances and Challenges; Spring: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Asinger, F. Paraffins: Chemistry and Technology; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Nawaz, Z. Light alkane dehydrogenation to light olefin technologies: A comprehensive review. Rev. Chem. Eng. 2015, 31, 413–436. [Google Scholar] [CrossRef]
- Gambo, Y.; Adamu, S.; Tanimu, G.; Abdullahi, I.M.; Lucky, R.A.; Ba-Shammakh, M.S.; Hossain, M.M. CO2-mediated oxidative dehydrogenation of light alkanes to olefins: Advances and perspectives in catalyst design and process improvement. Appl. Catal. A Gen. 2021, 623, 118273. [Google Scholar] [CrossRef]
- Sarathy, S.M.; Yeung, C.; Westbrook, C.K.; Pitz, W.J.; Mehl, M.; Thomson, M.J. An experimental and kinetic modeling study of n-octane and 2-methylheptane in an opposed-flow diffusion flame. Combust. Flame 2011, 158, 1277–1287. [Google Scholar] [CrossRef]
- Széchenyi, A.; Solymosi, F. n-Octane aromatization on Mo2C-containing catalysts. Appl. Catal. A Gen. 2006, 306, 149–158. [Google Scholar] [CrossRef]
- Hone, C.A.; Kappe, C.O. The use of molecular oxygen for liquid phase aerobic oxidations in continuous flow. Acc. Sustain. Flow Chem. 2020, 377, 67–110. [Google Scholar]
- Elkhalifa, E.A.; Friedrich, H.B. Dehydrocyclization of n-octane over boron-and barium-doped V-Mg-O catalysts: Influence of n-octane/oxygen ratios. Appl. Petrochem. Res. 2017, 7, 23–32. [Google Scholar] [CrossRef]
- Farahani, M.D.; Fadlalla, M.I.; Ezekiel, I.P.; Osman, N.S.; Moyo, T.; Claeys, M.; Friedrich, H.B. Nb2O5 as a radical modulator during oxidative dehydrogenation and as a Lewis acid promoter in CO2 assisted dehydrogenation of octane over confined 2D engineered NiO–Nb2O5–Al2O3. Catal. Sci. Technol. 2021, 11, 5321–5334. [Google Scholar] [CrossRef]
- Westbrook, C.K.; Pitz, W.J.; Herbinet, O.; Curran, H.J.; Silke, E.J. A comprehensive detailed chemical kinetic reaction mechanism for combustion of n-alkane hydrocarbons from n-octane to n-hexadecane. Combust. Flame 2009, 156, 181–199. [Google Scholar] [CrossRef]
- Oehlschlaeger, M.A.; Steinberg, J.; Westbrook, C.K.; Pitz, W.J. The autoignition of iso-cetane at high to moderate temperatures and elevated pressures: Shock tube experiments and kinetic modeling. Combust. Flame 2009, 156, 2165–2172. [Google Scholar] [CrossRef]
- Isazade, A.F. A short overview on oxidation of n-alkanes on various catalysts and C–H bond activation. Process. Petrochem. Oil Refin. 2022, 23, 99. [Google Scholar]
- Dasireddy, V.D.B.C. Oxidation and Oxidative Dehyrogenation of n-Octane Using V2O5 Supported on Hydroxyapatites. Ph.D. Thesis, University of KwaZulu-Natal, Durban, South Africa, 2012. [Google Scholar]
- Védrine, J.C.; Fechete, I. Heterogeneous partial oxidation catalysis on metal oxides. Comptes Rendus Chim. 2016, 19, 1203–1225. [Google Scholar] [CrossRef]
- Ruiz, P.; Karelovic, A.; Cortés Corberán, V. Unconventional oxidants for gas-phase oxidations. In Handbook of Advanced Methods and Processes in Oxidation Catalysis: From Laboratory to Industry; World Scientific: Singapore, 2014; pp. 877–920. [Google Scholar]
- Dasireddy, V.D.; Singh, S.; Friedrich, H.B. Activation of n-octane using vanadium oxide supported on alkaline earth hydroxyapatites. Appl. Catal. A Gen. 2013, 456, 105–117. [Google Scholar] [CrossRef]
- Boreskov, G. Forms of oxygen bonds on the surface of oxidation catalysts. Discuss. Faraday Soc. 1966, 41, 263–276. [Google Scholar] [CrossRef]
- Carley, A.F.; Davies, P.R.; Roberts, M.W. Activation of oxygen at metal surfaces. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2005, 363, 829–846. [Google Scholar] [CrossRef]
- Fadlalla, M.I.; Friedrich, H.B. The effect of the oxidation environment on the activity and selectivity to aromatics and octenes over cobalt molybdate in the oxidative dehydrogenation of n-octane. Catal. Sci. Technol. 2014, 4, 4378–4385. [Google Scholar] [CrossRef]
- Liu, P.; Zhang, L.; Li, M.; Sun, N.; Wei, W. Recent progress in Cr-based catalysts for oxidative dehydrogenation of light alkanes by employing CO2 as a soft oxidant. Clean Energy 2021, 5, 623–633. [Google Scholar] [CrossRef]
- Coleman, M.G.; Brown, A.N.; Bolton, B.A.; Guan, H. Iron-catalyzed Oppenauer-type oxidation of alcohols. Adv. Synth. Catal. 2010, 352, 967–970. [Google Scholar] [CrossRef]
- Moloi, S.; Farahani, M.D.; Mahomed, A.S.; Singh, S.; Friedrich, H.B. The role of monomeric VOx supported on anatase in catalytic dehydrogenation of n-octane assisted by CO2 addition. Mol. Catal. 2022, 530, 112578. [Google Scholar] [CrossRef]
- Adam, D.S.; Mahomed, A.S.; Bala, M.D.; Friedrich, H.B. The role of CO2 in the dehydrogenation of n-octane using Cr-Fe catalysts supported on MgAl2O4. Mol. Catal. 2021, 513, 111782. [Google Scholar] [CrossRef]
- Gaxiola, E.; Castillón, F.; Hernández, J.A.; Acosta, B.; Díaz de León, J.; Fuentes, S.; Zepeda, T. Oxidative dehydrogenation of n-octane over Mg-containing SBA-15 material. Mater. Res. Innov. 2018, 22, 247–253. [Google Scholar] [CrossRef]
- Yao, S.; Yan, B.; Jiang, Z.; Liu, Z.; Wu, Q.; Lee, J.H.; Chen, J.G. Combining CO2 reduction with ethane oxidative dehydrogenation by oxygen-modification of molybdenum carbide. ACS Catal. 2018, 8, 5374–5381. [Google Scholar] [CrossRef]
- Huš, M.; Kopač, D.; Bajec, D.; Likozar, B. Likozar, B. Effect of surface oxidation on oxidative propane dehydrogenation over chromia: An ab initio multiscale kinetic study. ACS Catal. 2021, 11, 11233–11247. [Google Scholar] [CrossRef] [PubMed]
- Dasireddy, V.D.; Huš, M.; Likozar, B. Effect of O2, CO2 and N2O on Ni–Mo/Al2O3 catalyst oxygen mobility in n-butane activation and conversion to 1,3-butadiene. Catal. Sci. Technol. 2017, 7, 3291–3302. [Google Scholar] [CrossRef]
- Mars, P.; van Krevelen, D.W. Oxidations carried out by means of vanadium oxide catalysts. Chem. Eng. Sci. 1954, 3, 41–59. [Google Scholar] [CrossRef]
- Védrine, J.C. Heterogeneous Partial (amm)Oxidation and Oxidative Dehydrogenation Catalysis on Mixed Metal Oxides. Catalysts 2016, 6, 22. [Google Scholar] [CrossRef]
- Hosono, Y.; Saito, H.; Higo, T.; Watanabe, K.; Ito, K.; Tsuneki, H.; Maeda, S.; Hashimoto, K.; Sekine, Y. Co–CeO2 Interaction Induces the Mars–van Krevelen Mechanism in Dehydrogenation of Ethane. J. Phys. Chem. C 2021, 125, 11411–11418. [Google Scholar] [CrossRef]
- Pan, Z.-Z.; Li, Y.; Zhao, Y.; Zhang, C.; Chen, H. Bulk phase charge transfer in focus—And in sequential along with surface steps. Catal. Today 2021, 364, 2–6. [Google Scholar] [CrossRef]
- Liang, R.; Hu, A.; Hatat-Fraile, M.; Zhou, N. Fundamentals on Adsorption, Membrane Filtration, and Advanced Oxidation Processes for Water Treatment. In Nanotechnology for Water Treatment and Purification; Hu, A., Apblett, A., Eds.; Springer International Publishing: Cham, Switzerland, 2014; pp. 1–45. [Google Scholar]
- Baxter, R.J.; Hu, P. Insight into why the Langmuir–Hinshelwood mechanism is generally preferred. J. Chem. Phys. 2002, 116, 4379–4381. [Google Scholar] [CrossRef]
- Mahata, A.; Nair, A.S.; Pathak, B. Recent advancements in Pt-nanostructure-based electrocatalysts for the oxygen reduction reaction. Catal. Sci. Technol. 2019, 9, 4835–4863. [Google Scholar] [CrossRef]
- Dasireddy, V.D.; Khan, F.B.; Bharuth-Ram, K.; Singh, S.; Friedrich, H.B. Non oxidative and oxidative dehydrogenation of n-octane using FePO4: Effect of different FePO4 phases on the product selectivity. Catal. Sci. Technol. 2020, 10, 7591–7600. [Google Scholar] [CrossRef]
- Elkhalifa, E.A.; Friedrich, H.B. Magnesium oxide as a catalyst for the dehydrogenation of n-octane. Arab. J. Chem. 2018, 11, 1154–1159. [Google Scholar] [CrossRef]
- Padayachee, D.; Mahomed, A.S.; Singh, S.; Friedrich, H.B. Selected metal oxides for CH bond activation of n-octane and propensity for COx formation: An empirical study. Mol. Catal. 2019, 464, 1–9. [Google Scholar] [CrossRef]
- Padayachee, D.; Mahomed, A.S.; Singh, S.; Friedrich, H.B. Effect of the TiO2 anatase/rutile ratio and interface for the oxidative activation of n-octane. ACS Catal. 2020, 10, 2211–2220. [Google Scholar] [CrossRef]
- Elkhalifa, E.A.; Friedrich, H.B. Oxidative dehydrogenation and aromatization of n-octane over VMgO catalysts obtained by using different MgO precursors and different precursor treatments. J. Mol. Catal. A Chem. 2014, 392, 22–30. [Google Scholar] [CrossRef]
- Gounden, N. The Oxidative Activation of n-Octane over Titania Supported Cobalt Catalysts. Ph.D. Thesis, University of KwaZulu-Natal, Durban, South Africa, 2013. [Google Scholar]
- Elkhalifa, E.; Friedrich, H. Effects of boron and barium dopants on VMgO catalysts employed in the oxidative dehydrogenation of n-octane. Kinet. Catal. 2015, 56, 212–221. [Google Scholar] [CrossRef]
- Golandaj, A.J.; Mahomed, A.S.; Singh, S.; Friedrich, H.B. Effect of different weight loadings of MoOx/SBA-15 on the oxidative dehydrogenation of n-octane. J. Porous Mater. 2015, 22, 787–796. [Google Scholar] [CrossRef]
- Elkhalifa, E.A.; Friedrich, H.B. Oxidative dehydrogenation of n-octane over a vanadium–magnesium oxide catalyst: Influence of the gas hourly space velocity. Arab. J. Chem. 2019, 12, 2464–2469. [Google Scholar] [CrossRef]
- Ntola, P.; Friedrich, H.B.; Singh, S.; Olivier, E.J.; Farahani, M.; Mahomed, A.S. Effect of the fuel on the surface VOx concentration, speciation and physico-chemical characteristics of solution combustion synthesised VOx/MgO catalysts for n-octane activation. Catal. Commun. 2023, 174, 106571. [Google Scholar] [CrossRef]
- Ndlela, S.S.; Friedrich, H.B.; Cele, M.N. Oxidative dehydrogenation of n-octane using Ba and Ga-modified faujasite type catalysts prepared by different methods. J. Porous Mater. 2021, 28, 593–603. [Google Scholar] [CrossRef]
- Yeo, B.R. Alkanes, Alkenes and Aromatics: The Oxidative Dehydrogenation of n-Octane Using Iron Molybdate Catalysts. Ph.D. Thesis, Cardiff University, Cardiff, Wales, 2014. [Google Scholar]
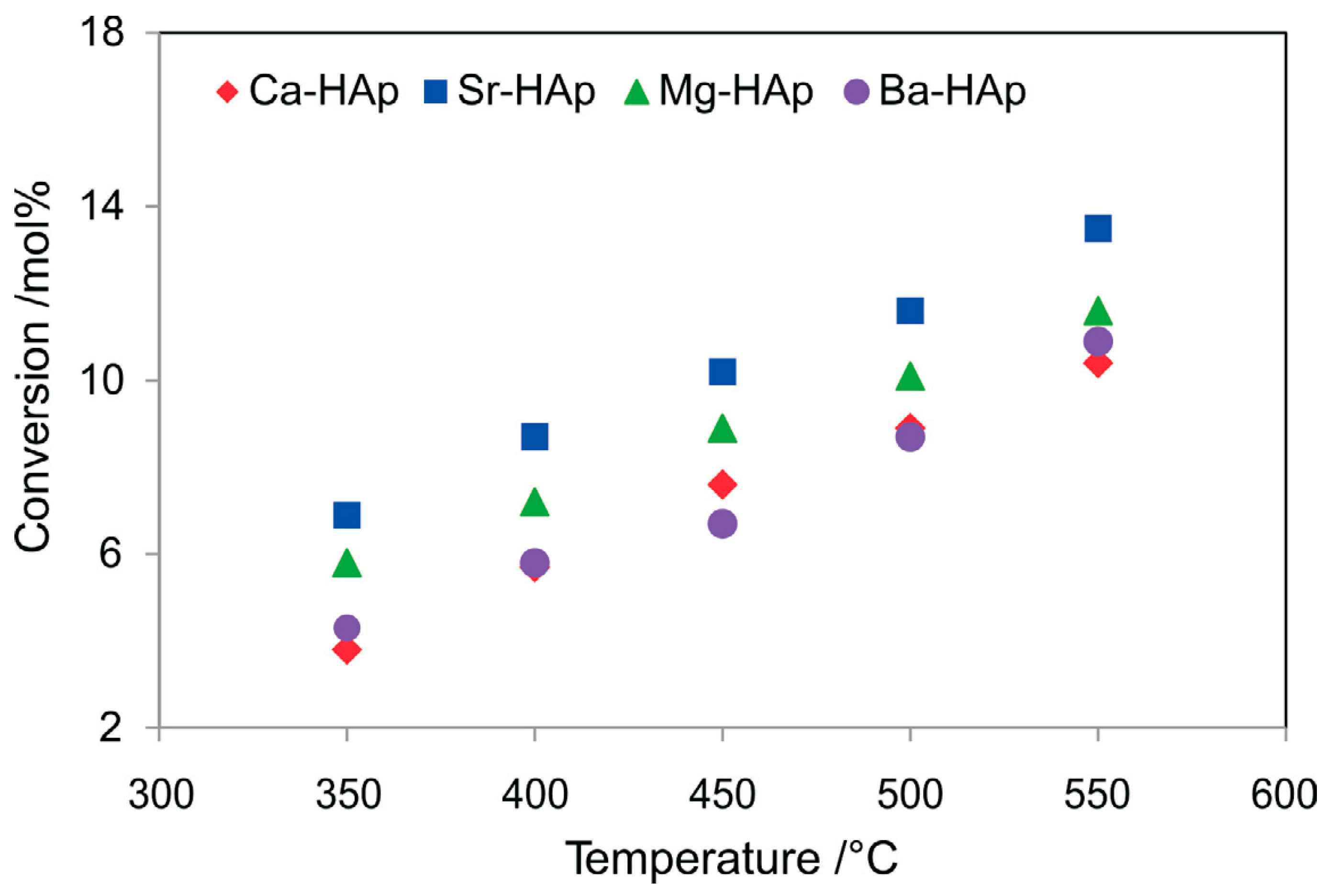
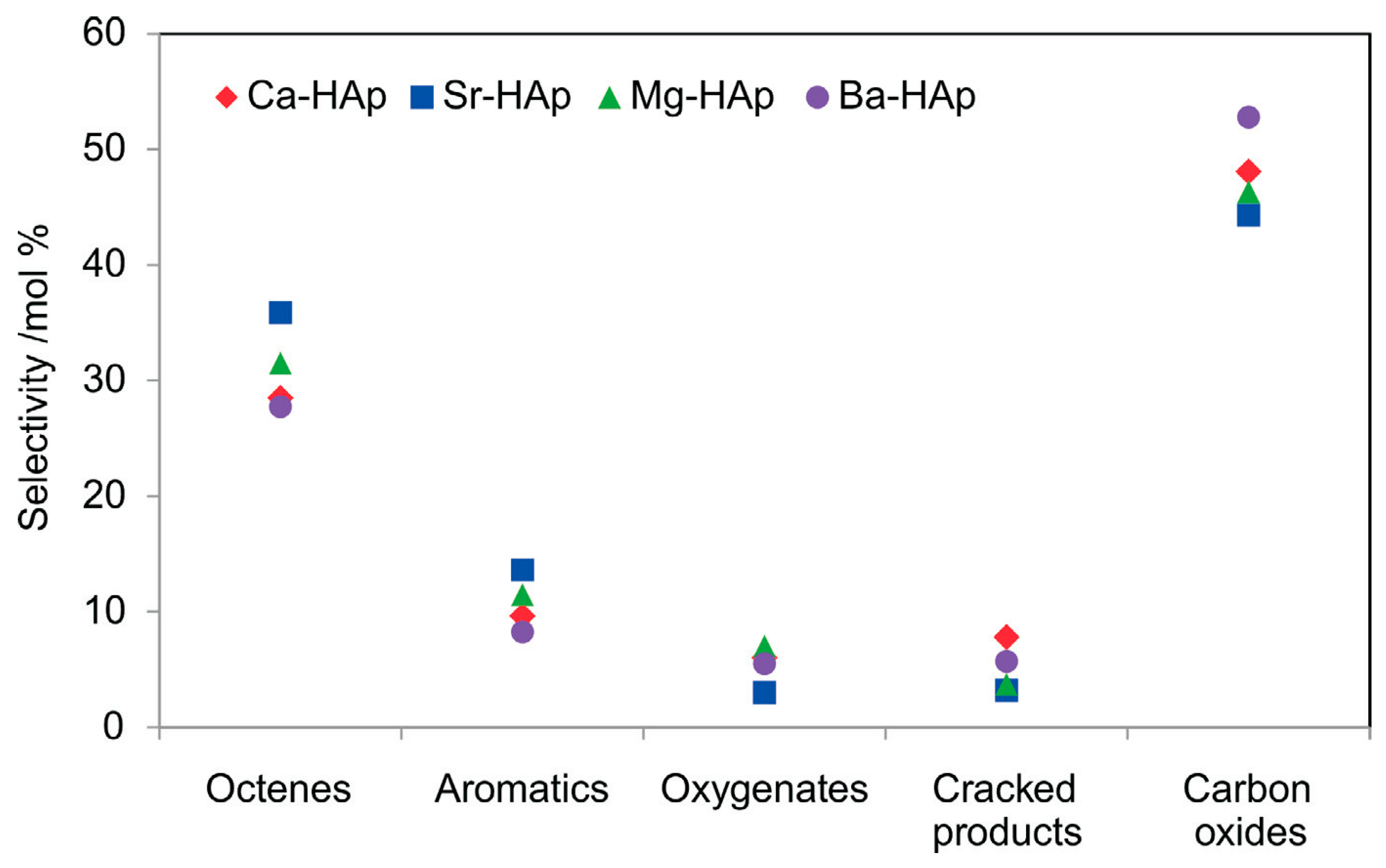
- Dasireddy, V.D.; Friedrich, H.B.; Singh, S. Studies towards a mechanistic insight into the activation of n-octane using vanadium supported on alkaline earth metal hydroxyapatites. Appl. Catal. A Gen. 2013, 467, 142–153. [Google Scholar] [CrossRef]
- Dasireddy, V.D.; Friedrich, H.B.; Singh, S. A kinetic insight into the activation of n-octane with alkaline-earth metal hydroxyapatites. S. Afr. J. Chem. 2015, 68, 195–200. [Google Scholar] [CrossRef][Green Version]
- Padayachee, D. The Catalysed Activation of n-Octane over Iron Modified Hydroxyapatites. Ph.D. Thesis, University of KwaZulu-Natal, Durban, South Africa, 2014. [Google Scholar]
- Fadlalla, M.I.; Farahani, M.D.; Friedrich, H.B. Three inter-linked active sites in the dehydrogenation of n-octane over magnesium molybdate based catalysts and their influences on coking and cracking side reactions. Mol. Catal. 2018, 461, 86–96. [Google Scholar] [CrossRef]
- Furukawa, S.; Shishido, T.; Teramura, K.; Tanaka, T. Reaction mechanism of selective photooxidation of hydrocarbons over Nb2O5. J. Phys. Chem. C 2011, 115, 19320–19327. [Google Scholar] [CrossRef]
- Friedrich, H.B.; Mahomed, A.S. The oxidative dehydrogenation of n-octane to styrene using catalysts derived from hydrotalcite-like precursors. Appl. Catal. A Gen. 2008, 347, 11–22. [Google Scholar] [CrossRef]
- Narayanappa, M.; Dasireddy, V.D.B.C.; Friedrich, H.B. Catalytic oxidation of n-octane over cobalt substituted ceria (Ce0.90Co0.10O2−δ) catalysts. Appl. Catal. A Gen. 2012, 447–448, 135–143. [Google Scholar] [CrossRef]
- Sugiyama, S.; Osaka, T.; Hirata, Y.; Sotowa, K.-I. Enhancement of the activity for oxidative dehydrogenation of propane on calcium hydroxyapatite substituted with vanadate. Appl. Catal. A Gen. 2006, 312, 52–58. [Google Scholar] [CrossRef]
- Monma, H. Catalytic behavior of calcium phosphates for decompositions of 2-propanol and ethanol. J. Catal. 1982, 75, 200–203. [Google Scholar] [CrossRef]
- Sugiyama, S.; Osaka, T.; Ueno, Y.; Sotowa, K.-I. Oxidative Dehydrogenation of Propane over Vanadate Catalysts Supported on Calcium and Strontium Hydroxyapatites. J. Jpn. Pet. Inst. 2008, 51, 50–57. [Google Scholar] [CrossRef]
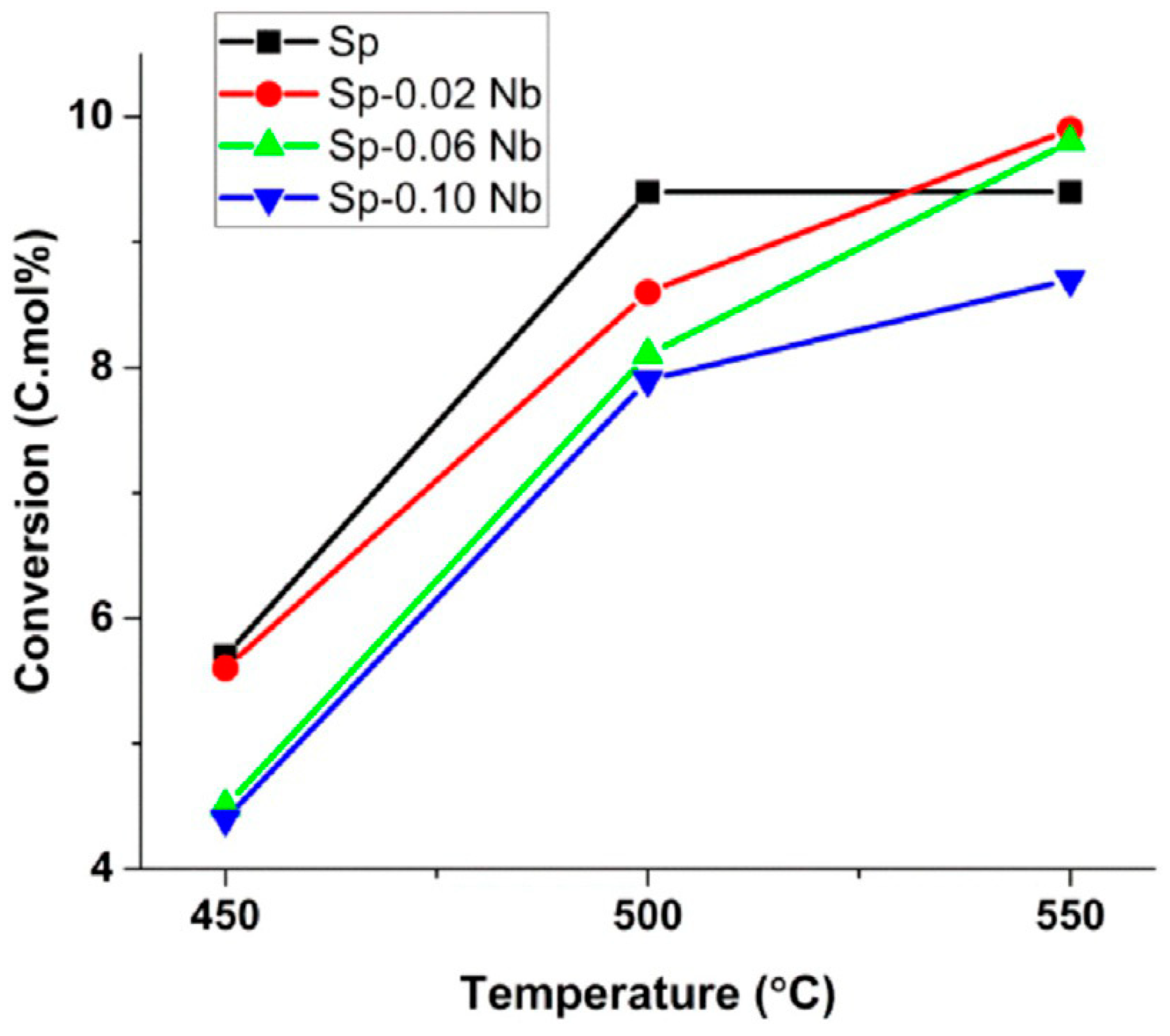
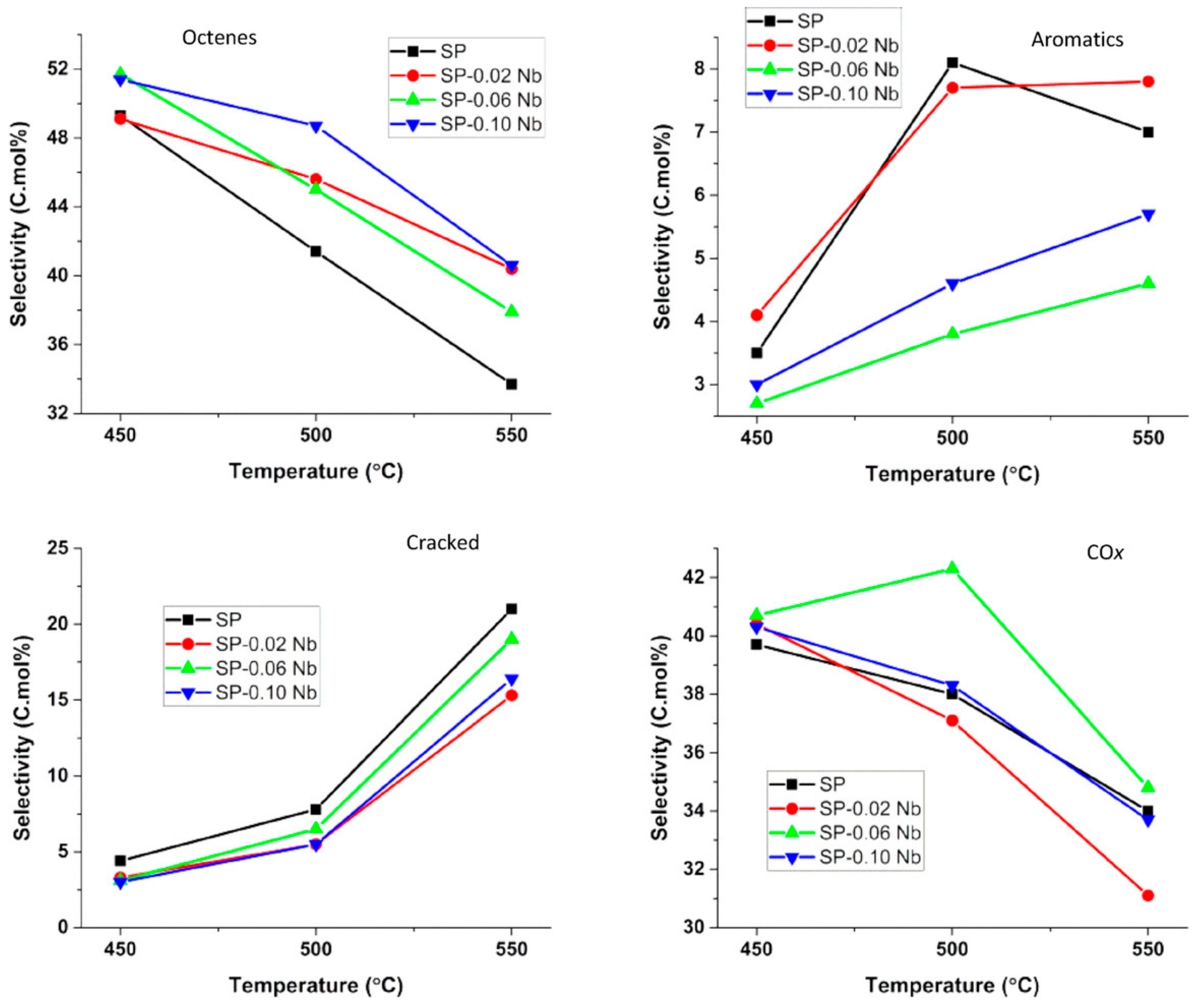
- Farahani, M.D.; Dasireddy, V.D.B.C.; Friedrich, H.B. Oxidative Dehydrogenation of n-Octane over Niobium-Doped NiAl2O4: An Example of Beneficial Coking in Catalysis over Spinel. ChemCatChem 2018, 10, 2059–2069. [Google Scholar] [CrossRef]
- Sun, X.; Li, B.; Metiu, H. Ethane Activation by Nb-Doped NiO. J. Phys. Chem. C 2013, 117, 23597–23608. [Google Scholar] [CrossRef]
- Popescu, I.; Skoufa, Z.; Heracleous, E.; Lemonidou, A.; Marcu, I.-C. A study by electrical conductivity measurements of the semiconductive and redox properties of Nb-doped NiO catalysts in correlation with the oxidative dehydrogenation of ethane. Phys. Chem. Chem. Phys. 2015, 17, 8138–8147. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, X.; Blume, R.; Zhang, A.; Schlögl, R.; Su, D.S. Surface-Modified Carbon Nanotubes Catalyze Oxidative Dehydrogenation of n-Butane. Science 2008, 322, 73–77. [Google Scholar] [CrossRef]
- Sattler, J.J.H.B.; Ruiz-Martinez, J.; Santillan-Jimenez, E.; Weckhuysen, B.M. Catalytic Dehydrogenation of Light Alkanes on Metals and Metal Oxides. Chem. Rev. 2014, 114, 10613–10653. [Google Scholar] [CrossRef]
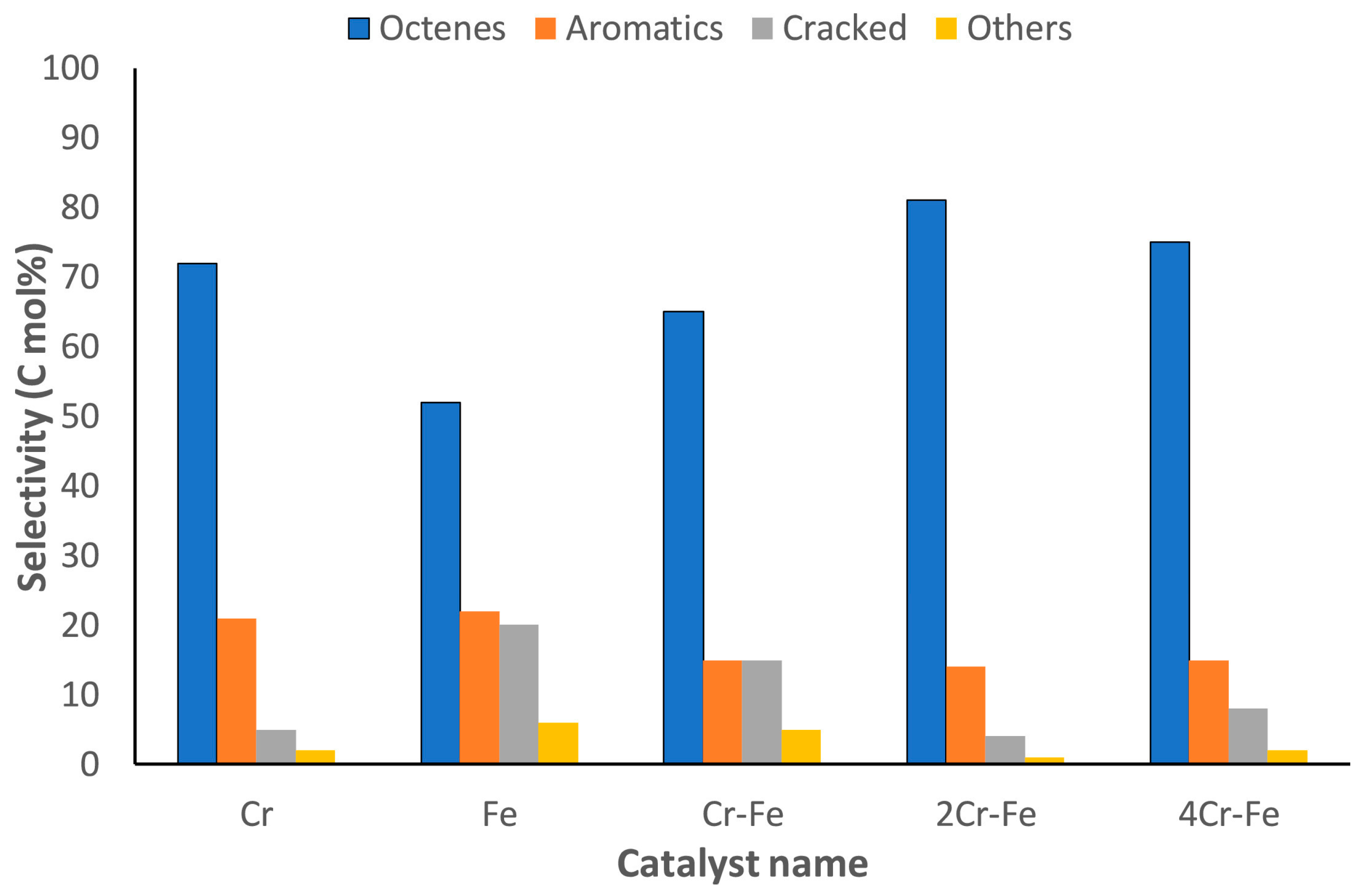
- Bandaru, H.; Mahomed, A.S.; Singh, S.; Friedrich, H.B. The effect of varying the metal ratio in a chromium molybdate catalysts for the oxidative dehydrogenation of n-octane. Mol. Catal. 2018, 460, 74–82. [Google Scholar] [CrossRef]
- Goncharov, D.V.; Belyaevskii, M.Y. Analysis and Mathematical Description of the Thermal-Oxidative Cracking of Naphtha. Chem. Pet. Eng. 2005, 41, 193–198. [Google Scholar] [CrossRef]
- Ntola, P.; Friedrich, H.B.; Mahomed, A.S.; Olivier, E.J.; Govender, A.; Singh, S. Exploring the role of fuel on the microstructure of VOx/MgO powders prepared using solution combustion synthesis. Mater. Chem. Phys. 2022, 278, 125602. [Google Scholar] [CrossRef]
- Zhou, Y.; Chai, Y.; Li, X.; Wu, Z.; Lin, J.; Han, Y.; Li, L.; Qi, H.; Gu, Y.; Kang, L. Defect-rich TiO2 in situ evolved from MXene for the enhanced oxidative dehydrogenation of ethane to ethylene. ACS Catal. 2021, 11, 15223–15233. [Google Scholar] [CrossRef]
- Stoian, M.; Rogé, V.; Lazar, L.; Maurer, T.; Védrine, J.C.; Marcu, I.-C.; Fechete, I. Total oxidation of methane on oxide and mixed oxide ceria-containing catalysts. Catalysts 2021, 11, 427. [Google Scholar] [CrossRef]
- Padayachee, D.; Dasireddy, V.D.; Singh, S.; Friedrich, H.B.; Bharuth-Ram, K.; Govender, A. An investigation of iron modified hydroxyapatites used in the activation of n-octane. Mol. Catal. 2017, 438, 256–266. [Google Scholar] [CrossRef]
- Dasireddy, V.D.; Singh, S.; Friedrich, H.B. Vanadium oxide supported on non-stoichiometric strontium hydroxyapatite catalysts for the oxidative dehydrogenation of n-octane. J. Mol. Catal. A Chem. 2014, 395, 398–408. [Google Scholar] [CrossRef]
- Grant, J.T.; Venegas, J.M.; McDermott, W.P.; Hermans, I. Aerobic oxidations of light alkanes over solid metal oxide catalysts. Chem. Rev. 2017, 118, 2769–2815. [Google Scholar] [CrossRef]
- Bugler, K. The Catalytic Oxidative Dehydrogenation of n-Octane Over Iron and Other Metal Molybdates. Ph.D. Thesis, Cardiff University, Cardiff, Wales, 2017. [Google Scholar]
- Naicker, K.; Mahomed, A.S.; Friedrich, H.B.; Singh, S. Influence of preparation method of high surface area MnOx/SBA-15 catalysts for the activation of n-octane. J. Porous Mater. 2019, 26, 301–309. [Google Scholar] [CrossRef]
- Blasco, T.; Nieto, J.L.; Dejoz, A.; Vazquez, M. Influence of the acid-base character of supported vanadium catalysts on their catalytic properties for the oxidative dehydrogenation of n-butane. J. Catal. 1995, 157, 271–282. [Google Scholar] [CrossRef]























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ethene | Polymers (PE), ethylene oxide, vinyl acetate, vinyl chloride, styrene |
| Propene | Propene polymers (PP), propene oxide, acetone, butanal, plasticizer alcohols, acrylonitrile, epichlorohydrin synthesis |
| 1-Butene | Butene polymers, solvents |
| Butadiene | Polymers, oligomers, adiponitrile, sulfolane, chloroprene, vinylcyclohexene, cyclododecatriene |
| 1-Hexene | Co-monomer in PE |
| 1-Heptene | Reagent and solvent in organic synthesis |
| 1-Octene | Co-monomer in PE |
| C10-C14 | Detergent alcohols |
| C14-C16 | Sulfates and sulfonates in detergents |
| Catalyst | Oxidant | Temperature/°C | C:Oxidant Ratio | Octane:O2 Ratio | Conversion/% | Octenes/% | Aromatics/% | COx/% | Reference |
|---|---|---|---|---|---|---|---|---|---|
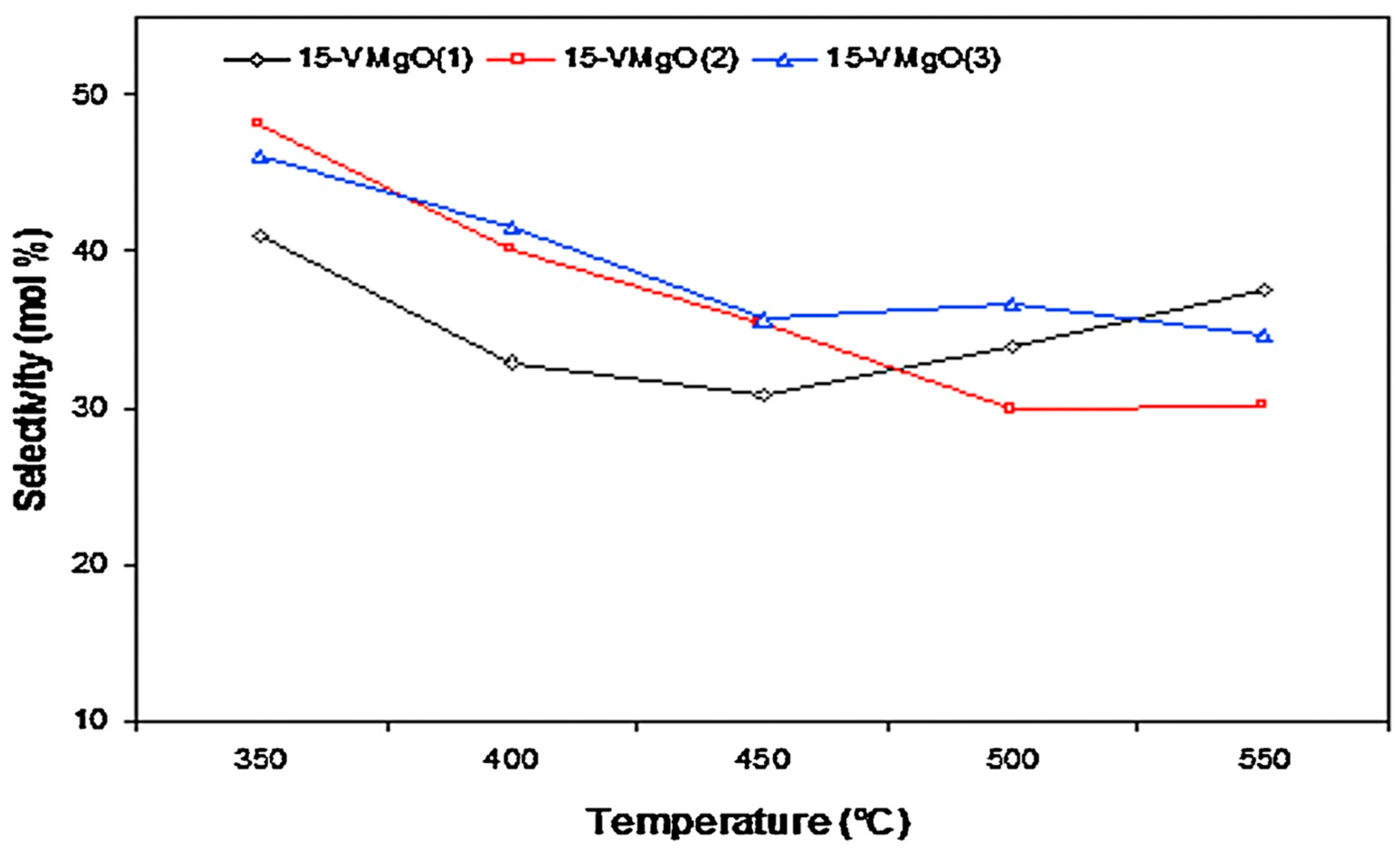
| V/MgO | O2 | 350–550 | - | 0.1–1.6 | 11.5–58.8 | 6.9–49.4 | 9.9–20.5 | 25.3–78 | [16] |
| Ba-V/MgO | O2 | 350–550 | - | 0.1–1.6 | 10.7–35.3 | 16.9–50.6 | 9.6–17.2 | 26.1–67.1 | [16] |
| B-V/MgO | O2 | 350–550 | - | 0.1–1.6 | 11–43.7 | 9.7–50 | 9.8–19 | 24.8–76 | [16] |
| 4 wt% VOx/TiO2 | CO2 | 550 | 8:7.5 | - | 5 | 68 | 10 | - | [30] |
| 9 wt% VOx/TiO2 | CO2 | 550 | 8:7.5 | - | 3 | 75 | 6 | - | [30] |
| V/MgO_1 * | O2 | 350–550 | - | 0.8 | 1.7–17 | 36–43 | 10–16 | 38–40 | [47] |
| V/MgO_2 * | O2 | 350–550 | - | 0.8 | 3–21 | 32–38 | 11–19 | 30–48 | [47] |
| V/MgO_3 * | O2 | 350–550 | - | 0.8 | 2–19 | 37–38 | 10–14 | 35–45 | [47] |
| V/MgO | O2 | 350–550 | - | 0.8 | 2–22 | 41–43 | 17–18 | 31–39 | [49] |
| B-V/MgO | O2 | 350–550 | - | 0.8 | 2–17 | 40–50 | 6–12 | 32–45 | [49] |
| Ba-V/MgO | O2 | 350–550 | - | 0.8 | 2–23 | 40–48 | 1–20 | 30–38 | [49] |
| 4wt%MnOx/SBA-15 | O2 | 450 | 8:3 | - | 9.8 | 32 | 5 | 45 | [50] |
| 7wt% MnOx/SBA-15 | O2 | 450 | 8:3 | - | 5 | 27 | 10 | 37 | [50] |
| 10wt% MnOx/SBA-15 | O2 | 450 | 8:3 | - | 3 | 27 | 8 | 46 | [50] |
| 18wt% MnOx/SBA-15 | O2 | 450 | 8:3 | - | 2 | 16 | 6 | 48 | [50] |
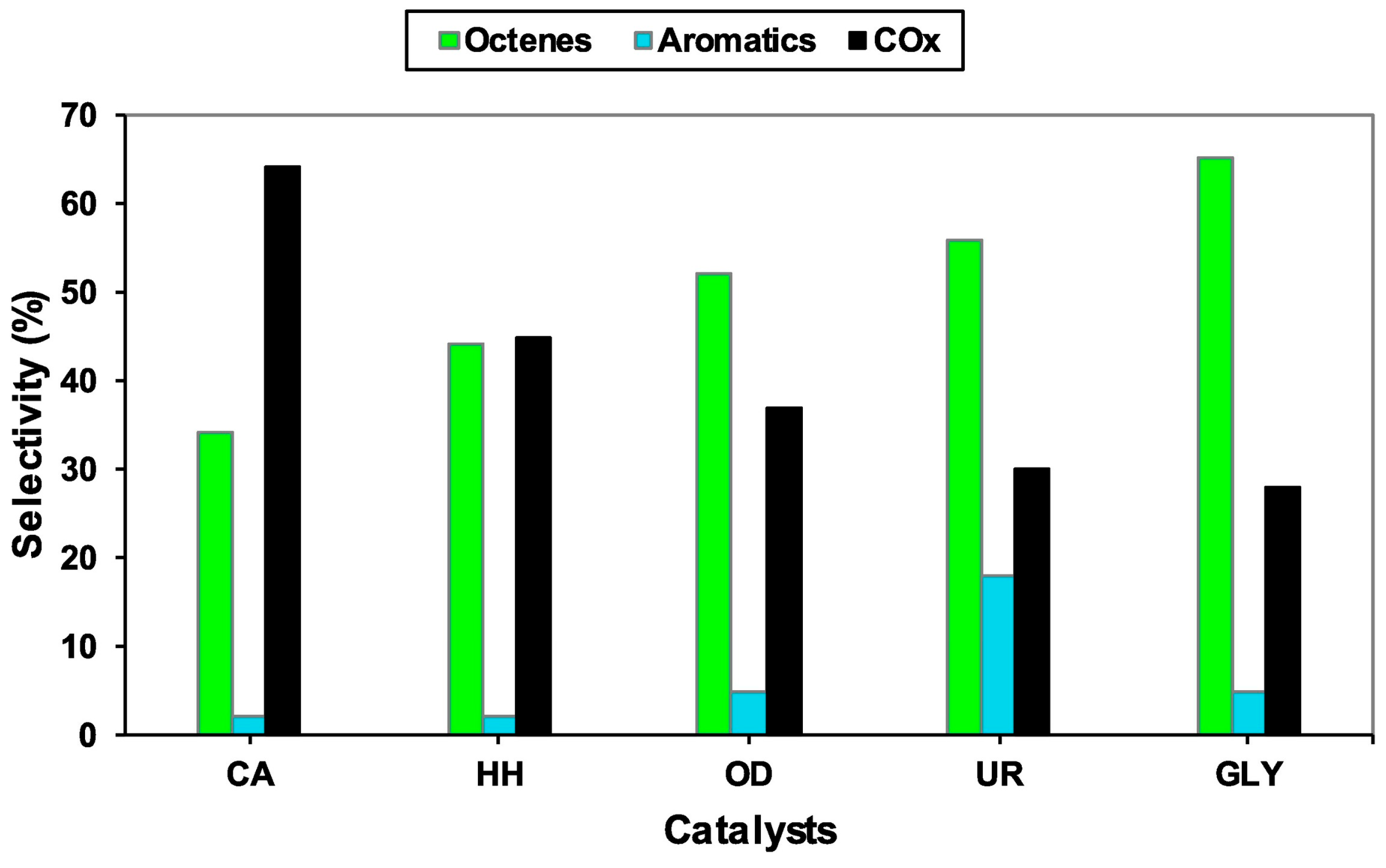
| VOx/MgO (citric acid) @ | O2 | 400 | 8:2 | - | 9 | 34 | 2 | 64 | [52] |
| VOx/MgO (oxalyldihydrazide) @ | O2 | 400 | 8:2 | - | 13 | 53 | 5 | 36 | [52] |
| VOx/MgO (hydrazine hydrate) @ | O2 | 400 | 8:2 | - | 10 | 44 | 2 | 44 | [52] |
| VOx/MgO (glycine) @ | O2 | 400 | 8:2 | - | 15 | 65 | 5 | 27 | [52] |
| VOx/MgO (urea) @ | O2 | 400 | 8:2 | - | 16 | 57 | 18 | 33 | [52] |
| 5%V/Sr-HAp | O2 | 350–550 | - | 1:1 | 16–28 | 68–75 | 10–14 | 10–13 | [76] |
| 7.5%V/Sr-HAp | O2 | 350–550 | - | 1:1 | 20–38 | 48–69 | 12–25 | 15–17 | [76] |
| 15%V/Sr-HAp | O2 | 350–550 | - | 1:1 | 24–46 | 40–62 | 20–33 | 13–15 | [76] |
| 2wt% MnOx/SBA-15 # | O2 | 450 | 8:2 | - | - | 12 | 5 | 75 | [79] |
| 9wt% MnOx/SBA-15 # | O2 | 450 | 8:2 | - | - | 28 | 1 | 70 | [79] |
| 18wt% MnOx/SBA-15 # | O2 | 450 | 8:2 | - | - | 26 | 3 | 72 | [79] |
| 2wt% MnOx/SBA-15 $ | O2 | 450 | 8:2 | - | - | 17 | 3 | 80 | [79] |
| 9wt% MnOx/SBA-15 $ | O2 | 450 | 8:2 | - | - | 38 | 4 | 55 | [79] |
| 18wt% MnOx/SBA-15 $ | O2 | 450 | 8:2 | - | - | 36 | 4 | 57 | [79] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ntola, P.; Shozi, M. Gas-Phase Oxidative Dehydrogenation of n-Octane over Metal Oxide Catalysts: A Review. Catalysts 2024, 14, 100. https://doi.org/10.3390/catal14020100
Ntola P, Shozi M. Gas-Phase Oxidative Dehydrogenation of n-Octane over Metal Oxide Catalysts: A Review. Catalysts. 2024; 14(2):100. https://doi.org/10.3390/catal14020100
Chicago/Turabian StyleNtola, Pinkie, and Mzamo Shozi. 2024. "Gas-Phase Oxidative Dehydrogenation of n-Octane over Metal Oxide Catalysts: A Review" Catalysts 14, no. 2: 100. https://doi.org/10.3390/catal14020100
APA StyleNtola, P., & Shozi, M. (2024). Gas-Phase Oxidative Dehydrogenation of n-Octane over Metal Oxide Catalysts: A Review. Catalysts, 14(2), 100. https://doi.org/10.3390/catal14020100