

Phosphate Coordination in a Water-Oxidizing Cobalt Oxide Electrocatalyst Revealed by X-ray Absorption Spectroscopy at the Phosphorus K-Edge


Abstract
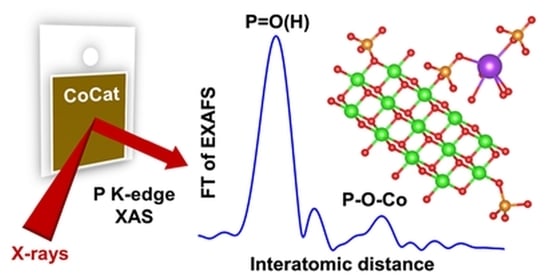
1. Introduction
2. Results and Discussion
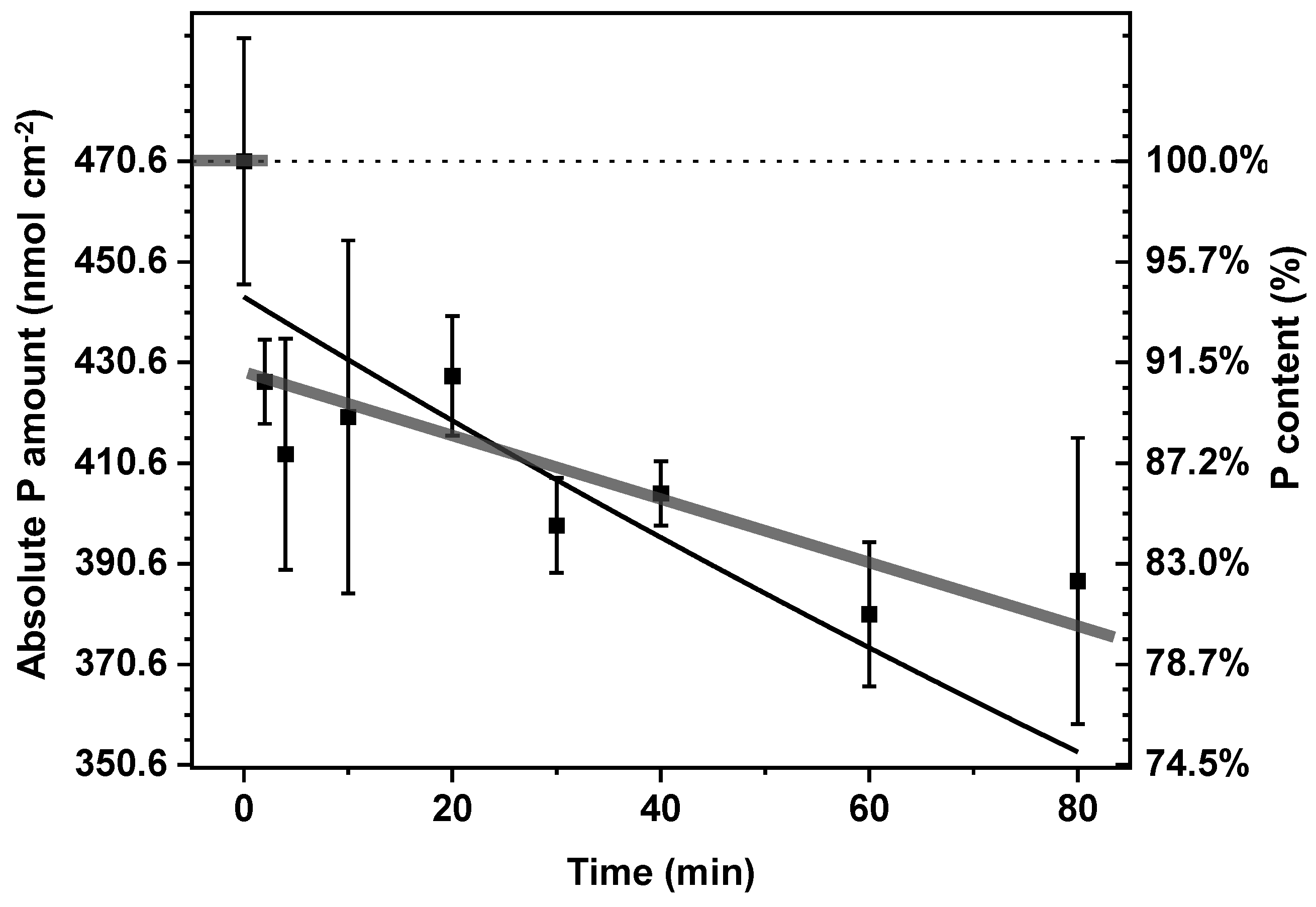
2.1. Slow Release of Phosphate from CoCat Film
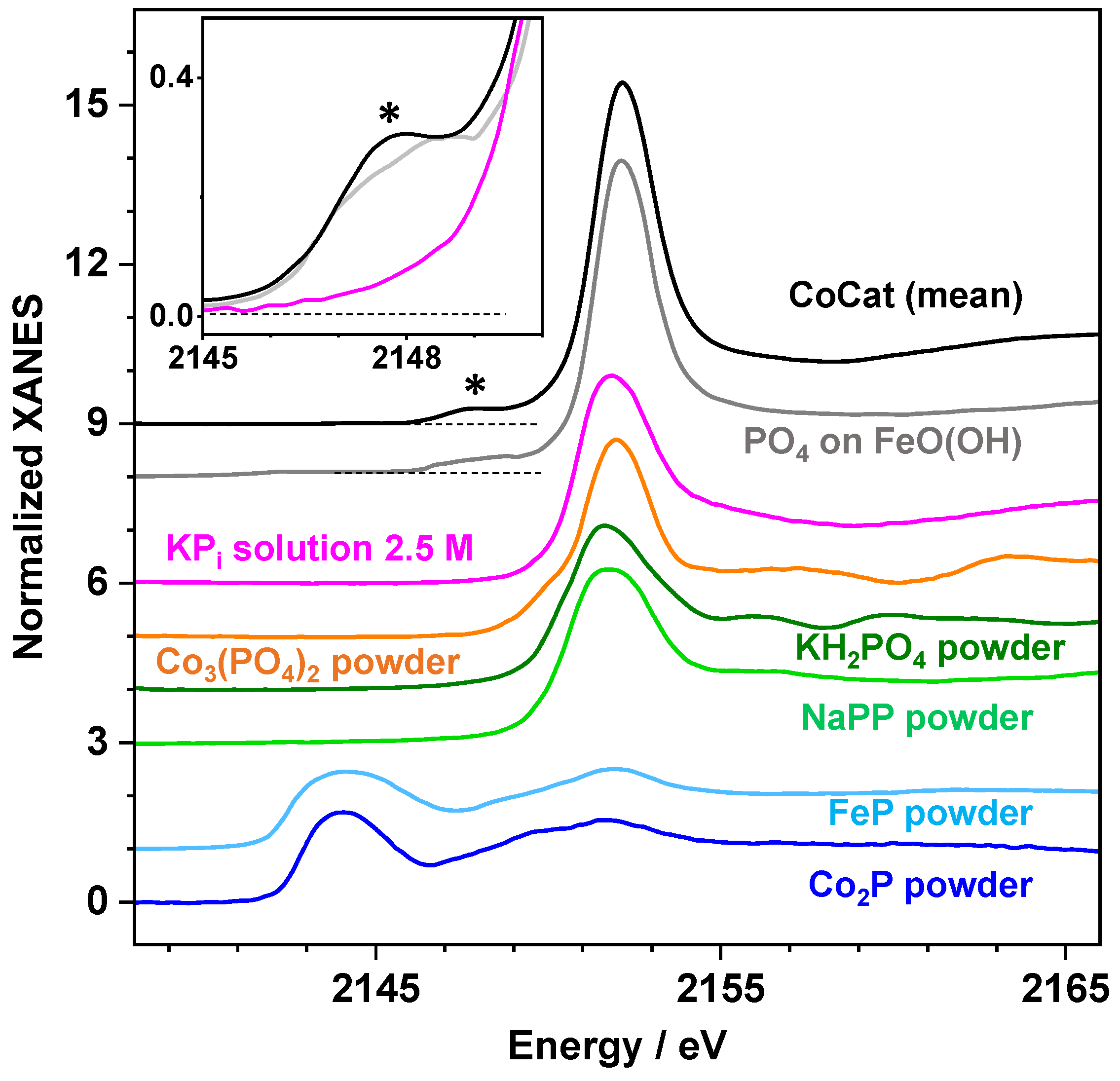
2.2. X-ray Absorption Spectroscopy at the P K-Edge Reveals Phosphate Binding Motifs
3. Materials and Methods
3.1. Malachite Green Assay for P Quantification
3.2. XAS Absorption Spectroscopy (XAS)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kanan, M.W.; Nocera, D.G. In situ formation of an oxygen-evolving catalyst in neutral water containing phosphate and Co2+. Science 2008, 321, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Klingan, K.; Ringleb, F.; Zaharieva, I.; Heidkamp, J.; Chernev, P.; Gonzalez-Flores, D.; Risch, M.; Fischer, A.; Dau, H. Water oxidation by amorphous cobalt-based oxides: Volume activity and proton transfer to electrolyte bases. ChemSusChem 2014, 7, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Kanan, M.W.; Yano, J.; Surendranath, Y.; Dinca, M.; Yachandra, V.K.; Nocera, D.G. Structure and valency of a cobalt-phosphate water oxidation catalyst determined by in situ X-ray spectroscopy. J. Am. Chem. Soc. 2010, 132, 13692–13701. [Google Scholar] [CrossRef] [PubMed]
- Risch, M.; Ringleb, F.; Kohlhoff, M.; Bogdanoff, P.; Chernev, P.; Zaharieva, I.; Dau, H. Water oxidation by amorphous cobalt-based oxides: In situ tracking of redox transitions and mode of catalysis. Energy Environ. Sci. 2015, 8, 661–674. [Google Scholar] [CrossRef]
- Ullman, A.M.; Brodsky, C.N.; Li, N.; Zheng, S.-l.; Nocera, D.G. Probing Edge Site Reactivity of Oxidic Cobalt Water Oxidation Catalysts. J. Am. Chem. Soc. 2016, 138, 4229–4236. [Google Scholar] [CrossRef] [PubMed]
- Du, P.W.; Kokhan, O.; Chapman, K.W.; Chupas, P.J.; Tiede, D.M. Elucidating the domain structure of the cobalt oxide water splitting catalyst by X-ray pair distribution function analysis. J. Am. Chem. Soc. 2012, 134, 11096–11099. [Google Scholar] [CrossRef]
- Eom, C.J.; Suntivich, J. In Situ Stimulated Raman Spectroscopy Reveals the Phosphate Network in the Amorphous Cobalt Oxide Catalyst and Its Role in the Catalyst Formation. J. Phys. Chem. C 2019, 123, 29284–29290. [Google Scholar] [CrossRef]
- Liu, Y.; Nocera, D.G. Spectroscopic Studies of Nanoparticulate Thin Films of a Cobalt-Based Oxygen Evolution Catalyst. J. Phys. Chem. C 2014, 118, 17060–17066. [Google Scholar] [CrossRef]
- Yoshida, M.; Mineo, T.; Mitsutomi, Y.; Yamamoto, F.; Kurosu, H.; Takakusagi, S.; Asakura, K.; Kondoh, H. Structural Relationship between CoO6 Cluster and Phosphate Species in a Cobalt–Phosphate Water Oxidation Catalyst Investigated by Co and P K-edge XAFS. Chem. Lett. 2015, 45, 277–279. [Google Scholar] [CrossRef]
- Harley, S.J.; Mason, H.E.; McAlpin, J.G.; Britt, R.D.; Casey, W.H. A 31P NMR investigation of the CoPi water-oxidation catalyst. Chem. Eur. J. 2012, 18, 10476–10479. [Google Scholar] [CrossRef]
- Pasquini, C.; D’Amario, L.; Zaharieva, I.; Dau, H. Operando Raman spectroscopy tracks oxidation-state changes in an amorphous Co oxide material for electrocatalysis of the oxygen evolution reaction. J. Chem. Phys. 2020, 152, 194202. [Google Scholar] [CrossRef]
- Risch, M.; Klingan, K.; Ringleb, F.; Chernev, P.; Zaharieva, I.; Fischer, A.; Dau, H. Water oxidation by electrodeposited cobalt oxides-role of anions and redox-inert cations in structure and function of the amorphous catalyst. ChemSusChem 2012, 5, 542–549. [Google Scholar] [CrossRef]
- Gerken, J.B.; Landis, E.C.; Hamers, R.J.; Stahl, S.S. Fluoride-modulated cobalt catalysts for electrochemical oxidation of water under non-alkaline conditions. ChemSusChem 2010, 3, 1176–1179. [Google Scholar] [CrossRef]
- Surendranath, Y.; Dinca, M.; Nocera, D.G. Electrolyte-dependent electrosynthesis and activity of cobalt-based water oxidation catalysts. J. Am. Chem. Soc. 2009, 131, 2615–2620. [Google Scholar] [CrossRef]
- Kanan, M.W.; Surendranath, Y.; Nocera, D.G. Cobalt–phosphate oxygen-evolving compound. Chem. Soc. Rev. 2009, 38, 109–114. [Google Scholar] [CrossRef]
- Lutterman, D.A.; Surendranath, Y.; Nocera, D.G. A self-healing oxygen-evolving catalyst. J. Am. Chem. Soc. 2009, 131, 3838–3839. [Google Scholar] [CrossRef]
- Surendranath, Y.; Lutterman, D.A.; Liu, Y.; Nocera, D.G. Nucleation, growth, and repair of a cobalt-based oxygen evolving catalyst. J. Am. Chem. Soc. 2012, 134, 6326–6336. [Google Scholar] [CrossRef]
- Liu, S.; Zaharieva, I.; D’Amario, L.; Mebs, S.; Kubella, P.; Yang, F.; Beyer, P.; Haumann, M.; Dau, H. Electrocatalytic Water Oxidation at Neutral pH–Deciphering the Rate Constraints for an Amorphous Cobalt-Phosphate Catalyst System. Adv. Energy Mater. 2022, 12, 2202914. [Google Scholar] [CrossRef]
- Pasquini, C.; Zaharieva, I.; Gonzalez-Flores, D.; Chernev, P.; Mohammadi, M.R.; Guidoni, L.; Smith, R.D.L.; Dau, H. H/D Isotope effects reveal factors controlling catalytic activity in Co-based oxides for water oxidation. J. Am. Chem. Soc. 2019, 141, 2938–2948. [Google Scholar] [CrossRef]
- Esswein, A.J.; Surendranath, Y.; Reece, S.Y.; Nocera, D.G. Highly active cobalt phosphate and borate based oxygen evolving catalysts operating in neutral and natural waters. Energy Environ. Sci. 2011, 4, 499–504. [Google Scholar] [CrossRef]
- Gerken, J.B.; McAlpin, J.G.; Chen, J.Y.C.; Rigsby, M.L.; Casey, W.H.; Britt, R.D.; Stahl, S.S. Electrochemical water oxidation with cobalt-based electrocatalysts from pH 0–14: The thermodynamic basis for catalyst structure, stability, and activity. J. Am. Chem. Soc. 2011, 133, 14431–14442. [Google Scholar] [CrossRef] [PubMed]
- Bediako, D.K.; Ullman, A.M.; Nocera, D.G. Catalytic Oxygen Evolution by Cobalt Oxido Thin Films. In Solar Energy for Fuels. Topics in Current Chemistry; Tüysüz, H., Chan, C.K., Eds.; Springer: Cham, Switzerland, 2016; Volume 371, pp. 173–213. [Google Scholar] [CrossRef]
- Khosravi, M.; Mohammadi, M.R. Trends and Progress in Application of Cobalt-Based Materials in Catalytic, Electrocatalytic, Photocatalytic, and Photoelectrocatalytic Water Splitting. Photosynth. Res. 2022, 154, 329–352. [Google Scholar] [CrossRef] [PubMed]
- Bediako, D.K.; Costentin, C.; Jones, E.C.; Nocera, D.G.; Savéant, J.-M. Proton–Electron Transport and Transfer in Electrocatalytic Films. Application to a Cobalt-Based O2-Evolution Catalyst. J. Am. Chem. Soc. 2013, 135, 10492–10502. [Google Scholar] [CrossRef]
- Farrow, C.L.; Bediako, D.K.; Surendranath, Y.; Nocera, D.G.; Billinge, S.J.L. Intermediate-Range Structure of Self-Assembled Cobalt-Based Oxygen-Evolving Catalyst. J. Am. Chem. Soc. 2013, 135, 6403–6406. [Google Scholar] [CrossRef]
- Surendranath, Y.; Kanan, M.W.; Nocera, D.G. Mechanistic studies of the oxygen evolution reaction by a cobalt-phosphate catalyst at neutral pH. J. Am. Chem. Soc. 2010, 132, 16501–16509. [Google Scholar] [CrossRef]
- Brodsky, C.N.; Bediako, D.K.; Shi, C.; Keane, T.P.; Costentin, C.; Billinge, S.J.L.; Nocera, D.G. Proton–Electron Conductivity in Thin Films of a Cobalt–Oxygen Evolving Catalyst. ACS Appl. Mater. Interfaces 2019, 2, 3–12. [Google Scholar] [CrossRef]
- Villalobos, J.; Gonzalez-Flores, D.; Klingan, K.; Chernev, P.; Kubella, P.; Urcuyo, R.; Pasquini, C.; Mohammadi, M.R.; Smith, R.D.L.; Montero, M.L.; et al. Structural and functional role of anions in electrochemical water oxidation probed by arsenate incorporation into cobalt-oxide materials. Phys. Chem. Chem. Phys. 2019, 21, 12485–12493. [Google Scholar] [CrossRef]
- Gonzalez-Flores, D.; Sanchez, I.; Zaharieva, I.; Klingan, K.; Heidkamp, J.; Chernev, P.; Menezes, P.W.; Driess, M.; Dau, H.; Montero, M.L. Heterogeneous water oxidation: Surface activity versus amorphization activation in cobalt phosphate catalysts. Angew. Chem. Int. Ed. 2015, 54, 2472–2476. [Google Scholar] [CrossRef]
- Costentin, C.; Porter, T.R.; Saveant, J.-M. Conduction and Reactivity in Heterogeneous-Molecular Catalysis: New Insights in Water Oxidation Catalysis by Phosphate Cobalt Oxide Films. J. Am. Chem. Soc. 2016, 138, 5615–5622. [Google Scholar] [CrossRef]
- Mohammadi, M.R.; Loos, S.; Chernev, P.; Pasquini, C.; Zaharieva, I.; Gonzalez-Flores, D.; Kubella, P.; Klingan, K.; Smith, R.D.L.; Dau, H. Exploring the Limits of Self-Repair in Cobalt Oxide Films for Electrocatalytic Water Oxidation. ACS Catal. 2020, 10, 7990–7999. [Google Scholar] [CrossRef]
- Costentin, C. Proton-Coupled Electron Transfer Catalyst: Heterogeneous Catalysis. Application to an Oxygen Evolution Catalyst. ACS Catal. 2020, 10, 7958–7967. [Google Scholar] [CrossRef]
- Hunter, B.M.; Hieringer, W.; Winkler, J.R.; Gray, H.B.; Müller, A.M. Effect of interlayer anions on [NiFe]-LDH nanosheet water oxidation activity. Energy Environ. Sci. 2016, 9, 1734–1743. [Google Scholar] [CrossRef]
- Yang, C.; Laberty-Robert, C.; Batuk, D.; Cibin, G.; Chadwick, A.V.; Pimenta, V.; Yin, W.; Zhang, L.; Tarascon, J.-M.; Grimaud, A. Phosphate Ion Functionalization of Perovskite Surfaces for Enhanced Oxygen Evolution Reaction. J. Phys. Chem. Lett. 2017, 8, 3466–3472. [Google Scholar] [CrossRef]
- Kwon, G.; Jang, H.; Lee, J.-S.; Mane, A.; Mandia, D.J.; Soltau, S.R.; Utschig, L.M.; Martinson, A.B.F.; Tiede, D.M.; Kim, H.; et al. Resolution of Electronic and Structural Factors Underlying Oxygen-Evolving Performance in Amorphous Cobalt Oxide Catalysts. J. Am. Chem. Soc. 2018, 140, 10710–10720. [Google Scholar] [CrossRef]
- Prietzel, J.; Klysubun, W. Phosphorus K -edge XANES spectroscopy has probably often underestimated iron oxyhydroxide-bound P in soils. J. Synchrotron Radiat. 2018, 25, 1736–1744. [Google Scholar] [CrossRef]
- Kim, H.; Park, J.; Park, I.; Jin, K.; Jerng, S.E.; Kim, S.H.; Nam, K.T.; Kang, K. Coordination tuning of cobalt phosphates towards efficient water oxidation catalyst. Nat. Commun. 2015, 6, 8253. [Google Scholar] [CrossRef]
- Ronge, E.; Ohms, J.; Roddatis, V.; Jones, T.; Sulzmann, F.; Knop-Gericke, A.; Schlögl, R.; Kurz, P.; Jooss, C.; Skorupska, K. Operation of calcium-birnessite water-oxidation anodes: Interactions of the catalyst with phosphate buffer anions. Sustain. Energy Fuels 2021, 5, 5535–5547. [Google Scholar] [CrossRef]
- Rudolph, W.W.; Irmer, G. Raman and infrared spectroscopic investigations on aqueous alkali metal phosphate solutions and density functional theory calculations of phosphate-water clusters. Appl. Spectrosc. 2007, 61, 1312–1327. [Google Scholar] [CrossRef]
- Preston, C.M.; Adams, W.A. A laser Raman spectroscopic study of aqueous orthophosphate salts. J. Phys. Chem. C 1979, 83, 814–821. [Google Scholar] [CrossRef]
- Mason, P.E.; Cruickshank, J.M.; Neilson, G.W.; Buchanan, P. Neutron scattering studies on the hydration of phosphate ions in aqueous solutions of K3PO4, K2HPO4 and KH2PO4. Phys. Chem. Chem. Phys. 2003, 5, 4686–4690. [Google Scholar] [CrossRef]
- Cade-Menun, B.; Liu, C.W. Solution Phosphorus-31 Nuclear Magnetic Resonance Spectroscopy of Soils from 2005 to 2013: A Review of Sample Preparation and Experimental Parameters. Soil Sci. Soc. Am. J. 2014, 78, 19–37. [Google Scholar] [CrossRef]
- Kruse, J.; Abraham, M.; Amelung, W.; Baum, C.; Bol, R.; Kühn, O.; Lewandowski, H.; Niederberger, J.; Oelmann, Y.; Rüger, C.; et al. Innovative methods in soil phosphorus research: A review. J. Plant. Nutr. Soil Sci. 2015, 178, 43–88. [Google Scholar] [CrossRef] [PubMed]
- Kizewski, F.; Liu, Y.T.; Morris, A.; Hesterberg, D. Spectroscopic Approaches for Phosphorus Speciation in Soils and Other Environmental Systems. J. Environ. Qual. 2011, 40, 751–766. [Google Scholar] [CrossRef] [PubMed]
- Khare, N.; Hesterberg, D.; Martin, J.D. XANES investigation of phosphate sorption in single and binary systems of iron and aluminum oxide minerals. Environ. Sci. Technol. 2005, 39, 2152–2160. [Google Scholar] [CrossRef]
- Khare, N.; Hesterberg, D.; Beauchemin, S.; Wang, S.-L. XANES Determination of Adsorbed Phosphate Distribution between Ferrihydrite and Boehmite in Mixtures. Soil Sci. Soc. Am. J. 2004, 68, 460–469. [Google Scholar] [CrossRef]
- Persson, I.; Trublet, M.; Klysubun, W. Structure Determination of Phosphoric Acid and Phosphate Ions in Aqueous Solution Using EXAFS Spectroscopy and Large Angle X-ray Scattering. J. Phys. Chem. A 2018, 122, 7413–7420. [Google Scholar] [CrossRef]
- Beauchemin, S.; Hesterberg, D.; Chou, J.; Beauchemin, M.; Simard, R.R.; Sayers, D.E. Speciation of Phosphorus in Phosphorus-Enriched Agricultural Soils Using X-Ray Absorption Near-Edge Structure Spectroscopy and Chemical Fractionation. J. Environ. Qual. 2003, 32, 1809–1819. [Google Scholar] [CrossRef]
- Luo, L.; Ma, Y.; Sanders, R.L.; Xu, C.; Li, J.; Myneni, S.C.B. Phosphorus speciation and transformation in long-term fertilized soil: Evidence from chemical fractionation and P K-edge XANES spectroscopy. Nutr. Cycl. Agroecosyst. 2017, 107, 215–226. [Google Scholar] [CrossRef]
- Hurtarte, L.C.C.; Souza-Filho, L.F.; Santos, W.O.; Vergütz, L.; Prietzel, J.; Hesterberg, D. Optimization of data processing minimizes impact of self-absorption on phosphorus speciation results by P K-edge XANES. Soil Systems 2019, 3, 61. [Google Scholar] [CrossRef]
- Menezes, P.W.; Panda, C.; Walter, C.; Schwarze, M.; Driess, M. A Cobalt-Based Amorphous Bifunctional Electrocatalysts for Water-Splitting Evolved from a Single-Source Lazulite Cobalt Phosphate. Adv. Func. Mater. 2019, 29, 1808632. [Google Scholar] [CrossRef]
- Gond, R.; Sada, K.; Senthilkumar, B.; Barpanda, P. Bifunctional Electrocatalytic Behavior of Sodium Cobalt Phosphates in Alkaline Solution. ChemElectroChem 2018, 5, 153–158. [Google Scholar] [CrossRef]
- Liu, M.; Qu, Z.; Yin, D.; Chen, X.; Zhang, Y.; Guo, Y.; Xiao, D. Cobalt–Iron Pyrophosphate Porous Nanosheets as Highly Active Electrocatalysts for the Oxygen Evolution Reaction. ChemElectroChem 2018, 5, 36–43. [Google Scholar] [CrossRef]
- Rouff, A.A.; Rabe, S.; Nachtegaal, M.; Vogel, F. X-ray absorption fine structure study of the effect of protonation on disorder and multiple scattering in phosphate solutions and solids. J. Phys. Chem. A 2009, 113, 6895–6903. [Google Scholar] [CrossRef]
- Dalba, G.; Fornasini, P.; Rocca, F.; Lagarde, P.; Vlaic, G. EXAFS study of the coordination of phosphorus in AgPO3 glass. J. Non-Cryst. Solids 1988, 106, 181–184. [Google Scholar] [CrossRef]
- Küper, G.; Hormes, J.; Sommer, K. In situ X-ray absorption spectroscopy at the K-edge of red phosphorus in polyamide 6,6 during a thermo-oxidative degradation. Macromol. Chem. Phys. 1994, 195, 1741–1753. [Google Scholar] [CrossRef]
- Liu, S.; Farhoosh, S.; Beyer, P.; Mebs, S.; Zaharieva, I.; Haumann, M.; Dau, H. Role of Potassium in Electrocatalytic Water Oxidation Investigated in a Volume-Active Cobalt Material at Neutral pH. Adv. Sustain. Syst. 2023, 7, 2300008. [Google Scholar] [CrossRef]
- Rao, A.S.; Reddy, K.S.; Takkar, P.N. Malachite green method compared to ascorbic acid for estimating small amounts of phosphorus in water, 0.01 M calcium chloride, and olsen soil extracts. Commun. Soil Sci. Plant Anal. 1997, 28, 589–601. [Google Scholar] [CrossRef]
- Feng, J.; Chen, Y.; Pu, J.; Yang, X.; Zhang, C.; Zhu, S.; Zhao, Y.; Yuan, Y.; Yuan, H.; Liao, F. An improved malachite green assay of phosphate: Mechanism and application. Anal. Biochem. 2011, 409, 144–149. [Google Scholar] [CrossRef]
- Rahutomo, S.; Kovar, J.L.; Thompson, M.L. Malachite Green Method for Determining Phosphorus Concentration in Diverse Matrices. Commun. Soil Sci. Plant Anal. 2019, 50, 1743–1752. [Google Scholar] [CrossRef]
- Franke, R. X-ray absorption and photoelectron spectroscopy investigation of binary Nickelphosphides. Spectrochim. Acta A Mol. Biomol. Spectrosc. 1997, 53, 933–941. [Google Scholar] [CrossRef]
- Yin, Z.; Kasrai, M.; Bancroft, G.M.; Tan, K.H.; Feng, X. X-ray-absorption spectroscopic studies of sodium polyphosphate glasses. Phys. Rev. B 1995, 51, 742–750. [Google Scholar] [CrossRef] [PubMed]
- Prietzel, J.; Dümig, A.; Wu, Y.; Zhou, J.; Klysubun, W. Synchrotron-based P K-edge XANES spectroscopy reveals rapid changes of phosphorus speciation in the topsoil of two glacier foreland chronosequences. Geochim. Cosmochim. Acta 2013, 108, 154–171. [Google Scholar] [CrossRef]
- Werner, F.; Prietzel, J. Standard Protocol and Quality Assessment of Soil Phosphorus Speciation by P K-Edge XANES Spectroscopy. Environ. Sci. Technol. 2015, 49, 10521–10528. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Rivard, C.; Wilken, V.; Muskolus, A.; Adam, C. Performance of secondary P-fertilizers in pot experiments analyzed by phosphorus X-ray absorption near-edge structure (XANES) spectroscopy. Ambio 2018, 47, 62–72. [Google Scholar] [CrossRef]
- Schmieder, F.; Gustafsson, J.P.; Klysubun, W.; Zehetner, F.; Riddle, M.; Kirchmann, H.; Bergström, L. Phosphorus speciation in cultivated organic soils revealed by P K-edge XANES spectroscopy. J. Plant. Nutr. Soil Sci. 2020, 183, 367–381. [Google Scholar] [CrossRef]
- Donahue, C.M.; Daly, S.R. Ligand K-Edge XAS Studies of Metal-Phosphorus Bonds: Applications, Limitations, and Opportunities. Comment. Inorg. Chem. 2018, 38, 54–78. [Google Scholar] [CrossRef]
- Popmintchev, D.; Galloway, B.R.; Chen, M.-C.; Dollar, F.; Mancuso, C.A.; Hankla, A.; Miaja-Avila, L.; O’Neil, G.; Shaw, J.M.; Fan, G.; et al. Near- and Extended-Edge X-Ray-Absorption Fine-Structure Spectroscopy Using Ultrafast Coherent High-Order Harmonic Supercontinua. Phys. Rev. Lett. 2018, 120, 093002. [Google Scholar] [CrossRef]
- Giusti, L.; Landaeta, V.R.; Vanni, M.; Kelly, J.A.; Wolf, R.; Caporali, M. Coordination chemistry of elemental phosphorus. Coord. Chem. Rev. 2021, 441, 213927. [Google Scholar] [CrossRef]
- Zabinsky, S.I.; Rehr, J.J.; Ankudinov, A.; Albers, R.C.; Eller, M.J. Multiple-scattering calculations of x-ray-absorption spectra. Phys. Rev. B 1995, 52, 2995–3009. [Google Scholar] [CrossRef]
- Dau, H.; Liebisch, P.; Haumann, M. X-ray absorption spectroscopy to analyze nuclear geometry and electronic structure of biological metal centers-potential and questions examined with special focus on the tetra-nuclear manganese complex of oxygenic photosynthesis. Anal. Bioanal. Chem. 2003, 376, 562–583. [Google Scholar] [CrossRef]
- Risch, M.; Khare, V.; Zaharieva, I.; Gerencser, L.; Chernev, P.; Dau, H. Cobalt-oxo core of a water-oxidizing catalyst film. J. Am. Chem. Soc. 2009, 131, 6936–6937. [Google Scholar] [CrossRef]
- The Materials Project. Materials Data on FeP by Materials Project; United States Department of Energy: Washington, DC, USA, 2020. [Google Scholar]
- Zheng, T.; Yang, Z.; Gui, D.; Liu, Z.; Wang, X.; Dai, X.; Liu, S.; Zhang, L.; Gao, Y.; Chen, L.; et al. Overcoming the crystallization and designability issues in the ultrastable zirconium phosphonate framework system. Nat. Commun. 2017, 8, 15369. [Google Scholar] [CrossRef]
- Hultgren, R.; Gingrich, N.S.; Warren, B.E. The Atomic Distribution in Red and Black Phosphorus and the Crystal Structure of Black Phosphorus. J. Chem. Phys. 1935, 3, 351–355. [Google Scholar] [CrossRef]
- Endo, S.; Chino, T.; Tsuboi, S.; Koto, K. Pressure-induced transition of the hydrogen bond in the ferroelectric compounds KH2PO4 and KD2PO4. Nature 1989, 340, 452–455. [Google Scholar] [CrossRef]
- Leung, K.Y.; Calvo, C. The Structure of Na4P2O7 at 22 °C. Can. J. Chem. 1972, 50, 2519–2526. [Google Scholar] [CrossRef]
- Wyckoff, R.W.G. Second edition. Interscience Publishers, New York, New York Note: Cadmium iodide structure. Cryst. Struct. 1963, 1, 239–444. [Google Scholar]
- Nord, A.G.; Stefanidis, T. Structure refinements of Co3(PO4)2. A note on the reliability of powder diffraction studies. Acta Chem. Scand. Ser. A 1983, 37, 715–721. [Google Scholar] [CrossRef][Green Version]
- Krupkova, R.; Fabry, J.; Vanek, P.; Cisarova, I. Two modifications of a KH2PO4.HF adduct. Acta Cryst. C 2003, 59, i79–i82. [Google Scholar] [CrossRef]
- Zhou, H.; Upreti, S.; Chernova, N.A.; Whittingham, M.S. Lithium cobalt(II) pyrophosphate, Li1.86CoP2O7, from synchrotron X-ray powder data. Acta Cryst. E 2011, 67, i58–i59. [Google Scholar] [CrossRef]
- Taxer, K.; Bartl, H. On the dimorphy between the variscite and clinovariscite group: Refined finestructural relationship of strengite and clinostrengite, Fe(PO4) 2H2O. Cryst. Res. Technol. 2004, 39, 1080–1088. [Google Scholar] [CrossRef]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. Commentary: The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Shell | N (per P) | R (Å) | 2σ2 (Å2) | RF (%) | |
|---|---|---|---|---|---|
| CoCat | P–O | 4 * | 1.55 ± 0.01 | 0.006 # | 11.7 |
| P–O–Oms | 6 * | 2.42 ± 0.01 | 0.006 # | ||
| P–Co | 0.1 ± 0.1 | 2.26 ± 0.03 | 0.006 # | ||
| P–Co | 0.7 ± 0.2 | 3.09 ± 0.03 | 0.006 # | ||
| P–K | 0.9 ± 0.3 | 3.30 ± 0.03 | 0.006 # |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Farhoosh, S.; Beyer, P.; Mebs, S.; Haumann, M.; Dau, H. Phosphate Coordination in a Water-Oxidizing Cobalt Oxide Electrocatalyst Revealed by X-ray Absorption Spectroscopy at the Phosphorus K-Edge. Catalysts 2023, 13, 1151. https://doi.org/10.3390/catal13081151
Liu S, Farhoosh S, Beyer P, Mebs S, Haumann M, Dau H. Phosphate Coordination in a Water-Oxidizing Cobalt Oxide Electrocatalyst Revealed by X-ray Absorption Spectroscopy at the Phosphorus K-Edge. Catalysts. 2023; 13(8):1151. https://doi.org/10.3390/catal13081151
Chicago/Turabian StyleLiu, Si, Shima Farhoosh, Paul Beyer, Stefan Mebs, Michael Haumann, and Holger Dau. 2023. "Phosphate Coordination in a Water-Oxidizing Cobalt Oxide Electrocatalyst Revealed by X-ray Absorption Spectroscopy at the Phosphorus K-Edge" Catalysts 13, no. 8: 1151. https://doi.org/10.3390/catal13081151
APA StyleLiu, S., Farhoosh, S., Beyer, P., Mebs, S., Haumann, M., & Dau, H. (2023). Phosphate Coordination in a Water-Oxidizing Cobalt Oxide Electrocatalyst Revealed by X-ray Absorption Spectroscopy at the Phosphorus K-Edge. Catalysts, 13(8), 1151. https://doi.org/10.3390/catal13081151