

Chiral Catalysts for the Enantioselective Carbon Dioxide-Based Cyclic Carbonates and Polycarbonates




Abstract
: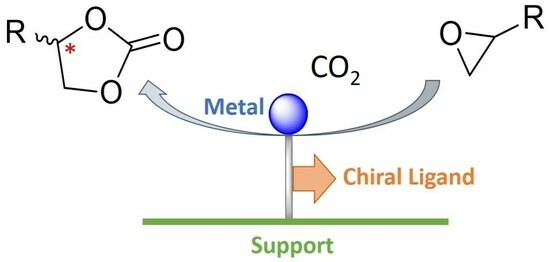
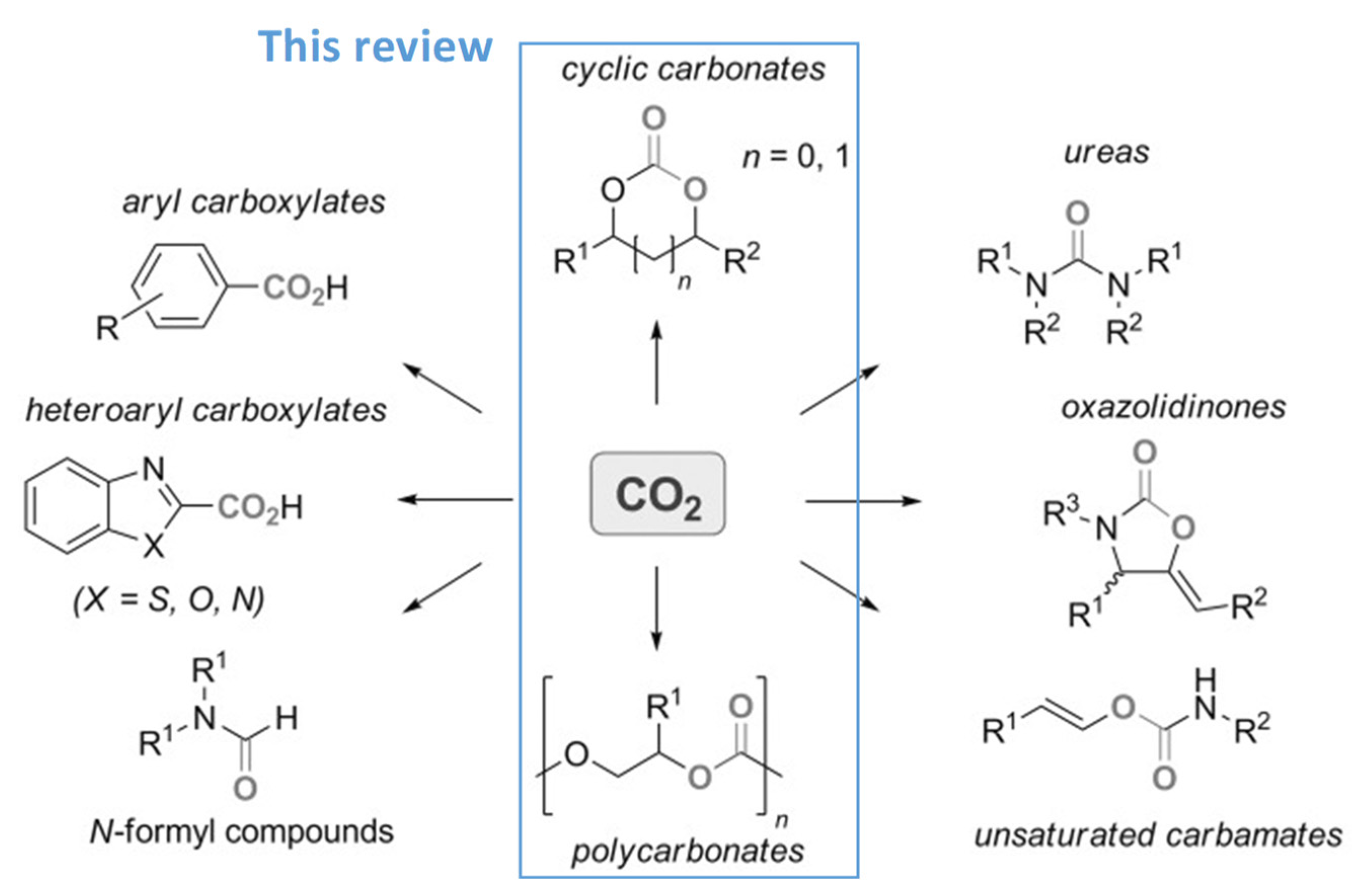
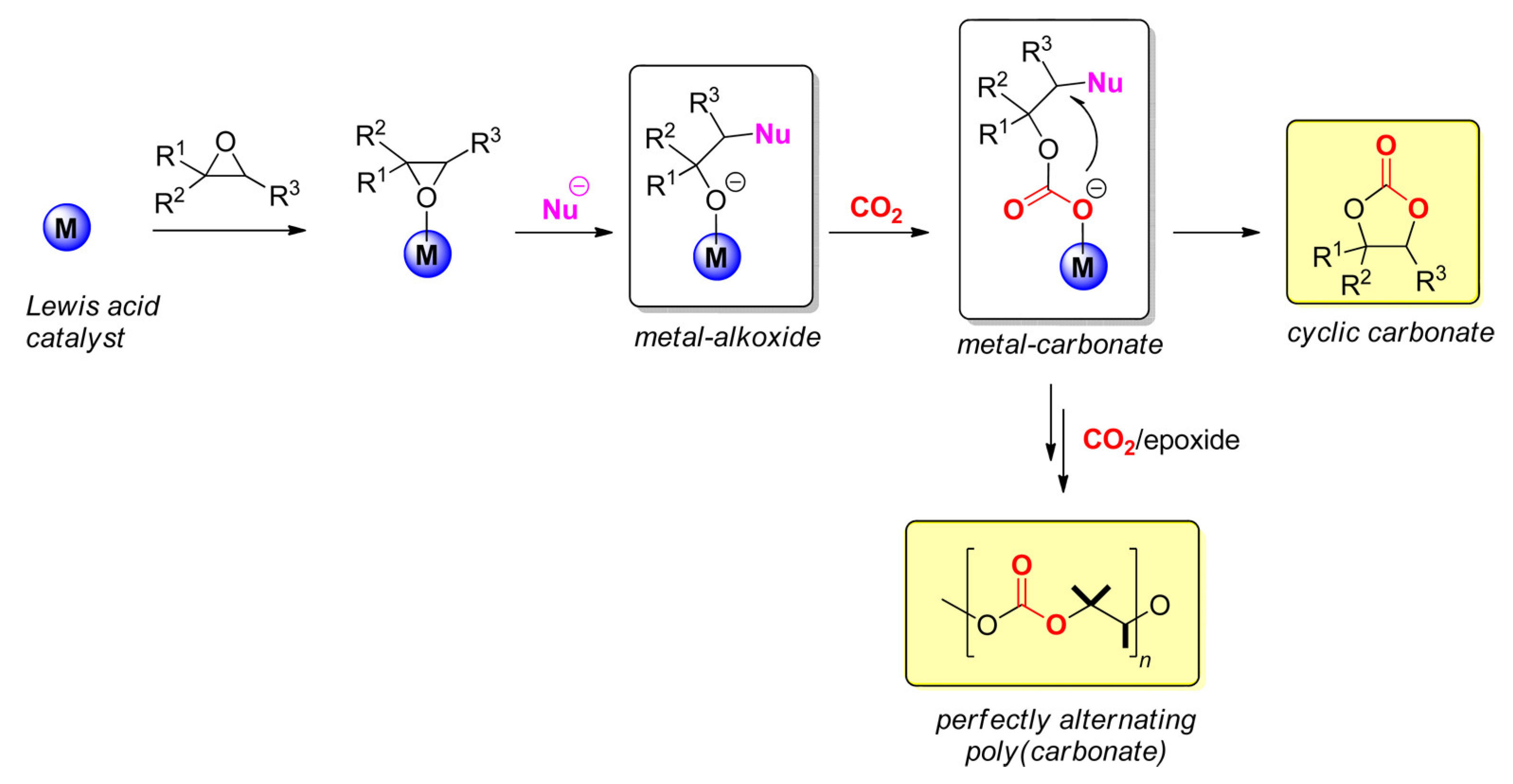
1. Introduction
2. Chiral Organometallic Complex Catalyst
2.1. Schiff-Base Metal Complexes
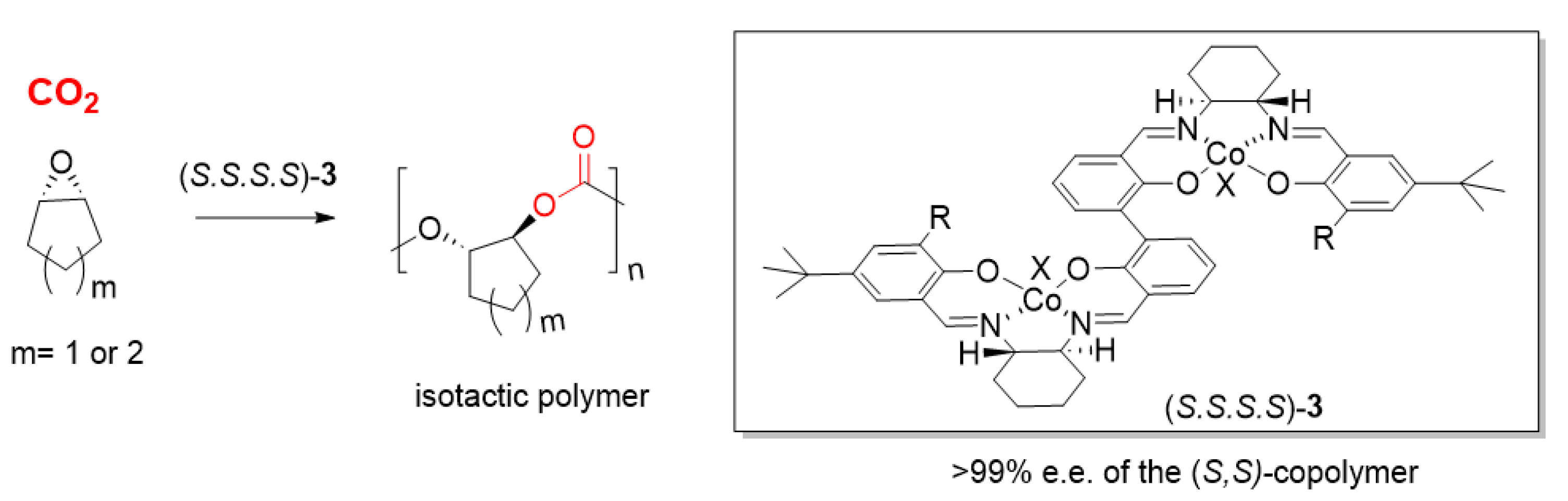
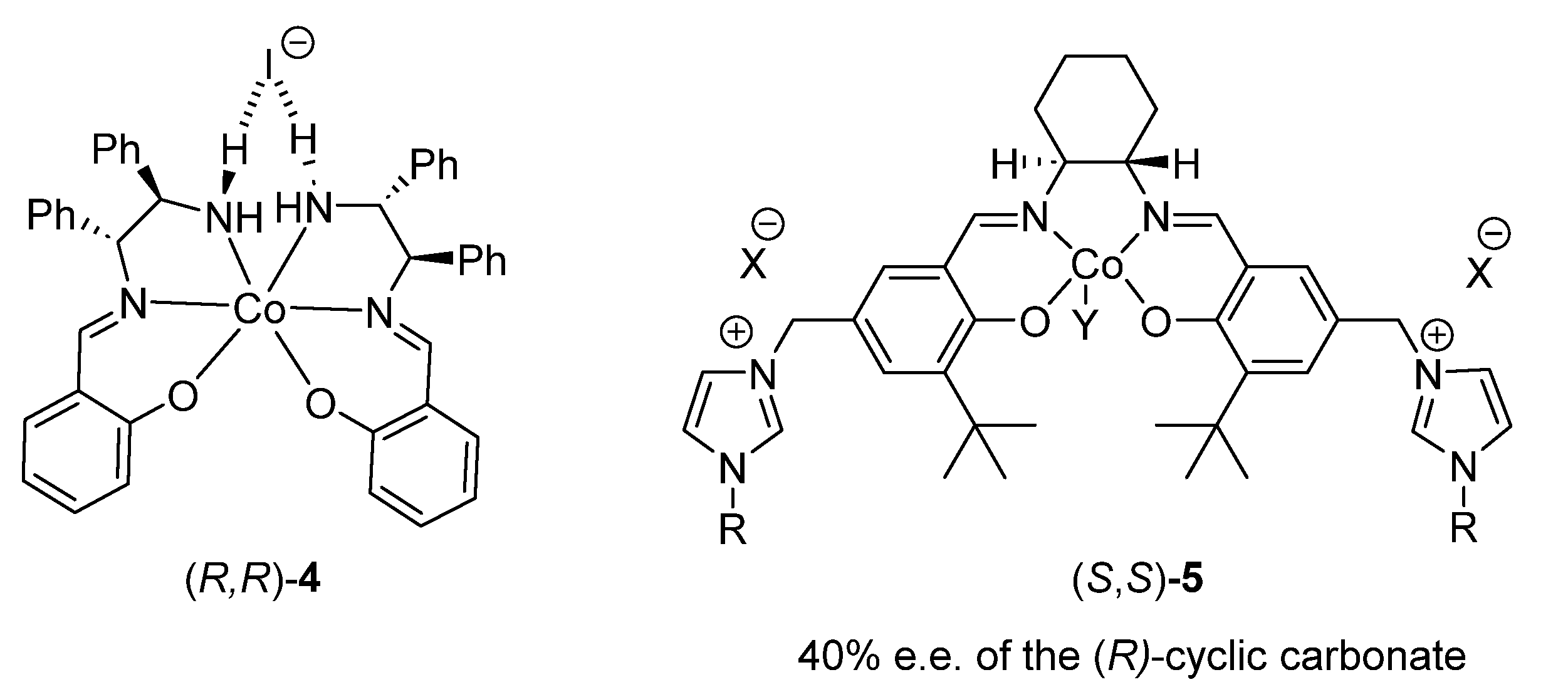
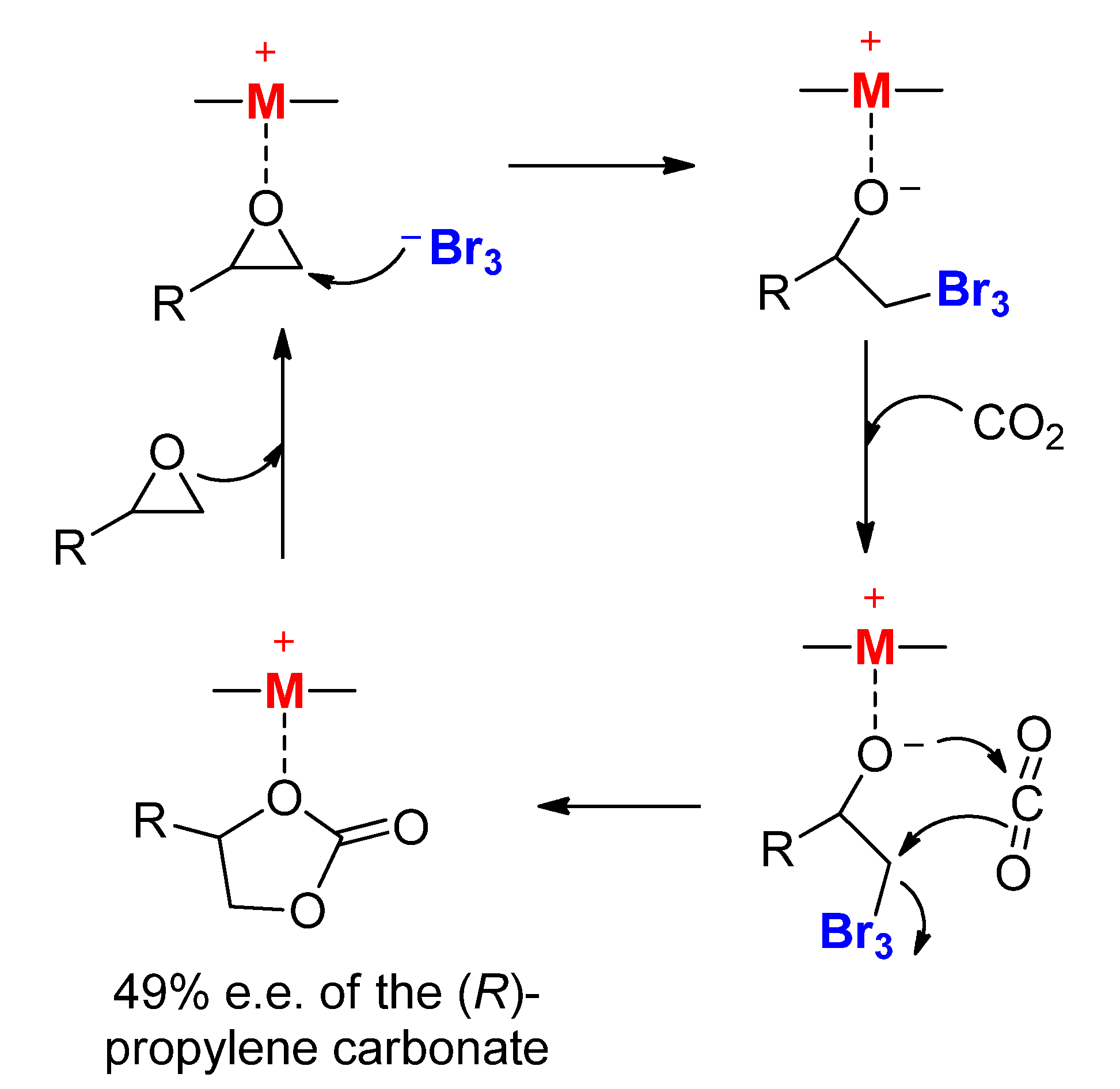
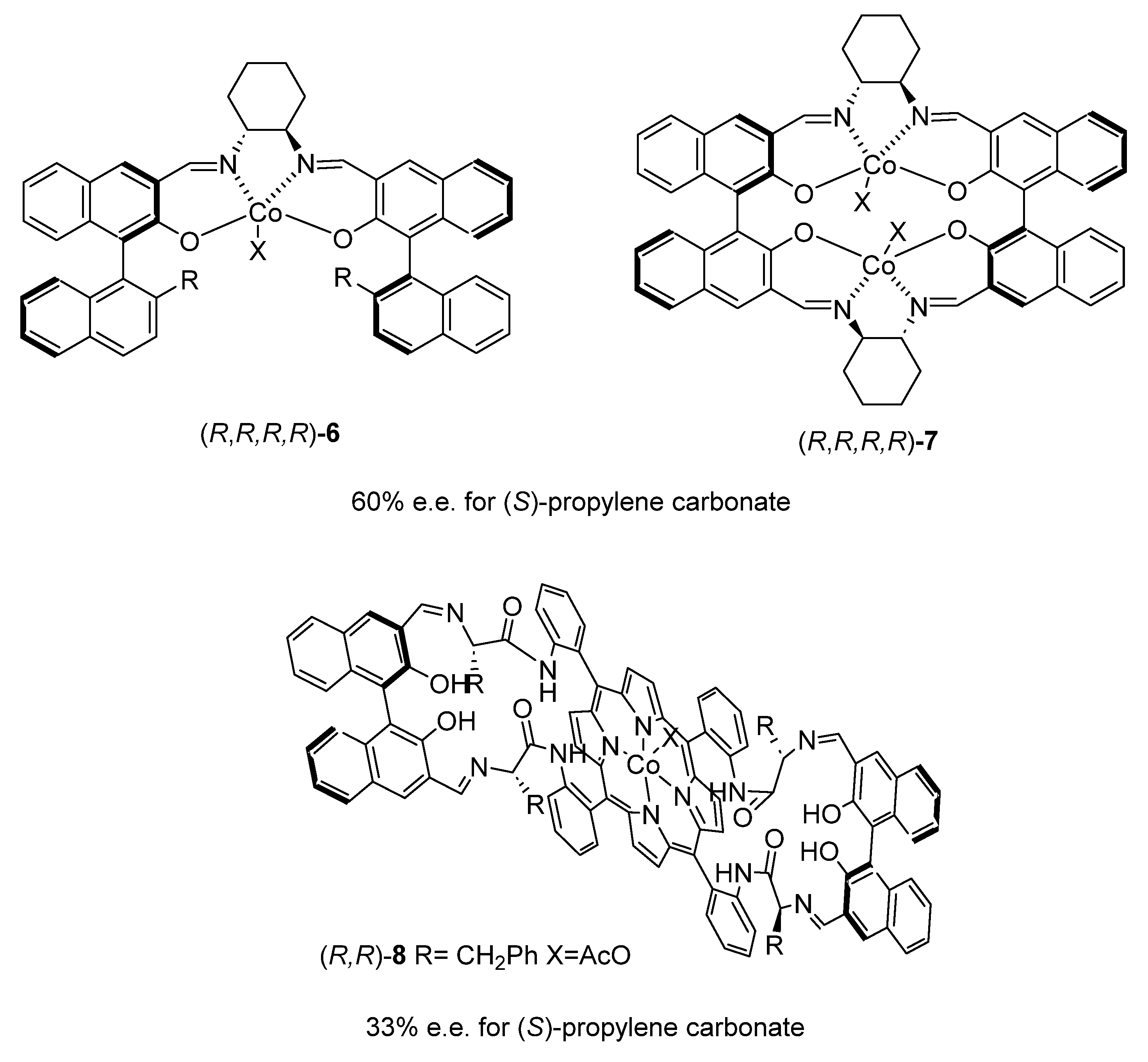
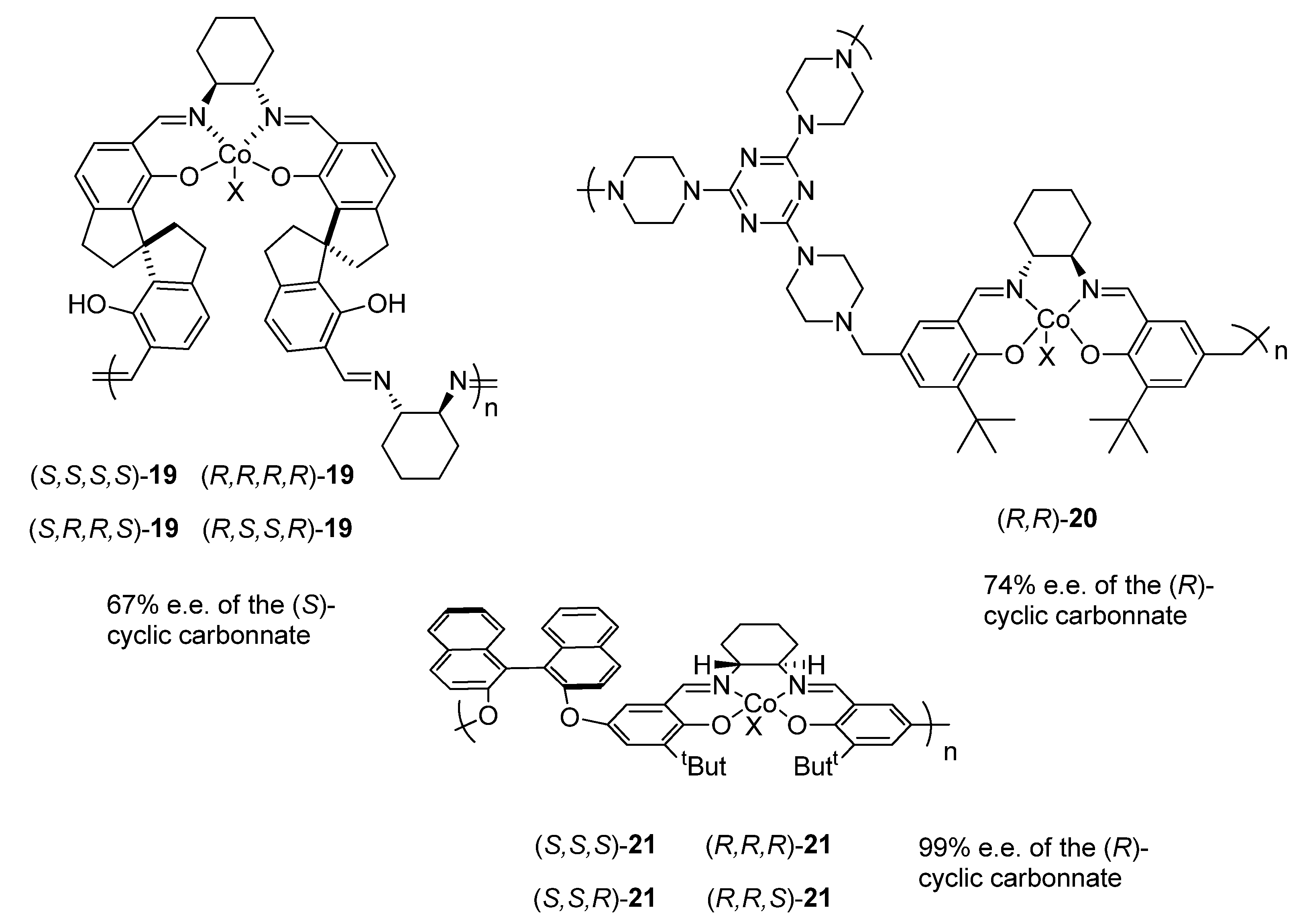
2.1.1. Cobalt Catalyst Based on Chiral Schiff-Base Metal Complexes
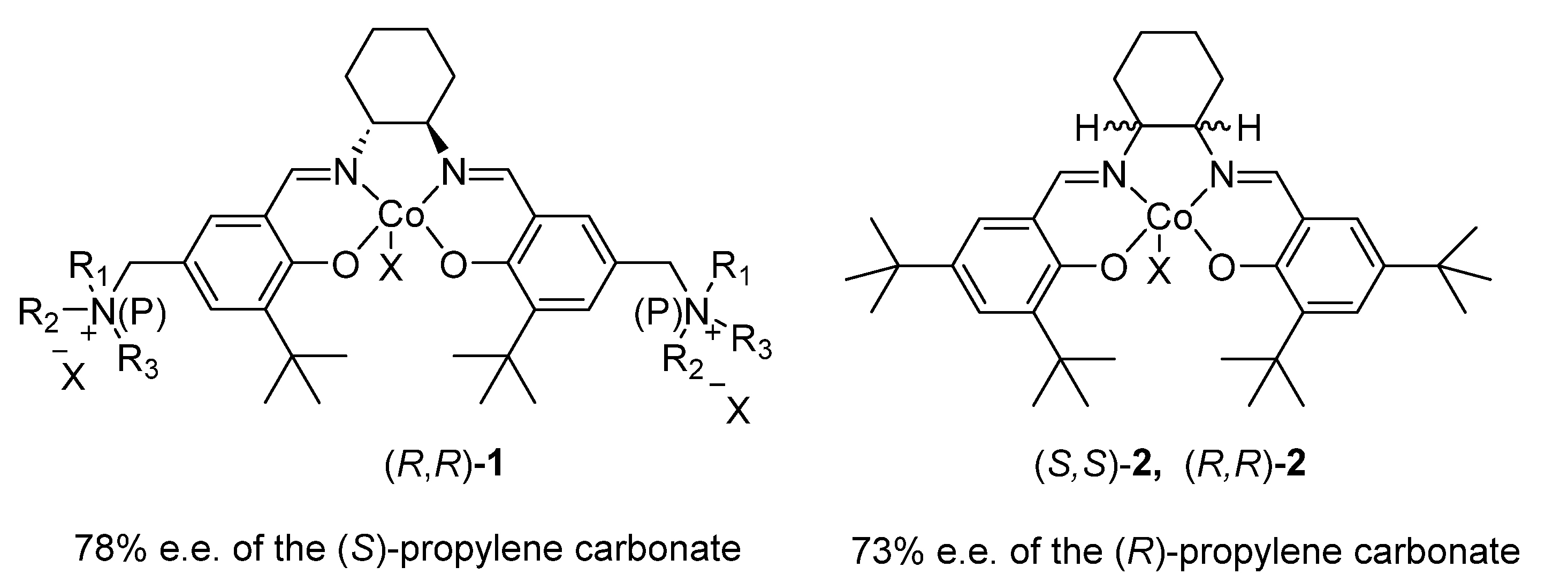
2.1.2. Ionic Liquid-Containing Cosalen Catalysts

| Entry | Catalyst | Co-Catalyst | Reaction Conditions | % Yield | % e.e. | Cycles | Ref |
|---|---|---|---|---|---|---|---|
| 1 | (R,R)-1 | none | 48 h, 0 °C, 6 bar | 24 | 78(S) | 5 | [41] |
| 2 | (S,S)-2 | N-benzylquininium chloride | 24 h, 20 °C, 8 bar | 8 | 73(R) | none | [42] |
| 3 | (R,R)-2 (X = Cl3CCO2) | [18-C-6K][L-Val]·0.5H2O | 5.5 h, 25 °C, 6 bar | 44 | 54(S) | 5 | [49] |
| 4 | (R,R)-2 (X = AcO) | [TBA]2[L-Tar] ionic liquid | 25 °C, 7 bar | 45 | 50(S) | 3 | [50] |
| 5 | (S,S)-5 (X = Br, Y = AcO, R = But) | none | 9 d, 25 °C, 12 bar | 6.2 | 57(R) | none | [51] |
| 6 | (R,R)-2 (X = p-MeC6H4SO3) | PTAT | 10 h, −5 °C, 7 bar | 43 | 49(R) | none | [52] |
2.1.3. BINOL-Cosalen Complexes
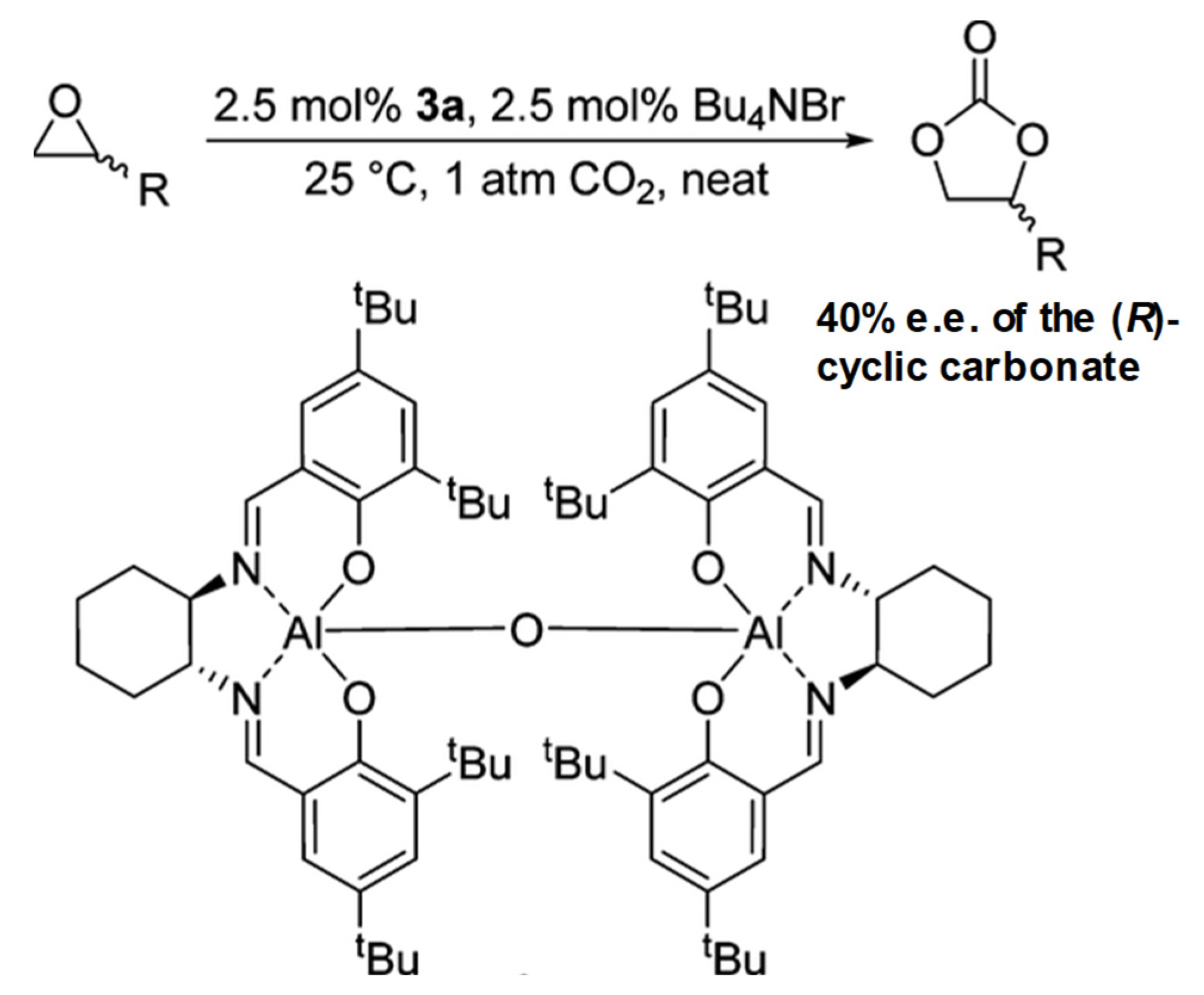
2.1.4. Al Salen Catalysts
2.1.5. Silver Salen Complexes
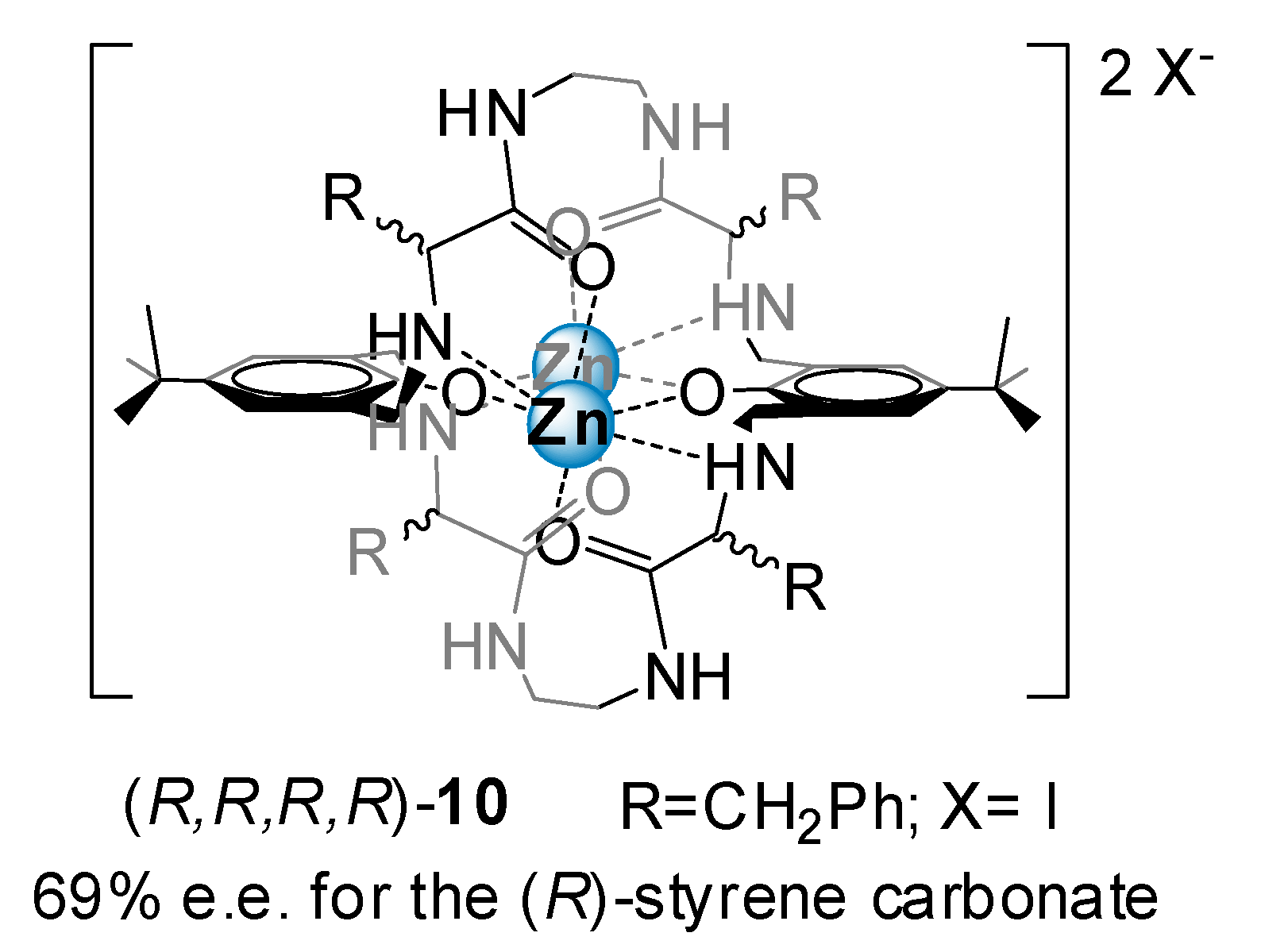
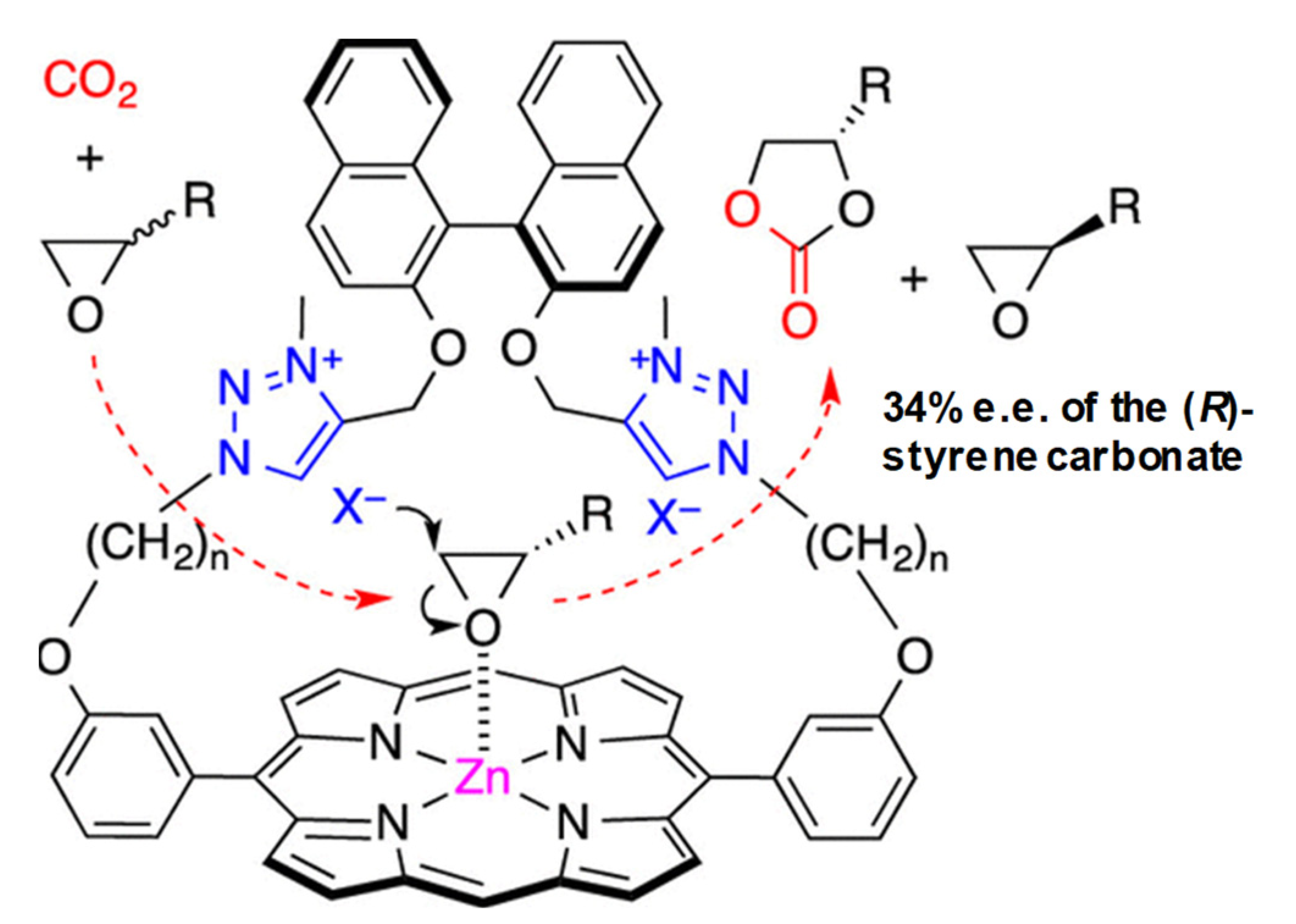
2.1.6. Zinc Salen and BINOL Complexes
2.1.7. Miscellaneous Salen/BINOL Metal Catalyst
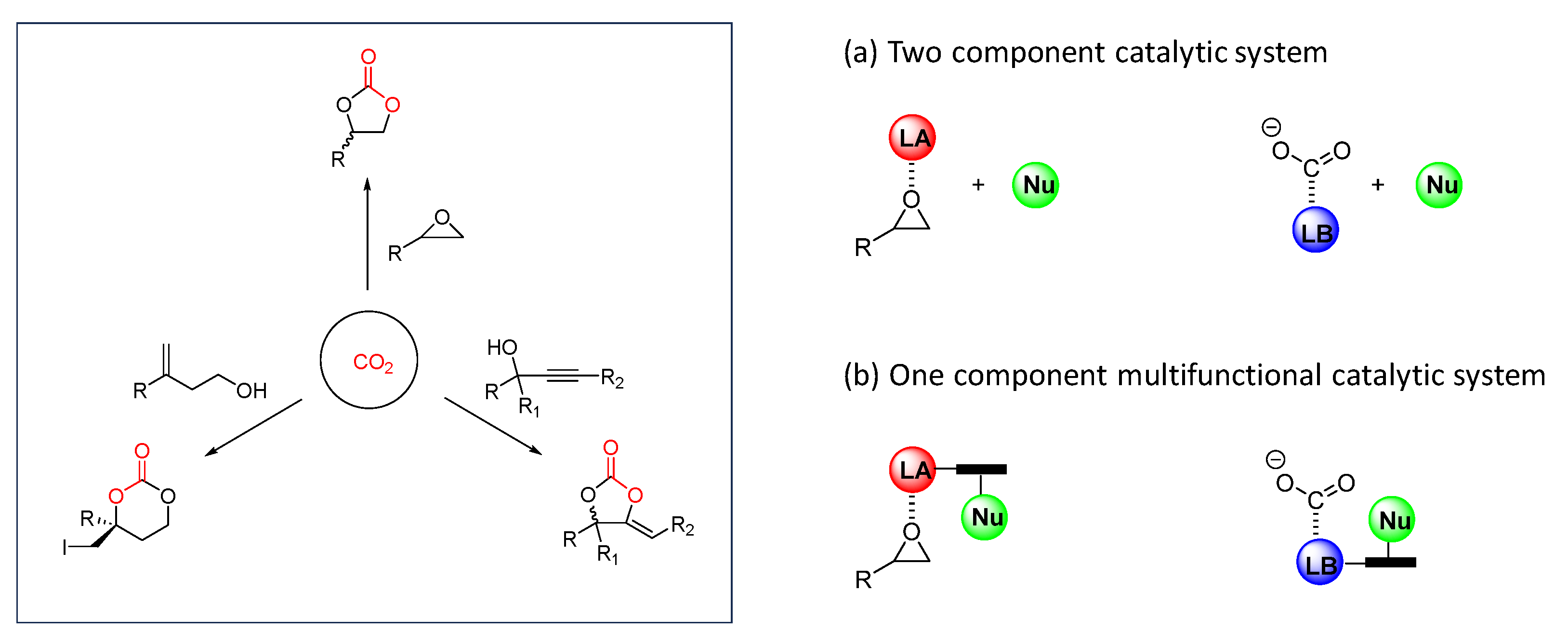
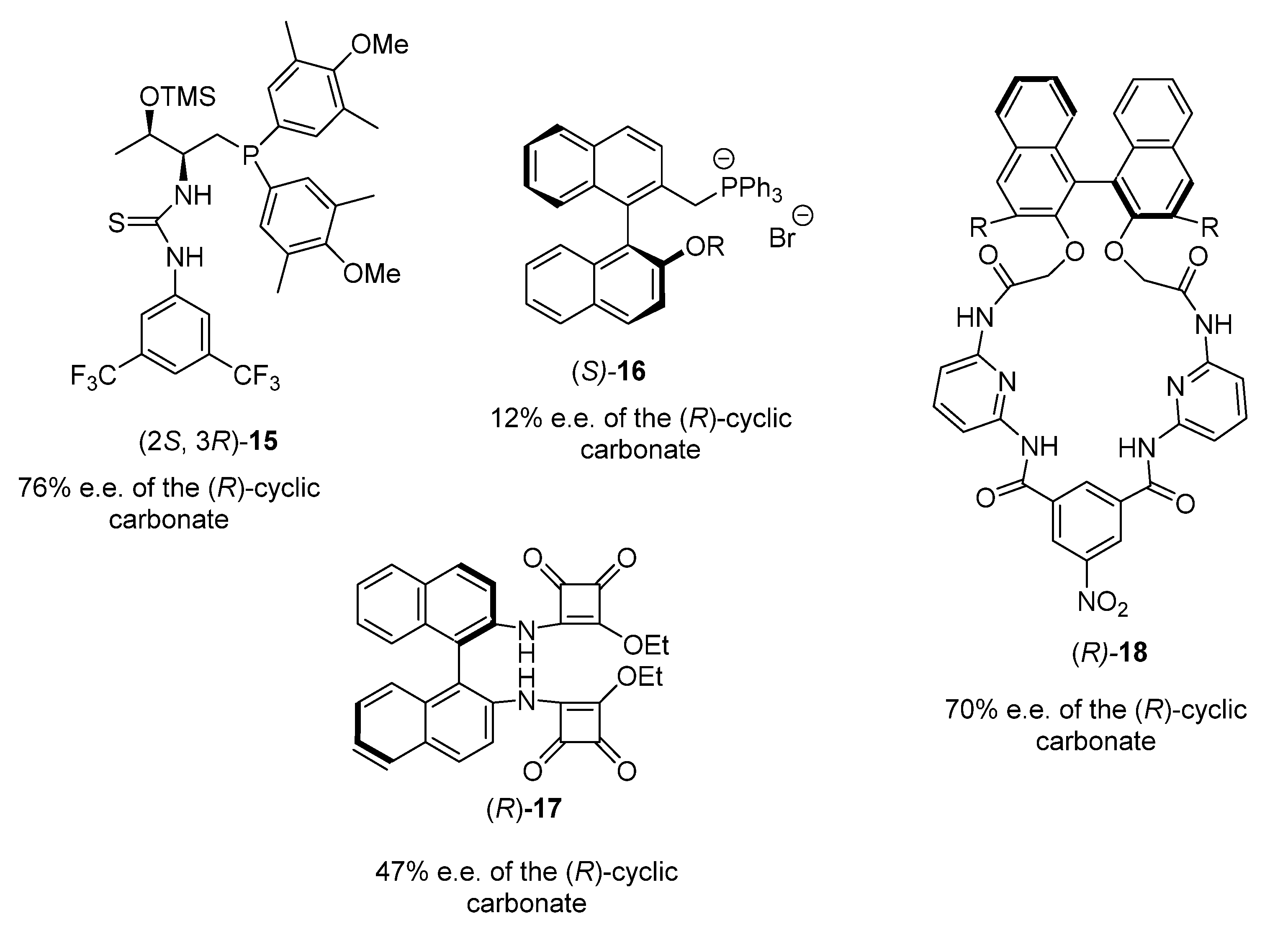
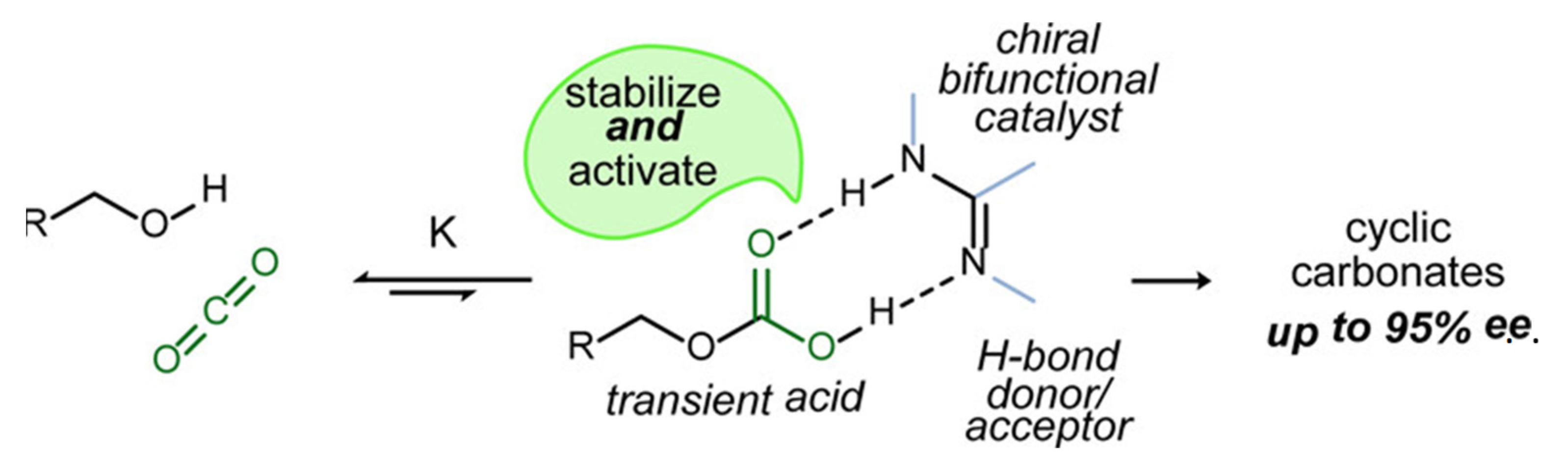
3. Chiral Organocatalyst
4. Immobilized Catalysts into Organic and Metal–Organic Chiral Polymers
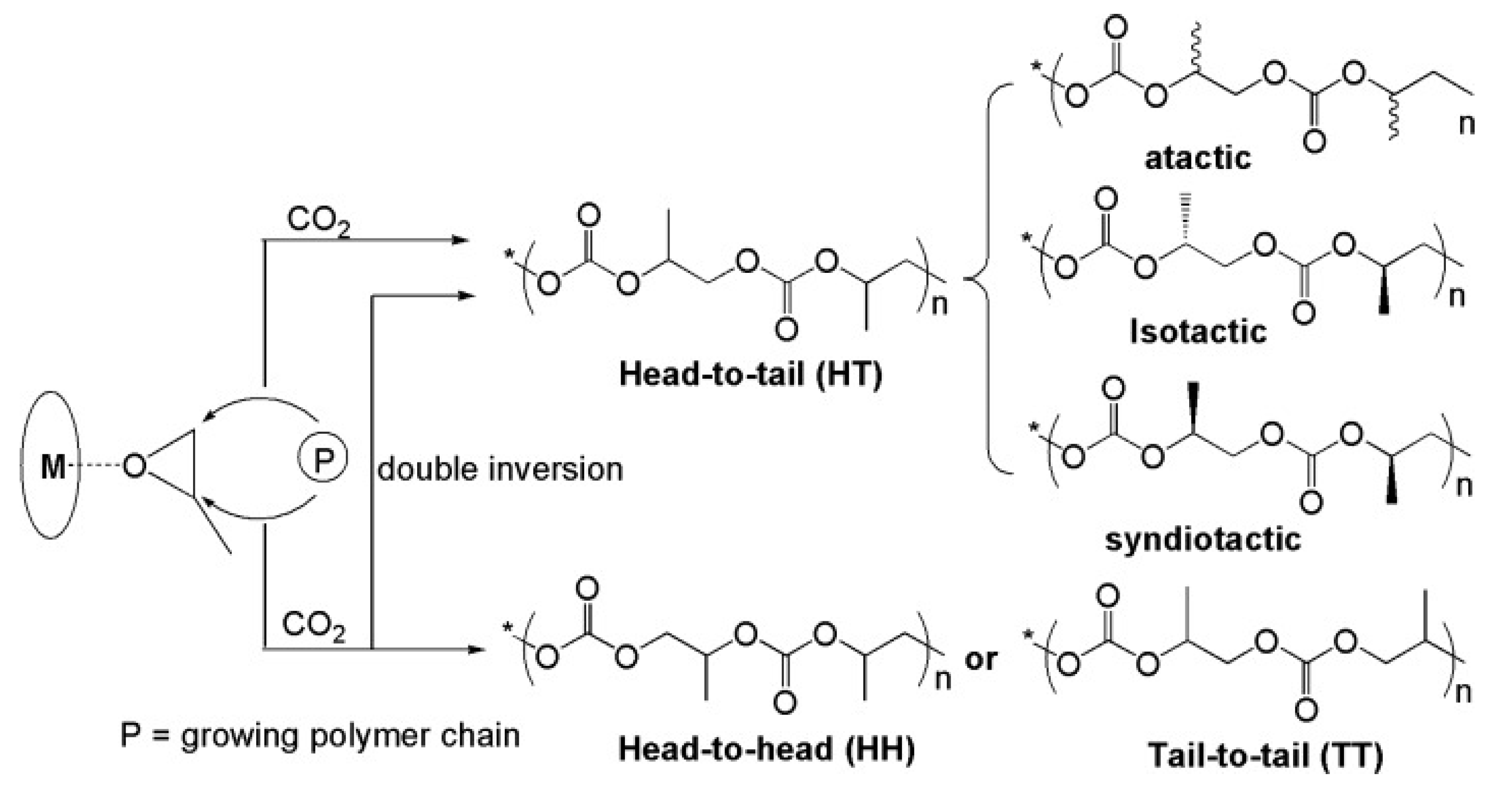
4.1. Cosalen Polymer Catalysts
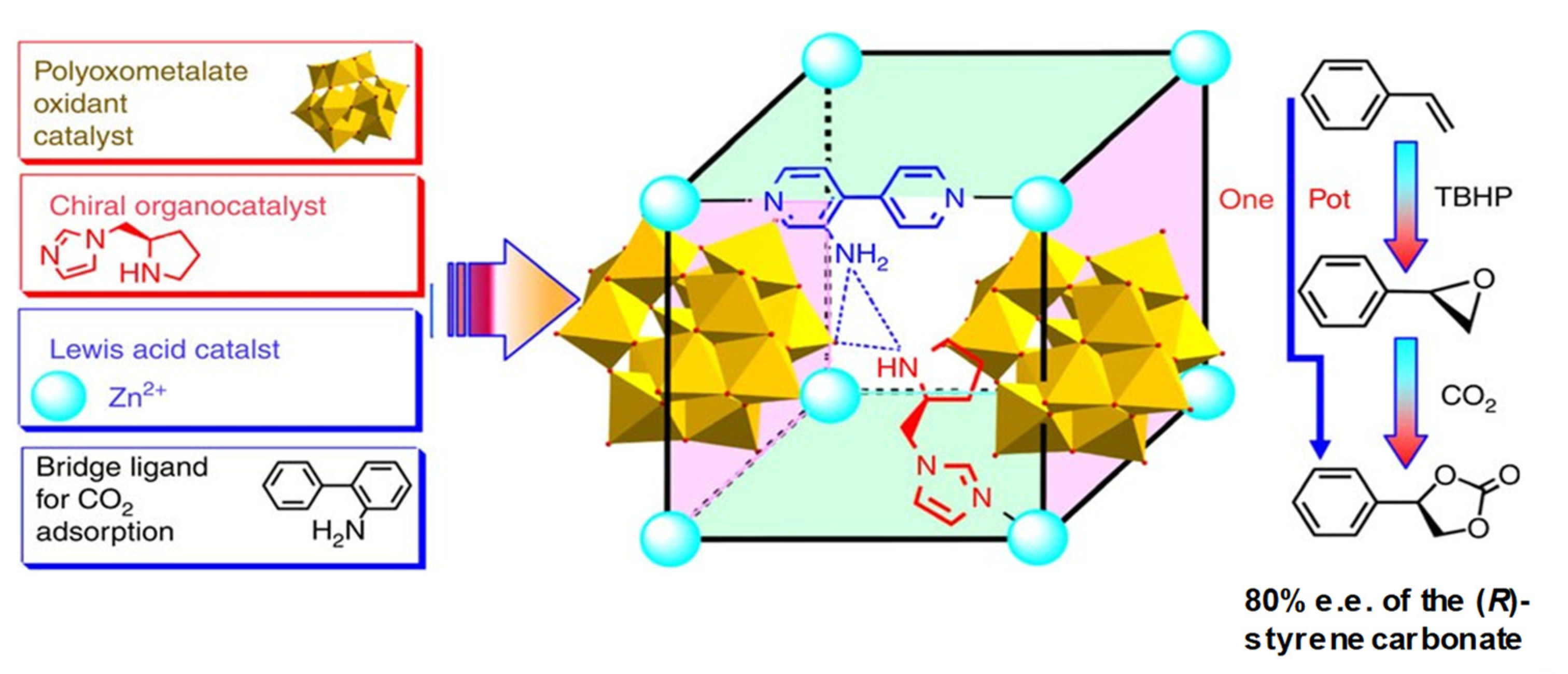
4.2. Chiral Metal–Organic Polymeric Catalysts: MOFs
5. Outlook and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Friedlingstein, P.; O’Sullivan, M.; Jones, M.W.; Andrew, R.M.; Gregor, L.; Hauck, J.; Le Quéré, C.; Luijkx, I.T.; Olsen, A.; Peters, G.P.; et al. Global Carbon Budget 2022. Earth Syst. Sci. Data 2022, 14, 4811–4900. [Google Scholar] [CrossRef]
- Liu, Q.; Wu, L.; Jackstell, R.; Beller, M. Using carbon dioxide as a building block in organic synthesis. Nat. Commun. 2015, 6, 5933. [Google Scholar] [CrossRef] [PubMed]
- Patra, B.R.; Gouda, S.P.; Pattnaik, F.; Nanda, S.; Dalai, A.K.; Naik, S. A brief overview of recent advancements in CO2 capture and valorization technologies. In Carbon Dioxide Capture and Conversion; Nanda, S., Vo, D.V.N., Nguyen, V.H., Eds.; Elsevier: Amsterdam, The Netherlands, 2022; pp. 1–16. [Google Scholar]
- Yaashikaa, P.R.; Kumar, P.S.; Varjani, S.J.; Saravanan, A. A review on photochemical, biochemical and electrochemical transformation of CO2 into value-added products. J. CO2 Util. 2019, 33, 131–147. [Google Scholar] [CrossRef]
- Mori, T.; Nishiyori, R.; Sumida, S.; Furuya, Y.; Shirakawa, S. Chiral Organocatalysts in Enantioselective CO2 Utilization Reactions. Eur. J. Org. Chem. 2023, 26, e202300551. [Google Scholar] [CrossRef]
- Kielland, N.; Whiteoak, C.J.; Kleij, A.W. Stereoselective Synthesis with Carbon Dioxide. Adv. Synth. Catal. 2013, 355, 2115–2138. [Google Scholar] [CrossRef]
- Childers, M.I.; Longo, J.M.; Van Zee, N.J.; LaPointe, A.M.; Coates, G.W. Stereoselective Epoxide Polymerization and Copolymerization. Chem. Rev. 2014, 114, 8129–8152. [Google Scholar] [CrossRef]
- Vaitla, J.; Guttormsen, Y.; Mannisto, J.K.; Nova, A.; Repo, T.; Bayer, A.; Hopmann, K.H. Enantioselective Incorporation of CO2: Status and Potential. ACS Catal. 2017, 7, 7231–7244. [Google Scholar] [CrossRef]
- Guo, X.; Wang, Y.; Chen, J.; Li, G.; Xia Chin, J.-B. Recent Advances of CO2 Fixation via Asymmetric Catalysis for the Direct Synthesis of Optically Active Small Molecules. Chin. J. Org. Chem. 2020, 40, 2208–2220. [Google Scholar] [CrossRef]
- Ran, C.-K.; Chen, X.-W.; Gui, Y.-Y.; Liu, J.; Song, L.; Ren, K.; Yu, D.-G. Recent advances in asymmetric synthesis with CO2. Sci. China Chem. 2020, 63, 1336–1351. [Google Scholar] [CrossRef]
- Wu, X.; Castro-Osma, J.A.; North, M. Synthesis of Chiral Cyclic Carbonates via Kinetic Resolution of Racemic Epoxides and Carbon Dioxide. Symmetry 2016, 8, 4. [Google Scholar] [CrossRef]
- Okuno, K.; Nishiyori, R.; Hiraki, M.; Shirakawa, S. Environmentally Benign Synthesis of Cyclic Carbonates from Epoxides and Carbon Dioxide Using Binary and Bifunctional Catalysts. Heterocycles 2021, 103, 94–109. [Google Scholar]
- Liao, X.; Su, Y.; Tang, X. Stereoselective synthesis of biodegradable polymers by salen-type metal catalysts. Sci. China Chem. 2022, 65, 2096–2121. [Google Scholar] [CrossRef]
- Schaus, S.E.; Brandes, B.D.; Larrow, J.F.; Tokunaga, M.; Hansen, K.B.; Gould, A.E.; Furrow, M.E.; Jacobsen, E.N. Highly Selective Hydrolytic Kinetic Resolution of Terminal Epoxides Catalyzed by Chiral (salen)CoIII Complexes. Practical Synthesis of Enantioenriched Terminal Epoxides and 1,2-Diols. J. Am. Chem. Soc. 2002, 124, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, E.N. Asymmetric catalysis of epoxide ring-opening reactions. Acc. Chem. Res. 2000, 33, 421–431. [Google Scholar] [CrossRef]
- Cavalleri, M.; Panza, N.; di Biase, A.; Tseberlidis, G.; Rizzato, S.; Abbiati, G.; Caselli, A. [Zinc(II)(Pyridine-Containing Ligand)] Complexes as Single-Component Efficient Catalyst for Chemical Fixation of CO2 with Epoxides. Eur. J. Org. Chem. 2021, 19, 2764–2771. [Google Scholar] [CrossRef]
- North, M.; Pasquale, R.; Young, C. Synthesis of cyclic carbonates from epoxides and CO2. Green Chem. 2010, 12, 1514–1539. [Google Scholar] [CrossRef]
- Martín, C.; Fiorani, G.; Kleij, A.W. Recent Advances in the Catalytic Preparation of Cyclic Organic Carbonates. ACS Catal. 2015, 5, 1353–1370. [Google Scholar] [CrossRef]
- Wang, M.-Y.; Song, Q.-W.; Ma, R.; Xie, J.-N.; He, L.-N. Efficient conversion of carbon dioxide at atmospheric pressure to 2-oxazolidinones promoted by bifunctional Cu(II)-substituted polyoxometalate-based ionic liquids. Green Chem. 2016, 18, 282–287. [Google Scholar] [CrossRef]
- Shaikh, R.R.; Pornpraprom, S.; D’Elia, V. Catalytic Strategies for the Cycloaddition of Pure, Diluted, and Waste CO2 to Epoxides under Ambient Conditions. ACS Catal. 2018, 8, 419–450. [Google Scholar] [CrossRef]
- Shi, Y.; Pan, B.W.; Zhou, Y.; Zhou, J.; Liu, Y.L.; Zhou, F. Catalytic enantioselective synthesis using carbon dioxide as a C1 synthon. Org. Biomol. Chem. 2020, 18, 8597–8619. [Google Scholar] [CrossRef]
- Nakano, K.; Hiyama, T.; Nozaki, K. Asymmetric amplification in asymmetric alternating copolymerization of cyclohexene oxide and carbon dioxide. Chem. Commun. 2005, 1871–1873. [Google Scholar] [CrossRef] [PubMed]
- Abbina, S.; Du, G.D. Chiral Amido-Oxazolinate Zinc Complexes for Asymmetric Alternating Copolymerization of CO2 and Cyclohexene Oxide. Organometallics 2012, 31, 7394–7403. [Google Scholar] [CrossRef]
- Xiao, Y.; Wang, Z.; Ding, K. Copolymerization of Cyclohexene Oxide with CO2 by Using Intramolecular Dinuclear Zinc Catalysts. Chem. Eur. J. 2005, 11, 3668–3678. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.P.; Ren, W.M.; Luo, Y.; Li, B.; Zhang, W.Z.; Lu, X.B. Enhanced Asymmetric Induction for the Copolymerization of CO2 and Cyclohexene Oxide with Unsymmetric Enantiopure SalenCo(III) Complexes: Synthesis of Crystalline CO2-Based Polycarbonate. J. Am. Chem. Soc. 2012, 134, 5682–5688. [Google Scholar] [CrossRef]
- Martinez, L.E.; Leighton, J.L.; Carsten, D.H.; Jacobsen, E.N. Highly Enantioselective Ring Opening of Epoxides Catalyzed by (salen)Cr(III) Complexes. J. Am. Chem. Soc. 1995, 117, 5897–5898. [Google Scholar] [CrossRef]
- Konsler, R.G.; Karl, J.; Jacobsen, E.N. Cooperative Asymmetric Catalysis with Dimeric Salen Complexes. J. Am. Chem. Soc. 1998, 120, 10780–10781. [Google Scholar] [CrossRef]
- Nakano, K.; Hashimoto, S.; Nozaki, K. Bimetallic mechanism operating in the copolymerization of propylene oxide with carbon dioxide catalyzed by cobalt–salen complexes. Chem. Sci. 2010, 1, 369–373. [Google Scholar] [CrossRef]
- Vagin, S.I.; Reichardt, R.; Klaus, S.; Rieger, B.A. Stable Doubly Hydrogen-Bridged Butterfly Shaped Diborane(4) Compound. J. Am. Chem. Soc. 2010, 132, 14367–14369. [Google Scholar] [CrossRef]
- Klaus, S.; Lehenmeier, M.W.; Anderson, C.E.; Rieger, B. Recent advances in CO2/epoxide copolymerization—New strategies and cooperative mechanisms. Coord. Chem. Rev. 2011, 255, 1460–1479. [Google Scholar] [CrossRef]
- Kember, M.R.; Knight, P.D.; Reung, P.T.R.; Williams, C.K. Highly Active Dizinc Catalyst for the Copolymerization of Carbon Dioxide and Cyclohexene Oxide at One Atmosphere Pressure. Angew. Chem. 2009, 121, 949–951. [Google Scholar] [CrossRef]
- Jutz, F.; Buchard, A.; Kember, M.R.; Fredriksen, S.B.; Williams, C.K. Mechanistic Investigation and Reaction Kinetics of the Low-Pressure Copolymerization of Cyclohexene Oxide and Carbon Dioxide Catalyzed by a Dizinc Complex. J. Am. Chem. Soc. 2011, 133, 17395–17405. [Google Scholar] [CrossRef]
- Kember, M.R.; Williams, C.K. Efficient Magnesium Catalysts for the Copolymerization of Epoxides and CO2; Using Water to Synthesize Polycarbonate Polyols. J. Am. Chem. Soc. 2012, 134, 15676–15679. [Google Scholar] [CrossRef] [PubMed]
- Kember, M.R.; Jutz, F.; Buchard, A.; White, A.J.P.; Williams, C.K. Di-cobalt(II) catalysts for the copolymerisation of CO2 and cyclohexene oxide: Support for a dinuclear mechanism? Chem. Sci. 2012, 3, 1245–1255. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Wildeson, J.R.; Yarbrough, J.C.; Reibenspies, J.H. Bis 2,6-difluorophenoxide Dimeric Complexes of Zinc and Cadmium and Their Phosphine Adducts: Lessons Learned Relative to Carbon Dioxide/Cyclohexene Oxide Alternating Copolymerization Processes Catalyzed by Zinc Phenoxides. J. Am. Chem. Soc. 2000, 122, 12487–12496. [Google Scholar] [CrossRef]
- Moore, D.R.; Cheng, M.; Lobkovsky, E.B.; Coates, G.W. Mechanism of the Alternating Copolymerization of Epoxides and CO2 Using β-Diiminate Zinc Catalysts: Evidence for a Bimetallic Epoxide Enchainment. J. Am. Chem. Soc. 2003, 125, 11911–11924. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.Y.; Kwon, H.Y.; Na, S.J.; Han, S.; Yun, H.; Lee, H.; Park, Y.W. Bimetallic Anilido-Aldimine Zinc Complexes for Epoxide/CO2 Copolymerization. J. Am. Chem. Soc. 2005, 127, 3031–3037. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.-M.; Duan, W.-L.; Shi, M. Chemical fixation of carbon dioxide catalyzed by binaphthyldiamino Zn, Cu, and Co salen-type complexes. J. Org. Chem. 2003, 68, 1559–1562. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.-B.; Liang, B.; Zhang, Y.-J.; Tian, Y.-Z.; Wang, Y.-M.; Bai, C.-X.; Wang, H.; Zhang, R. Asymmetric Catalysis with CO2: Direct Synthesis of Optically Active Propylene Carbonate from Racemic Epoxides. J. Am. Chem. Soc. 2004, 126, 3732. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Qin, X.; Wang, J.; Ma, L.; Chen, T. Metal complex catalysts broaden bioorthogonal reactions. Sci. China Chem. 2023. [Google Scholar] [CrossRef]
- Chang, T.; Jin, L.; Jing, H. Bifunctional Chiral Catalyst for the Synthesis of Chiral Cyclic Carbonates from Carbon Dioxide and Epoxides. ChemCatChem 2009, 1, 379–383. [Google Scholar] [CrossRef]
- Suling, Z.; Yingying, S.; Huanwang, J.; Peng, Y.; Qiang, C. Cinchona-Derived Quaternary Ammonium Salts-Improved Asymmetric Cycloaddition of CO2 to Epoxides. Chin. J. Catal. 2009, 30, 1255–1260. [Google Scholar]
- Liu, Y.; Ren, W.M.; Liu, J.; Lu, X.B. Asymmetric Copolymerization of CO2 with meso-Epoxides Mediated by Dinuclear Cobalt(III) Complexes: Unprecedented Enantioselectivity and Activity. Angew. Chem. Int. Ed. 2013, 52, 11594–11598. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ren, W.M.; He, K.K.; Lu, X.-B. Crystalline-gradient polycarbonates prepared from enantioselective terpolymerization of meso-epoxides with CO2. Nat. Commun. 2014, 5, 5687. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ren, W.M.; Liu, C.; Fu, S.; Wang, M.; He, K.K.; Li, R.R.; Zhang, R.; Lu, X.B. Highly Regioselective and Alternating Copolymerization of Racemic Styrene Oxide and Carbon Dioxide via Heterogeneous Double Metal Cyanide Complex Catalyst. Macromolecules 2014, 47, 7775–7788. [Google Scholar] [CrossRef]
- Liu, Y.; Ren, W.M.; He, K.K.; Zhang, W.Z.; Li, W.B.; Wang, M.; Lu, X.B.J. CO2-Mediated Formation of Chiral Carbamates from meso-Epoxides via Polycarbonate Intermediates. Org. Chem. 2016, 81, 8959–8966. [Google Scholar] [CrossRef] [PubMed]
- Li, W.B.; Liu, Y.; Lu, X.B. Bimetallic Cobalt Complex-Mediated Enantioselective Terpolymerizations of Carbon Dioxide, Cyclohexene Oxide, and β-Butyrolactone. Organometallics 2020, 39, 1628–1633. [Google Scholar] [CrossRef]
- Emelyanov, M.A.; Stoletova, N.V.; Smolyakov, A.F.; Ilin, M.M.; Maleev, V.I.; Larionov, V.A. Synthesis and a Catalytic Study of Diastereomeric Cationic Chiral-at Cobalt Complexes Based on (R, R)-1,2-Diphenylethylenediamine. Inorg. Chem. 2021, 60, 13960–13967. [Google Scholar] [CrossRef]
- Ying, S.Y.; Ru, J.Q.; Ling, Z.S.; Wang, J.H.; Qian, Z.Q. Chiral metal-containing ionic liquid: Synthesis and applications in the enantioselective cycloaddition of carbon dioxide to epoxides. Sci. China Chem. 2011, 54, 1044. [Google Scholar]
- Zhang, S.; Huang, Y.; Jing, H.; Yao, W.; Yan, P. Chiral ionic liquids improved the asymmetric cycloaddition of CO2 to epoxides. Green Chem. 2009, 11, 935–938. [Google Scholar] [CrossRef]
- Duan, S.; Jing, X.; Li, D.; Jing, H. Catalytic asymmetric cycloaddition of CO2 to epoxides via chiral bifunctional ionic liquids. J. Mol. Catal. A 2016, 411, 34–39. [Google Scholar] [CrossRef]
- Chang, T.; Jing, H.; Jin, L.; Qiu, W. Quaternary onium tribromide catalyzed cyclic carbonate synthesis from carbon dioxide and epoxides. J. Mol. Catal. A 2007, 264, 241–247. [Google Scholar]
- Jin, L.; Huang, Y.; Jing, H.; Chang, T.; Yan, P. Chiral catalysts for the asymmetric cycloaddition of carbon dioxide with epoxides. Tetrahedron Asymmetry 2008, 19, 1947–1953. [Google Scholar] [CrossRef]
- Ren, W.-M.; Wu, G.-P.; Lin, F.; Jiang, J.-Y.; Liu, C.; Luo, Y.; Lu, X.-B. Role of the co-catalyst in the asymmetric coupling of racemic epoxides with CO2 using multichiral Co(III) complexes: Product selectivity and enantioselectivity. Chem. Sci. 2012, 3, 2094–2102. [Google Scholar] [CrossRef]
- Fu, X.; Jing, X.; Jin, L.; Zhang, L.; Zhang, X.; Hu, B.; Jing, H. Chiral basket-handle porphyrin-Co complexes for the catalyzed asymmetric cycloaddition of CO2 to epoxides. Chin. J. Catal. 2018, 39, 997–1003. [Google Scholar] [CrossRef]
- Yang, H.Q.; Chen, Z.Q. Theoretical investigation on conversion of CO2 with epoxides to cyclic carbonates by bifunctional metal-salen complexes bearing ionic liquid substituents. Mol. Catal. 2021, 511, 111733. [Google Scholar] [CrossRef]
- North, M.; Quek, S.C.Z.; Pridmore, N.E.; Whitwood, A.C.; Wu, X. Aluminum(salen) Complexes as Catalysts for the Kinetic Resolution of Terminal Epoxides via CO2 Coupling. ACS Catal. 2015, 5, 3398–3402. [Google Scholar] [CrossRef]
- Xie, S.; Gao, X.; Zhou, F.; Wu, H.; Zhou, J. Enantioselective carboxylative cyclization of propargylic alcohol with carbon dioxide under mild conditions. J. Chin. Chem Lett. 2020, 31, 324–328. [Google Scholar] [CrossRef]
- Yoshida, S.; Fukui, K.; Kikuchi, S.; Yamada, T. Silver-Catalyzed Enantioselective Carbon Dioxide Incorporation into Bispropargylic Alcohols. J. Am. Chem. Soc. 2010, 132, 4072–4073. [Google Scholar] [CrossRef]
- Esteve, F.; Porcar, R.; Bolte, M.; Altava, B.; Luis, S.V.; García-Verdugo, E. A bioinspired approach toward efficient supramolecular catalysts for CO2 conversion. Chem. Catal. 2023, 3, 100482. [Google Scholar] [CrossRef]
- Maeda, K.; Ogawa, K.; Sadanaga, K.; Takaishi, K.; Ema, T. Chiroptical and catalytic properties of doubly binaphthyl-strapped chiral porphyrins. Chem. Commun. 2019, 55, 1064–1106. [Google Scholar] [CrossRef]
- Ema, T. Chiral Bifunctional Metalloporphyrin Catalysts for Kinetic Resolution of Epoxides with Carbon Dioxide. Org. Lett. 2019, 21, 1853. [Google Scholar]
- Brunner, M.; Mußmann, L.; Vogt, D. Kinetic Resolution of Oxiranes by Use of Chiral Lewis Acid Catalysts. Synlett 1993, 12, 893–894. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A.; Gianfrate, L.; Pastore, C. Enantioselective synthesis of organic carbonates promoted by Nb(IV) and Nb(V) catalysts. Appl. Catal. A 2003, 255, 5–11. [Google Scholar] [CrossRef]
- Qin, M.J.; Larionov, V.A.; Harms, K.; Meggers, E. Kinetic Resolution of Epoxides with CO2 Catalyzed by a Chiral-at-Iridium Complex. ChemSusChem 2019, 12, 320–325. [Google Scholar] [CrossRef]
- Li, B.; Wu, G.-P.; Ren, W.-M.; Wang, Y.-M.; Rao, D.-Y.; Lu, X.-B. Asymmetric, Regio- and Stereo-Selective Alternating Copolymerization of CO2 and Propylene Oxide Catalyzed by Chiral Chromium Salan Complexes. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 6102–6113. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, H.; Mu, S.; Zhang, W.Z.; Ren, W.M.; Lu, X.B. Highly regio- and stereoselective synthesis of cyclic carbonates from biomass-derived polyols via organocatalytic cascade reaction. Green Chem. 2019, 21, 6335–6341. [Google Scholar] [CrossRef]
- Schörgenhumer, J.; Tifner, M.; Waser, M. (Thio)urea containing quaternary ammonium salts for the CO2-fixation with epoxides. Monatshefte Für Chem.—Chem. Mon. 2019, 150, 789–794. [Google Scholar] [CrossRef]
- Sun, Y.L.; Wei, Y.; Shi, M. Phosphine-catalyzed fixation of CO2 with γ-hydroxyl alkynone under ambient temperature and pressure: Kinetic resolution and further conversion. Org. Chem. Front. 2019, 6, 2420. [Google Scholar] [CrossRef]
- Vara, B.A.; Struble, T.J.; Wang, W.; Dobish, M.C.; Johnston, J.N. Enantioselective Small Molecule Synthesis by Carbon Dioxide Fixation using a Dual Brønsted Acid/Base Organocatalyst. J. Am. Chem. Soc. 2015, 137, 7302–7305. [Google Scholar] [CrossRef]
- Liu, S.; Suematsu, N.; Maruoka, K.; Shirakawa, S. Design of bifunctional quaternary phosphonium salt catalysts for CO2 fixation reaction with epoxides under mild conditions. Green Chem. 2016, 18, 4611–4615. [Google Scholar] [CrossRef]
- Kazuto Takaishi, Takafumi Okuyama, Shota Kadosaki, Masanobu Uchiyama, and Tadashi Ema, Hemisquaramide Tweezers as Organocatalysts: Synthesis of Cyclic Carbonates from Epoxides and CO2. Org. Lett. 2019, 21, 1397–1401. [CrossRef] [PubMed]
- Ema, T.; Yokoyama, M.; Watanabe, S.; Sasaki, S.; Ota, H.; Takaishi, K. Chiral Macrocyclic Organocatalysts for Kinetic Resolution of Disubstituted Epoxides with Carbon Dioxide. Org. Lett. 2017, 19, 4070–4073. [Google Scholar] [CrossRef] [PubMed]
- Song, C.E.; Lee, S.G. Supported chiral catalysts on inorganic materials. Chem. Rev. 2002, 102, 3495–3524. [Google Scholar] [CrossRef] [PubMed]
- Altava, B.; Burguete, M.I.; García-Verdugo, E.; Luis, S.V. Chiral catalysts immobilized on achiral polymers: Effect of the polymer support on the performance of the catalyst. Chem. Soc. Rev. 2018, 47, 2722–2771. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, Y.; Wang, K.; Fu, X.; Chen, F.; Jing, H. Chiral oligomers of spiro-salencobalt(III)X for catalytic asymmetric cycloaddition of epoxides with CO2. Catal. Commun. 2016, 81, 50–53. [Google Scholar] [CrossRef]
- Roy, T.; Kureshy, R.I.; Khan, N.U.H.; Abdi, S.H.R.; Bajaj, H.C. Asymmetric cycloaddition of CO2 and an epoxide using recyclable bifunctional polymeric Co(III) salen complexes under mild conditions. Catal. Sci. Technol. 2013, 3, 2661–2667. [Google Scholar] [CrossRef]
- Yan, P.; Jing, H. Catalytic Asymmetric Cycloaddition of Carbon Dioxide and Propylene Oxide Using Novel Chiral Polymers of BINOL-Salen- Cobalt(III) Salts. Adv. Synth. Catal. 2009, 351, 1325–1332. [Google Scholar] [CrossRef]
- Ren, Y.; Cheng, X.; Yang, S.; Qi, C.; Jiang, H.; Mao, Q. A chiral mixed metal–organic framework based on a Ni(saldpen) metalloligand: Synthesis, characterization and catalytic performances. Dalton Trans. 2013, 42, 9930–9937. [Google Scholar] [CrossRef]
- Li, J.; Fan, Y.; Ren, Y.; Liao, J.; Qi, C.; Jiang, H. Development of Isostructural Porphyrin–Salen Chiral Metal–Organic Frameworks through Postsynthetic Metalation Based on Single-Crystal to Single-Crystal Transformation. Inorg. Chem. 2018, 57, 1203–1212. [Google Scholar] [CrossRef]
- Han, Q.; Qi, B.; Ren, W.; He, C.; Niu, J.; Duan, C. Polyoxometalate-based homochiral metal-organic frameworks for tandem asymmetric transformation of cyclic carbonates from olefins. Nat. Commun. 2015, 6, 10007. [Google Scholar] [CrossRef]
- Guo, L.; Lamb, K.J.; North, M. Recent developments in organocatalysed transformations of epoxides and carbon dioxide into cyclic carbonates. Green Chem. 2021, 23, 77–118. [Google Scholar] [CrossRef]
- Martín, N.; Portillo, A.; Ateka, A.; Cirujano, F.G.; Oar-Arteta, L.; Aguayo, A.; Dusselier, M. MOF-derived/zeolite hybrid catalyst for the production of light olefins from CO2. ChemCatChem 2020, 12, 5750–5758. [Google Scholar] [CrossRef]
- Martín, N.; Cirujano, F.G. Multifunctional heterogeneous catalysts for the tandem CO2 hydrogenation-Fischer Tropsch synthesis of gasoline. J. CO2 Util. 2022, 65, 102176. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altava, B.; Cirujano, F.G.; García-Verdugo, E. Chiral Catalysts for the Enantioselective Carbon Dioxide-Based Cyclic Carbonates and Polycarbonates. Catalysts 2023, 13, 1441. https://doi.org/10.3390/catal13111441
Altava B, Cirujano FG, García-Verdugo E. Chiral Catalysts for the Enantioselective Carbon Dioxide-Based Cyclic Carbonates and Polycarbonates. Catalysts. 2023; 13(11):1441. https://doi.org/10.3390/catal13111441
Chicago/Turabian StyleAltava, Belén, Francisco G. Cirujano, and Eduardo García-Verdugo. 2023. "Chiral Catalysts for the Enantioselective Carbon Dioxide-Based Cyclic Carbonates and Polycarbonates" Catalysts 13, no. 11: 1441. https://doi.org/10.3390/catal13111441
APA StyleAltava, B., Cirujano, F. G., & García-Verdugo, E. (2023). Chiral Catalysts for the Enantioselective Carbon Dioxide-Based Cyclic Carbonates and Polycarbonates. Catalysts, 13(11), 1441. https://doi.org/10.3390/catal13111441