
Catalytic Ozonation of Formaldehyde with an Oxygen-Vacancy-Rich MnOx/γ-Al2O3 Catalyst at Room Temperature



Abstract
:
1. Introduction
2. Results and Discussion
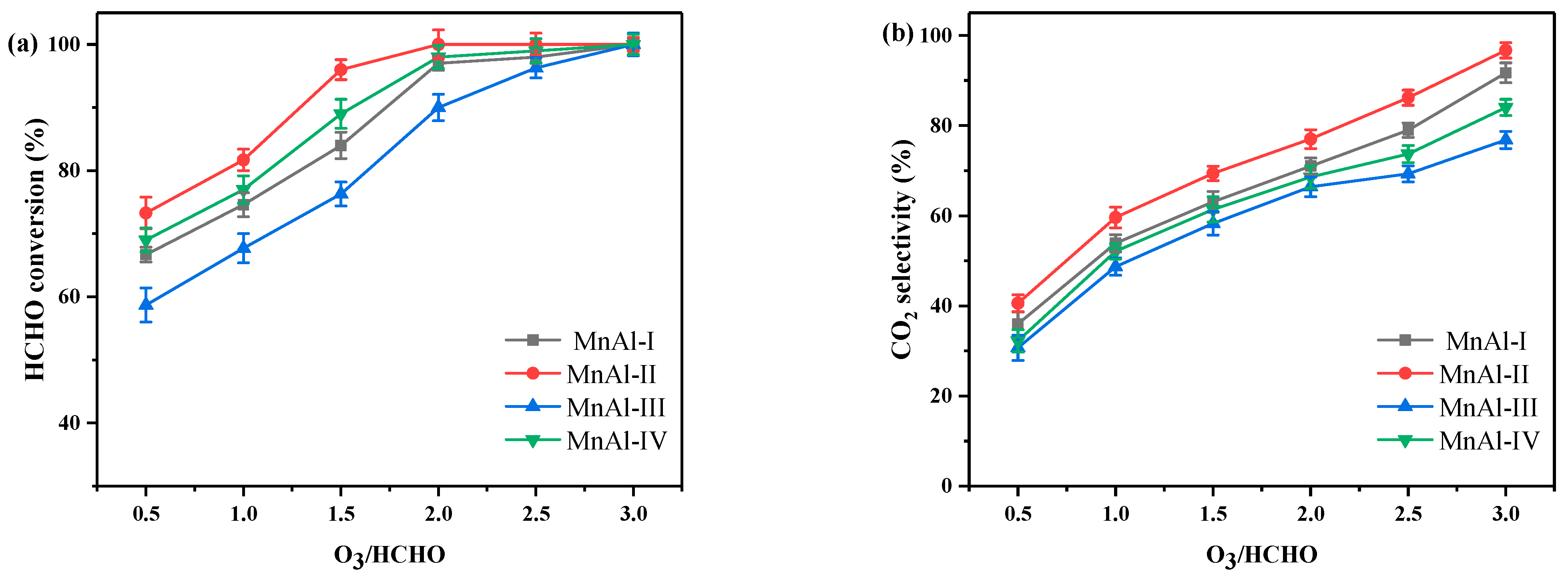
2.1. Catalytic Ozonation of HCHO
2.2. Catalyst Characterization
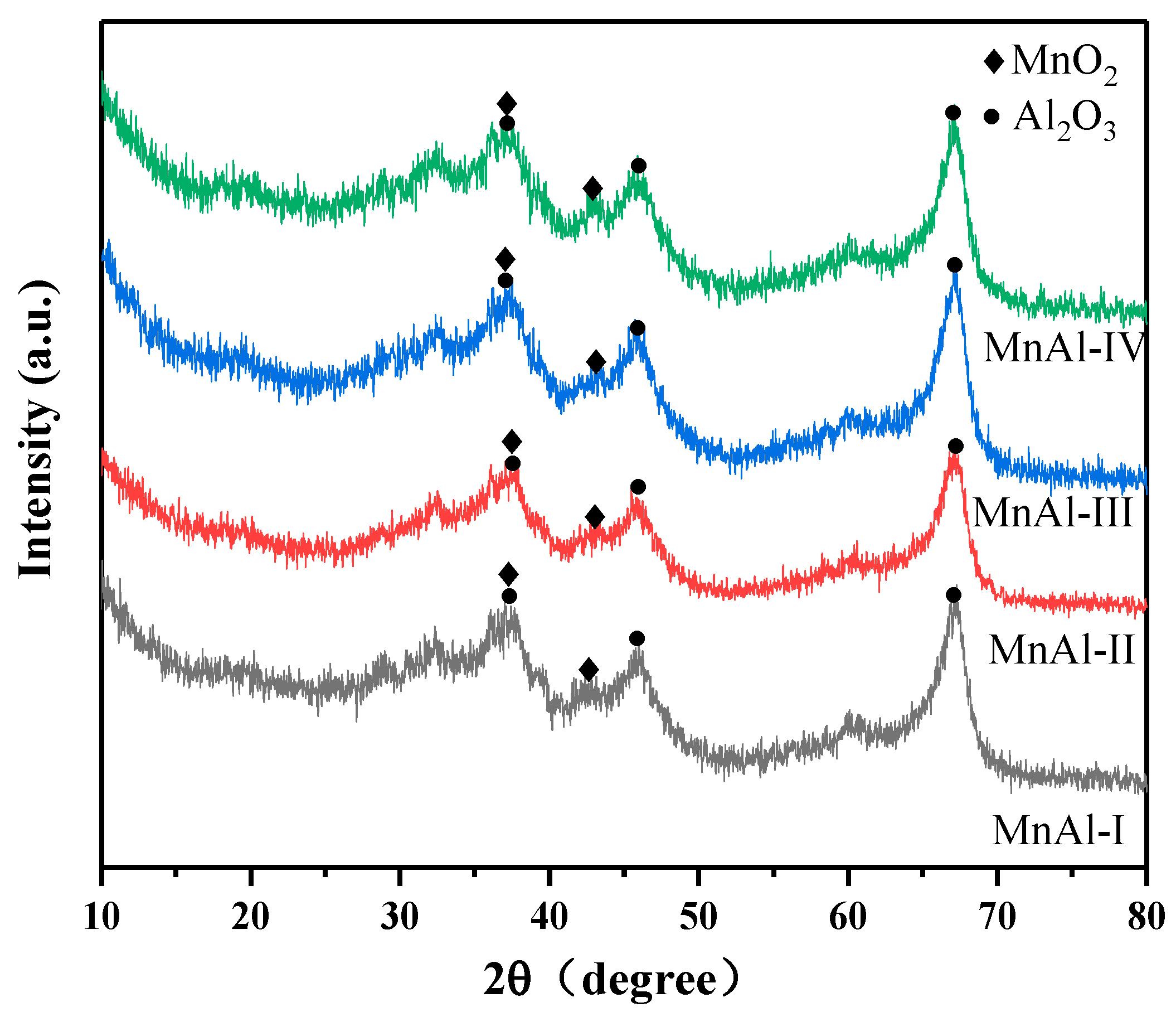
2.2.1. Crystalline Structures
2.2.2. Textual Properties
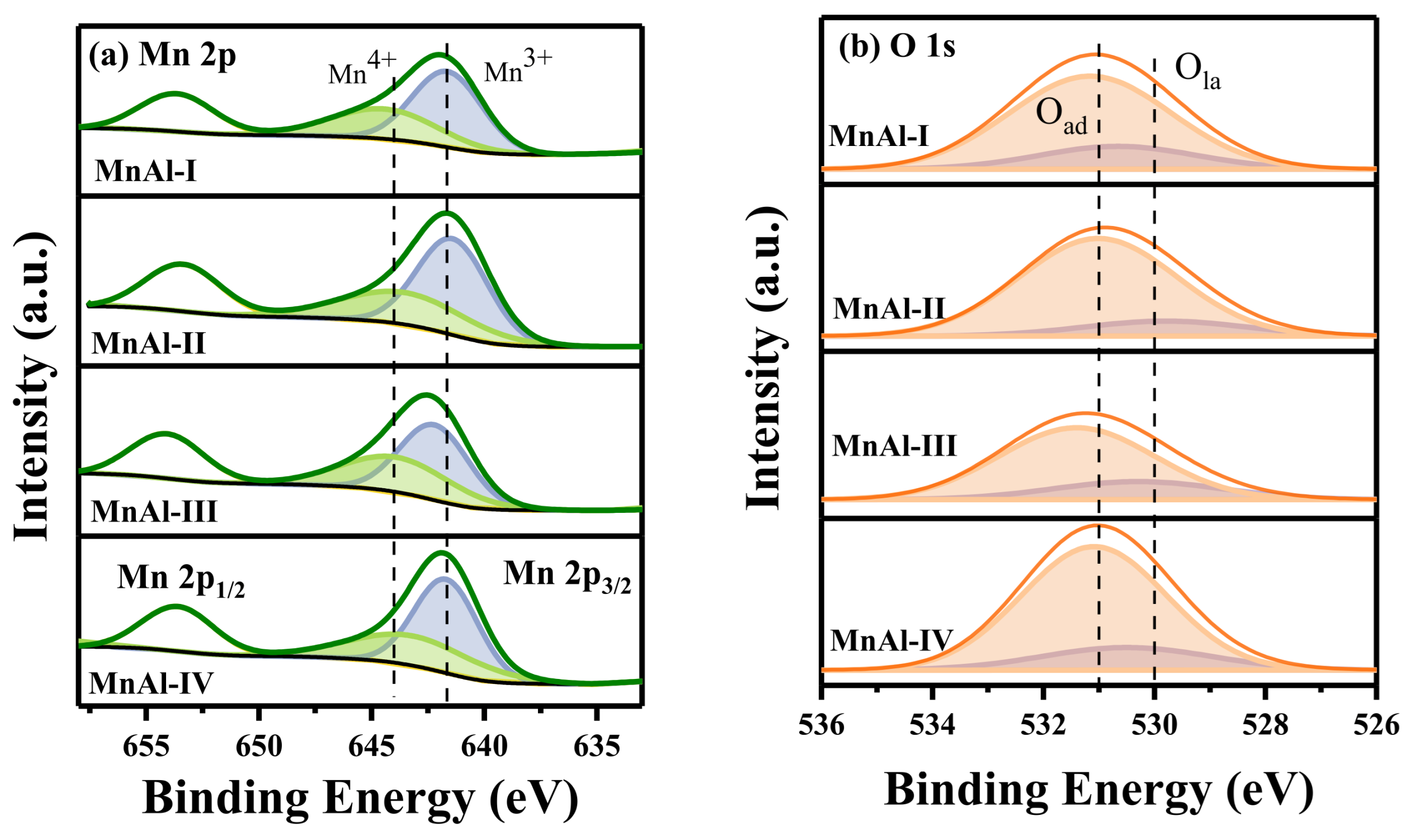
2.2.3. Surface Properties
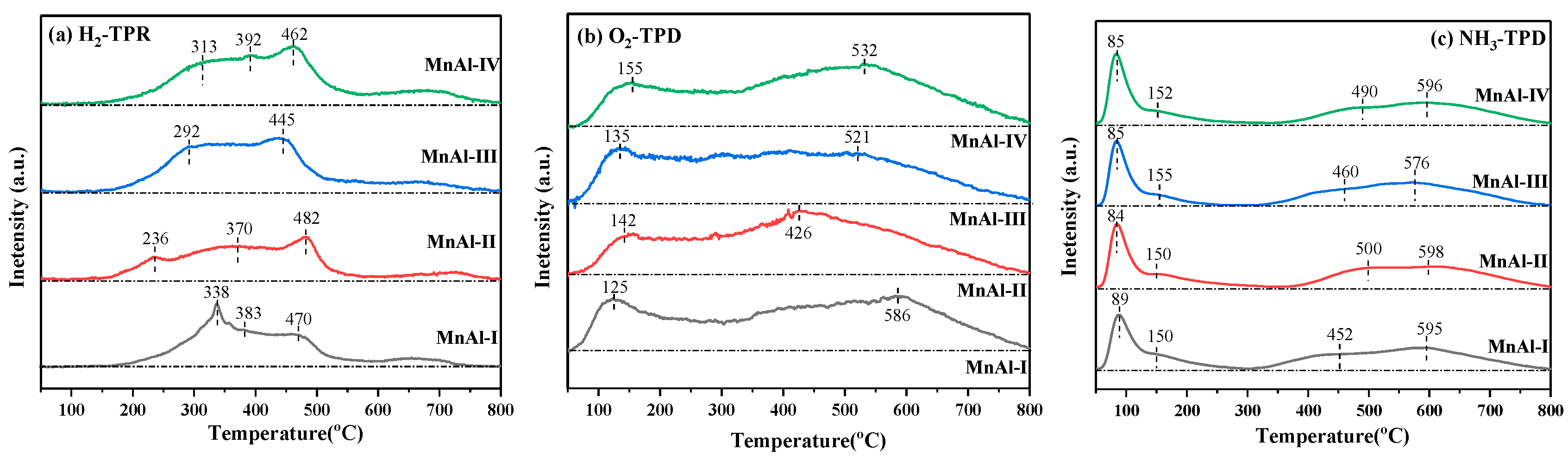
2.2.4. Temperature-Programmed Studies
2.3. Effect of H2O
2.4. In Situ DRIFTS Measurement
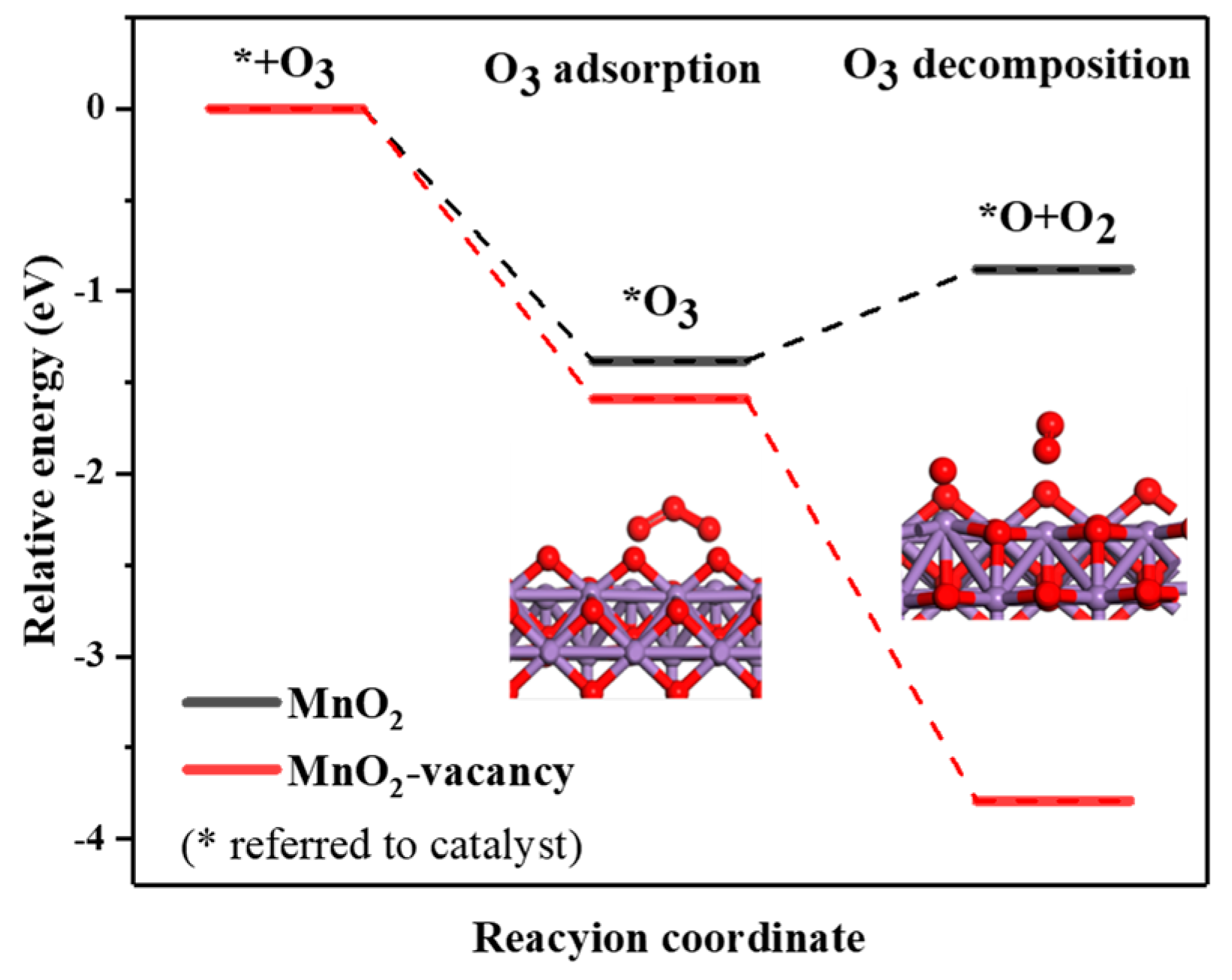
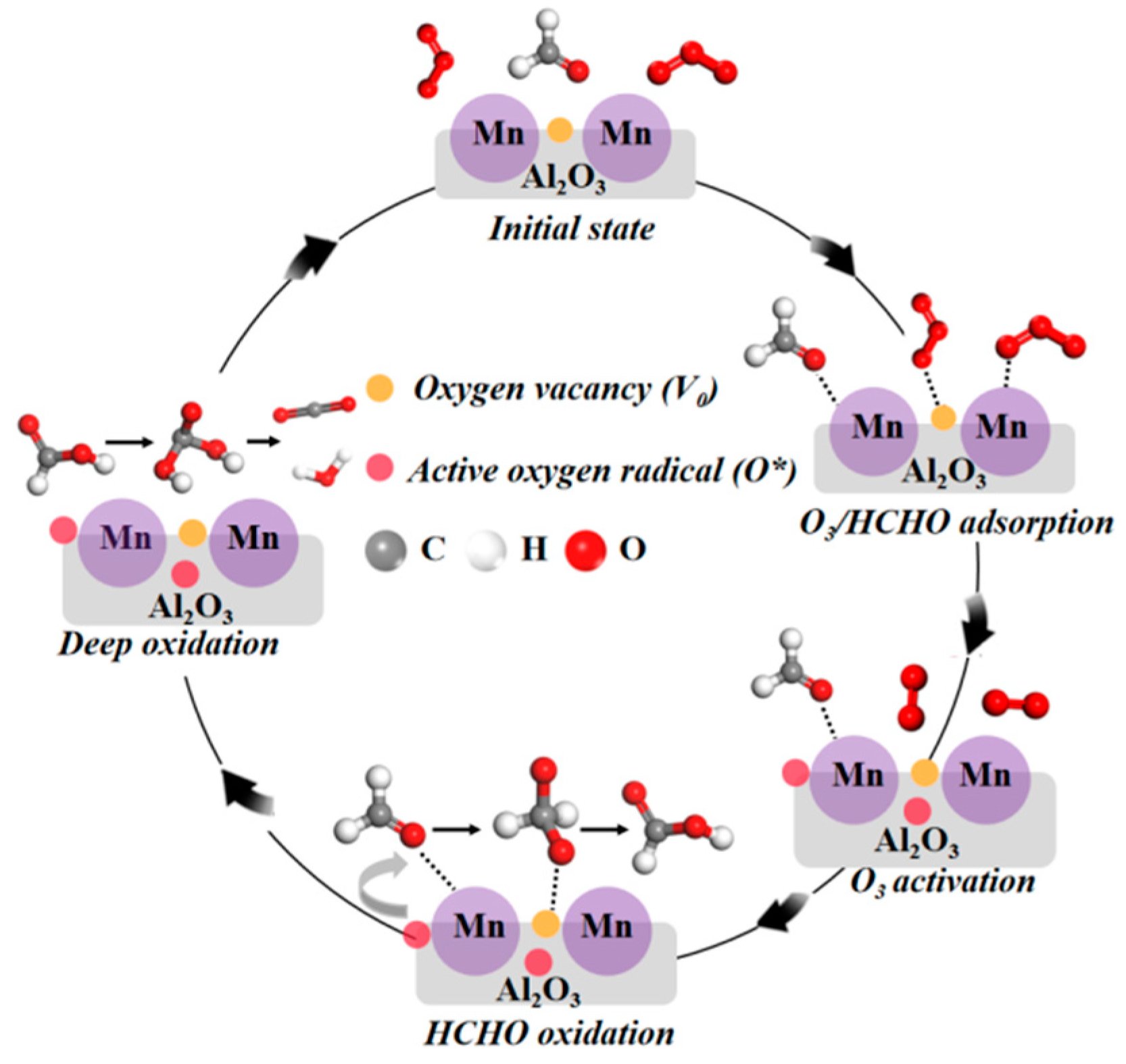
2.5. Theoretical Calculation and Reaction Mechanism
3. Materials and Methods
3.1. Catalyst Preparation
3.2. Catalytic Activity
3.3. Catalyst Characterization
3.4. DFT Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Liang, W.; Yang, S.; Yang, X. Long-term formaldehyde emissions from medium-density fiberboard in a full-scale experimental room: Emission characteristics and the effects of temperature and humidity. Environ. Sci. Technol. 2015, 49, 10349–10356. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Li, D.; Zhang, P.; Li, J.; Wang, J.; Yu, J. Formaldehyde and volatile organic compound (VOC) emissions from particleboard: Identification of odorous compounds and effects of heat treatment. Build. Environ. 2017, 117, 118–126. [Google Scholar] [CrossRef]
- Salthammer, T. Formaldehyde sources, formaldehyde concentrations and air exchange rates in European housings. Build. Environ. 2019, 150, 219–232. [Google Scholar] [CrossRef]
- Ji, J.; Lu, X.; Chen, C.; He, M.; Huang, H. Potassium-modulated δ-MnO2 as robust catalysts for formaldehyde oxidation at room temperature. Appl. Catal. B Environ. 2020, 260, 118210. [Google Scholar] [CrossRef]
- Robert, B.; Nallathambi, G. Indoor formaldehyde removal by catalytic oxidation, adsorption and nanofibrous membranes: A review. Environ. Chem. Lett. 2021, 19, 2551–2579. [Google Scholar] [CrossRef]
- Shao, J.; Lin, F.; Wang, Z.; Liu, P.; Tang, H.; He, Y.; Cen, K. Low temperature catalytic ozonation of toluene in flue gas over Mn-based catalysts: Effect of support property and SO2/water vapor addition. Appl. Catal. B Environ. 2020, 266, 118662. [Google Scholar] [CrossRef]
- He, C.; Wang, Y.; Li, Z.; Huang, Y.; Liao, Y.; Xia, D.; Lee, S. Facet engineered α-MnO2 for efficient catalytic ozonation of odor CH3SH: Oxygen vacancy-induced active centers and catalytic mechanism. Environ. Sci. Technol. 2020, 54, 12771–12783. [Google Scholar] [CrossRef]
- Ge, Y.; Fu, K.; Zhao, Q.; Ji, N.; Song, C.; Ma, D.; Liu, Q. Performance study of modified Pt catalysts for the complete oxidation of acetone. Chem. Eng. Sci. 2019, 206, 499–506. [Google Scholar] [CrossRef]
- Zhang, C.; He, H.; Tanaka, K.I. Perfect catalytic oxidation of formaldehyde over a Pt/TiO2 catalyst at room temperature. Catal. Commun. 2005, 6, 211–214. [Google Scholar] [CrossRef]
- Zhang, C.; He, H.; Tanaka, K.I. Catalytic performance and mechanism of a Pt/TiO2 catalyst for the oxidation of formaldehyde at room temperature. Appl. Catal. B Environ. 2006, 65, 37–43. [Google Scholar] [CrossRef]
- Huang, H.; Leung, D.Y. Complete oxidation of formaldehyde at room temperature using TiO2 supported metallic Pd nanoparticles. ACS Catal. 2011, 1, 348–354. [Google Scholar] [CrossRef]
- Yu, X.; He, J.; Wang, D.; Hu, Y.; Tian, H.; He, Z. Facile controlled synthesis of Pt/MnO2 nanostructured catalysts and their catalytic performance for oxidative decomposition of formaldehyde. J. Phys. Chem. C 2012, 116, 851–860. [Google Scholar] [CrossRef]
- Guo, Y.; Wen, M.; Li, G.; An, T. Recent advances in VOC elimination by catalytic oxidation technology onto various nanoparticles catalysts: A critical review. Appl. Catal. B Environ. 2021, 281, 119447. [Google Scholar] [CrossRef]
- Sekine, Y. Oxidative decomposition of formaldehyde by metal oxides at room temperature. Atmos. Environ. 2002, 36, 5543–5547. [Google Scholar] [CrossRef]
- Zhang, Y.; Shi, J.; Fang, W.; Chen, M.; Zhang, Z.; Jiang, Z.; Shangguan, W.; Einaga, H. Simultaneous catalytic elimination of formaldehyde and ozone over one-dimensional rod-like manganese dioxide at ambient temperature. J. Chem. Technol. Biotechnol. 2019, 94, 2305–2317. [Google Scholar] [CrossRef]
- Xiang, L.; Lin, F.; Cai, B.; Li, G.; Zhang, L.; Wang, Z.; Yan, B.; Wang, Y.; Chen, G. Catalytic ozonation of CH2Cl2 over hollow urchin-like MnO2 with regulation of active oxygen by catalyst modification and ozone promotion. J. Hazard. Mater. 2022, 436, 129217. [Google Scholar] [CrossRef]
- Xi, Y.; Reed, C.; Lee, Y.K.; Oyama, S.T. Acetone oxidation using ozone on manganese oxide catalysts. J. Phys. Chem. B 2005, 109, 17587–17596. [Google Scholar] [CrossRef]
- Ghavami, M.; Soltan, J.; Chen, N.J.C.L. Synthesis of MnOx/Al2O3 catalyst by Polyol method and its application in room temperature ozonation of toluene in air. Catal. Lett. 2021, 151, 1418–1432. [Google Scholar] [CrossRef]
- Yang, Y.; Si, W.; Peng, Y.; Chen, J.; Wang, Y.; Chen, D.; Tian, Z.; Wang, J.; Li, J. Oxygen vacancy engineering on copper-manganese spinel surface for enhancing toluene catalytic combustion: A comparative study of acid treatment and alkali treatment. Appl. Catal. B Environ. 2024, 340, 123142. [Google Scholar] [CrossRef]
- Zhao, D.Z.; Shi, C.; Li, X.S.; Zhu, A.M.; Jang, B.W.L. Enhanced effect of water vapor on complete oxidation of formaldehyde in air with ozone over MnOx catalysts at room temperature. J. Hazard. Mater. 2012, 239, 362–369. [Google Scholar] [CrossRef]
- Wang, H.; Huang, Z.; Jiang, Z.; Jiang, Z.; Zhang, Y.; Zhang, Z.; Shangguan, W. Trifunctional C@ MnO catalyst for enhanced stable simultaneously catalytic removal of formaldehyde and ozone. ACS Catal. 2018, 8, 3164–3180. [Google Scholar] [CrossRef]
- Li, J.W.; Pan, K.L.; Yu, S.J.; Yan, S.Y.; Chang, M.B. Removal of formaldehyde over MnxCe1−xO2 catalysts: Thermal catalytic oxidation versus ozone catalytic oxidation. J. Environ. Sci. 2014, 26, 2546–2553. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, G.; Deng, W.; Han, J.; Qin, L.; Zhao, B.; Guo, L.; Xing, F. Study on the structure-activity relationship of Fe-Mn oxide catalysts for chlorobenzene catalytic combustion. Chem. Eng. J. 2020, 395, 125172. [Google Scholar] [CrossRef]
- Zhang, Z.; Xiang, L.; Lin, F.; Wang, Z.; Yan, B.; Chen, G. Catalytic deep degradation of Cl-VOCs with the assistance of ozone at low temperature over MnO2 catalysts. Chem. Eng. J. 2021, 426, 130814. [Google Scholar] [CrossRef]
- Esmaeilirad, M.; Zabihi, M.; Shayegan, J.; Khorasheh, F. Oxidation of toluene in humid air by metal oxides supported on γ-alumina. J. Hazard. Mater. 2017, 333, 293–307. [Google Scholar] [CrossRef]
- Liu, P.; Tang, H.; Shao, J.; He, Y.; Zhu, Y.; Alegria, E.C.; Wang, Z.; Pombeiro, A.J. Catalytic ozonation of multi-VOCs mixtures over Cr-based bimetallic oxides catalysts from simulated flue gas: Effects of NO/SO2/H2O. Chemosphere 2023, 340, 139851. [Google Scholar] [CrossRef]
- Liu, P.; Chen, L.; Tang, H.; Shao, J.; Lin, F.; He, Y.; Zhu, Y.; Wang, Z. Low temperature ozonation of acetone by transition metals derived catalysts: Activity and sulfur/water resistance. Catalysts 2022, 12, 1090. [Google Scholar] [CrossRef]
- Jia, J.; Zhang, P.; Chen, L. Catalytic decomposition of gaseous ozone over manganese dioxides with different crystal structures. Appl. Catal. B Environ. 2016, 189, 210–218. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, W.; Zhang, P.; Zhang, J. Nitric acid-treated birnessite-type MnO2: An efficient and hydrophobic material for humid ozone decomposition. Appl. Surf. Sci. 2018, 442, 640–649. [Google Scholar] [CrossRef]
- Zhang, Z.; Lin, F.; Xiang, L.; Yu, H.; Wang, Z.; Yan, B.; Chen, G. Synergistic effect for simultaneously catalytic ozonation of chlorobenzene and NO over MnCoOx catalysts: Byproducts formation under practical conditions. Chem. Eng. J. 2022, 427, 130929. [Google Scholar] [CrossRef]
- Shao, J.; Lin, F.; Huang, Y.; Wang, Z.; Li, Y.; Chen, G.; Cen, K. MnOx fabrication with rational design of morphology for enhanced activity in NO oxidation and SO2 resistance. Appl. Surf. Sci. 2020, 503, 144064. [Google Scholar] [CrossRef]
- Yang, J.; Huang, Y.; Chen, Y.W.; Xia, D.; Mou, C.Y.; Hu, L.; Zeng, J.; He, C.; Wong, P.K.; Zhu, H.Y. Active site-directed tandem catalysis on CuO/VO-MnO2 for efficient and stable catalytic ozonation of S-VOCs under mild condition. Nano Today 2020, 35, 100944. [Google Scholar] [CrossRef]
- Dong, L.; Tang, Y.; Li, B.; Zhou, L.; Gong, F.; He, H.; Sun, B.; Tang, C.; Gao, F.; Dong, L. Influence of molar ratio and calcination temperature on the properties of TixSn1−xO2 supporting copper oxide for CO oxidation. Appl. Catal. B Environ. 2016, 180, 451–462. [Google Scholar] [CrossRef]
- Fernandes, A.; Gągol, M.; Makoś, P.; Khan, J.A.; Boczkaj, G. Integrated photocatalytic advanced oxidation system (TiO2/UV/O3/H2O2) for degradation of volatile organic compounds. Sep. Purif. Technol. 2019, 224, 1–14. [Google Scholar] [CrossRef]
- Sun, P.; Wang, W.; Dai, X.; Weng, X.; Wu, Z. Mechanism study on catalytic oxidation of chlorobenzene over MnxCe1−xO2/H-ZSM5 catalysts under dry and humid conditions. Appl. Catal. B Environ. 2016, 198, 389–397. [Google Scholar] [CrossRef]
- Weng, X.; Sun, P.; Long, Y.; Meng, Q.; Wu, Z. Catalytic oxidation of chlorobenzene over MnxCe1−xO2/HZSM-5 catalysts: A study with practical implications. Environ. Sci. Technol. 2017, 51, 8057–8066. [Google Scholar] [CrossRef]
- Tian, M.; Guo, X.; Dong, R.; Guo, Z.; Shi, J.; Yu, Y.; Cheng, M.; Albilali, R.; He, C. Insight into the boosted catalytic performance and chlorine resistance of nanosphere-like meso-macroporous CrOx/MnCo3Ox for 1, 2-dichloroethane destruction. Appl. Catal. B Environ. 2019, 259, 118018. [Google Scholar] [CrossRef]
- Tang, H.; Wang, Z.; Shao, J.; Lin, F.; Liu, P.; He, Y.; Zhu, Y. Catalytic decomposition of residual ozone over cactus-like MnO2 nanosphere: Synergistic mechanism and SO2/H2O interference. ACS Omega 2022, 7, 9818–9833. [Google Scholar] [CrossRef]
- Sun, S.; Ding, J.; Bao, J.; Gao, C.; Qi, Z.; Li, C. Photocatalytic oxidation of gaseous formaldehyde on TiO2: An in situ DRIFTS study. Catal. Lett. 2010, 137, 239–246. [Google Scholar] [CrossRef]
- Chen, B.B.; Zhu, X.B.; Crocker, M.; Wang, Y.; Shi, C. FeOx-supported gold catalysts for catalytic removal of formaldehyde at room temperature. Appl. Catal. B Environ. 2014, 154, 73–81. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, J.; Rong, S.; Wang, H.; Zhang, P. Cerium modified birnessite-type MnO2 for gaseous formaldehyde oxidation at low temperature. Appl. Catal. B Environ. 2017, 211, 212–221. [Google Scholar] [CrossRef]
- Soria, J.; Sanz, J.; Sobrados, I.; Coronado, J.M.; Hernández-Alonso, M.D.; Fresno, F. Water-hydroxyl interactions on small anatase nanoparticles prepared by the hydrothermal route. J. Phys. Chem. C 2010, 114, 16534–16540. [Google Scholar] [CrossRef]
- Hernández-Alonso, M.D.; García-Rodríguez, S.; Suárez, S.; Portela, R.; Sánchez, B.; Coronado, J.M. Operando DRIFTS study of the role of hydroxyls groups in trichloroethylene photo-oxidation over titanate and TiO2 nanostructures. Catal. Today 2013, 206, 32–39. [Google Scholar] [CrossRef]
- Yamamoto, M.; Yoshida, T.; Yamamoto, N.; Nomoto, T.; Yamamoto, Y.; Yagi, S.; Yoshida, H. Photocatalytic reduction of CO2 with water promoted by Ag clusters in Ag/Ga2O3 photocatalysts. J. Mater. Chem. A 2015, 3, 16810–16816. [Google Scholar] [CrossRef]
- Cheng, Y.; Chen, Z.; Yan, P.; Shen, J.; Kang, J.; Wang, S.; Duan, X. Synergistic catalytic ozonation mediated by dual active sites of oxygen vacancies and defects in biomass-derived composites for long-lasting water decontamination. ACS Catal. 2024, 14, 4040–4052. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.; Probert, M.A.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717. [Google Scholar] [CrossRef]
- Luo, J.; Meng, X.; Crittenden, J.; Qu, J.; Hu, C.; Liu, H.; Peng, P. Arsenic adsorption on α-MnO2 nanofibers and the significance of (1 0 0) facet as compared with (1 1 0). Chem. Eng. J. 2018, 331, 492–500. [Google Scholar] [CrossRef]
- Li, G.; Zhao, P.; Zheng, H.; Yang, L.; Lu, S.; Peng, P. Research on the removal mechanism of antimony on α-MnO2 nanorod in aqueous solution: DFT+ U method. J. Hazard. Mater. 2018, 354, 8–16. [Google Scholar] [CrossRef]
- Luo, J.; Hu, C.; Meng, X.; Crittenden, J.; Qu, J.; Peng, P. Antimony removal from aqueous solution using novel α-MnO2 nanofibers: Equilibrium, kinetic, and density functional theory studies. ACS Sustain. Chem. Eng. 2017, 5, 2255–2264. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Reaction Mixture | GHSV (h−1) | Temperature (°C) | Conversion (%) | Reference |
|---|---|---|---|---|---|
| MnOx | HCHO: 82 ppm | 200,000 | 25 | ~100 | [20] |
| Ozone: 720 ppm | |||||
| α-MnO2 | HCHO: 60 ppm | / | Ambient temperature | 80 | [15] |
| Ozone: 230 ppm | |||||
| MnO@C | HCHO: 60 ppm | / | 30 | 100 | [21] |
| Ozone: 180 ppm | |||||
| Mn0.5Ce0.5O2 | HCHO: 61 ppm | 10,000 | 25 | ~83 | [22] |
| Ozone: 203 ppm | |||||
| MnAl-II | HCHO: 30 ppm | 50,000 | 30 | 100 | This work |
| Ozone: 60 ppm |
| Catalyst | BET Surface Area /m2·g−1 | Pore Volume a /cm3·g−1 | Average Pore Diameter b/nm |
|---|---|---|---|
| MnAl-I | 183.5 | 0.35 | 6.0 |
| MnAl-II | 188.3 | 0.34 | 5.9 |
| MnAl-III | 169.4 | 0.32 | 5.9 |
| MnAl-IV | 174.5 | 0.33 | 5.9 |
| Catalysts | Mn 2p3/2 | O 1s | Mn3+/Mn4+ | Oad/Ola | ||
|---|---|---|---|---|---|---|
| Mn4+ (%) | Mn3+ (%) | Ola (%) | Oad (%) | |||
| MnAl-I | 37.9 | 62.1 | 19.3 | 80.7 | 1.64 | 4.18 |
| MnAl-II | 37.7 | 62.3 | 13.7 | 86.3 | 1.65 | 6.30 |
| MnAl-III | 42.3 | 57.7 | 21.4 | 78.6 | 1.36 | 3.67 |
| MnAl-IV | 38.9 | 61.1 | 17.7 | 82.3 | 1.57 | 4.65 |
| Catalyst | H2 Uptake /(mmol·gcat−1) | O2 Desorption /(a.u.·gcat−1) | NH3 Desorption /(mmol·gcat−1) |
|---|---|---|---|
| MnAl-I | 4.03 | 3.60 | 3.26 |
| MnAl-II | 4.05 | 3.75 | 3.29 |
| MnAl-III | 5.28 | 3.36 | 3.28 |
| MnAl-IV | 5.15 | 3.68 | 3.22 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.; Zhang, Y.; Hou, B.; He, Y.; Weng, W.; Zhu, Y.; Wang, Z. Catalytic Ozonation of Formaldehyde with an Oxygen-Vacancy-Rich MnOx/γ-Al2O3 Catalyst at Room Temperature. Catalysts 2024, 14, 885. https://doi.org/10.3390/catal14120885
Sun Y, Zhang Y, Hou B, He Y, Weng W, Zhu Y, Wang Z. Catalytic Ozonation of Formaldehyde with an Oxygen-Vacancy-Rich MnOx/γ-Al2O3 Catalyst at Room Temperature. Catalysts. 2024; 14(12):885. https://doi.org/10.3390/catal14120885
Chicago/Turabian StyleSun, Yulin, Yiwei Zhang, Baoqing Hou, Yong He, Wubin Weng, Yanqun Zhu, and Zhihua Wang. 2024. "Catalytic Ozonation of Formaldehyde with an Oxygen-Vacancy-Rich MnOx/γ-Al2O3 Catalyst at Room Temperature" Catalysts 14, no. 12: 885. https://doi.org/10.3390/catal14120885
APA StyleSun, Y., Zhang, Y., Hou, B., He, Y., Weng, W., Zhu, Y., & Wang, Z. (2024). Catalytic Ozonation of Formaldehyde with an Oxygen-Vacancy-Rich MnOx/γ-Al2O3 Catalyst at Room Temperature. Catalysts, 14(12), 885. https://doi.org/10.3390/catal14120885