

Identifying the Active Phase of RuO2 in the Catalytic CO Oxidation Reaction, Employing Operando CO Infrared Spectroscopy and Online Mass Spectrometry

Abstract
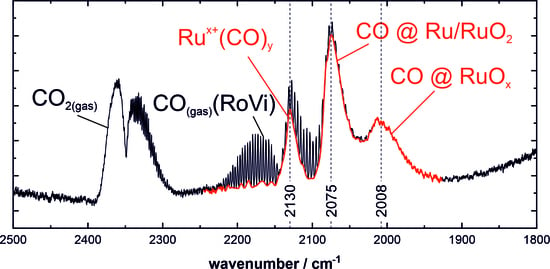
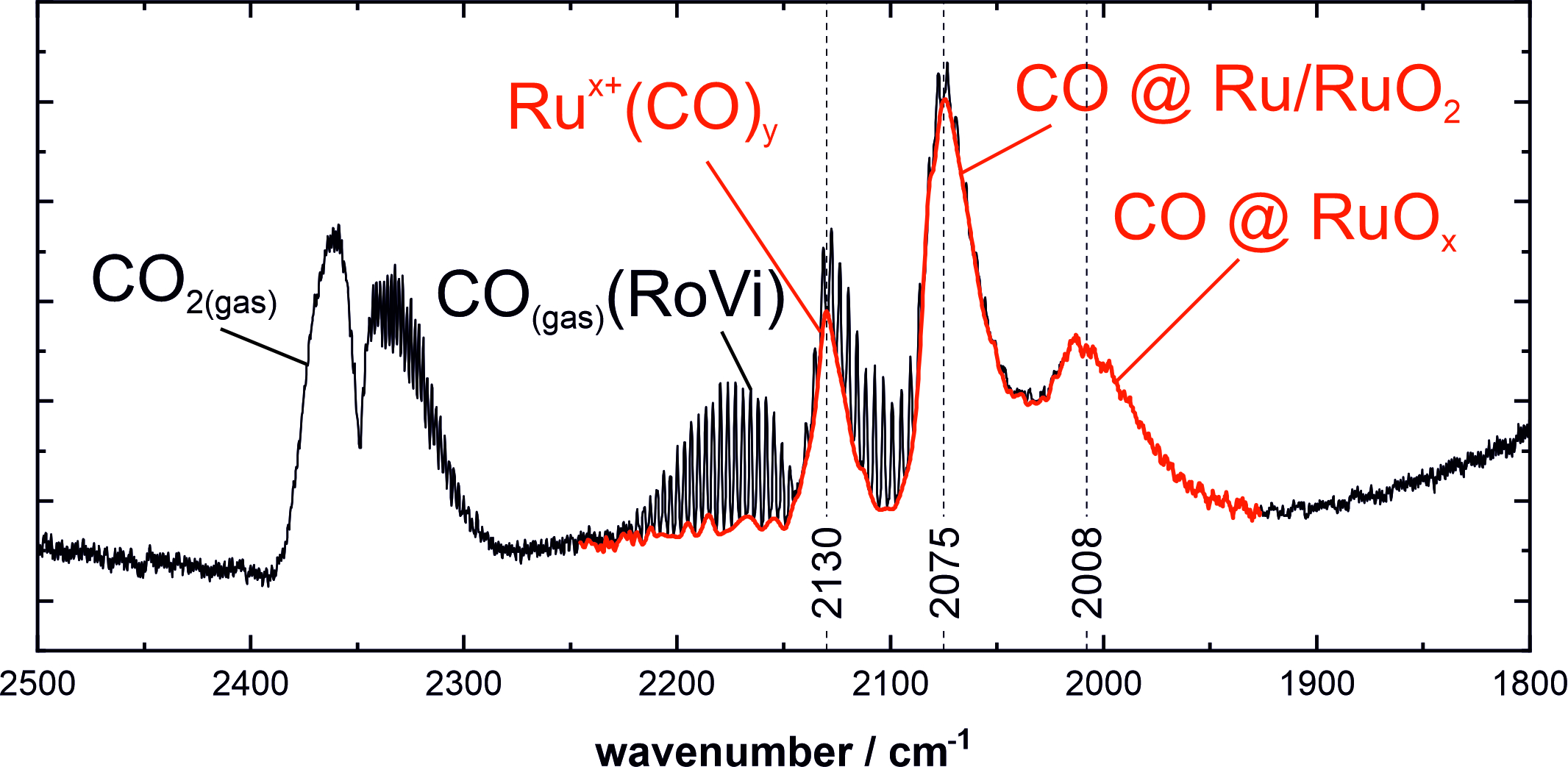
1. Introduction
2. Experimental Results
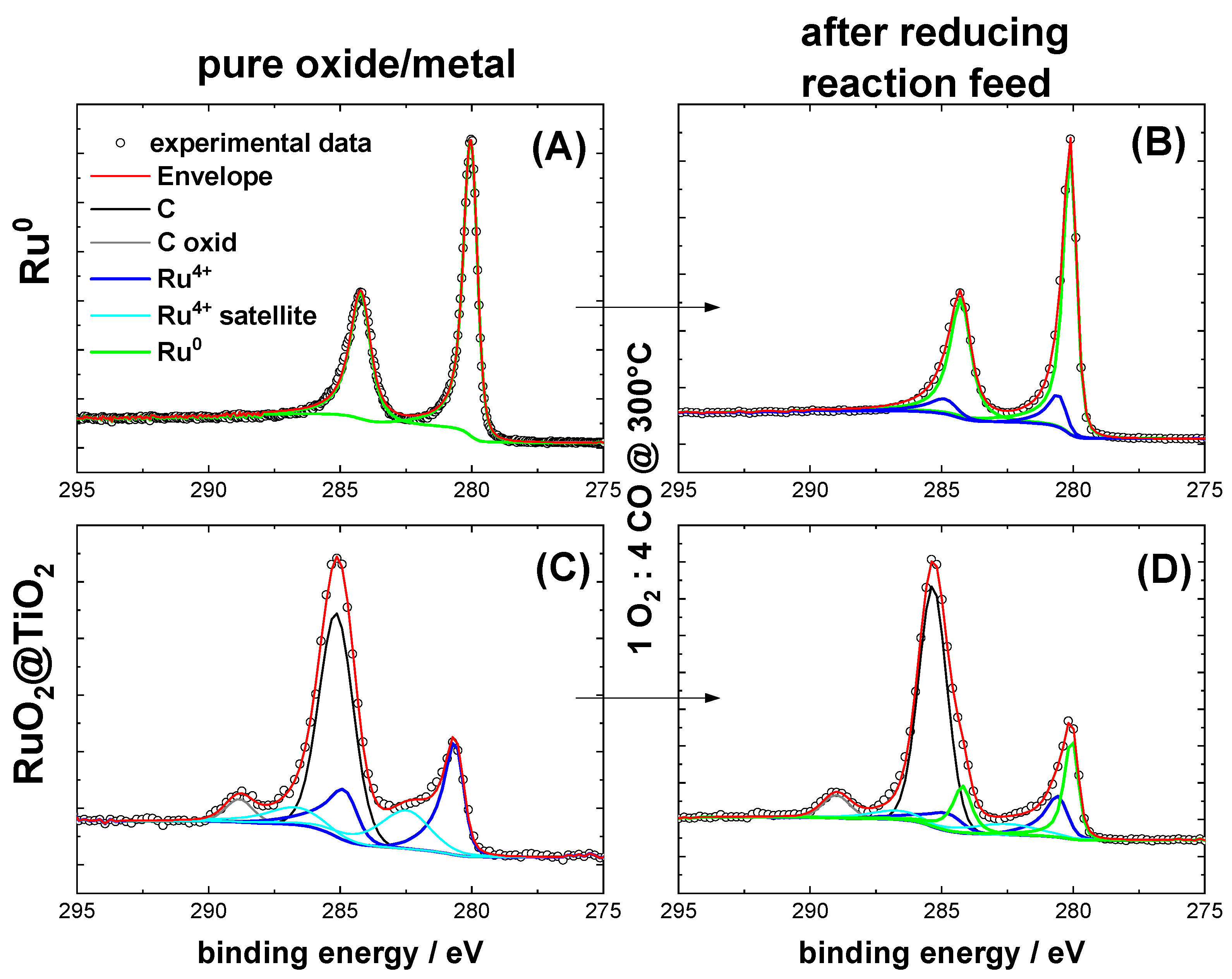
2.1. Characterization of Pre-Oxidized and Pre-Reduced RuO2@TiO2 and Ru0 + TiO2 Samples
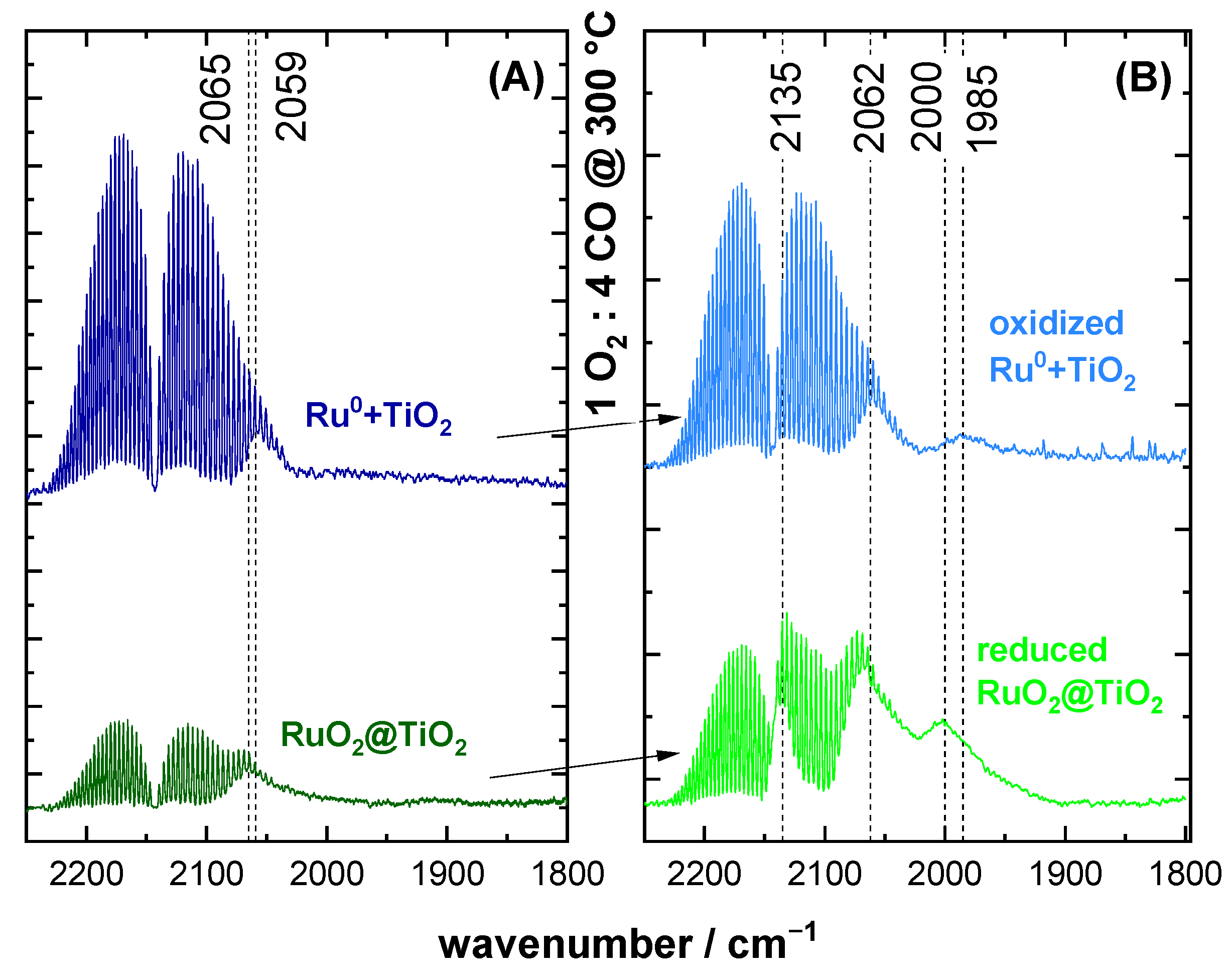
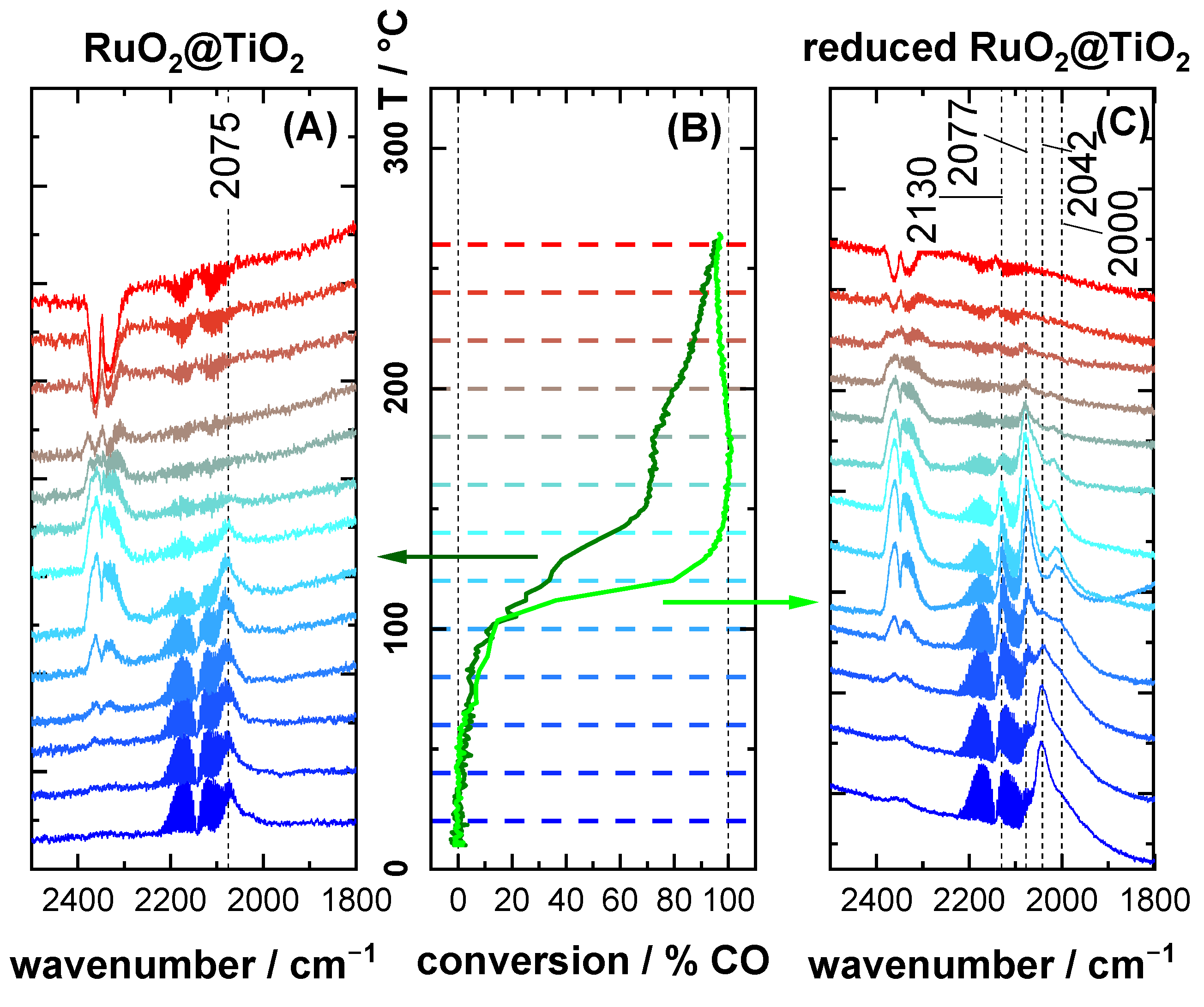
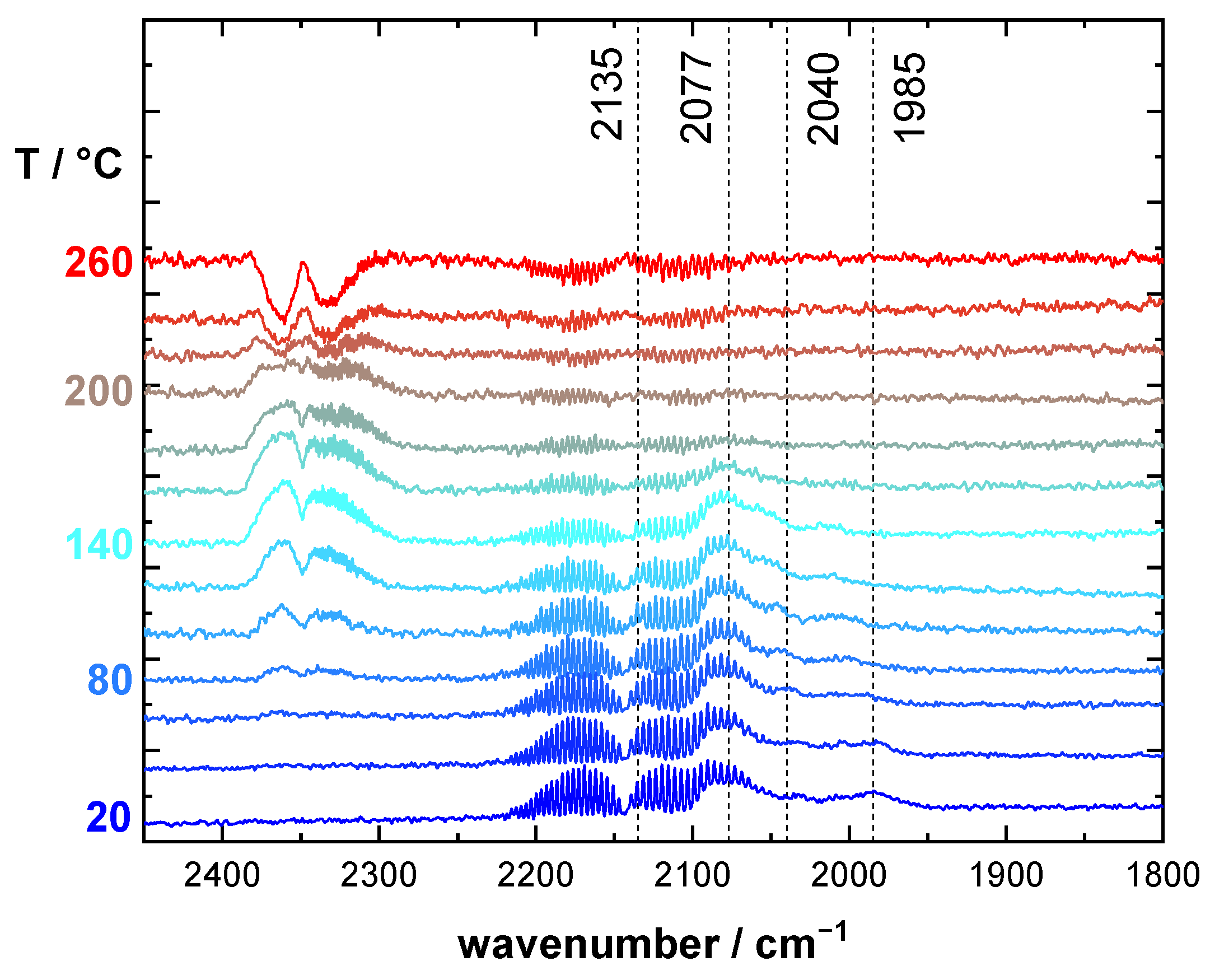
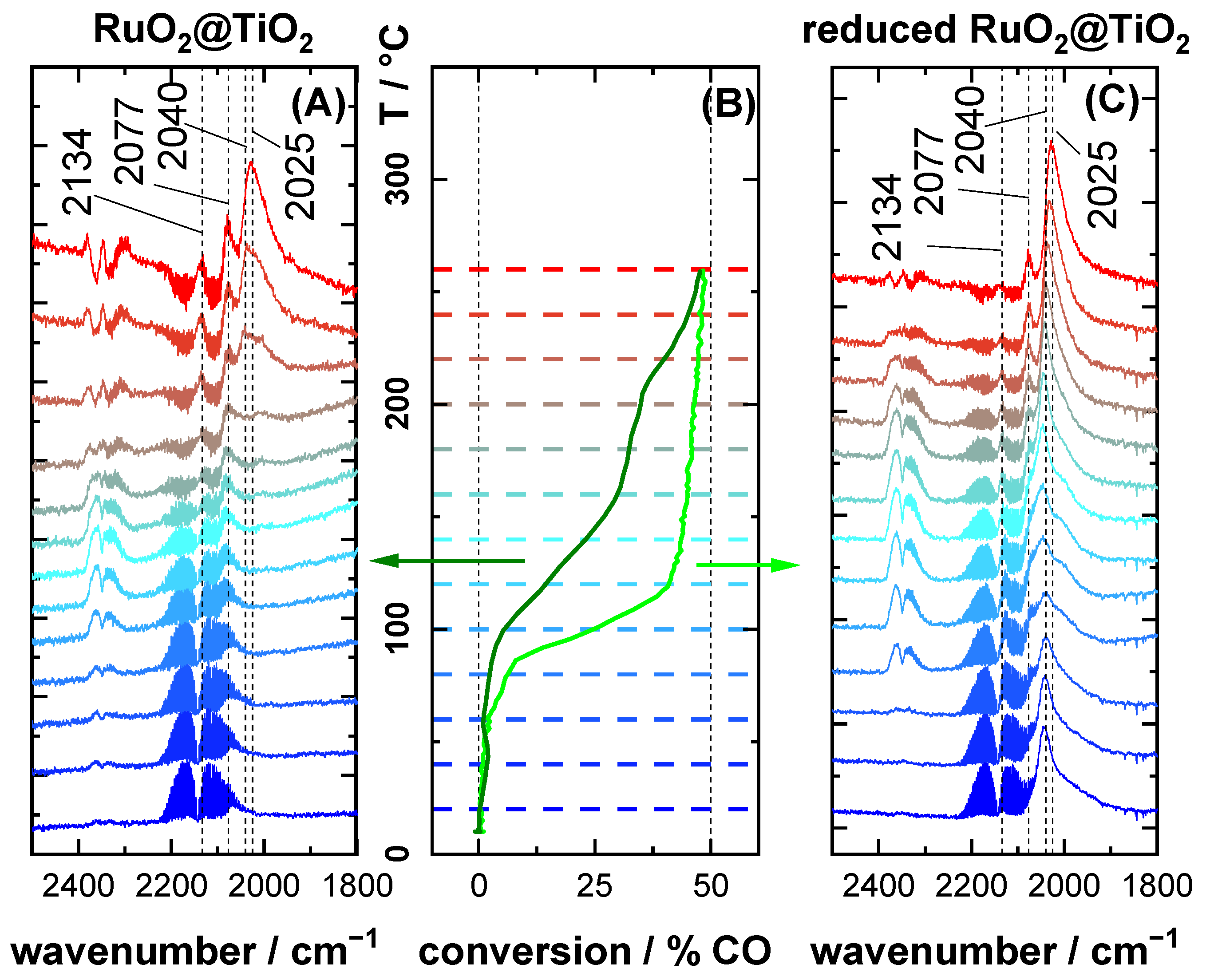
2.2. CO DRIFTS Experiments of Oxidized and Reduced RuO2@TiO2 and Ru0 + TiO2 Samples
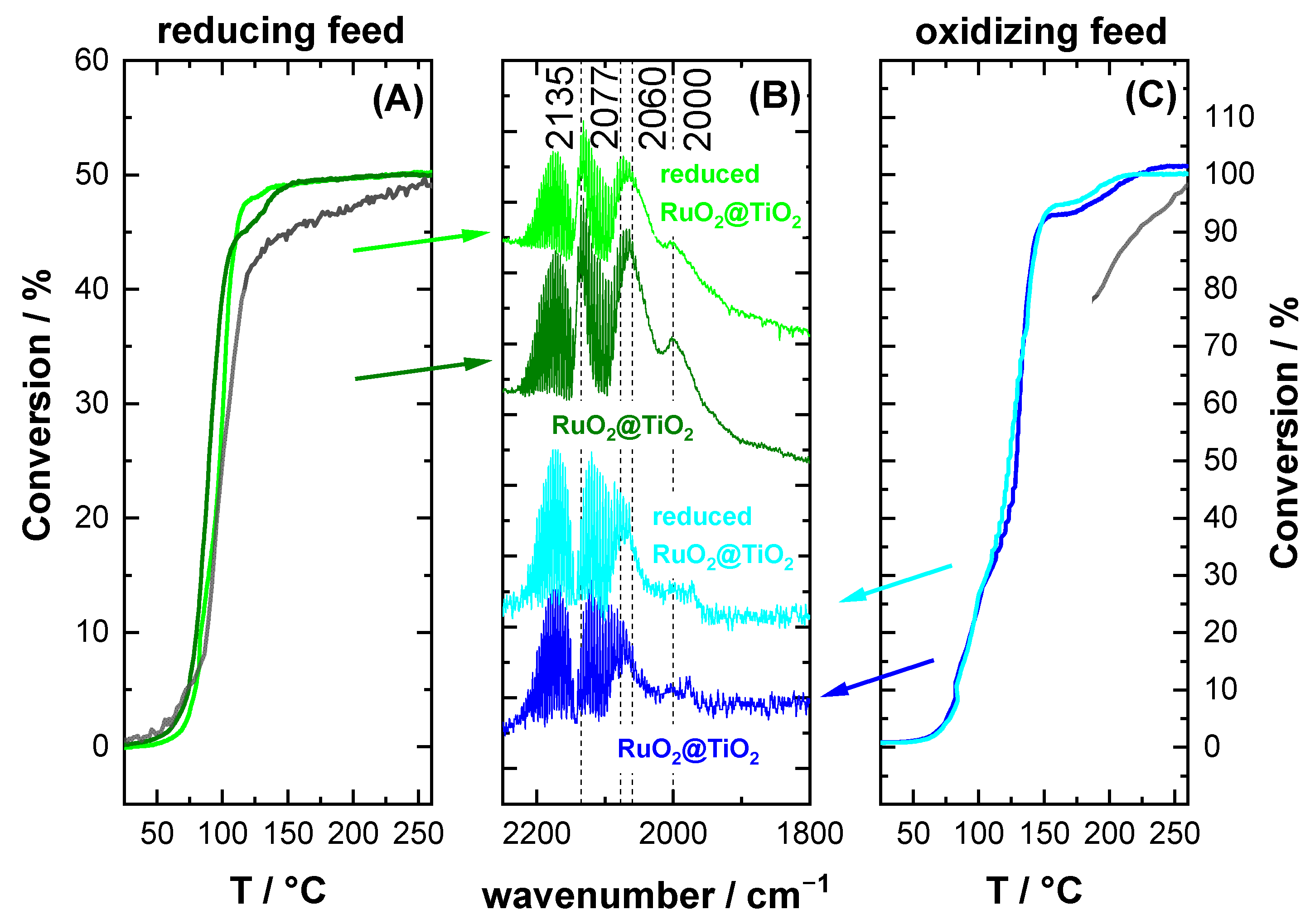
2.3. CO Oxidation Experiments of RuO2@TiO2 Samples
3. Discussion
3.1. Stretching Vibrations of Adsorbed CO Probing the Actual Surface Oxidation State of RuO2
3.2. CO Oxidation as Case Study
- I.
- 1985–2030 cm−1: CO on partially reduced RuO2 with shape and position changing according to surface composition
- II.
- 2040 cm−1: CO sitting possibly in boundary region between surface phases with the highest activity.
- III.
- 2075 cm−1: CO on oxide RuO2
- IV.
4. Experimental Details
4.1. Sample Preparation and Characterization
4.2. Reaction Conditions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schlögl, R. Heterogeneous Catalysis. Angew. Chem. Int. Ed. 2015, 54, 3465–3520. [Google Scholar] [CrossRef]
- Over, H.; Balmes, O.; Lundgren, E. Direct Comparison of the Reactivity of the Non-Oxidic Phase of Ru(0001) and the RuO2 Phase in the CO Oxidation Reaction. Surf. Sci. 2009, 603, 298–303. [Google Scholar] [CrossRef]
- Gao, F.; Goodman, D.W. CO Oxidation over Ruthenium: Identification of the Catalytically Active Phases at near-Atmospheric Pressures. Phys. Chem. Chem. Phys. 2012, 14, 6688–6697. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Pan, L.; Li, T.; Diulus, J.T.; Asthagiri, A.; Weaver, J.F. CO Oxidation on PdO(101) during Temperature-Programmed Reaction Spectroscopy: Role of Oxygen Vacancies. J. Phys. Chem. C 2014, 118, 28647–28661. [Google Scholar] [CrossRef]
- Weaver, J.F.; Choi, J.; Mehar, V.; Wu, C. Kinetic Coupling among Metal and Oxide Phases during CO Oxidation on Partially Reduced PdO(101): Influence of Gas-Phase Composition. ACS Catal. 2017, 7, 7319–7331. [Google Scholar] [CrossRef]
- Van Spronsen, M.A.; Frenken, J.W.M.; Groot, I.M.N. Surface Science under Reaction Conditions: CO Oxidation on Pt and Pd Model Catalysts. Chem. Soc. Rev. 2017, 46, 4347–4374. [Google Scholar] [CrossRef]
- Gustafson, J.; Balmes, O.; Zhang, C.; Shipilin, M.; Schaefer, A.; Hagman, B.; Merte, L.R.; Martin, N.M.; Carlsson, P.A.; Jankowski, M.; et al. The Role of Oxides in Catalytic CO Oxidation over Rhodium and Palladium. ACS Catal. 2018, 8, 4438–4445. [Google Scholar] [CrossRef]
- Martin, R.; Kim, M.; Lee, C.J.; Mehar, V.; Albertin, S.; Hejral, U.; Merte, L.R.; Asthagiri, A.; Weaver, J.F. Isothermal Reduction of IrO2(110) Films by Methane Investigated Using in Situ X-ray Photoelectron Spectroscopy. ACS Catal. 2021, 11, 5004–5016. [Google Scholar] [CrossRef]
- Abb, M.J.S.; Weber, T.; Langsdorf, D.; Koller, V.; Gericke, S.M.; Pfaff, S.; Busch, M.; Zetterberg, J.; Preobrajenski, A.; Grönbeck, H.; et al. Thermal Stability of Single-Crystalline IrO2(110) Layers: Spectroscopic and Adsorption Studies. J. Phys. Chem. C 2020, 124, 15324–15336. [Google Scholar] [CrossRef]
- Timmer, P.; Weber, T.; Glatthaar, L.; Over, H. Operando CO Infrared Spectroscopy and On-Line Mass Spectrometry for Studying the Active Phase of IrO2 in the Catalytic CO Oxidation Reaction. Inorganics 2023, 11, 102. [Google Scholar] [CrossRef]
- Weckhuysen, B.M. Determining the Active Site in a Catalytic Process: Operando Spectroscopy Is More than a Buzzword. Phys. Chem. Chem. Phys. 2003, 5, 4351–4360. [Google Scholar] [CrossRef]
- Grunwaldt, J.D.; Baiker, A. In Situ Spectroscopic Investigation of Heterogeneous Catalysts and Reaction Media at High Pressure. Phys. Chem. Chem. Phys. 2005, 7, 3526–3539. [Google Scholar] [CrossRef] [PubMed]
- Ryczkowski, J. IR Spectroscopy in Catalysis. Catal. Today 2001, 68, 263–381. [Google Scholar] [CrossRef]
- Over, H.; Kim, Y.D.; Seitsonen, A.P.; Wendt, S.; Leudgren, E.; Schmidt, M.; Varga, P.; Morgante, A.; Ertl, G. Atomic-Scale Structure and Catalytic Reactivity of the RuO2(110) Surface. Science 2000, 287, 1474–1476. [Google Scholar] [CrossRef] [PubMed]
- Goodman, D.W.; Peden, C.H.F.; Chen, M.S. CO Oxidation on Ruthenium: The Nature of the Active Catalytic Surface. Surf. Sci. 2007, 601, 18–20. [Google Scholar] [CrossRef]
- Over, H.; Muhler, M.; Seitsonen, A.P. Comment on “CO Oxidation on Ruthenium: The Nature of the Active Catalytic Surface” by D.W. Goodman, C.H.F. Peden, M.S. Chen. Surf. Sci. 2007, 601, 5659–5662. [Google Scholar] [CrossRef]
- Rosenthal, D.; Girgsdies, F.; Timpe, O.; Weinberg, G.; Schlögl, R. Oscillatory Behavior in the CO-Oxidation over Bulk Ruthenium Dioxide–The Effect of the CO/O2 Ratio. Z. Phys. Chem. 2011, 225, 57–68. [Google Scholar] [CrossRef]
- Seki, K. Development of RuO2/Rutile-TiO2 Catalyst for Industrial HCl Oxidation Process. Catal. Surv. Asia 2010, 14, 168–175. [Google Scholar] [CrossRef]
- Over, H.; Schomäcker, R. What Makes a Good Catalyst for the Deacon Process? ACS Catal. 2013, 3, 1034–1046. [Google Scholar] [CrossRef]
- Pérez-Ramírez, J.; Mondelli, C.; Schmidt, T.; Schlüter, O.F.K.; Wolf, A.; Mleczko, L.; Dreier, T. Sustainable Chlorine Recycling via Catalysed HCl Oxidation: From Fundamentals to Implementation. Energy Environ. Sci. 2011, 4, 4786–4799. [Google Scholar] [CrossRef]
- Over, H. Atomic Scale Insights into Electrochemical versus Gas Phase Oxidation of HCl over RuO2-Based Catalysts: A Comparative Review. Electrochim. Acta 2013, 93, 314–333. [Google Scholar] [CrossRef]
- Fabbri, E.; Habereder, A.; Waltar, K.; Kötz, R.; Schmidt, T.J. Developments and Perspectives of Oxide-Based Catalysts for the Oxygen Evolution Reaction. Catal. Sci. Technol. 2014, 4, 3800–3821. [Google Scholar] [CrossRef]
- Over, H. Fundamental Studies of Planar Single-Crystalline Oxide Model Electrodes (RuO2, IrO2) for Acidic Water Splitting. ACS Catal. 2021, 11, 8848–8871. [Google Scholar] [CrossRef]
- Drochner, A.; Fehlings, M.; Krauß, K.; Vogel, H. A New DRIFTS Cell for the In-Situ Investigation of Heterogeneously Catalyzed Reactions. Chem. Eng. Technol. 2000, 23, 319–322. [Google Scholar] [CrossRef]
- Meunier, F.C. Pitfalls and Benefits of: In Situ and Operando Diffuse Reflectance FT-IR Spectroscopy (DRIFTS) Applied to Catalytic Reactions. React. Chem. Eng. 2016, 1, 134–141. [Google Scholar] [CrossRef]
- Freund, H.J.; Meijer, G.; Scheffler, M.; Schlögl, R.; Wolf, M. CO Oxidation as a Prototypical Reaction for Heterogeneous Processes. Angew. Chem. Int. Ed. 2011, 50, 10064–10094. [Google Scholar] [CrossRef]
- Meunier, F.C. Relevance of IR Spectroscopy of Adsorbed CO for the Characterization of Heterogeneous Catalysts Containing Isolated Atoms. J. Phys. Chem. C 2021, 125, 21810–21823. [Google Scholar] [CrossRef]
- Zaera, F. Infrared Absorption Spectroscopy of Adsorbed CO: New Applications in Nanocatalysis for an Old Approach. ChemCatChem 2012, 4, 1525–1533. [Google Scholar] [CrossRef]
- Morgan, D.J. Resolving Ruthenium: XPS Studies of Common Ruthenium Materials. Surf. Interface Anal. 2015, 47, 1072–1079. [Google Scholar] [CrossRef]
- Peden, C.H.F.; Goodman, D.W.; Weisel, M.D.; Hoffmann, F.M. In-Situ FT-IRAS Study of the CO Oxidation Reaction over Ru(001). I. Evidence for an Eley-Rideal Mechanism at High Pressures? Surf. Sci. 1991, 253, 44–58. [Google Scholar] [CrossRef]
- Farkas, A.; Mellau, G.C.; Over, H. Novel Insight in the CO Oxidation on RuO2(110) by in Situ Reflection-Absorption Infrared Spectroscopy. J. Phys. Chem. C 2009, 113, 14341–14355. [Google Scholar] [CrossRef]
- Chin, S.Y.; Williams, C.T.; Amiridis, M.D. FTIR Studies of CO Adsorption on Al2O3- and SiO2-Supported Ru Catalysts. J. Phys. Chem. B 2006, 110, 871–882. [Google Scholar] [CrossRef]
- Binsted, N.; Evans, J.; Greaves, G.N.; Price, R.J. Characterization of Supported Rhodium and Ruthenium Carbonyl Clusters by EXAFS Spectroscopy. Organometallics 1989, 8, 613–620. [Google Scholar] [CrossRef]
- Bogdan, P.L.; Weitz, E. A Transient Infrared Spectroscopy Study of Coordinatively Unsaturated Ruthenium Carbonyls. J. Am. Chem. Soc. 1989, 111, 3163–3167. [Google Scholar] [CrossRef]
- Schiffer, A.; Jakob, P.; Menzel, D. The (2CO+O)(2×2)/Ru(001) Layer: Preparation, Characterization, and Analysis of Interaction Effects in Vibrational Spectra. Surf. Sci. 1997, 389, 116–130. [Google Scholar] [CrossRef]
- Farkas, A.; Hess, F.; Over, H. “First-Principles” Kinetic Monte Carlo Simulations Revisited: CO Oxidation over RuO2(110). J. Comput. Chem. 2011, 33, 757–766. [Google Scholar] [CrossRef]
- Kiss, J.T.; Gonzalez, R.D. Catalytic Oxidation of Carbon Monoxide over Ru/SiO2. An In Situ Infrared and Kinetic Study. J. Phys. Chem. 1984, 88, 892–897. [Google Scholar] [CrossRef]
- Assmann, J.; Narkhede, V.; Khodeir, L.; Löffler, E.; Hinrichsen, O.; Birkner, A.; Over, H.; Muhler, M. On the Nature of the Active State of Supported Ruthenium Catalysts Used for the Oxidation of Carbon Monoxide: Steady-State and Transient Kinetics Combined with in Situ Infrared Spectroscopy. J. Phys. Chem. B 2004, 108, 14634–14642. [Google Scholar] [CrossRef]
- Goodman, D.W.; Peden, C.H.F.; Chen, M.S. Reply to Comment on “CO Oxidation on Ruthenium: The Nature of the Active Catalytic Surface” by H. Over, M. Muhler, A.P. Seitsonen. Surf. Sci. 2007, 601, 5663–5665. [Google Scholar] [CrossRef]
- Gao, F.; Goodman, D.W. Reaction Kinetics and Polarization Modulation Infrared Reflection Absorption Spectroscopy Investigations of CO Oxidation over Planar Pt-Group Model Catalysts. Langmuir 2010, 26, 16540–16551. [Google Scholar] [CrossRef]
- Miller, B.K.; Crozier, P.A. Linking Changes in Reaction Kinetics and Atomic-Level Surface Structures on a Supported Ru Catalyst for CO Oxidation. ACS Catal. 2021, 11, 1456–1463. [Google Scholar] [CrossRef]
- Over, H.; Knapp, M.; Lundgren, E.; Seitsonen, A.P.; Schmid, M.; Varga, P. Visualization of Atomic Processes on Ruthenium Dioxide Using Scanning Tunneling Microscopy. ChemPhysChem 2004, 5, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Over, H.; Balmes, O.; Lundgren, E. In Situ Structure-Activity Correlation Experiments of the Ruthenium Catalyzed CO Oxidation Reaction. Catal. Today 2009, 145, 236–242. [Google Scholar] [CrossRef]
- Hess, F.; Sack, C.; Langsdorf, D.; Over, H. Probing the Activity of Different Oxygen Species in the CO Oxidation over RuO2(110) by Combining Transient Reflection-Absorption Infrared Spectroscopy with Kinetic Monte Carlo Simulations. ACS Catal. 2017, 7, 8420–8428. [Google Scholar] [CrossRef]
- Madey, T.E.; Albert Engelhardt, H.; Menzel, D. Adsorption of Oxygen and Oxidation of CO on the Ruthenium (001) Surface. Surf. Sci. 1975, 48, 304–328. [Google Scholar] [CrossRef]
- Narloch, B.; Held, G.; Menzel, D. Structural Rearrangement by Coadsorption: A LEED IV Determination of the Ru(001)-p(2 × 2)(2O + CO) Structure. Surf. Sci. 1994, 317, 131–142. [Google Scholar] [CrossRef]
- Martynova, Y.; Yang, B.; Yu, X.; Boscoboinik, J.A.; Shaikhutdinov, S.; Freund, H.J. Low Temperature CO Oxidation on Ruthenium Oxide Thin Films at Near-Atmospheric Pressures. Catal. Lett. 2012, 142, 657–663. [Google Scholar] [CrossRef]
- Seitsonen, A.P.; Kim, Y.D.; Knapp, M.; Wendt, S.; Over, H. CO Adsorption on the Reduced (Formula Presented) Surface: Energetics and Structure. Phys. Rev. B Condens. Matter. Mater. Phys. 2002, 65, 035413-1–035413-9. [Google Scholar] [CrossRef]
- Kim, Y.K.; Morgan, G.A.; Yates, J.T. Role of Atomic Step Defect Sites on the Catalytic Oxidation of Carbon Monoxide: Comparison between Ru(001) and Ru(109) Single-Crystal Surfaces. J. Phys. Chem. C 2007, 111, 3366–3368. [Google Scholar] [CrossRef]
- Šljivančanin, Ž.; Hammer, B. CO Oxidation on Fully Oxygen Covered Ru(0001): Role of Step Edges. Phys. Rev. B Condens. Matter. Mater. Phys. 2010, 81, 121413-1–121413-4. [Google Scholar] [CrossRef]
- Aßmann, J.; Löffler, E.; Birkner, A.; Muhler, M. Ruthenium as Oxidation Catalyst: Bridging the Pressure and Material Gaps between Ideal and Real Systems in Heterogeneous Catalysis by Applying DRIFT Spectroscopy and the TAP Reactor. Catal. Today 2003, 85, 235–249. [Google Scholar] [CrossRef]
- Assmann, J.; Narkhede, V.; Breuer, N.A.; Muhler, M.; Seitsonen, A.P.; Knapp, M.; Crihan, D.; Farkas, A.; Mellau, G.; Over, H. Heterogeneous Oxidation Catalysis on Ruthenium: Bridging the Pressure and Materials Gaps and Beyond. J. Phys. Condens. Matter 2008, 20, 184017. [Google Scholar] [CrossRef]
- Narkhede, V.; Aßmann, J.; Muhler, M. Structure-Activity Correlations for the Oxidation of CO over Polycrystalline RuO2 Powder Derived from Steady-State and Transient Kinetic Experiments. Z. Phys. Chem. 2005, 219, 979–995. [Google Scholar] [CrossRef]
- Rosenthal, D.; Girgsdies, F.; Timpe, O.; Blume, R.; Weinberg, G.; Teschner, D.; Schlögl, R. On the CO-Oxidation over Oxygenated Ruthenium. Z. Phys. Chem. 2009, 223, 183–207. [Google Scholar] [CrossRef]
- Blume, R.; Hävecker, M.; Zafeiratos, S.; Teschner, D.; Kleimenov, E.; Knop-Gericke, A.; Schlögl, R.; Barinov, A.; Dudin, P.; Kiskinova, M. Catalytically Active States of Ru(0001) Catalyst in CO Oxidation Reaction. J. Catal. 2006, 239, 354–361. [Google Scholar] [CrossRef]
- Over, H.; Seitsonen, A.P.; Lundgren, E.; Schmid, M.; Varga, P. Experimental and Simulated STM Images of Stoichiometric and Partially Reduced RuO2(110) Surfaces Including Adsorbates. Surf. Sci. 2002, 515, 143–156. [Google Scholar] [CrossRef]
- Khalid, O.; Spriewald Luciano, A.; Drazic, G.; Over, H. Mixed RuxIr1−xO2 Supported on Rutile TiO2: Catalytic Methane Combustion, a Model Study. ChemCatChem 2021, 13, 3983–3994. [Google Scholar] [CrossRef]
- He, Y.; Langsdorf, D.; Li, L.; Over, H. Versatile Model System for Studying Processes Ranging from Heterogeneous to Photocatalysis: Epitaxial RuO2(110) on TiO2(110). J. Phys. Chem. C 2015, 119, 2692–2702. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Band Position/cm−1 | Adsorbed Species | Substrate | Ref. | Remarks |
|---|---|---|---|---|
| 1860–1970 | CO | Mildly reduced RuO2(110) | [31] | Bridging CO being the majority species |
| 2070–2080 | CO | Mildly reduced RuO2(110) | [31] | On-top and bridging CO couple to a single band |
| 2000–2050 | [Ru0-CO]-linear | Ru/Al2O3 | [32] | Terminal CO |
| 2010–2070 | terminal CO | Ru-carbonyl | [33,34] | Depends on the carbonyl size |
| 2048 | CO | Ru(001) | [30] | Measured after exposure to oxidizing conditions; likely partially oxidized |
| 2068 | (CO+O)(2 × 2) | Ru(001) | [35] | -- |
| 2080 | CO | Ru(001) | [30] | Measured under strongly oxidizing conditions at 500 K, likely oxidized |
| 2100–2123 | CO | Stoichiometric RuO2(110) | [31] | On-top CO on stoichiometric RuO2(110) |
| 2100–2135 | CO/O | RuO2(110) | [36] | On-top CO being the majority species |
| 2100–2150 | CO/O | RuO2(110) | [31] | O being the majority species; on-top CO embedded in O matrix |
| 2070, 2130 | Rux+-(CO)3 | Ru/Al2O3 | [32,37] | x = 1–3 |
| 2134 | Rux+(CO)y | Ru/SiO2 | [38] | Linked to oxidation of Ru |
| 2135 | Rux+(CO)y | Reduced RuO2(110) | [3] | x and y undetermined; linked to surface roughening due to reduction |
| Reaction Conditions | Ar/% | O2/% | CO/% |
|---|---|---|---|
| Oxidizing | 96 | 2 | 2 |
| Reducing | 95 | 1 | 4 |
| CO only | 96 | - | 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timmer, P.; Glatthaar, L.; Weber, T.; Over, H. Identifying the Active Phase of RuO2 in the Catalytic CO Oxidation Reaction, Employing Operando CO Infrared Spectroscopy and Online Mass Spectrometry. Catalysts 2023, 13, 1178. https://doi.org/10.3390/catal13081178
Timmer P, Glatthaar L, Weber T, Over H. Identifying the Active Phase of RuO2 in the Catalytic CO Oxidation Reaction, Employing Operando CO Infrared Spectroscopy and Online Mass Spectrometry. Catalysts. 2023; 13(8):1178. https://doi.org/10.3390/catal13081178
Chicago/Turabian StyleTimmer, Phillip, Lorena Glatthaar, Tim Weber, and Herbert Over. 2023. "Identifying the Active Phase of RuO2 in the Catalytic CO Oxidation Reaction, Employing Operando CO Infrared Spectroscopy and Online Mass Spectrometry" Catalysts 13, no. 8: 1178. https://doi.org/10.3390/catal13081178
APA StyleTimmer, P., Glatthaar, L., Weber, T., & Over, H. (2023). Identifying the Active Phase of RuO2 in the Catalytic CO Oxidation Reaction, Employing Operando CO Infrared Spectroscopy and Online Mass Spectrometry. Catalysts, 13(8), 1178. https://doi.org/10.3390/catal13081178