


Recent Advances in Electrocatalysts for Ammonia Oxidation Reaction



Abstract
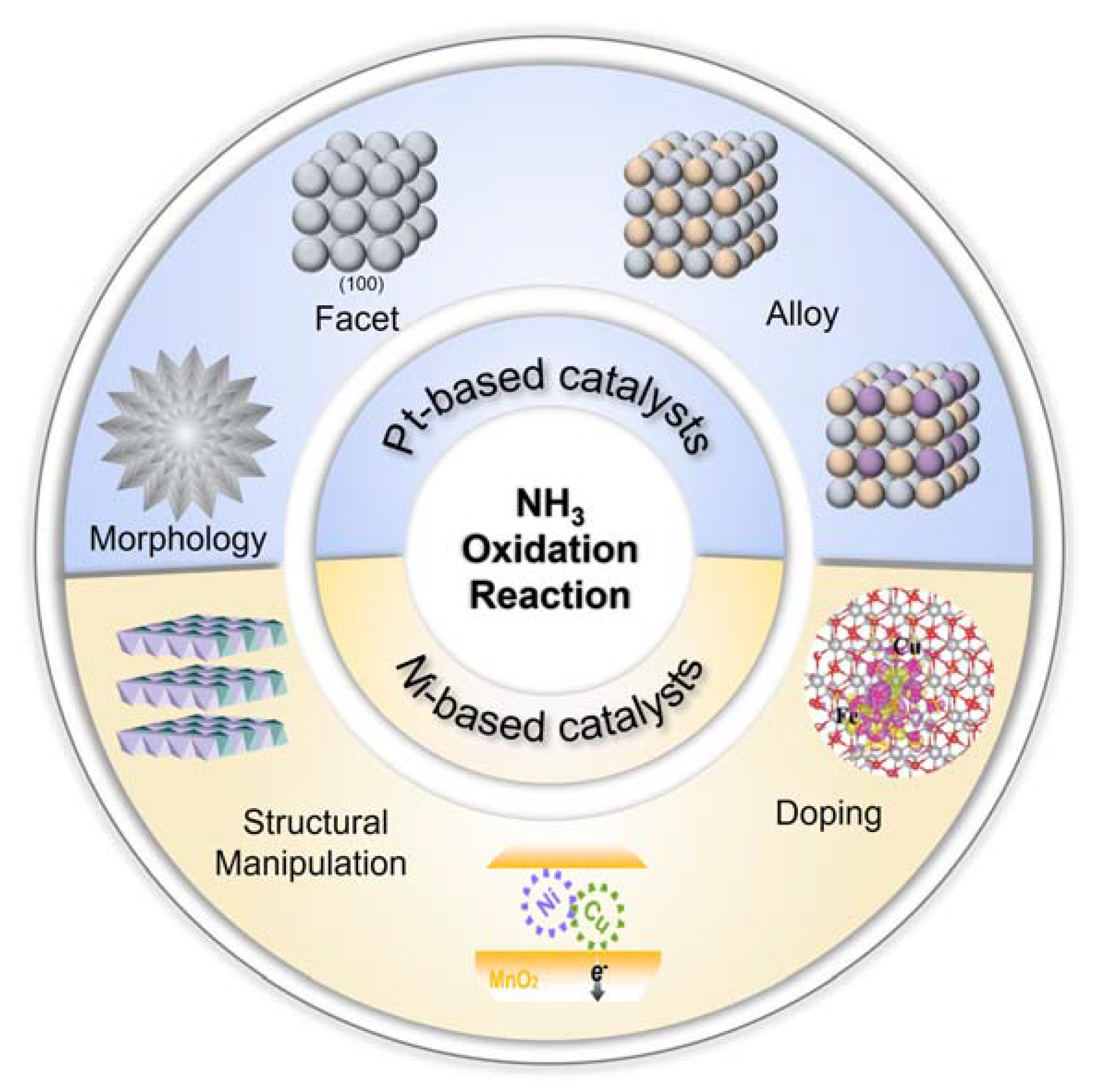
1. Introduction
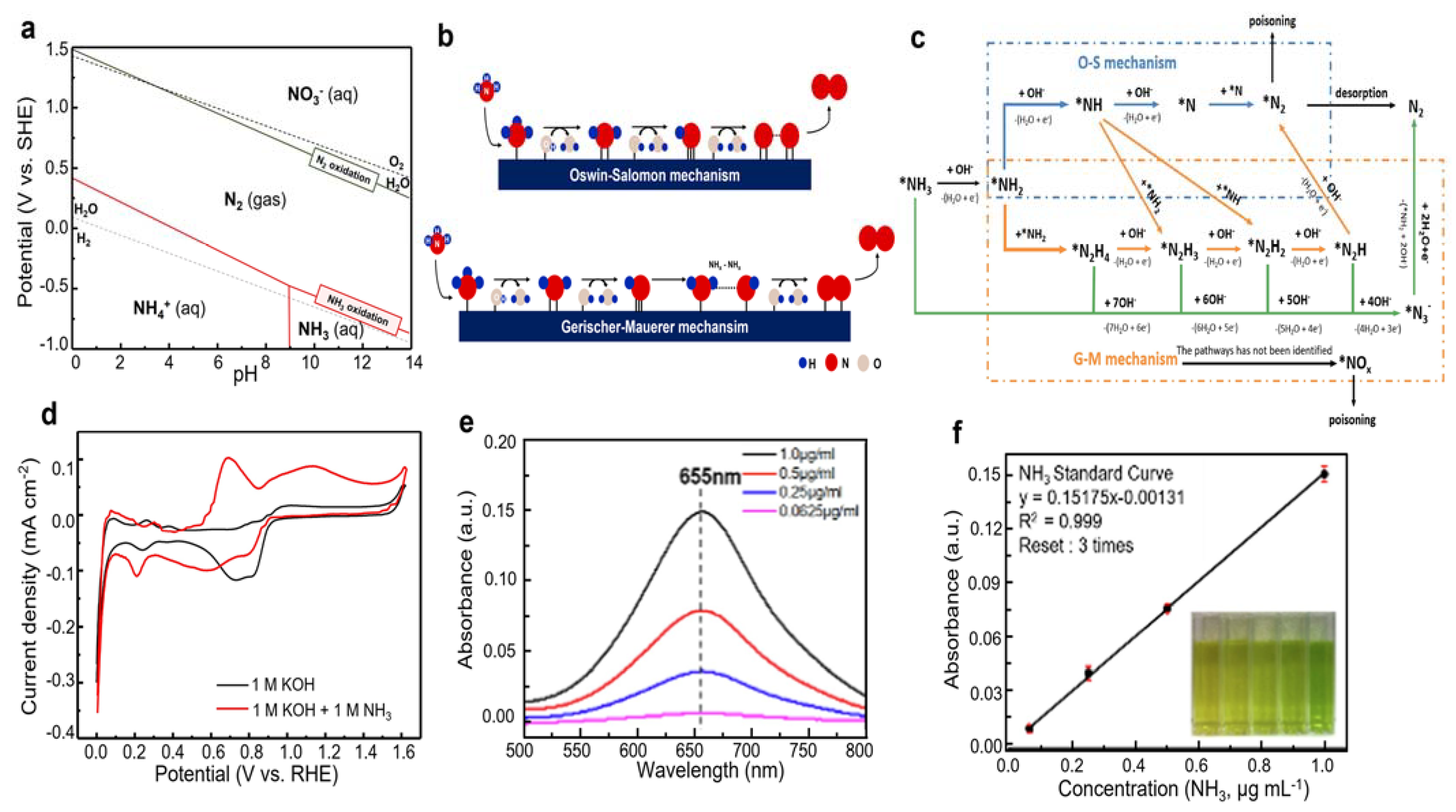
2. Fundamentals of Ammonia Oxidation

3. Electrocatalysts for Ammonia Oxidation
3.1. Pt-Based Electrocatalysts
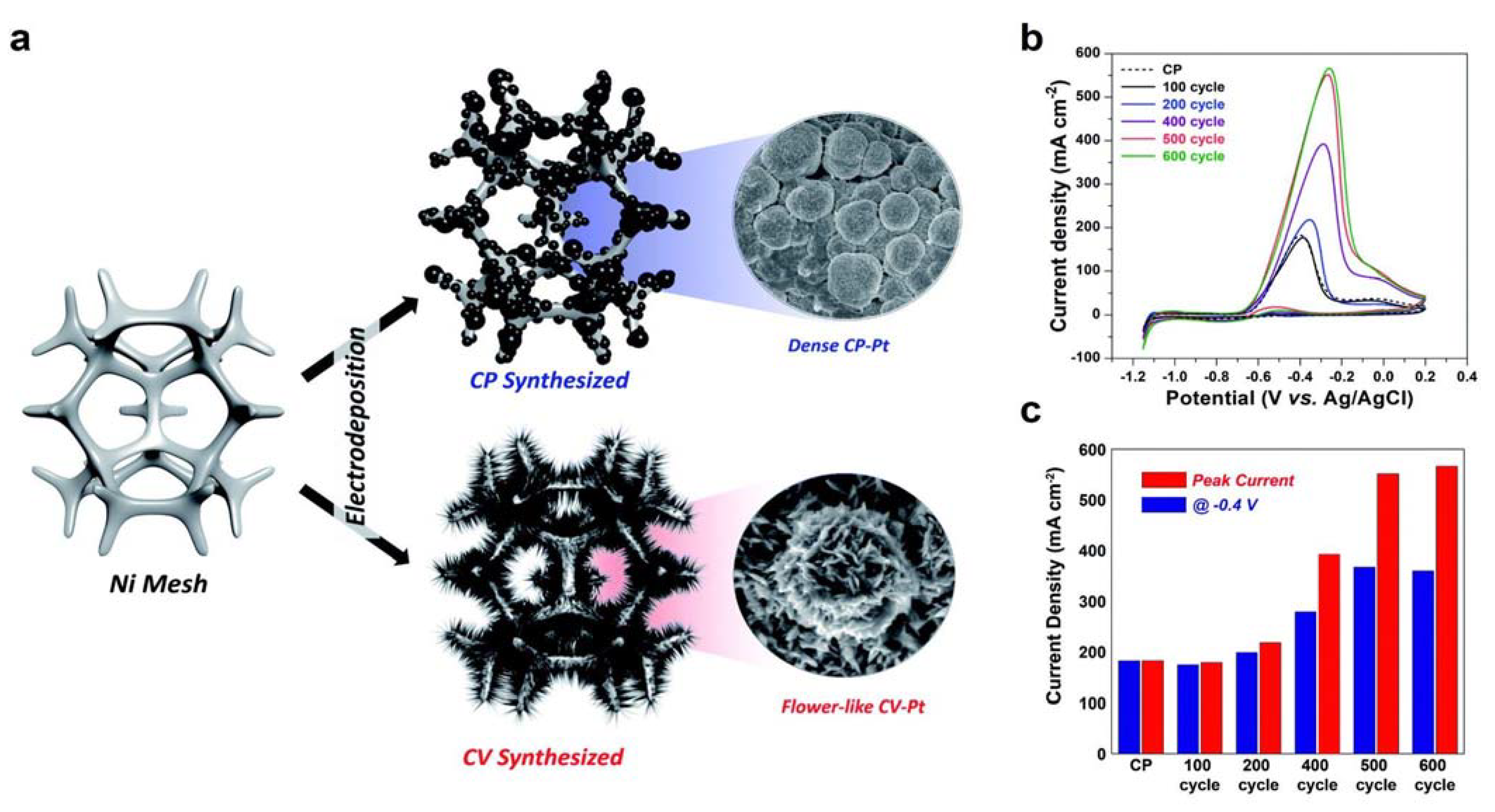
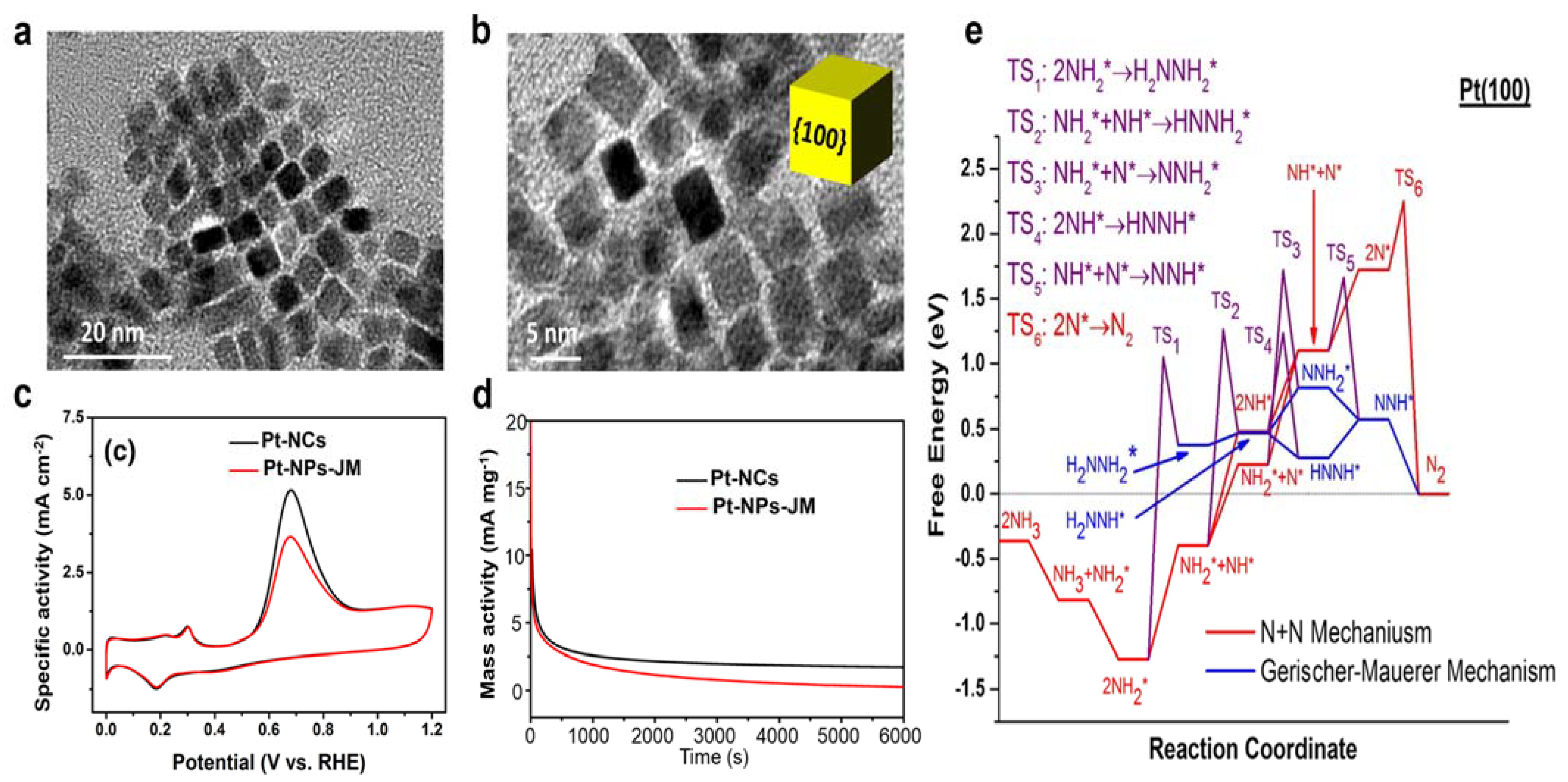
3.1.1. Pure Pt Electrocatalysts

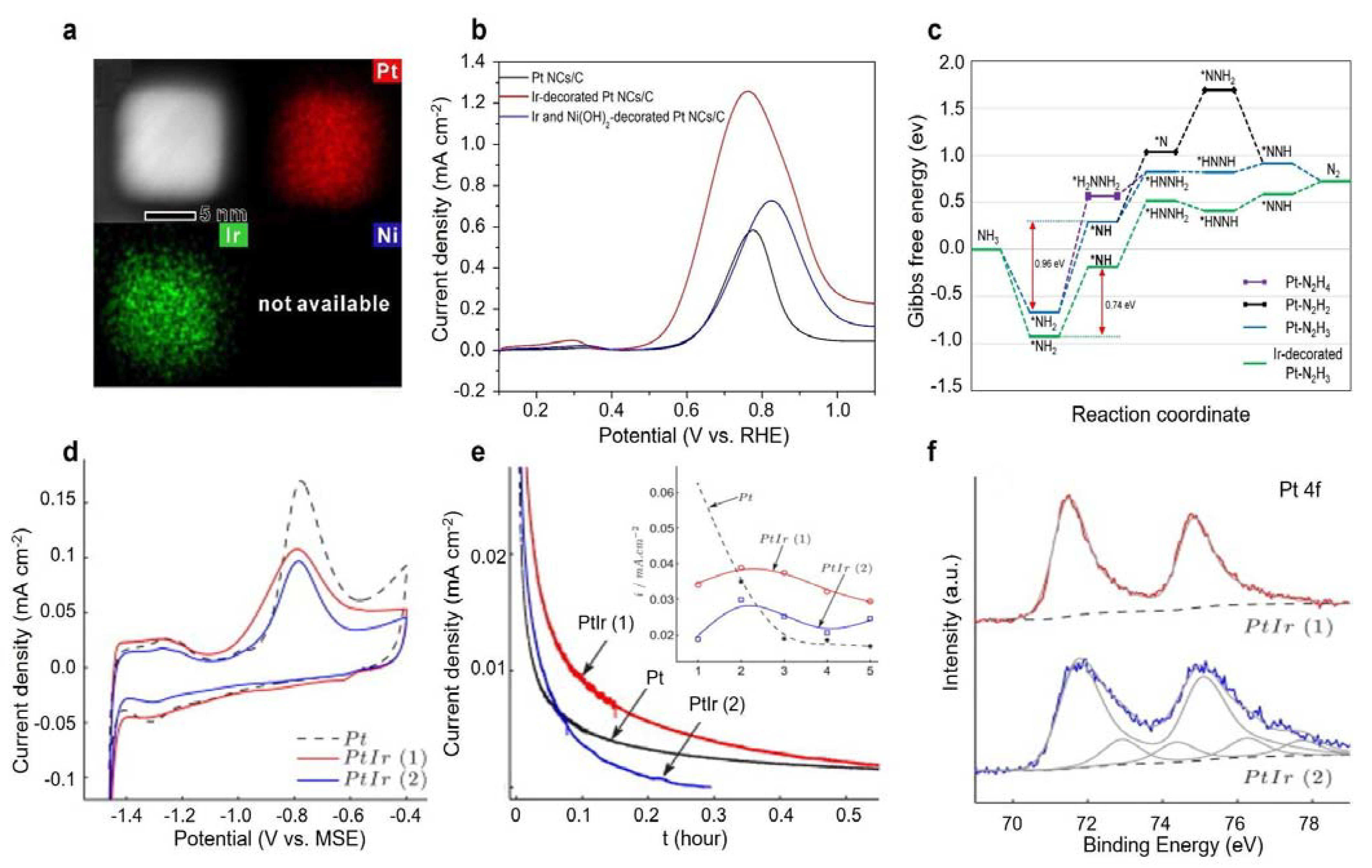
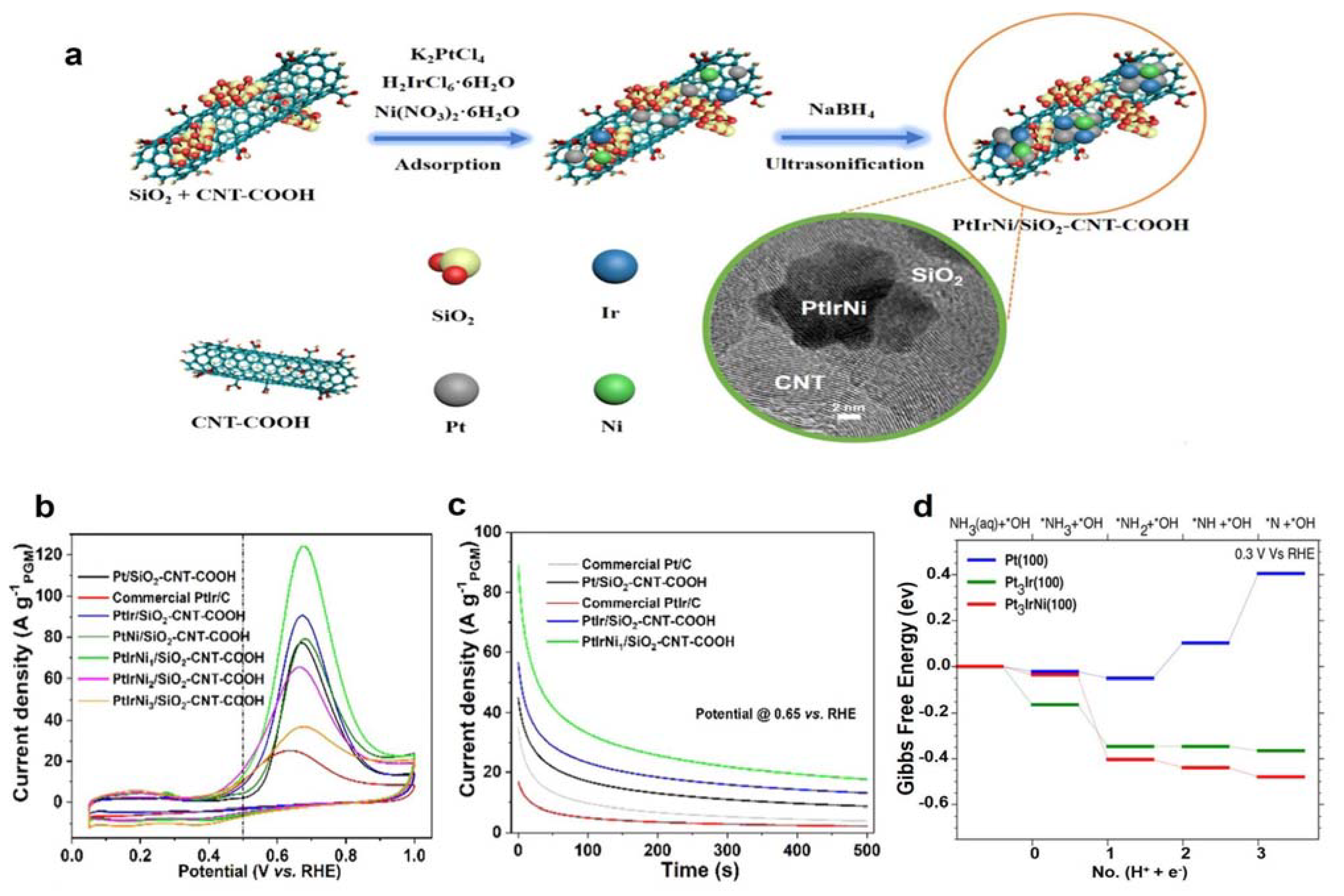
3.1.2. Pt-Ir Bimetal Electrocatalysts
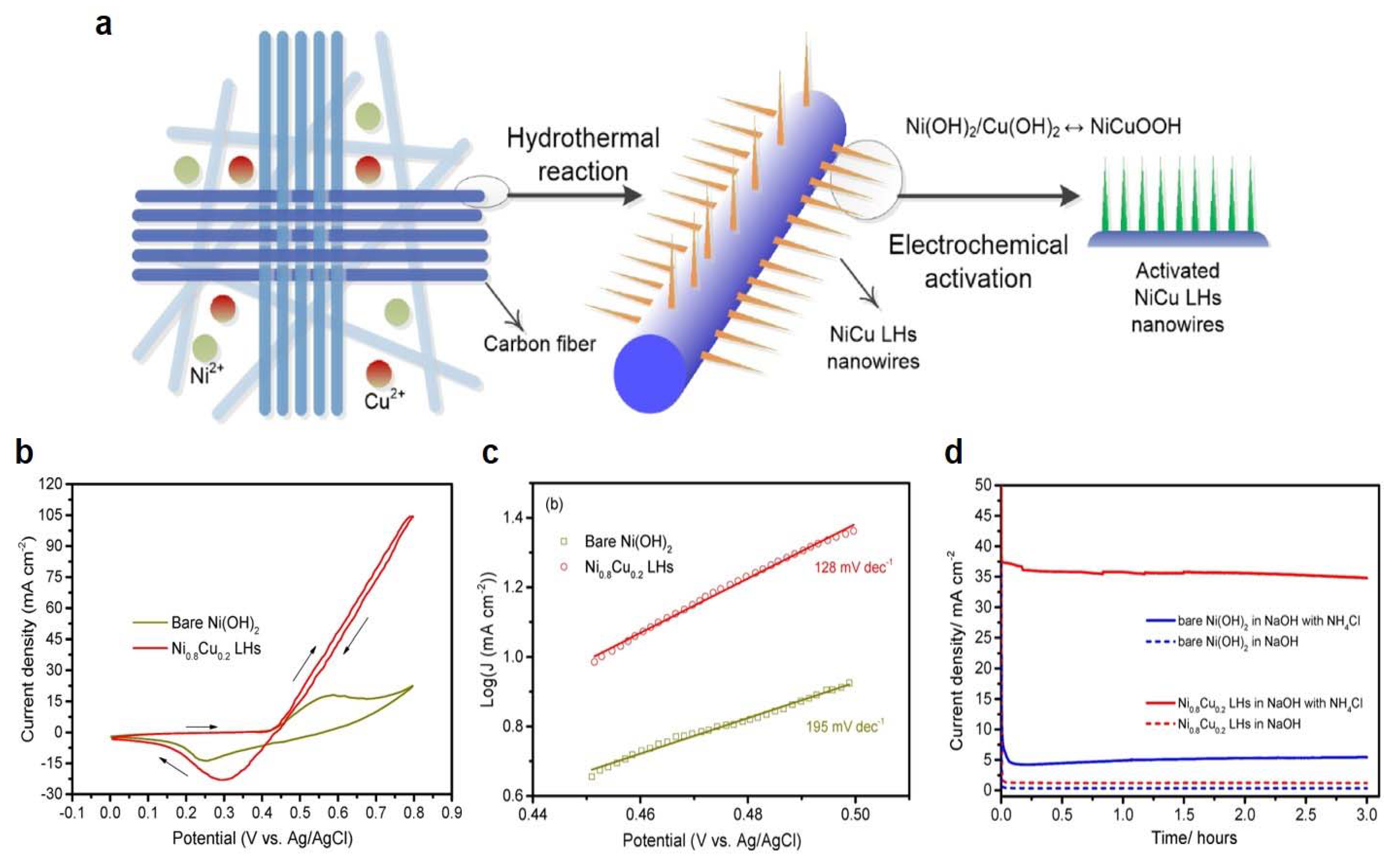
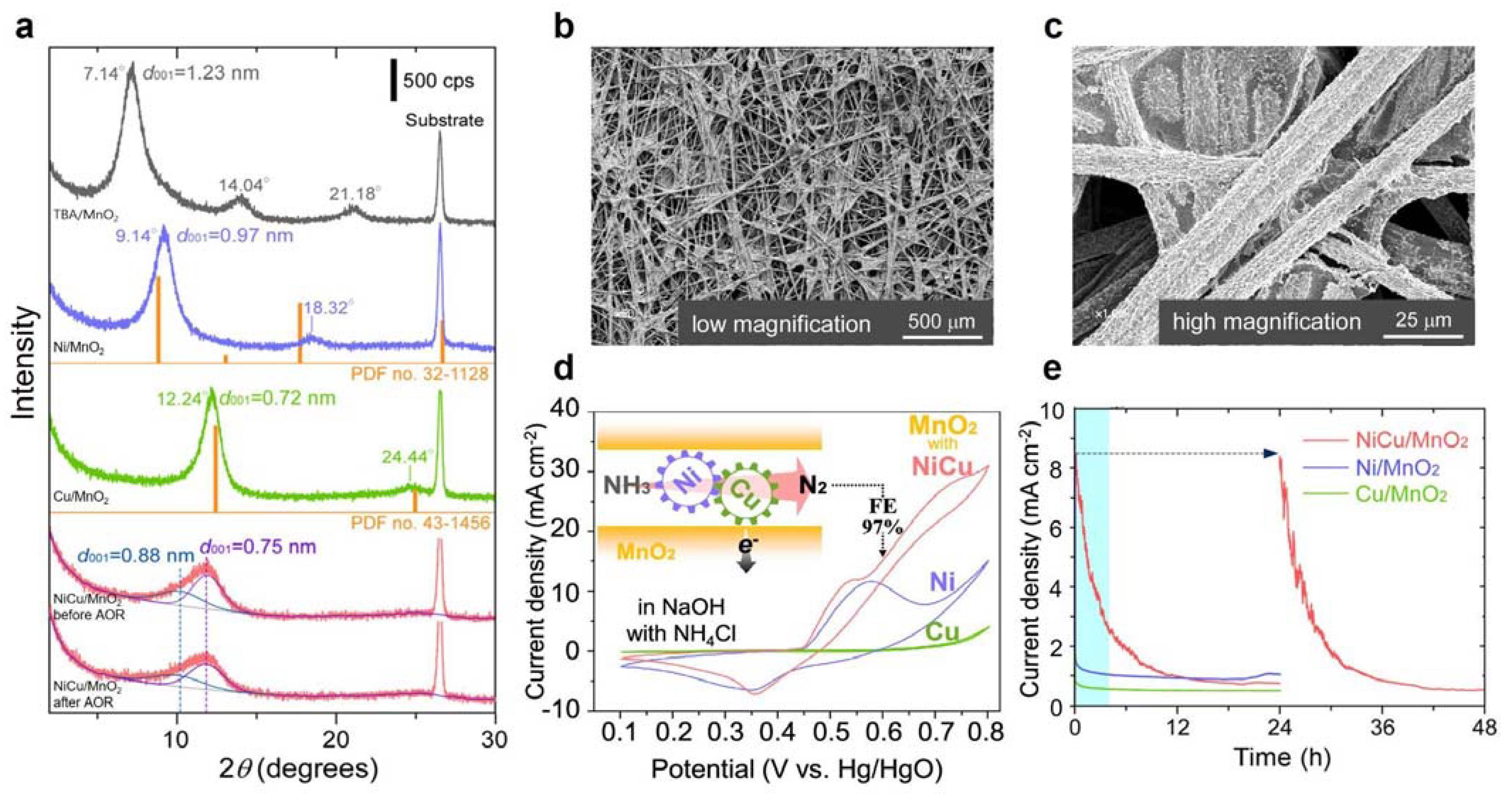
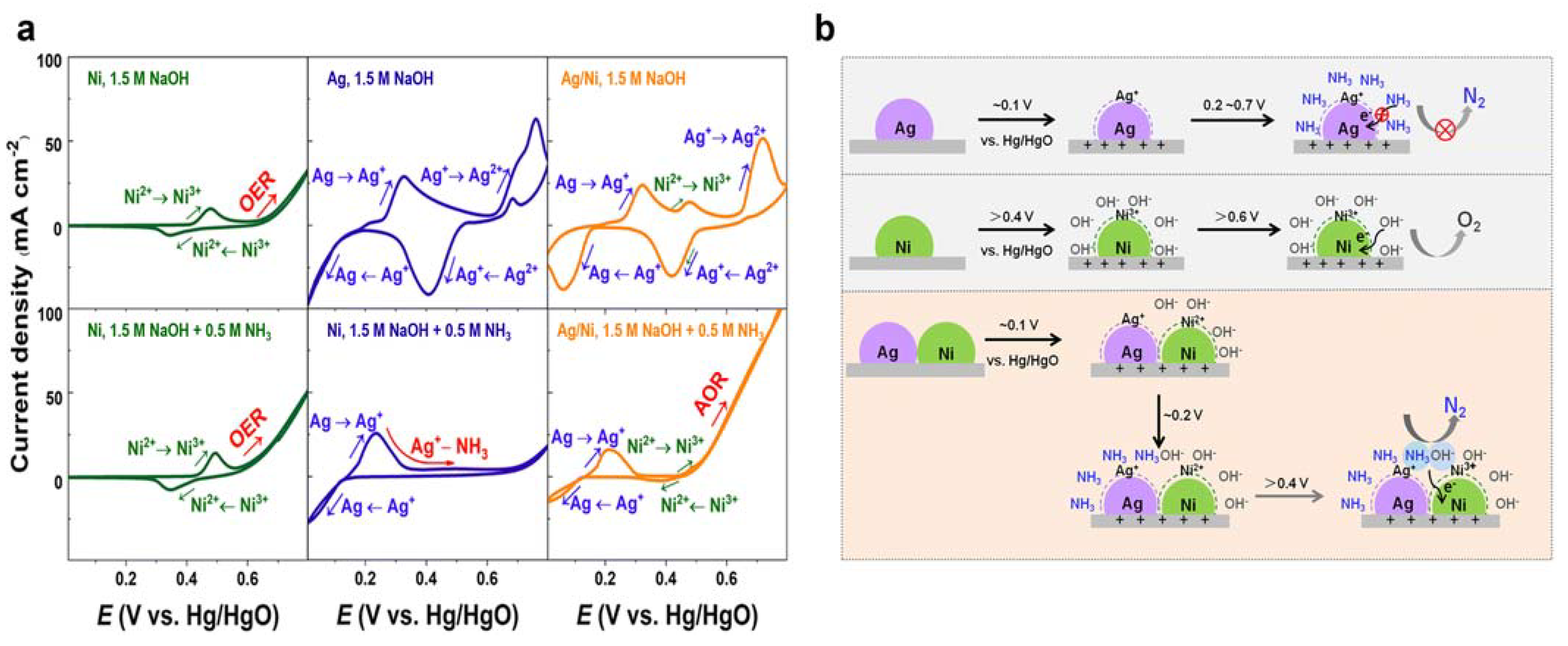
3.2. Ni-Based Metal Electrocatalysts
4. Summary and Perspective
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Matsui, T.; Suzuki, S.; Katayama, Y.; Yamauchi, K.; Okanishi, T.; Muroyama, H.; Eguchi, K. In Situ Attenuated Total Reflection Infrared Spectroscopy on Electrochemical Ammonia Oxidation over Pt Electrode in Alkaline Aqueous Solutions. Langmuir 2015, 31, 11717–11723. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Yang, W.; Lee, W.H.; Han, M.H.; Moon, J.; Jeon, C.; Kim, D.; Ji, S.G.; Chae, K.H.; Lee, K.-S.; et al. Operando Stability of Platinum Electrocatalysts in Ammonia Oxidation Reactions. ACS Catal. 2020, 10, 11674–11684. [Google Scholar] [CrossRef]
- Lamb, K.E.; Dolan, M.D.; Kennedy, D.F. Ammonia for hydrogen storage; A review of catalytic ammonia decomposition and hydrogen separation and purification. Int. J. Hydrogen Energy 2019, 44, 3580–3593. [Google Scholar] [CrossRef]
- Lumley, M.A.; Radmilovic, A.; Jang, Y.J.; Lindberg, A.E.; Choi, K.S. Perspectives on the Development of Oxide-Based Photocathodes for Solar Fuel Production. J. Am. Chem. Soc. 2019, 141, 18358–18369. [Google Scholar] [CrossRef]
- Jang, Y.J.; Choi, K.-S. Enabling electrochemical N2 reduction to NH3 in the low overpotential region using non-noble metal Bi electrodes via surface composition modification. J. Mater. Chem. A 2020, 8, 13842–13851. [Google Scholar] [CrossRef]
- Choe, S.; Kim, S.M.; Lee, Y.; Seok, J.; Jung, J.; Lee, J.S.; Jang, Y.J. Rational design of photocatalysts for ammonia production from water and nitrogen gas. Nano Converg. 2021, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.J.; Evans, T.A.; Samanta, B.; Zeng, K.; Toroker, M.C.; Choi, K.-S. A comparative study of Bi, Sb, and BiSb for electrochemical nitrogen reduction leading to a new catalyst design strategy. J. Mater. Chem. A 2021, 9, 20453–20465. [Google Scholar] [CrossRef]
- Lee, C.; Kim, H.; Jang, Y.J. Three Phase Boundary Engineering Using Hydrophilic–Hydrophobic Poly(N-isopropylacrylamide) with Oxygen-Vacant TiO2 Photocatalysts for Photocatalytic N2 Reduction. ACS Appl. Energy Mater. 2022, 5, 11018–11024. [Google Scholar] [CrossRef]
- Moon, Y.H.; Kim, N.Y.; Kim, S.M.; Jang, Y.J. Recent Advances in Electrochemical Nitrogen Reduction Reaction to Ammonia from the Catalyst to the System. Catalysts 2022, 12, 1015. [Google Scholar] [CrossRef]
- Morales-Navas, C.; Martinez-Rodriguez, R.A.; Vidal-Iglesias, F.J.; Pena, A.; Soto-Perez, J.J.; Trinidad, P.; Solla-Gullon, J.; Tzvetkov, T.; Doan, J.; Smotkin, E.S.; et al. Autonomous electrochemical system for ammonia oxidation reaction measurements at the International Space Station. NPJ Micrograv. 2023, 9, 20. [Google Scholar] [CrossRef]
- Habibzadeh, F.; Miller, S.L.; Hamann, T.W.; Smith, M.R., 3rd. Homogeneous electrocatalytic oxidation of ammonia to N2 under mild conditions. Proc. Natl. Acad. Sci. USA 2019, 116, 2849–2853. [Google Scholar] [CrossRef] [PubMed]
- Dunn, P.L.; Cook, B.J.; Johnson, S.I.; Appel, A.M.; Bullock, R.M. Oxidation of Ammonia with Molecular Complexes. J. Am. Chem. Soc. 2020, 142, 17845–17858. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Liu, J.; Song, C.; Ogata, Y.; Rao, H.; Zhao, X.; Xu, H.; Chen, Y. Different Reaction Mechanisms of Ammonia Oxidation Reaction on Pt/Al2O3 and Pt/CeZrO2 with Various Pt States. ACS Appl. Mater. Interfaces 2019, 11, 23102–23111. [Google Scholar] [CrossRef] [PubMed]
- Ruan, C.; Wang, X.; Wang, C.; Zheng, L.; Li, L.; Lin, J.; Liu, X.; Li, F.; Wang, X. Selective catalytic oxidation of ammonia to nitric oxide via chemical looping. Nat. Commun. 2022, 13, 718. [Google Scholar] [CrossRef]
- Zott, M.D.; Peters, J.C. Enhanced Ammonia Oxidation Catalysis by a Low-Spin Iron Complex Featuring Cis Coordination Sites. J. Am. Chem. Soc. 2021, 143, 7612–7616. [Google Scholar] [CrossRef]
- Zhao, Y.; Setzler, B.P.; Wang, J.; Nash, J.; Wang, T.; Xu, B.; Yan, Y. An Efficient Direct Ammonia Fuel Cell for Affordable Carbon-Neutral Transportation. Joule 2019, 3, 2472–2484. [Google Scholar] [CrossRef]
- Guo, Y.; Pan, Z.; An, L. Carbon-free sustainable energy technology: Direct ammonia fuel cells. J. Power Sources 2020, 476, 228454. [Google Scholar] [CrossRef]
- Gwak, J.; Choun, M.; Lee, J. Alkaline Ammonia Electrolysis on Electrodeposited Platinum for Controllable Hydrogen Production. ChemSusChem 2016, 9, 403–408. [Google Scholar] [CrossRef]
- Xu, W.; Du, D.; Lan, R.; Humphreys, J.; Miller, D.N.; Walker, M.; Wu, Z.; Irvine, J.T.S.; Tao, S. Electrodeposited NiCu bimetal on carbon paper as stable non-noble anode for efficient electrooxidation of ammonia. Appl. Catal. B Environ. 2018, 237, 1101–1109. [Google Scholar] [CrossRef]
- Herron, J.A.; Ferrin, P.; Mavrikakis, M. Electrocatalytic Oxidation of Ammonia on Transition-Metal Surfaces: A First-Principles Study. J. Phys. Chem. C 2015, 119, 14692–14701. [Google Scholar] [CrossRef]
- Katayama, Y.; Okanishi, T.; Muroyama, H.; Matsui, T.; Eguchi, K. Enhancement of ammonia oxidation activity over Y2O3-modified platinum surface: Promotion of NH2,ad dimerization process. J. Catal. 2016, 344, 496–506. [Google Scholar] [CrossRef]
- Rosca, V.; Koper, M.T. Electrocatalytic oxidation of ammonia on Pt(111) and Pt(100) surfaces. Phys. Chem. Chem. Phys. 2006, 8, 2513–2524. [Google Scholar] [CrossRef]
- Vidal-Iglesias, F.J.; Solla-Gullón, J.; Montiel, V.; Feliu, J.M.; Aldaz, A. Screening of electrocatalysts for direct ammonia fuel cell: Ammonia oxidation on PtMe (Me: Ir, Rh, Pd, Ru) and preferentially oriented Pt(100) nanoparticles. J. Power Sources 2007, 171, 448–456. [Google Scholar] [CrossRef]
- Xu, W.; Lan, R.; Du, D.; Humphreys, J.; Walker, M.; Wu, Z.; Wang, H.; Tao, S. Directly growing hierarchical nickel-copper hydroxide nanowires on carbon fibre cloth for efficient electrooxidation of ammonia. Appl. Catal. B Environ. 2017, 218, 470–479. [Google Scholar] [CrossRef]
- Tsai, M.-H.; Chen, T.-C.; Juang, Y.; Hua, L.-C.; Huang, C. High catalytic performance of CuCo/nickel foam electrode for ammonia electrooxidation. Electrochem. Commun. 2020, 121, 106875. [Google Scholar] [CrossRef]
- Nagita, K.; Yuhara, Y.; Fujii, K.; Katayama, Y.; Nakayama, M. Ni- and Cu-co-Intercalated Layered Manganese Oxide for Highly Efficient Electro-Oxidation of Ammonia Selective to Nitrogen. ACS Appl. Mater. Interfaces 2021, 13, 28098–28107. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Yang, Y.; Xi, S.; Diao, C.; Yu, Z.; Lee, W.S.V.; Xue, J. Deciphering NH3 Adsorption Kinetics in Ternary Ni-Cu-Fe Oxyhydroxide toward Efficient Ammonia Oxidation Reaction. Small 2021, 17, e2005616. [Google Scholar] [CrossRef]
- Zhang, S.; Yan, L.; Jiang, H.; Yang, L.; Zhao, Y.; Yang, X.; Wang, Y.; Shen, J.; Zhao, X. Facile Fabrication of a Foamed Ag3CuS2 Film as an Efficient Self-Supporting Electrocatalyst for Ammonia Electrolysis Producing Hydrogen. ACS Appl. Mater. Interfaces 2022, 14, 9036–9045. [Google Scholar] [CrossRef]
- Finkelstein, D.A.; Bertin, E.; Garbarino, S.; Guay, D. Mechanistic Similarity in Catalytic N2 Production from NH3 and NO2− at Pt(100) Thin Films: Toward a Universal Catalytic Pathway for Simple N-Containing Species, and Its Application to in Situ Removal of NH3 Poisons. J. Phys. Chem. C 2015, 119, 9860–9878. [Google Scholar] [CrossRef]
- Vidal-Iglesias, F.J.; Solla-Gullón, J.; Feliu, J.M.; Baltruschat, H.; Aldaz, A. DEMS study of ammonia oxidation on platinum basal planes. J. Electroanal. Chem. 2006, 588, 331–338. [Google Scholar] [CrossRef]
- Oswin, H.G.; Salomon, M. The anodic oxidation of ammonia at platinum black electrodes in aqueous koh electrolyte. Can. J. Chem. 1963, 41, 1686–1694. [Google Scholar] [CrossRef]
- Gerischer, H.; Mauerer, A. Untersuchungen Zur anodischen Oxidation von Ammoniak an Platin-Elektroden. J. Electroanal. Chem. Interfacial Electrochem. 1970, 25, 421–433. [Google Scholar] [CrossRef]
- Gootzen, J.F.E.; Wonders, A.H.; Visscher, W.H.M.; Van Santen, R.A.; van Veen, J.A.R. A DEMS and cyclic voltammetry study of NH3 oxidation on platinized platinum. Electrochim. Acta 1998, 43, 1851–1861. [Google Scholar] [CrossRef]
- Xi, X.; Fan, Y.; Zhang, K.; Liu, Y.; Nie, F.; Guan, H.; Wu, J. Carbon-free sustainable energy technology: Electrocatalytic ammonia oxidation reaction. Chem. Eng. J. 2022, 435, 134818. [Google Scholar] [CrossRef]
- Ye, J.-Y.; Lin, J.-L.; Zhou, Z.-Y.; Hong, Y.-H.; Sheng, T.; Rauf, M.; Sun, S.-G. Ammonia electrooxidation on dendritic Pt nanostructures in alkaline solutions investigated by in-situ FTIR spectroscopy and online electrochemical mass spectroscopy. J. Electroanal. Chem. 2018, 819, 495–501. [Google Scholar] [CrossRef]
- Wei, R.-L.; Liu, Y.; Chen, Z.; Jia, W.-S.; Yang, Y.-Y.; Cai, W.-B. Ammonia oxidation on iridium electrode in alkaline media: An in situ ATR-SEIRAS study. J. Electroanal. Chem. 2021, 896, 115254. [Google Scholar] [CrossRef]
- Kim, H.; Chung, M.W.; Choi, C.H. NO-induced deactivation of Pt electrocatalysis towards the ammonia oxidation reaction. Electrochem. Commun. 2018, 94, 31–35. [Google Scholar] [CrossRef]
- Kutz, R.B.; Braunschweig, B.; Mukherjee, P.; Dlott, D.D.; Wieckowski, A. Study of Ethanol Electrooxidation in Alkaline Electrolytes with Isotope Labels and Sum-Frequency Generation. J. Phys. Chem. Lett. 2011, 2, 2236–2240. [Google Scholar] [CrossRef]
- Ma, L.; Chu, D.; Chen, R. Comparison of ethanol electro-oxidation on Pt/C and Pd/C catalysts in alkaline media. Int. J. Hydrogen Energy 2012, 37, 11185–11194. [Google Scholar] [CrossRef]
- Shih, Y.-J.; Huang, Y.-H.; Huang, C.P. Electrocatalytic ammonia oxidation over a nickel foam electrode: Role of Ni(OH)2(s)-NiOOH(s) nanocatalysts. Electrochim. Acta 2018, 263, 261–271. [Google Scholar] [CrossRef]
- Lee, H.; Lee, J.-H.; Lee, Y.; Cho, E.-B.; Jang, Y.J. Boosting solar-driven N2 to NH3 conversion using defect-engineered TiO2/CuO heterojunction photocatalyst. Appl. Surf. Sci. 2023, 620, 156812. [Google Scholar] [CrossRef]
- Yang, Y.; Kim, J.; Jo, H.; Seong, A.; Lee, M.; Min, H.-K.; Seo, M.-g.; Choi, Y.; Kim, G. A rigorous electrochemical ammonia electrolysis protocol with in operando quantitative analysis. J. Mater. Chem. A 2021, 9, 11571–11579. [Google Scholar] [CrossRef]
- Zhang, X.; Kong, R.M.; Du, H.; Xia, L.; Qu, F. Highly efficient electrochemical ammonia synthesis via nitrogen reduction reactions on a VN nanowire array under ambient conditions. Chem. Commun. 2018, 54, 5323–5325. [Google Scholar] [CrossRef] [PubMed]
- Siddharth, K.; Alam, P.; Hossain, M.D.; Xie, N.; Nambafu, G.S.; Rehman, F.; Lam, J.W.Y.; Chen, G.; Cheng, J.; Luo, Z.; et al. Hydrazine Detection during Ammonia Electro-oxidation Using an Aggregation-Induced Emission Dye. J. Am. Chem. Soc. 2021, 143, 2433–2440. [Google Scholar] [CrossRef]
- Li, L.; Tang, C.; Yao, D.; Zheng, Y.; Qiao, S.-Z. Electrochemical Nitrogen Reduction: Identification and Elimination of Contamination in Electrolyte. ACS Energy Lett. 2019, 4, 2111–2116. [Google Scholar] [CrossRef]
- de Vooys, A.C.A.; Koper, M.T.M.; Van Santen, R.A.; van Veen, J.A.R. The role of adsorbates in the electrochemical oxidation of ammonia on noble and transition metal electrodes. J. Electroanal. Chem. 2001, 506, 127–137. [Google Scholar] [CrossRef]
- Vidal-Iglesias, F.J.; Solla-Gullón, J.; Rodríguez, P.; Herrero, E.; Montiel, V.; Feliu, J.M.; Aldaz, A. Shape-dependent electrocatalysis: Ammonia oxidation on platinum nanoparticles with preferential (100) surfaces. Electrochem. Commun. 2004, 6, 1080–1084. [Google Scholar] [CrossRef]
- Zhang, C.; Hwang, S.Y.; Peng, Z. Shape-enhanced ammonia electro-oxidation property of a cubic platinum nanocrystal catalyst prepared by surfactant-free synthesis. J. Mater. Chem. A 2013, 1, 14402–14408. [Google Scholar] [CrossRef]
- Liu, J.; Zhong, C.; Yang, Y.; Wu, Y.T.; Jiang, A.K.; Deng, Y.D.; Zhang, Z.; Hu, W.B. Electrochemical preparation and characterization of Pt particles on ITO substrate: Morphological effect on ammonia oxidation. Int. J. Hydrogen Energy 2012, 37, 8981–8987. [Google Scholar] [CrossRef]
- Sun, H.-Y.; Xu, G.-R.; Li, F.-M.; Hong, Q.-L.; Jin, P.-J.; Chen, P.; Chen, Y. Hydrogen generation from ammonia electrolysis on bifunctional platinum nanocubes electrocatalysts. J. Energy Chem. 2020, 47, 234–240. [Google Scholar] [CrossRef]
- Martinez-Rodriguez, R.A.; Vidal-Iglesias, F.J.; Solla-Gullon, J.; Cabrera, C.R.; Feliu, J.M. Synthesis of Pt Nanoparticles in Water-in-Oil Microemulsion: Effect of HCl on Their Surface Structure. J. Am. Chem. Soc. 2014, 136, 1280–1283. [Google Scholar] [CrossRef] [PubMed]
- Katsounaros, I.; Figueiredo, M.C.; Calle-Vallejo, F.; Li, H.; Gewirth, A.A.; Markovic, N.M.; Koper, M.T.M. On the mechanism of the electrochemical conversion of ammonia to dinitrogen on Pt(100) in alkaline environment. J. Catal. 2018, 359, 82–91. [Google Scholar] [CrossRef]
- Pillai, H.S.; Xin, H. New Insights into Electrochemical Ammonia Oxidation on Pt(100) from First Principles. Ind. Eng. Chem. Res. 2019, 58, 10819–10828. [Google Scholar] [CrossRef]
- Elnabawy, A.O.; Herron, J.A.; Karraker, S.; Mavrikakis, M. Structure sensitivity of ammonia electro-oxidation on transition metal surfaces: A first-principles study. J. Catal. 2021, 397, 137–147. [Google Scholar] [CrossRef]
- Lomocso, T.L.; Baranova, E.A. Electrochemical oxidation of ammonia on carbon-supported bi-metallic PtM (M = Ir, Pd, SnOx) nanoparticles. Electrochim. Acta 2011, 56, 8551–8558. [Google Scholar] [CrossRef]
- Sacré, N.; Duca, M.; Garbarino, S.; Imbeault, R.; Wang, A.; Hadj Youssef, A.; Galipaud, J.; Hufnagel, G.; Ruediger, A.; Roué, L.; et al. Tuning Pt–Ir Interactions for NH3 Electrocatalysis. ACS Catal. 2018, 8, 2508–2518. [Google Scholar] [CrossRef]
- Liu, Z.; Li, Y.; Zhang, X.; Rao, S.; Li, J.; Wang, W.; Sun, Z.; Yang, J. Surface Structure Engineering of PtPd Nanoparticles for Boosting Ammonia Oxidation Electrocatalysis. ACS Appl. Mater. Interfaces 2022, 14, 28816–28825. [Google Scholar] [CrossRef] [PubMed]
- Endo, K.; Nakamura, K.; Katayama, Y.; Miura, T. Pt–Me (Me = Ir, Ru, Ni) binary alloys as an ammonia oxidation anode. Electrochim. Acta 2004, 49, 2503–2509. [Google Scholar] [CrossRef]
- Siddharth, K.; Hong, Y.; Qin, X.; Lee, H.J.; Chan, Y.T.; Zhu, S.; Chen, G.; Choi, S.-I.; Shao, M. Surface engineering in improving activity of Pt nanocubes for ammonia electrooxidation reaction. Appl. Catal. B Environ. 2020, 269, 118821. [Google Scholar] [CrossRef]
- Allagui, A.; Oudah, M.; Tuaev, X.; Ntais, S.; Almomani, F.; Baranova, E.A. Ammonia electro-oxidation on alloyed PtIr nanoparticles of well-defined size. Int. J. Hydrogen Energy 2013, 38, 2455–2463. [Google Scholar] [CrossRef]
- Li, Y.; Li, X.; Pillai, H.S.; Lattimer, J.; Mohd Adli, N.; Karakalos, S.; Chen, M.; Guo, L.; Xu, H.; Yang, J.; et al. Ternary PtIrNi Catalysts for Efficient Electrochemical Ammonia Oxidation. ACS Catal. 2020, 10, 3945–3957. [Google Scholar] [CrossRef]
- Du, X.T.; Yang, Y.; Liu, J.; Liu, B.; Liu, J.B.; Zhong, C.; Hu, W.B. Surfactant-free and template-free electrochemical approach to prepare well-dispersed Pt nanosheets and their high electrocatalytic activities for ammonia oxidation. Electrochim. Acta 2013, 111, 562–566. [Google Scholar] [CrossRef]
- Bertin, E.; Garbarino, S.; Guay, D.; Solla-Gullón, J.; Vidal-Iglesias, F.J.; Feliu, J.M. Electrodeposited platinum thin films with preferential (100) orientation: Characterization and electrocatalytic properties for ammonia and formic acid oxidation. J. Power Sources 2013, 225, 323–329. [Google Scholar] [CrossRef]
- Okanishi, T.; Katayama, Y.; Muroyama, H.; Matsui, T.; Eguchi, K. SnO2-modified Pt electrocatalysts for ammonia–fueled anion exchange membrane fuel cells. Electrochim. Acta 2015, 173, 364–369. [Google Scholar] [CrossRef]
- Katayama, Y.; Okanishi, T.; Muroyama, H.; Matsui, T.; Eguchi, K. Electrochemical Oxidation of Ammonia over Rare Earth Oxide Modified Platinum Catalysts. J. Phys. Chem. C 2015, 119, 9134–9141. [Google Scholar] [CrossRef]
- Silva, J.C.M.; Piasentin, R.M.; Spinacé, E.V.; Neto, A.O.; Baranova, E.A. The effect of antimony-tin and indium-tin oxide supports on the catalytic activity of Pt nanoparticles for ammonia electro-oxidation. Mater. Chem. Phys. 2016, 180, 97–103. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, G.; Yu, M.; Wang, X.; Lv, J.; Yang, F. Free-Standing 3D Porous N-Doped Graphene Aerogel Supported Platinum Nanocluster for Efficient Hydrogen Production from Ammonia Electrolysis. ACS Sustain. Chem. Eng. 2018, 6, 8437–8446. [Google Scholar] [CrossRef]
- Le Vot, S.; Roué, L.; Bélanger, D. Synthesis of Pt–Ir catalysts by coelectrodeposition: Application to ammonia electrooxidation in alkaline media. J. Power Sources 2013, 223, 221–231. [Google Scholar] [CrossRef]
- Zhong, C.; Liu, J.; Ni, Z.; Deng, Y.; Chen, B.; Hu, W. Shape-controlled synthesis of Pt-Ir nanocubes with preferential (100) orientation and their unusual enhanced electrocatalytic activities. Sci. China Mater. 2014, 57, 13–25. [Google Scholar] [CrossRef]
- Morita, S.; Kudo, E.; Shirasaka, R.; Yonekawa, M.; Nagai, K.; Ota, H.; Mikka, N.; Shiroishi, H. Electrochemical oxidation of ammonia by multi-wall-carbon-nanotube-supported Pt shell–Ir core nanoparticles synthesized by an improved Cu short circuit deposition method. J. Electroanal. Chem. 2016, 762, 29–36. [Google Scholar] [CrossRef]
- Jiang, J. Promotion of PtIr and Pt catalytic activity towards ammonia electrooxidation through the modification of Zn. Electrochem. Commun. 2017, 75, 52–55. [Google Scholar] [CrossRef]
- Lin, X.; Zhang, X.; Wang, Z.; Zhu, X.; Zhu, J.; Chen, P.; Lyu, T.; Li, C.; Tian, Z.Q.; Shen, P.K. Hyperbranched concave octahedron of PtIrCu nanocrystals with high-index facets for efficiently electrochemical ammonia oxidation reaction. J. Colloid Interface Sci. 2021, 601, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Pillai, H.S.; Wang, T.; Hwang, S.; Zhao, Y.; Qiao, Z.; Mu, Q.; Karakalos, S.; Chen, M.; Yang, J.; et al. High-performance ammonia oxidation catalysts for anion-exchange membrane direct ammonia fuel cells. Energy Environ. Sci. 2021, 14, 1449–1460. [Google Scholar] [CrossRef]
- Silva, J.C.M.; Ntais, S.; Teixeira-Neto, É.; Spinacé, E.V.; Cui, X.; Neto, A.O.; Baranova, E.A. Evaluation of carbon supported platinum–ruthenium nanoparticles for ammonia electro-oxidation: Combined fuel cell and electrochemical approach. Int. J. Hydrogen Energy 2017, 42, 193–201. [Google Scholar] [CrossRef]
- Xue, Q.; Zhao, Y.; Zhu, J.; Ding, Y.; Wang, T.; Sun, H.; Li, F.; Chen, P.; Jin, P.; Yin, S.; et al. PtRu nanocubes as bifunctional electrocatalysts for ammonia electrolysis. J. Mater. Chem. A 2021, 9, 8444–8451. [Google Scholar] [CrossRef]
- Kapałka, A.; Cally, A.; Neodo, S.; Comninellis, C.; Wächter, M.; Udert, K.M. Electrochemical behavior of ammonia at Ni/Ni(OH)2 electrode. Electrochem. Commun. 2010, 12, 18–21. [Google Scholar] [CrossRef]
- Zhang, S.; Zhao, Y.; Yan, L.; Jiang, H.; Yang, X.; Wang, Y.; Song, H.; Zhao, X. Electrochemical ammonia oxidation reaction on defect-rich TiO nanofibers: Experimental and theoretical studies. Int. J. Hydrogen Energy 2021, 46, 39208–39215. [Google Scholar] [CrossRef]
- Zott, M.D.; Garrido-Barros, P.; Peters, J.C. Electrocatalytic Ammonia Oxidation Mediated by a Polypyridyl Iron Catalyst. ACS Catalysis 2019, 9, 10101–10108. [Google Scholar] [CrossRef]
- Choueiri, R.M.; Tatarchuk, S.W.; Klinkova, A.; Chen, L.D. Mechanism of ammonia oxidation to dinitrogen, nitrite, and nitrate on β-Ni(OH)2 from first-principles simulations. Electrochem. Sci. Adv. 2021, 2, 2100142. [Google Scholar] [CrossRef]
- Allagui, A.; Sarfraz, S.; Baranova, E.A. NixPd1- (x = 0.98, 0.93, and 0.58) nanostructured catalysts for ammonia electrooxidation in alkaline media. Electrochim. Acta 2013, 110, 253–259. [Google Scholar] [CrossRef]
- Wang, R.; Liu, H.; Zhang, K.; Zhang, G.; Lan, H.; Qu, J. Ni(II)/Ni(III) redox couple endows Ni foam-supported Ni2P with excellent capability for direct ammonia oxidation. Chem. Eng. J. 2021, 404, 126795. [Google Scholar] [CrossRef]
- Jiang, K.; Li, K.; Liu, Y.-Q.; Lin, S.; Wang, Z.; Wang, D.; Ye, Y. Nickel-cobalt nitride nanoneedle supported on nickel foam as an efficient electrocatalyst for hydrogen generation from ammonia electrolysis. Electrochim. Acta 2022, 403, 139700. [Google Scholar] [CrossRef]
- Jin, Y.; Chen, X.; Wang, J. From inert to active: A cocktail-like mediation of an Ag/Ni mixture for electrocatalytic ammonia oxidation reaction. Chem. Commun. 2022, 58, 10631–10634. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Chen, Z.; Cai, J.; Jin, Y.; Wang, T.; Wang, J. Activating copper oxide for stable electrocatalytic ammonia oxidation reaction via in-situ introducing oxygen vacancies. Nano Res. 2022, 15, 5987–5994. [Google Scholar] [CrossRef]
- Hou, J.; Cheng, Y.; Pan, H.; Kang, P. CuSn Double-Metal Hydroxides for Direct Electrochemical Ammonia Oxidation to Dinitrogen. ChemElectroChem 2022, 9, e202101301. [Google Scholar] [CrossRef]
- Huang, J.; Cai, J.; Wang, J. Nanostructured Wire-in-Plate Electrocatalyst for High-Durability Production of Hydrogen and Nitrogen from Alkaline Ammonia Solution. ACS Appl. Energy Mater. 2020, 3, 4108–4113. [Google Scholar] [CrossRef]
- Yang, A.; Wang, J.; Su, K.; Lei, W.; Qiu, X.; Tang, Y. Modulating Hydroxyl-Rich Interfaces on Nickel-Copper Double Hydroxide Nanotyres to Pre-activate Alkaline Ammonia Oxidation Reactivity. Chemistry 2021, 27, 4869–4875. [Google Scholar] [CrossRef]
- Song, J.; Yinhai, Y.; Jia, Y.; Wang, T.; Wei, J.; Wang, M.; Zhou, S.; Li, Z.; Hou, Y.; Lei, L.; et al. Improved NH3-N conversion efficiency to N2 activated by BDD substrate on NiCu electrocatalysis process. Sep. Purif. Technol. 2021, 276, 119350. [Google Scholar] [CrossRef]
- Wang, H.; Tong, X.; Zhou, L.; Wang, Y.; Liao, L.; Ouyang, S.; Zhang, H. Unique three-dimensional nanoflower-like NiCu electrodes constructed by Co, S co-doping for efficient ammonia oxidation reaction. Sep. Purif. Technol. 2022, 303, 122293. [Google Scholar] [CrossRef]
- Almomani, F.; Bhosale, R.; Khraisheh, M.; Kumar, A.; Tawalbeh, M. Electrochemical oxidation of ammonia on nickel oxide nanoparticles. Int. J. Hydrogen Energy 2020, 45, 10398–10408. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, W.; Wang, H.; Tong, X.; Wang, Y.; Yang, X.; Wu, Z.; Liu, Z. A core-shell NiCu@NiCuOOH 3D electrode induced by surface electrochemical reconstruction for the ammonia oxidation reaction. Int. J. Hydrogen Energy 2022, 47, 16080–16091. [Google Scholar] [CrossRef]
- Pillai, H.S.; Li, Y.; Wang, S.H.; Omidvar, N.; Mu, Q.; Achenie, L.E.K.; Abild-Pedersen, F.; Yang, J.; Wu, G.; Xin, H. Interpretable design of Ir-free trimetallic electrocatalysts for ammonia oxidation with graph neural networks. Nat. Commun. 2023, 14, 792. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Liang, Z.; Ma, Z.; Zhang, Y.; Chen, J.; Adzic, R.R.; Wang, J.X. Temperature-Dependent Kinetics and Reaction Mechanism of Ammonia Oxidation on Pt, Ir, and PtIr Alloy Catalysts. J. Electrochem. Soc. 2018, 165, J3095–J3100. [Google Scholar] [CrossRef]
- Fang, H.; Liao, C.; Ying, Y.; Cheng, J.; Wang, Q.; Huang, H.; Luo, Y.; Jiang, L. Creating metal-carbide interactions to boost ammonia oxidation activity for low-temperature direct ammonia fuel cells. J. Catal. 2023, 417, 129–139. [Google Scholar] [CrossRef]
- Ma, H.; Schneider, W.F. Structure- and Temperature-Dependence of Pt-Catalyzed Ammonia Oxidation Rates and Selectivities. ACS Catal. 2019, 9, 2407–2414. [Google Scholar] [CrossRef]
- Katsounaros, I.; Chen, T.; Gewirth, A.A.; Markovic, N.M.; Koper, M.T. Evidence for Decoupled Electron and Proton Transfer in the Electrochemical Oxidation of Ammonia on Pt(100). J. Phys. Chem. Lett. 2016, 7, 387–392. [Google Scholar] [CrossRef]
- Skachkov, D.; Venkateswara Rao, C.; Ishikawa, Y. Combined First-Principles Molecular Dynamics/Density Functional Theory Study of Ammonia Electrooxidation on Pt(100) Electrode. J. Phys. Chem. C 2013, 117, 25451–25466. [Google Scholar] [CrossRef]
- Jiang, X.; Ying, D.; Liu, X.; Liu, M.; Zhou, S.; Guo, C.; Zhao, G.; Wang, Y.; Jia, J. Identification of the role of Cu site in Ni-Cu hydroxide for robust and high selective electrochemical ammonia oxidation to nitrite. Electrochim. Acta 2020, 345, 136157. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Electrolyte | Onset Potential (VRHE) | Peak Current Density | Ref | |
|---|---|---|---|---|---|
| Pt-based catalysts | 500 CV-Pt | 1 M NH3 + 5 M KOH | 0.42 | 0.27 mA cmECSA−2 | [42] |
| Flower-like Pt | 0.1 M NH3 + 1 M KOH | 0.5 | 0.48 mA cmECSA−2 | [49] | |
| Pt nanocubes (Pt-NCs) | 0.1 M NH4OH + 1 M KOH | 0.5 | 5.1 mA cmECSA−2 | [50] | |
| Pt nanoparticles (Pt NPs) | 0.1 M NH3 + 0.2 M NaOH | 0.55 | 1.96 mA cmECSA−2 | [51] | |
| Ir-decorated Pt | 0.1 M NH3 + 0.1 M KOH | 0.43 | 1.26 mA cmECSA−2 | [59] | |
| PtIr nanoparticle | 0.5 M NH4OH + 1 M KOH | 0.4 | 0.11 mA cmECSA−2 | [60] | |
| PtIrNi/SiO2-CNT-COOH | 0.1 M NH3 + 1 M KOH | 0.4 | 2.48 mA cm−2 | [61] | |
| PtNC/C | 0.1 M NH3 + 1 M KOH | 0.48 | 3.89 mA cmECSA−2 | [48] | |
| Pt nanosheet | 0.1 M NH3 + 1 M KOH | 0.57 | 0.32 mA cmECSA−2 | [62] | |
| Pt thin film | 0.1 M NH3 + 0.2 M NaOH | 0.5 | 0.212 mA cmECSA−2 | [63] | |
| Pt nanofilm | 0.1 M NH3 + 1 M KOH | 0.45 | 2.15 mA cm−2 | [1] | |
| C-Pt/SnO2 | 0.1 M NH3 + 1 M KOH | 0.45 | 1.6 mA cm−2 | [64] | |
| Pt/PBI/MWNT-CeO2 | 0.1 M NH3 + 1 M KOH | 0.45 | 0.26 mA cmECSA−2 | [65] | |
| Pt/C-ITO | 1 M NH4OH + 1 M KOH | 0.54 | 1.35 mA cmECSA−2 | [66] | |
| Y2O3-modified Pt/Si | 0.1 M NH3 + 1 M KOH | 0.45 | 0.19 mA cmECSA−2 | [21] | |
| Pt/NGA | 0.1 M NH3 + 1 M KOH | 0.5 | 0.64 mA cmECSA−2 | [67] | |
| Pt90Ir10 | 0.1 M NH3 + 1 M KOH | 0.58 | 0.37 mA cm−2 | [68] | |
| Pt-Ir nanocubes | 0.1 M NH3 + 1 M KOH | 0.45 | 1.32 mA cm−2 ECSA | [69] | |
| Pt/Ir/MWCNT | 0.1 M NH3 + 0.1 M KOH | 0.38 | 0.23 mA cm−2 ECSA | [70] | |
| PtIrZn | 0.1 M NH3 + 0.5 M NaOH | 0.3 | 0.56 mA cm−2 | [71] | |
| PtIrCu HCOND | 0.1 M NH3 + 1 M KOH | 0.35 | 122.9 A g−1 Pt | [72] | |
| PtIrZn2/CeO2-ZIF-8 | 0.1 M NH3 + 1 M KOH | 0.35 | 0.64 mA cm−2 | [73] | |
| Pt90Ru10/C | 1 M NH4OH + 1 M KOH | 0.5 | 0.91 mA cmECSA−2 | [74] | |
| Pt6Ru-NCs | 0.1 M NH3 + 1 M KOH | 0.5 | 1.02 mA cmECSA−2 | [75] | |
| Pt85Pd15/rGO | 0.1 M NH3 + 1 M KOH | 0.47 | 1.46 mA cm−2 | [57] |
| Catalysts | Electrolyte | Onset Potential (VRHE) | Stability | N2 Selectivity (%) | Removal Efficiency (%) | Ref | |
|---|---|---|---|---|---|---|---|
| Ni-based catalysts | Ni0.8Cu0.2 LHs | 0.55 M NH4Cl + 0.5 M NaOH | 1.39 | 35 mA cm−2 for 3 h | - | 84 | [24] |
| NiCu/MnO2 | 0.055 M NH4Cl +0.5 M NaOH | 1.6 | 3 mA cm−2 for 4 h | 97.4 | - | [26] | |
| NiCuFe | 0.055 M NH4Cl + 0.5 M NaOH | 1.43 | 20 mA cm−2 for 12 h | - | 89.4 | [27] | |
| Ag/Ni | 0.5 M NH3 + 1.5 M NaOH | 1.37 | 10 mA cm−2 for 350 h | 59.8 | - | [83] | |
| NiCu/CP | 0.055 M NH4Cl + 0.5 M NaOH | 1.47 | 8 mA cm−2 for 2 h | - | 78 | [19] | |
| Wire-in-plate nanostructured Ni(OH)2-Cu2O@CuO | 1 M NH3 + 1 M KOH | 1.37 | 60 mA cm−2 for 32 h | - | - | [86] | |
| NiCu DHTs | 0.05 M NH4OH + 0.1 M NaOH | 1.31 | 0.5 mA cm−2 for 1 h | - | - | [87] | |
| NiCu/BDD | 0.5 M NH3 + 0.5 M NaOH | 1.35 | 5 mA cm−2 for 7 h | 88.6 | 44.8 | [88] | |
| NiCuCo-S-T/CP | 0.2 M NH4Cl + 1 M NaOH | 1.24 | 105 mA cm−2 for 12 h | 81.2 | 98 | [89] | |
| NiO-TiO2 | 0.2 M NH4NO3 + 1 M NaOH | 1.53 | - | 90 | 96.4 | [90] | |
| Cu-based catalysts | CuO | 1 M NH3 + 1 M KOH | 1.19 | 200 mA cm−2 for 400 h | 62.8 | - | [84] |
| CuSn(OH)6 | 10 mM NH3 +0.5 M K2SO4 | 1.56 | - | 84.5 | - | [85] | |
| Ti-based catalyst | TiO | 0.1 M NH3 + 0.5 M NaOH | 0.4 | 0.01 mA cm−2 for 2 h | - | - | [77] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, J.H.; Park, S.Y.; Youn, D.H.; Jang, Y.J. Recent Advances in Electrocatalysts for Ammonia Oxidation Reaction. Catalysts 2023, 13, 803. https://doi.org/10.3390/catal13050803
Jang JH, Park SY, Youn DH, Jang YJ. Recent Advances in Electrocatalysts for Ammonia Oxidation Reaction. Catalysts. 2023; 13(5):803. https://doi.org/10.3390/catal13050803
Chicago/Turabian StyleJang, Ji Hee, So Young Park, Duck Hyun Youn, and Youn Jeong Jang. 2023. "Recent Advances in Electrocatalysts for Ammonia Oxidation Reaction" Catalysts 13, no. 5: 803. https://doi.org/10.3390/catal13050803
APA StyleJang, J. H., Park, S. Y., Youn, D. H., & Jang, Y. J. (2023). Recent Advances in Electrocatalysts for Ammonia Oxidation Reaction. Catalysts, 13(5), 803. https://doi.org/10.3390/catal13050803