


Atom Transfer Radical Addition via Dual Photoredox/Manganese Catalytic System

Abstract
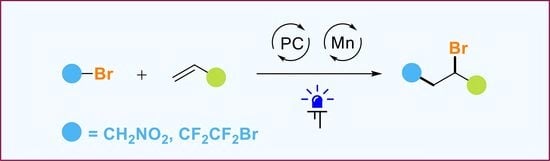
1. Introduction
2. Results
3. Materials and Methods
3.1. General Information
3.1.1. Synthesis of 2-Cyclopropylpent-4-en-2-ol (1s) [52]
3.1.2. Synthesis of 1-Phenylbut-3-en-1-yl 2,2,2-Trifluoroacetate (1t) [53]
3.2. ATRA Reaction of Bromides 2a-b with Alkenes (General Procedure)
3.3. Gram-Scale Synthesis of 1,4-Dibromo-1,1,2,2-tetrafluorodecane (3u)
3.4. Synthesis of 1-Bromo-1,1,2,2-tetrafluorodec-3-ene (4)
3.5. Synthesis of 9-Bromo-6,6,7,7-tetrafluoropentadecan-1-ol (5)
3.6. Synthesis of 1,1,2,2-Tetrafluorodecane (6)
3.7. Radical Clock Experiment, Synthesis of Dimethyl 3-(3-Bromo-2,2,3,3-tetrafluoropropyl)-4-(bromomethyl)cyclopentane-1,1-dicarboxylate (7)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Munoz-Molina, J.M.; Belderrain, T.R.; Perez, P.J. Atom Transfer Radical Reactions as a Tool for Olefin Functionalization—On the Way to Practical Applications. Eur. J. Inorg. Chem. 2011, 2011, 3155–3164. [Google Scholar] [CrossRef]
- Reiser, O. Shining Light on Copper: Unique Opportunities for Visible-Light-Catalyzed Atom Transfer Radical Addition Reactions and Related Processes. Acc. Chem. Res. 2016, 49, 1990–1996. [Google Scholar] [CrossRef] [PubMed]
- Eckenhoff, W.T.; Pintauer, T. Copper Catalyzed Atom Transfer Radical Addition (ATRA) and Cyclization (ATRC) Reactions in the Presence of Reducing Agents. Catal. Rev. 2010, 52, 1–59. [Google Scholar] [CrossRef]
- Pintauer, T. Catalyst Regeneration in Transition-Metal-Mediated Atom-Transfer Radical Addition (ATRA) and Cyclization (ATRC) Reactions. Eur. J. Inorg. Chem. 2010, 2010, 2449–2460. [Google Scholar] [CrossRef]
- Engl, S.; Reiser, O. Copper-photocatalyzed ATRA reactions: Concepts, applications, and opportunities. Chem. Soc. Rev. 2022, 51, 5287–5299. [Google Scholar] [CrossRef]
- Pintauer, T.; Matyjaszewski, K. Atom transfer radical addition and polymerization reactions catalyzed by ppm amounts of copper complexes. Chem. Soc. Rev. 2008, 37, 1087–1097. [Google Scholar] [CrossRef]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef]
- Tucker, J.W.; Stephenson, C.R.J. Shining Light on Photoredox Catalysis: Theory and Synthetic Applications. J. Org. Chem. 2012, 77, 1617–1622. [Google Scholar] [CrossRef]
- Marzo, L.; Pagire, S.K.; Reiser, O.; König, B. Visible-Light Photocatalysis: Does It Make a Difference in Organic Synthesis? Angew. Chem. Int. Ed. 2018, 57, 10034–10072. [Google Scholar] [CrossRef]
- Candish, L.; Collins, K.D.; Cook, G.C.; Douglas, J.J.; Gómez-Suárez, A.; Jolit, A.; Keess, S. Photocatalysis in the Life Science Industry. Chem. Rev. 2022, 122, 2907–2980. [Google Scholar] [CrossRef]
- Schultz, D.M.; Yoon, T.P. Solar Synthesis: Prospects in Visible Light Photocatalysis. Science 2014, 343, 1239176. [Google Scholar] [CrossRef] [PubMed]
- Wallentin, C.-J.; Nguyen, J.D.; Finkbeiner, P.; Stephenson, C.R.J. Visible Light-Mediated Atom Transfer Radical Addition via Oxidative and Reductive Quenching of Photocatalysts. J. Am. Chem. Soc. 2012, 134, 8875–8884. [Google Scholar] [CrossRef] [PubMed]
- Barata-Vallejo, S.; Cooke, M.V.; Postigo, A. Radical Fluoroalkylation Reactions. ACS Catal. 2018, 8, 7287–7307. [Google Scholar] [CrossRef]
- Chernov, G.I.; Levin, V.V.; Dilman, A.D. Photocatalytic reactions of fluoroalkyl iodides with alkenes. Russ. Chem. Bull. 2023, 72, 61–72. [Google Scholar] [CrossRef]
- Rosso, C.; Williams, J.D.; Filippini, G.; Prato, M.; Kappe, C.O. Visible-Light-Mediated Iodoperfluoroalkylation of Alkenes in Flow and Its Application to the Synthesis of a Key Fulvestrant Intermediate. Org. Lett. 2019, 21, 5341–5345. [Google Scholar] [CrossRef]
- Helmecke, L.; Spittler, M.; Baumgarten, K.; Czekelius, C. Metal-Free Activation of C–I Bonds and Perfluoroalkylation of Alkenes with Visible Light Using Phosphine Catalysts. Org. Lett. 2019, 21, 7823–7827. [Google Scholar] [CrossRef] [PubMed]
- Mao, T.; Ma, M.-J.; Zhao, L.; Xue, D.-P.; Yu, Y.; Gu, J.; He, C.-Y. A general and green fluoroalkylation reaction promoted via noncovalent interactions between acetone and fluoroalkyl iodides. Chem. Commun. 2020, 56, 1815–1818. [Google Scholar] [CrossRef]
- Li, D.; Mao, T.; Huang, J.; Zhu, Q. Copper-Catalyzed Bromodifluoroacetylation of Alkenes with Ethyl Bromodifluoroacetate. J. Org. Chem. 2018, 83, 10445–10452. [Google Scholar] [CrossRef]
- Granados, A.; Dhungana, R.K.; Sharique, M.; Majhi, J.; Molander, G.A. From Styrenes to Fluorinated Benzyl Bromides: A Photoinduced Difunctionalization via Atom Transfer Radical Addition. Org. Lett. 2022, 24, 4750–4755. [Google Scholar] [CrossRef]
- Fedorov, O.V.; Scherbinina, S.I.; Levin, V.V.; Dilman, A.D. Light-Mediated Dual Phosphine-/Copper-Catalyzed Atom Transfer Radical Addition Reaction. J. Org. Chem. 2019, 84, 11068–11079. [Google Scholar] [CrossRef]
- Matsuo, K.; Yamaguchi, E.; Itoh, A. Atom-Transfer Radical Addition Photocatalysis Using a Heteroleptic Copper Complex. Asian J. Org. Chem. 2018, 7, 2435–2438. [Google Scholar] [CrossRef]
- Földesi, T.; Sipos, G.; Adamik, R.; Nagy, B.; Tóth, B.L.; Bényei, A.; Szekeres, K.J.; Láng, G.G.; Demeter, A.; Peelen, T.J.; et al. Design and application of diimine-based copper(i) complexes in photoredox catalysis. Org. Biomol. Chem. 2019, 17, 8343–8347. [Google Scholar] [CrossRef] [PubMed]
- Kostromitin, V.S.; Zemtsov, A.A.; Kokorekin, V.A.; Levin, V.V.; Dilman, A.D. Atom-transfer radical addition of fluoroalkyl bromides to alkenes via a photoredox/copper catalytic system. Chem. Commun. 2021, 57, 5219–5222. [Google Scholar] [CrossRef] [PubMed]
- Kostromitin, V.S.; Zemtsov, A.A.; Levin, V.V.; Dilman, A.D. Photocatalytic Atom-Transfer Radical Addition of Activated Chlorides to Alkenes. Adv. Synth. Cat. 2021, 363, 5336–5340. [Google Scholar] [CrossRef]
- Fishwick, B.R.; Rowles, D.K.; Stirling, C.J.M. Bromonitromethane—A versatile electrophile. J. Chem. Soc. Perkin Trans. 1986, 1, 1171–1179. [Google Scholar] [CrossRef]
- Dmowski, W. 1,2-Dibromotetrafluoroethane (Freon 114B2) as a building block for fluorine compounds. J. Fluor. Chem. 2012, 142, 6–13. [Google Scholar] [CrossRef]
- Reichle, A.; Koch, M.; Sterzel, H.; Großkopf, L.-J.; Floss, J.; Rehbein, J.; Reiser, O. Copper(I) Photocatalyzed Bromonitroalkylation of Olefins: Evidence for Highly Efficient Inner-Sphere Pathways. Angew. Chem. Int. Ed. 2023, 62, e202219086. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Onai, R.; Uraguchi, D.; Ooi, T. Redox-regulated divergence in photocatalytic addition of α-nitro alkyl radicals to styrenes. Chem. Commun. 2020, 56, 11014–11017. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.-H.; Huang, W.-Y. Studies on sulfinatodehalogenation: The addition reaction of halocarbons with olefins initiated by sodium dithionite. J. Fluor. Chem. 2001, 110, 59–61. [Google Scholar] [CrossRef]
- Hu, C.-M.; Qiu, Y.-L. Addition of 1,2-dihaloperfluoroalkanes to alkenes initiated by dichlorobis(π-cyclopentadienyl)titanium and iron. J. Fluor. Chem. 1991, 55, 109–111. [Google Scholar] [CrossRef]
- Vaclavik, J.; Klimankova, I.; Budinska, A.; Beier, P. Advances in the Synthesis and Application of Tetrafluoroethylene- and 1,1,2,2-Tetrafluoroethyl-Containing Compounds. Eur. J. Org. Chem. 2018, 2018, 3554–3593. [Google Scholar] [CrossRef]
- Bian, K.-J.; Nemoto, D., Jr.; Kao, S.-C.; He, Y.; Li, Y.; Wang, X.-S.; West, J.G. Modular Difunctionalization of Unactivated Alkenes through Bio-Inspired Radical Ligand Transfer Catalysis. J. Am. Chem. Soc. 2022, 144, 11810–11821. [Google Scholar] [CrossRef] [PubMed]
- Speckmeier, E.; Fischer, T.G.; Zeitler, K. A Toolbox Approach To Construct Broadly Applicable Metal-Free Catalysts for Photoredox Chemistry: Deliberate Tuning of Redox Potentials and Importance of Halogens in Donor-Acceptor Cyanoarenes. J. Am. Chem. Soc. 2018, 140, 15353–15365. [Google Scholar] [CrossRef]
- Shang, T.-Y.; Lu, L.-H.; Cao, Z.; Liu, Y.; He, W.-M.; Yu, B. Recent advances of 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN) in photocatalytic transformations. Chem. Commun. 2019, 55, 5408–5419. [Google Scholar] [CrossRef] [PubMed]
- Kostromitin, V.S.; Sorokin, A.O.; Levin, V.V.; Dilman, A.D. Aminals as powerful XAT-reagents: Activation of fluorinated alkyl chlorides. Chem. Sci. 2023, 14, 3229–3234. [Google Scholar] [CrossRef] [PubMed]
- Kostromitin, V.S.; Levin, V.V.; Dilman, A.D. Organophotoredox-Catalyzed Reductive Tetrafluoroalkylation of Alkenes. J. Org. Chem. 2023, 88, 6523–6531. [Google Scholar] [CrossRef]
- Panferova, L.I.; Levin, V.V.; Struchkova, M.I.; Dilman, A.D. Light-mediated copper-catalyzed phosphorus/halogen exchange in 1,1-difluoroalkylphosphonium salts. Chem. Commun. 2019, 55, 1314–1317. [Google Scholar] [CrossRef]
- Kosobokov, M.D.; Zubkov, M.O.; Levin, V.V.; Kokorekin, V.A.; Dilman, A.D. Fluoroalkyl sulfides as photoredox-active coupling reagents for alkene difunctionalization. Chem. Commun. 2020, 56, 9453–9456. [Google Scholar] [CrossRef]
- Kawamoto, A.M.; Wills, M. Enantioselective synthesis of β-hydroxy amines and aziridines using asymmetric transfer hydrogenation of α-amino ketones. J. Chem. Soc. Perkin Trans. 2001, 1, 1916–1928. [Google Scholar] [CrossRef]
- Yoshida, M.; Higuchi, M.; Shishido, K. Stereoselective Construction of Substituted Chromans by Palladium-Catalyzed Cyclization of Propargylic Carbonates with 2-(2-Hydroxyphenyl)acetates. Org. Lett. 2009, 11, 4752–4755. [Google Scholar] [CrossRef]
- Sušnik, P.; Hilt, G. Homoallylpinacolboronic Ester as Alkene Component in Cobalt-Catalyzed Alder Ene Reactions. Organometallics 2014, 33, 5907–5910. [Google Scholar] [CrossRef]
- Beckwith, A.L.J.; Moad, G. Cyclization of 3-allylhex-5-enyl radical: Mechanism, and implications concerning the structures of cyclopolymers. J. Chem. Soc. Perkin Trans. 1975, 2, 1726–1733. [Google Scholar] [CrossRef]
- Burns, J.M.; Krenske, E.H.; McGeary, R.P. Aromatic Claisen Rearrangements of Benzyl Ketene Acetals: Conversion of Benzylic Alcohols to (ortho-Tolyl)acetates. Eur. J. Org. Chem. 2017, 2017, 252–256. [Google Scholar] [CrossRef]
- Voutyritsa, E.; Nikitas, N.F.; Apostolopoulou, M.K.; Gerogiannopoulou, A.D.D.; Kokotos, C.G. Photoorganocatalytic Atom Transfer Radical Addition of Bromoacetonitrile to Aliphatic Olefins. Synthesis 2018, 50, 3395–3401. [Google Scholar] [CrossRef]
- Scheller, M.E.; Frei, B. Syntheses of Cyclopropyl Silyl Ketones. Helv. Chim. Acta 1986, 69, 44–52. [Google Scholar] [CrossRef]
- Chen, C.; Dugan, T.R.; Brennessel, W.W.; Weix, D.J.; Holland, P.L. Z-Selective Alkene Isomerization by High-Spin Cobalt(II) Complexes. J. Am. Chem. Soc. 2014, 136, 945–955. [Google Scholar] [CrossRef]
- Lu, Z.; Zeng, X.; Hammond, G.B.; Xu, B. Widely Applicable Hydrofluorination of Alkenes via Bifunctional Activation of Hydrogen Fluoride. J. Am. Chem. Soc. 2017, 139, 18202–18205. [Google Scholar] [CrossRef]
- Fan, B.-Z.; Hiasa, H.; Lv, W.; Brody, S.; Yang, Z.-Y.; Aldrich, C.; Cushman, M.; Liang, J.-H. Design, synthesis and structure-activity relationships of novel 15-membered macrolides: Quinolone/quinoline-containing sidechains tethered to the C-6 position of azithromycin acylides. Eur. J. Med. Chem. 2020, 193, 112222. [Google Scholar] [CrossRef]
- Huang, H.; Li, X.; Yu, C.; Zhang, Y.; Mariano, P.S.; Wang, W. Visible-Light-Promoted Nickel- and Organic-Dye-Cocatalyzed Formylation Reaction of Aryl Halides and Triflates and Vinyl Bromides with Diethoxyacetic Acid as a Formyl Equivalent. Angew. Chem. Int. Ed. 2017, 56, 1500–1505. [Google Scholar] [CrossRef]
- Lux, M.; Klussmann, M. Additions of Aldehyde-Derived Radicals and Nucleophilic N-Alkylindoles to Styrenes by Photoredox Catalysis. Org. Lett. 2020, 22, 3697–3701. [Google Scholar] [CrossRef]
- Hayashi, Y.; Gotoh, H.; Tamura, T.; Yamaguchi, H.; Masui, R.; Shoji, M. Cysteine-Derived Organocatalyst in a Highly Enantioselective Intramolecular Michael Reaction. J. Am. Chem. Soc. 2005, 127, 16028–16029. [Google Scholar] [CrossRef] [PubMed]
- Curry, M.J.; Stevens, I.D.R. Preparation and stereochemistry of some 1,1-disubstituted buta-1,3-dienes. J. Chem. Soc. Perkin Trans. 1980, 1, 1756–1760. [Google Scholar] [CrossRef]
- Creary, X.; O’Donnel, B.D.; Vervaeke, M. Homoallyl−Cyclopropylcarbinyl Cation Manifold. Trimethylsilyl versus Aryl Stabilization. J. Org. Chem. 2007, 72, 3360–3368. [Google Scholar] [CrossRef] [PubMed]
- Schulz, G.; George, V.; Taser, D.; Kirschning, A. Taming Bromine Azide for Use in Organic Solvents─Radical Bromoazidations and Alcohol Oxidations. J. Org. Chem. 2023, 88, 3781–3786. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
| Entry | Deviation from Stand. Cond. | Conv. of 1a, % 1 | Y. of 3a, % 2 | Y. of 4a, % 2 |
| 1 | None | >99 | 70 (60) 3 | 20 |
| 2 | MeCN as solv., 24 h | >99 | 56 | 19 |
| 3 | DMF as solv., 24 h | 97 | 64 | 21 |
| 4 | 3DPAFIPN as PC, 24 h | 91 | 32 | 25 |
| 5 | Conc. 0.125 M | >99 | 61 | 13 |
| 6 | 60 W LED, 2 h | >99 | 68 | 14 |
| 7 | 1.2 equiv. of 2a, 8 h | >99 | 68 | 17 |
| 8 | L1 (11%) | >99 | 71 | 18 |
| 9 | L2 (11%) | 85 | 57 | 20 |
| 10 | L3 (11%) | 95 | 48 | 22 |
| 11 | no MnBr2 | >99 | 42 | 13 |
| 12 | Imes·CuBr instead of MnBr2 | 1 | n.d. | n.d. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kostromitin, V.S.; Levin, V.V.; Dilman, A.D. Atom Transfer Radical Addition via Dual Photoredox/Manganese Catalytic System. Catalysts 2023, 13, 1126. https://doi.org/10.3390/catal13071126
Kostromitin VS, Levin VV, Dilman AD. Atom Transfer Radical Addition via Dual Photoredox/Manganese Catalytic System. Catalysts. 2023; 13(7):1126. https://doi.org/10.3390/catal13071126
Chicago/Turabian StyleKostromitin, Vladislav S., Vitalij V. Levin, and Alexander D. Dilman. 2023. "Atom Transfer Radical Addition via Dual Photoredox/Manganese Catalytic System" Catalysts 13, no. 7: 1126. https://doi.org/10.3390/catal13071126
APA StyleKostromitin, V. S., Levin, V. V., & Dilman, A. D. (2023). Atom Transfer Radical Addition via Dual Photoredox/Manganese Catalytic System. Catalysts, 13(7), 1126. https://doi.org/10.3390/catal13071126