


Effect of Alkoxy Substituents on the Regioselectivity of Catalytic C-H Activation in Benzoic Acids: Experimental and DFT Study



Abstract
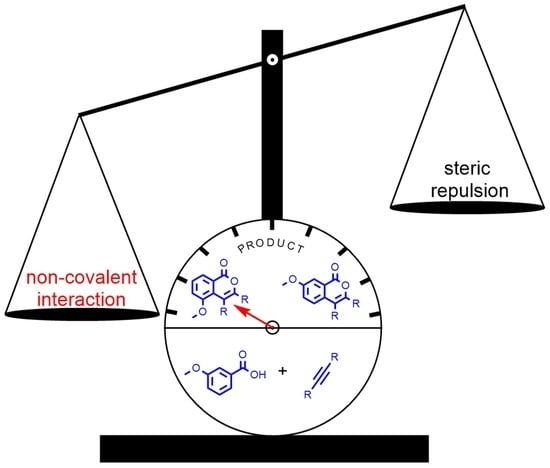
:
1. Introduction
2. Results and Discussion
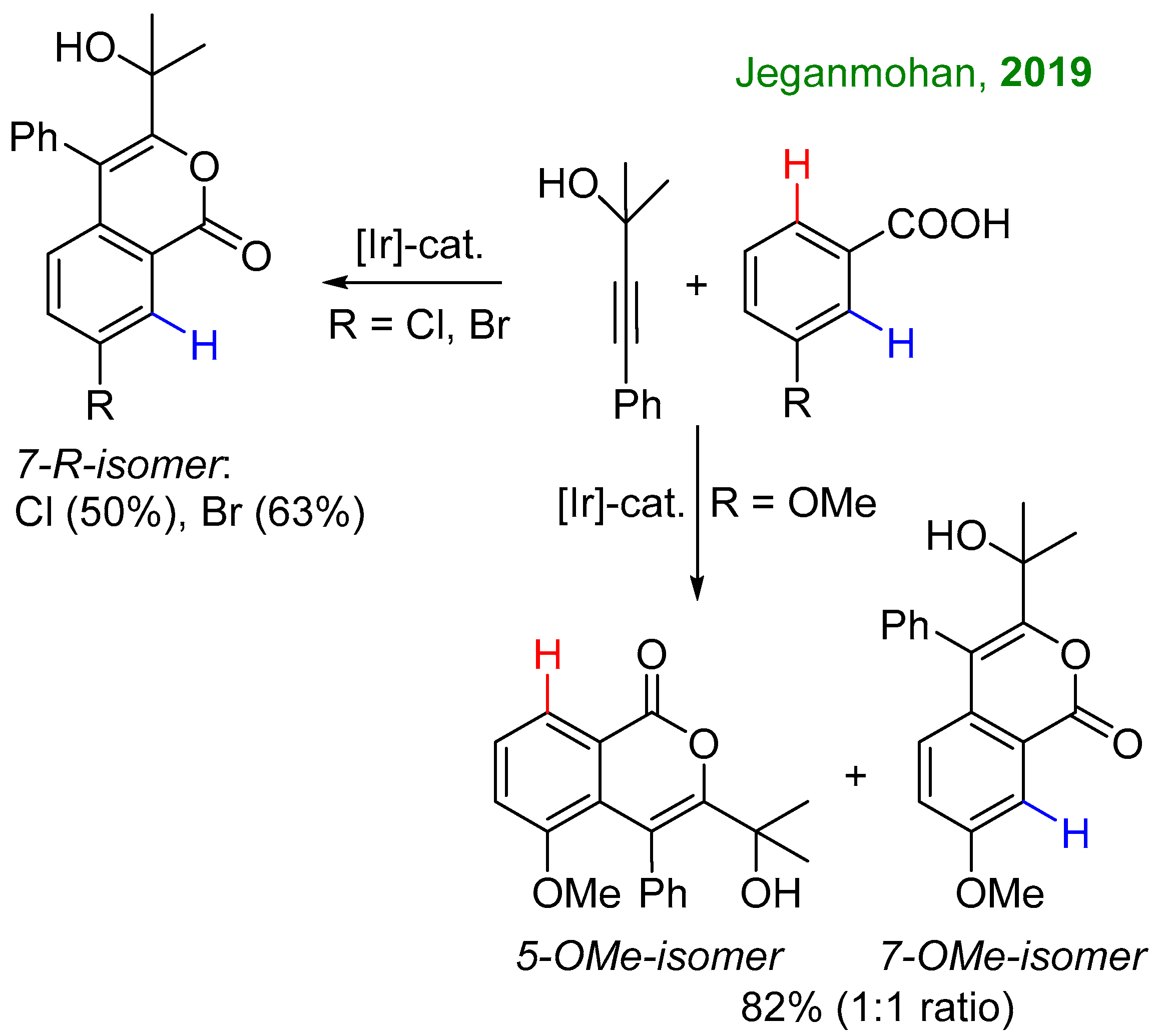
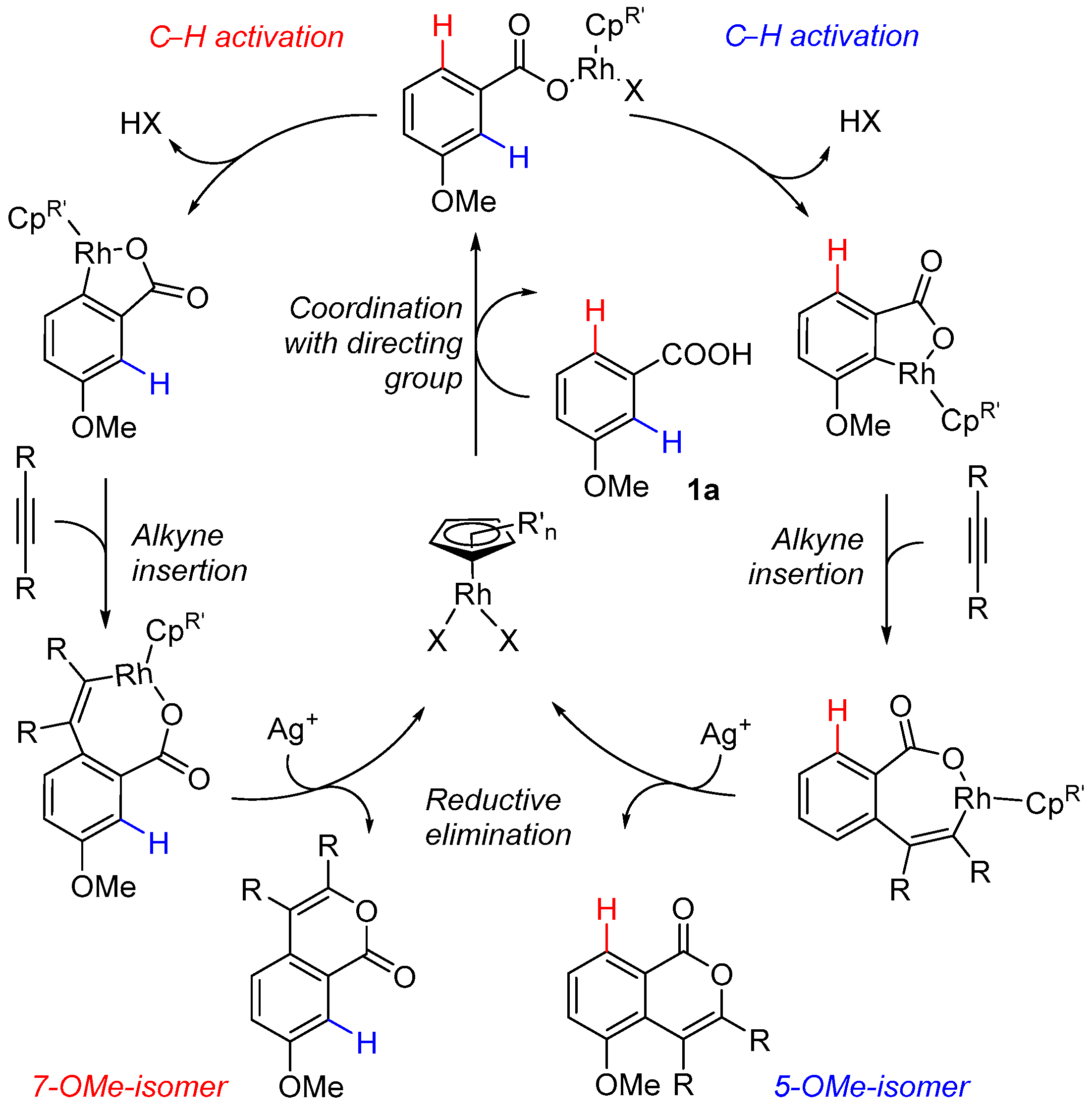
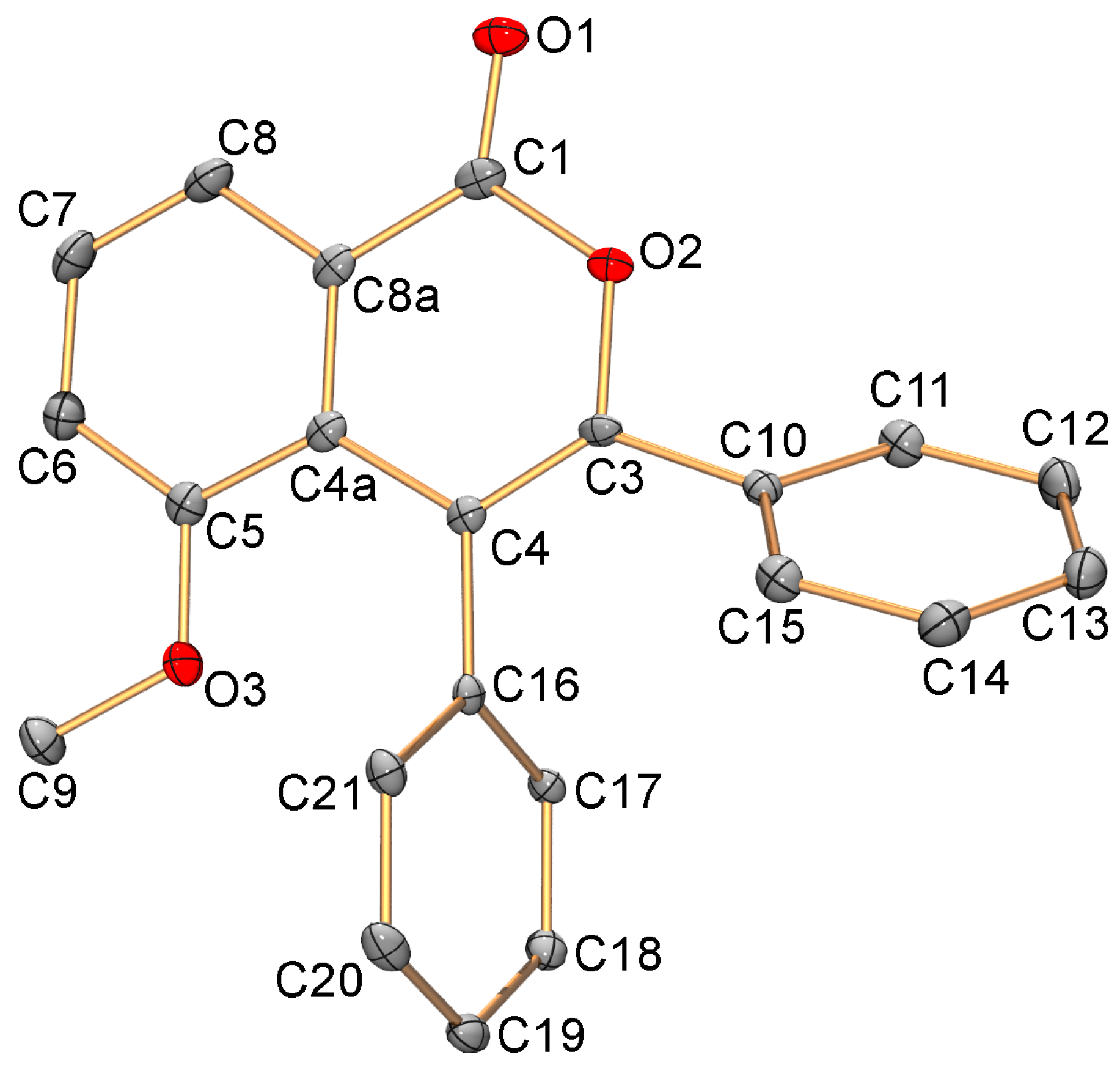
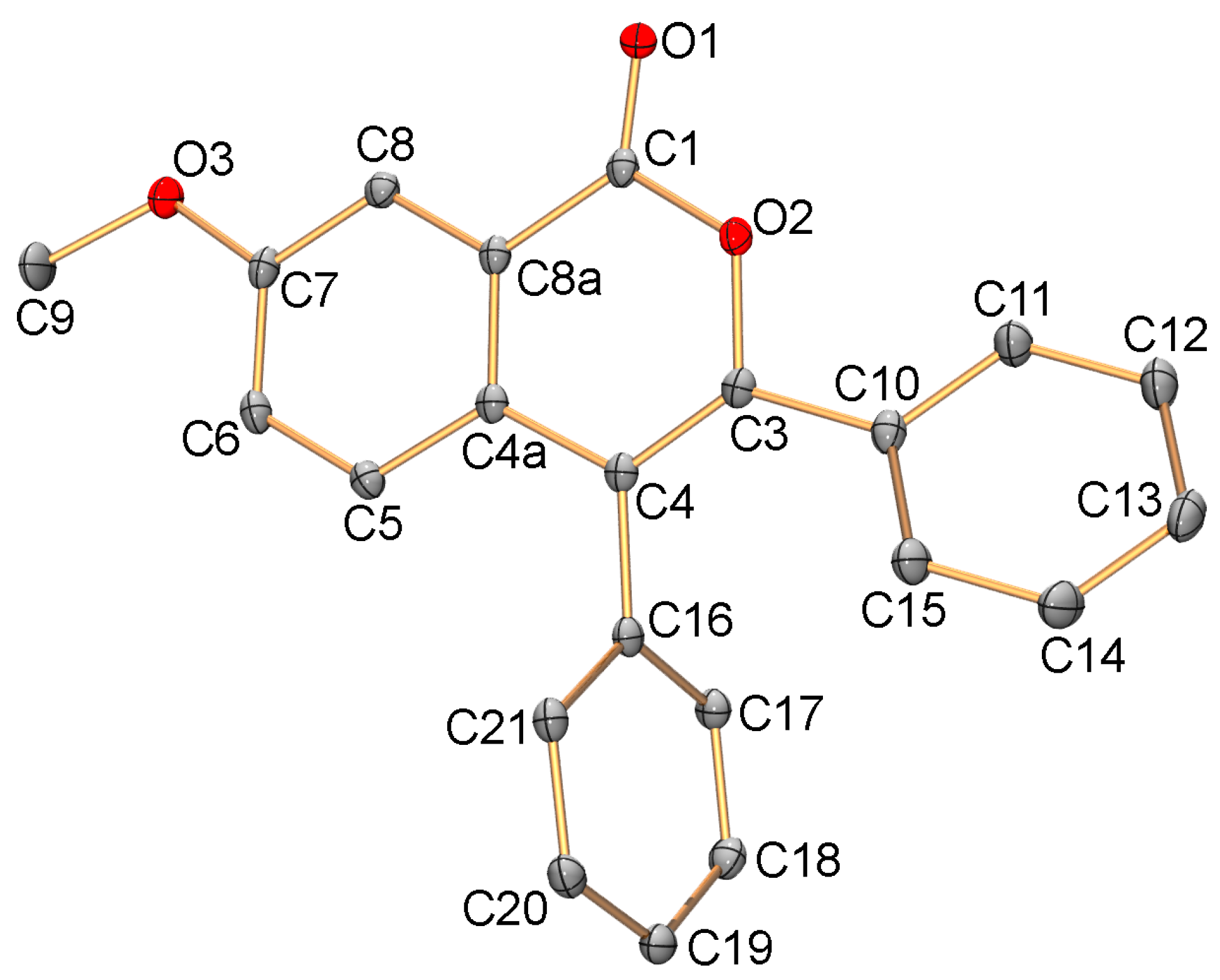
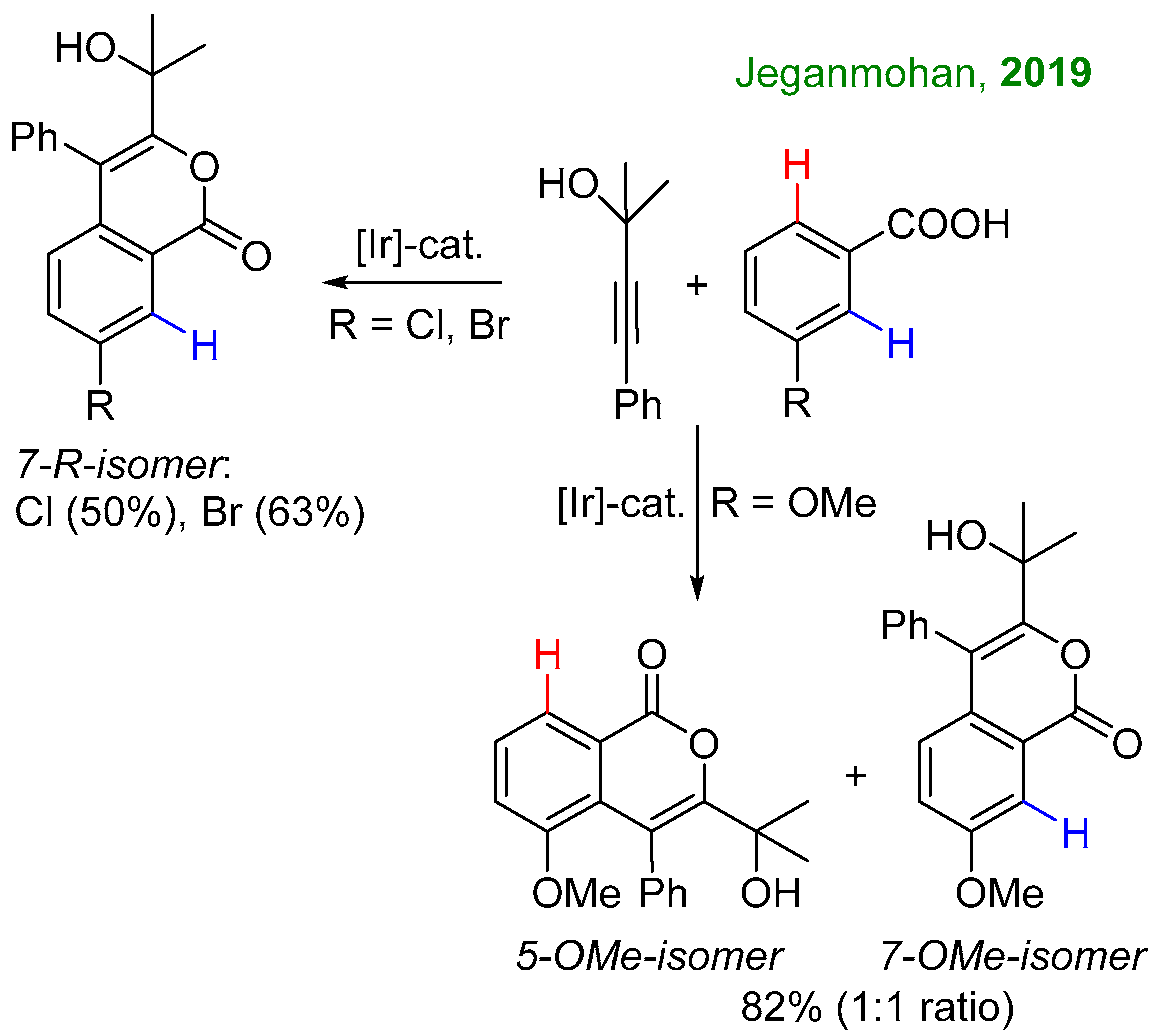
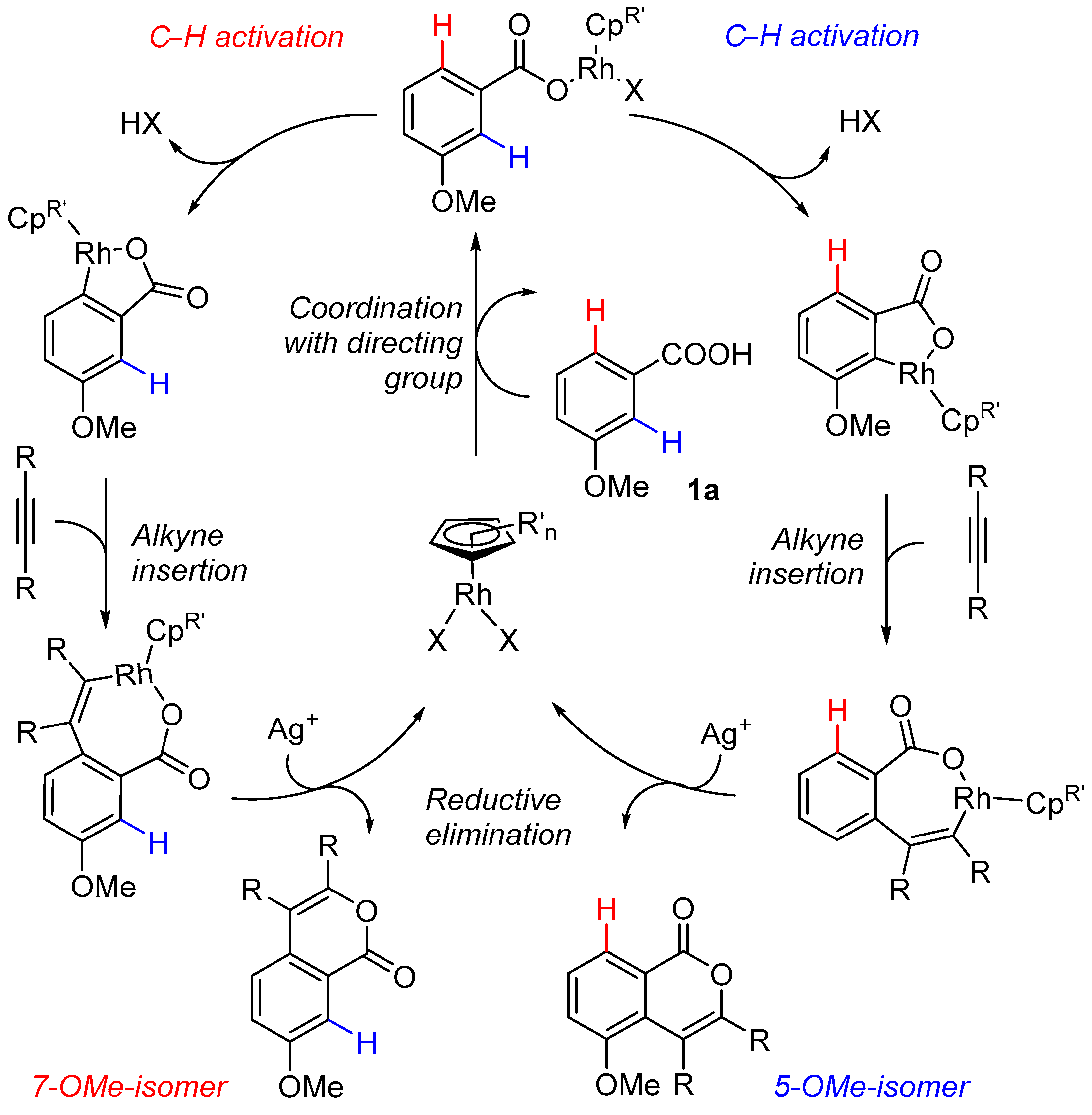
2.1. C-H Activation of 3-Methoxybenzoic Acid
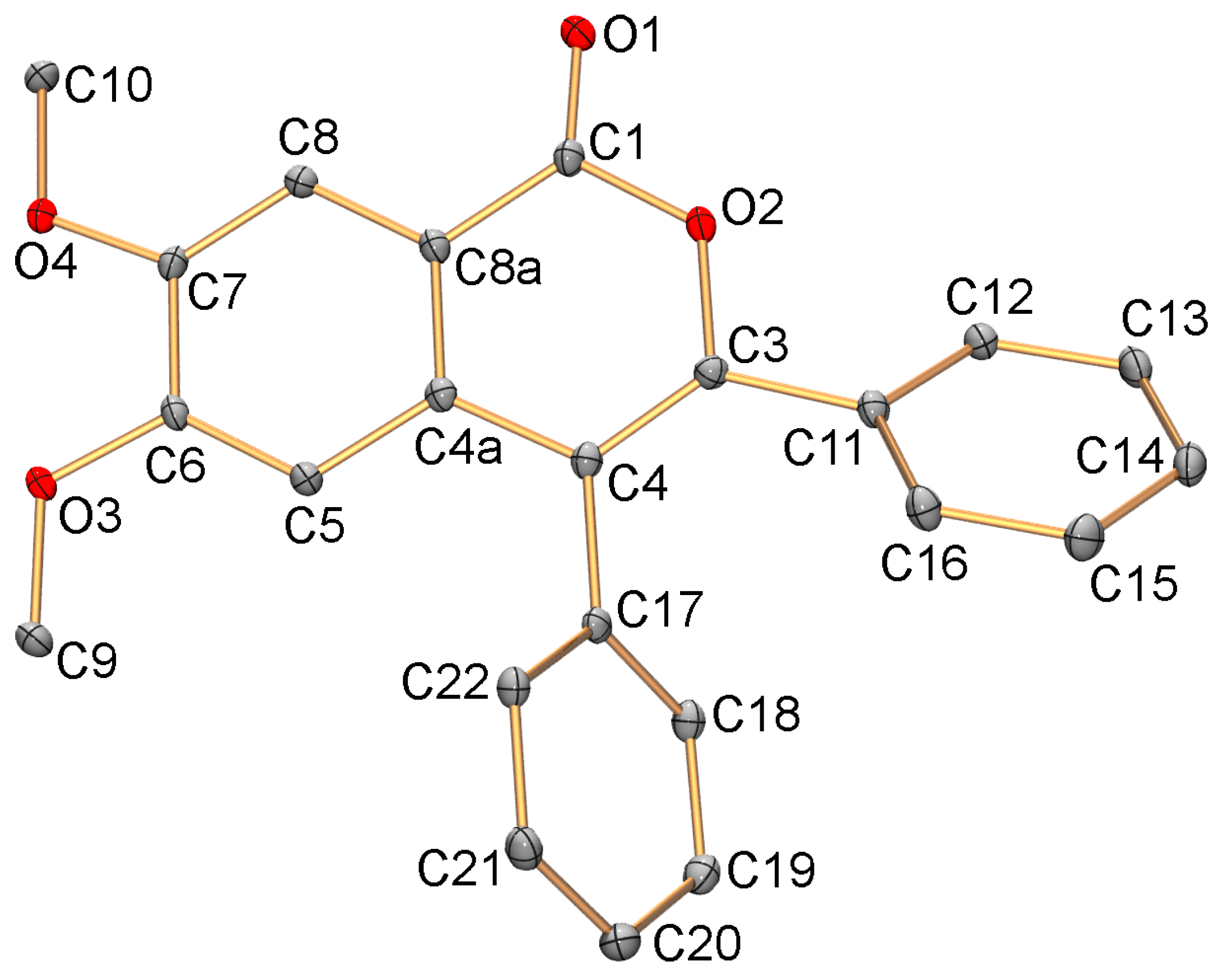
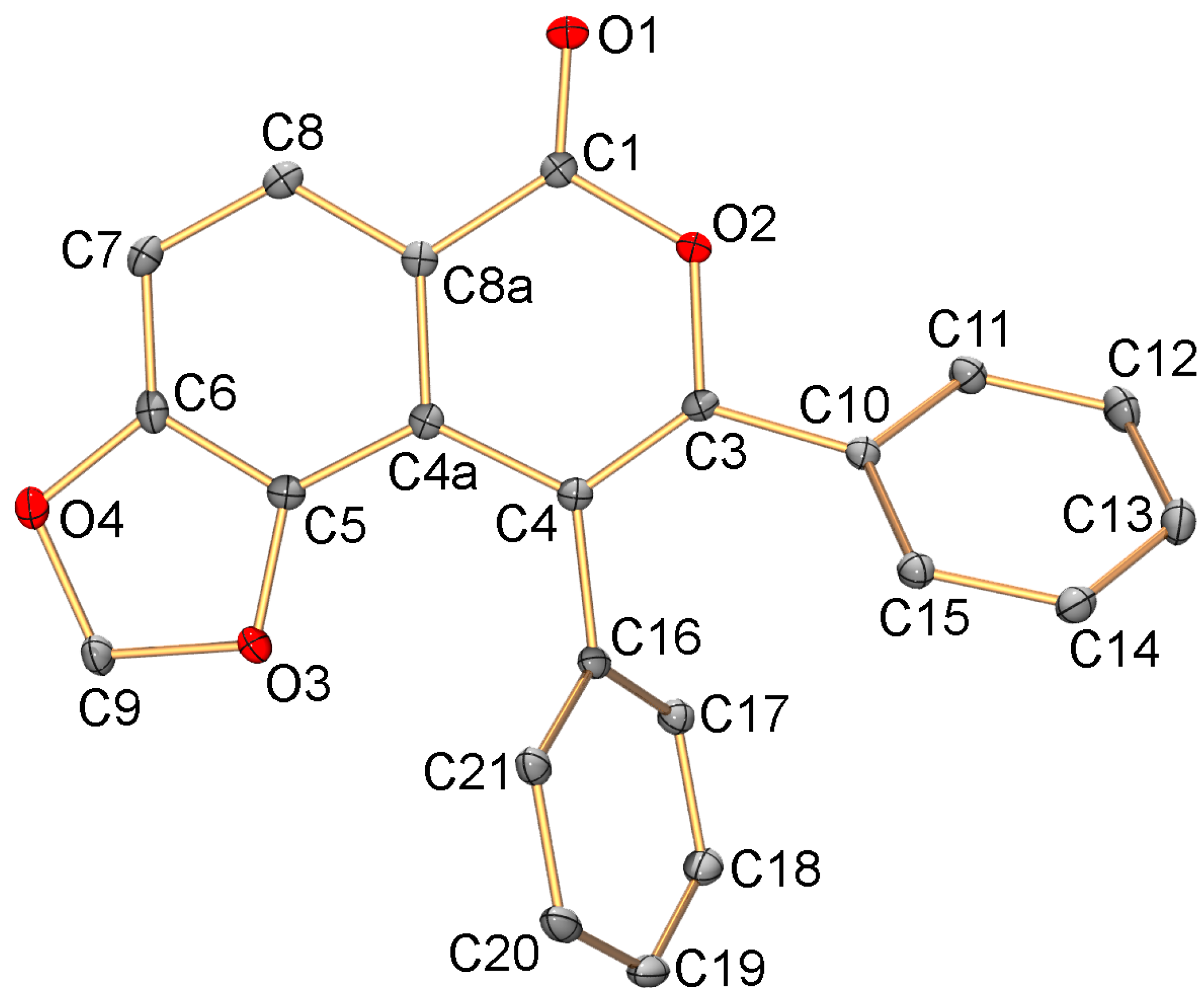
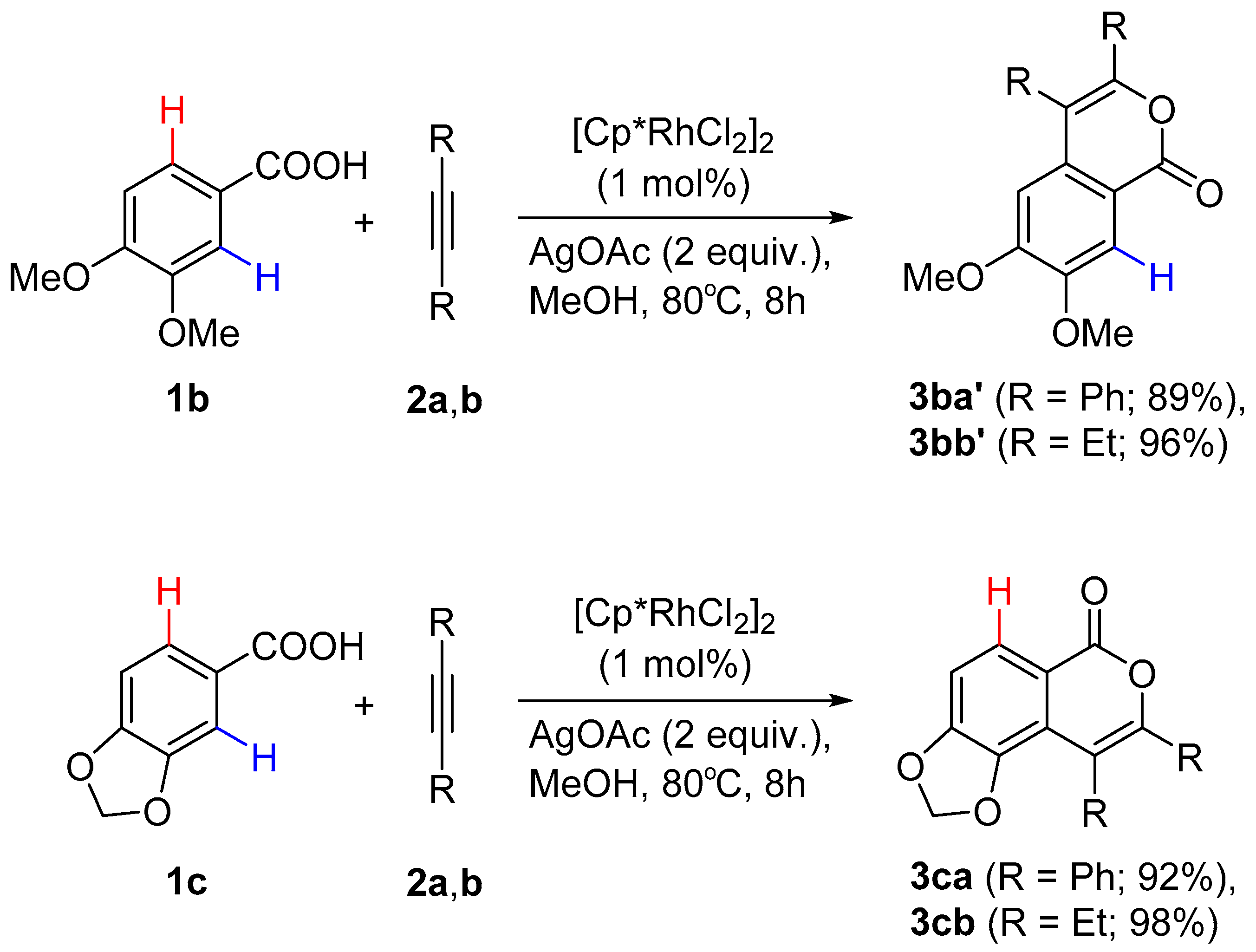
2.2. C-H Activation of 3,4-Dimethoxybenzoic and Piperonylic Acids
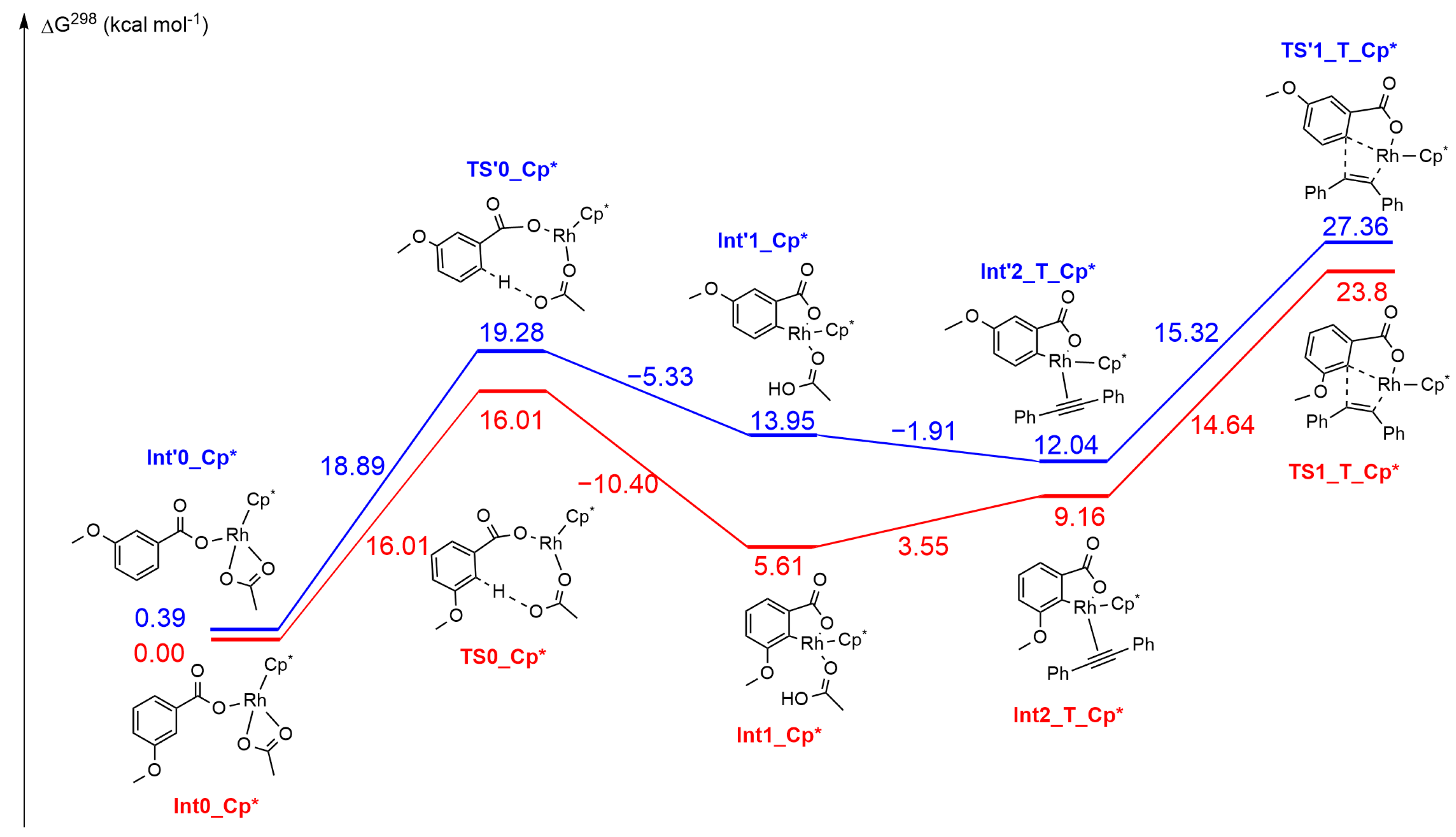
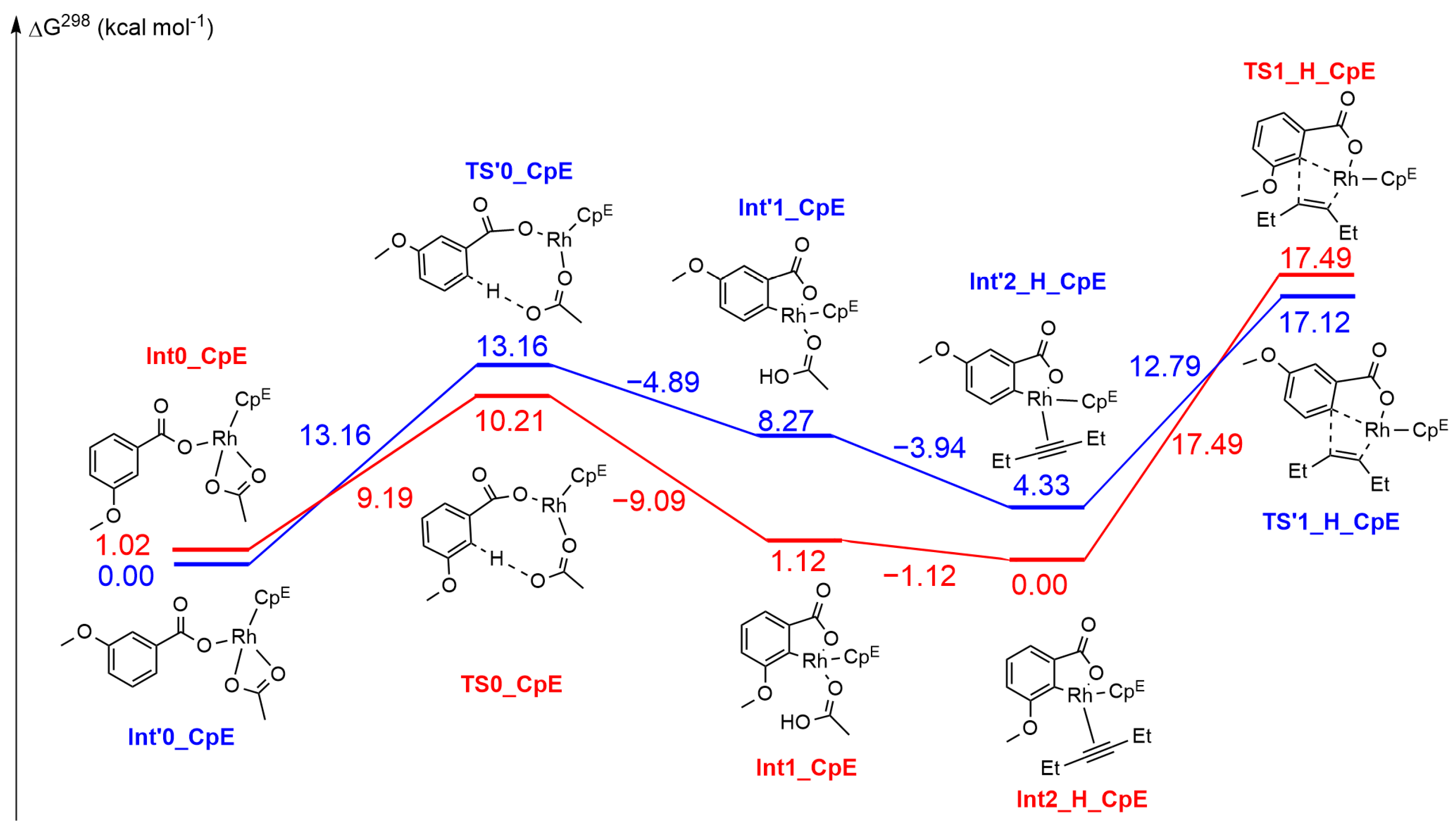
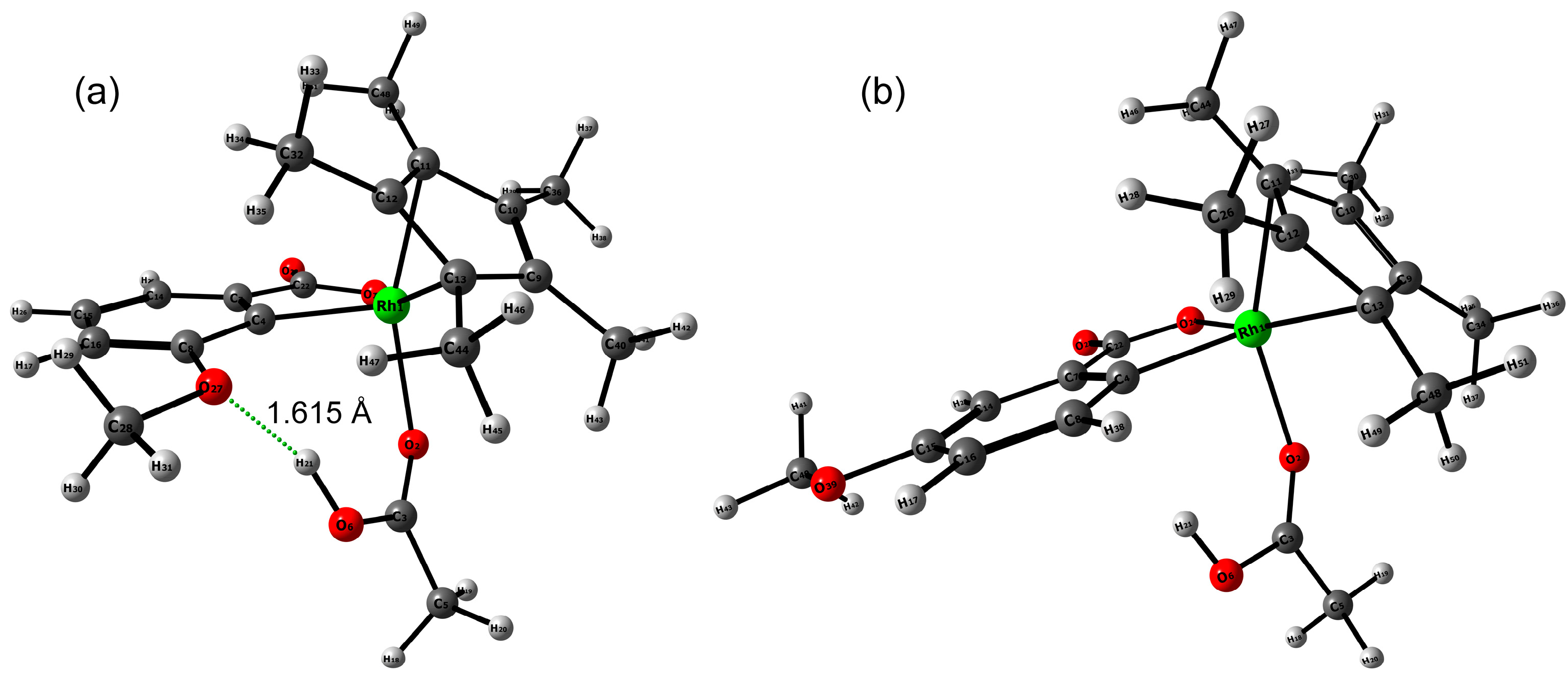
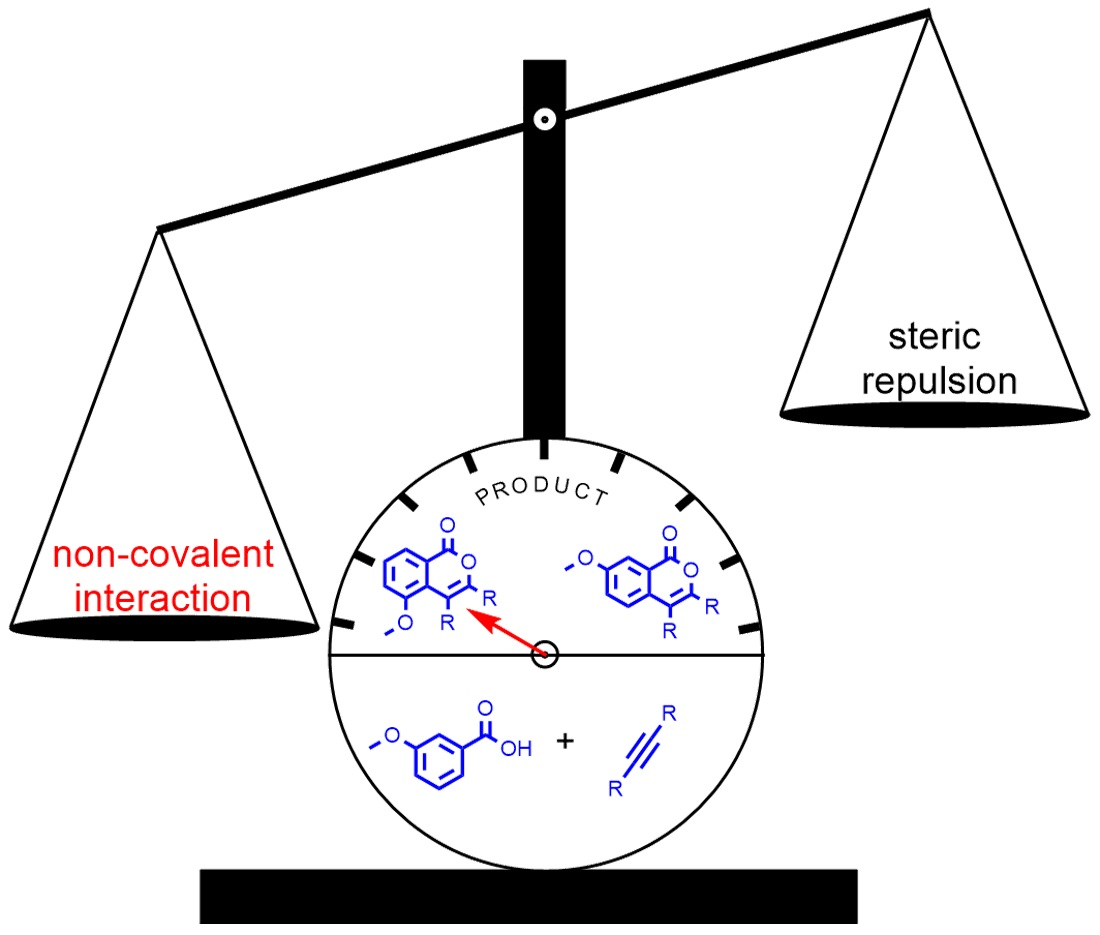
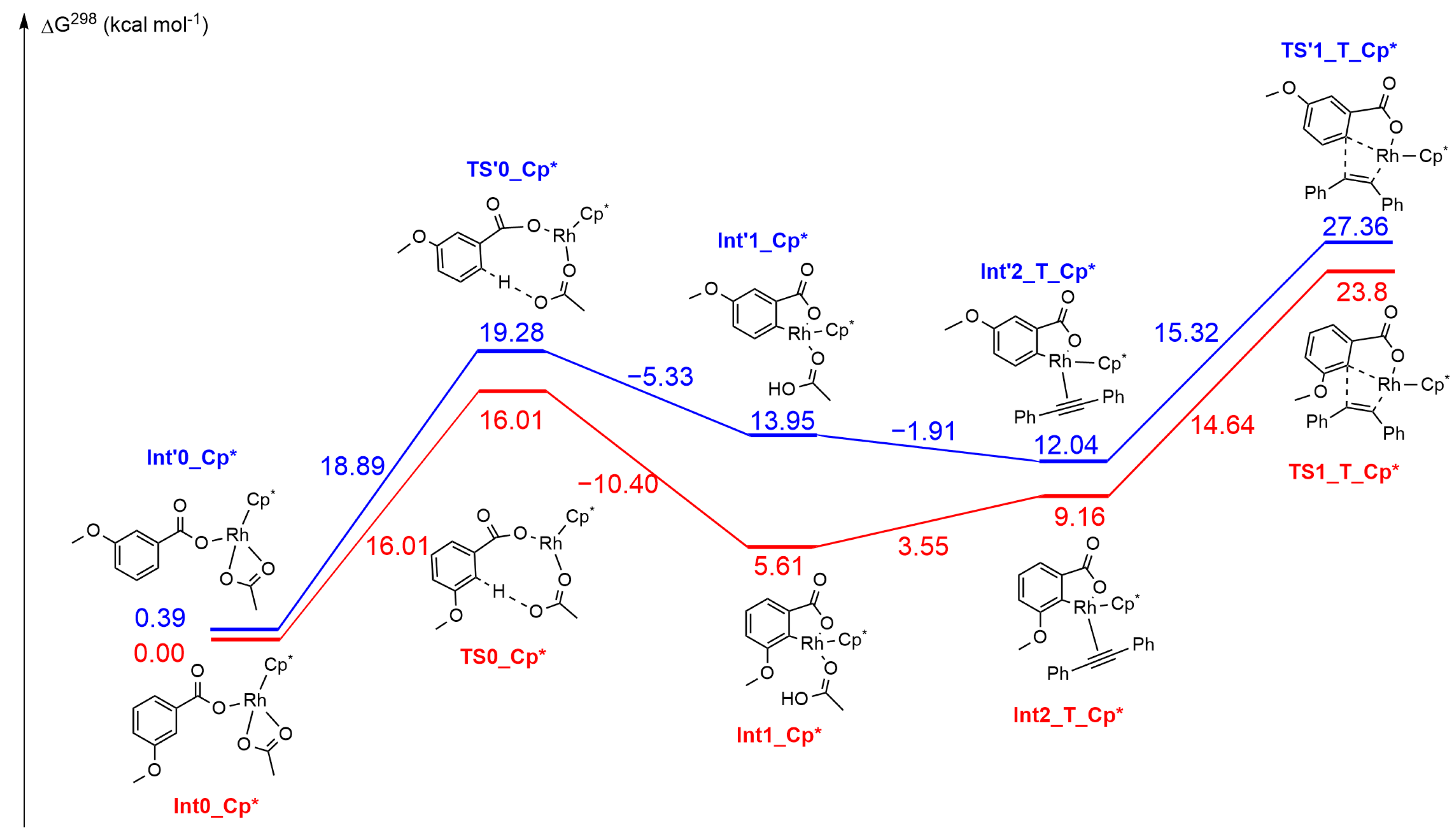
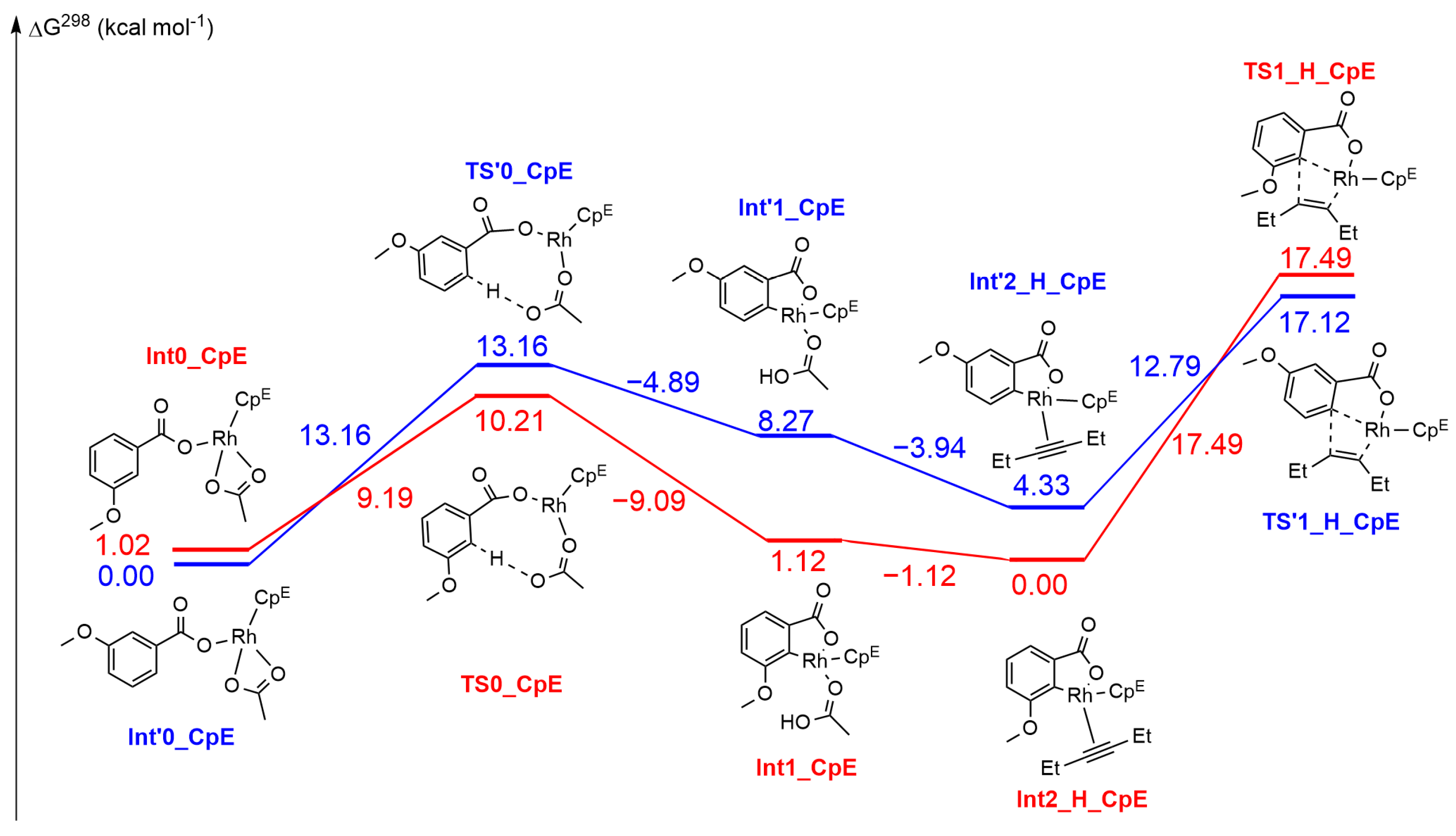
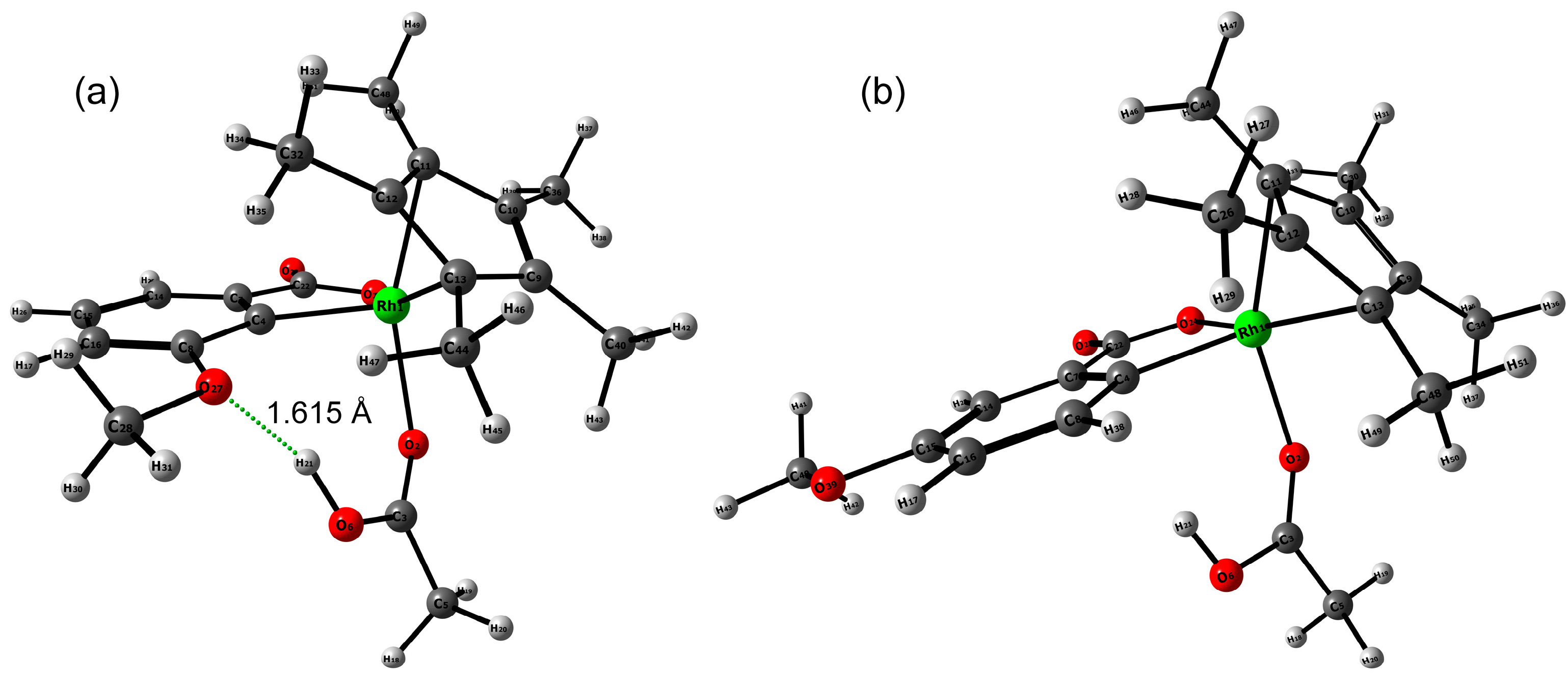
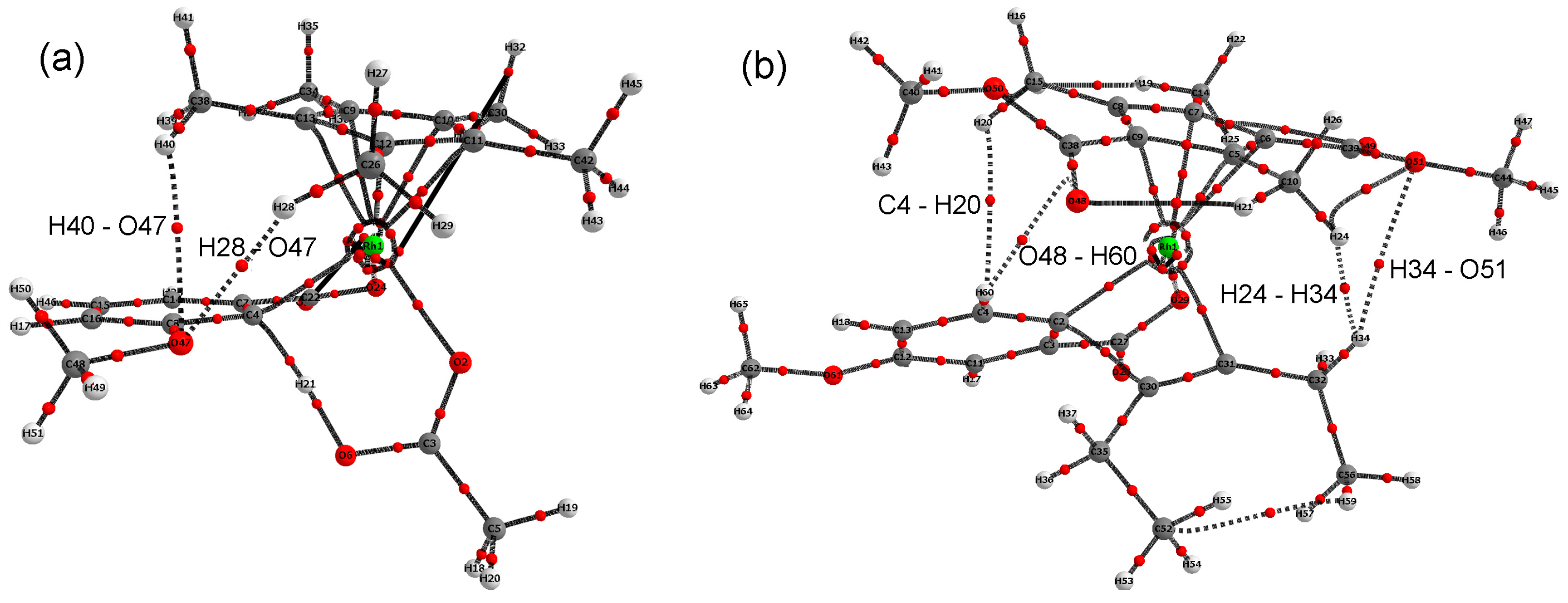
2.3. DFT Investigation
3. Materials and Methods
3.1. General Information
3.2. Reactions of 3-Methoxybenzoic Acid 1a with Alkynes
3.3. Reactions of 3,4-Methoxybenzoic Acid 1b with Alkynes
3.4. Reactions of Piperonylic Acid 1c with Alkynes
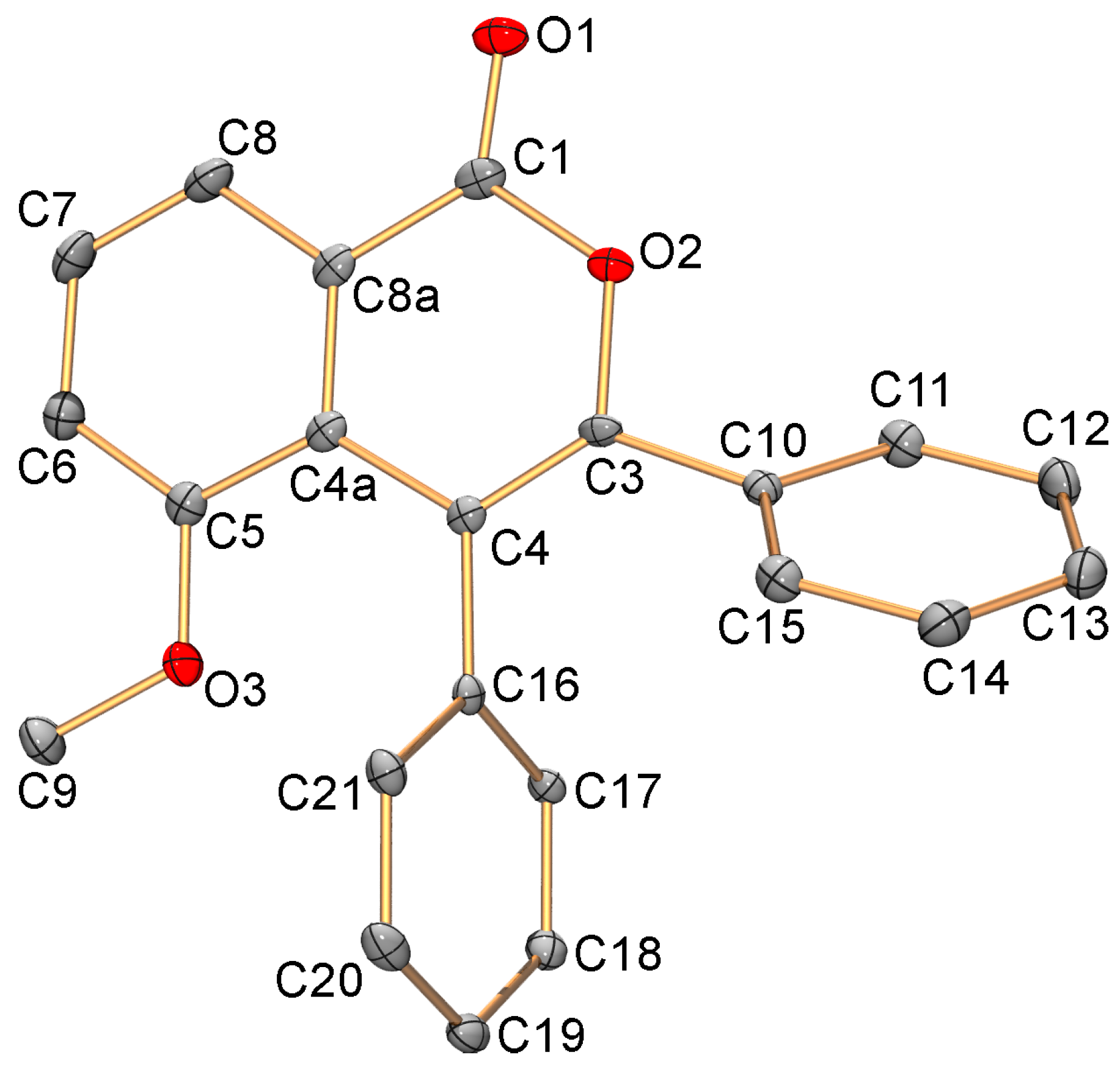
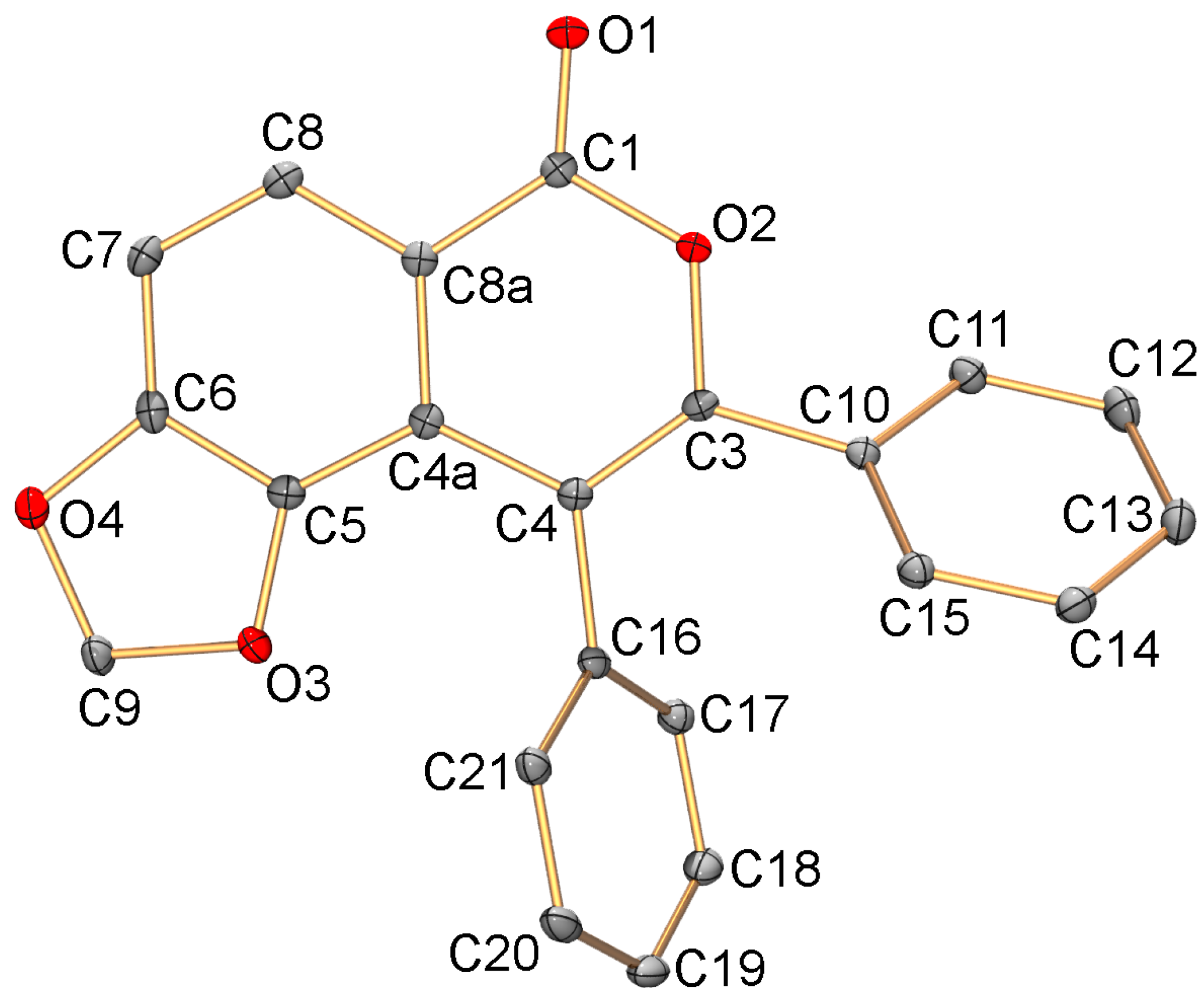
3.5. X-ray Diffraction Study
3.6. DFT Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shabir, G.; Saeed, A.; El-Seedi, H.R. Natural isocoumarins: Structural styles and biological activities, the revelations carry on …. Phytochemistry 2021, 181, 112568. [Google Scholar] [CrossRef]
- Lichota, A.; Gwozdzinski, K. Anticancer Activity of Natural Compounds from Plant and Marine Environment. Int. J. Mol. Sci. 2018, 19, 3533. [Google Scholar] [CrossRef] [PubMed]
- Cramer, J.; Sager, C.P.; Ernst, B. Hydroxyl Groups in Synthetic and Natural-Product-Derived Therapeutics: A Perspective on a Common Functional Group. J. Med. Chem. 2019, 62, 8915–8930. [Google Scholar] [CrossRef] [PubMed]
- Kharitonov, V.B.; Muratov, D.V.; Loginov, D.A. Cyclopentadienyl complexes of group 9 metals in the total synthesis of natural products. Coord. Chem. Rev. 2022, 471, 214744. [Google Scholar] [CrossRef]
- Jardim, G.A.M.; de Carvalho, R.L.; Nunes, M.P.; Machado, L.A.; Almeida, L.D.; Bahou, K.A.; Bower, J.F.; da Silva Júnior, E.N. Looking deep into C-H functionalization: The synthesis and application of cyclopentadienyl and related metal catalysts. Chem. Commun. 2022, 58, 3101–3121. [Google Scholar] [CrossRef]
- Loginov, D.A.; Konoplev, V.E. Oxidative coupling of benzoic acids with alkynes: Catalyst design and selectivity. J. Organomet. Chem. 2018, 867, 14–24. [Google Scholar] [CrossRef]
- Song, G.; Li, X. Substrate activation strategies in rhodium(III)-catalyzed selective functionalization of arenes. Acc. Chem. Res. 2015, 48, 1007–1020. [Google Scholar] [CrossRef]
- Satoh, T.; Miura, M. Oxidative coupling of aromatic substrates with alkynes and alkenes under rhodium catalysis. Chem. Eur. J. 2010, 16, 11212–11222. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, C.; Wang, Y.; Dong, L.; Sun, J. Total synthesis of Sparstolonin B, a potent anti-inflammatory agent. RSC Adv. 2015, 5, 12354–12357. [Google Scholar] [CrossRef]
- Li, X.; Huang, T.; Song, Y.; Qi, Y.; Li, L.; Li, Y.; Xiao, Q.; Zhang, Y. Co(III)-catalyzed annulative vinylene transfer via C-H activation: Three-step total synthesis of 8-oxopseudopalmatine and oxopalmatine. Org. Lett. 2020, 22, 5925–5930. [Google Scholar] [CrossRef]
- Song, L.; Tian, G.; Van der Eycken, E.V. Rhodium(III)-catalyzed intermolecular cascade annulation through CH activation: Concise synthesis of rosettacin. Mol. Catal. 2018, 459, 129–134. [Google Scholar] [CrossRef]
- Xu, X.; Liu, Y.; Park, C.-M. Rhodium(III)-catalyzed intramolecular annulation through C-H activation: Total synthesis of (±)-antofine, (±)-septicine, (±)-tylophorine, and rosettacin. Angew. Chem. Int. Ed. 2012, 51, 9372–9376. [Google Scholar] [CrossRef]
- Jayakumar, J.; Parthasarathy, K.; Cheng, C.-H. One-pot synthesis of isoquinolinium salts by rhodium-catalyzed C-H bond activation: Application to the total synthesis of oxychelerythrine. Angew. Chem. Int. Ed. 2012, 51, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Du, J.; Yang, Y.; Li, Y. Rhodium(III)-catalyzed intramolecular redoxneutral annulation of tethered alkynes: Formal total synthesis of (±)-goniomitine. Chem. Eur. J. 2014, 20, 12768–12772. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, R.L.; Almeida, R.G.; Murali, K.; Machado, L.A.; Pedrosa, L.F.; Dolui, P.; Maiti, D.; da Silva Júnior, E.N. Removal and modification of directing groups used in metal-catalyzed C-H functionalization: The magical step of conversion into ‘conventional’ functional groups. Org. Biomol. Chem. 2021, 19, 525–547. [Google Scholar] [CrossRef] [PubMed]
- Murali, K.; Machado, L.A.; Carvalho, R.L.; Pedrosa, L.F.; Mukherjee, R.; da Silva Júnior, E.N.; Maiti, D. Decoding directing groups and their pivotal role in C-H activation. Chem. Eur. J. 2021, 27, 12453–12508. [Google Scholar] [CrossRef]
- Sambiagio, C.; Schönbauer, D.; Blieck, R.; Dao-Huy, T.; Pototschnig, G.; Schaaf, P.; Wiesinger, T.; Zia, M.F.; Wencel-Delord, J.; Besset, T.; et al. A comprehensive overview of directing groups applied in metal-catalysed CH functionalisation chemistry. Chem. Soc. Rev. 2018, 47, 6603–6743. [Google Scholar] [CrossRef]
- Drapeau, M.P.; Goossen, L.J. Carboxylic acids as directing groups for C-H bond functionalization. Chem. Eur. J. 2016, 22, 18654–18677. [Google Scholar] [CrossRef]
- Sihag, P.; Jeganmohan, M. Regioselective Synthesis of Isocoumarins via Iridium(III)-Catalyzed Oxidative Cyclization of Aromatic Acids with Propargyl Alcohols. J. Org. Chem. 2019, 84, 2699–2712. [Google Scholar] [CrossRef]
- Luo, F.; He, S.; Gou, Q.; Chen, J.; Zhang, M. Rhodium-catalyzed oxidative coupling of benzoic acids with propargyl alcohols: An efficient access to isocoumarins. Tetrahedron Lett. 2021, 64, 152724. [Google Scholar] [CrossRef]
- Vorobyeva, D.V.; Petropavlovskikh, D.A.; Godovikov, I.A.; Nefedov, S.E.; Osipov, S.N. Rh(III)-Catalyzed C-H Activation/Annulation of Aryl Hydroxamates with CF3-Containing α-Propargyl α-Amino Acid Derivatives. Eur. J. Org. Chem. 2021, 2021, 1883–1890. [Google Scholar] [CrossRef]
- Kudo, E.; Shibata, Y.; Yamazaki, M.; Masutomi, K.; Miyauchi, Y.; Fukui, M.; Sugiyama, H.; Uekusa, H.; Satoh, T.; Miura, M.; et al. Oxidative Annulation of Arenecarboxylic and Acrylic Acids with Alkynes under Ambient Conditions Catalyzed by an Electron-Deficient Rhodium(III) Complex. Chem. Eur. J. 2016, 22, 14190–14194. [Google Scholar] [CrossRef] [PubMed]
- Hyster, T.K.; Rovis, T. Rhodium(III)-Catalyzed C–H Activation Mediated Synthesis of Isoquinolones from Amides and Cyclopropenes. Synlett 2013, 24, 1842–1844. [Google Scholar] [CrossRef] [PubMed]
- Unoh, Y.; Hirano, K.; Satoh, T.; Miura, M. Synthesis of highly substituted isocoumarins by rhodium-catalyzed annulation of readily available benzoic acids. Tetrahedron 2013, 69, 4454–4458. [Google Scholar] [CrossRef]
- Chinnagolla, R.K.; Jeganmohan, M. Regioselective synthesis of isocoumarins by ruthenium-catalyzed aerobic oxidative cyclization of aromatic acids with alkynes. Chem. Commun. 2012, 48, 2030–2032. [Google Scholar] [CrossRef]
- Kharitonov, V.B.; Muratov, D.V.; Nelyubina, Y.V.; Loginov, D.A. Formation of a Naphthalene Framework by Rhodium(III)-Catalyzed Double C–H Functionalization of Arenes with Alkynes: Impact of a Supporting Ligand and an Acid Additive. Synthesis 2022, 54, 5119–5127. [Google Scholar]
- Inai, Y.; Usuki, Y.; Satoh, T. Synthesis of Benzo-Fused Cyclic Compounds via Rhodium-Catalyzed Decarboxylative Coupling of Aromatic Carboxylic Acids with Alkynes. Synthesis 2021, 53, 3029–3036. [Google Scholar]
- Honjo, Y.; Shibata, Y.; Kudo, E.; Namba, T.; Masutomi, K.; Tanaka, K. Room Temperature Decarboxylative and Oxidative [2+2+2] Annulation of Benzoic Acids with Alkynes Catalyzed by an Electron-Deficient Rhodium(III) Complex. Chem. Eur. J. 2018, 24, 317–321. [Google Scholar] [CrossRef]
- Kharitonov, V.B.; Runikhina, S.A.; Nelyubina, Y.V.; Muratov, D.V.; Chusov, D.; Loginov, D.A. Easy Access to Versatile Catalytic Systems for C-H Activation and Reductive Amination Based on Tetrahydrofluorenyl Rhodium(III) Complexes. Chem. Eur. J. 2021, 27, 10903–10912. [Google Scholar] [CrossRef]
- Datsenko, V.P.; Nelyubina, Y.V.; Smol’yakov, A.F.; Loginov, D.A. Cyclooctadiene iridium complexes [Cp*Ir(COD)X]+ (X = Cl, Br, I): Synthesis and application for oxidative coupling of benzoic acid with alkynes. J. Organomet. Chem. 2018, 874, 7–12. [Google Scholar] [CrossRef]
- Frasco, D.A.; Lilly, C.P.; Boyle, P.D.; Ison, E.A. Cp*IrIII-Catalyzed Oxidative Coupling of Benzoic Acids with Alkynes. ACS Catal. 2013, 3, 2421–2429. [Google Scholar] [CrossRef]
- Arsenov, M.A.; Fedorov, Y.V.; Muratov, D.V.; Nelyubina, Y.V.; Loginov, D.A. Synthesis of isocoumarins and PAHs with electron-withdrawing substituents: Impact of the substituent nature on the photophysical behavior. Dye. Pigment. 2022, 206, 110653. [Google Scholar] [CrossRef]
- Arsenov, M.A.; Loginov, D.A. Recent Advances in the Synthesis of Isocoumarins and Polyaromatic Hydrocarbons for Photoactive Materials. INEOS OPEN 2021, 4, 133–139. [Google Scholar] [CrossRef]
- Molotkov, A.P.; Arsenov, M.A.; Kapustin, D.A.; Muratov, D.V.; Shepel’, N.E.; Fedorov, Y.V.; Smol’yakov, A.F.; Knyazeva, E.I.; Lypenko, D.A.; Dmitriev, A.V.; et al. Effect of Cp-Ligand Methylation on Rhodium(III)-Catalyzed Annulations of Aromatic Carboxylic Acids with Alkynes: Synthesis of Isocoumarins and PAHs for Organic Light-Emitting Devices. ChemPlusChem 2020, 85, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, M.M.; Loginov, D.A. Rhoda- and iridacarborane halide complexes: Synthesis, structure and application in homogeneous catalysis. J. Organomet. Chem. 2020, 910, 121135. [Google Scholar] [CrossRef]
- Loginov, D.A.; Muratov, D.V.; Nelyubina, Y.V.; Laskova, J.; Kudinov, A.R. µ-Borole triple-decker complexes as catalysts for oxidative coupling of benzoic acid with alkynes. Structure of a hybrid rhodacyclopentadienyl/borole triple-decker complex. J. Mol. Catal. A 2017, 426, 393–397. [Google Scholar] [CrossRef]
- White, C.; Yates, A.; Maitlis, P.M. (η5-Pentamethylcyclopentadienyl)Rhodium and -Iridium Compounds. Inorg. Synth. 1992, 29, 228–234. [Google Scholar]
- Ueura, K.; Satoh, T.; Miura, M. Rhodium- and Iridium-Catalyzed Oxidative Coupling of Benzoic Acids with Alkynes via Regioselective C-H Bond Cleavage. J. Org. Chem. 2007, 72, 5362–5367. [Google Scholar] [CrossRef]
- Kharitonov, V.B.; Muratov, D.V.; Nelyubina, Y.V.; Shutkov, I.A.; Nazarov, A.A.; Loginov, D.A. Triphenylcyclopentadienyl Rhodium Complexes in Catalytic C-H Annulations. Application for Synthesis of Natural Isocoumarins. J. Org. Chem. 2023. [Google Scholar] [CrossRef]
- Loginov, D.A.; Vinogradov, M.M.; Starikova, Z.A.; Petrovskii, P.V.; Kudinov, A.R. Arene complexes [(η-C5H5)M(η-C6R6)]2+ (M = Rh, Ir). Russ. Chem. Bull. 2004, 53, 1949–1953. [Google Scholar] [CrossRef]
- Loginov, D.A.; Belova, A.O.; Kudinov, A.R. Rhodacarboranes as catalysts for oxidative coupling of benzoic acid with diphenylacetylene. Russ. Chem. Bull. 2014, 63, 983–986. [Google Scholar] [CrossRef]
- Lin, W.; Li, W.; Lu, D.; Su, F.; Wen, T.-B.; Zhang, H.-J. Dual Effects of Cyclopentadienyl Ligands on Rh(III)-Catalyzed Dehydrogenative Arylation of Electron-Rich Alkenes. ACS Catal. 2018, 8, 8070–8076. [Google Scholar] [CrossRef]
- Shibata, Y.; Tanaka, K. Catalytic [2+2+1] Cross-Cyclotrimerization of Silylacetylenes and Two Alkynyl Esters To Produce Substituted Silylfulvenes. Angew. Chem. Int. Ed. 2011, 50, 10917–10921. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, Y.; Ishigaki, S.; Tanaka, J.; Tanaka, K. Acceleration Mechanisms of C–H Bond Functionalization Catalyzed by Electron-Deficient CpRh(III) Complexes. ACS Catal. 2021, 11, 13591–13602. [Google Scholar] [CrossRef]
- Araujo Dias, A.J.; Takahashi, H.; Nogami, J.; Nagashima, Y.; Tanaka, K. Oxidative [4 + 2] annulation of 1-naphthols with alkynes accelerated by an electron-deficient rhodium(III) catalysts. Org. Biomol. Chem. 2022, 20, 1008–1012. [Google Scholar] [CrossRef]
- Wang, D.; Astruc, D. The Golden Age of Transfer Hydrogenation. Chem. Rev. 2015, 115, 6621–6686. [Google Scholar] [CrossRef]
- Biriukov, K.O.; Afanasyev, O.I.; Godovikova, M.I.; Loginov, D.A.; Chusov, D.A. Osmium-catalyzed reduction processes. Russ. Chem. Rev. 2022, 91, RCR5045. [Google Scholar] [CrossRef]
- Kozuch, S.; Martin, J.M.L. The Rate-Determining Step is Dead. Long Live the Rate-Determining State! ChemPhysChem 2011, 12, 1413–1418. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Gurbanov, A.V.; Guseinov, F.I.; Guedes da Silva, M.F.C. Noncovalent interactions in metal complex catalysis. Coord. Chem. Rev. 2019, 387, 32–46. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Kopylovich, M.N.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. (Eds.) Noncovalent Interactions in Catalysis; Royal Society of Chemistry: Cambridge, UK, 2019. [Google Scholar]
- MacMillan, D.W.C. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Kumar, N.; Saha, S.; Sastry, G.N. Towards developing a criterion to characterize non-covalent bonds: A quantum mechanical study. Phys. Chem. Chem. Phys. 2021, 23, 8478–8488. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. A Description of the Chemical Bond in Terms of Local Properties of Electron Density and Energy. Croat. Chem. Acta 1984, 56, 1259–1281. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, 71 Pt A, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, 64 Pt A, 112–122. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system—Version 5.0. WIREs Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll, Version 19.10.12; TK Gristmill Software: Overland Park, KS, USA, 2019. Available online: aim.tkgristmill.com(accessed on 12 February 2021).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | R | Catalyst | Total Yield, % b | Ratio of 5/7-Isomers c |
| 1 | Ph | [Cp*RhCl2]2 | 90 | 2.4:1 |
| 2 | Ph | [CpPh3RhI2]2 | 66 | 2.2:1 |
| 3 | Ph | [CpRhI2]n | 91 | 1.85:1 |
| 4 | Ph | [CpERhCl2]2 | 20 | 1.72:1 |
| 5 | Et | [Cp*RhCl2]2 | 97 | 1.40:1 |
| 6 | Et | [CpPh3RhI2]2 | 97 | 1.25:1 |
| 7 | Et | [CpRhI2]n | 94 | 1.10:1 |
| 8 | Et | [CpERhCl2]2 | 95 | 0.90:1 |
| 3aa | 3aa’ | 3ba’ | 3ca | |
|---|---|---|---|---|
| Formula unit | C22H16O3 | C22H16O3 | C23H18O4 | C22H14O4 |
| Molecular weight | 328.35 | 328.35 | 358.37 | 342.33 |
| Crystal system | Monoclinic | Triclinic | Monoclinic | Triclinic |
| Space group | P21/c | P-1 | P21/n | P-1 |
| Z | 8 | 4 | 8 | 2 |
| a (Å) | 9.3656(9) | 9.3355(2) | 10.2837(5) | 6.8314(2) |
| b (Å) | 15.4672(19) | 14.0312(4) | 33.7768(15) | 9.6009(2) |
| c (Å) | 22.587(2) | 14.1181(4) | 10.3573(4) | 12.8243(3) |
| α (deg) | 90 | 60.4420(10) | 90 | 71.7570(10) |
| β (deg) | 93.808(5) | 87.511(2) | 100.700(2) | 79.6350(10) |
| γ (deg) | 90 | 82.039(2) | 90 | 85.8540(10) |
| V (Å3) | 3264.8(6) | 1592.46(7) | 3535.1(3) | 785.69(3) |
| Dcalc (g cm–3) | 1.336 | 1.370 | 1.347 | 1.447 |
| Linear absorption μ (cm–1) | 0.88 | 0.91 | 0.92 | 1.00 |
| F(000) | 1376 | 688 | 1504 | 356 |
| 2θmax (deg) | 54 | 56 | 56 | 56 |
| Reflections collected | 34,022 | 19,932 | 27,437 | 9892 |
| Independent reflections | 6973 | 7685 | 8518 | 3782 |
| Observed reflections (I > 2σ(I)) | 3653 | 5218 | 6190 | 3027 |
| Number of parameters | 453 | 453 | 491 | 235 |
| R1 | 0.0705 | 0.0575 | 0.0485 | 0.0415 |
| wR2 | 0.1615 | 0.1499 | 0.1133 | 0.1034 |
| GOOF | 1.030 | 1.021 | 1.022 | 1.023 |
| Δρmax/Δρmin (e Å−3) | 0.313/−0.235 | 0.541/−0.330 | 0.286/−0.235 | 0.305/−0.237 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kharitonov, V.B.; Muratov, D.V.; Nelyubina, Y.V.; Loginov, D.A. Effect of Alkoxy Substituents on the Regioselectivity of Catalytic C-H Activation in Benzoic Acids: Experimental and DFT Study. Catalysts 2023, 13, 389. https://doi.org/10.3390/catal13020389
Kharitonov VB, Muratov DV, Nelyubina YV, Loginov DA. Effect of Alkoxy Substituents on the Regioselectivity of Catalytic C-H Activation in Benzoic Acids: Experimental and DFT Study. Catalysts. 2023; 13(2):389. https://doi.org/10.3390/catal13020389
Chicago/Turabian StyleKharitonov, Vladimir B., Dmitry V. Muratov, Yulia V. Nelyubina, and Dmitry A. Loginov. 2023. "Effect of Alkoxy Substituents on the Regioselectivity of Catalytic C-H Activation in Benzoic Acids: Experimental and DFT Study" Catalysts 13, no. 2: 389. https://doi.org/10.3390/catal13020389
APA StyleKharitonov, V. B., Muratov, D. V., Nelyubina, Y. V., & Loginov, D. A. (2023). Effect of Alkoxy Substituents on the Regioselectivity of Catalytic C-H Activation in Benzoic Acids: Experimental and DFT Study. Catalysts, 13(2), 389. https://doi.org/10.3390/catal13020389