

Copper-Catalyzed/Hypervalent Iodine-Mediated Functionalization of Unactivated Compounds



Abstract
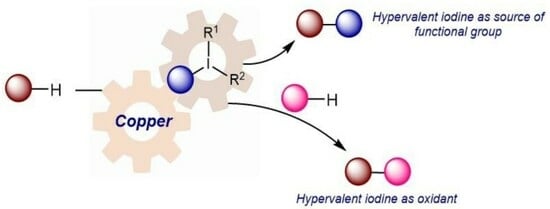
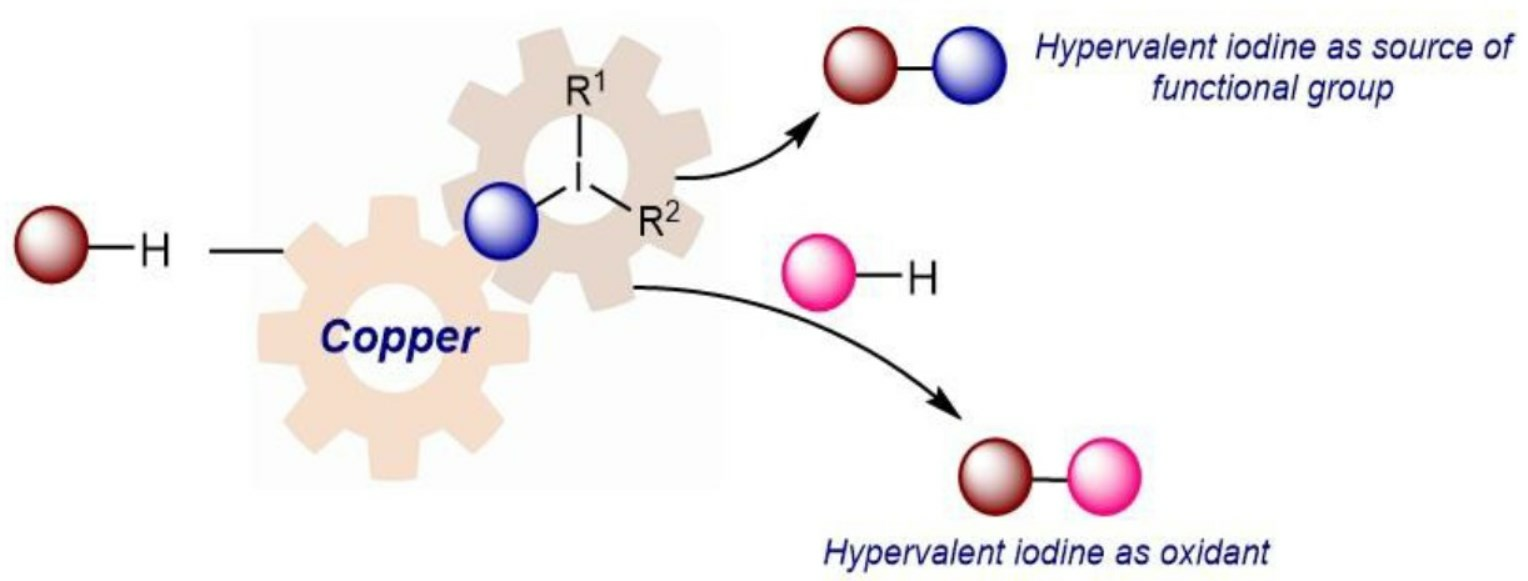{kind=link}
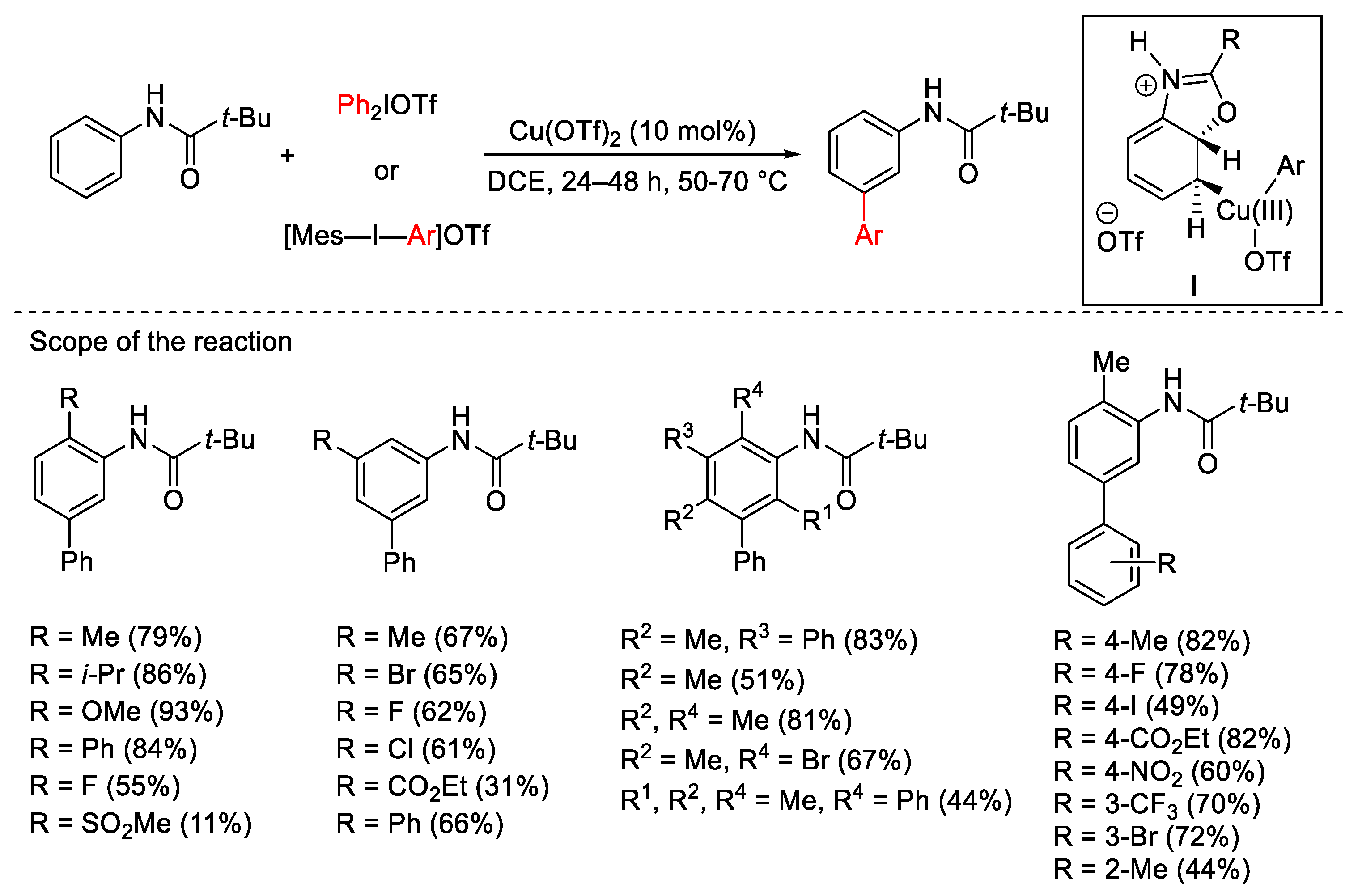{kind=link}
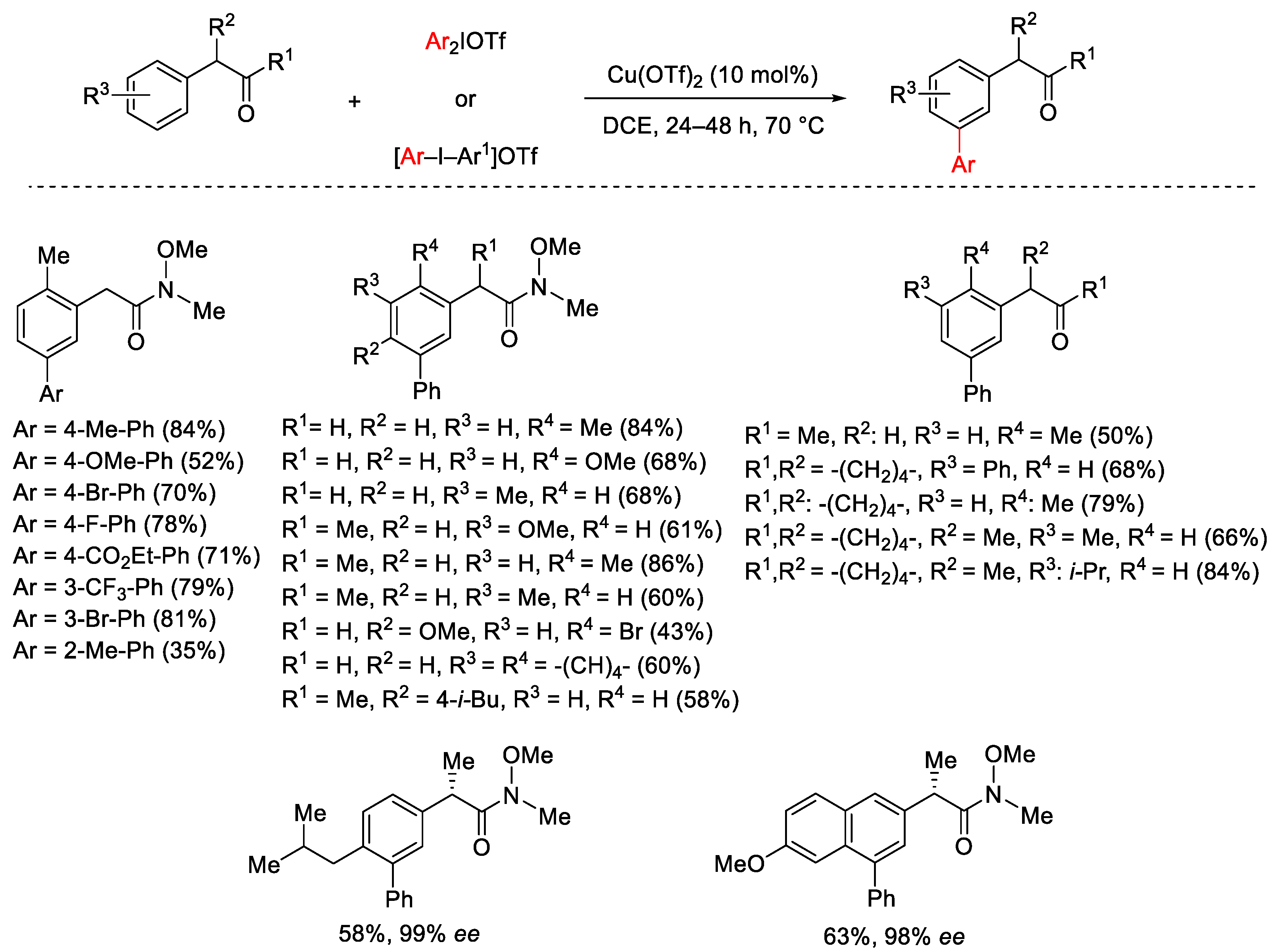{kind=link}
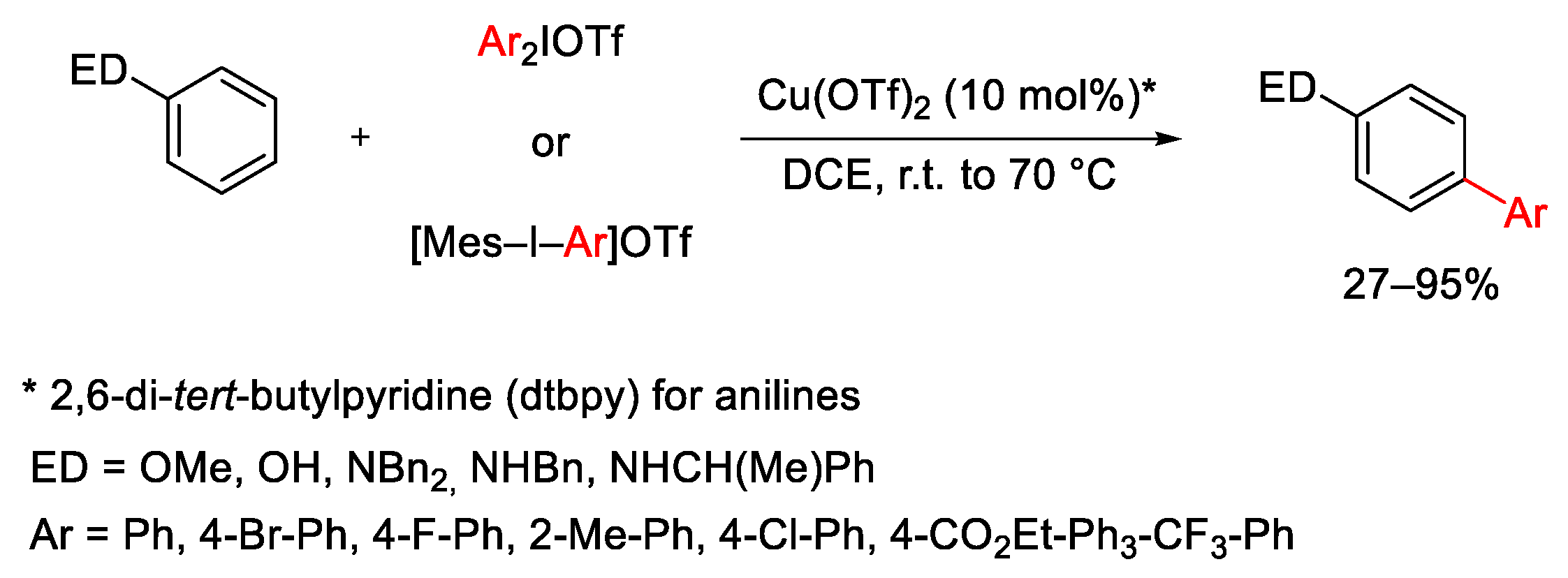{kind=link}
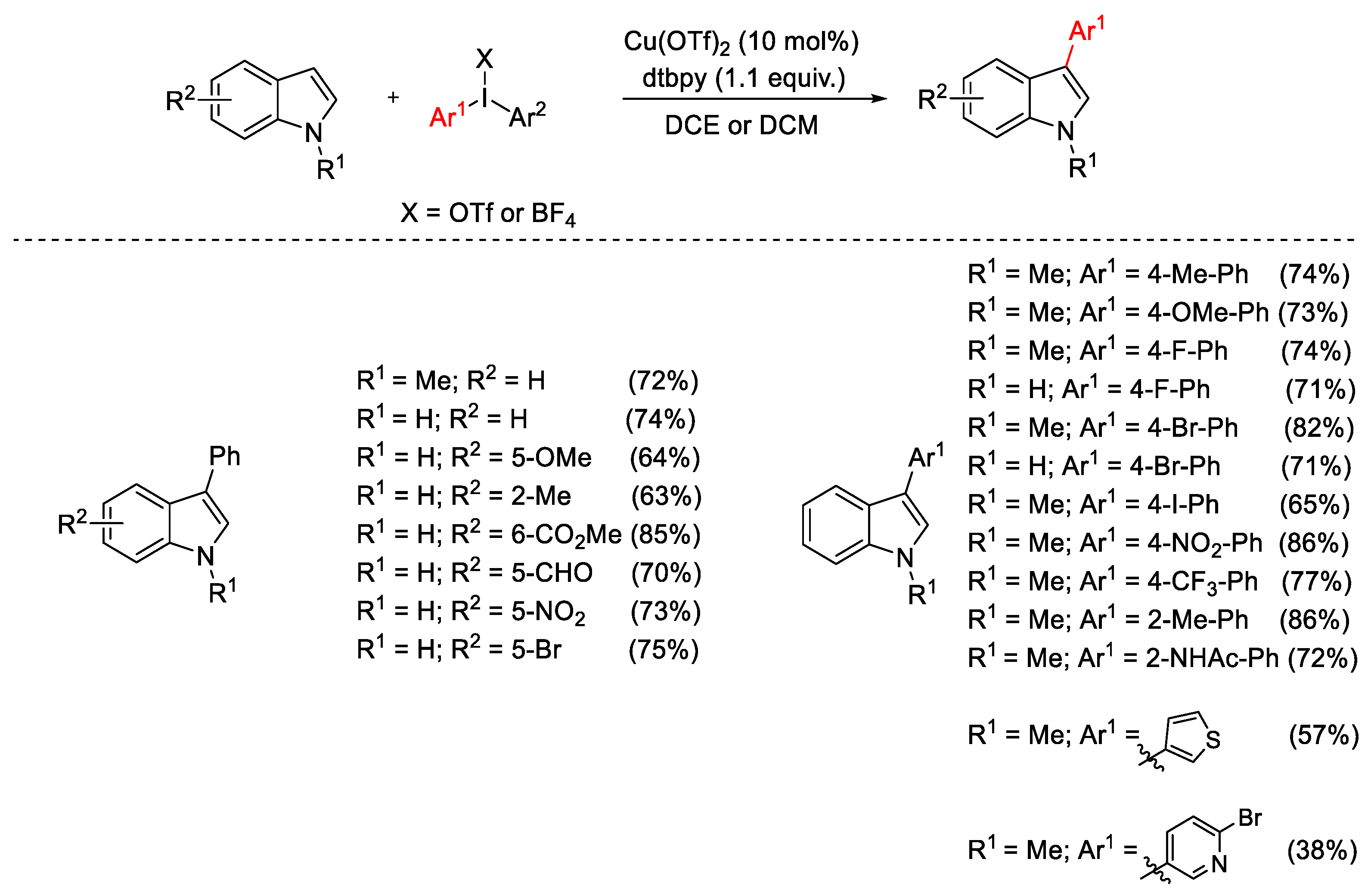{kind=link}
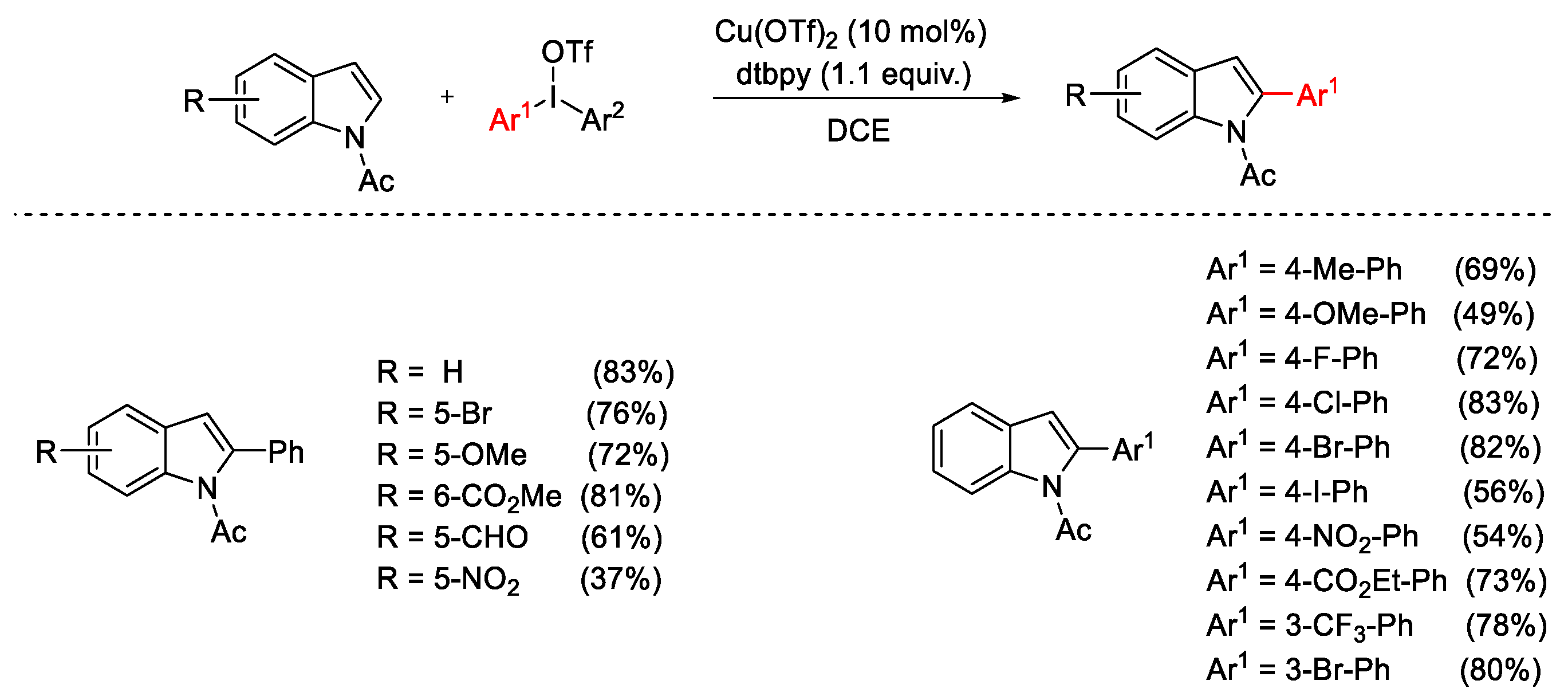{kind=link}
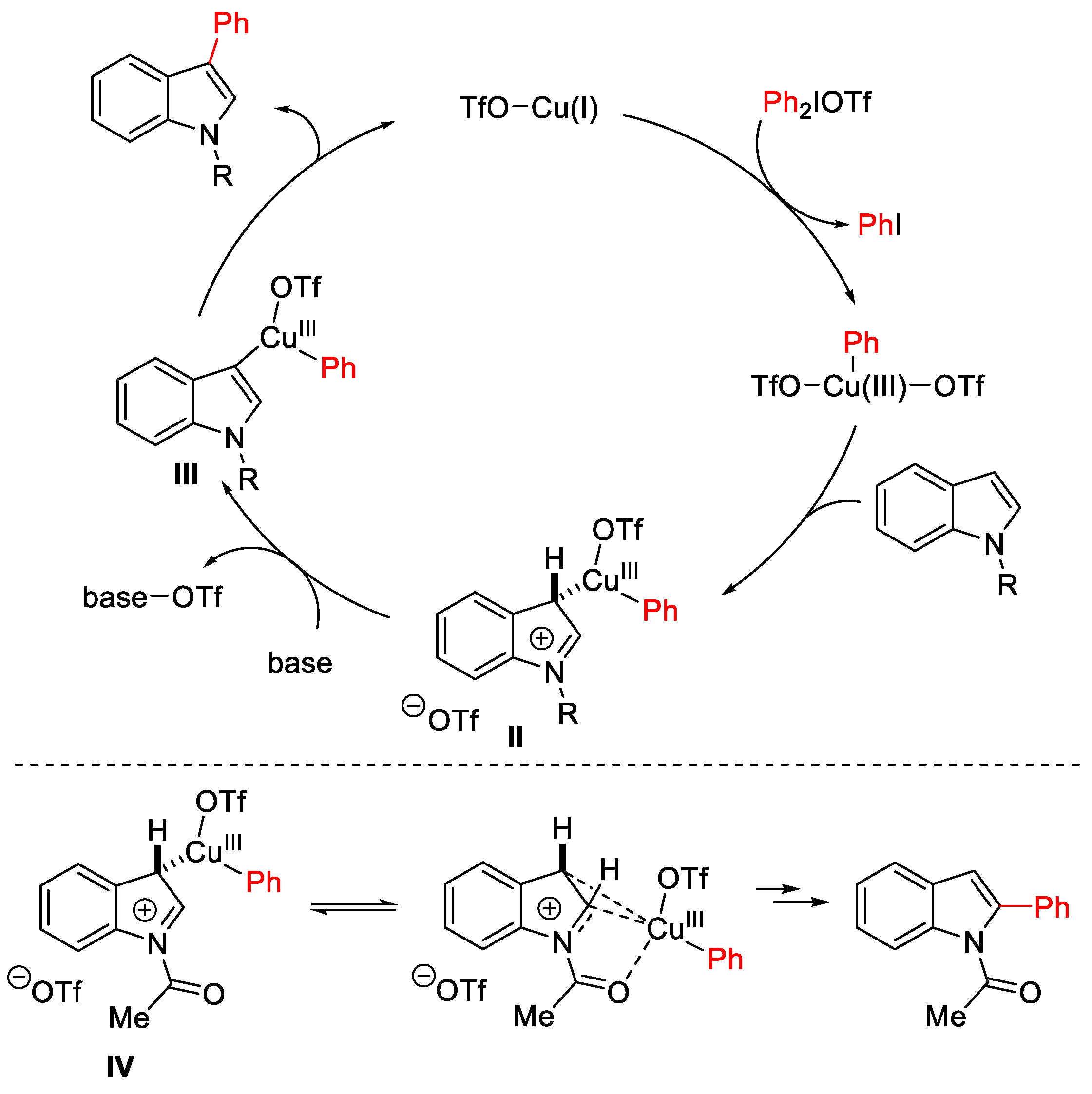{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
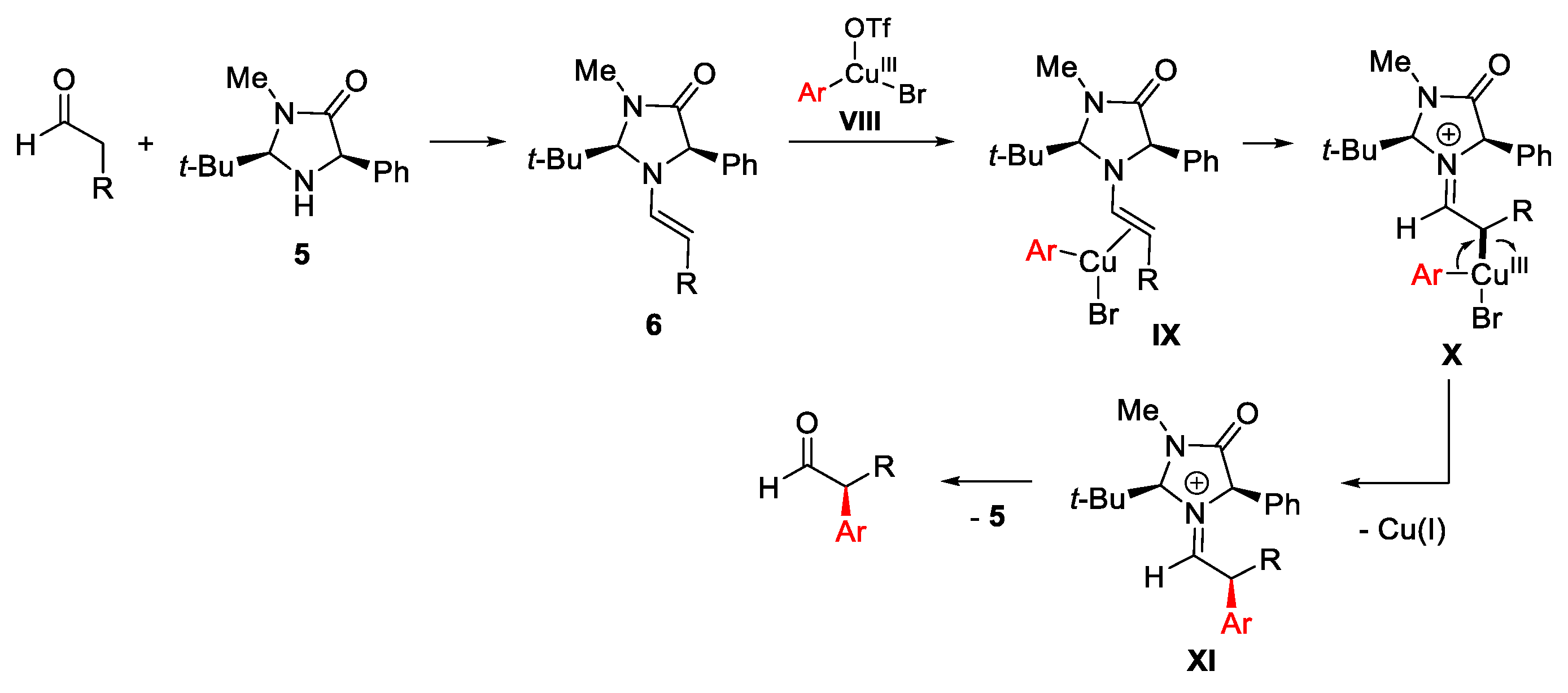{kind=link}
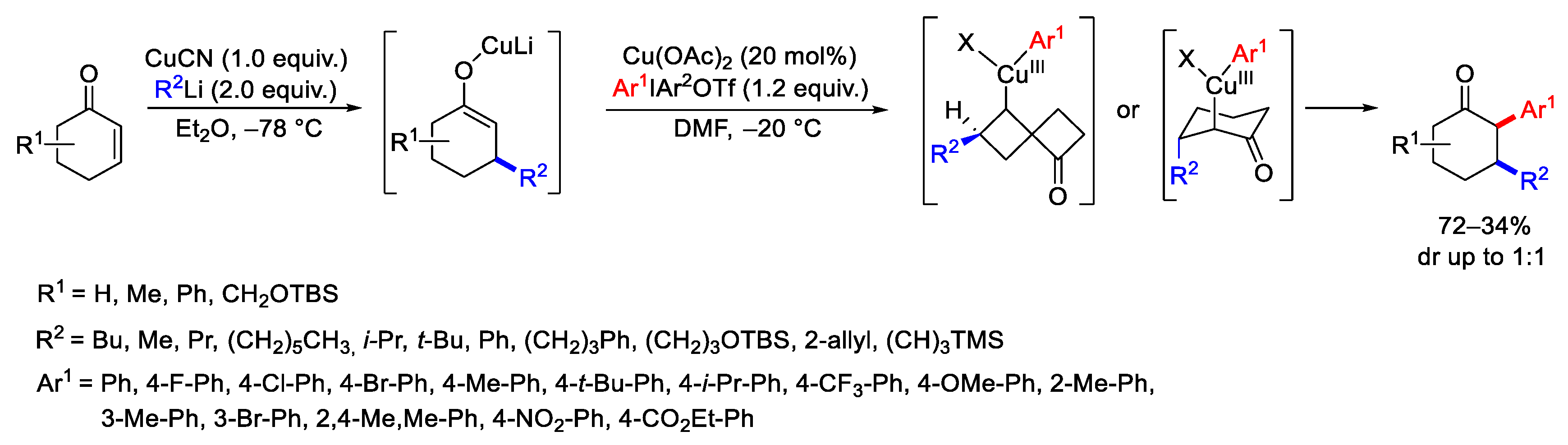{kind=link}
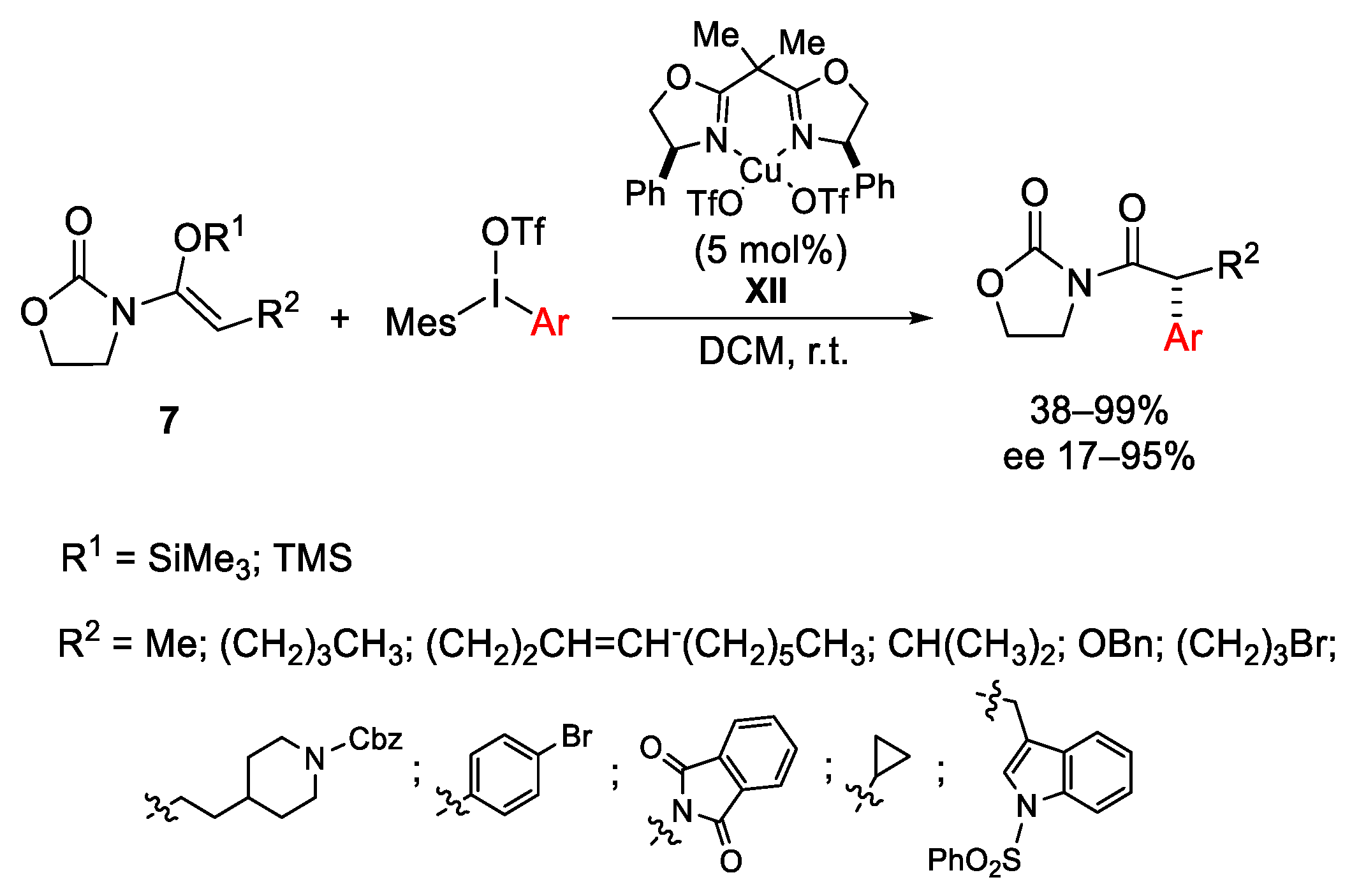{kind=link}
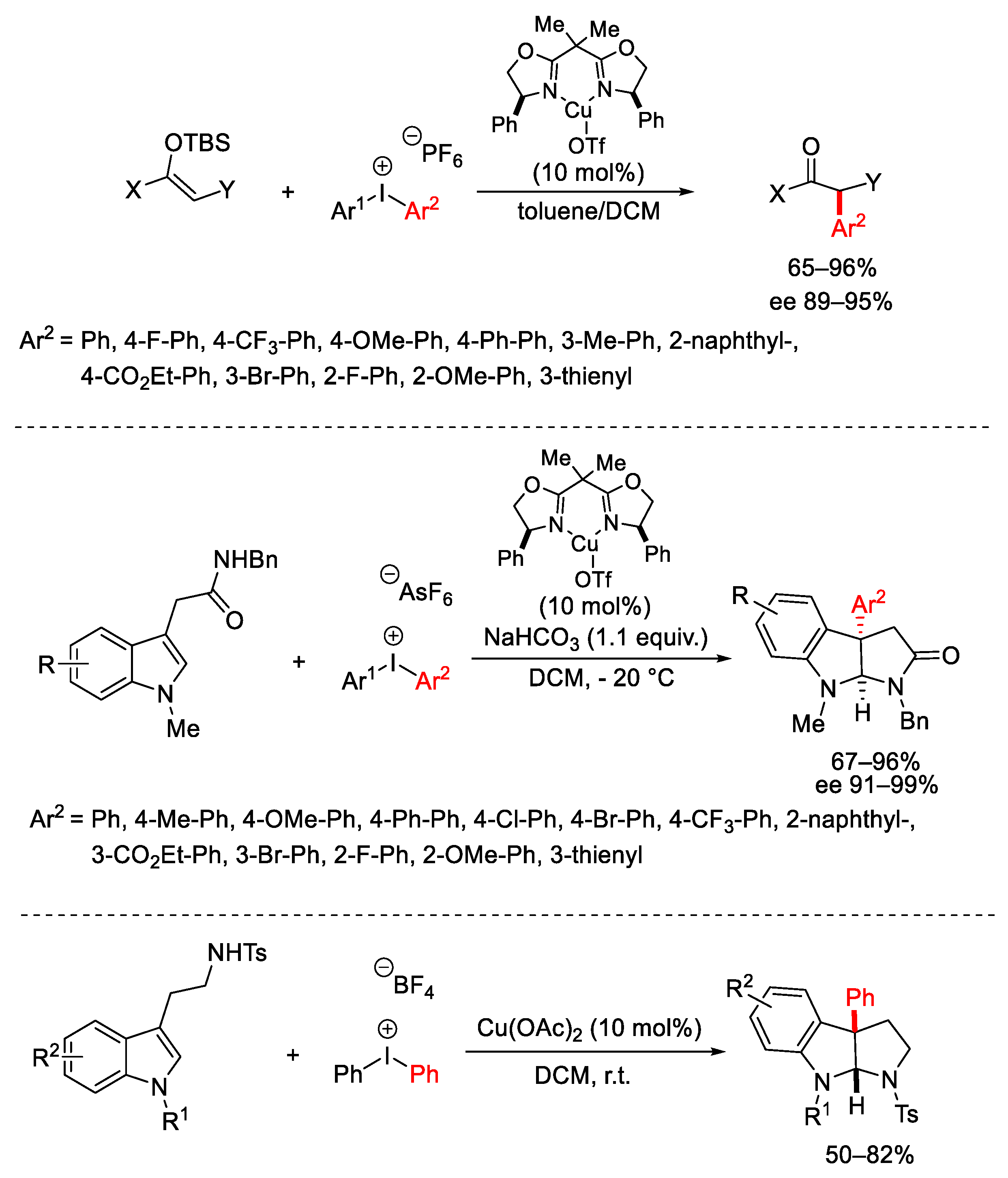{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
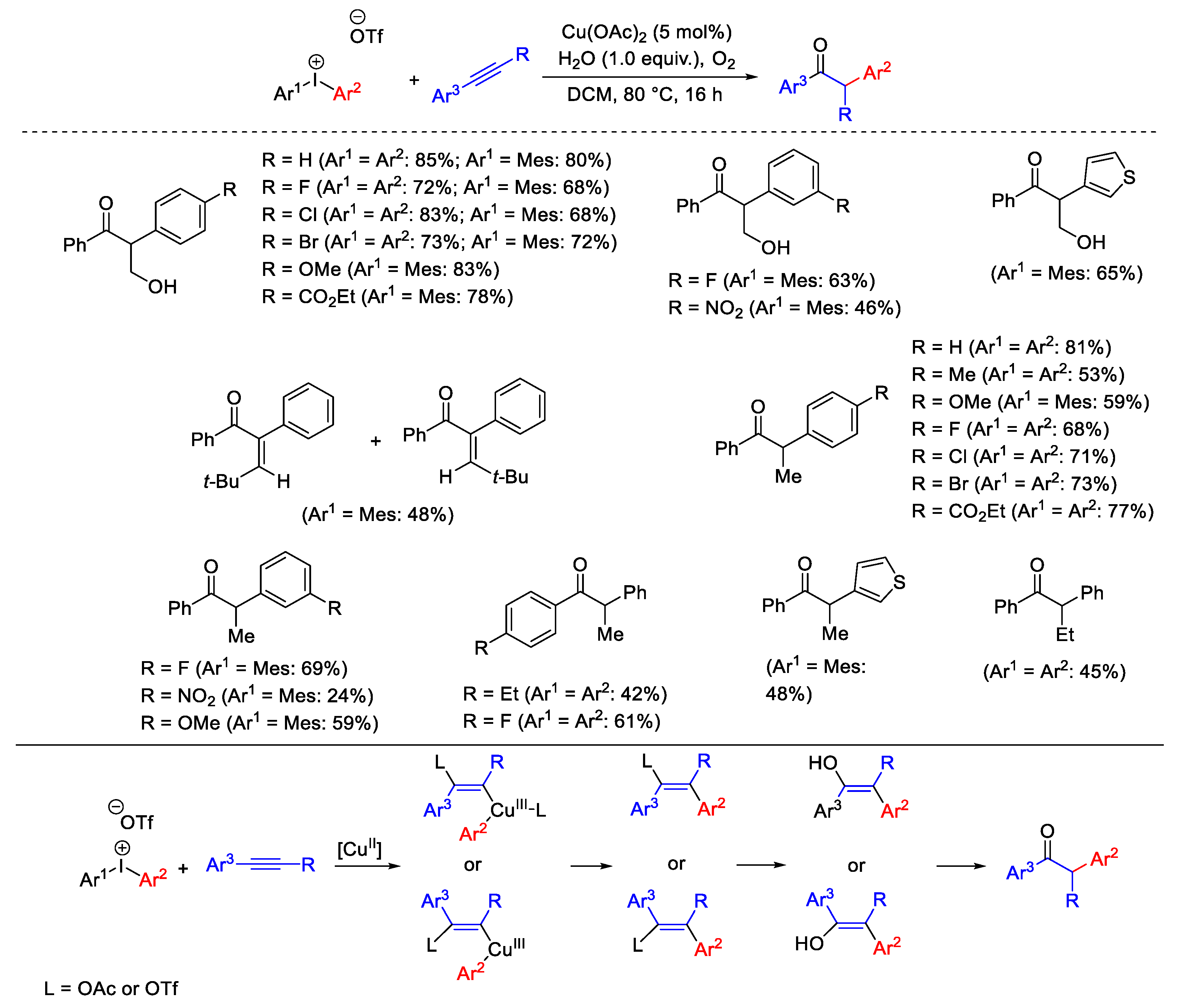{kind=link}
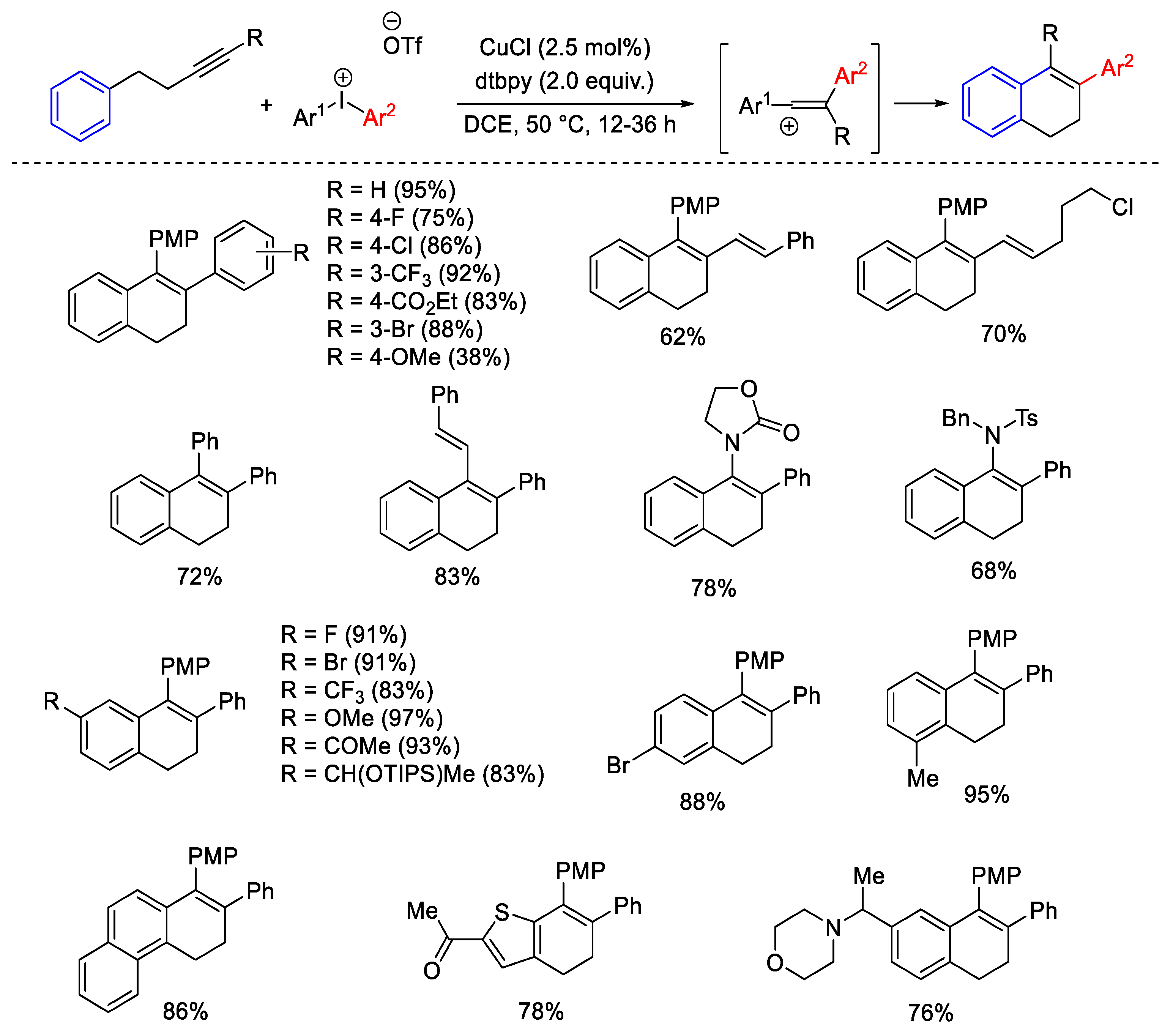{kind=link}
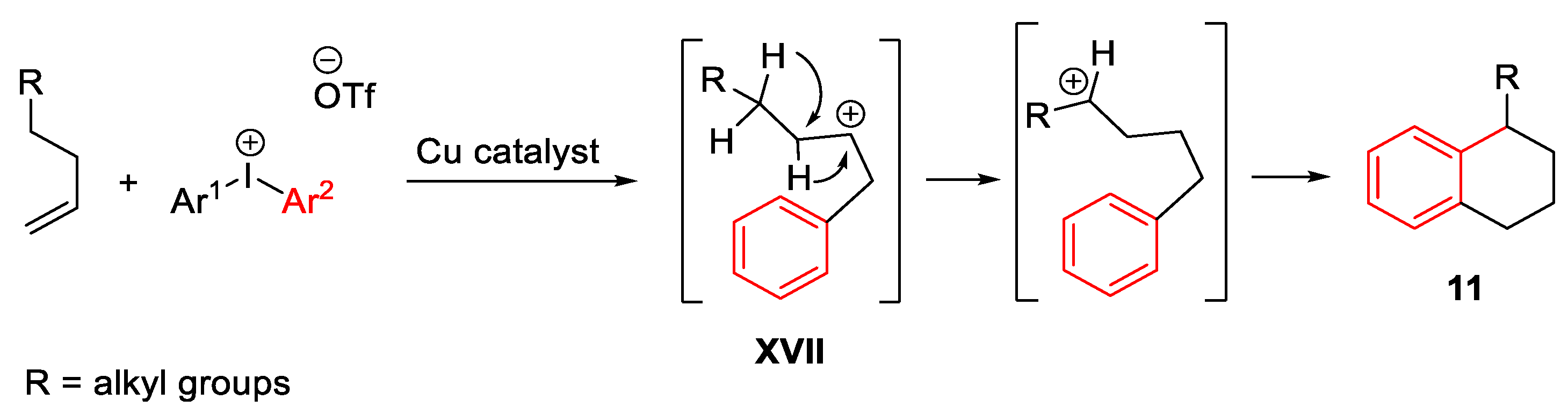{kind=link}
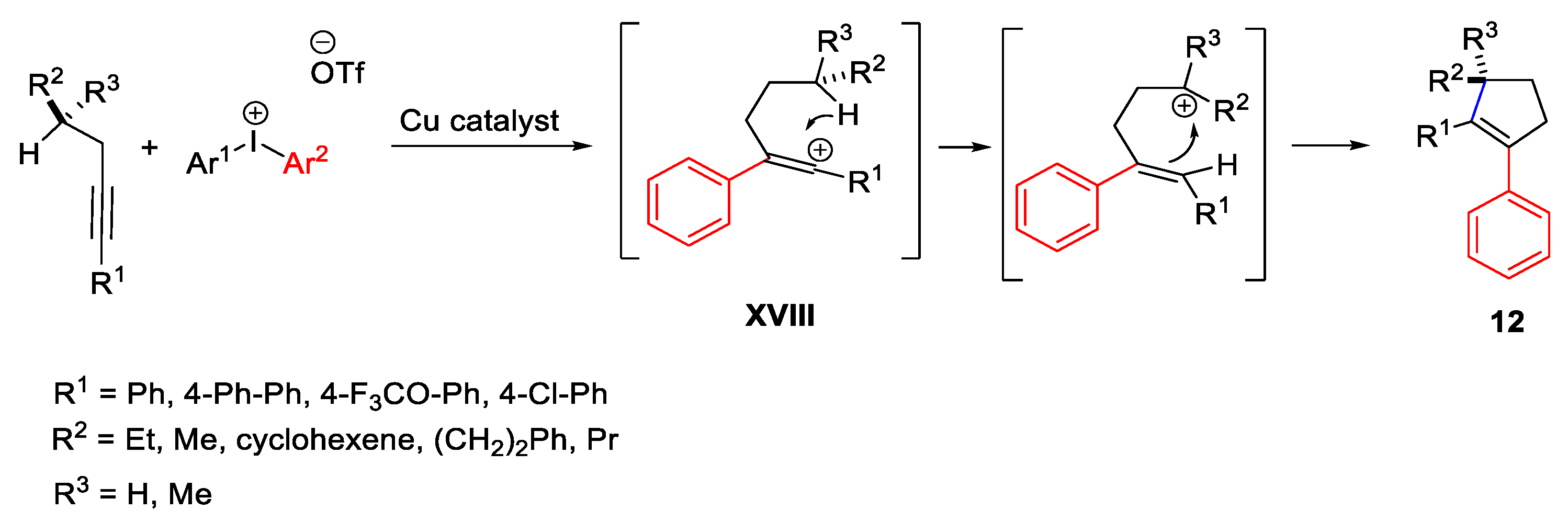{kind=link}
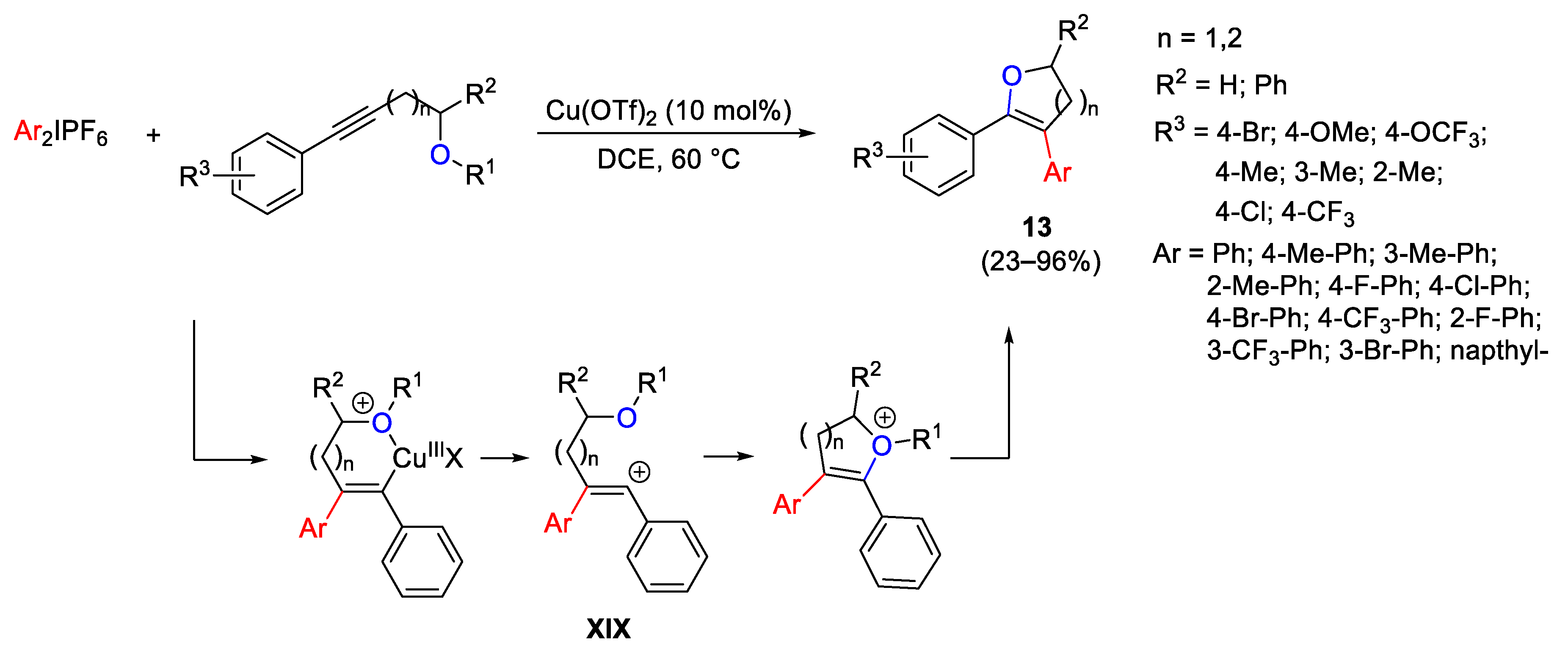{kind=link}
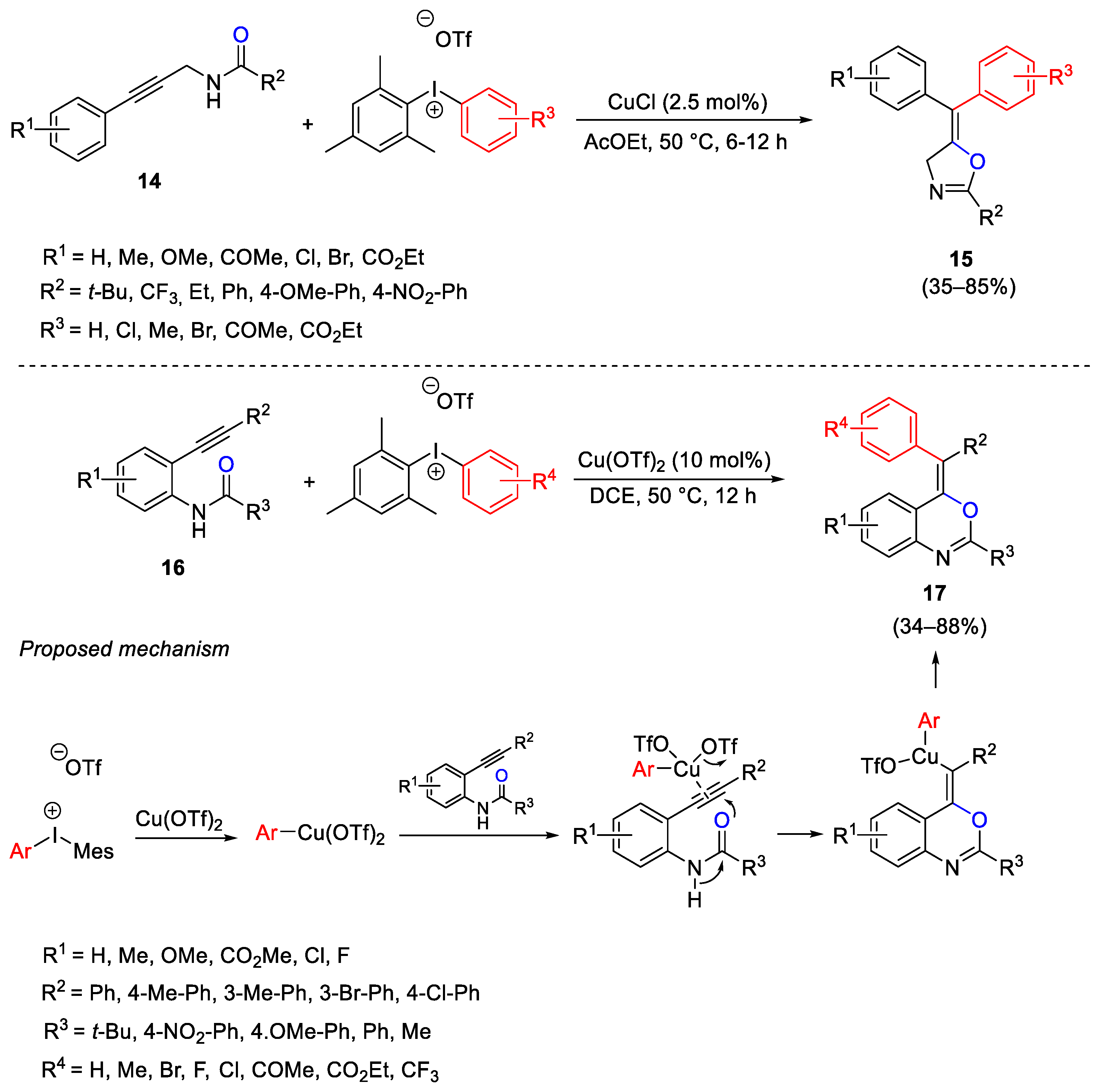{kind=link}
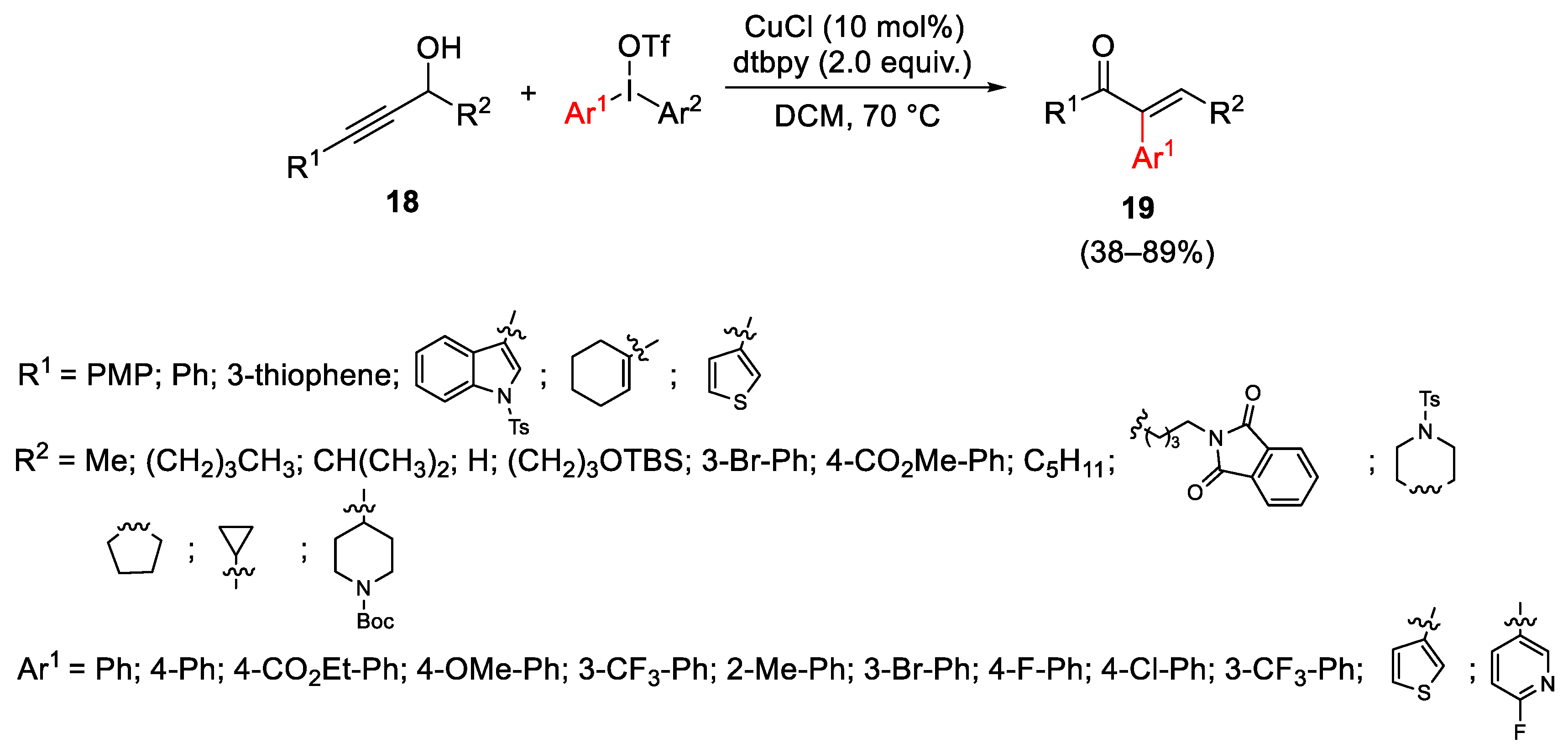{kind=link}
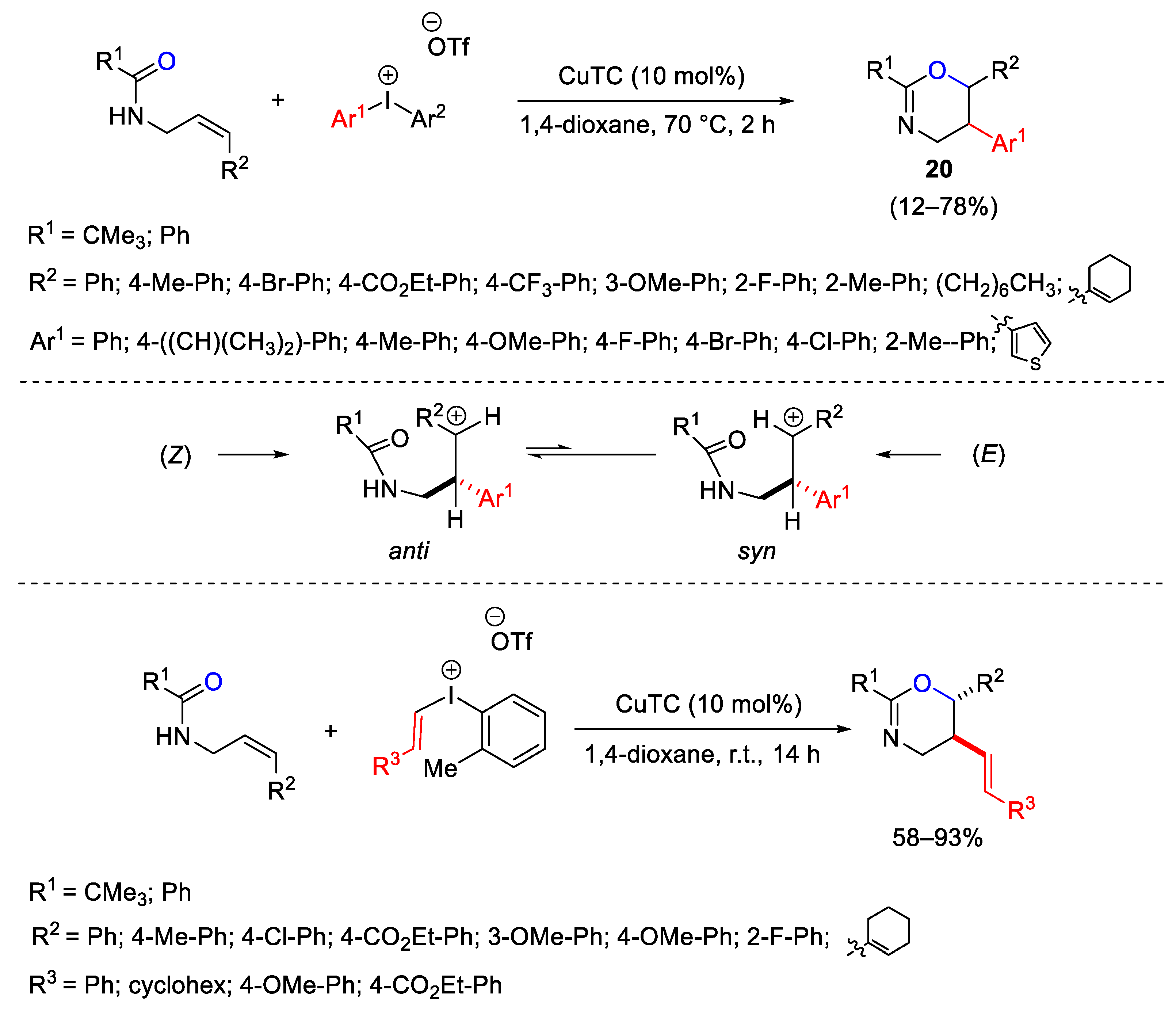{kind=link}
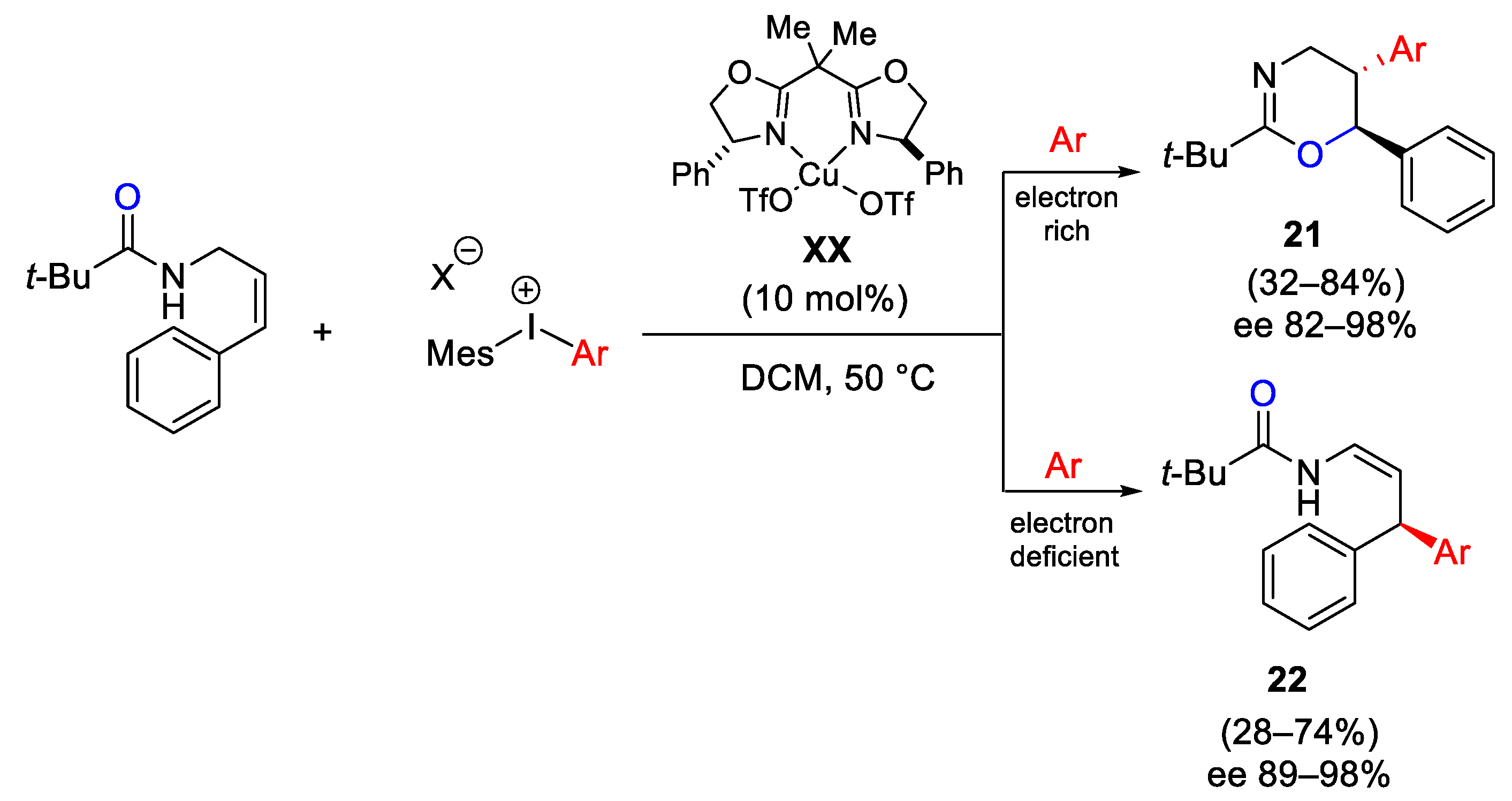{kind=link}
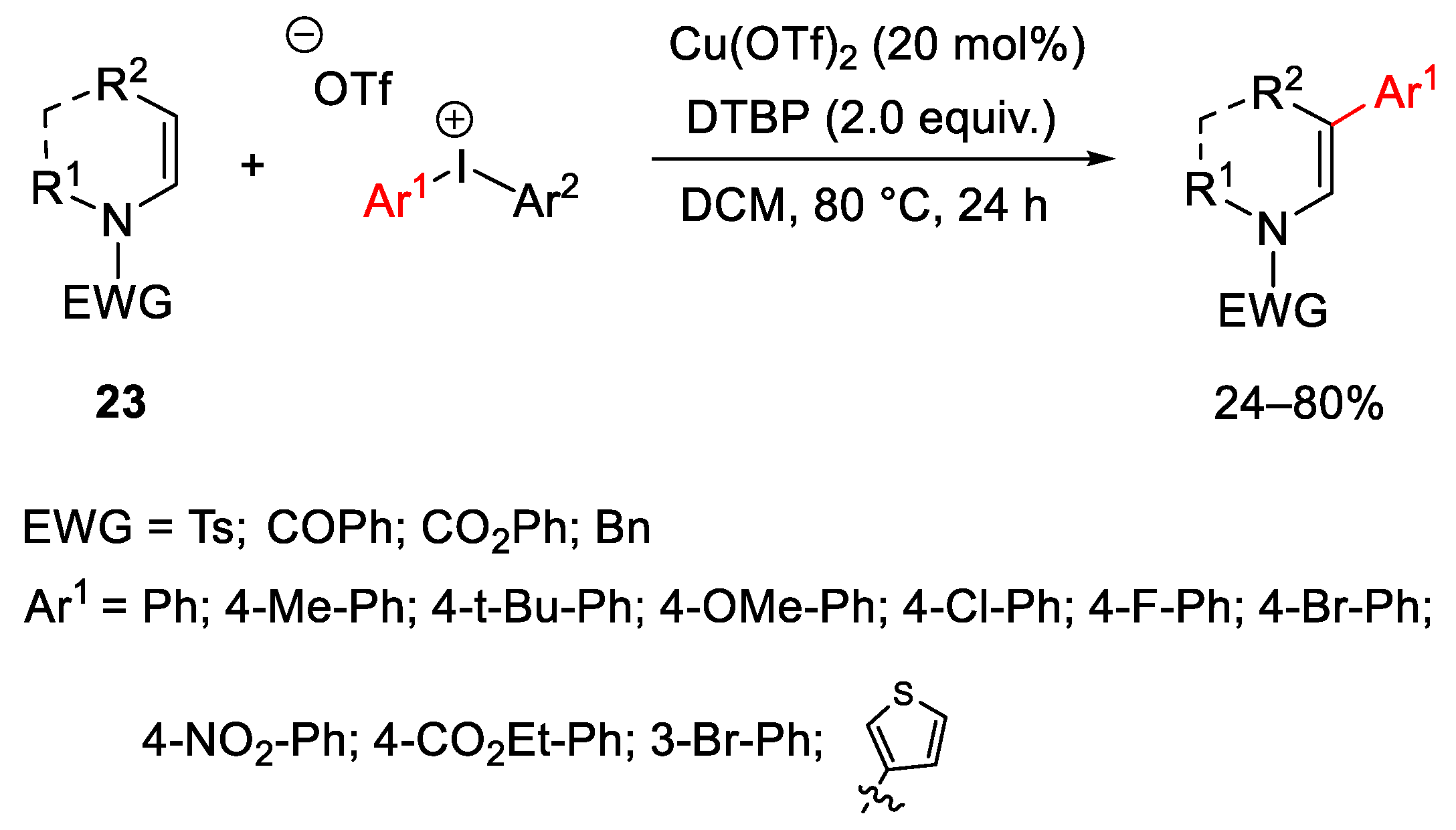{kind=link}
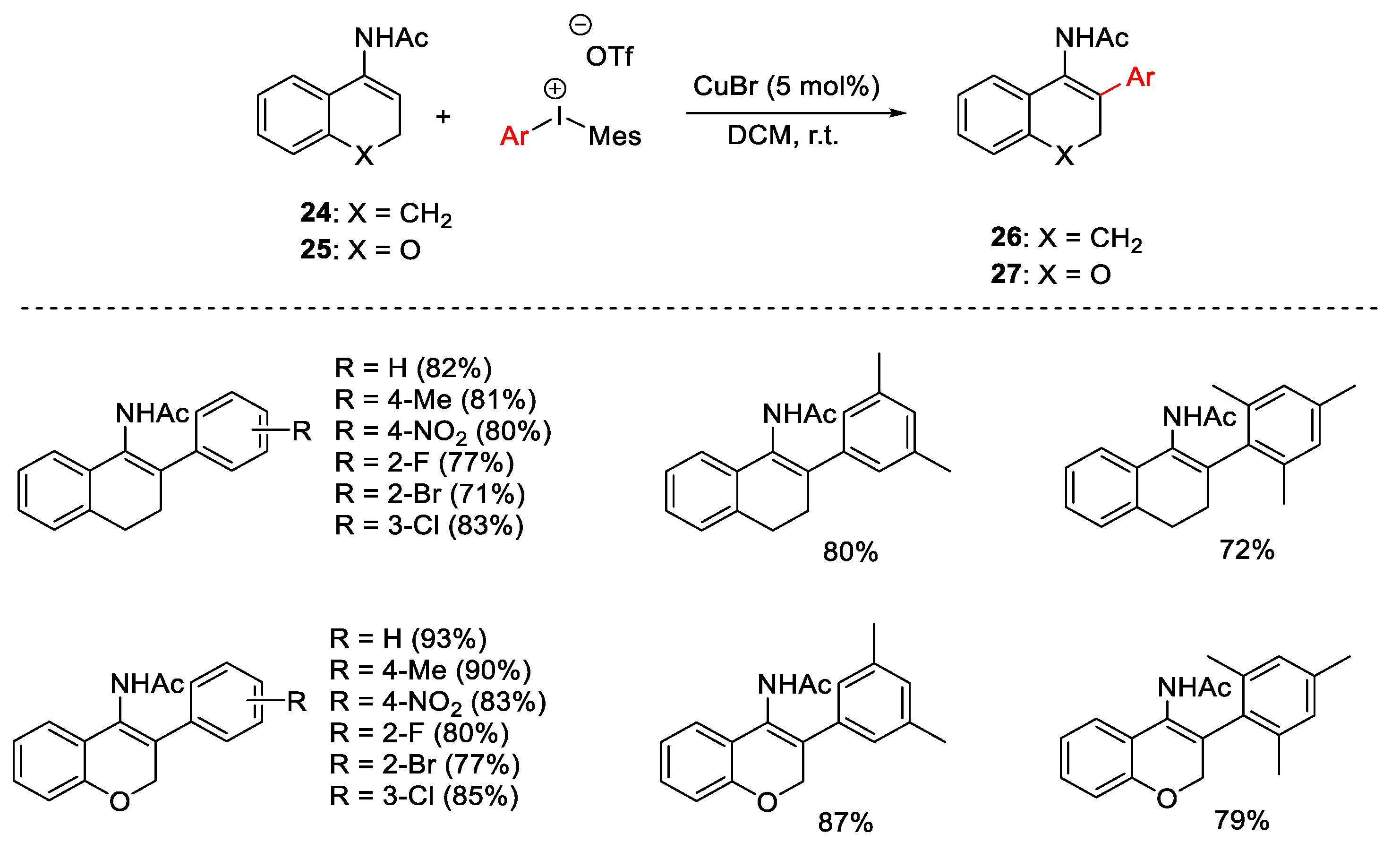{kind=link}
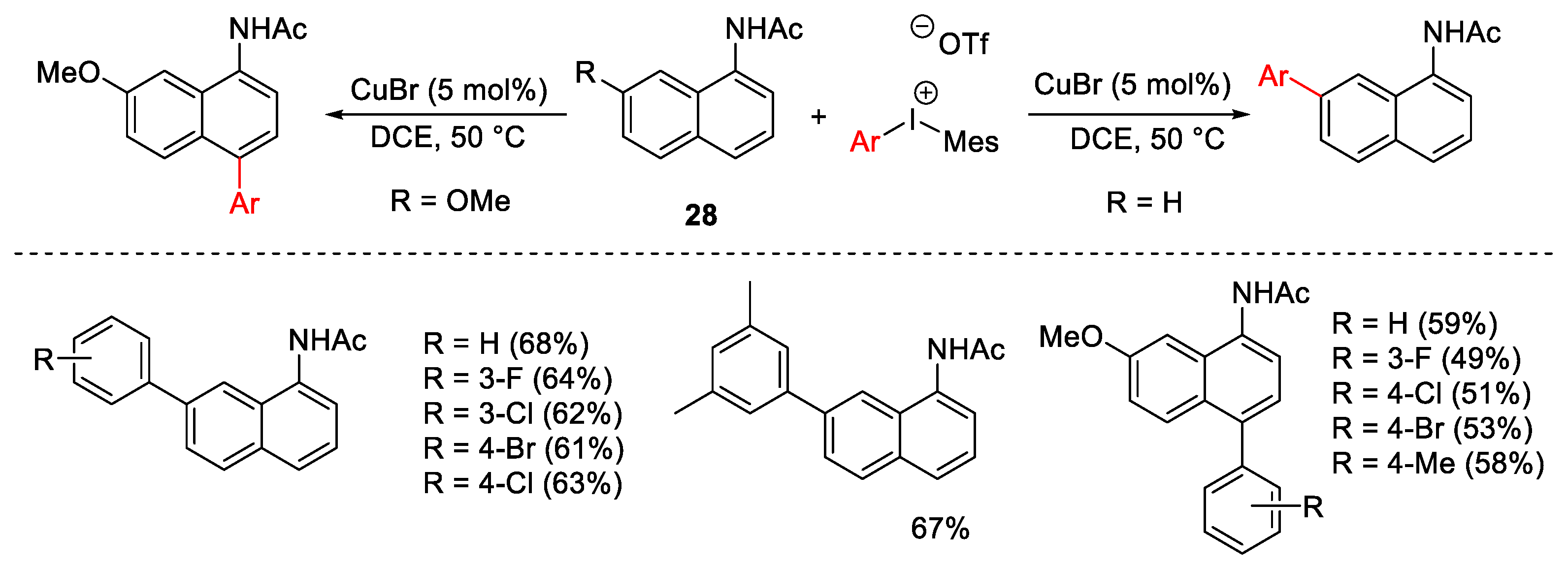{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
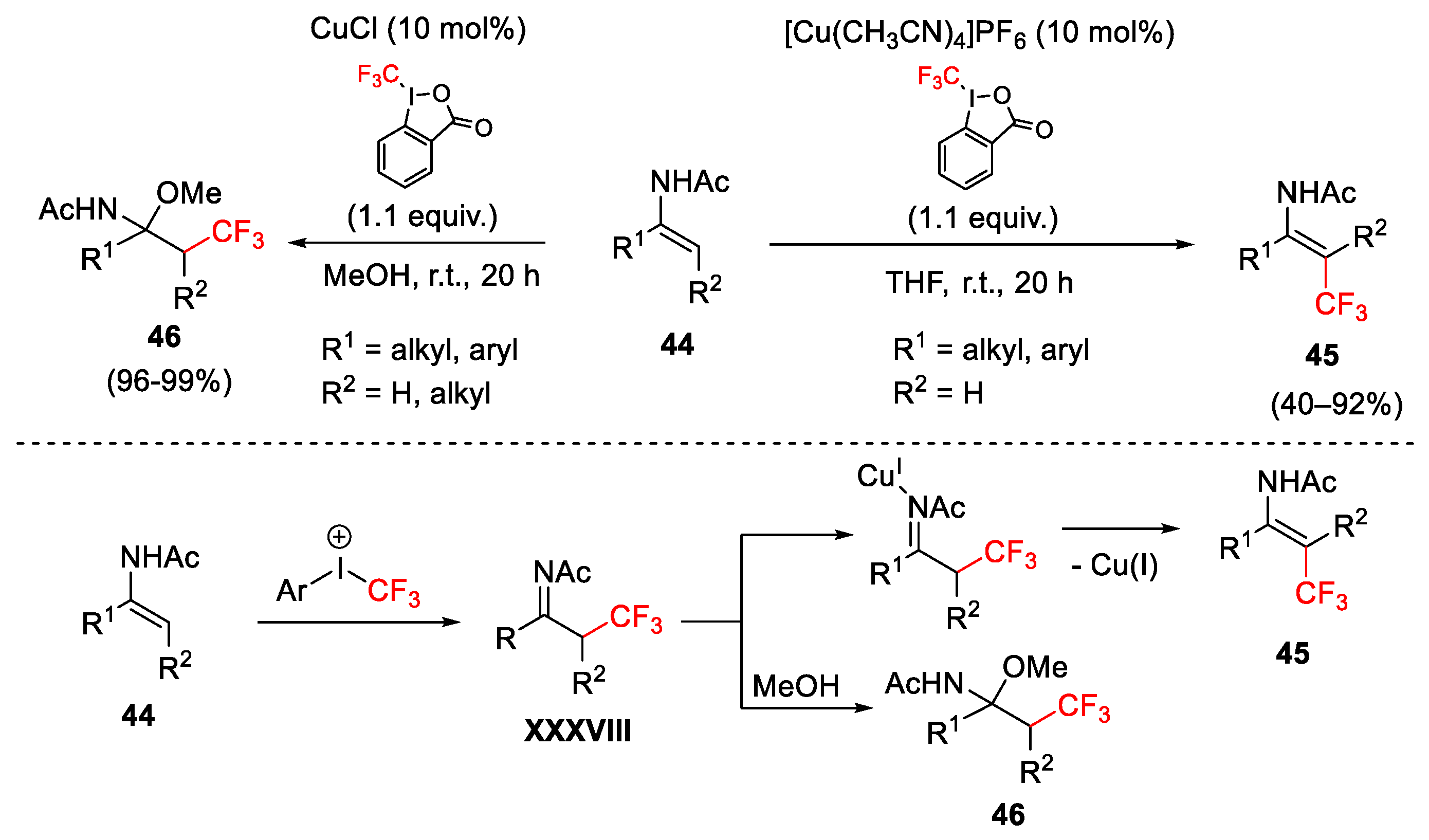{kind=link}
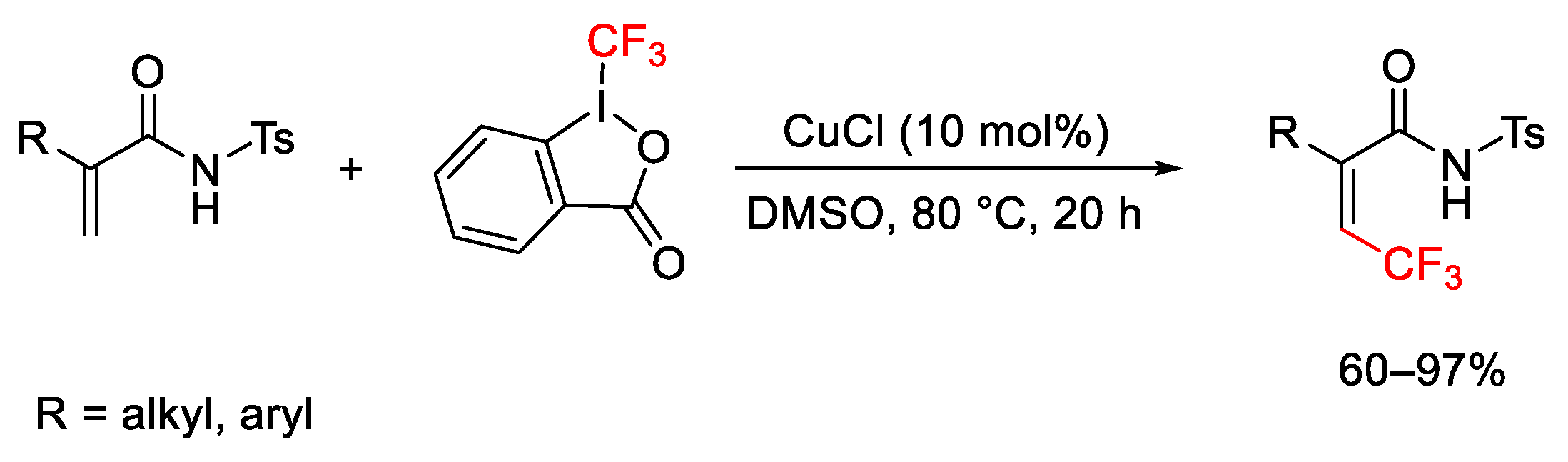{kind=link}
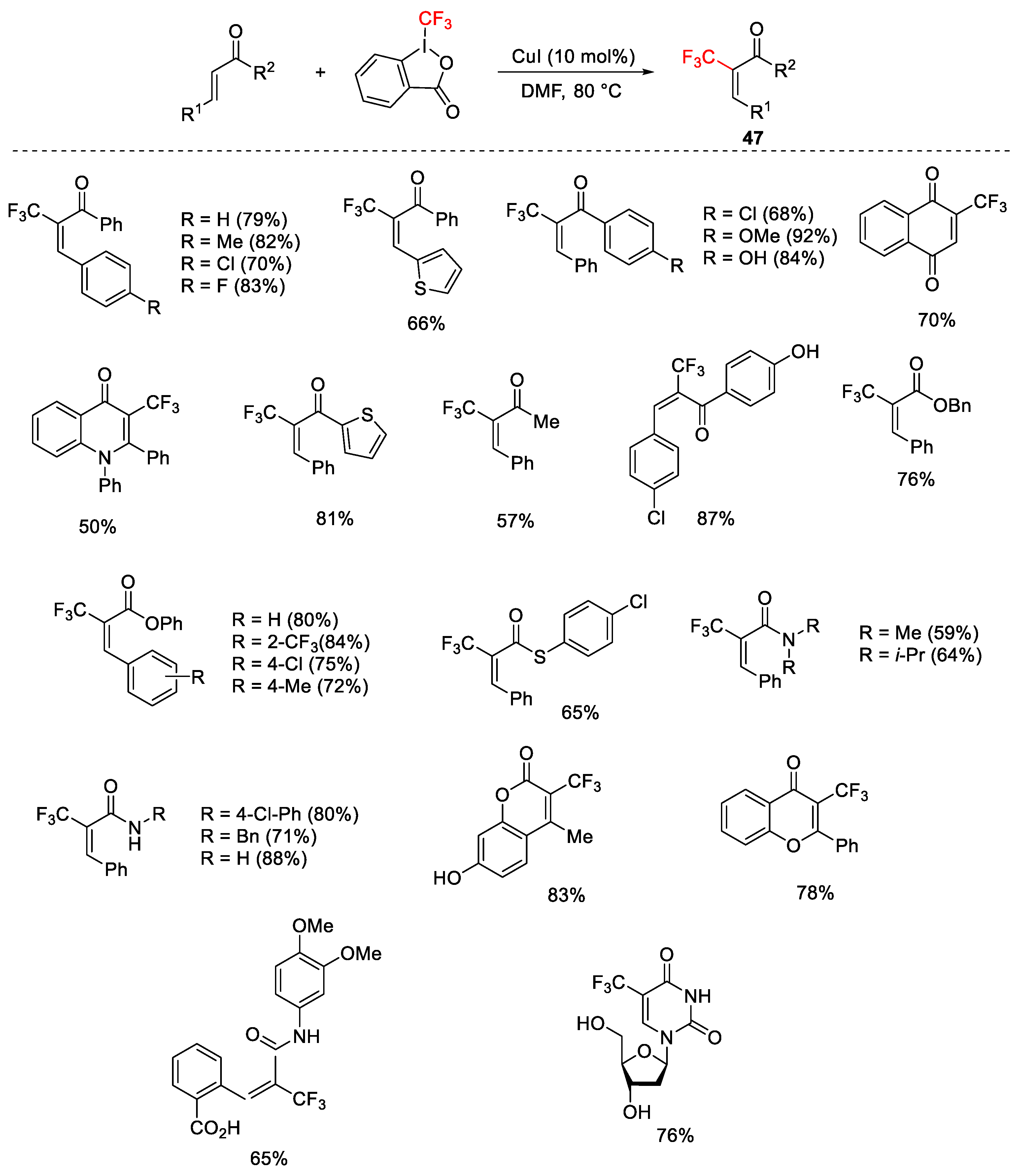{kind=link}
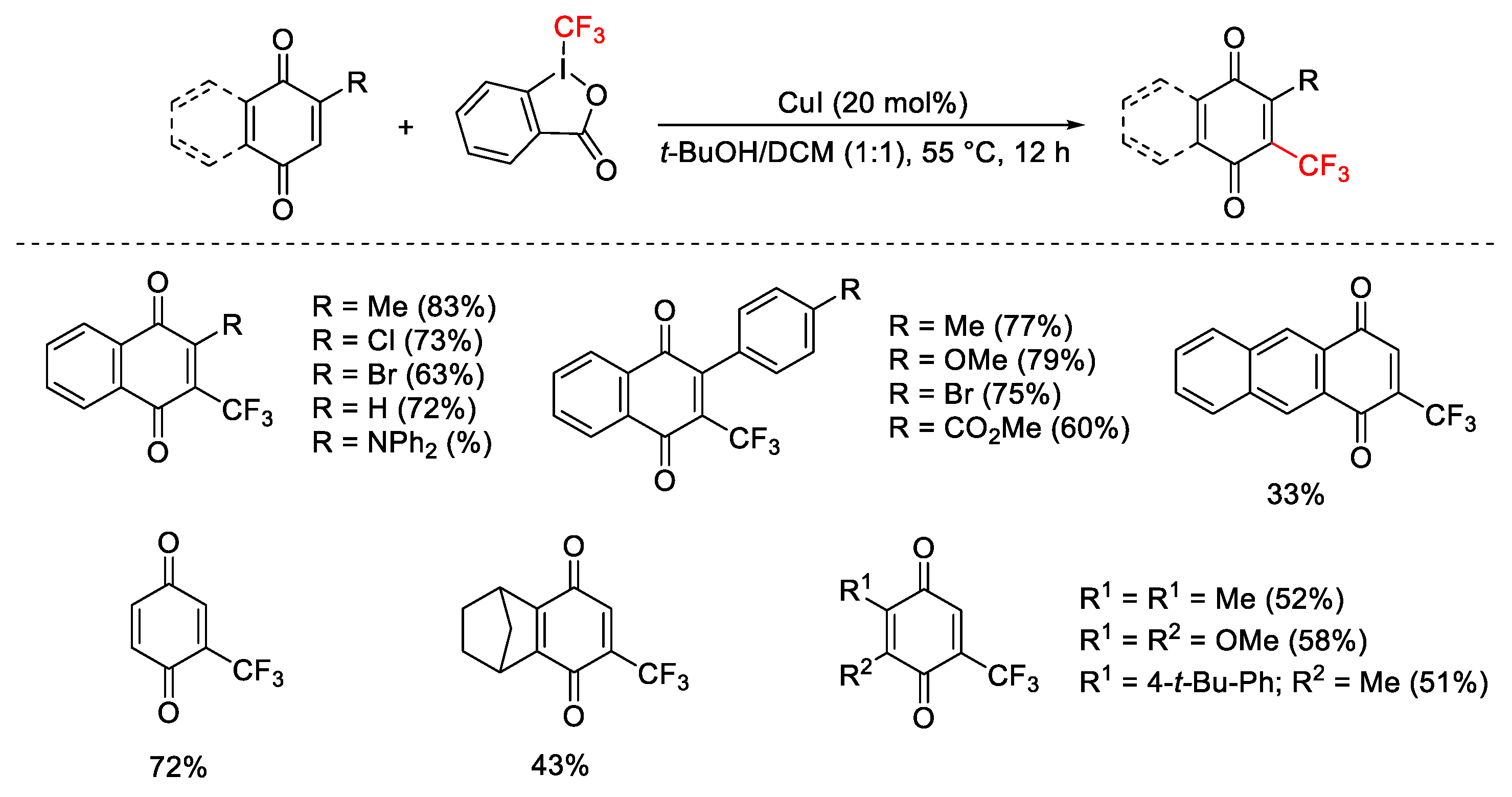{kind=link}
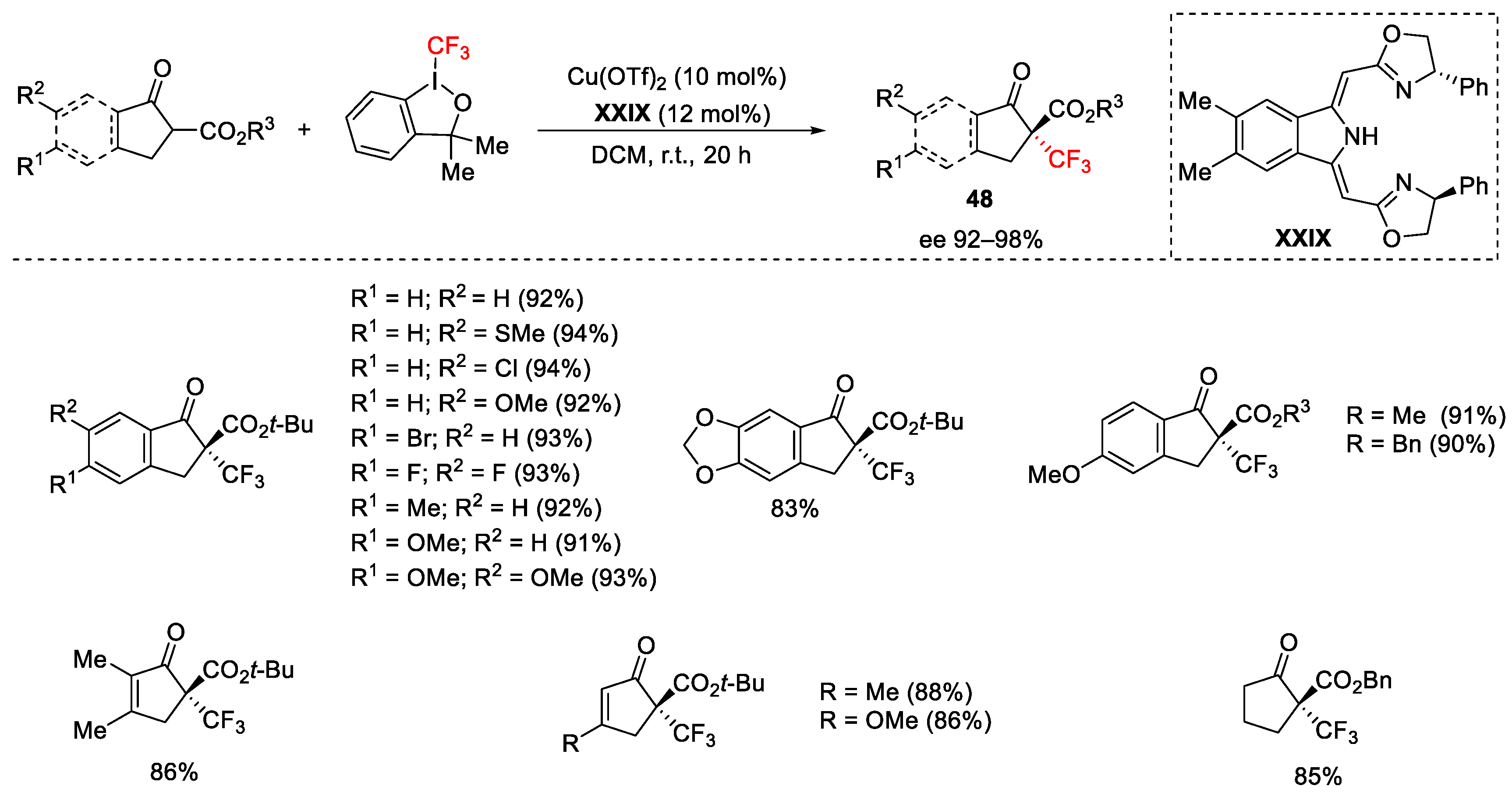{kind=link}
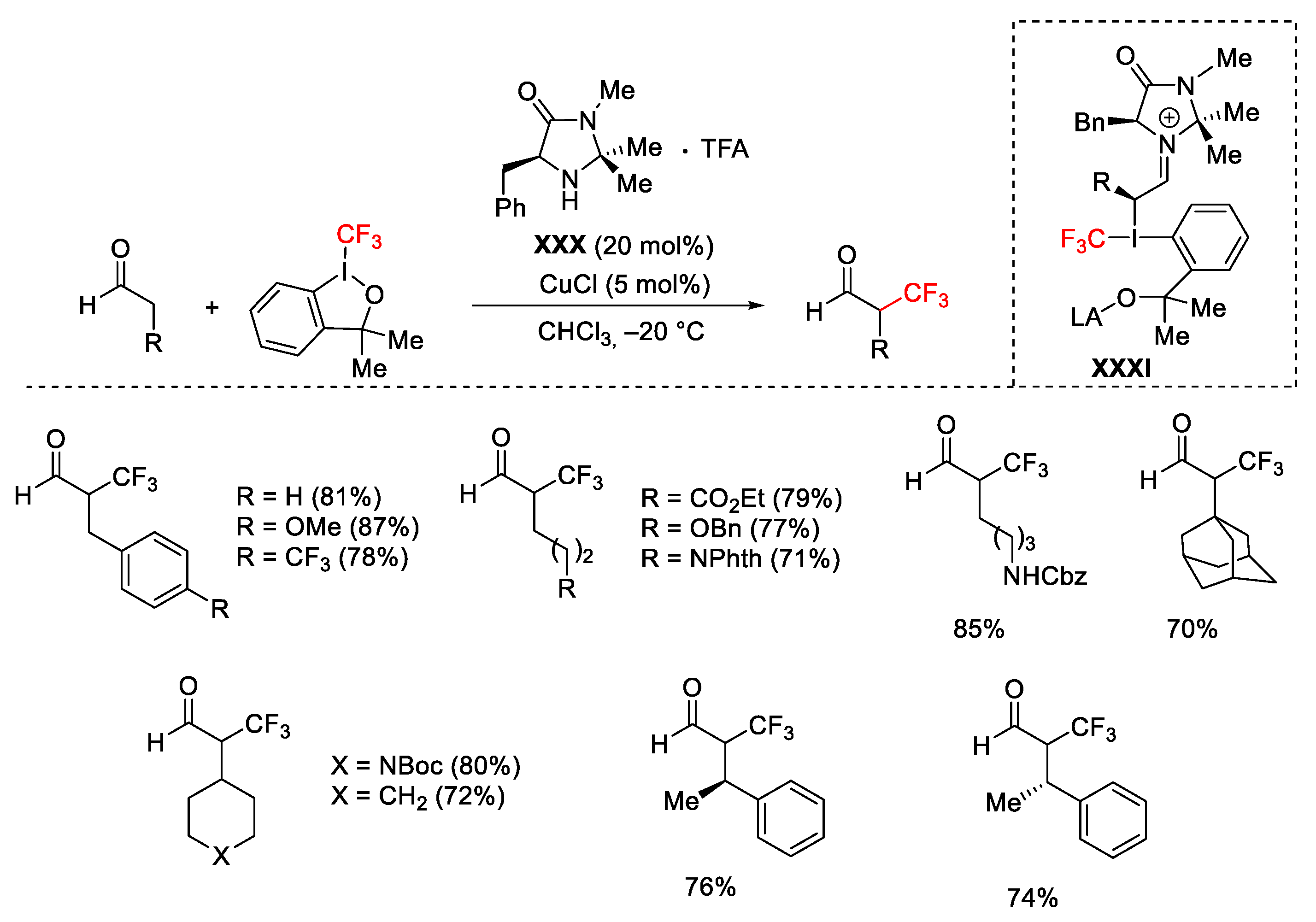{kind=link}
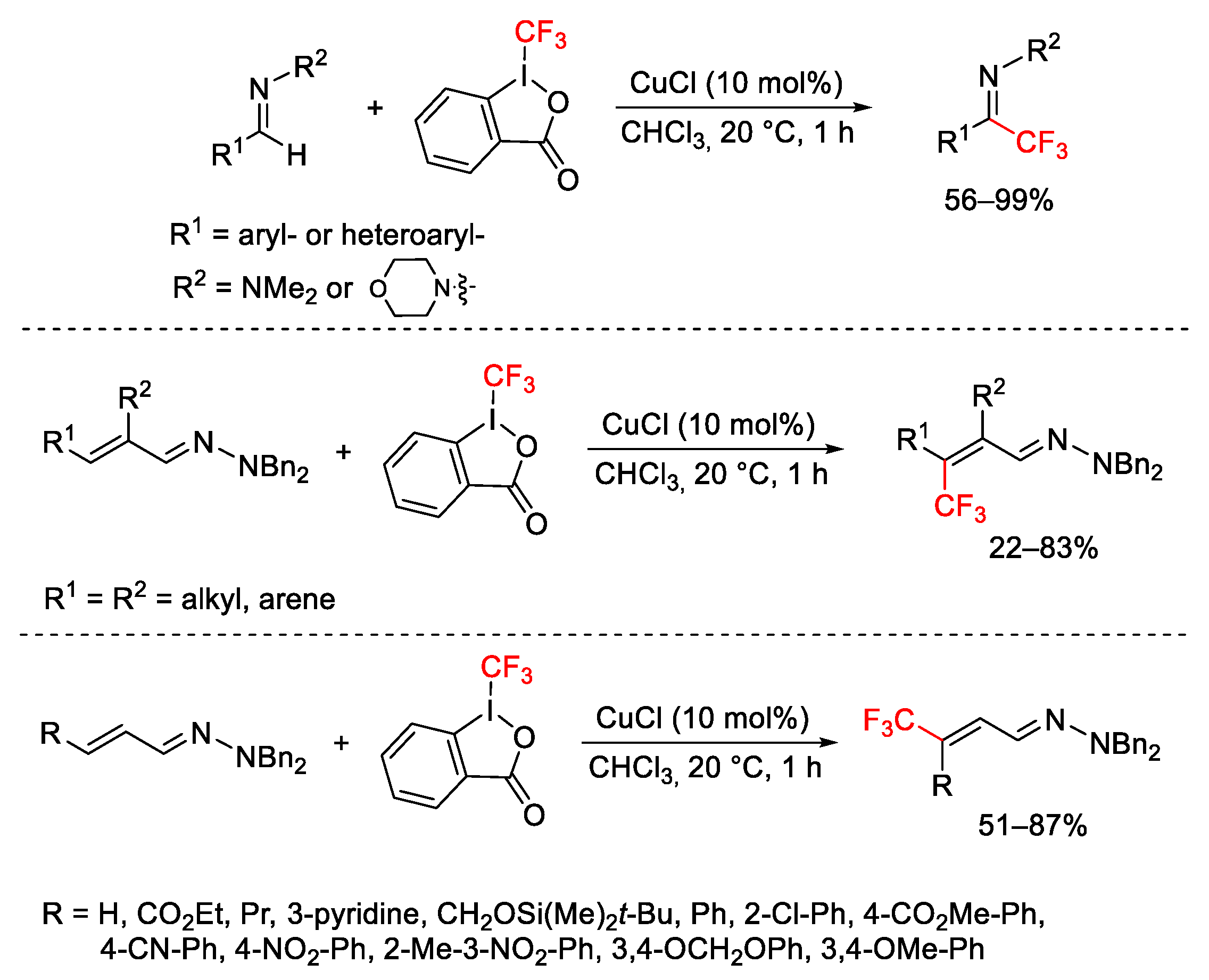{kind=link}
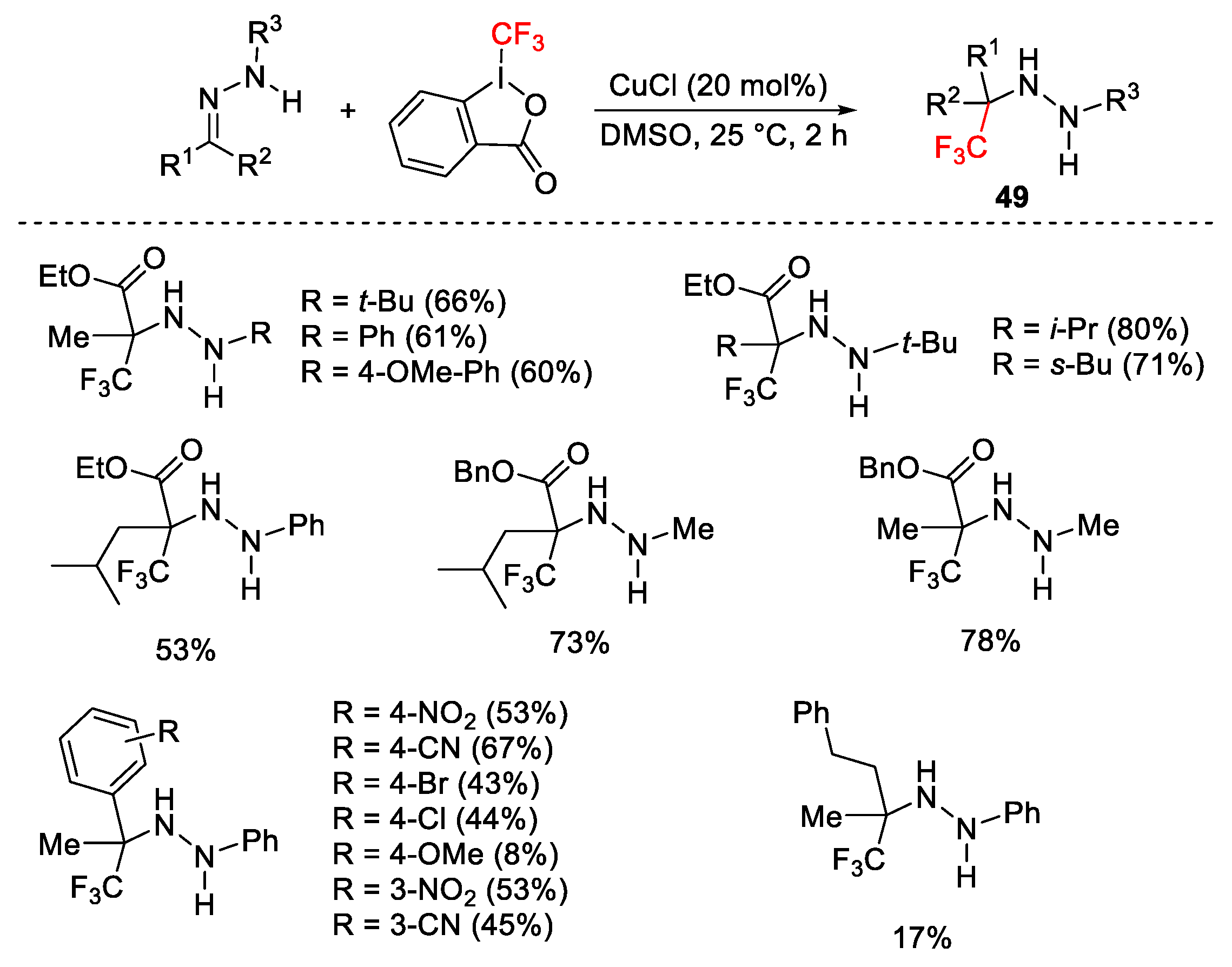{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
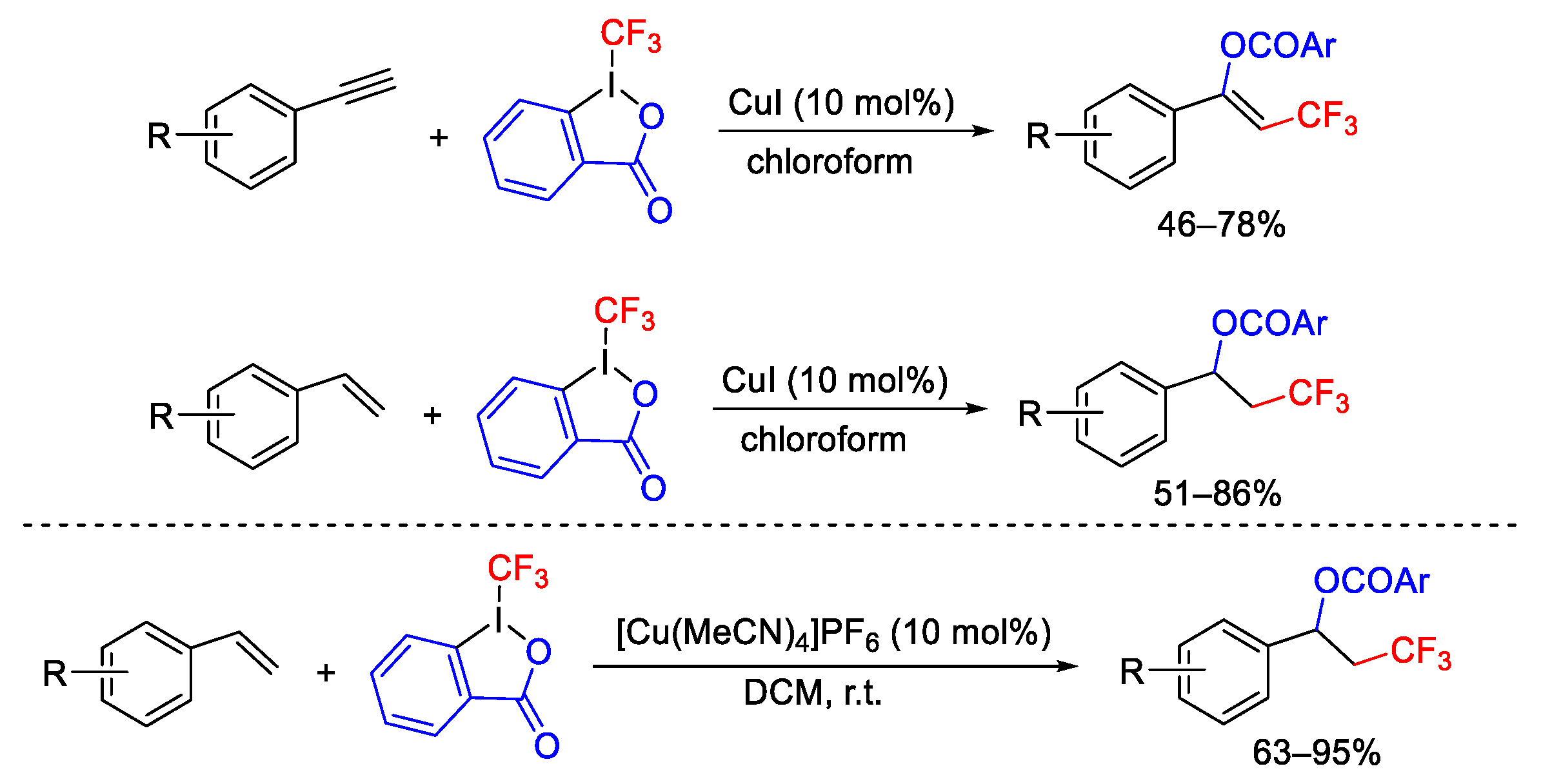{kind=link}
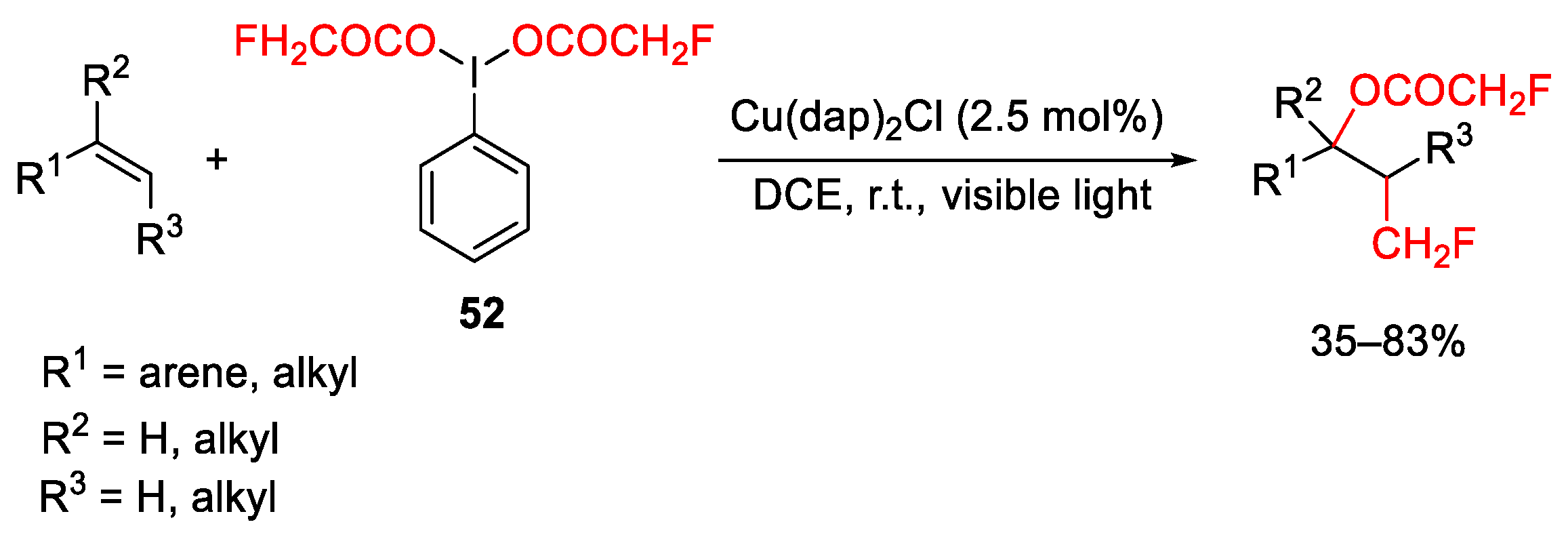{kind=link}
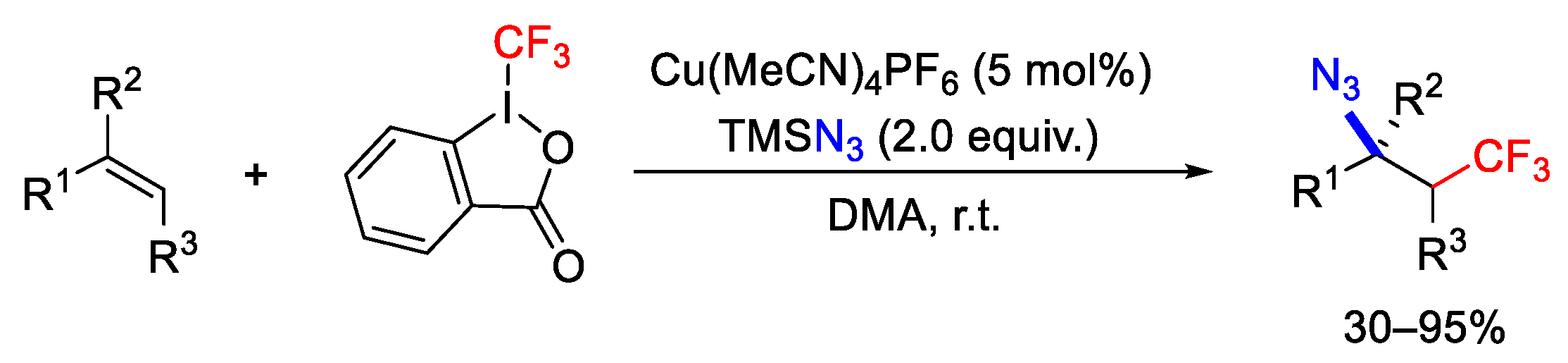{kind=link}
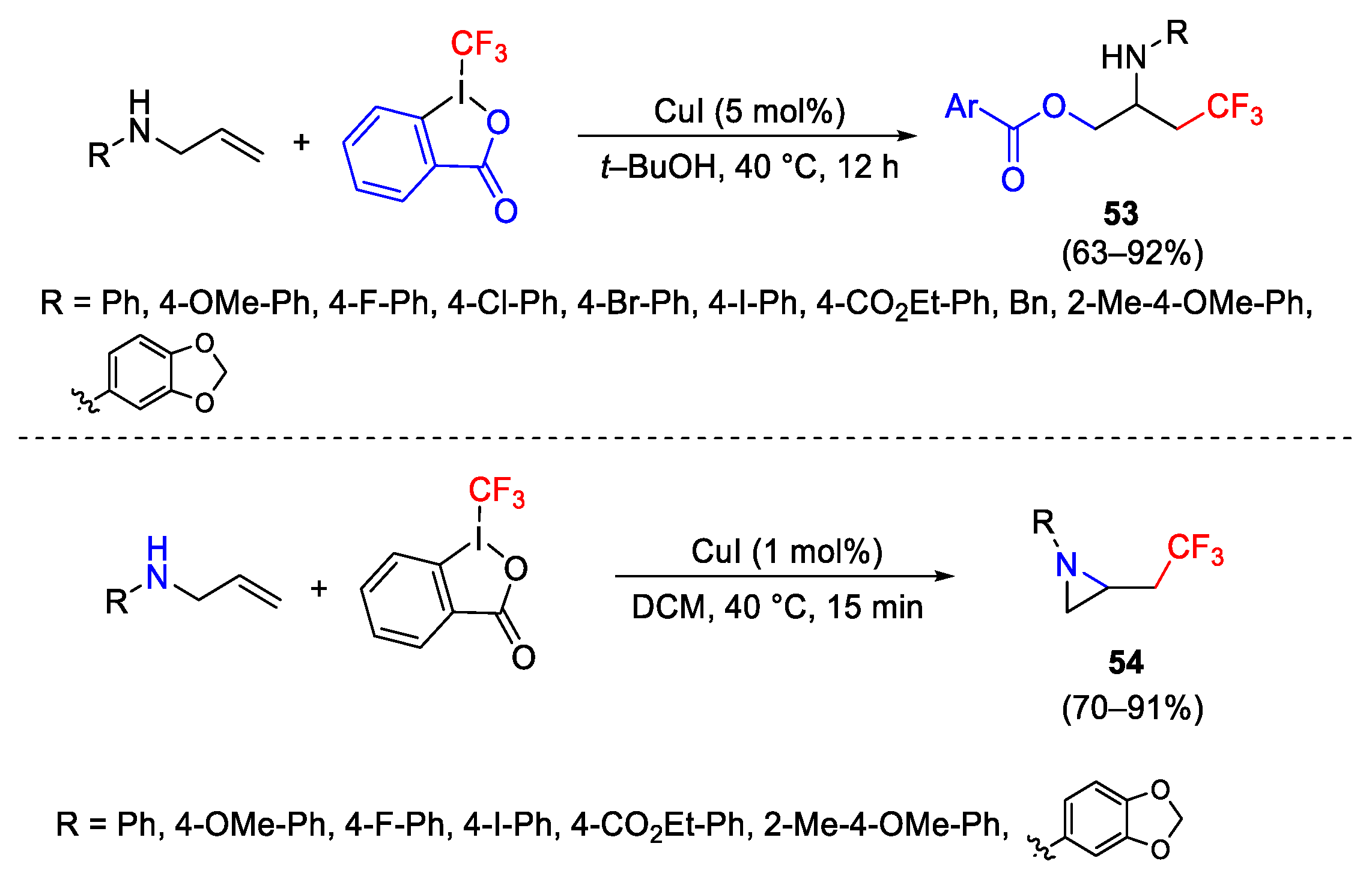{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
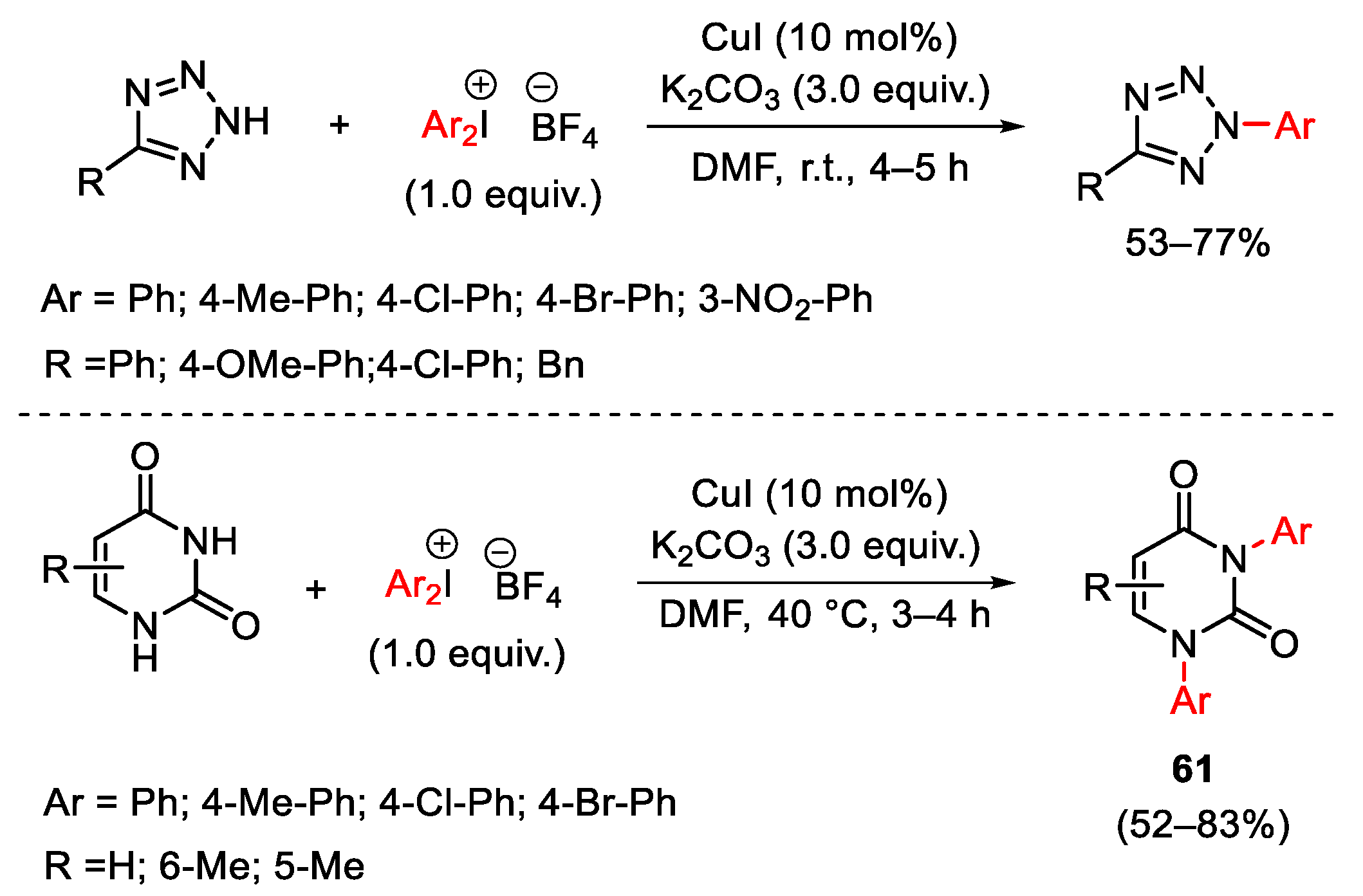{kind=link}
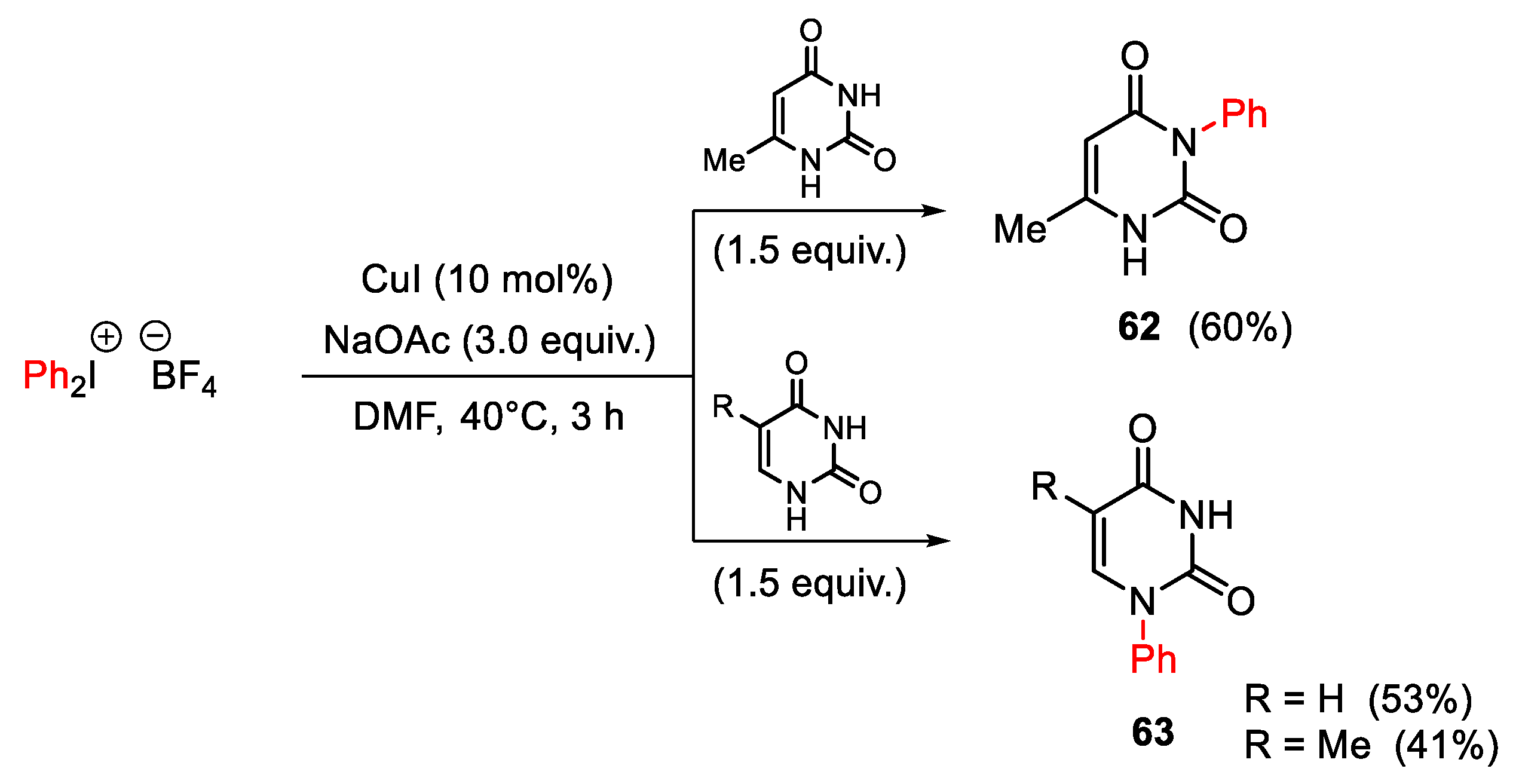{kind=link}
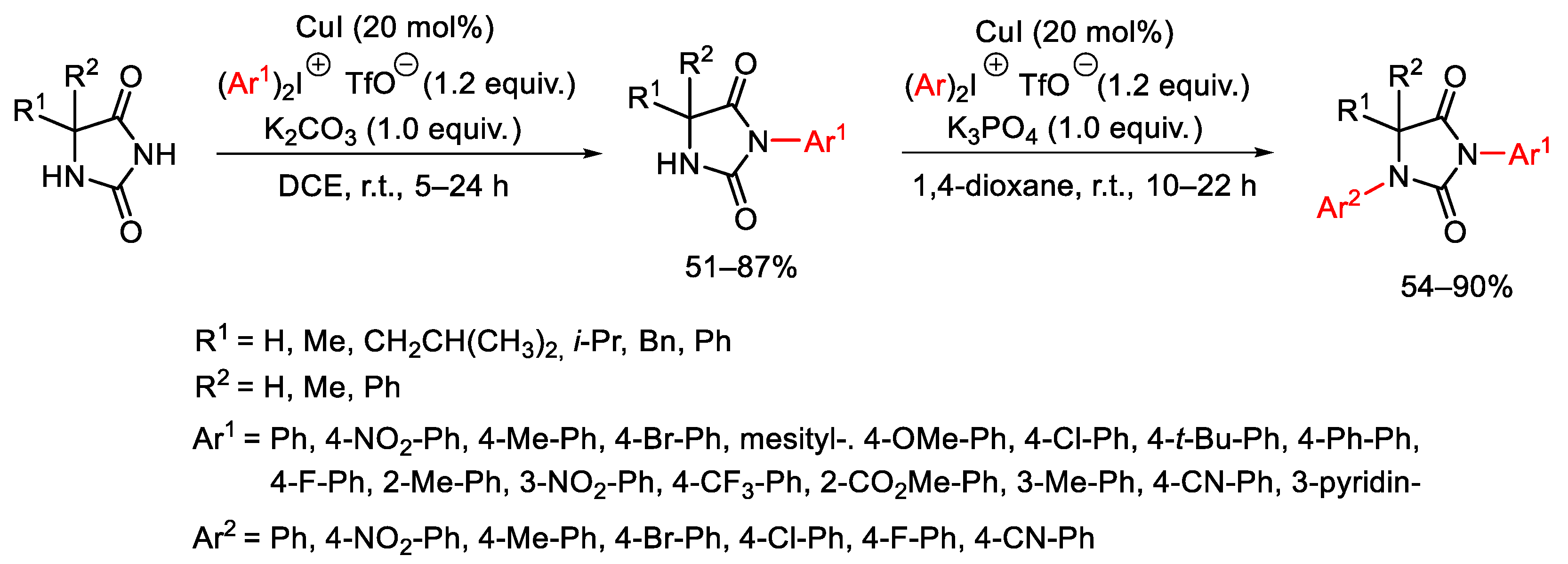{kind=link}
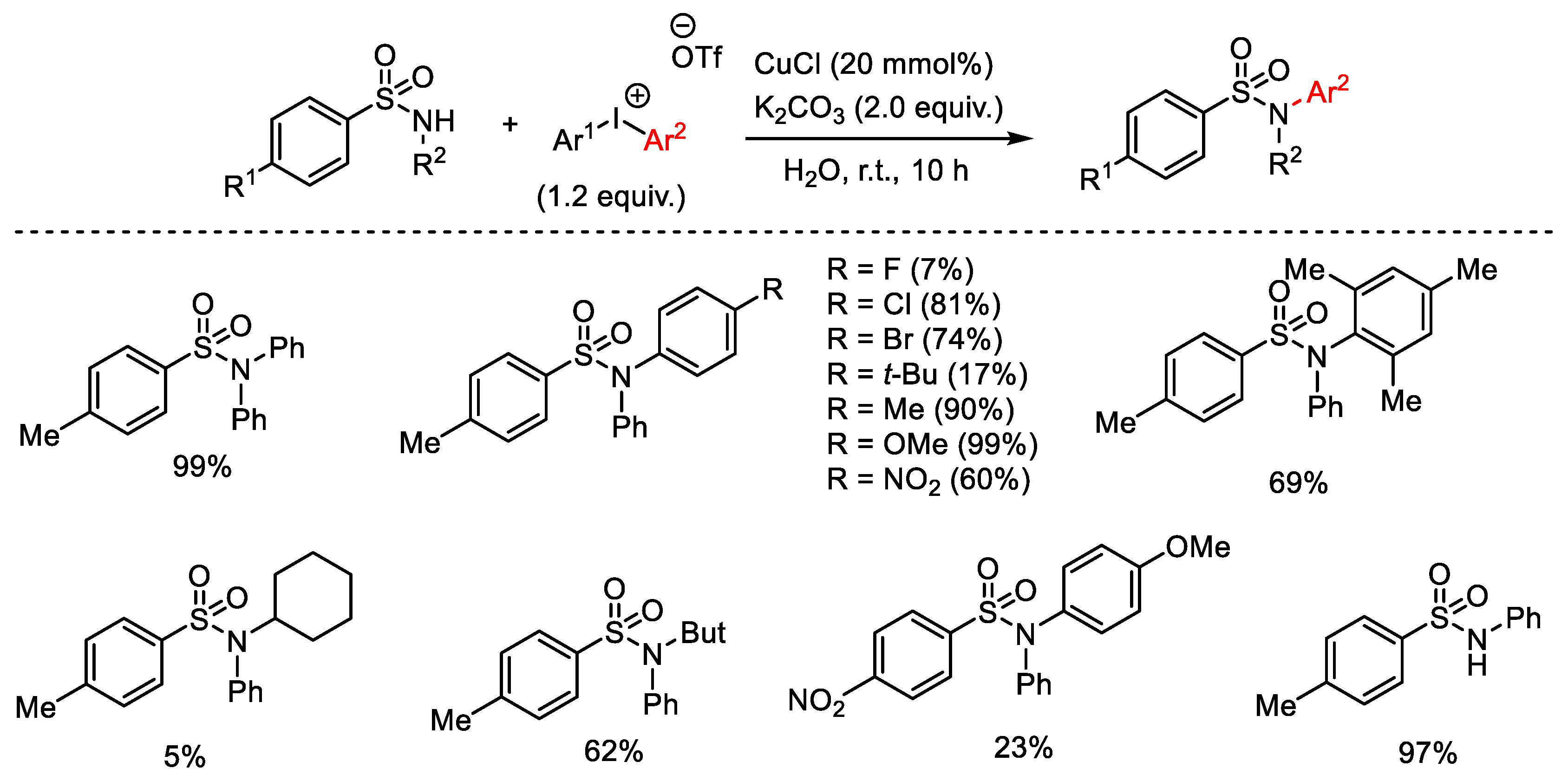{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
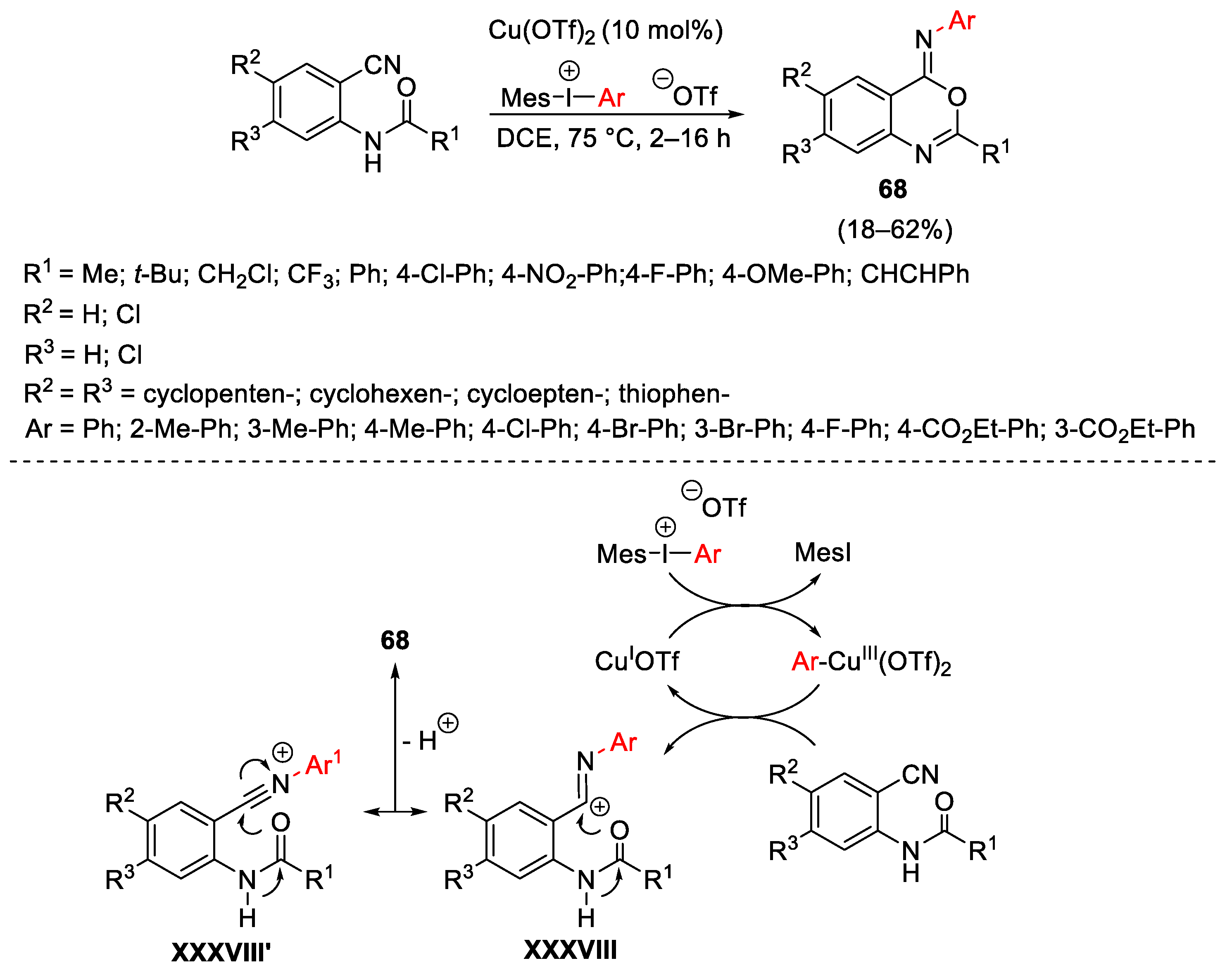{kind=link}
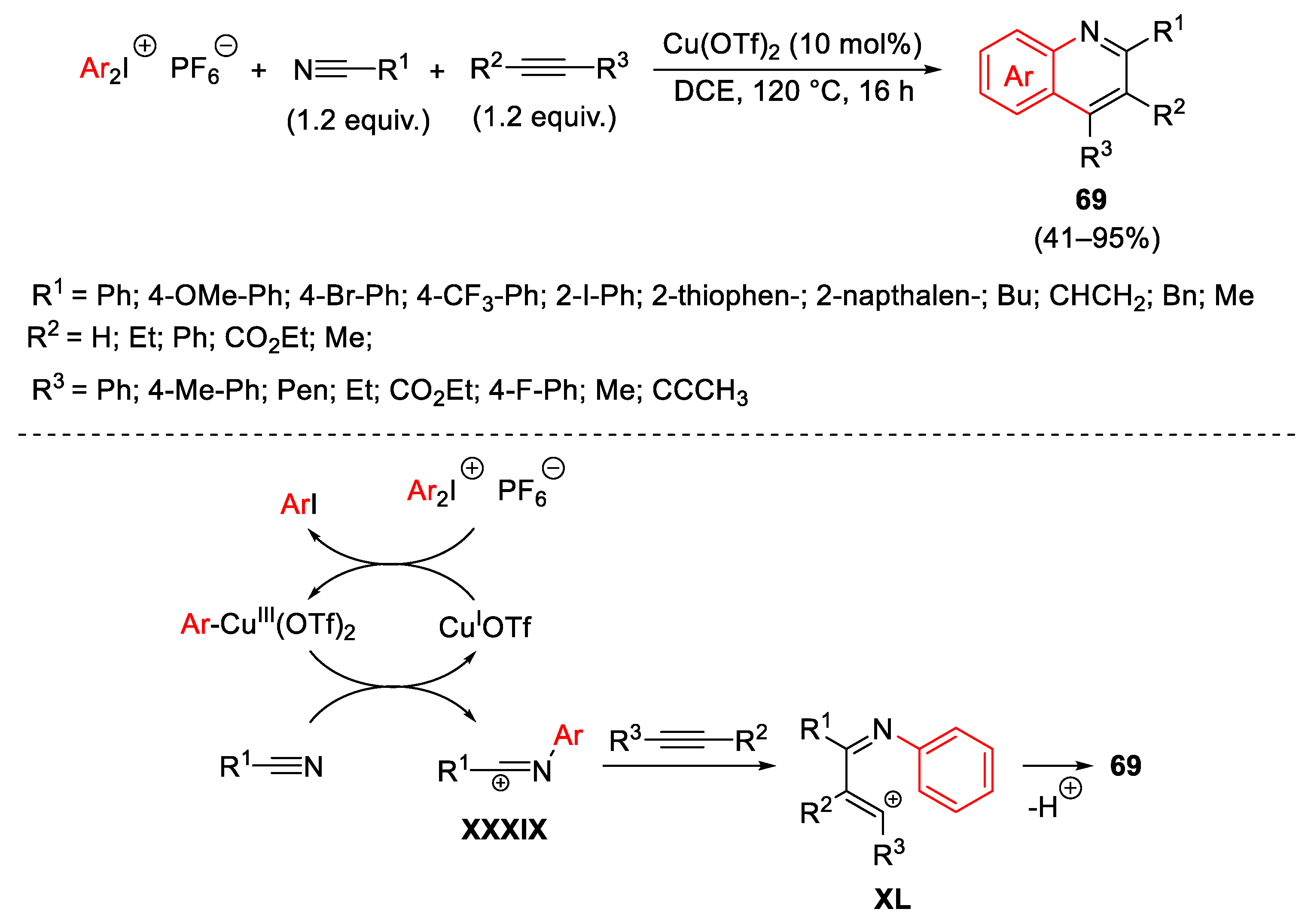{kind=link}
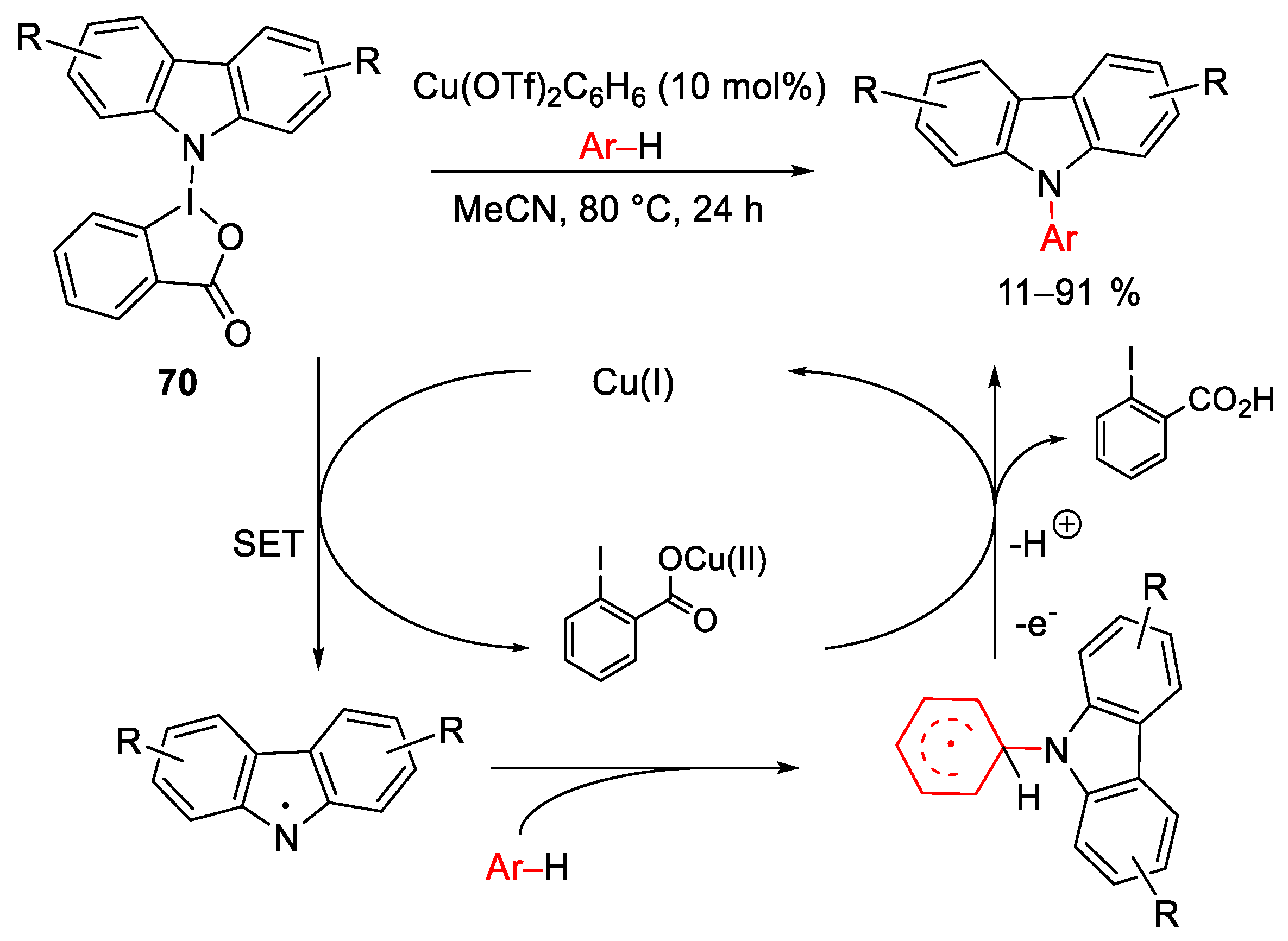{kind=link}
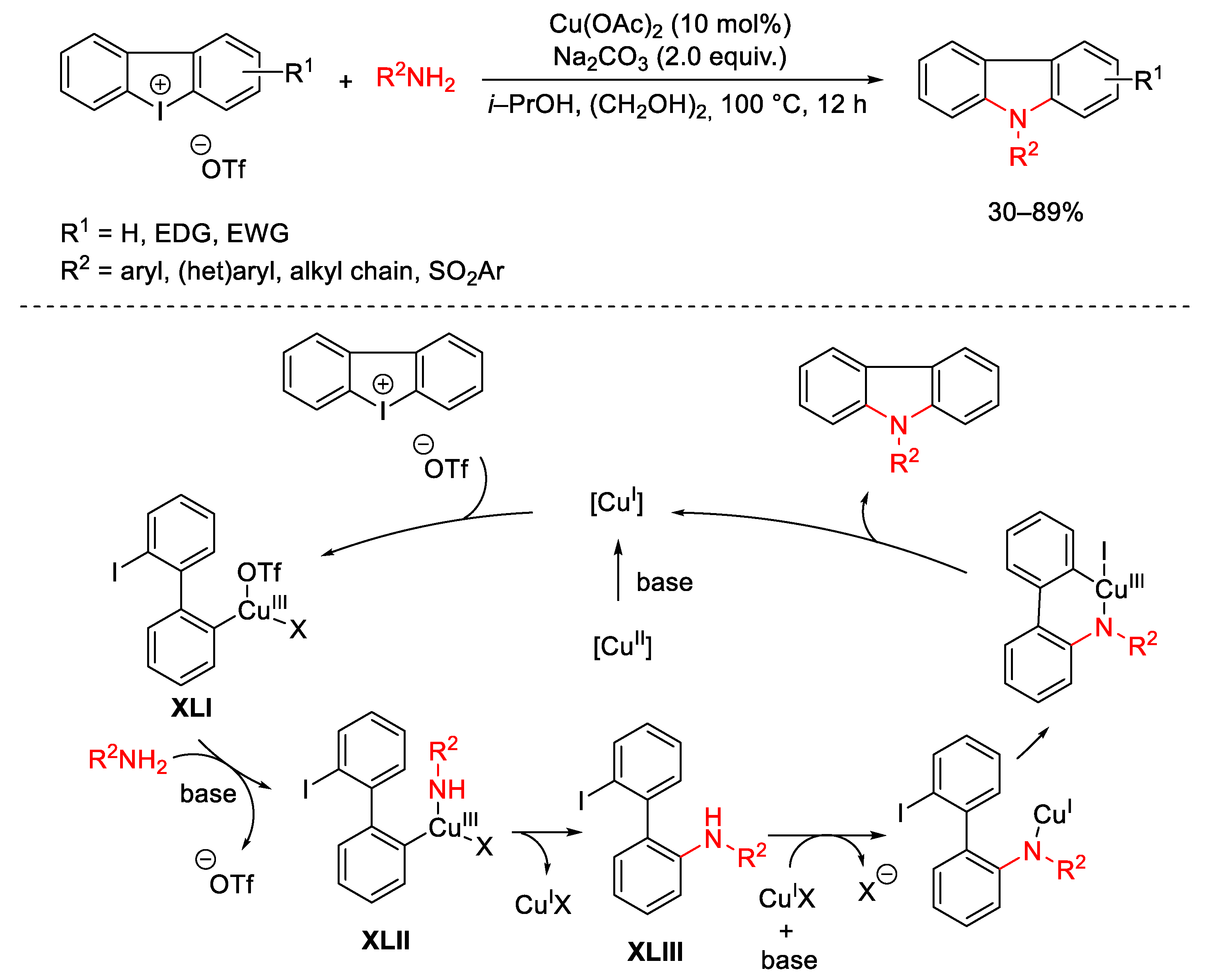{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
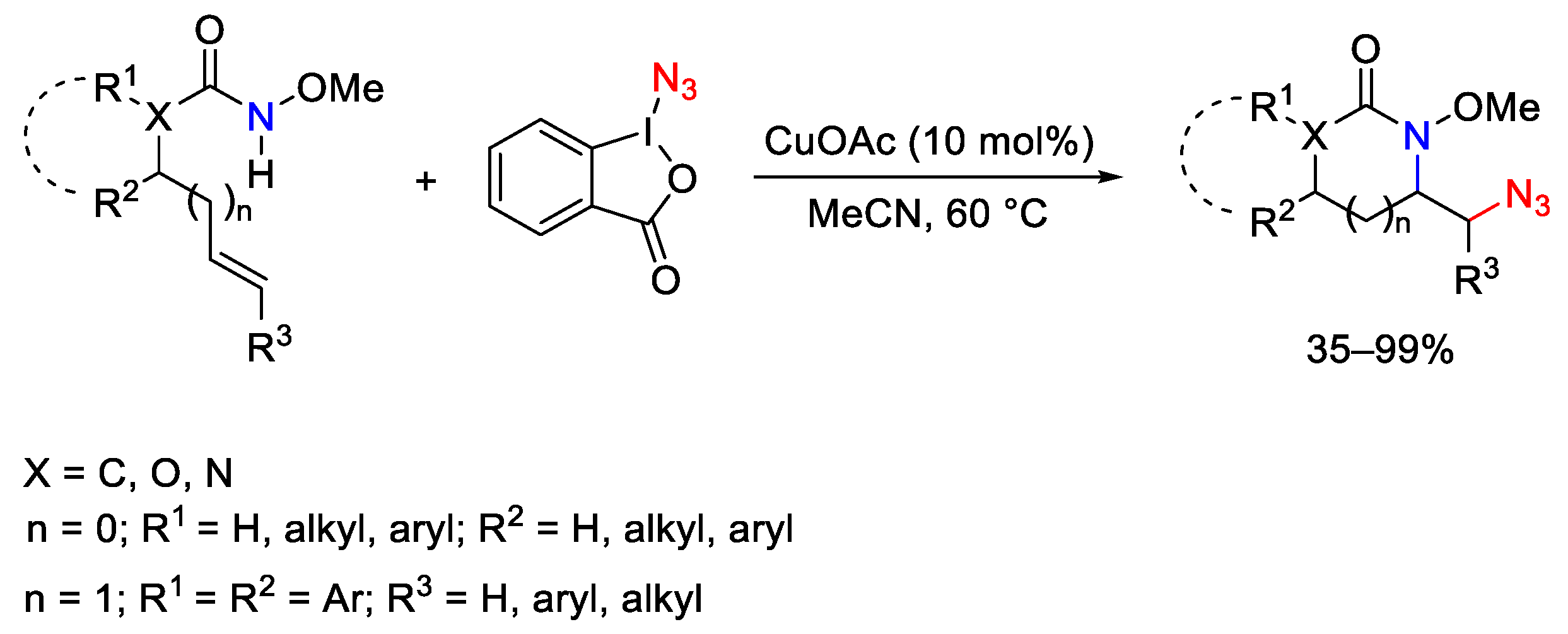{kind=link}
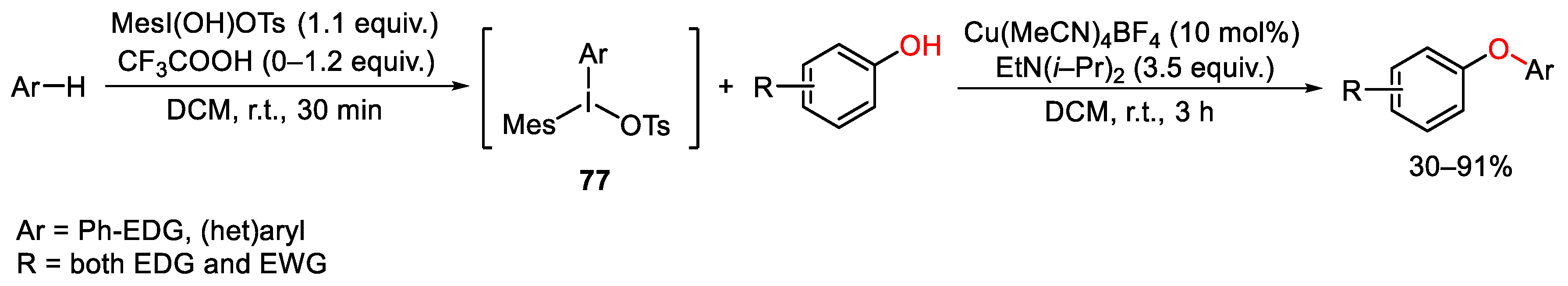{kind=link}
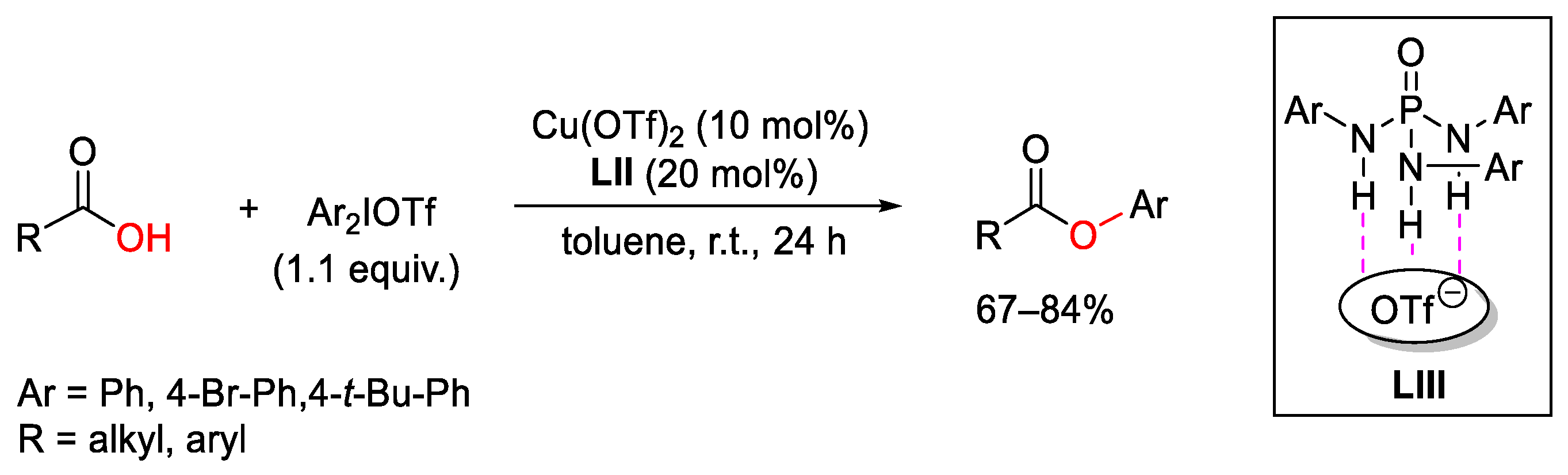{kind=link}
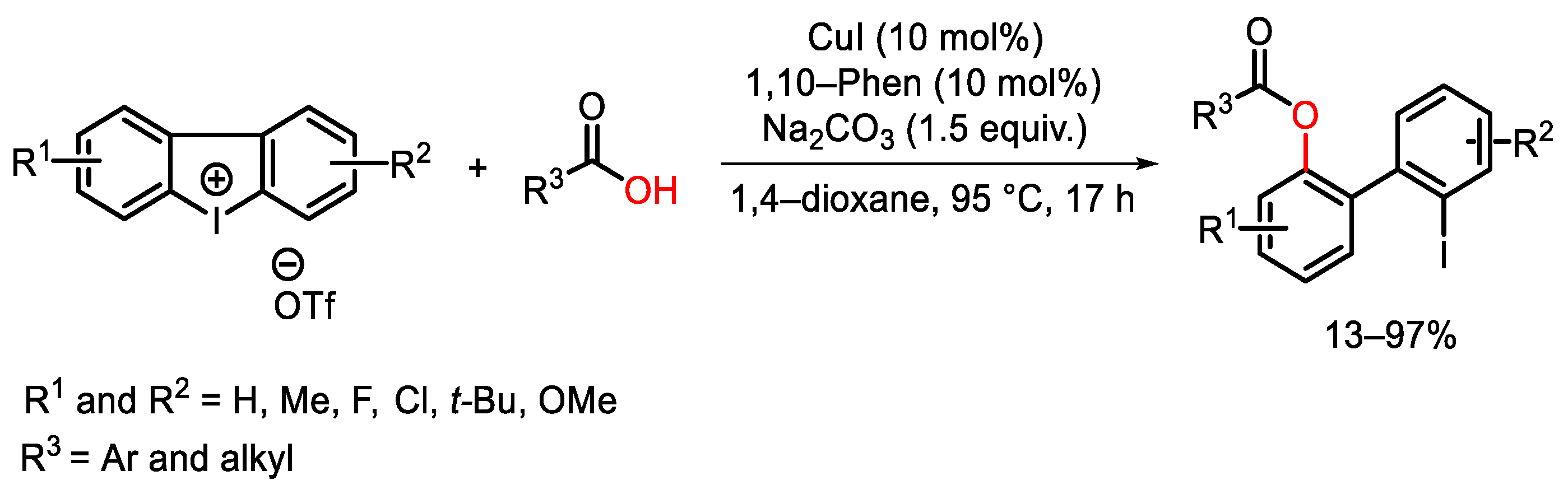{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
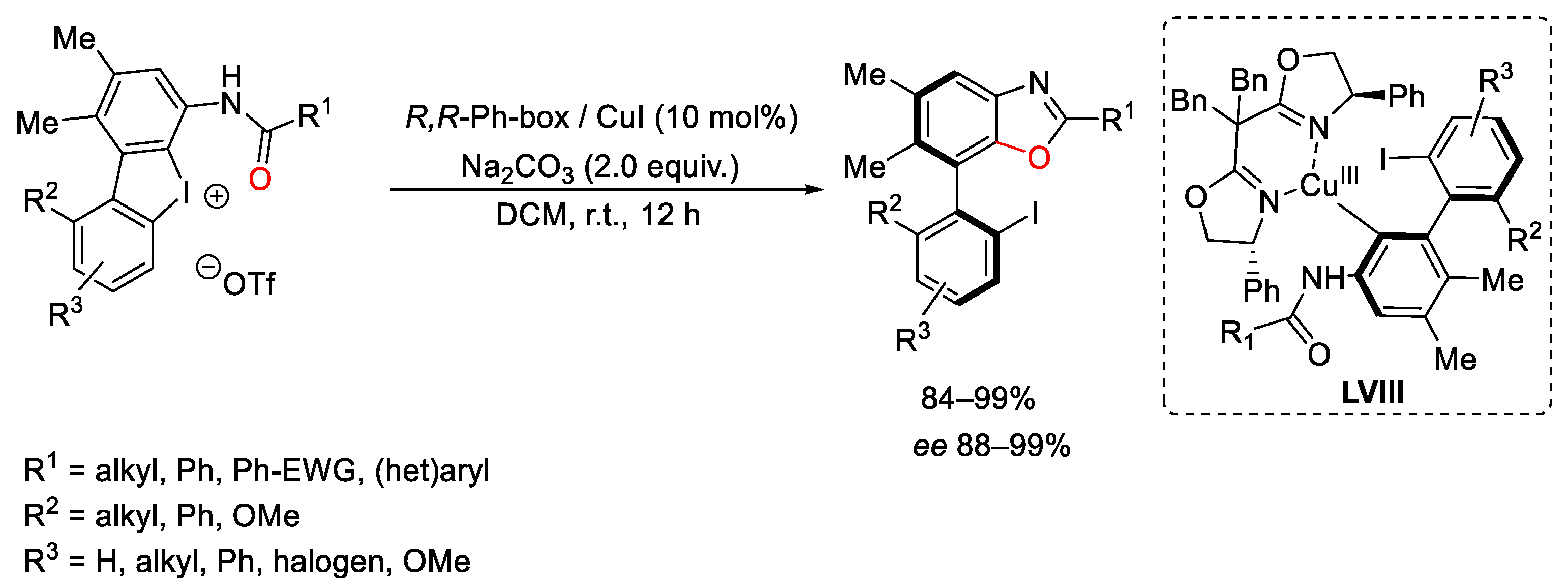{kind=link}
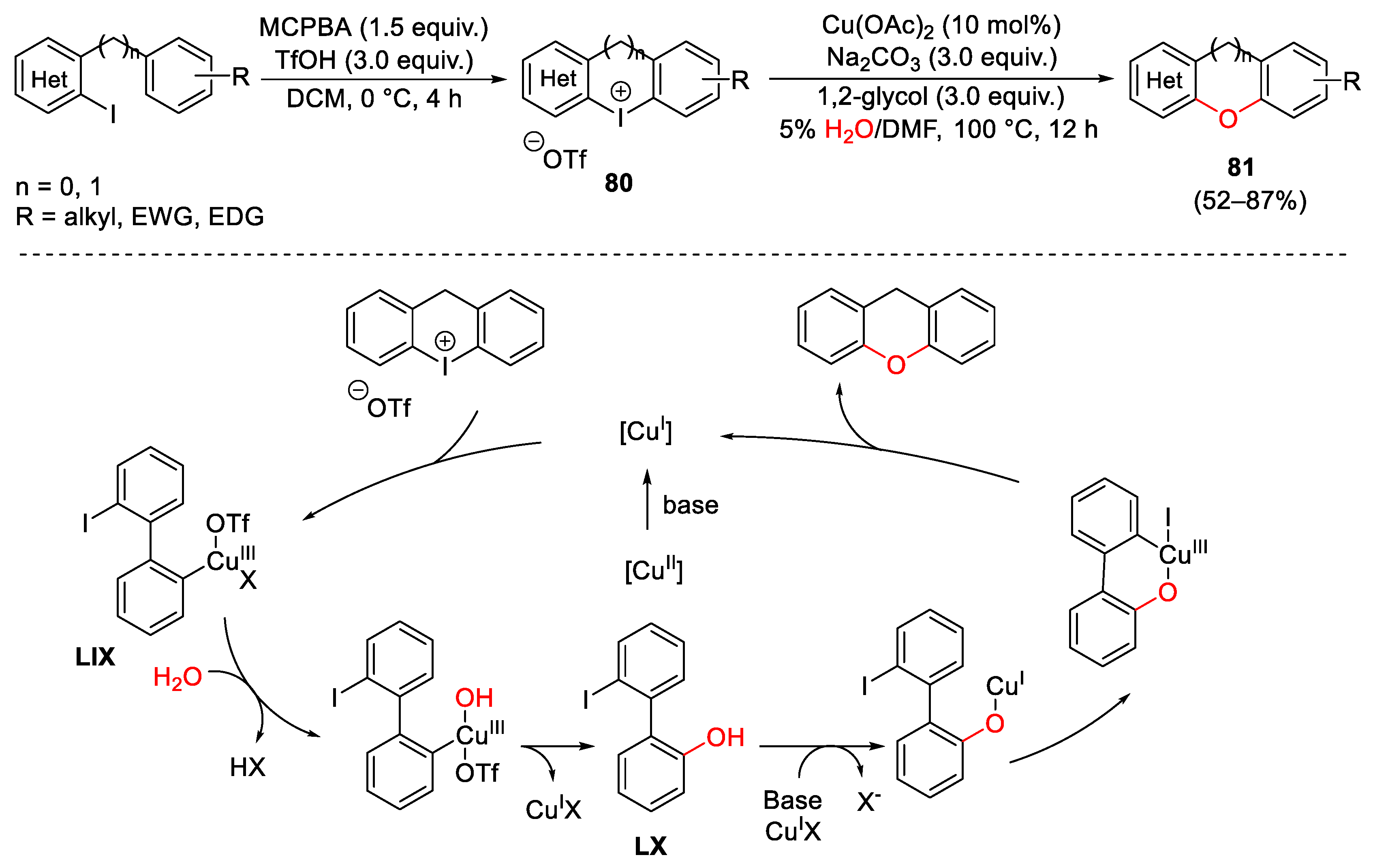{kind=link}
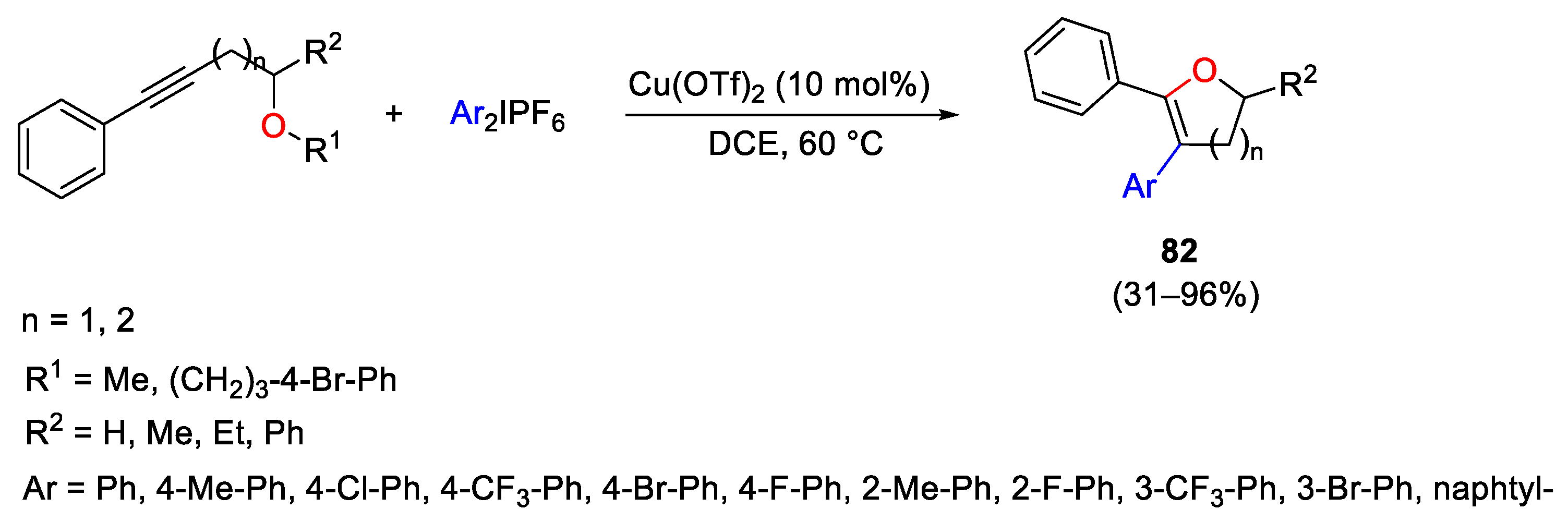{kind=link}
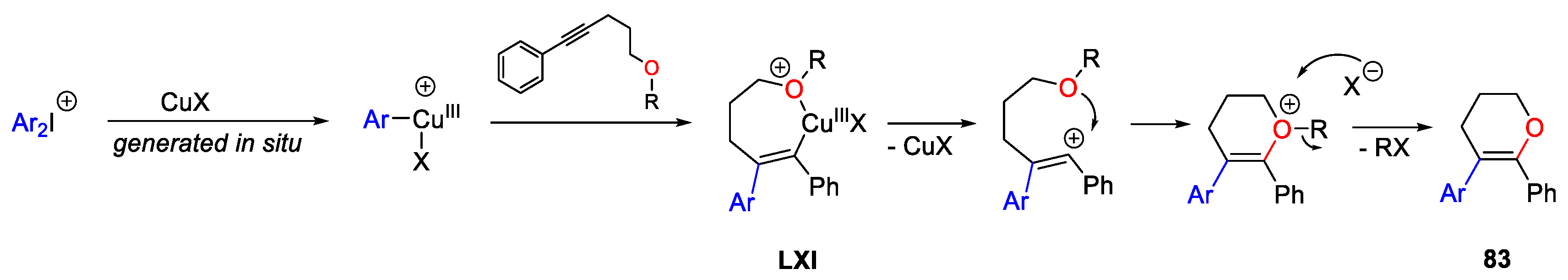{kind=link}
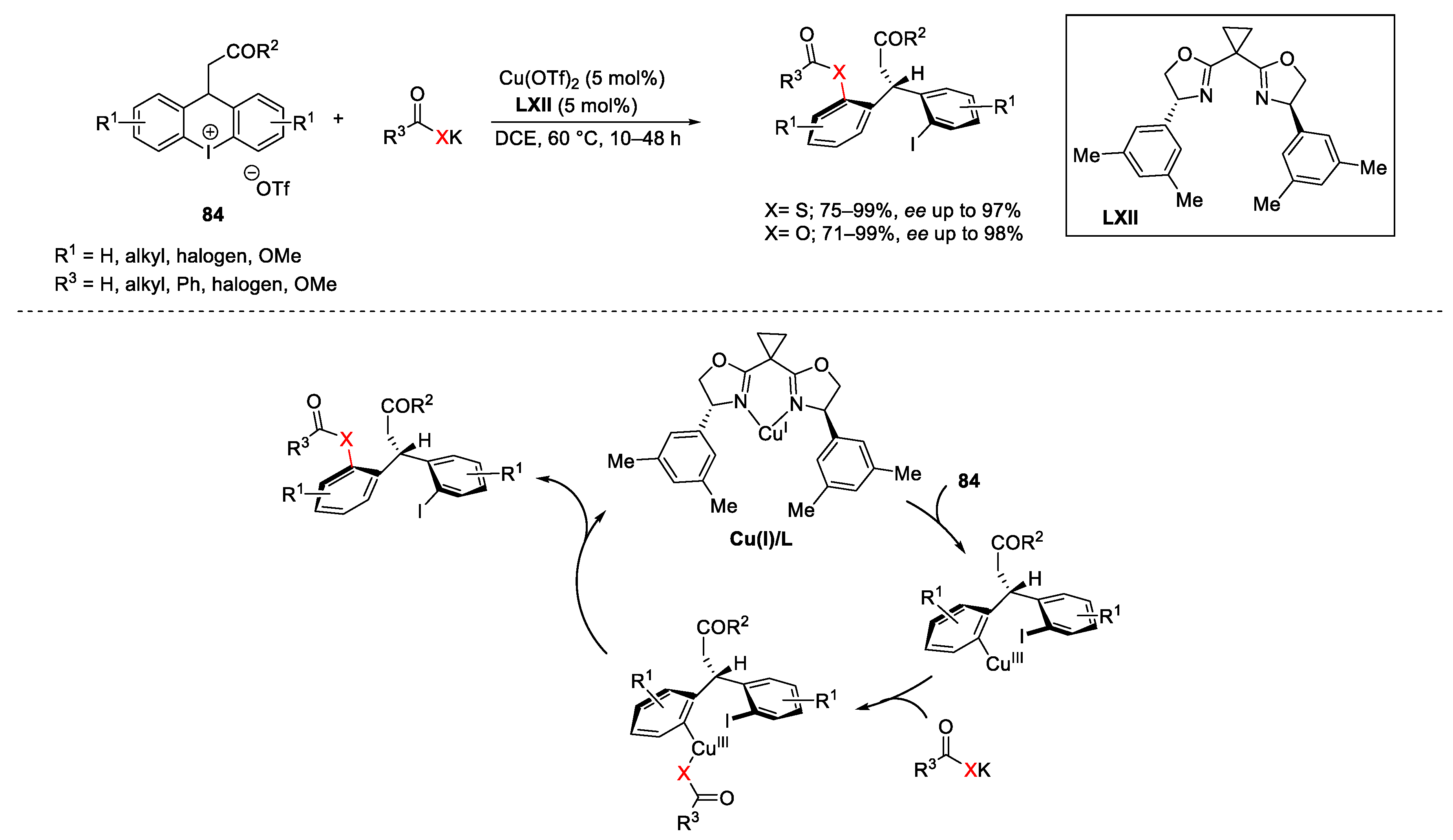{kind=link}
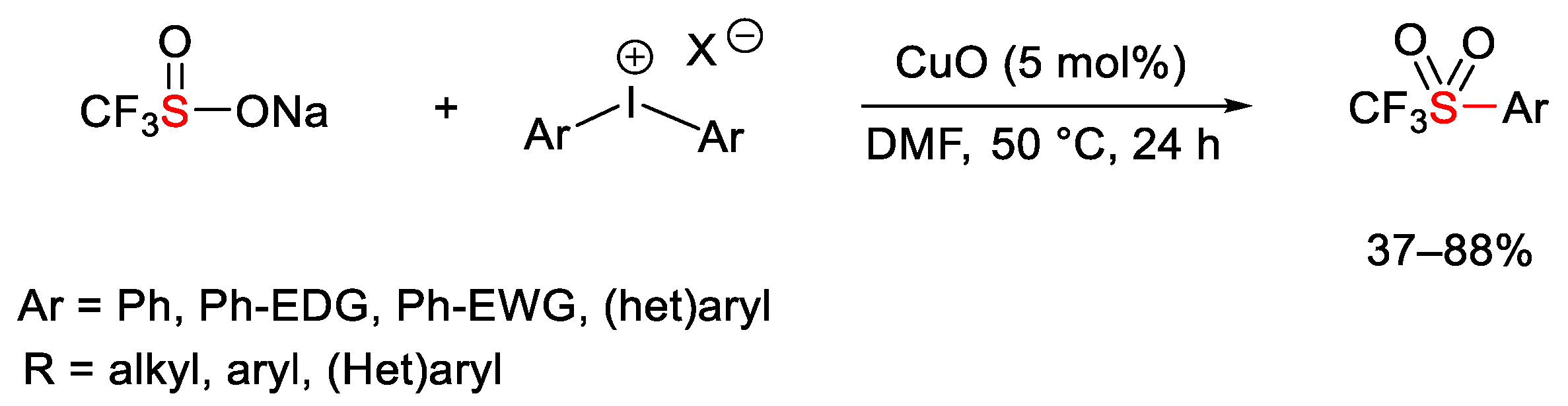{kind=link}
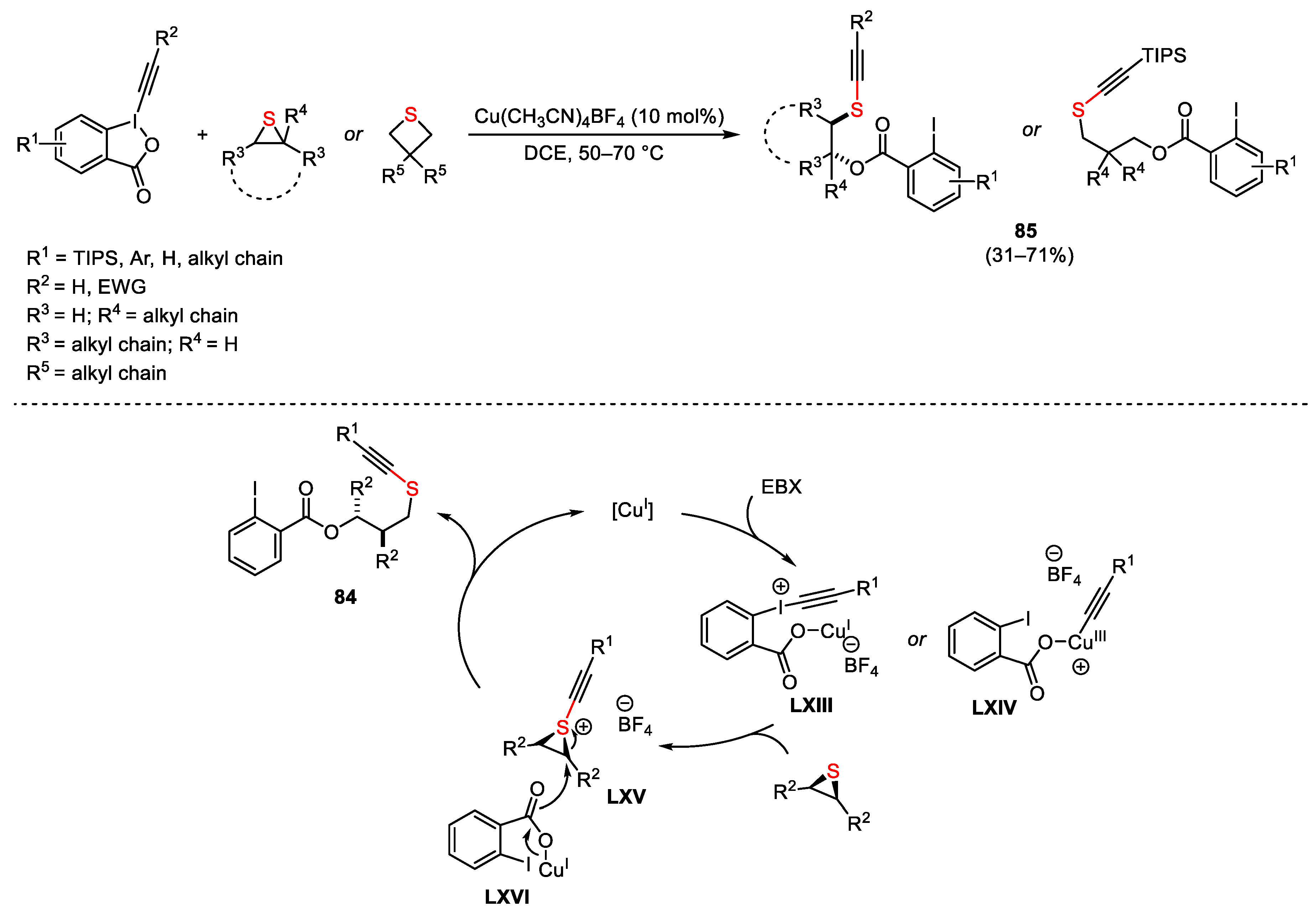{kind=link}
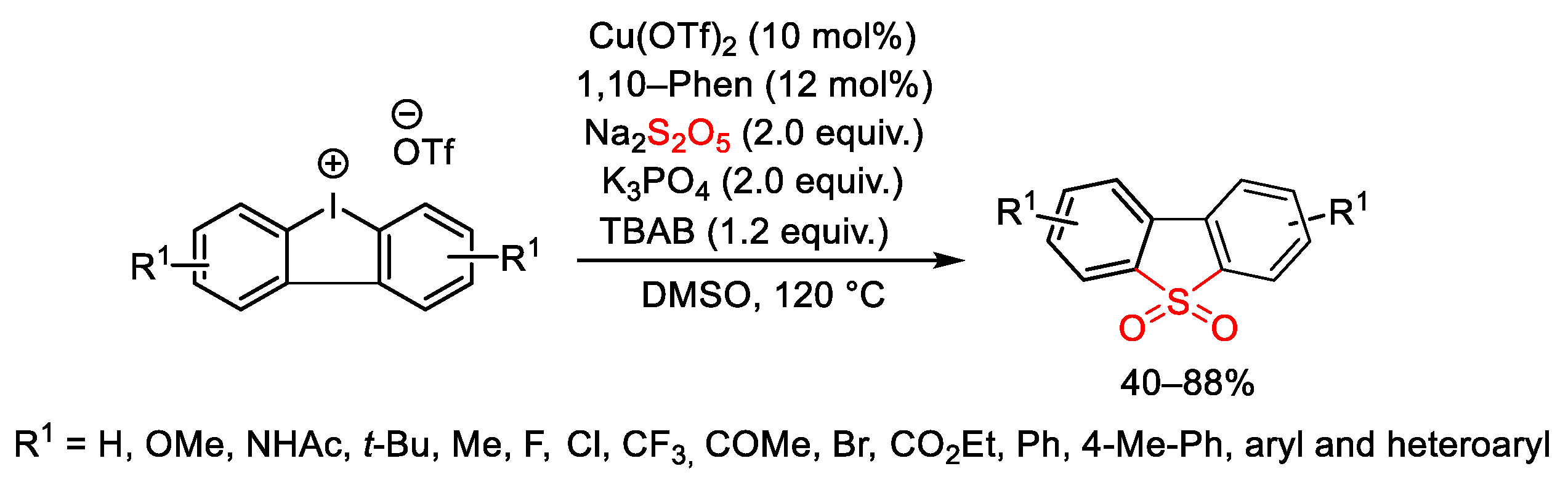{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
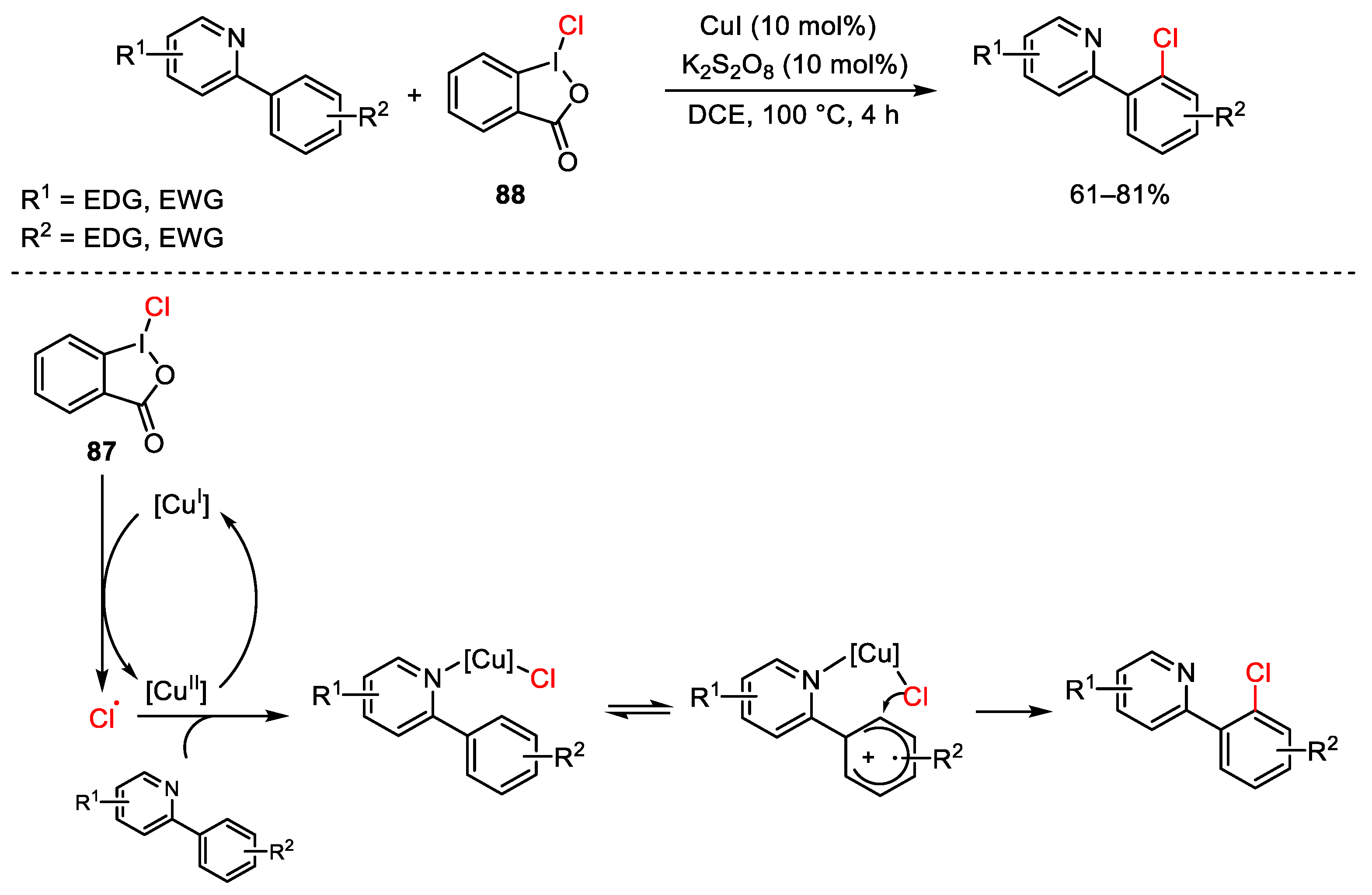{kind=link}
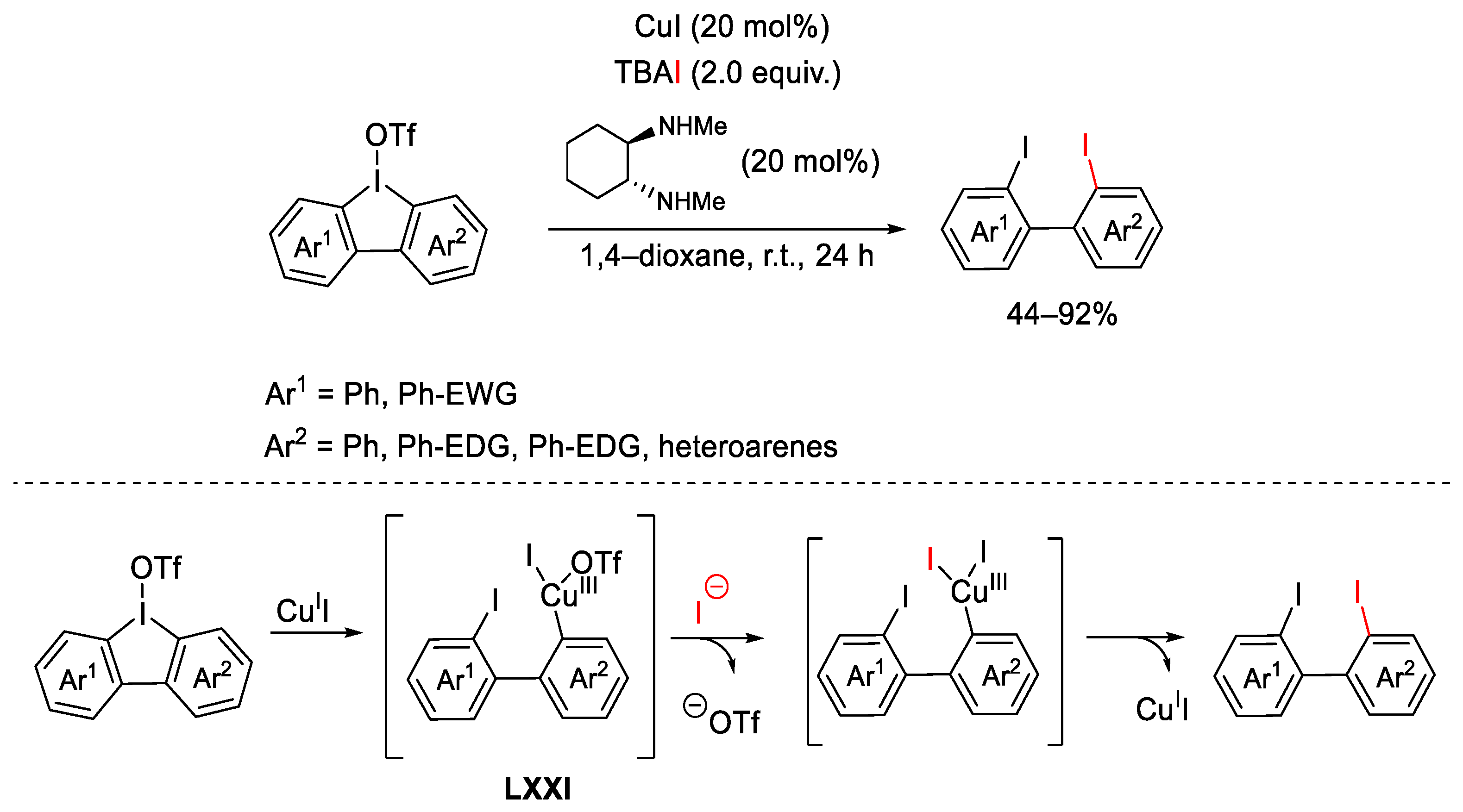{kind=link}
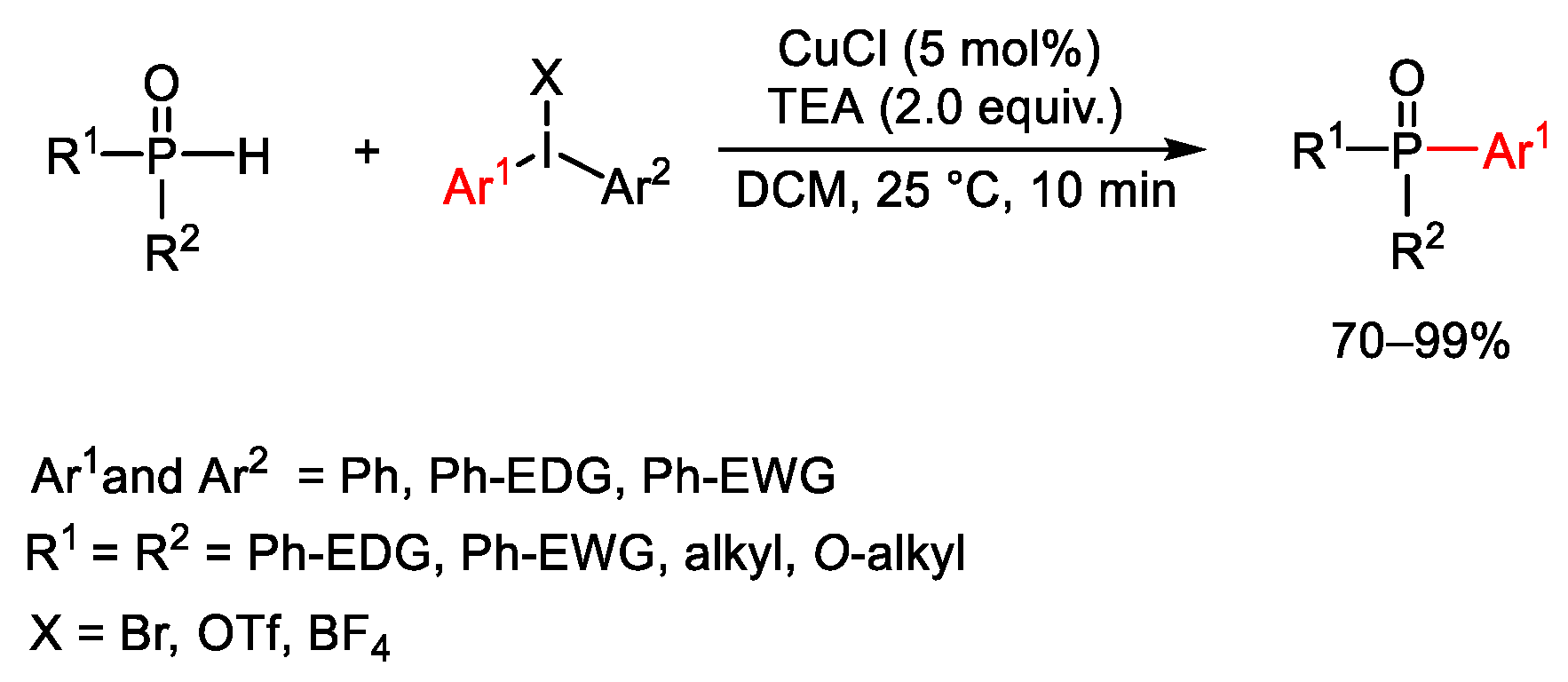{kind=link}
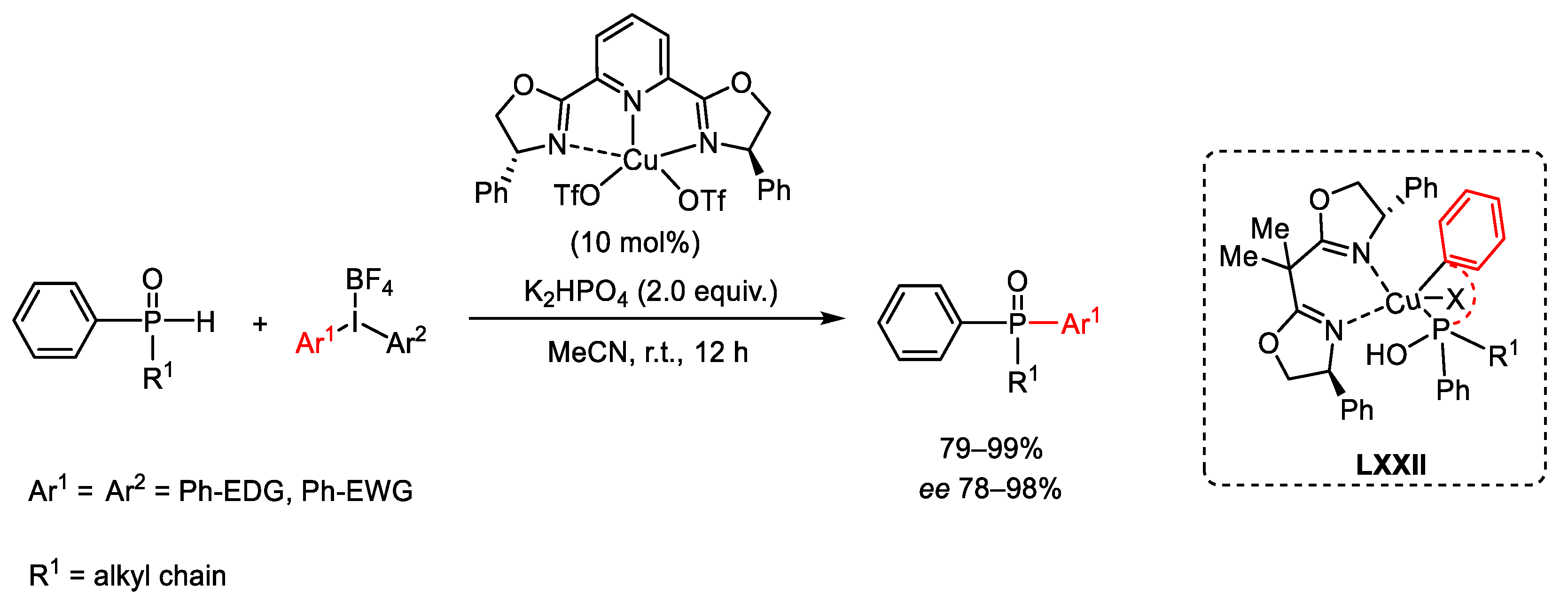{kind=link}
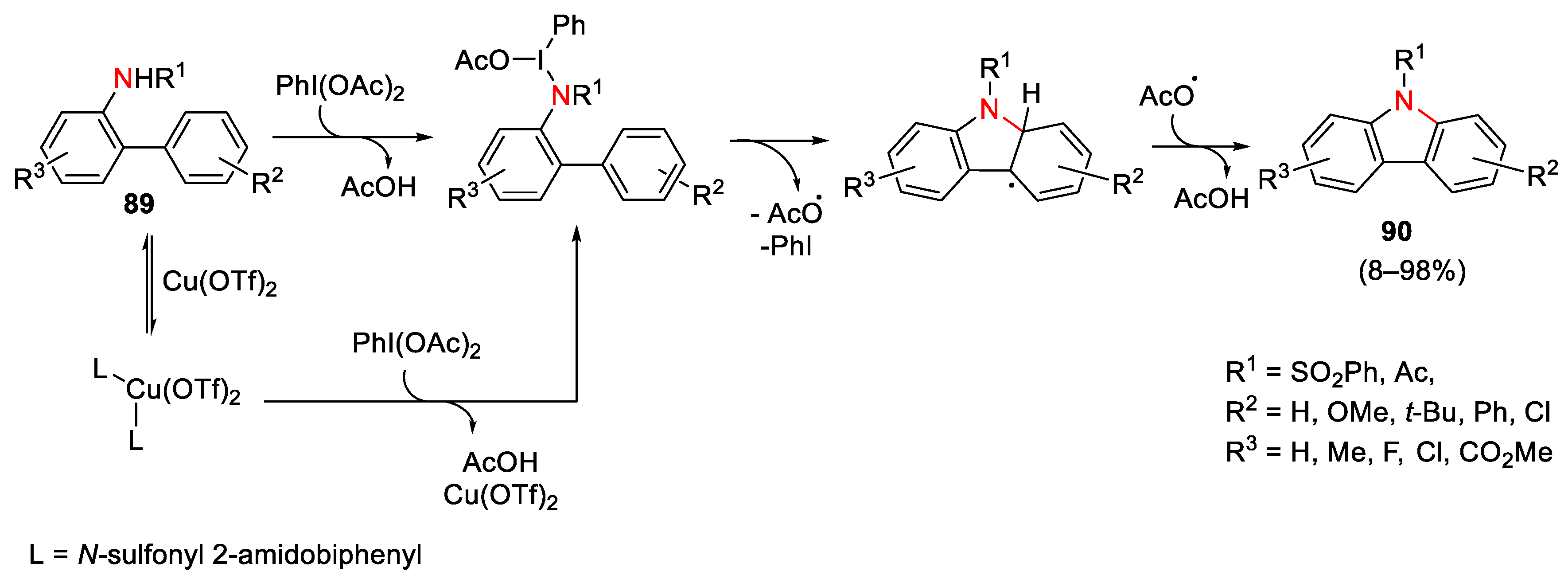{kind=link}
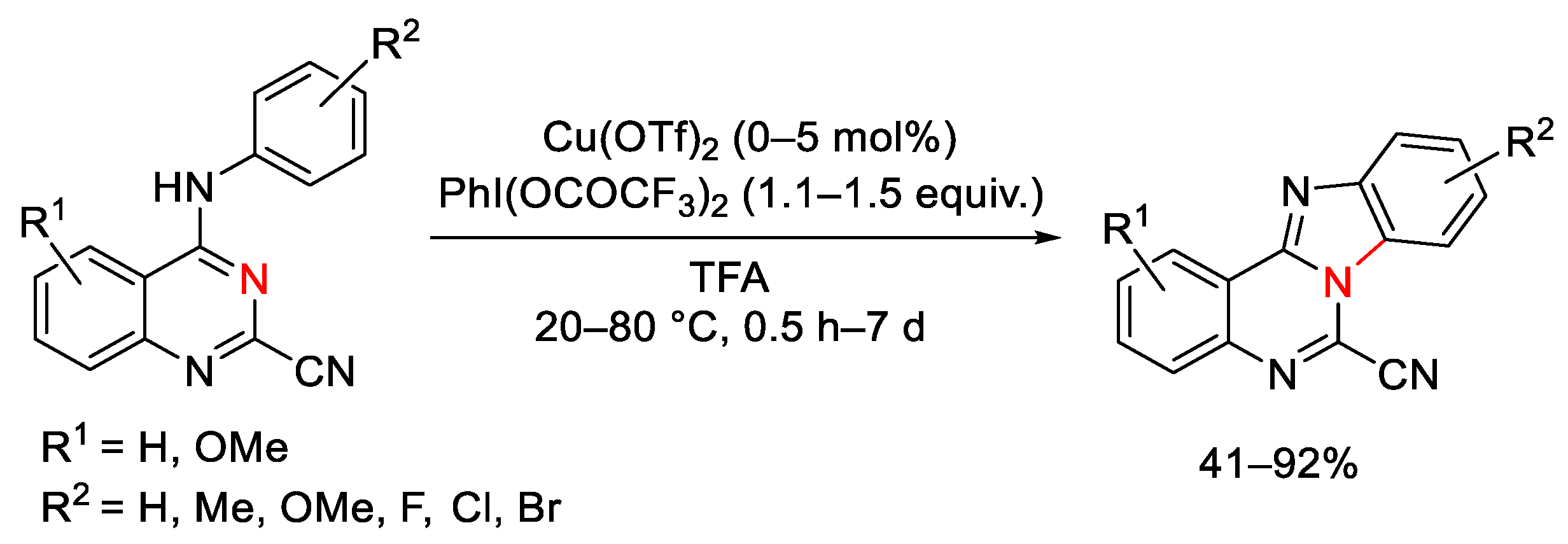{kind=link}
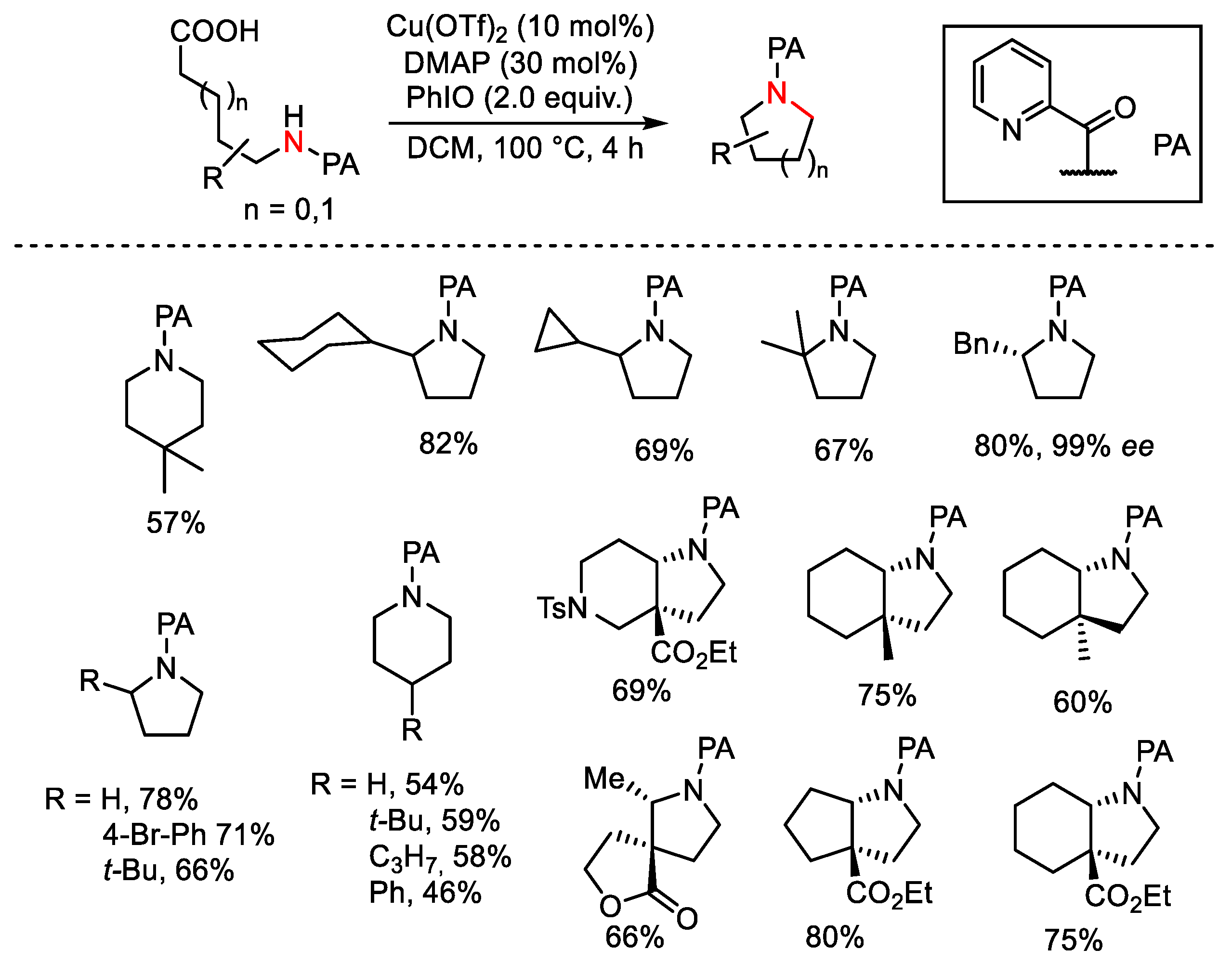{kind=link}
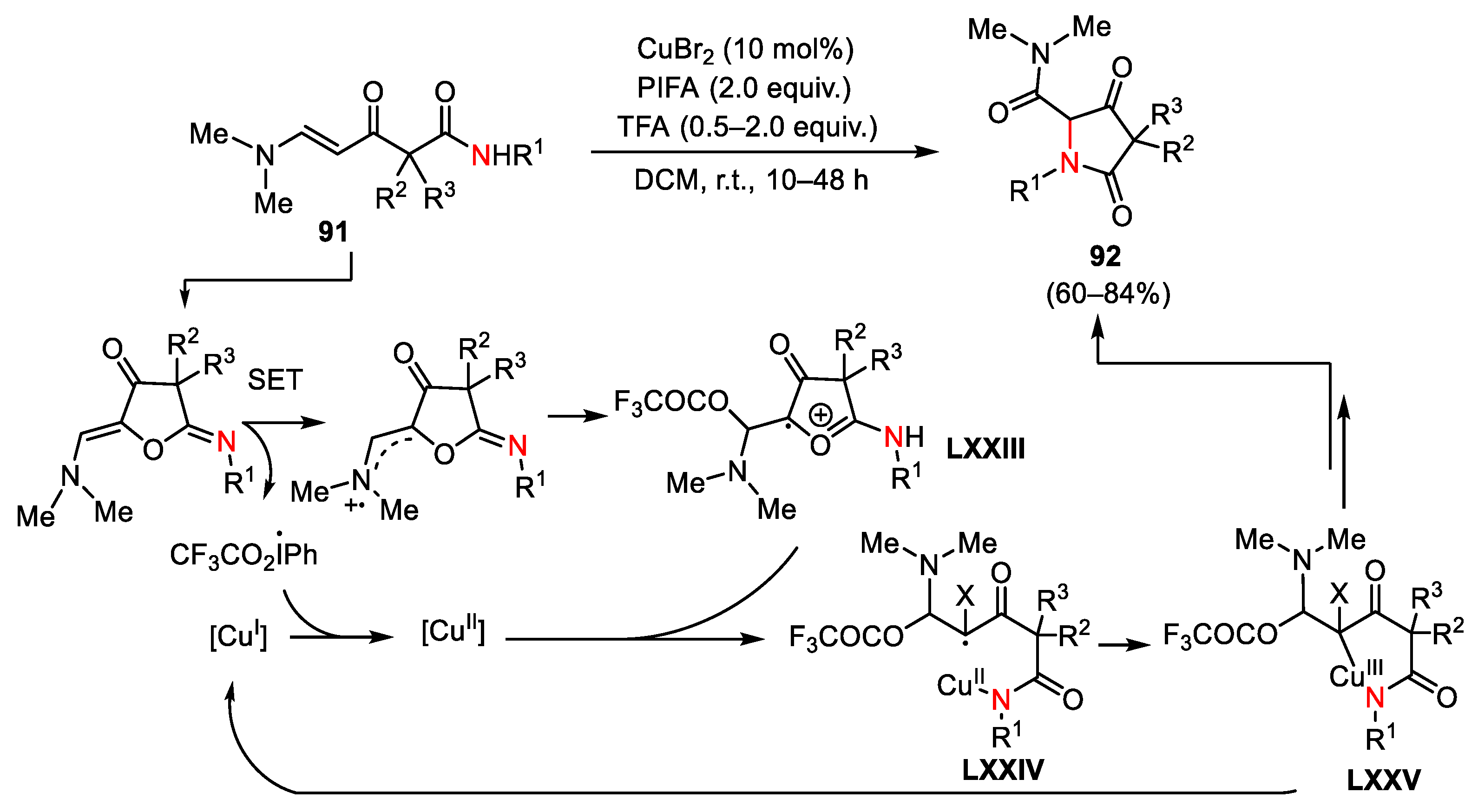{kind=link}
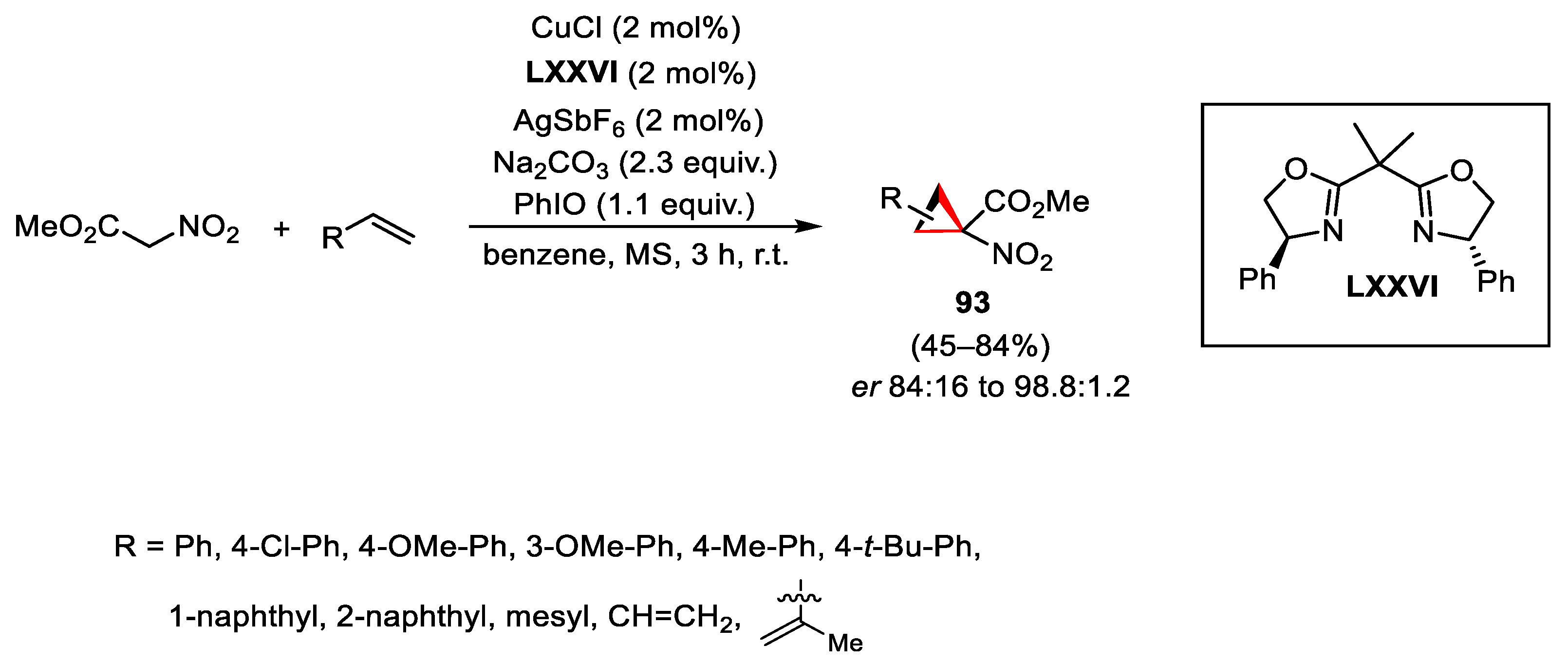{kind=link}
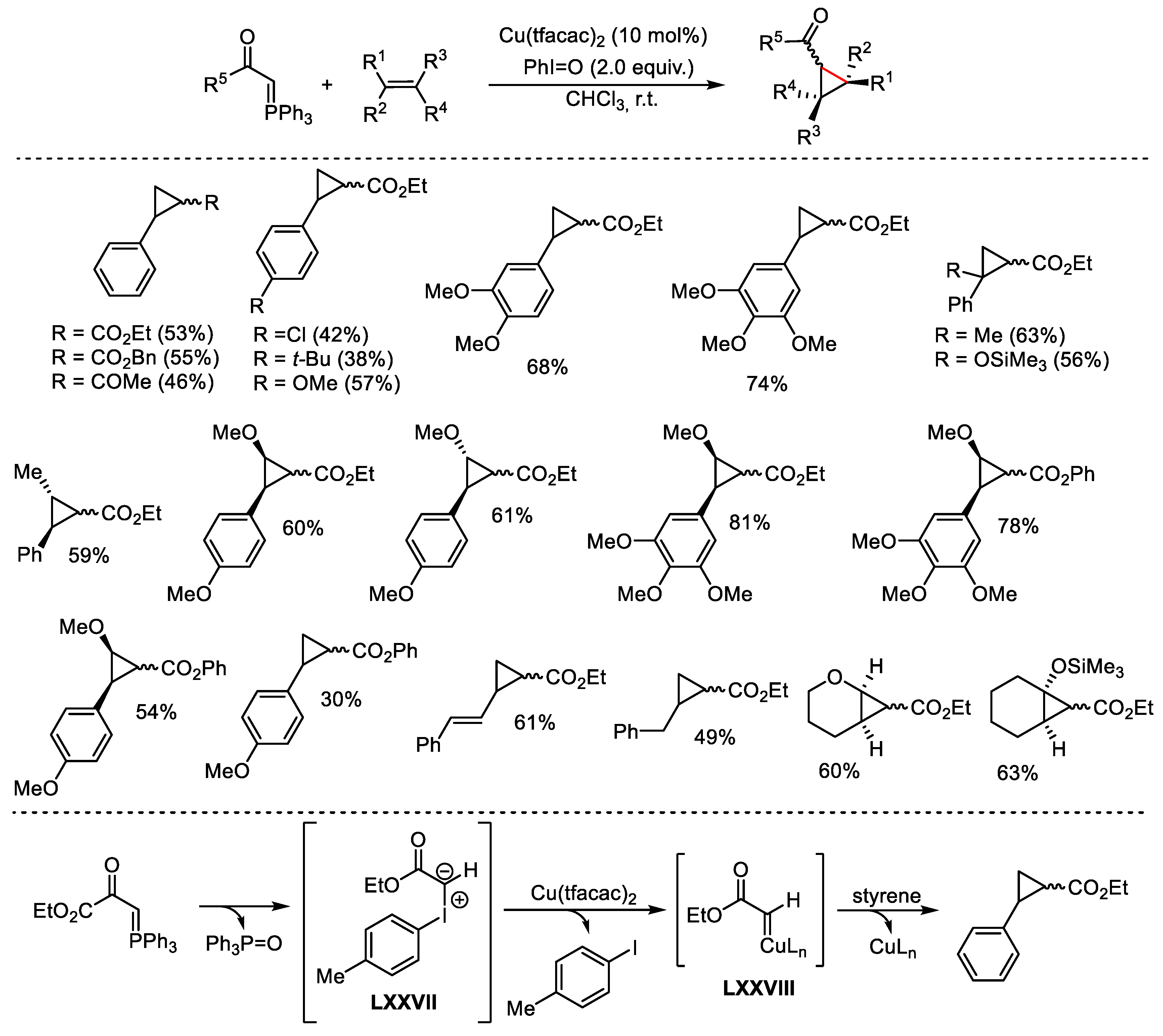{kind=link}
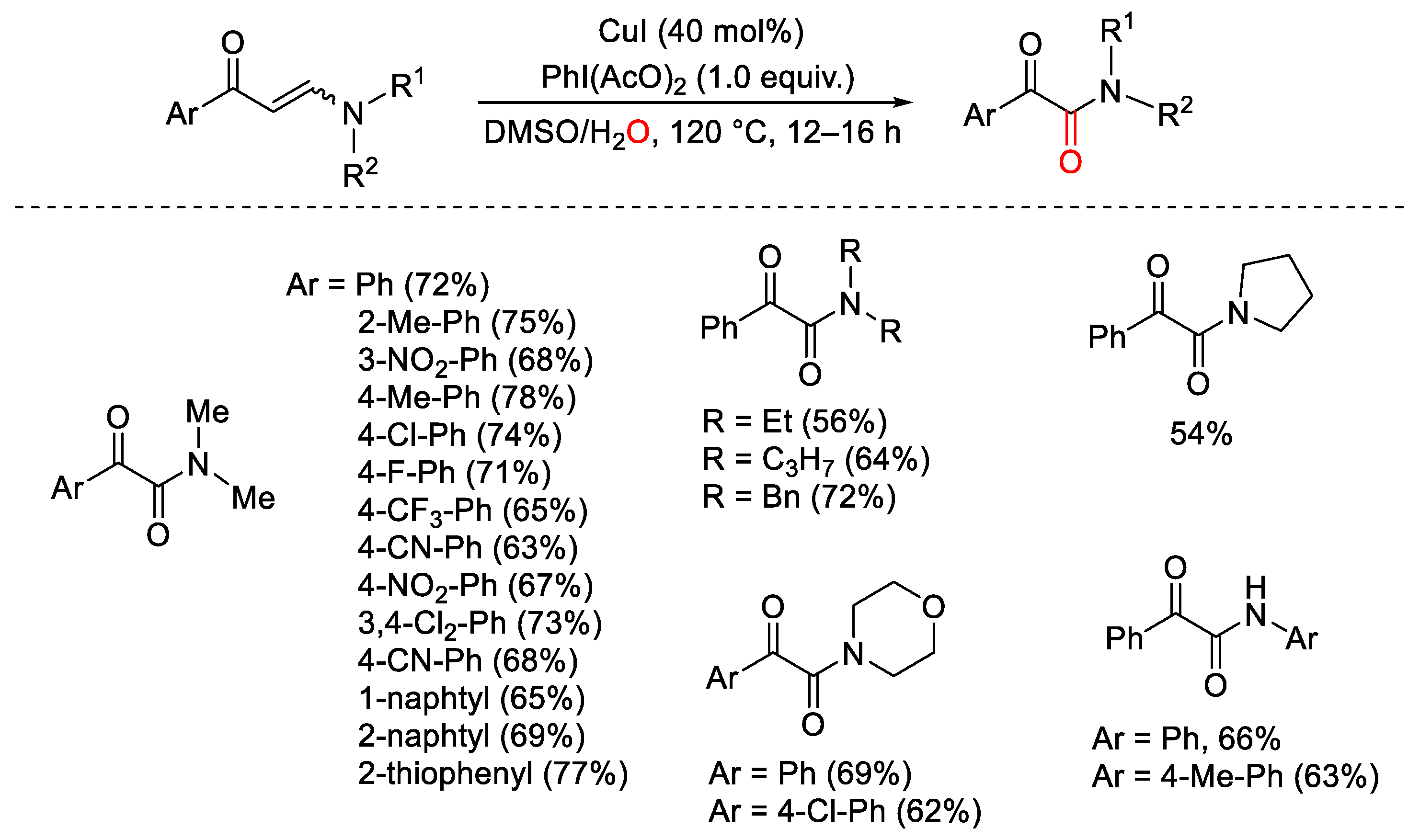{kind=link}
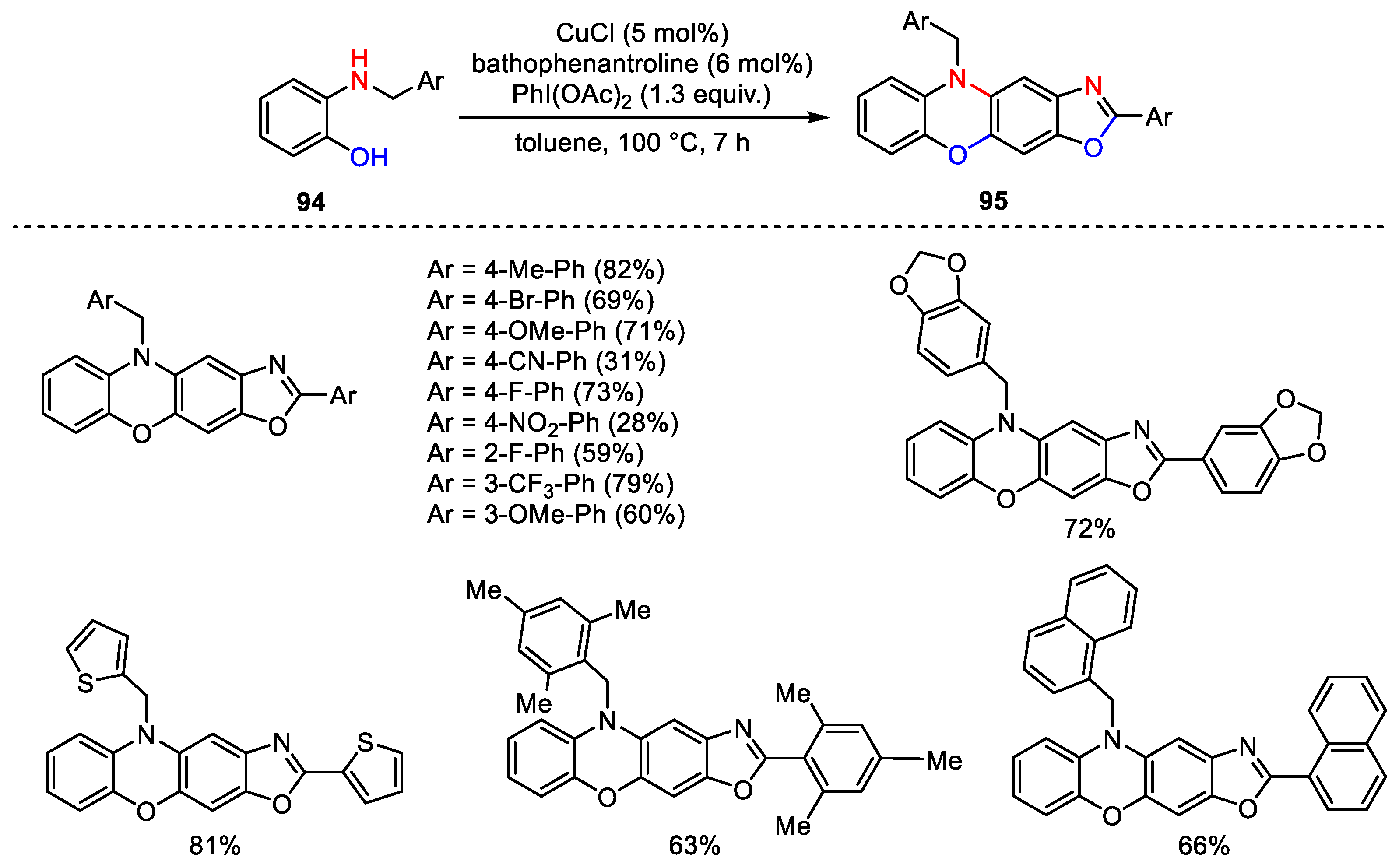{kind=link}
{kind=link}
{kind=link}
1. Introduction
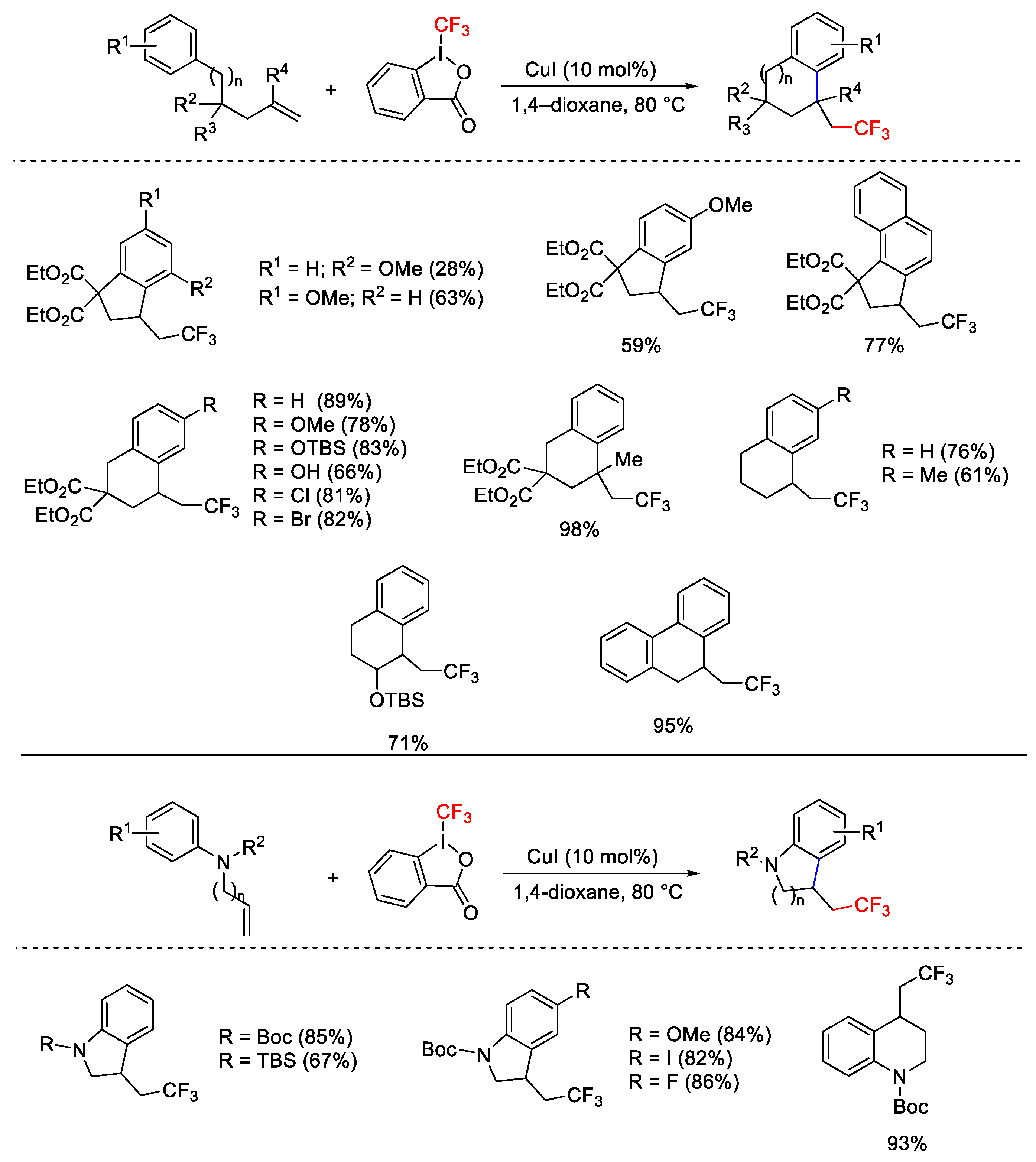
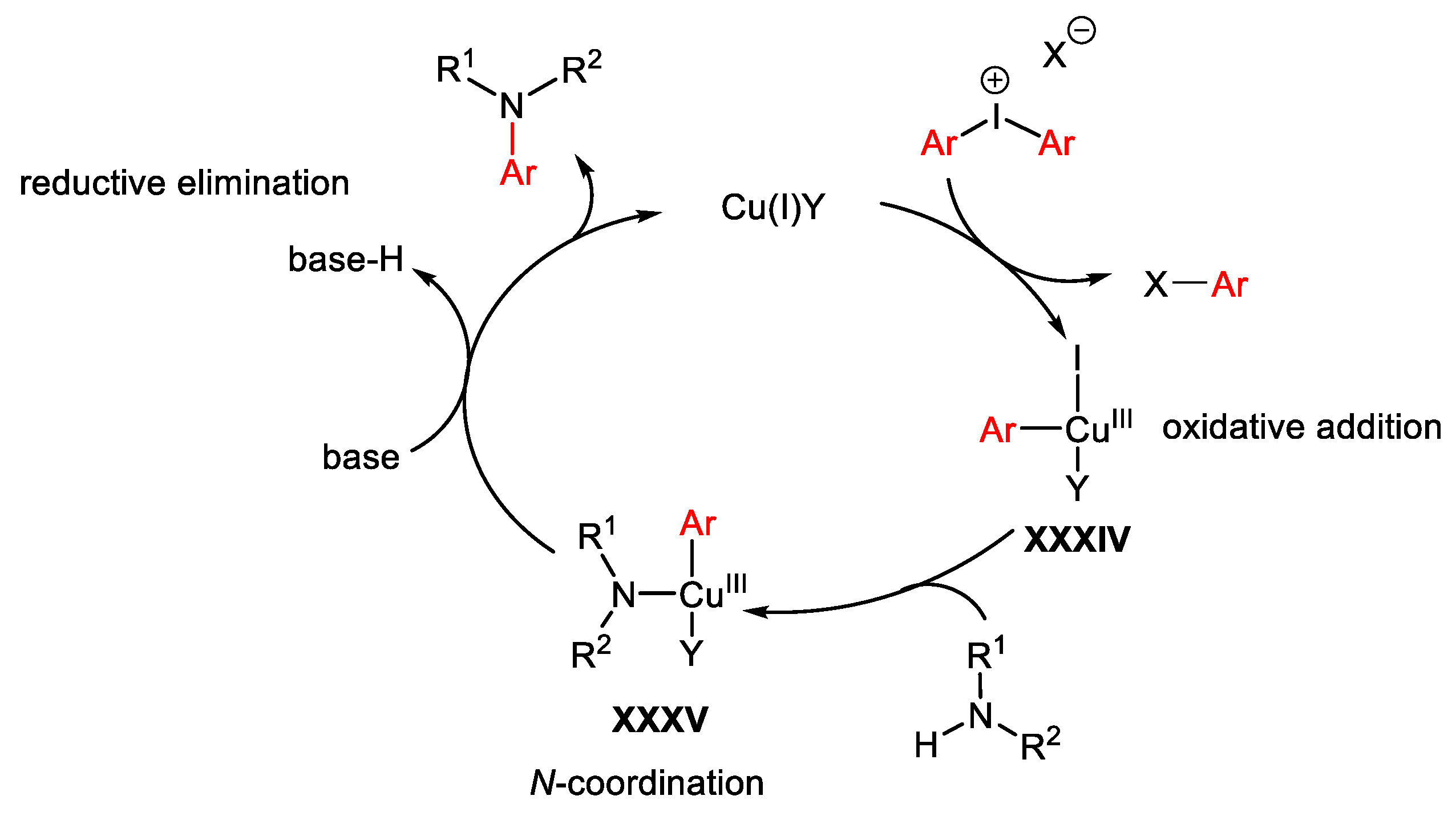
2. Hypervalent Iodine as Reagent in C-C Bond Formation
2.1. Arylation Reactions
2.2. Alkynylation Reactions
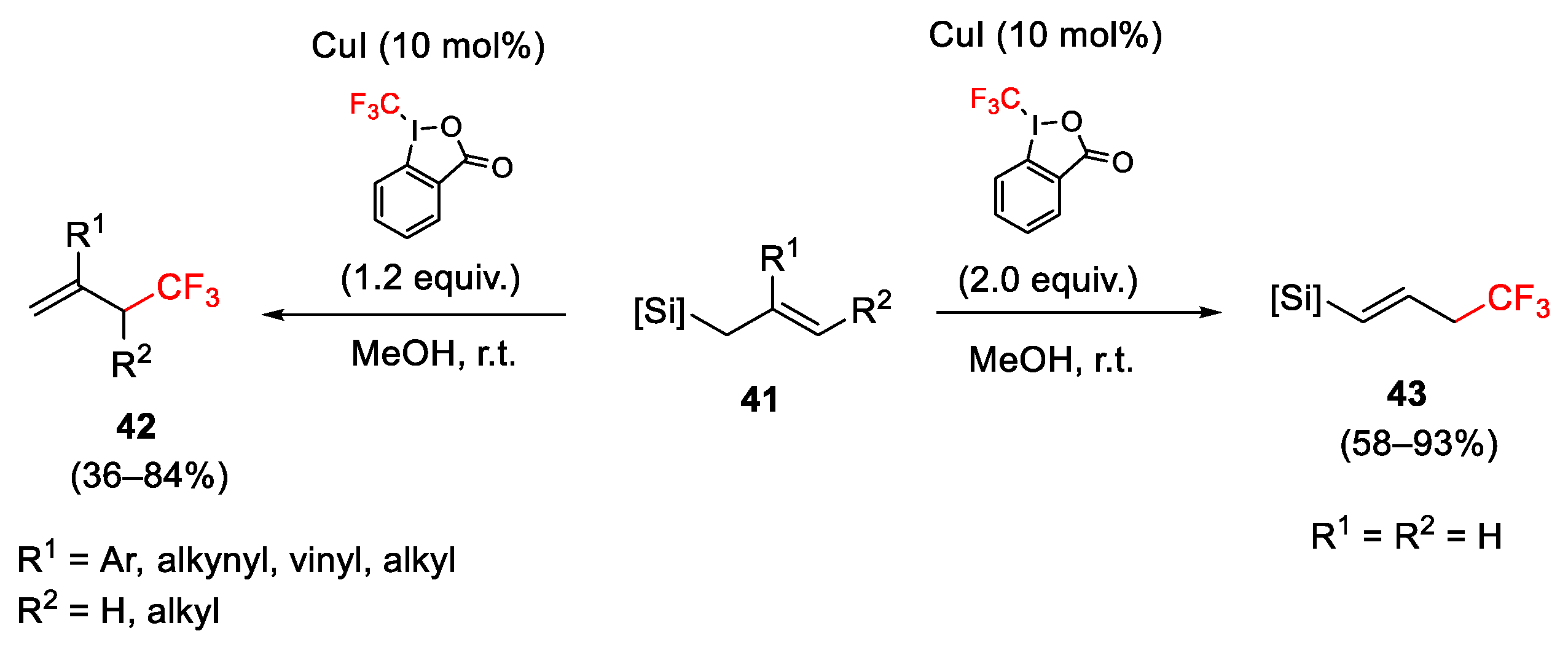
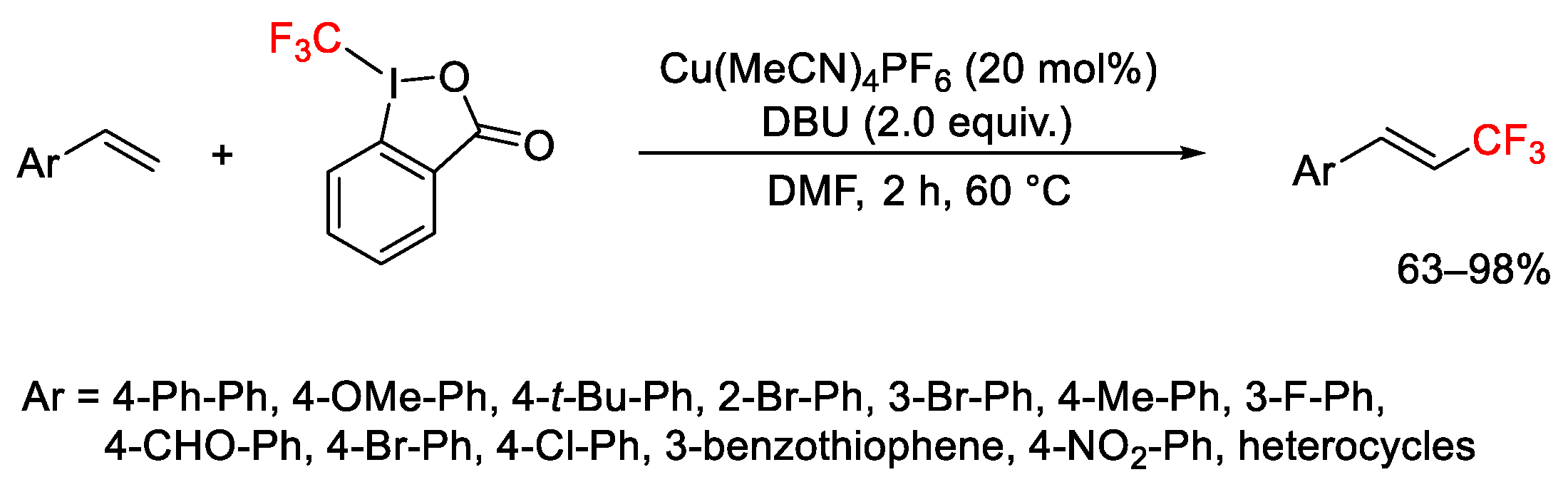
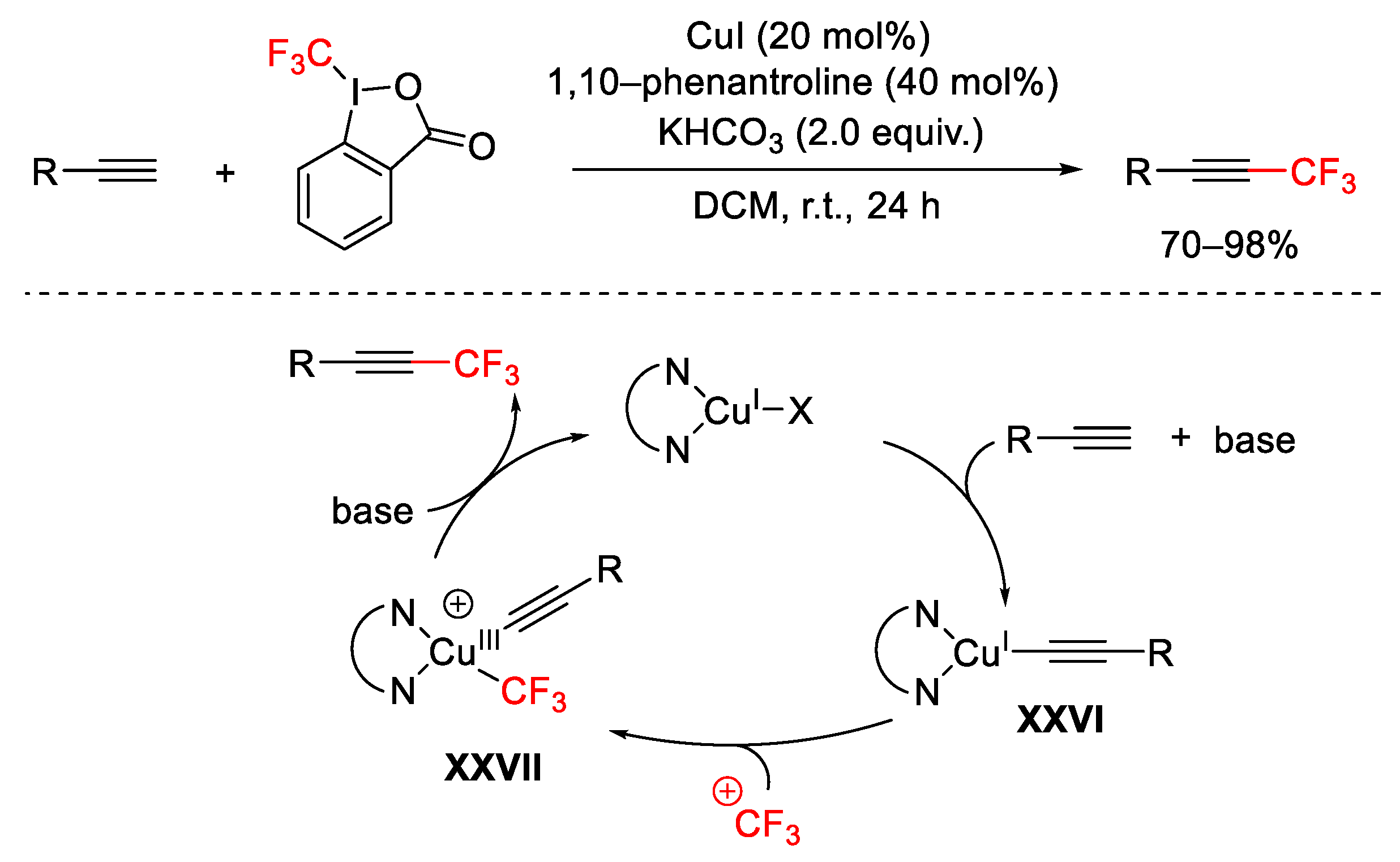
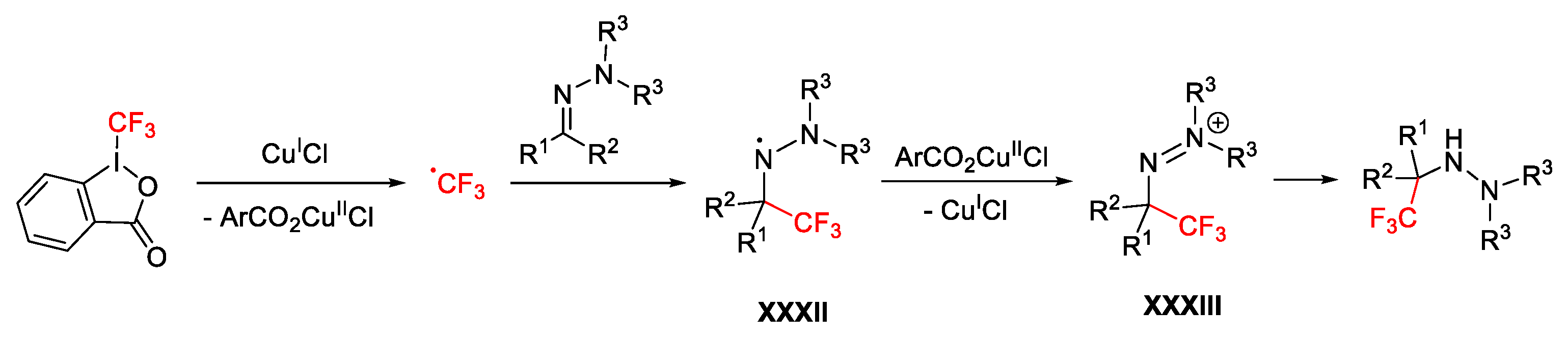
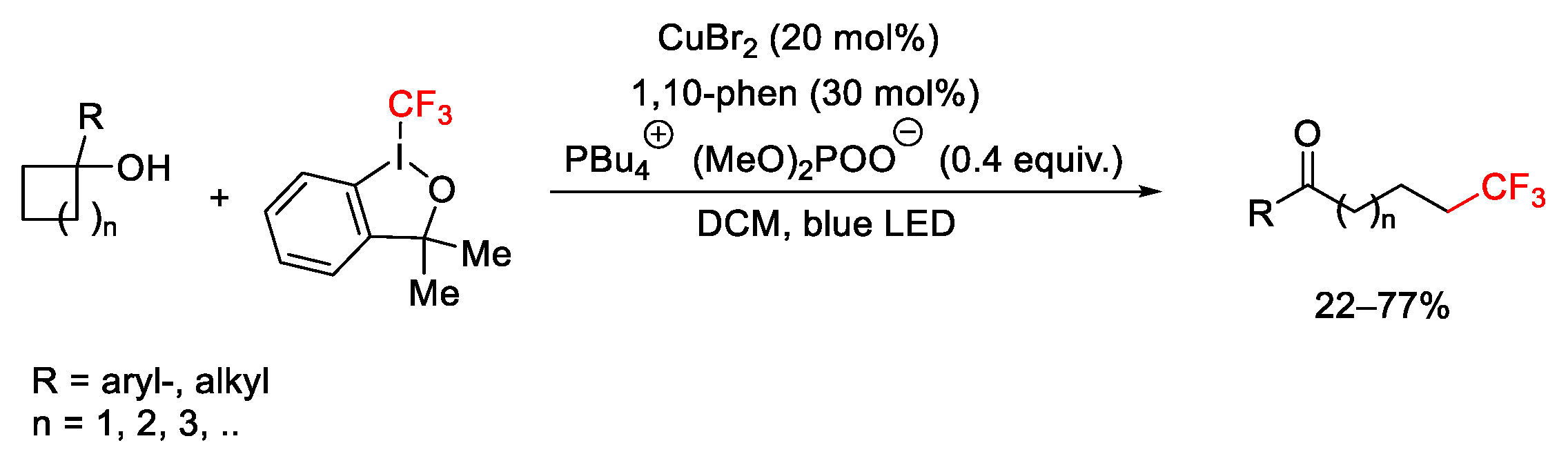
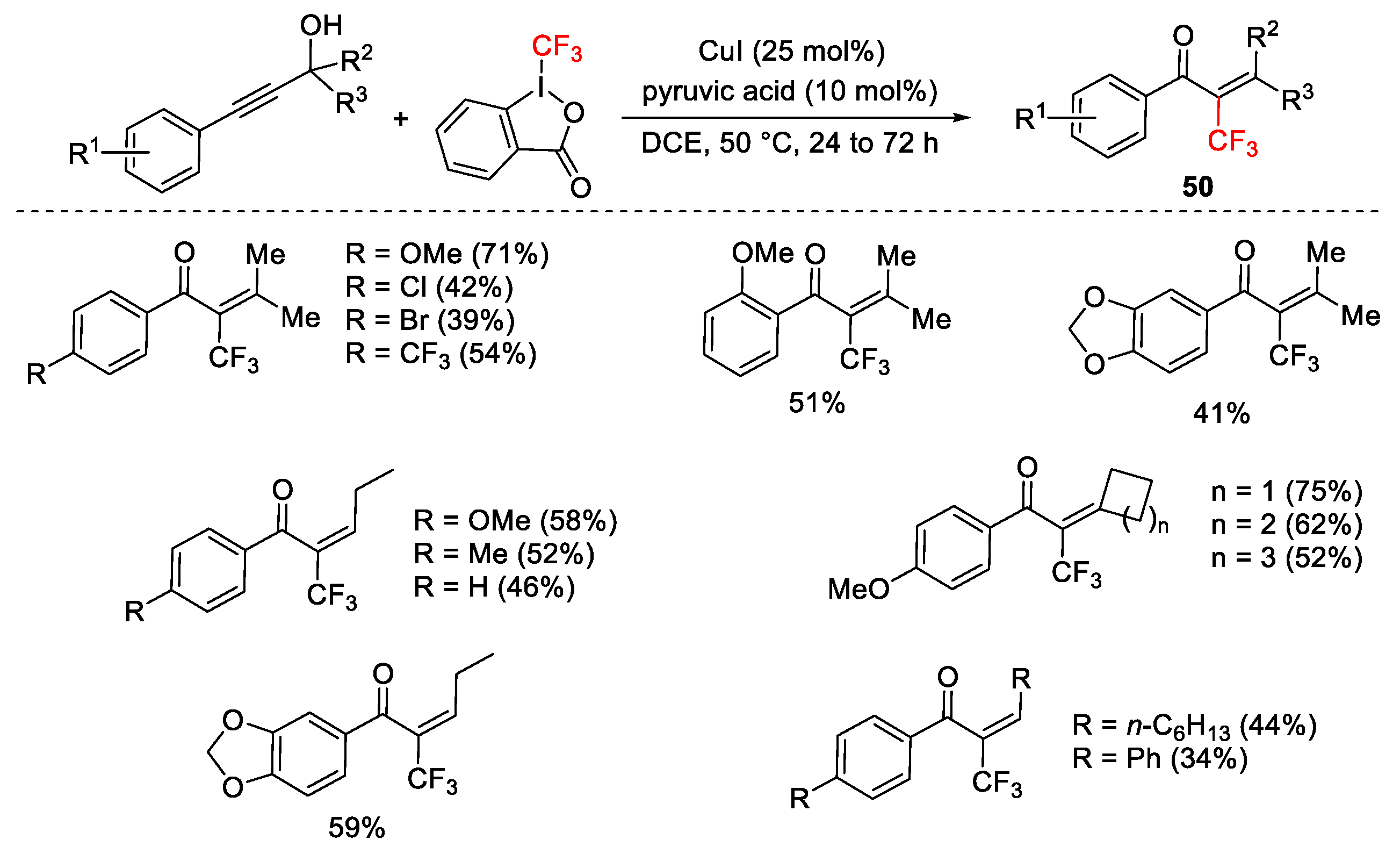
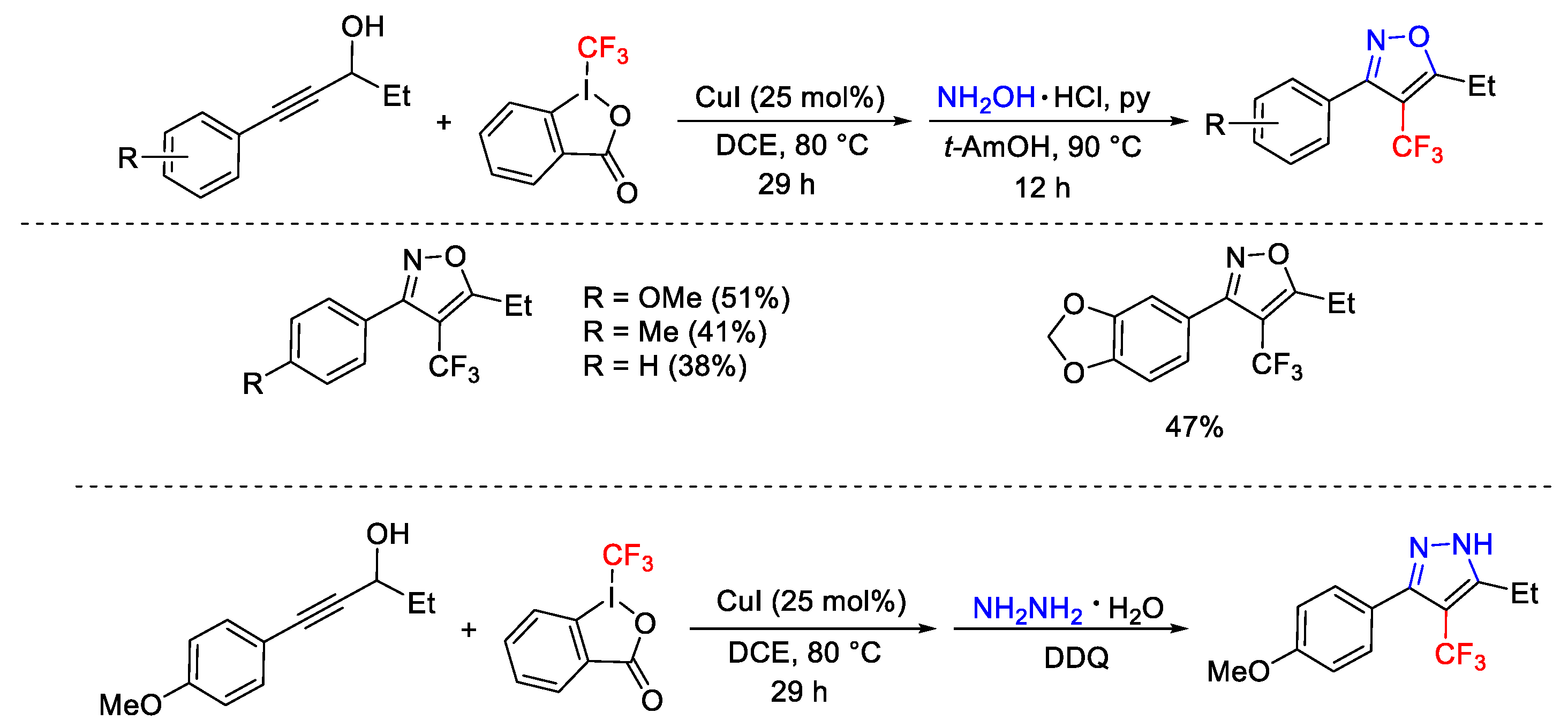
2.3. Trifluoromethylation Reactions
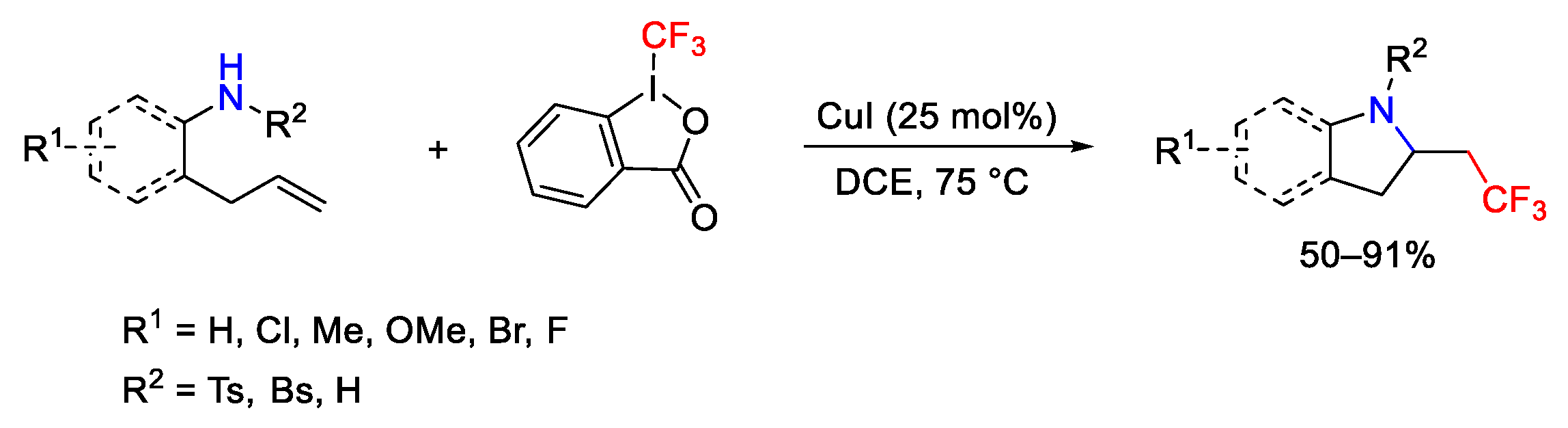
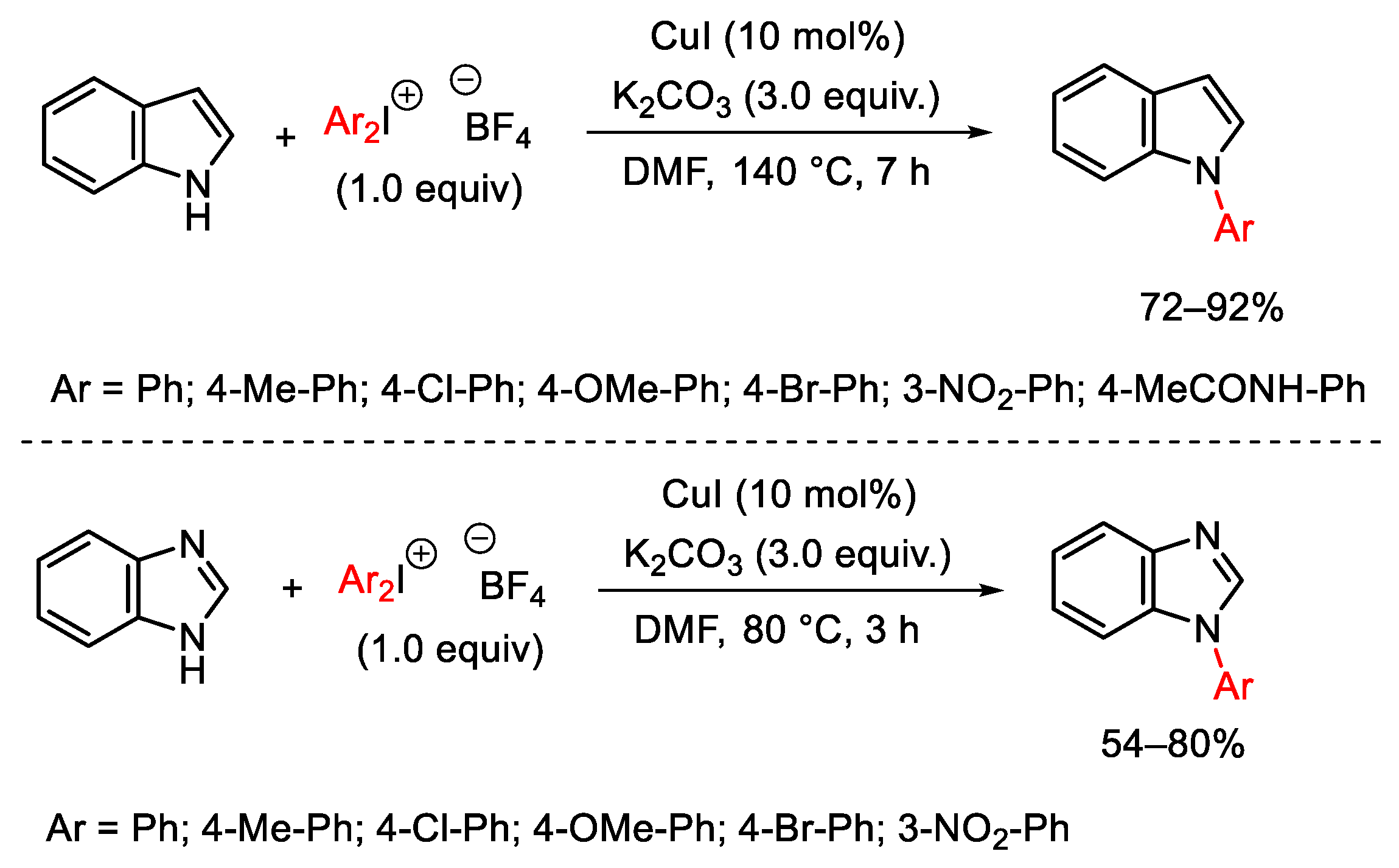
3. Hypervalent Iodine as Reagent in C-N Bond Formation
3.1. Arylation Reactions
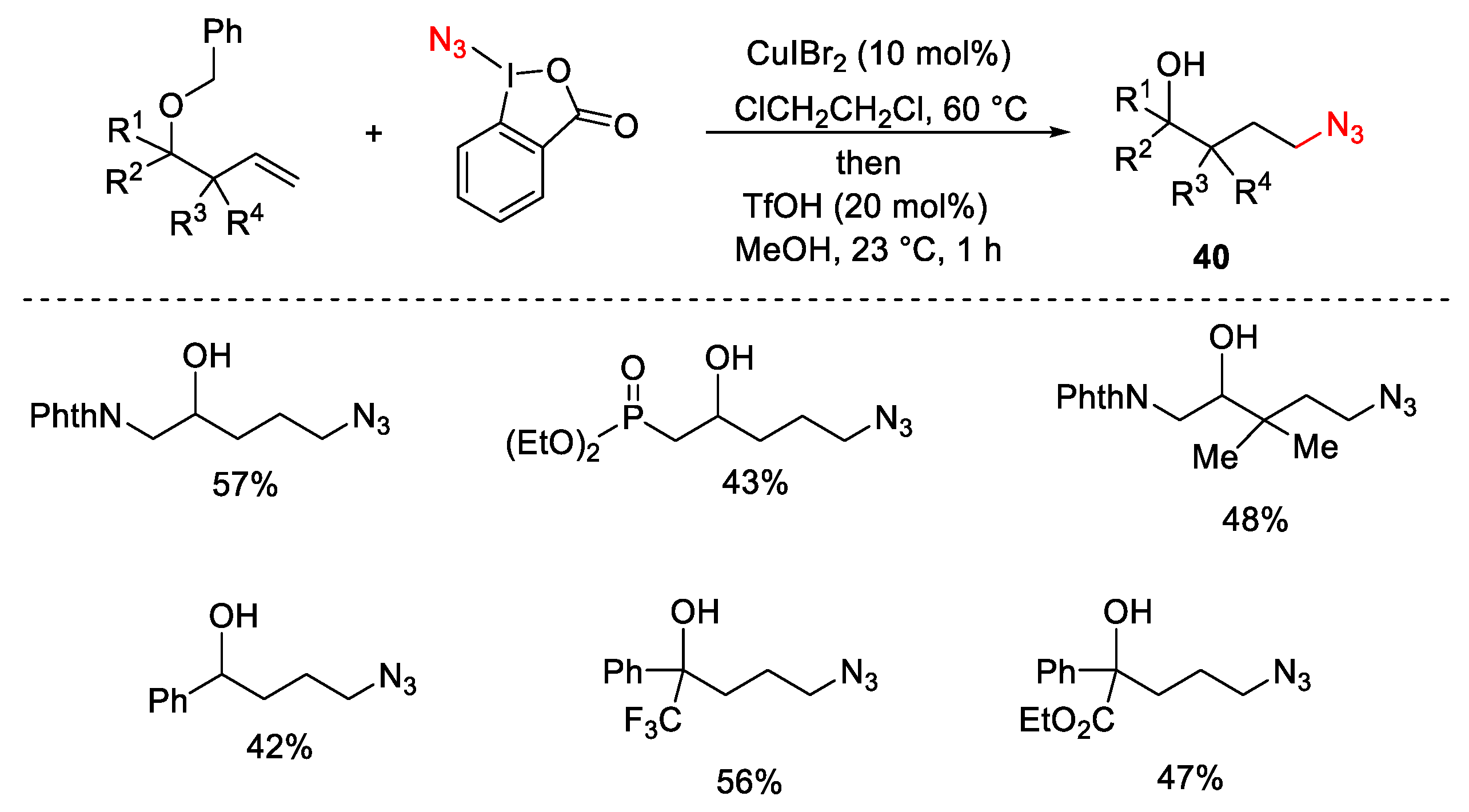
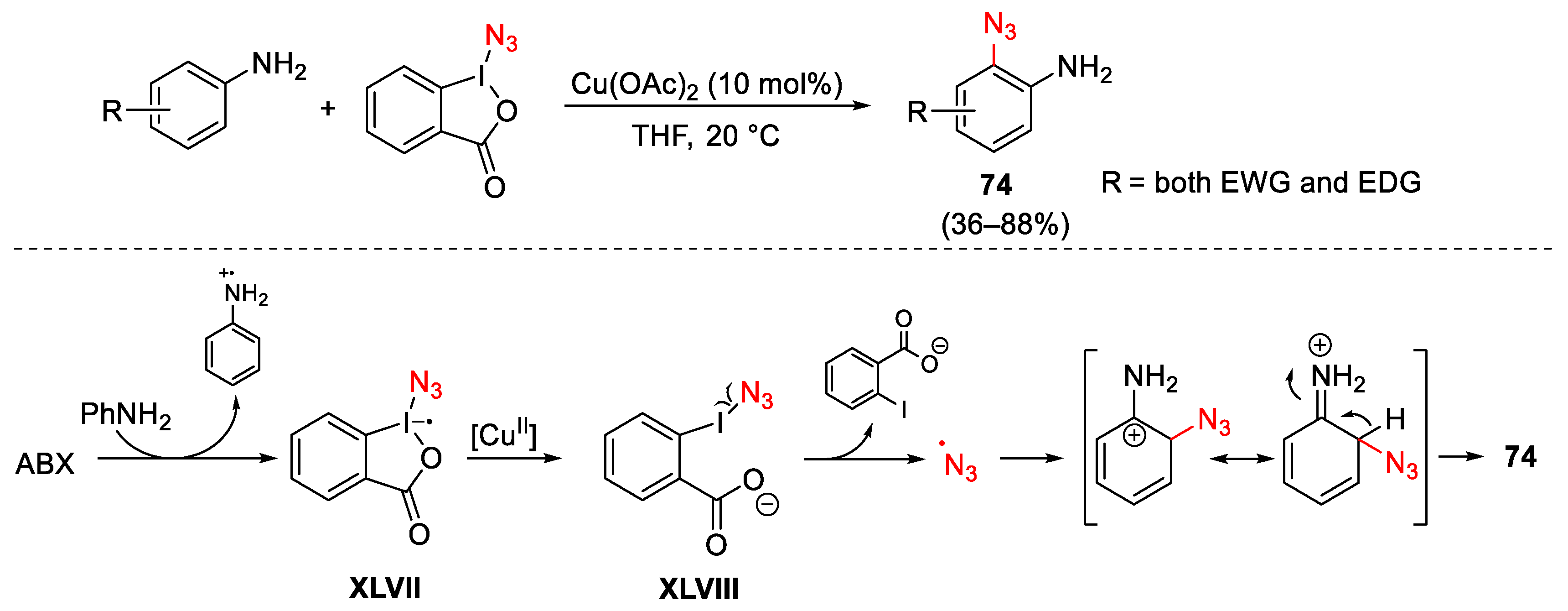
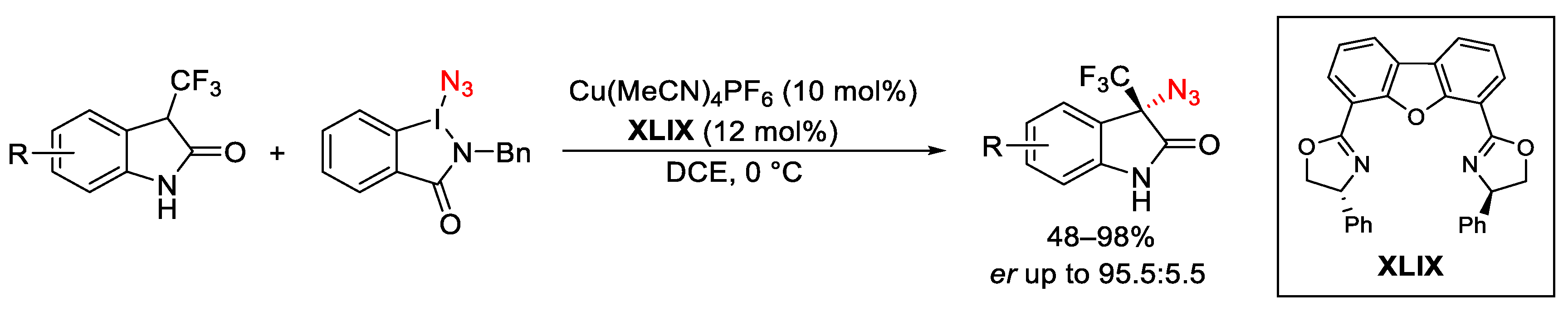
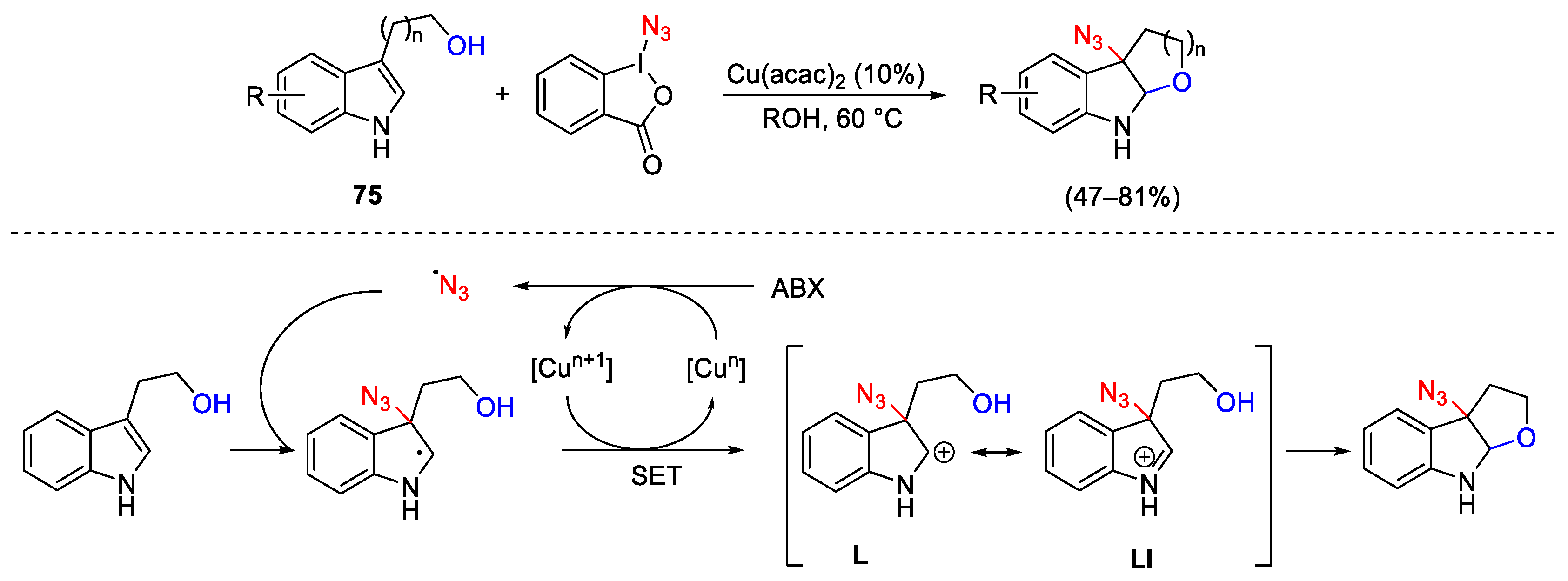
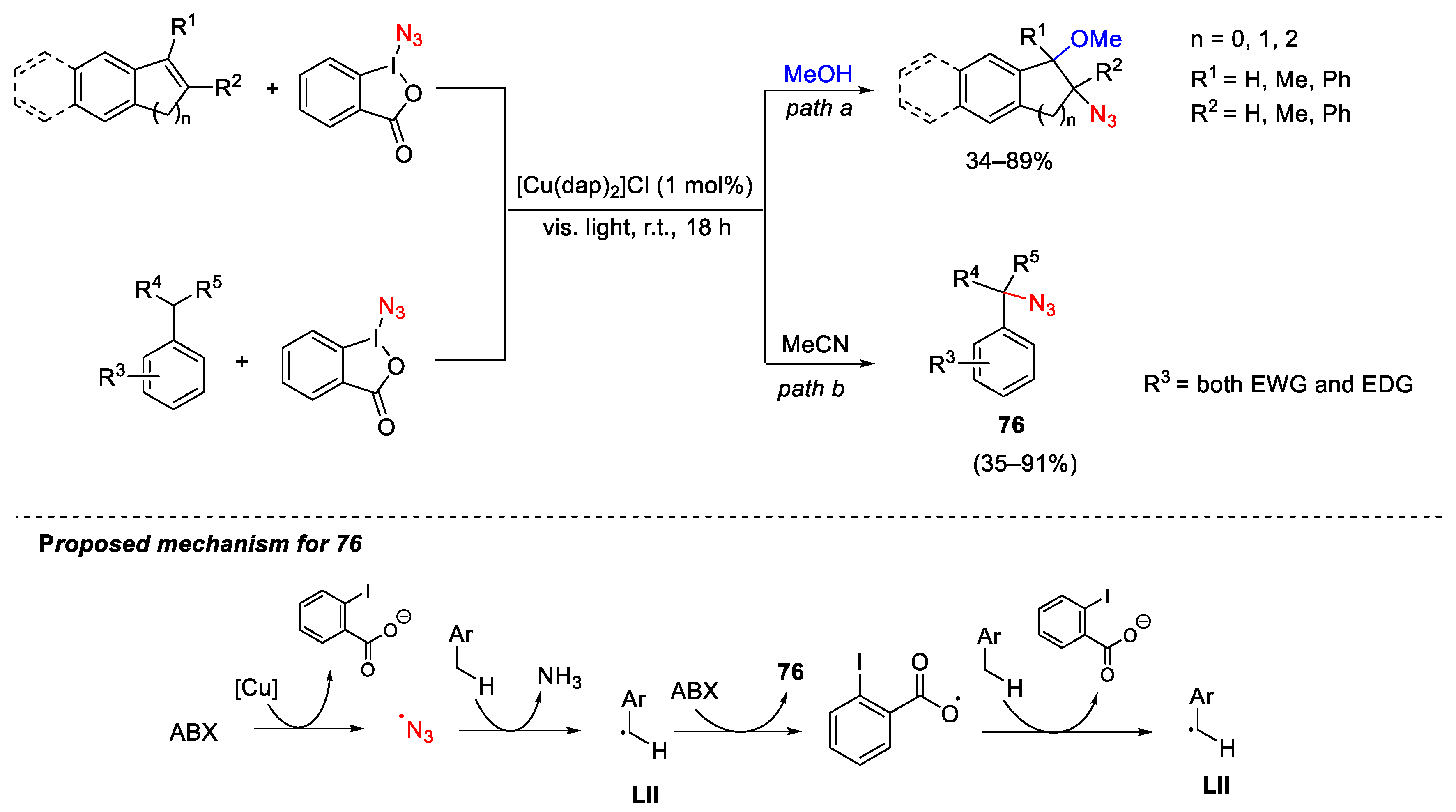
3.2. Azidation Reactions
4. Hypervalent Iodine as Reagent in C-O Bond Formation
5. Hypervalent Iodine as Reagent in C-S Bond Formation
6. Hypervalent Iodine as Reagent in C-X Bond Formation
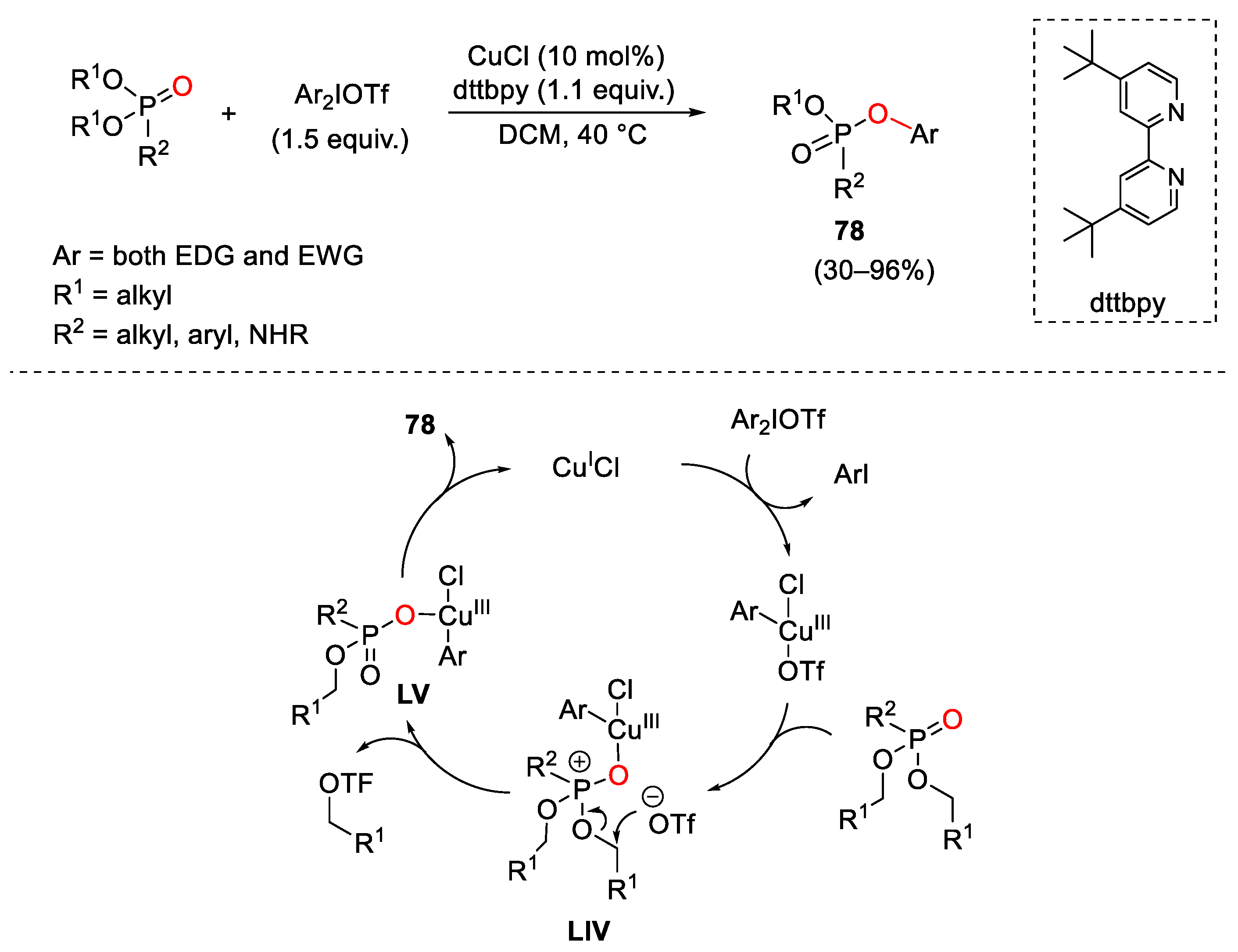
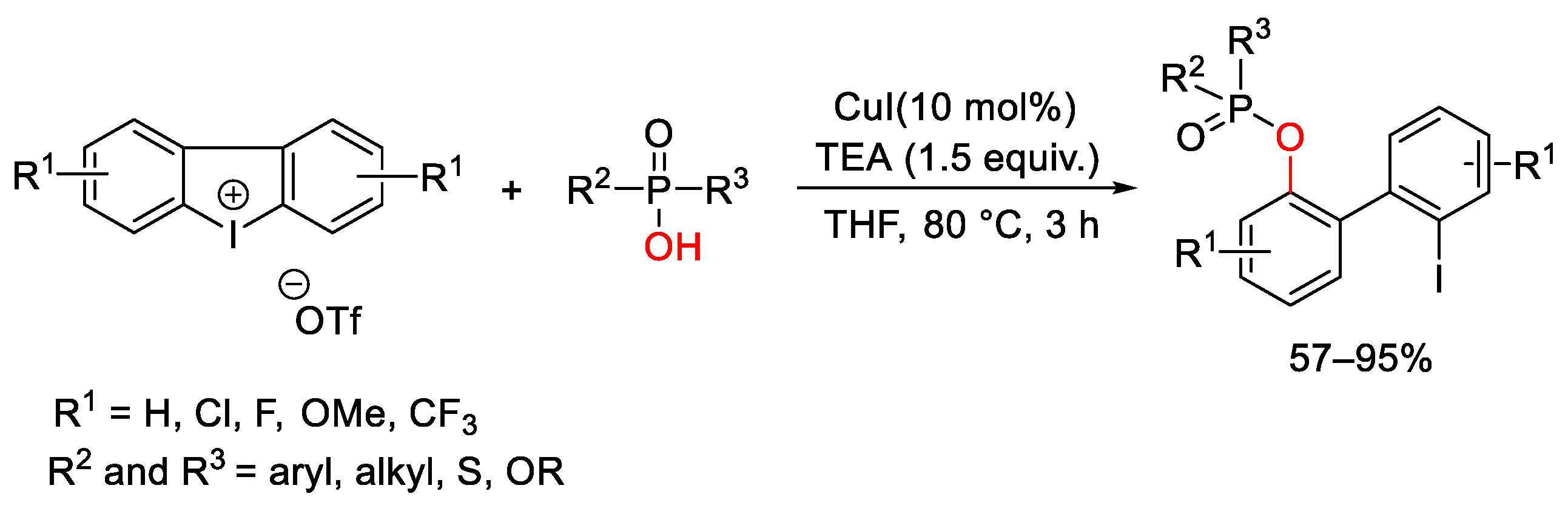
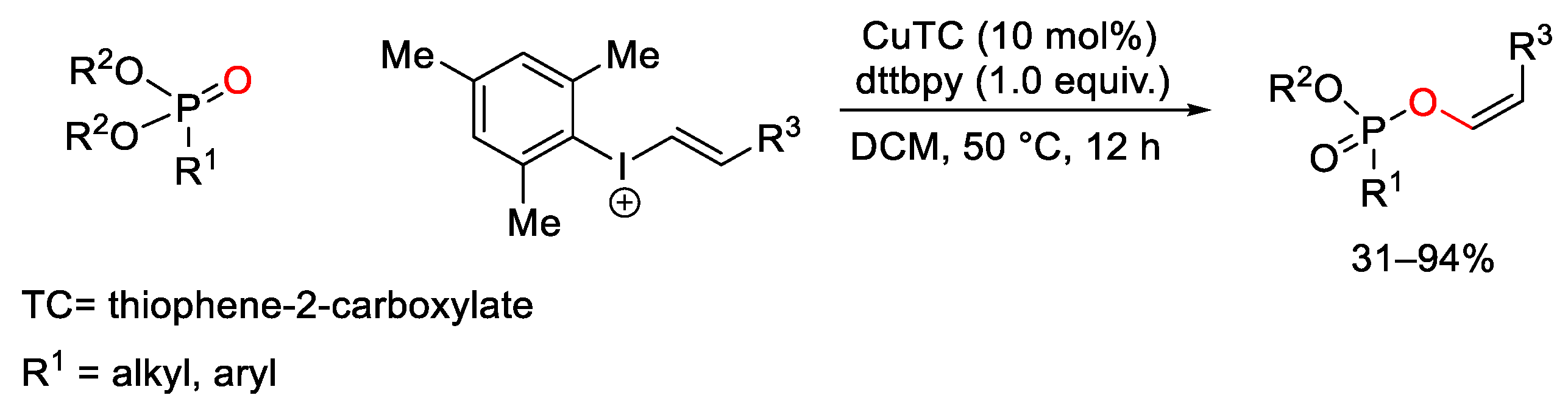
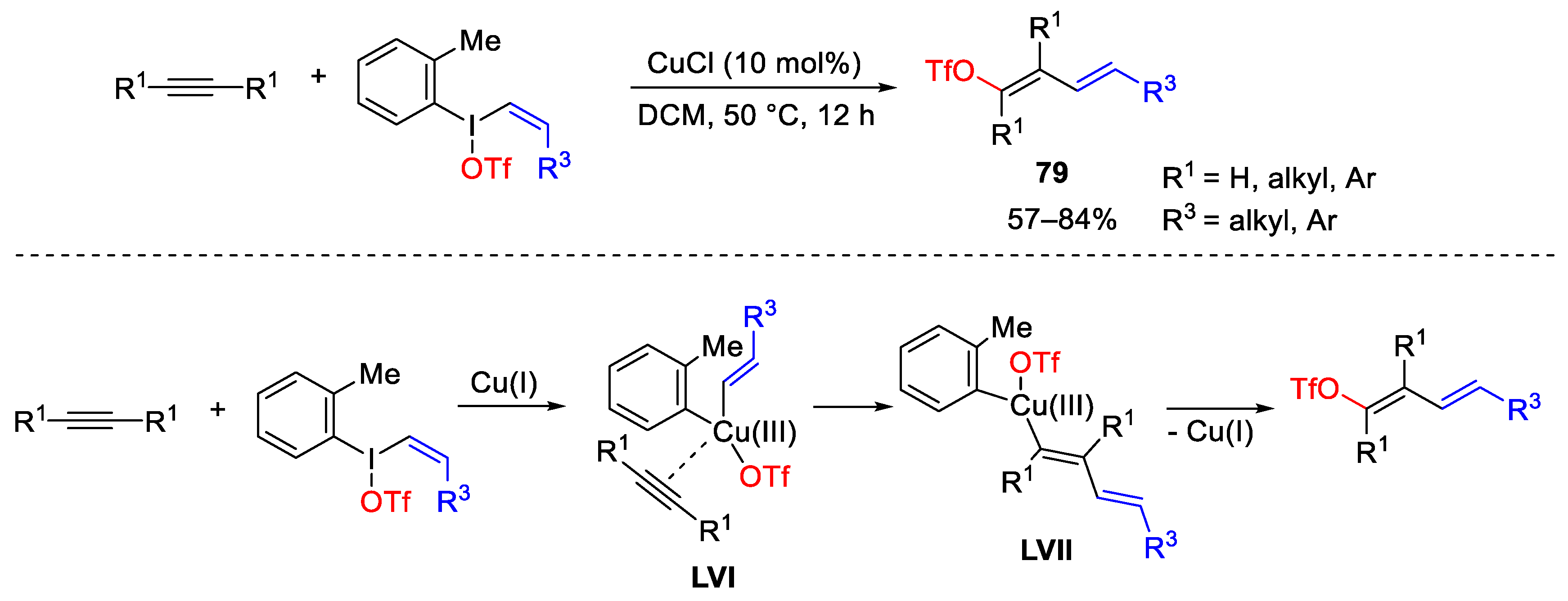
7. Hypervalent Iodine as Reagent in C-P Bond Formation
8. Hypervalent Iodines as Oxidant
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zhdankin, V.V. Hypervalent Iodine Chemistry: Preparation, Structure, and Synthetic Applications of Polyvalent Iodine Compounds; Wiley: Chichester, UK, 2013. [Google Scholar]
- Richardson, R.D.; Wirth, T. Hypervalent Iodine Goes Catalytic. Angew. Chem. Int. Ed. 2006, 45, 4402–4404. [Google Scholar] [CrossRef] [PubMed]
- Wirth, T. Hypervalent Iodine Chemistry in Synthesis: Scope and New Directions. Angew. Chem. Int. Ed. 2005, 44, 3656–3665. [Google Scholar] [CrossRef]
- Wirth, T. Hypervalent Iodine Chemistry; Springer: Berlin/Heidelberg, Germany, 2003. [Google Scholar]
- Varvoglis, A. Hypervalent Iodine in Organic Synthesis; Academic Press: San Diego, CA, USA, 1996. [Google Scholar]
- Le Du, E.; Waser, J. Recent progress in alkynylation with hypervalent iodine reagents. Chem. Commun. 2023, 59, 1589–1604. [Google Scholar] [CrossRef]
- Simonet-Davin, R.; Waser, J. Azidation with Hypervalent Iodine Reagents. Synthesis 2023, 55, 1652–1661. [Google Scholar] [CrossRef]
- Yoshimura, A.; Zhdanlin, V.V. Advances in Synthetic Applications of Hypervalent Iodine Compounds. Chem. Rev. 2016, 116, 3328–3435. [Google Scholar] [CrossRef]
- Shetgaonkar, S.E.; Singh, F.V. Catalytic stereoselective synthesis involving hypervalent iodine-based chiral auxiliaries. Org. Biomol. Chem. 2023, 21, 4163–4180. [Google Scholar] [CrossRef]
- Singh, F.V.; Shetgaonkar, S.E. Progress in organocatalysis with hypervalent iodine cataysts. Chem. Soc. Rev. 2022, 51, 8102–8139. [Google Scholar] [CrossRef]
- Rani, N.; Soni, R.; Sihag, M.; Kinger, M.; Aneja, D.K. Combined Approach of Hypervalent Iodine Reagents and Transition Metals in Organic Reactions. Adv. Synth. Catal. 2022, 364, 1798–1848. [Google Scholar] [CrossRef]
- Shetgaonkar, S.E.; Mamgain, R.; Kikushima, K.; Dohi, T.; Singh, F.V. Palladium-Catalyzed Organic Reactions Involving Hypervalent Iodine Reagents. Molecules 2022, 27, 3900. [Google Scholar] [CrossRef]
- Sun, T.-Y.; Wang, X.; Geng, H.; Xie, Y.; Wu, Y.-D.; Zhang, X.; Schaefer, H.F. III Why does Togni’s Reagent I exist in the High-Energy Hypervalent Iodine Form? Re-Evaluation of Benziodoxole Based Hypervalent Iodine Reagents. Chem. Commun. 2016, 52, 5371–5374. [Google Scholar] [CrossRef]
- Zhdankin, V.V. Benziodoxole-Based Hypervalent Iodine Reagents in Organic Synthesis. Curr. Org. Synth. 2005, 2, 121–145. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Mathison, C.J.N.; Montagnon, T. o-Iodoxybenzoic Acid (IBX) as a Viable Reagent in the Manipulation of Ni-trogen- and Sulfur-Containing Substrates: Scope, Generality, and Mechanism of IBX-Mediated Amine Oxidations and Dithiane Deprotections. J. Am. Chem. Soc. 2004, 126, 5192–5201. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Baran, P.S.; Zhong, Y.-L.; Barluenga, S.; Hunt, K.W.; Kranich, R.; Vega, J.A. Iodine(V) Reagents in Organic Synthesis. Part 3. New Routes to Heterocyclic Compounds via o-Iodoxybenzoic Acid-Mediated Cyclizations: Generality, Scope, and Mechanism. J. Am. Chem. Soc. 2002, 124, 2233–2244. [Google Scholar] [CrossRef] [PubMed]
- Zhdankin, V.V.; Stang, P.J. Chemistry of Polyvalent Iodine. Chem. Rev. 2008, 108, 5299–5358. [Google Scholar] [CrossRef]
- Meyer, S.D.; Schreiber, S.L. Accelerationof the Dess-Martin oxidation by water. J. Org. Chem. 1994, 59, 7549–7552. [Google Scholar] [CrossRef]
- Duschek, A.; Kirh, S.F. 2-Iodoxybenzoic Acid—A Simple Oxidant with a Dazzling Array of Potential Applications. Angew. Chem. Int. Ed. 2011, 50, 1524–1552. [Google Scholar] [CrossRef]
- Wirth, T. IBX—New Reactions with an Old Reagent. Angew. Chem. Int. Ed. 2001, 40, 2812–2814. [Google Scholar] [CrossRef]
- Ochiai, M.; Sueda, T.; Miyamoto, K.; Kriprof, P.; Zhdankin, V.V. Trans Influences on Hypervalent Bonding of Aryl λ3-Iodanes: Their Stabilities and Isodesmic Reactions of Benziodoxolones and Benziodazolones. Angew. Chem. Int. Ed. 2006, 45, 8203–8206. [Google Scholar] [CrossRef]
- Zhdankin, V.V.; Kuehl, C.J.; Krasunsky, A.P.; Bolz, J.T.; Simonsen, A.J. 1-(Organosulfonyloxy)-3(1H)-1,2-benziodoxoles: Preparation and Reactions with Alkynyltrimethylsilanes. J. Org. Chem. 1996, 61, 6547–6551. [Google Scholar] [CrossRef]
- Ochiai, M.; Masaki, Y.; Shiro, M. Synthesis and Structure of 1-Alkynyl-1,2-Benziodoxol-3(1H)-Ones. J. Org. Chem. 1991, 56, 5511–5513. [Google Scholar] [CrossRef]
- Zhdankin, V.V.; Krasutsky, A.P.; Kuehl, C.J.; Simonsen, A.J.; Woodward, J.K.; Mismash, B.; Bolz, J.T. Preparation, X-Ray Crystal Structure, and Chemistry of Stable Azidoiodinanes Derivatives of Benziodoxole. J. Am. Chem. Soc. 1996, 118, 5192–5197. [Google Scholar] [CrossRef]
- Zhdankin, V.V.; Kuehl, C.J.; Krasutsky, A.P.; Bolz, J.T.; Mismash, B.; Woodward, J.K.; Simonsen, A.J. 1-Cyano-3-(1H)-1,2-Benziodoxols: Stable Cyanoiodinanes and Efficient Reagents for Direct N-Alkyl Cyanation of N,N-Dialkylarylamines. Tetrahedron Lett. 1995, 36, 7975–7978. [Google Scholar] [CrossRef]
- Daugulis, O.; Do, H.-Q.; Shabashov, D. Palladium- and Copper-Catalyzed Arylation of Carbon-Hydrogen Bonds. Acc. Chem. Res. 2009, 42, 1074–1086. [Google Scholar] [CrossRef] [PubMed]
- Fañanás-Mastral, M. Copper-Catalyzed Arylation with Diaryliodonium Salts. Synthesis 2017, 49, 1905–1930. [Google Scholar] [CrossRef]
- Loro, C.; Oble, J.; Foschi, F.; Papis, M.; Beccalli, E.M.; Giofrè, S.; Poli, G.; Broggini, G. Acid-mediated decarboxylatiove C-H coupling between arenes and O-allyl carbamates. Org. Chem. Front. 2022, 9, 1711–1718. [Google Scholar] [CrossRef]
- Phipps, R.J.; Gaunt, M.J. A Meta-Selective Copper-Catalyzed C-H Bond Arylation. Science 2009, 323, 1593–1597. [Google Scholar] [CrossRef]
- Chen, B.; Hou, X.-L.; Li, Y.-X.; Wu, Y.-D. Mechanistic Understanding of the Unespected Meta Selectivity in Copper-Catalyzed Anilide C-H Bond Arylation. J. Am. Chem. Soc. 2011, 133, 7668–7671. [Google Scholar] [CrossRef]
- Duong, H.A.; Gilligan, R.E.; Cooke, M.L.; Phipps, R.J.; Gaunt, M.J. Copper(II)-Catalyzed meta-Selective Direct Arylation of α-Aryl Carbonyl Compounds. Angew. Chem. Int. Ed. 2011, 50, 463–466. [Google Scholar] [CrossRef]
- Ciana, C.-L.; Phipps, R.J.; Brandt, J.R.; Meyer, F.-M.; Gaunt, M.J. A Highly Para-Selective Copper(II)-Catalyzed Direct Arylation of Aniline and Phenol Derivatives. Angew. Chem. Int. Ed. 2011, 50, 458–462. [Google Scholar] [CrossRef]
- Phipps, R.J.; Grimster, R.P.; Gaunt, M.J. Cu(II)-Catalyzed Direct and Site-Selective Arylation of Indoles Under Mild Conditions. J. Am. Chem. Soc. 2008, 130, 8172–8174. [Google Scholar] [CrossRef] [PubMed]
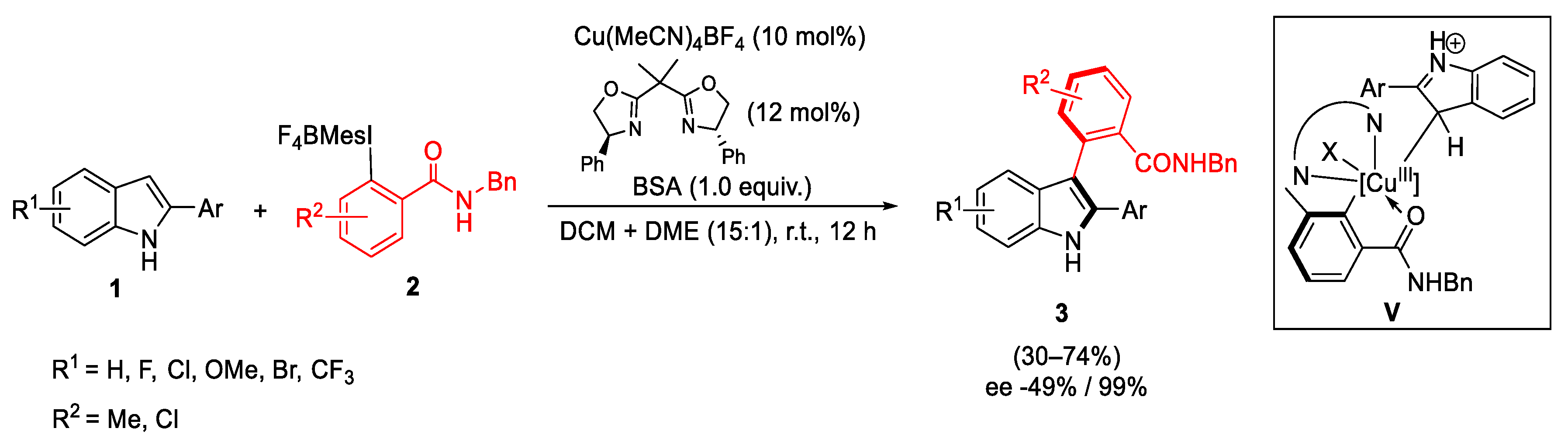
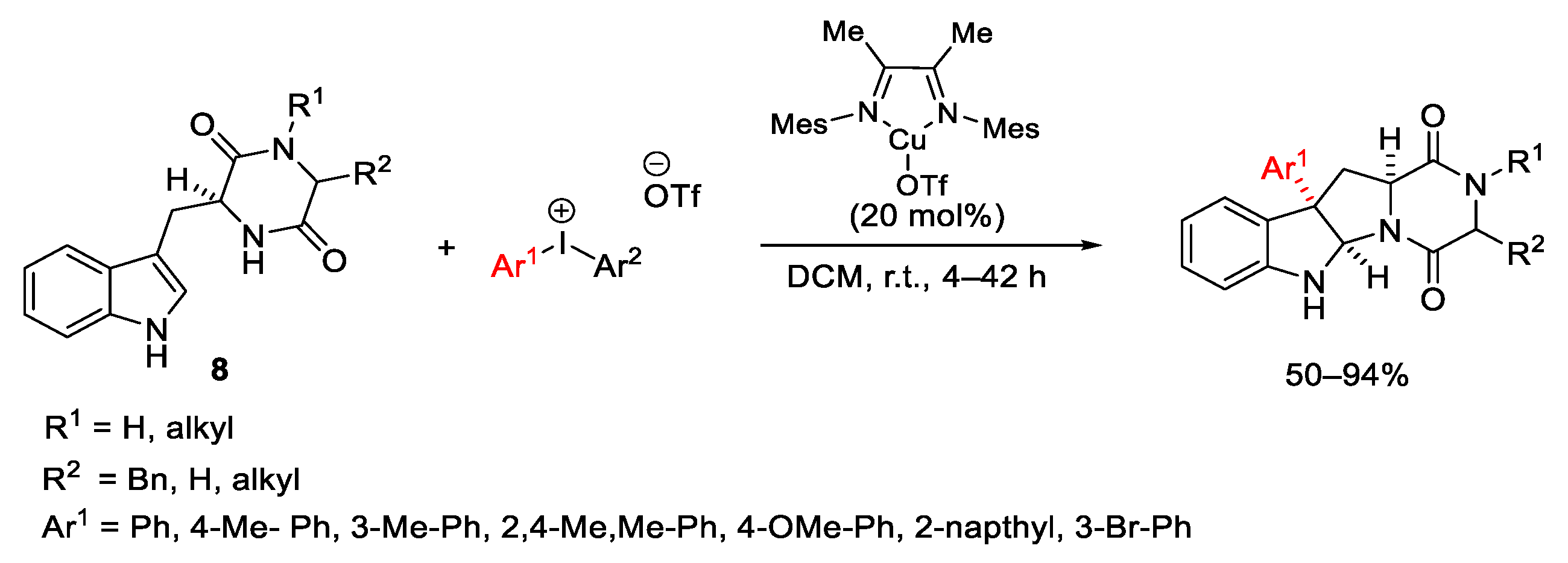
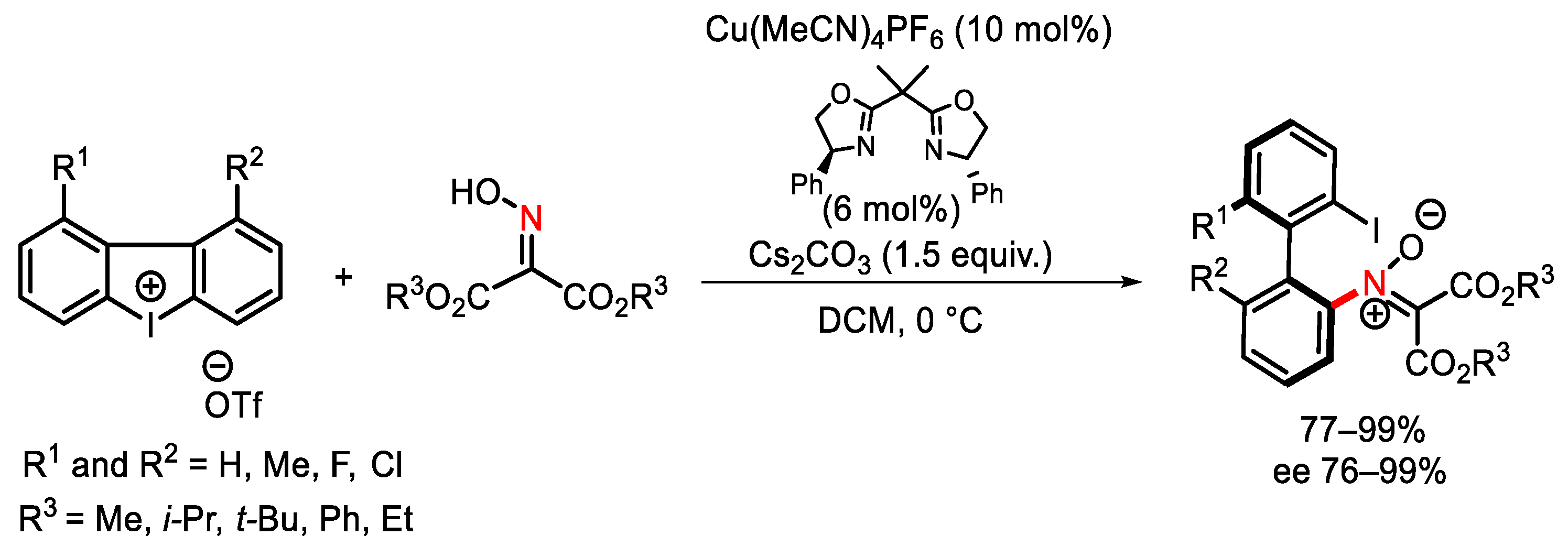
- Liang, H.; Zhu, G.; Pu, X.; Qiu, L. Copper-Catalyzed Enantioselective C-H Arylation between 2-Arylindoles and Hypervalent Iodine Reagents. Org. Lett. 2021, 23, 9246–9250. [Google Scholar] [CrossRef]
- Pitts, A.K.; O’Hara, F.; Snell, R.H.; Gaunt, M.J. A Concise and Scalable Strategy for the Total Synthesis of Dictyodendrin B Based on Sequential C-H- Functionalization. Angew. Chem. Int. Ed. 2015, 54, 5451–5455. [Google Scholar] [CrossRef] [PubMed]
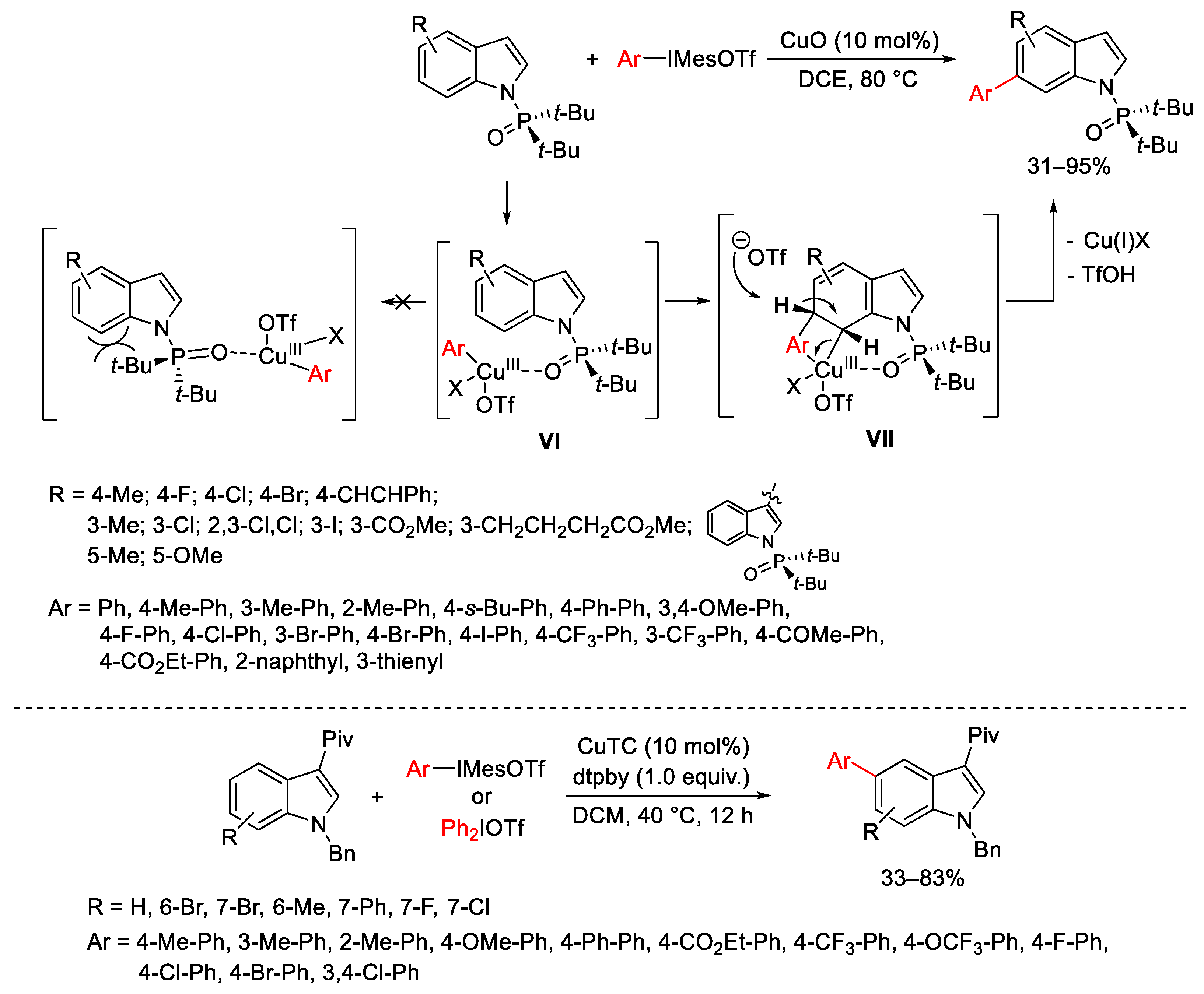
- Yang, Y.; Gao, P.; Zhao, Y.; Shi, Z. Regiocontrolled Direct C-H Arylation of Indoles at the C4 and C5 Positions. Angew. Chem. Int. Ed. 2017, 56, 3966–3971. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, R.; Zhao, Y.; Zhao, D.; Shi, Z. Cu-Catalyzed Direct C6-Arylation of Indoles. J. Am. Chem. Soc. 2016, 138, 8734–8737. [Google Scholar] [CrossRef]
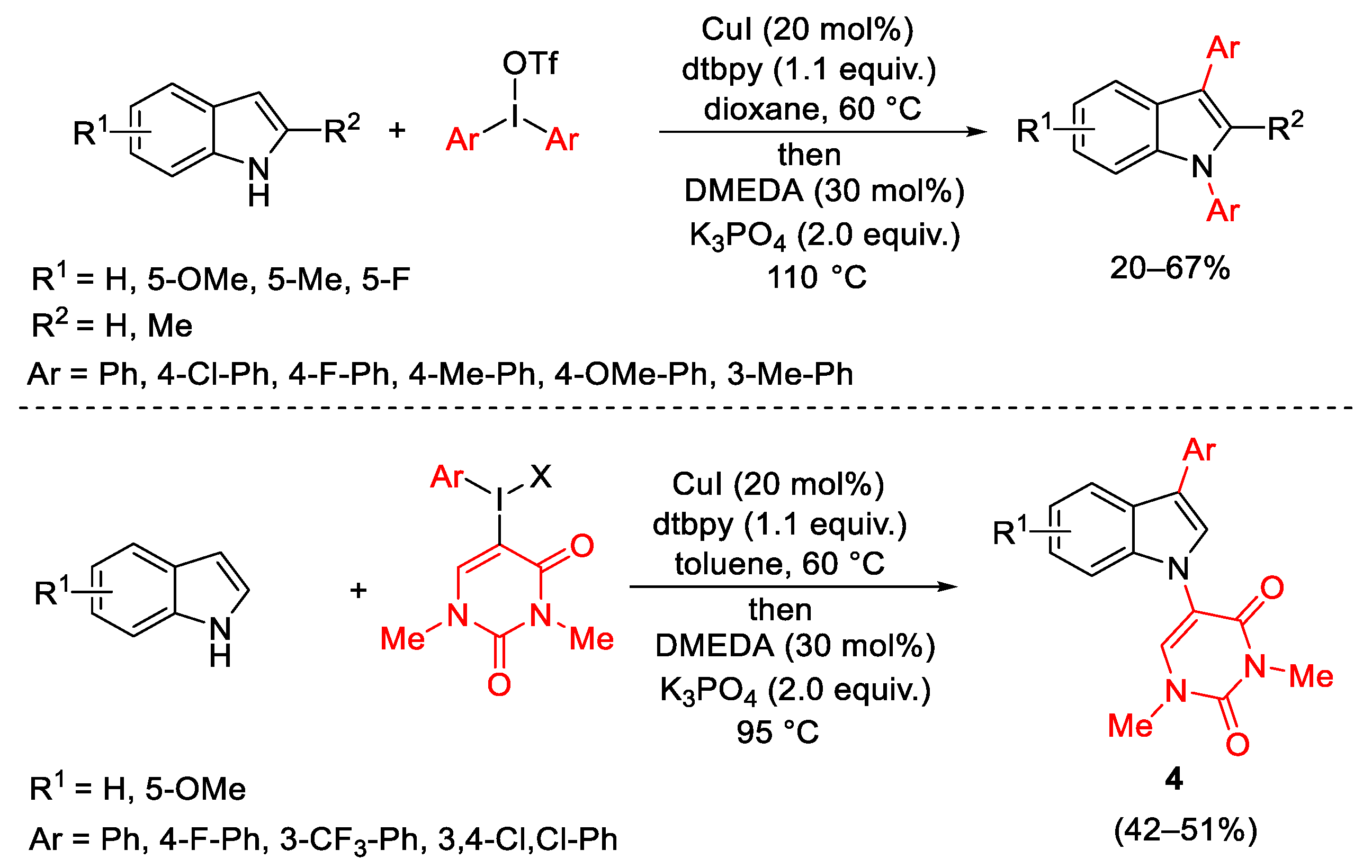
- Modha, S.G.; Greaney, M.F. Atom-Economical Transformation od Diaryliodoniu, Salts: Tandem C-H and N-H Arylation of Indoles. J. Am. Chem. Soc. 2015, 137, 1416–1419. [Google Scholar] [CrossRef]
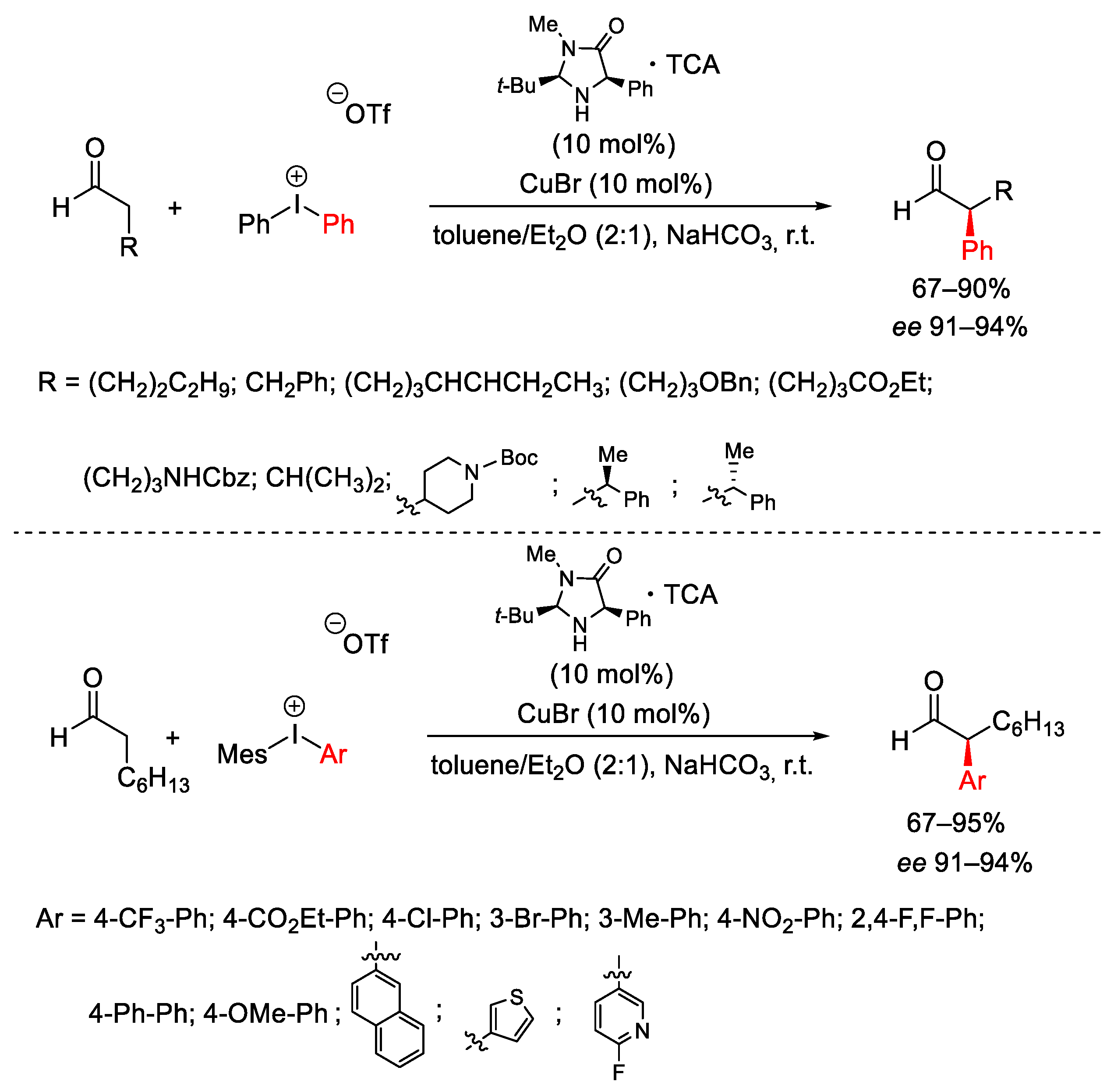
- Allen, A.E.; MacMillan, D.W.C. Enantioselective α-Arylation of Aldehydes via the Productive Merger of Iodonium Salts and Organocatalysis. J. Am. Chem. Soc. 2011, 133, 4260–4263. [Google Scholar] [CrossRef]
- Pan, J.-L.; Chen, T.; Zhang, Z.-Q.; Li, Y.-F.; Zhang, X.-M.; Zhang, F.-M. A Cu-mediated one-pot Michael addition/α-arylation strategy using a diaryliodonium salt: A direct and efficient approach to α-aryl-β-substituted cyclic ketone scaffolds. Chem. Commun. 2016, 52, 2382–2385. [Google Scholar] [CrossRef] [PubMed]
- Bigot, A.; Williamson, A.E.; Gaunt, M.J. Enantioselective α-Arylation of N-Acyloxazolidinones with Copper(II)-bisoxazoline Catalysts and Diaryliodonium Salts. J. Am. Chem. Soc. 2011, 133, 13778–13781. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.S.; Simonovich, S.P.; Jamison, C.R.; MacMillan, D.W.C. Enantioselective α-Arylation od Carbonyls via Cu(I)-Bisoxazoline Catalysis. J. Am. Chem. Soc. 2011, 133, 13782–13785. [Google Scholar] [CrossRef]
- Zhu, S.; MacMillan, D.W.C. Enantioselective Copper-Catalyzed Construction of Aryl Pyrrolindolines via an Aryla-tion-Cyclization Cascade. J. Am. Chem. Soc. 2012, 134, 10815–10818. [Google Scholar] [CrossRef]
- Kieffer, M.E.; Chuang, K.V.; Reisman, S.E. A copper-catalyzed arylation of tryptamines for the direct synthesis of aryl pyr-roloindolines. Chem. Sci. 2012, 3, 3170–3174. [Google Scholar] [CrossRef] [PubMed]
- Kieffer, M.E.; Chuangm, K.V.; Reinsman, S.E. Copper-Catalyzed Diasteroselective Arylation of Tryptophan Derivatives: Total Synthesis of (+)-Naseseazines A and B. J. Am. Chem. Soc. 2013, 135, 5557–5560. [Google Scholar] [CrossRef] [PubMed]
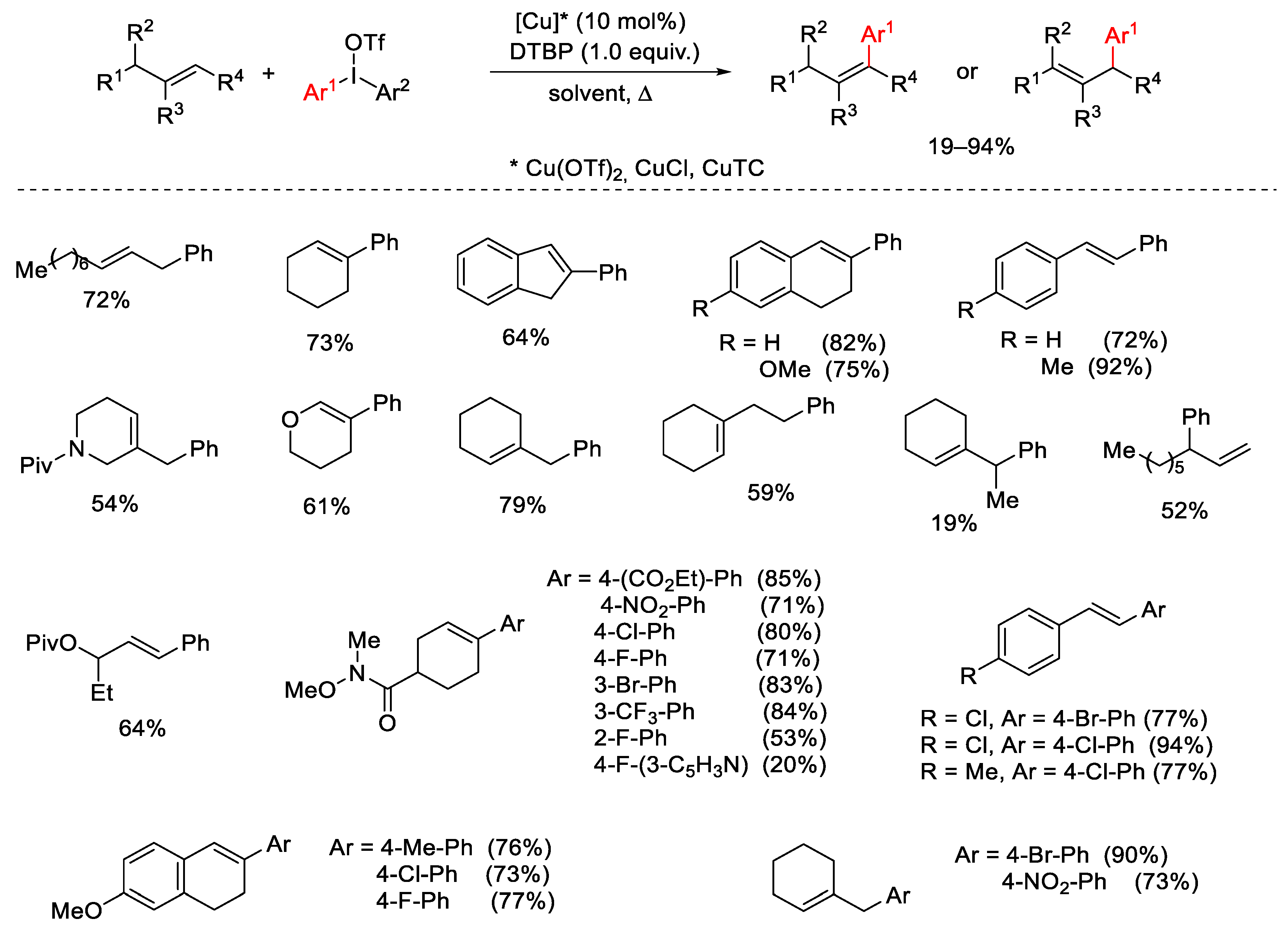
- Phipps, R.J.; McMurray, L.; Ritter, S.; Doung, H.A.; Gaunt, M.J. Copper-Catalyzed Alkene Arylation with Diaryliodonium Salts. J. Am. Chem. Soc. 2012, 134, 10773–10776. [Google Scholar] [CrossRef] [PubMed]
- Almasalma, A.A.; Mejia, E. 1-Phenyl-1,2-benziodoxol-3-(1H)-one as Synthon for Phthalide Synthesis through Pd-Free, Base-Free, Sonoganshira-Type Couple Cyclization Reaction. Eur. J. Org. Chem. 2018, 2018, 188–195. [Google Scholar] [CrossRef]
- Xu, Z.-F.; Cai, C.-X.; Liu, J.-T. Copper-Catalyzed Regioselective Reaction of Internal Alkenes and Diaryliodonium Salts. Org. Lett. 2013, 15, 2096–2099. [Google Scholar] [CrossRef]
- Walkinshaw, A.J.; Xu, W.; Suero, M.G.; Gaunt, M.J. Copper-Catalyzed Carboarylation of Alkynes via Vinyl Cations. J. Am. Chem. Soc. 2013, 135, 12532–12535. [Google Scholar] [CrossRef]
- Peng, J.; Chen, C.; Chen, J.; Su, X.; Xi, C.; Chen, H. Cu-Catalyzed Arylcarbocyclization of Alkynes with Diaryliodonium Salts through C-C Bond Formation on Inert C(sp3)-H Bond. Org. Lett. 2014, 16, 3776–3779. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Das, S.; Walkingshaw, A.J.; Casitas, A.; Taylor, M.; Suero, M.G.; Gaunt, M.J. Cu-Catalyzed Cascades to Carbocyles: Union of Diaryliodonium Salts with Alkenes or Alkynes Exploiting Remote Carbocations. J. Am. Chem. Soc. 2014, 136, 8851–8854. [Google Scholar] [CrossRef]
- Chen, J.; Chen, C.; Chen, J.; Wang, G.; Qu, H. Cu-catalyzed intramolecular aryl-etherification reactions of alkoxyl alkynes with diaryliodonium salts via cleavage of a stable C–O bond. Chem. Commun. 2015, 51, 1356–1359. [Google Scholar] [CrossRef]
- Sinai, A.; Vangel, D.; Gati, T.; Bombincz, P.; Novak, Z. Utilization of Copper-Catalyzed Carboarylation-Ring Closure for the Synthesis of New Oxazoline Derivatives. Org. Lett. 2015, 17, 4136–4139. [Google Scholar] [CrossRef]
- Sinai, A.; Meszaros, A.; Gati, T.; Kudar, V.; Pallo, A.; Novak, Z. Copper-Catalyzed Oxidative Ring Closure and Carboarylation of 2-Ethynylanilides. Org. Lett. 2013, 15, 5654–5657. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Collins, B.S.L.; Suero, M.G.; Gaunt, M.J. Copper-Catalyzed Arylative Meyer-Schuster Rearrangement of Propargylic Alcohols to Complex Enones Using Diaryliodonium Salts. Angew. Chem. Int. Ed. 2013, 52, 5799–5802. [Google Scholar] [CrossRef]
- Cahard, E.; Bremeyer, N.; Gaunt, M.J. Copper-Catalyzed Intramolecular Electrophilic Carbofunctionalization of Allylic Amides. Angew. Chem. Int. Ed. 2013, 52, 9284–9288. [Google Scholar] [CrossRef] [PubMed]
- Cahard, E.; Male, H.P.J.; Tissot, M.; Gaunt, M.J. Enantioselective and Regiodivergent Copper-Catalyzed Electrophilic Arylation of Allylic Amides with Diaryliodonium Salts. J. Am. Chem. Soc. 2015, 137, 7986–7989. [Google Scholar] [CrossRef] [PubMed]
- Gigant, N.; Chausset-Boissarie, L.; Belhomme, M.-C.; Poisson, T.; Pannecoucke, X.; Gillaizeau, I. Copper-Catalyzed Direct Ar-ylation of Cyclic Enamides Using Diaryliodonium Salts. Org. Lett. 2013, 15, 278–281. [Google Scholar] [CrossRef] [PubMed]
- Prakash, M.; Muthusamy, S.; Kesavan, V. Copper(I) Bromide Catalyzed Arylation of Cyclic Enamides and Naph-thyl-1-acetamides Using Diaryliodonium Salts. J. Org. Chem. 2014, 79, 7836–7843. [Google Scholar] [CrossRef]
- Kumar, D.; Pilania, M.; Arun, V.; Pooniya, S. C-H arylation of azaheterocycles: A direct ligand-free and Cu-catalyzed approach using diaryliodonium salts. Org. Biomol. Chem. 2014, 12, 6340–6344. [Google Scholar] [CrossRef]
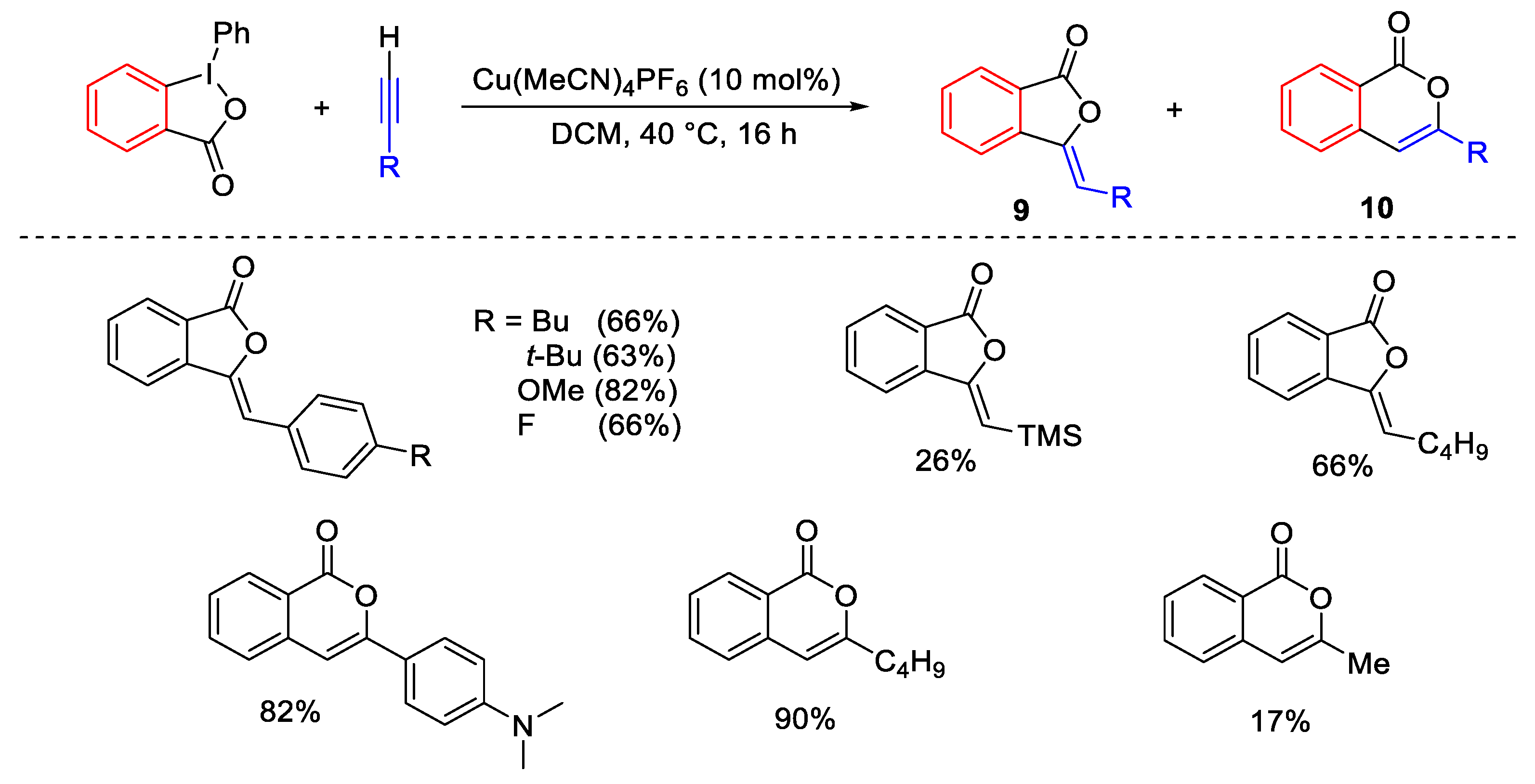
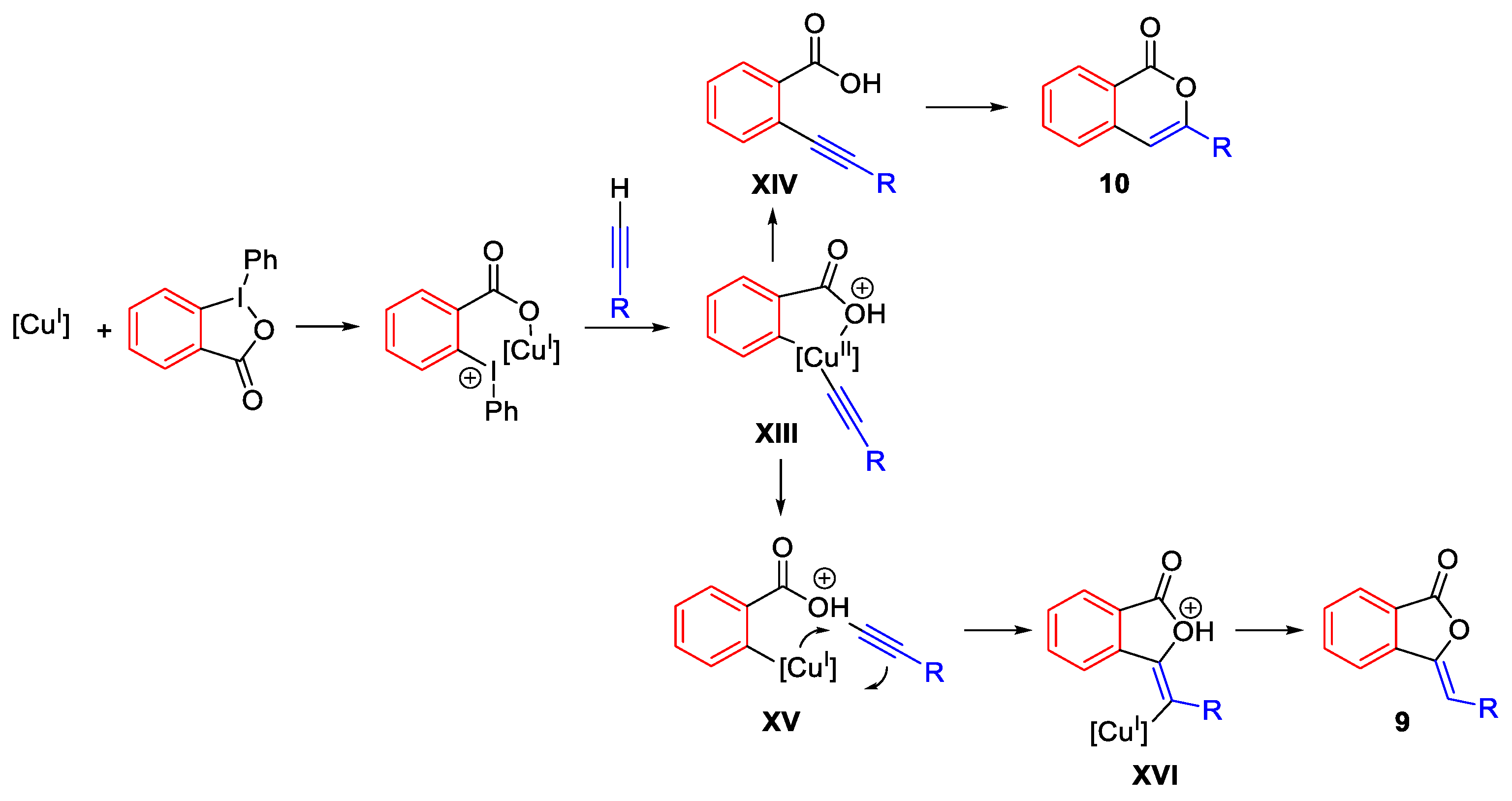
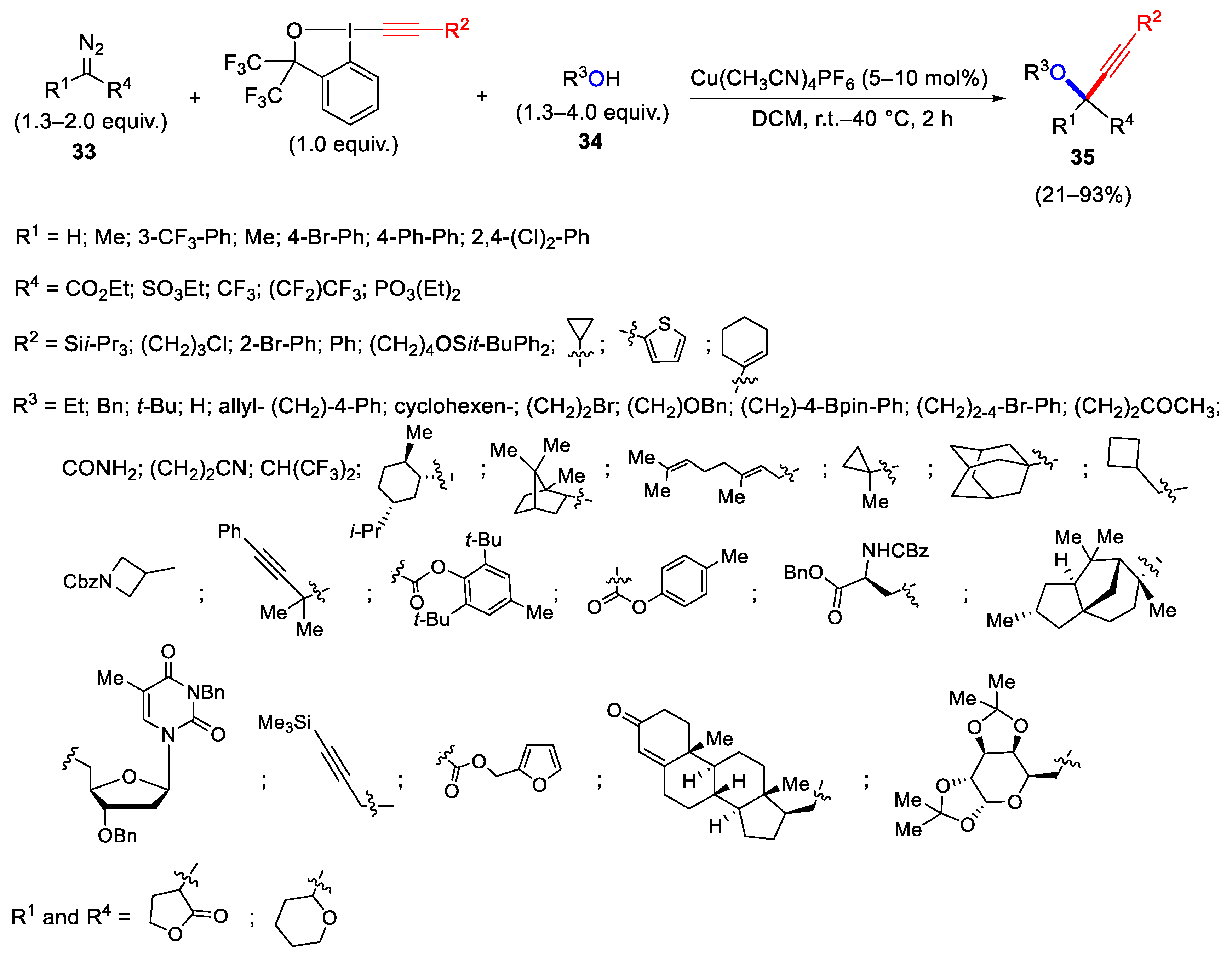
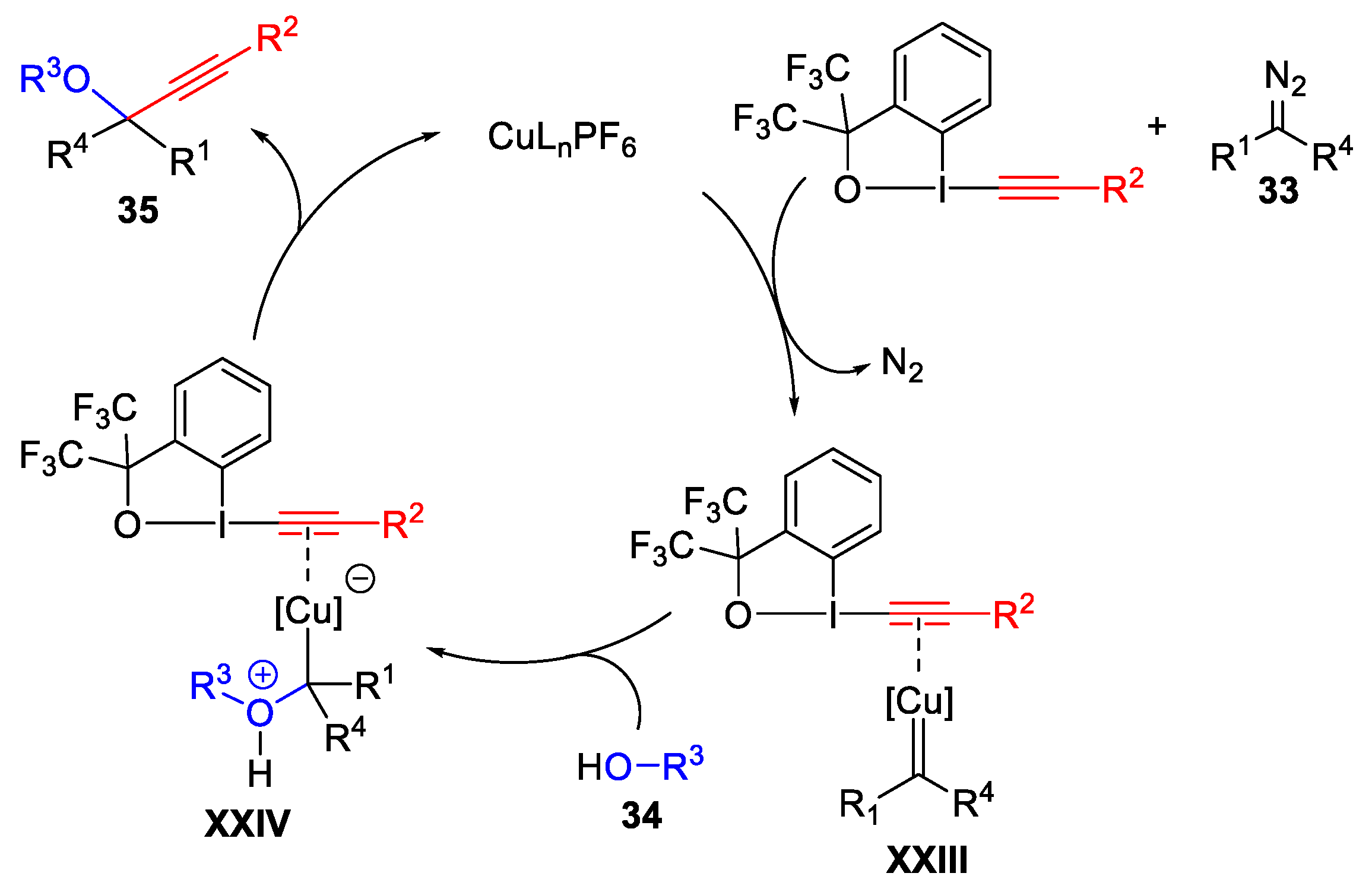
- Hari, D.P.; Waser, J. Copper-Catalyzed Oxy-Alkynylation of Diazo Compounds with Hypervalent Iodine Reagents. J. Am. Chem. Soc. 2016, 138, 2190–2193. [Google Scholar] [CrossRef]
- Hari, D.P.; Schouwey, L.; Baber, V.; Scopelliti, R.; Fadaei-Tirani, F.; Waser, J. Ethynylbenziodazolones (EBZ) as Electrophilic Alkynylation Reagents for the Highly Enantioselective Copper-Catalyzed Oxyalkynylation of Diazo Compounds. Chem. Eur. J. 2019, 25, 9522–9528. [Google Scholar] [CrossRef]
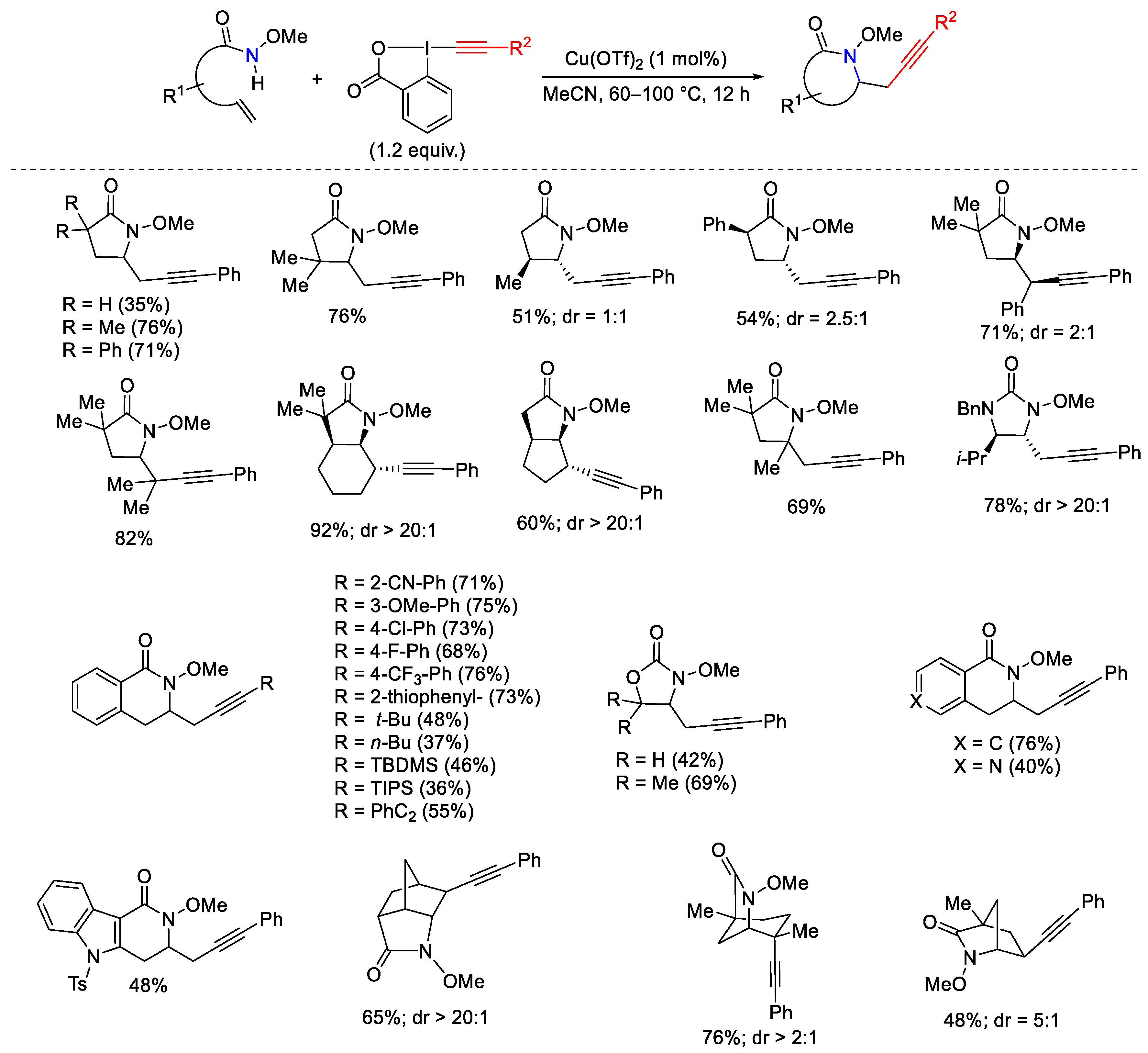
- Pisella, G.; Gagnebin, A.; Waser, J. Three-Component Reaction for the Synthesis of Highly Functionalized Propargyl Ethers. Chem. Eur. J. 2020, 26, 10199–10204. [Google Scholar] [CrossRef]
- Shen, K.; Wang, Q. Copper-catalyzed aminoalkynylation of alkenes with hypervalent iodine reagents. Chem. Sci. 2017, 8, 8265–8270. [Google Scholar] [CrossRef]
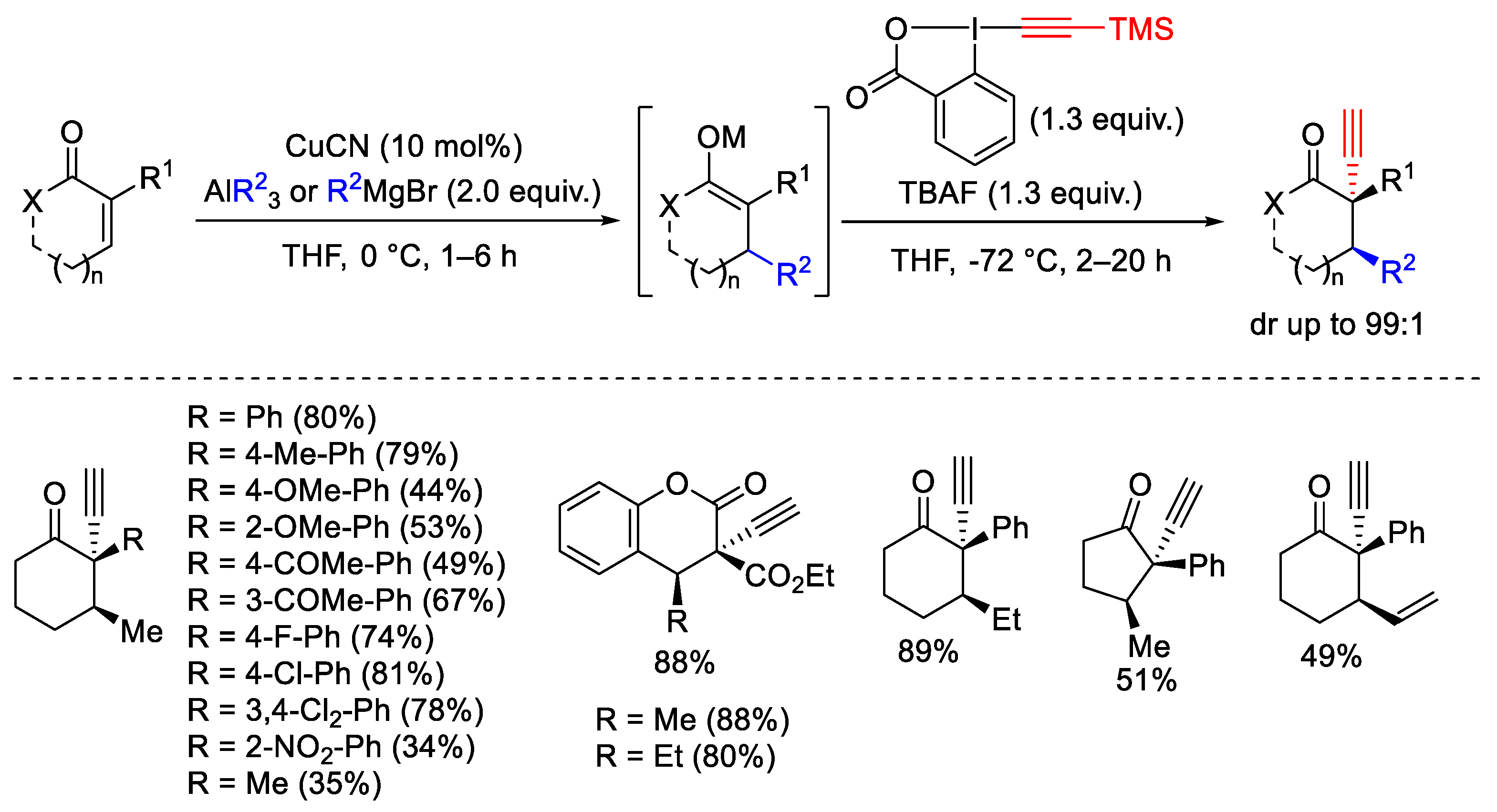
- Teodoro, B.V.M.; Silva, L.F.J. Sequential Michael Addition/Electrophilic Alkynylation: Synthesis of α-Alkynyl-β-Substituted Ketones and Chromanones. J. Org. Chem. 2018, 83, 13604–13611. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Guo, X.-Q.; Chen, L.-M.; Kang, T.-R. Synthesis, Characterization of Spirocyclic λ3-Iodanes and Their Application to Prepare 4,1-Benzoxazepine-2,5-diones and 1,3-Diynes. Chem. Eur. J. 2021, 27, 4312–4316. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ye, Y.; Zhang, S.; Feng, J.; Xu, Y.; Zhang, Y.; Wang, J. Copper-Catalyzed C(sp3)-C(sp3) Bond Formation Using a Hypervalent Iodine Reagent: An Efficient Allylic Trifluoromethylation. J. Am. Chem. Soc. 2011, 133, 16410–16413. [Google Scholar] [CrossRef] [PubMed]
- Parsons, A.T.; Buchwald, S.L. Copper-Catalyzed Trifluoromethylation of Unactivated Olefins. Angew. Chem. Int. Ed. 2011, 50, 9120–9123. [Google Scholar] [CrossRef]
- Lonca, G.H.; Ong, D.Y.; Tran, T.M.H.; Tejo, C.; Chiba, S.; Gagosz, F. Anti-Markovinkov Hydrofunctionalization of Alkenes: Usa of a Benzyl Group as a Traceless Redox-Active Hydrogen Donor. Angew. Chem. Int. Ed. 2017, 56, 11440–11444. [Google Scholar] [CrossRef]
- Shimizu, R.; Egami, H.; Hamashima, Y.; Sodeoka, M. Copper-Catalyzed Trifluoromethylation of Allylsilanes. Angew. Chem. Int. Ed. 2012, 51, 4577–4580. [Google Scholar] [CrossRef]
- Wang, X.-P.; Lin, J.-H.; Zhang, C.-P.; Xiao, J.-C.; Zheng, X. Copper-catalyzed trifluoromethylation of alkenes with an electrophilic trifluoromethylating reagent. Beilstein J. Org. Chem. 2013, 9, 2635–2640. [Google Scholar] [CrossRef]
- Weng, Z.; Li, H.; He, W.; Yao, L.-F.; Tan, J.; Chen, J.; Yuan, Y.; Huan, K.-W. Mild copper-catalyzed trifluoromethylation of terminal alkynes using an electrophilic trifluoromethylating reagent. Tetrahedron 2012, 68, 2527–2531. [Google Scholar] [CrossRef]
- Feng, C.; Loh, T.-P. Copper-catalyzed olefinic trifluoromethylation of enamides at room temperature. Chem. Sci. 2012, 3, 3458–3462. [Google Scholar] [CrossRef]
- Feng, C.; Loh, T.-P. Directing-Group-Assisted Copper-Catalyzed Olefinic Trifluoromethylation of Electron-Deficient Alkenes. Angew. Chem. Int. Ed. 2013, 52, 12414–12417. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Ning, Y.; Mi, P.; Liao, P.; Bi, X. Catalytic C-H α-Trifluoromethylation of α,β-Unsaturated Carbonyl Compounds. Org. Lett. 2014, 16, 1522–1525. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ye, Y.; Ji, G.; Xu, Y.; Zhang, S.; Feng, J.; Zhang, Y.; Wang, J. Copper-Catalyzed Direct C-H Trifluoromethylation of Quinones. Org. Lett. 2013, 15, 3730–3733. [Google Scholar] [CrossRef]
- Deng, Q.-H.; Wadepohl, H.; Gade, L.H. Highly Enantioselective Copper-Catalyzed Electrophilic Trifluoromethylation of b-Ketoesters. J. Am. Chem. Soc. 2012, 134, 10769–10772. [Google Scholar] [CrossRef]
- Allen, A.E.; MacMillan, D.W.C. The Productive Merger of Iodonium Salts and Organocatalysis: A Non-photolytic Approach to the Enantioselective α-Trifluoromethylation of Aldehydes. J. Am. Chem. Soc. 2010, 132, 4986–4987. [Google Scholar] [CrossRef] [PubMed]
- Pair, E.; Monteiro, N.; Bouyssi, D.; Baudoin, O. Copper-Catalyzed Trifluoromethylation of N,N-Dialkylhydrazones. Angew. Chem. Int. Ed. 2013, 52, 5346–5349. [Google Scholar] [CrossRef]
- Prieto, A.; Jearmet, E.; Monteiro, N.; Bouyssi, D.; Baudoin, O. Copper-Catalyzed b-Trifluoromethylation of Conjugated Hydrazones. Org. Lett. 2014, 16, 4770–4773. [Google Scholar] [CrossRef]
- Prieto, A.; Bouyssi, D.; Monteiro, N. Copper-Catalyzed Trifluoromethylation od Hydrazones Leading to the Formation of Quaternary α-Trifluoromethyl Diazenes. Asian J. Org. Chem. 2016, 5, 742–745. [Google Scholar] [CrossRef]
- Wu, S.; Li, J.; He, R.; Jia, K.; Chen, Y. Terminal Trifluoromethylation of Ketones via Selective C-C Cleavage of Cycloalkanols Enabled by Hypervalent Iodine Reagents. Org. Lett. 2021, 23, 9204–9209. [Google Scholar] [CrossRef]
- Xiong, Y.-P.; Wu, M.-Y.; Zhang, X.-Y.; Ma, C.-L.; Huang, L.; Zhao, L.-J.; Tan, B.; Liu, X.-Y. Direct Access to α-Trifluoromethyl Enones via Efficient Copper-Catalyzed Trifluoromethylation of Meyer-Schuster Rearrangement. Org. Lett. 2014, 16, 1000–1003. [Google Scholar] [CrossRef]
- Cai, S.; Chen, C.; Sun, Z.; Xi, C. CuCl-catalyzed ortho trifluoromethylation of arenes and heteroarenes with a pivalamido directing group. Chem. Commun. 2013, 49, 4552–4554. [Google Scholar] [CrossRef]
- Liu, T.; Shen, Q. Copper-Catalyzed Trifluoromethylation of Aryl and Vinyl Boronic Acids with An Electrophilic Trifluoro-methylating Reagent. Org. Lett. 2011, 13, 2342–2345. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Fang, X.; Lin, X.; Li, H.; He, W.; Huang, K.-W.; Yuan, Y.; Weng, Z. Room-temperature base-free copper-catalyzed trifluoromethylation of organotrifluoroborates to trifluoromethylarenes. Tetrahedron 2012, 68, 9949–9953. [Google Scholar] [CrossRef]
- Zheng, H.; Huang, Y.; Wang, Z.; Li, H.; Huang, K.-W.; Yuan, Y.; Weng, Z. Synthesis of trifluoromethylated acetylenes via copper-catalyzed trifluoromethylation of alkynyltrifluoroborates. Tetrahedron Lett. 2012, 53, 6646–6649. [Google Scholar] [CrossRef]
- Shimizu, R.; Egami, H.; Nagi, T.; Chae, J.; Hamashima, Y.; Sodeoka, M. Direct C2-trifluoromethylation of indole derivatives catalyzed by copper acetate. Tetrahedron Lett. 2010, 51, 5947–5949. [Google Scholar] [CrossRef]
- Janson, P.G.; Ghoneim, I.; Ilchenko, N.O.; Szabó, K.J. Electrophilic Trifluoromethylation by Copper-Catalyzed Addition of CF3-Transfer Reagents to Alkenes and Alkynes. Org. Lett. 2012, 14, 2882–2885. [Google Scholar] [CrossRef]
- Egami, H.; Shimizu, R.; Sodeoka, M. Oxytrifluoromethylation of multiple bonds using copper catalyst under mild conditions. Tetrahedron Lett. 2012, 53, 5503–5506. [Google Scholar] [CrossRef]
- Ramkumar, N.; Baumane, L.; Zacs, D.; Veliks, J. Merging Copper(I) Photoredox Catalysis and Iodine(III) Chemistry for the Oxy-monofluoromethylation of Alkenes. Angew. Chem. Int. Ed. 2023, 62, e202219027. [Google Scholar] [CrossRef]
- Wang, F.; Qi, X.; Liang, Z.; Chen, P.; Liu, G. Copper-Catalyzed Intermolecular Trifluoromethylazidation of Alkenes: Convenient Access to CF3-Containing Alkyl Azides. Angew. Chem. Int. Ed. 2014, 53, 1881–1886. [Google Scholar] [CrossRef]
- Ilchenko, N.O.; Janson, P.G.; Szabo, K.J. Copper-Mediated Cyanotrifluoromethylation of Styrenes Using the Togni Reagent. J. Org. Chem. 2013, 78, 11087–11091. [Google Scholar] [CrossRef]
- Egami, H.; Kawamura, S.; Miyazaki, A.; Sodeaoka, M. Trifluoromethylation Reactions for the Synthesis of β-Trifluoromethylamines. Angew. Chem. Int. Ed. 2013, 125, 7995–7998. [Google Scholar] [CrossRef]
- Kawamura, S.; Egami, H.; Sodeoka, M. Aminotrifluoromethylation of Olefins via Cyclic Amine Formation: Mechanistic Study and Application to Synthesis of Trifluoromethylated Pyrrolidines. J. Am. Chem. Soc. 2015, 137, 4865–4873. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.; Wang, Q. Copper-catalyzed aminotrifluoromethylayion of alkenes: A facile synthesis of CF3-containing lactams. Org. Chem. Front. 2016, 3, 222–226. [Google Scholar] [CrossRef]
- Zhu, R.; Buchwald, S.L. Copper-Catalyzed Oxytrifluoromethylation of Unactivated Alkenes. J. Am. Chem. Soc. 2012, 134, 12462–12465. [Google Scholar] [CrossRef]
- Zhu, R.; Buchwald, S.L. Enantioselective Functionalization of Radical Intermediates in Redox Catalysis: Copper-Catalyzed Asymmetric Oxytrifluoromethylation of Alkenes. Angew. Chem. Int. Ed. 2013, 52, 12655–12658. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.-F.; Zhu, C.-L.; Xu, H. Copper(I)-catalyzed diastereoselective hydroxytrifluoromethylation of dienes accelerated by phosphine ligands. Chem. Sci. 2013, 4, 2478–2482. [Google Scholar] [CrossRef]
- Yu, Q.; Ma, S. Copper-Catalyzed Cyclic Oxytrifluoromethylation of 2,3-Allenoic Acids to Trifluoromethylated Butenolides. Chem. Eur. J. 2013, 19, 13304–13308. [Google Scholar] [CrossRef]
- He, Y.-T.; Li, L.-H.; Yang, Y.-F.; Wang, Y.-Q.; Luo, J.-Y.; Liu, X.-Y.; Liang, Y.-M. Copper-catalyzed synthesis od trifluoromethyl-substituted isoxazolines. Chem. Commun. 2013, 49, 5687–5689. [Google Scholar] [CrossRef]
- Gao, P.; Yan, X.-B.; Tao, T.; Yang, F.; He, T.; Song, Z.-R.; Liu, X.-L.; Liang, Y.-M. Copper-Catalyzed Trifluoromethylation-Cyclization of Enynes: Highly Regioselective Construction of Trifluoromethylated Carbocycles and Heterocycles. Chem. Eur. J. 2013, 19, 14420–14424. [Google Scholar] [CrossRef]
- Egami, H.; Shimizu, R.; Kawamura, S.; Sodeoka, M. Alkene Trifluoromethylation Coupled with C-C Bond Formation: Con-struction of Trifluoromethylated Carbocycles and Heterocycles. Angew. Chem. Int. Ed. 2013, 52, 4000–4003. [Google Scholar] [CrossRef]
- Lin, J.-S.; Xiong, Y.-P.; Ma, C.-L.; Zhao, L.-J.; Tan, B.; Liu, X.-Y. Efficient Copper-Catalyzed Direct Intramolecular Aminotrifluoromethylation of Unactivated Alkenes with Diverse Nitrogen-Based Nucleophiles. Chem. Eur. J. 2014, 20, 1332–1340. [Google Scholar] [CrossRef]
- Zhou, T.; Chen, Z.-C. Hypervalent iodine in synthesis. 77. An efficient method for the synthesis of N-arylindoles by the cop-per-catalyzed N-arylation of indole with diaryliodonium salts. Synth. Commun. 2002, 32, 903–907. [Google Scholar] [CrossRef]
- Zhou, T.; Chen, Z.-C. Hypervalent Iodine in Synthesis 85: An Efficient Method for the Synthesis of N-Arylbenzimidazoles by the Copper- Catalyzed N-Arylation of Benzimidazole with Diaryliodonium Salts. Heteroat. Chem. 2002, 13, 617–619. [Google Scholar] [CrossRef]
- Zhou, T.; Chen, Z.C. Hypervalent iodine in synthesis 87: The synthesis of 2,5-diaryl-2H-tetrazoles. J. Chem. Res. 2004, 2004, 404–405. [Google Scholar] [CrossRef]
- Zhou, T.; Li, T.C.; Chen, Z.-C. Hypervalent Iodine in Synthesis. Part 86. Helv. Chim. Acta 2005, 88, 290–296. [Google Scholar] [CrossRef]
- Saikia, R.A.; Barman, D.; Dutta, A.; Thakur, A.J. N1- and N3-Arylations of Hydantoins Employing Diaryliodonium Salts via Copper(I) Catalysis at Room Temperature. Eur. J. Org. Chem. 2021, 2021, 400–410. [Google Scholar] [CrossRef]
- Geng, X.; Mao, S.; Chen, L.; Yu, J.; Han, J.; Hua, J.; Wang, L. Copper-catalyzed direct N-arylsulfonamides using diaryliodonium salts in water. Tetrahedron Lett. 2014, 55, 3856–3859. [Google Scholar] [CrossRef]
- Vaddula, B.; Leazer, J.; Varma, R.S. Copper-Catalyzed Ultrasound-Expedited N-Arylation of Sulfoximines using Diaryliodonium Salts. Adv. Synth. Catal. 2012, 354, 986–990. [Google Scholar] [CrossRef]
- Watanabe, K.; Moriyama, K. Copper-Catalyzed Indole-Selective C–N Coupling Reaction of Indolyl(2-alkoxy-phenyl)iodonium Imides: Effect of Substituent on Iodoarene as Dummy Ligand. J. Org. Chem. 2018, 83, 14827–14833. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Moriyama, K. Cu-Catalyzed Oxidative 3-Amination of Indoles via Formation of Indolyl(aryl)iodonium Imides Using o-Substituted (Diacetoxyiodo)arene as a High-Performance Hypervalent Iodine Compound. Molecules 2019, 24, 1147. [Google Scholar] [CrossRef]
- Sokolovs, I.; Lubriks, D.; Suna, E. Copper-Catalyzed Intermolecular C–H Amination of (Hetero)arenes via Transient Unsymmetrical λ3-Iodanes. J. Am. Chem. Soc. 2014, 136, 6920–6928. [Google Scholar] [CrossRef]
- Berzina, B.; Sokolovs, I.; Suna, E. Copper-Catalyzed para-Selective C–H Amination of Electron-Rich Arenes. ACS Catal. 2015, 5, 7008–7014. [Google Scholar] [CrossRef]
- Wu, Y.; Izquierdo, S.; Vidossich, P.; Lledýs, A.; Shafir, A. NH-Heterocyclic Aryliodonium Salts and their Selective Conversion into N1-Aryl-5-iodoimidazoles. Angew. Chem. Int. Ed. 2016, 55, 7152–7156. [Google Scholar] [CrossRef]
- Lv, T.; Wang, Z.; You, J.; Lan, J.; Gao, G. Copper-Catalyzed Direct Aryl Quaternization of N-Substituted Imidazoles to Form Imidazolium Salts. J. Org. Chem. 2013, 78, 5723–5730. [Google Scholar] [CrossRef] [PubMed]
- Reus, C.; Stolar, M.; Vanderkley, J.; Nebauer, J.; Baumgartner, T. A Convenient N-Arylation Route for Electron-Deficient Pyridines: The Case of π-Extended Electrochromic Phosphaviologens. J. Am. Chem. Soc. 2015, 137, 11710–11717. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Cheng, G.; Zhang, H.; Xu, X.; Gao, J.; Cui, X. Copper-Catalyzed One-Pot Synthesis of Unsymmetrical Arylurea Derivatives via Tandem Reaction of Diaryliodonium Salts with N-Arylcyanamide. J. Org. Chem. 2014, 79, 8156–8162. [Google Scholar] [CrossRef]
- Peng, X.; Sun, Z.; Kuang, P.; Li, L.; Chen, J.; Chemn, J. Copper-Catalyzed Selective Arylation of Nitriles with Cyclic Diaryl Iodonium Salts: Direct Access to Structurally Diversified Diarylmethane Amides with Potential Neuroprotective and Anticancer Activities. Org. Lett. 2020, 22, 5789–5795. [Google Scholar] [CrossRef]
- Aradia, K.; Novak, Z. Copper-Catalyzed Oxidative Ring Closure of ortho-Cyanoanilides with Hypervalent Iodonium Salts: Arylation–Ring Closure Approach to Iminobenzoxazines. Adv. Synth. Catal. 2015, 357, 371–376. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, C.; Peng, J.; Li, J. Copper(II)-Catalyzed Three-Component Cascade Annulation of Diaryliodoniums, Nitriles, and Alkynes: A Regioselective Synthesis of Multiply Substituted Quinolines. Angew. Chem. Int. Ed. 2013, 52, 5323–5327. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, J.; Lan, T.; Cheng, S.; Liu, W.; Chen, C. Preparation, Characterization, and Reactivity of Aliphatic Amino Iodane(III) Reagents. Eur. J. Org. Chem. 2021, 2021, 436–442. [Google Scholar] [CrossRef]
- Tianlei, L.; Yue, Z.; Wei, L.; Chanjuan, X.; Chao, C. Carbazolation Study of Active Arenes with Carbazole-Containing Hypervalent Iodine(III) Reagents. Chin. J. Org. Chem. 2019, 39, 2166–2174. [Google Scholar] [CrossRef]
- Zhu, D.; Liu, Q.; Luo, B.; Chen, M.; Pi, R.; Huang, P.; Wen, S. Synthesis of Carbazoles via One-Pot Copper-Catalyzed Amine Insertion into Cyclic Diphenyleneiodoniums as a Strategy to Generate a Drug-Like Chemical Library. Adv. Synth. Catal. 2013, 355, 2172–2178. [Google Scholar] [CrossRef]
- Xu, S.; Zhao, K.; Gu, Z. Copper-Catalyzed Asymmetric Ring-Opening of Cyclic Diaryliodonium with Benzylic and Aliphatic Amines. Adv. Synth. Catal. 2018, 360, 3877–3883. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, K.; Li, N.; Yu, J.; Gong, L.-Z.; Gu, Z. Atroposelective Ring Opening of Cyclic Diaryliodonium Salts with Bulky Anilines Controlled by a Chiral Cobalt(III) Anion. Angew. Chem. Int. Ed. 2020, 59, 19899–19904. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhang, M.; Zhan, S.; Gu, Z. Copper-Catalyzed Enantioselective Ring-Opening of Cyclic Diaryliodoniums and O-Alkylhydroxylamines. Org. Lett. 2019, 21, 6374–6377. [Google Scholar] [CrossRef]
- Chao, Z.; Ma, M.; Gu, Z. Cu-Catalyzed Site-Selective and Enantioselective Ring Opening of Cyclic Diaryliodoniums with 1,2,3-Triazoles. Org. Lett. 2020, 22, 6441–6446. [Google Scholar] [CrossRef]
- Guo, W.; Gu, J.; Gu, Z. Catalytic Asymmteric Synthesis of Atropisomeric Nitrones: Versatile Intermediate for Axially Chiral Biaryls. Org. Lett. 2020, 22, 7622–7628. [Google Scholar] [CrossRef]
- Hu, Y.; Zheng, S.; Fan, W.; Yuana, W. Copper-Catalysed Electrophilic Amination of Aryl(alkenyl) Boronic Acids with Nitrogen-Containing Hypervalent Iodine (III) Reagent. Adv. Synth. Catal. 2021, 363, 4701–4707. [Google Scholar] [CrossRef]
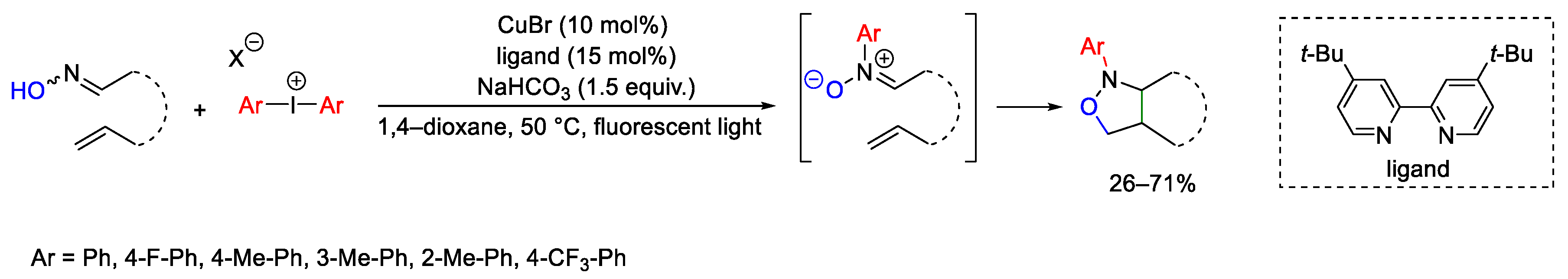
- Tanaka, K.; Yoshida, M.; Yamamoto, A.; Hashimoto, Y.; Morita, N.; Tamura, O. Synthesis of N-Aryl Isoxazolidines by Photo-Promoted N-Selective Arylation of Oximes and Cyclization Using Hypervalent Iodine Reagents and Copper Catalyst. Adv. Synth. Catal. 2023, 365, 1419–1424. [Google Scholar] [CrossRef]
- Sivaguru, P.; Ning, Y.; Bi, X. New Strategies for the Synthesis of Aliphatic Azides. Chem. Rev. 2021, 121, 4253–4307. [Google Scholar] [CrossRef]
- Sala, R.; Loro, C.; Foschi, F.; Broggini, G. Transition Metal Catalyzed Azidation Reactions. Catalysts 2020, 10, 1173. [Google Scholar] [CrossRef]
- Foschi, F.; Loro, C.; Sala, R.; Oble, J.; Lo Presti, L.; Beccalli, E.M.; Poli, G.; Broggini, G. Intramolecular Aminoazidation of Unactivated Terminal Alkenes by Palladium-Catalyzed Reactions with Hydrogen Peroxide as the Oxidant. Org. Lett. 2020, 22, 1402–1406. [Google Scholar] [CrossRef] [PubMed]
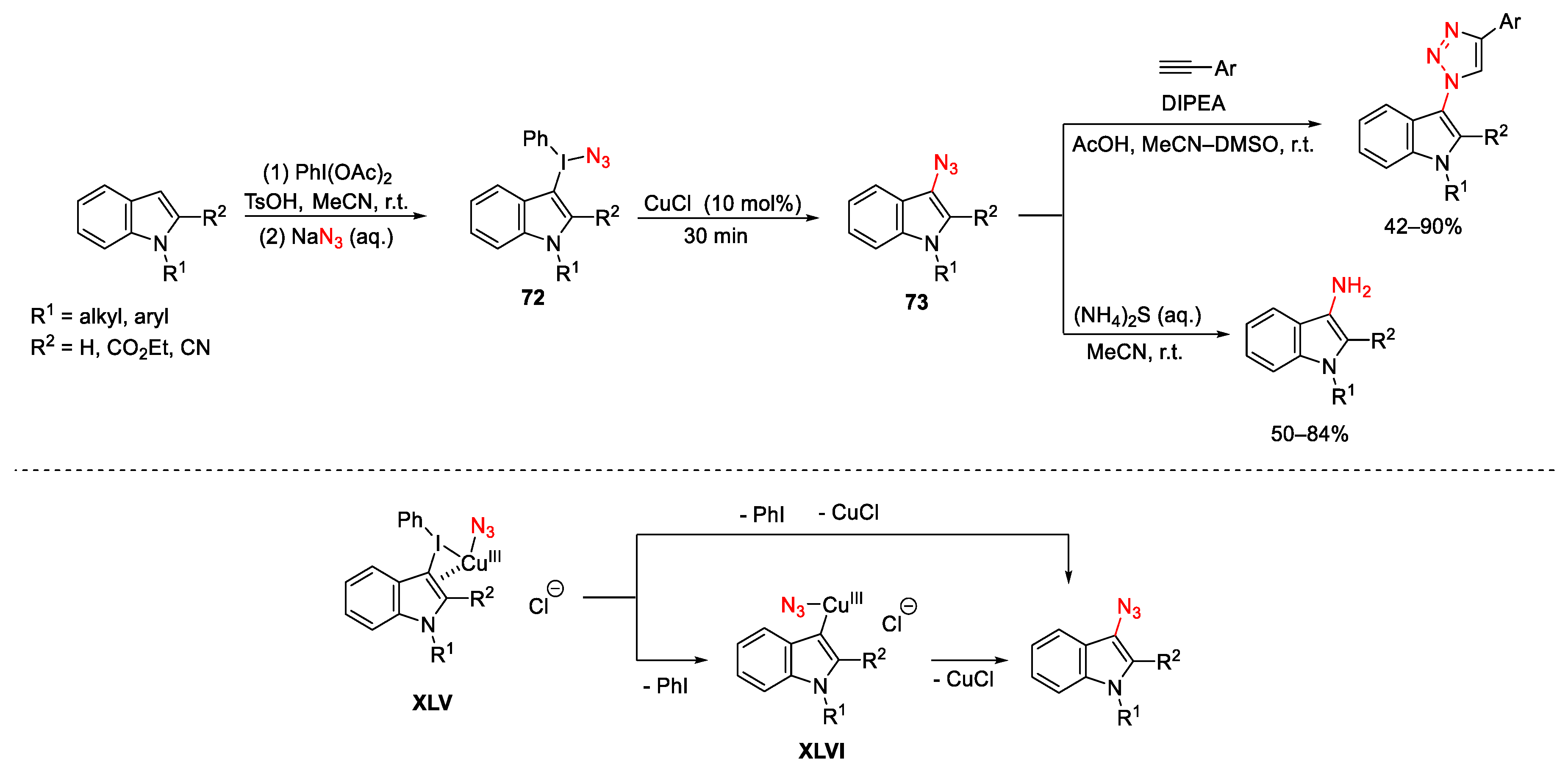
- Lubriks, D.; Sokolovs, I.; Suna, E. Indirect C–H Azidation of Heterocycles via Copper-Catalyzed Regioselective Fragmentation of Unsymmetrical λ3-Iodanes. J. Am. Chem. Soc. 2012, 134, 15436–15442. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Wan, W.; Ma, G.; Gao, W.; Jiang, H.; Zhuc, S.; Hao, J. Room-Temperature Cu(ii)-Catalyzed Aromatic C–H Azidation for the Synthesis of Ortho-Azido Anilines with Excellent Regioselectivity. Chem. Commun. 2014, 50, 5733–5736. [Google Scholar] [CrossRef]
- Chen, Y.-X.; Huo, T.; Yin, Q.; Jiang, L.-F.; Cheng, X.; Ma, H.-X.; Jiang, Y.-X.; Sun, M.-Z.; Deng, Q.-H. Azidobenziodazolones as Azido Sources for the Enantioselective Copper-Catalyzed Azidation of N-Unprotected 3-Trifluoromethylated Oxindoles. Org. Lett. 2023, 25, 2739–2744. [Google Scholar] [CrossRef]
- Yin, H.; Wang, T.; Jiao, N. Copper-Catalyzed Oxoazidation and Alkoxyazidation of Indoles. Org. Lett. 2014, 16, 2302–2305. [Google Scholar] [CrossRef]
- Fumagalli, G.; Rabet, P.T.G.; Boyd, S.; Greaney, M.F. Three-Component Azidation of Styrene-Type Double Bonds: Light-Switchable Behavior of a Copper Photoredox Catalyst. Angew. Chem. Int. Ed. 2015, 54, 11481–11484. [Google Scholar] [CrossRef]
- Rabet, P.T.R.; Fumagalli, G.; Boyd, S.; Greaney, M.F. Benzylic C–H Azidation Using the Zhdankin Reagent and a Copper Photoredox Catalyst. Org. Lett. 2016, 18, 1646–1649. [Google Scholar] [CrossRef]
- Shen, K.; Wang, Q. Copper-Catalyzed Alkene Aminoazidation as a Rapid Entry to 1,2-Diamines and Installation of an Azide Reporter onto Azahetereocycles. J. Am. Chem. Soc. 2017, 139, 13110–13116. [Google Scholar] [CrossRef]
- Sokolovs, I.; Suna, E. Para-Selective Cu-Catalyzed C–H Aryloxylation of Electron-Rich Arenes and Heteroarenes. J. Org. Chem. 2016, 81, 371–379. [Google Scholar] [CrossRef]
- Bhattarai, B.; Tay, J.-H.; Nagorny, P. Thiophosphoramides as Cooperative Catalysts for Copper-Catalyzed Arylation of Carboxylates with Diaryliodonium Salts. Chem. Commun. 2015, 51, 5398–5401. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Yang, S.; Zhang, C.; Ding, M.; Liu, M.; Guo, J.; Zhang, F. Copper-Catalyzed Selective Diphenylation of Carboxylic Acids with Cycli Diaryliodonium Salts. J. Org. Chem. 2017, 82, 5250–5262. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Xu, K.; Fang, Q.; Wang, Y.; Tang, B.; Zhang, F. Enantioselective Synthesis of Axially Chiral Biaryls via Cu-Catalyzed Acyloxylation of Cyclic Diaryliodonium Salts. ACS Catal. 2019, 9, 4951–4957. [Google Scholar] [CrossRef]
- Fañanás-Mastral, M.; Feringa, B.L. Copper-Catalyzed Synthesis of Mixed Alkyl Aryl Phosphonates. J. Am. Chem. Soc. 2014, 136, 9894–9897. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Xiong, B.; Zhou, C.; Liu, C.; Xu, W.; Yang, C.-A.; Tang, K.-W.; Wong, W.-Y. Copper-Catalyzed Diphenylation of P(O)-OH Bonds with Cyclic Diaryliodonium Salts. Chem. Asian J. 2019, 14, 4365–4374. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Zhao, K.; Wang, Z.; Zhang, F.-L.; Gu, Z. Enantioselective Ring-Opening/Oxidative Phosphorylation and P-Transfer Reaction of Cyclic Diaryliodoniums. ACS Catal. 2019, 9, 9852–9858. [Google Scholar] [CrossRef]
- Vázquez-Galiñanes, N.; Andón-Rodríguez, M.; Gómez-Roibás, P.; Fañanás-Mastral, M. Copper-catalyzed O-alkenylation of phosphonates. Beilstein J. Org. Chem. 2020, 16, 611–615. [Google Scholar] [CrossRef]
- Suero, M.G.; Bayle, E.D.; Collins, B.S.L.; Gaunt, M.J. Copper-Catalyzed Electrophilic Carbofunctionalization of Alkynes to Highly Functionalized Tetrasubstituted Alkenes. J. Am. Chem. Soc. 2013, 135, 5332–5335. [Google Scholar] [CrossRef]
- Zhu, D.; Sun, Y.; Hui, P.; Li, H.; Yan, Y.; Kuang, H. Enantioselective Synthesis of Axially Chiral Oxazole Biaryls by Copper-Catalyzed Oxidation of Cyclic Diaryliodoniums. Eur. J. Org. Chem. 2022, 2022, e202101449. [Google Scholar] [CrossRef]
- Zhu, D.; Liu, P.; Lu, W.; Wang, H.; Luo, B.; Hu, Y.; Huang, P.; Wen, S. Relayed Regioselective Alkynylation/Olefination of Unsymmetrical Cyclic Diaryliodonium Species Catalyzed by Cu and Pd: Affording Fluorescent Cytotoxic Benzoxazoles. Chem. Eur. J. 2015, 21, 18915–18920. [Google Scholar] [CrossRef]
- Zhu, D.; Wu, Z.; Luo, B.; Du, Y.; Liu, P.; Chen, Y.; Hu, Y.; Huang, P.; Wen, S. Heterocyclic Iodoniums for the Assembly of Oxygen-Bridged Polycyclic Heteroarenes with Water as the Oxygen Source. Org. Lett. 2018, 20, 4815–4818. [Google Scholar] [CrossRef]
- Zhu, D.; Li, M.; Wu, Z.; Du, Y.; Luo, B.; Huang, P.; Wen, S. Copper-Catalyzed One-Pot Synthesis of Dibenzofurans, Xanthenes, and Xanthones from Cyclic Diphenyl Iodoniums. Eur. J. Org. Chem. 2019, 2019, 4566–4571. [Google Scholar] [CrossRef]
- Li, J.; Xu, Q.-N.; Wang, Z.-B.; Li, Y.; Liu, L. Synthesis of Dibenzofurans from Cyclic Diaryliodonium Triflates and Water via Oxygen–Iodine Exchange Approach. ACS Omega 2018, 3, 12923–12929. [Google Scholar] [CrossRef]
- Luo, B.; Cui, Q.; Luo, H.; Hu, Y.; Huang, P.; Wen, S. N-Benzyldithiocarbamate Salts as Sulfur Sources to Access Tricyclic Thioheterocycles Mediated by Copper Species. Adv. Synth. Catal. 2016, 358, 2733–2738. [Google Scholar] [CrossRef]
- Wang, M.; Wei, J.; Fan, Q.; Jiang, X. Cu(II)-catalyzed sulfide construction: Both aryl groups utilization of intermolecular and intramolecular diaryliodonium salt. Chem. Commun. 2017, 53, 2918–2921. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, K.; Gu, Z. Transition Metal-Catalyzed Biaryl Atropisomer Synthesis via a Torsional Strain Promoted Ring-Opening Reaction. Acc. Chem. Res. 2022, 55, 1620–1633. [Google Scholar] [CrossRef] [PubMed]
- Hou, M.; Deng, R.; Gu, Z. Cu-Catalyzed Enantioselective Atropisomer Synthesis via Thiolative Ring Opening of Five-Membered Cyclic Diaryliodoniums. Org. Lett. 2018, 20, 5779–5783. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Chao, Z.; Li, C.; Gu, Z. Cu-Catalyzed Enantioselective Ring Opening of Cyclic Diaryliodoniums toward the Synthesis of Chiral Diarylmethanes. J. Am. Chem. Soc. 2018, 140, 9400–9403. [Google Scholar] [CrossRef] [PubMed]
- Cullen, S.C.; Shekhar, S.; Nere, N.K. Cu-Catalyzed Couplings of Aryl Iodonium Salts with Sodium Trifluoromethanesulfinate. J. Org. Chem. 2013, 78, 12194–12201. [Google Scholar] [CrossRef]
- Borrel, J.; Pisella, G.; Waser, J. Copper-Catalyzed Oxyalkynylation of C–S Bonds in Thiiranes and Thiethanes with Hypervalent Iodine Reagents. Org. Lett. 2020, 22, 422–427. [Google Scholar] [CrossRef]
- Wang, M.; Chen, S.; Jiang, X. Construction of Functionalized Annulated Sulfone via SO2/I Exchange of Cyclic Diaryliodonium Salts. Org. Lett. 2017, 19, 4916–4919. [Google Scholar] [CrossRef]
- Yang, Y.-D.; Azuma, A.; Tokunaga, E.; Yamasaki, M.; Shiro, M.; Shibata, N. Trifluoromethanesulfonyl Hypervalent Iodonium Ylide for Copper-Catalyzed Trifluoromethylthiolation of Enamines, Indoles, and β-Keto Esters. J. Am. Chem. Soc. 2013, 135, 8782–8785. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Wang, Z.; Zhao, K.; Gu, Z. Enantioselective preparation of atropisomeric biaryl trifluoromethylsulfanes via ring-opening of cyclic diaryliodoniums. Chem. Commun. 2021, 57, 3881. [Google Scholar] [CrossRef]
- Khan, S.; Volla, C.M.R. Cu-catalyzed Cascade Cyclization of Isothiocyanates, Alkynes, and Diaryliodonium Salts: Access to Diversely Functionalized Quinolines. Chem. Eur. J. 2017, 23, 12462–12466. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhou, J.; Wang, C.; Zhang, C.; Zhang, X.-Q.; Wang, Y. Design, development and applications of copper-catalyzed regioselective (4+2) annulations between diaryliodonium salts and alkynes. Commun. Chem. 2022, 5, 145. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhu, D.; Luo, B.; Zhang, N.; Liu, Q.; Hu, Y.; Pi, R.; Huang, P.; Wen, S. Mild Cu(I)-Catalyzed Cascade Reaction of Cyclic Diaryliodoniums, Sodium Azide, and Alkynes: Efficient Synthesis of Trazolophenanthridines. Org. Lett. 2014, 16, 5600–5603. [Google Scholar] [CrossRef]
- Urones, B.; Martínez, A.M.; Rodríguez, N.; Arrayás, R.G.; Carretero, J.C. Copper-catalyzed ortho-halogenation of protected anilines. Chem. Commun. 2013, 49, 11044–11046. [Google Scholar] [CrossRef]
- Giofré, S.; Loro, C.; Molteni, L.; Castellano, C.; Contini, A.; Nava, D.; Broggini, G.; Beccalli, E.M. Copper(II)-Catalyzed Aminohalogenation of Alkynyl Carbamates. Eur. J. Org. Chem. 2021, 2021, 1750–1757. [Google Scholar] [CrossRef]
- Hao, W.; Liu, Y. C-H bond halogenation catalyzed or mediated by copper: An overview. Beilstein J. Org. Chem. 2015, 11, 2132–2144. [Google Scholar] [CrossRef]
- Yuan, W.; Szabò, K.J. Catalytic Intramolecular Aminofluorination, Oxyfluorination, and Carbofluorination with a Stable and Versatile Hypervalent Fluoroiodine Reagent. Angew. Chem. Int. Ed. 2015, 54, 8533–8537. [Google Scholar] [CrossRef]
- Parvathaneni, S.P.; Perumgani, P.C. Regioselective Chlorination of Aryl C-H bonds with Hypervalent Iodine(III) Reagent 1-Chloro-1,2-benziodoxol-3-one. Asian J. Org. Chem. 2018, 7, 324–327. [Google Scholar] [CrossRef]
- Wu, B.; Yoshikai, N. Conversion of 2-Iodobiaryls into 2,2’-Diiodobiaryls via Oxidation- Iodination Sequences: A Versatile Route to Ladder-Type Heterofluorenes. Angew. Chem. Int. Ed. 2015, 54, 8736–8739. [Google Scholar] [CrossRef]
- Ke, J.; Zu, B.; Guo, Y.; Li, Y.; He, C. Hexafluoroisopropanol-Enabled Copper-Catalyzed Asymmetric Halogenation of Cyclic Diaryliodoniums for the Synthesis of Axially Chiral 2,2’-Dihalobiaryls. Org. Lett. 2021, 23, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, P.; Gao, Y.; Chen, Y.; Tang, G.; Zhao, Y. Copper-Catalyzed P-Arylation via Direct Coupling of Diaryliodonium Salts with Phosphorus Nucleophiles at Room Temperature. J. Org. Chem. 2013, 78, 8176–8183. [Google Scholar] [CrossRef]
- Beaud, R.; Phipps, R.J.; Gaunt, M.J. Enantioselective Cu-Catalyzed Arylation of Secondary Phosphine Oxides with Diaryliodonium Salts toward the Synthesis of P-Chiral Phosphines. J. Am. Chem. Soc. 2016, 138, 13183–13186. [Google Scholar] [CrossRef] [PubMed]
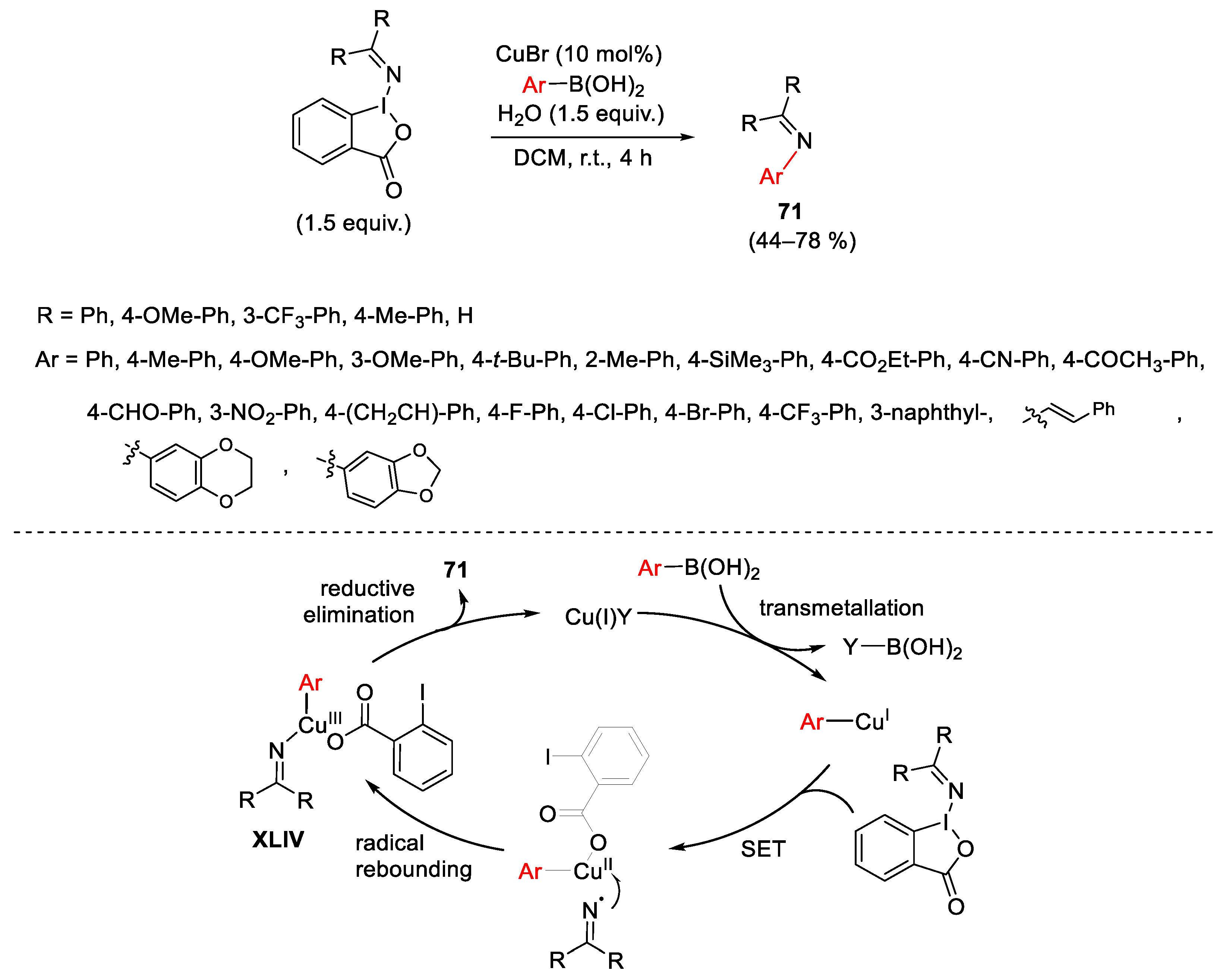
- Cho, S.H.; Yoon, J.; Chang, S. Intramolecular Oxidative C-N Bond for the Synthesis of Carbazoles: Comparison of Reactivity between the Copper-Catalyzed and Metal-Free Conditions. J. Am. Chem. Soc. 2011, 133, 5996–6005. [Google Scholar] [CrossRef]
- Mirallai, S.I.; Koutentis, P.A. The Conversion of 4-Anilinquinazoline- and 3-Aryl-4-imino-3,4-dihydro-quinazoline-2-carbonitriles into Benzo[4,5]imidazo[1,3-c]quinazoline-6-carbonitriles via Oxidative and Nonoxidative C-N Couplings. J. Org. Chem. 2015, 80, 8329–8340. [Google Scholar] [CrossRef]
- Liu, Z.-J.; Lu, X.; Wang, G.; Li, L.; Jiang, W.-T.; Wang, Y.-D.; Xiao, B.; Fu, Y. Directing Group in Decarboxylative Cross-Coupling: Copper-Catalyzed Site-Selective C-N Bond Formation from Nonactivated Aliphatic Carboxylic Acids. J. Am. Chem. Soc. 2016, 138, 9714–9719. [Google Scholar] [CrossRef]
- Yang, Y.-N.; Jiang, J.-L.; Shi, J. Mechanistic Study of Copper-Catalyzed Decarboxylative C-N Cross-Coupling with Hypervalent Iodine Oxidant. Organometallics 2017, 36, 2081–2087. [Google Scholar] [CrossRef]
- Yuan, J.; Rao, C.B.; Liang, Y.; Zhang, R.; Zhang, Q.; Hou, L.; Dong, D. Copper-Catalyzed Regioselective Oxidation Cycloamidation of α-[(β–Dimethylamino)propenoyl]-Alkylamides: Synthetic Route to Substituted Pyrrolidine-2,4—Diones. Adv. Synth. Catal. 2019, 361, 160–169. [Google Scholar] [CrossRef]
- Moreau, B.; Alberico, D.; Lindsay, V.N.G.; Charette, A.B. Catalytic asymmetric synthesis of nitrocyclopropane carboxylates. Tetrahedron 2012, 68, 3487–3496. [Google Scholar] [CrossRef]
- Chidley, T.; Murphy, G.K. Cyclopropanation of alkenes with metallocarbenes generated from monocarbonyl iodonium ylides. Org. Biomol. Chem. 2018, 16, 8486–8490. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.-P.; Lin, Y.; Cao, X.; Liu, Y.; Wei, L. Copper-catalyzed, hypervalent iodine mediated C=C bond activation of enaminones for the synthesis of α-keto-amides. Chem. Commun. 2016, 52, 1270–1273. [Google Scholar] [CrossRef] [PubMed]
- Loro, C.; Molteni, L.; Papis, M.; Beccalli, E.M.; Nava, D.; Lo Presti, L.; Brenna, S.; Colombo, G.; Foschi, F.; Broggini, G. Direct Synthesis of Fluorescent Oxazolo-phenoxazines by Copper-Catalyzed/Hypervalent Iodine(III)-Mediated Dimerization/Cyclization of 2-Benzylamino-phenols. J. Org. Chem. 2022, 87, 1032–1042. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Huang, F.; Lou, J.; Wang, Q.; Yu, Z. Copper-promoted direct C-H alkoxylation of S,S-functionalized internal olefins with alcohols. Org. Biomol. Chem. 2017, 15, 5535–5540. [Google Scholar] [CrossRef] [PubMed]








































































































































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papis, M.; Foschi, F.; Colombo, S.; Beccalli, E.M.; Loro, C.; Broggini, G. Copper-Catalyzed/Hypervalent Iodine-Mediated Functionalization of Unactivated Compounds. Catalysts 2023, 13, 1243. https://doi.org/10.3390/catal13091243
Papis M, Foschi F, Colombo S, Beccalli EM, Loro C, Broggini G. Copper-Catalyzed/Hypervalent Iodine-Mediated Functionalization of Unactivated Compounds. Catalysts. 2023; 13(9):1243. https://doi.org/10.3390/catal13091243
Chicago/Turabian StylePapis, Marta, Francesca Foschi, Sara Colombo, Egle Maria Beccalli, Camilla Loro, and Gianluigi Broggini. 2023. "Copper-Catalyzed/Hypervalent Iodine-Mediated Functionalization of Unactivated Compounds" Catalysts 13, no. 9: 1243. https://doi.org/10.3390/catal13091243
APA StylePapis, M., Foschi, F., Colombo, S., Beccalli, E. M., Loro, C., & Broggini, G. (2023). Copper-Catalyzed/Hypervalent Iodine-Mediated Functionalization of Unactivated Compounds. Catalysts, 13(9), 1243. https://doi.org/10.3390/catal13091243