2.2.1. Unassisted Syntheses of Heterobimetallic NHC Complexes and Their Catalytic Properties
In this section, we review the synthetic methods of those NHC complexes in which—in most cases—the formation of the heterobimetallic compounds was not assisted by other interactions (e.g., cyclometalation) or coordination to other donor groups (anchors) of the ligand.
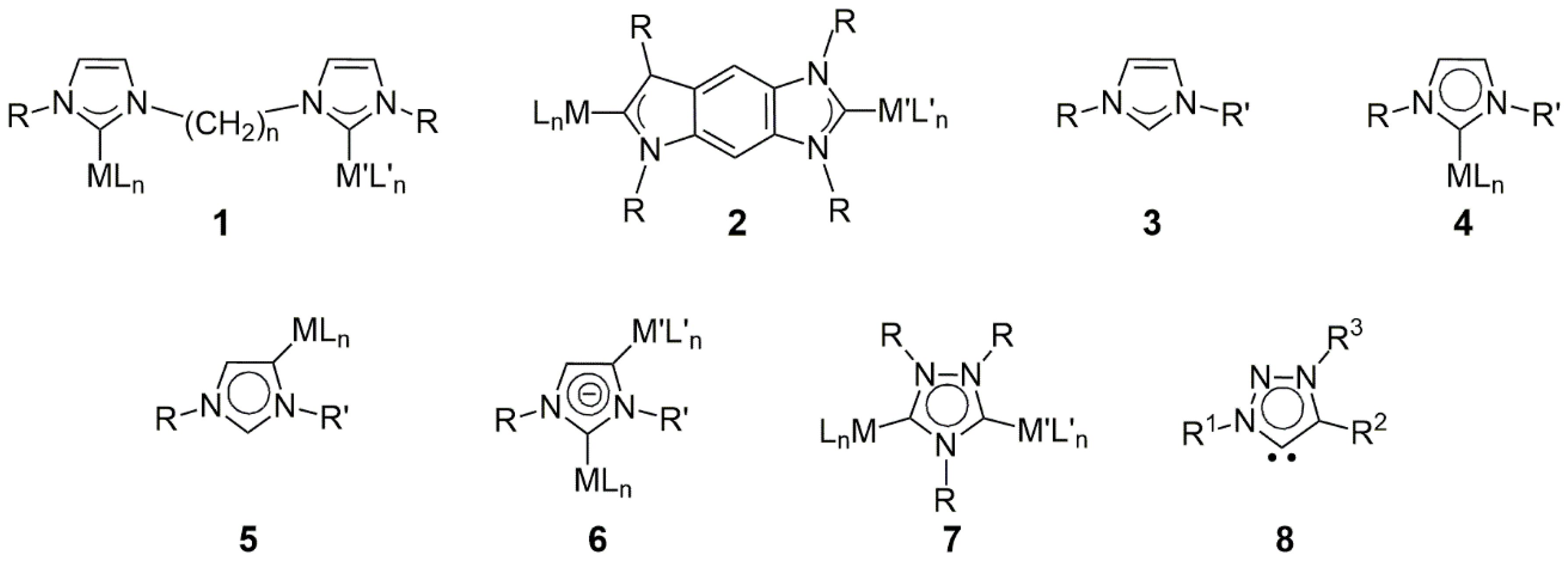
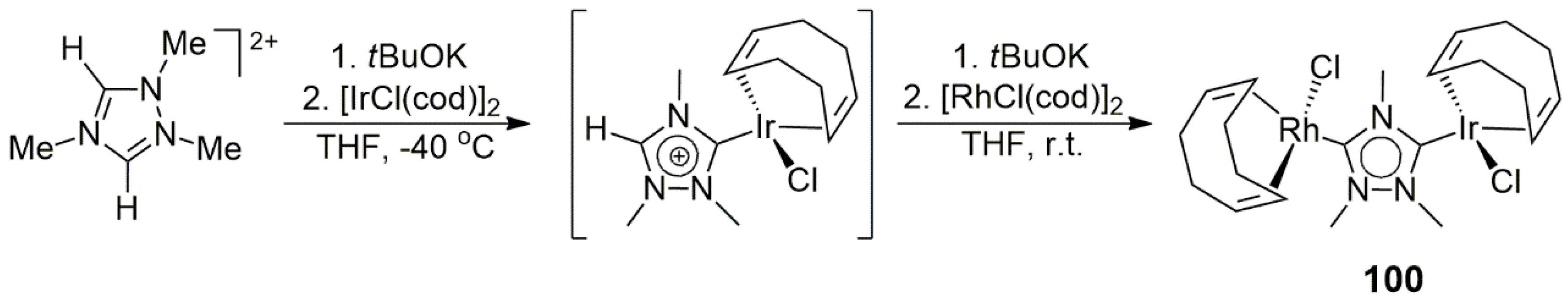
The simplest way to obtain a bis(imidazole-ylidene) carbene ligand precursor is to link two imidazolium units by a suitable alkyl, aryl, or other kind of bridge between the two unsubstituted nitrogens [
21,
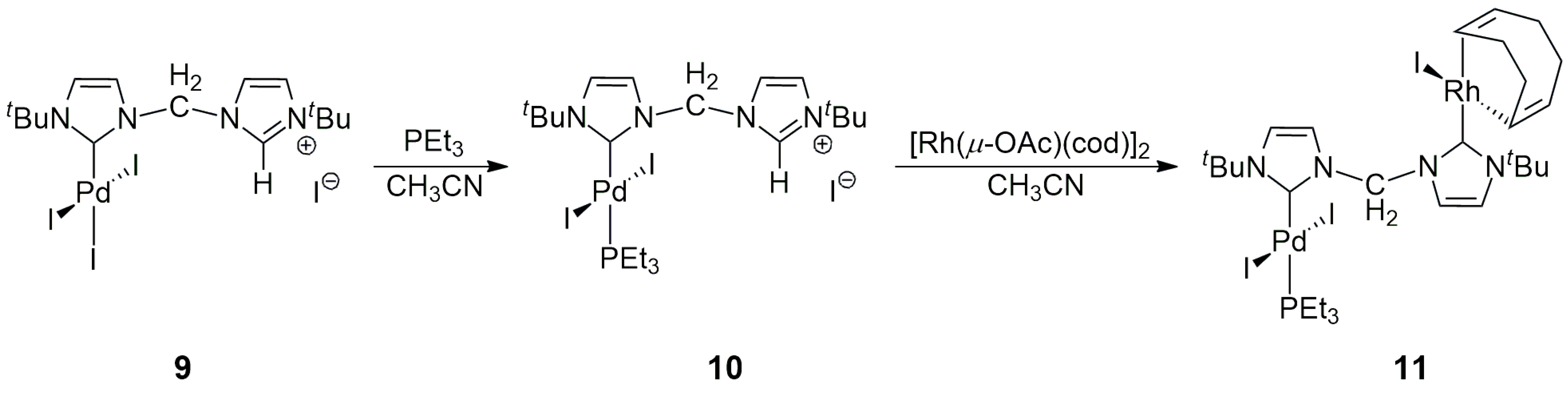
22]. This technique was used by Cowie and co-workers for the synthesis of
11 (
Scheme 2) [
23]. One of the C2 carbenes was coordinated to Pd, while the remaining imidazolium moiety was metalated by [Rh(μ-OAc)(cod)]
2 to yield the Pd/Rh heterobimetallic NHC complex
11 (the acetate served as an internal base). The cod ligand (1,5-cyclooctadiene) could be easily replaced by CO, affording the dicarbonyl product. The analogous Pd/Ir(cod), Pd/Ir(CO)
2, and Ir/Rh complexes were also prepared and characterized. However, no catalysis by these heterobimetallic NHC complexes was reported.
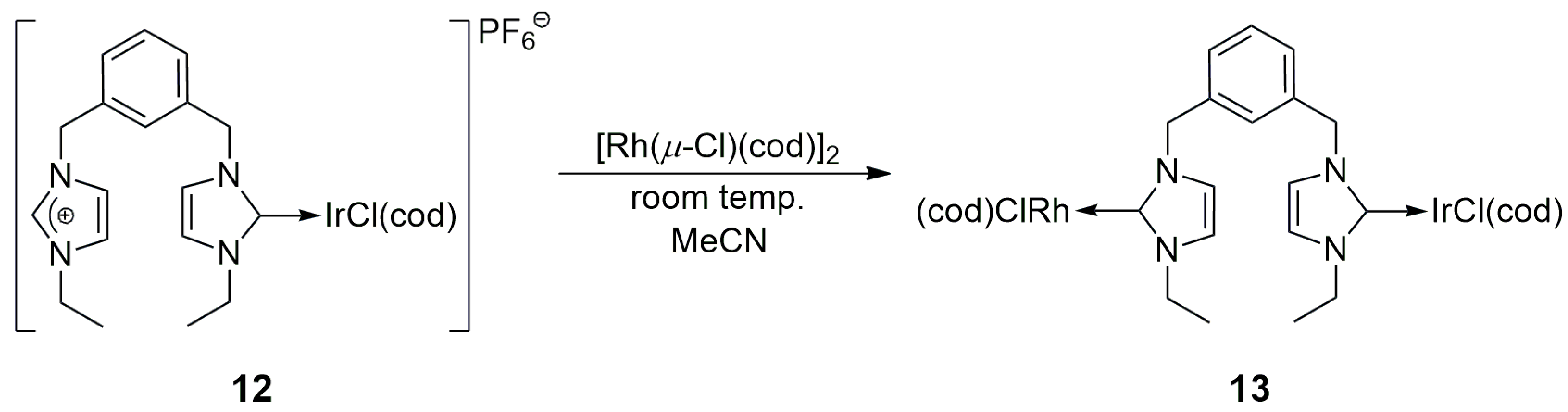
Braunstein and co-workers reported the synthesis of the bis(N-ethylimidazolium) compound
12, where the two imidazolium units were attached to an
m-xylyl linker (
Scheme 3) [
24]. The deprotonation of
12 under the effect of LiN(SiMe
3)
2 afforded the free dicarbene. Interestingly, the reaction of this dicarbene with [Ir(μ-Cl)(cod)]
2 did not lead to the formation of a chelate Ir complex; instead, the reaction gave either the classical mono- or bis-carbene Ir-NHC complexes. This observation opened the way for sequential metalation, in which the free dicarbene was first reacted with [Ir(μ-Cl)(cod)]
2 with the formation of
12, followed by another metalation step using [Rh(μ-Cl)(cod)]
2, which led to the heterobimetallic product
13. The new NHC complexes were not applied as catalysts.
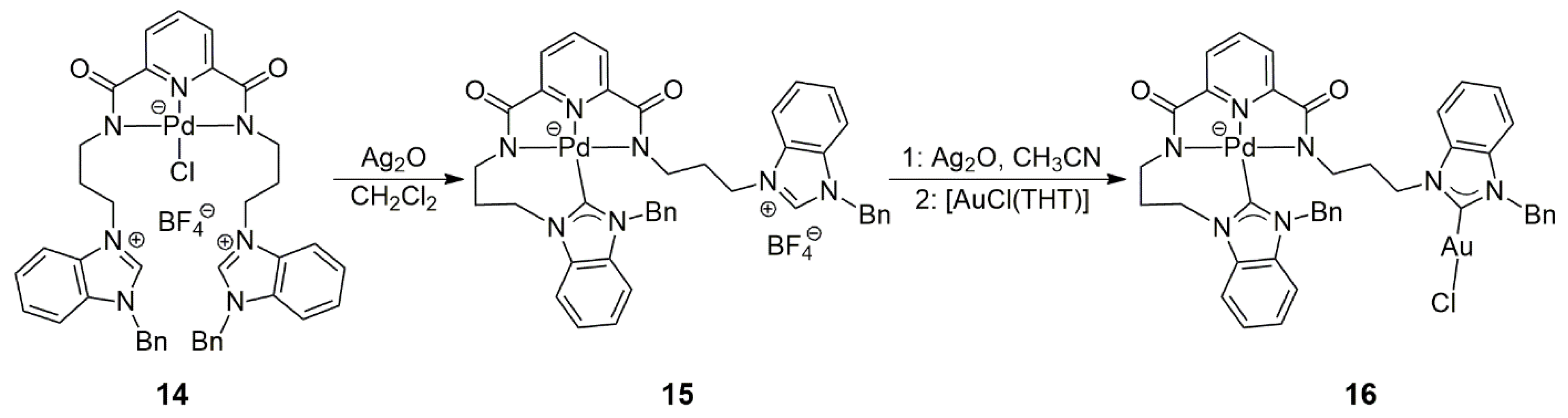
Teng and Vinh Huyn prepared a bis(carbene) precursor that contained an N,N,N-pincer linker between two alkylbenzimidazolium moieties [
25]. Capitalizing on the ‘borderline’ hard/soft coordination properties of Pd(II), they synthesized
14, in which the palladium ion is coordinated only by hard donor atoms (
Scheme 4). In reaction with a half equivalent of Ag
2O, chloride abstraction from Pd led to
15. In addition to the three pincer N donor atoms, in this complex, Pd is also bound to a benzimidazole-ylidene unit, while the molecule still contains a non-metalated benzimidazolium part. The metalation of the latter with the use of Ag
2O and [AuCl(THT)] (THT = tetrahydrothiophene) afforded the heterobimetallic Pd/Au complex
16. The analogous Pd/Pd complex was also prepared. Although the potential use of this ligand for the preparation of cooperative heterobimetallic bis(NHC)-complex catalysts was mentioned, no such experiments were described in the paper.
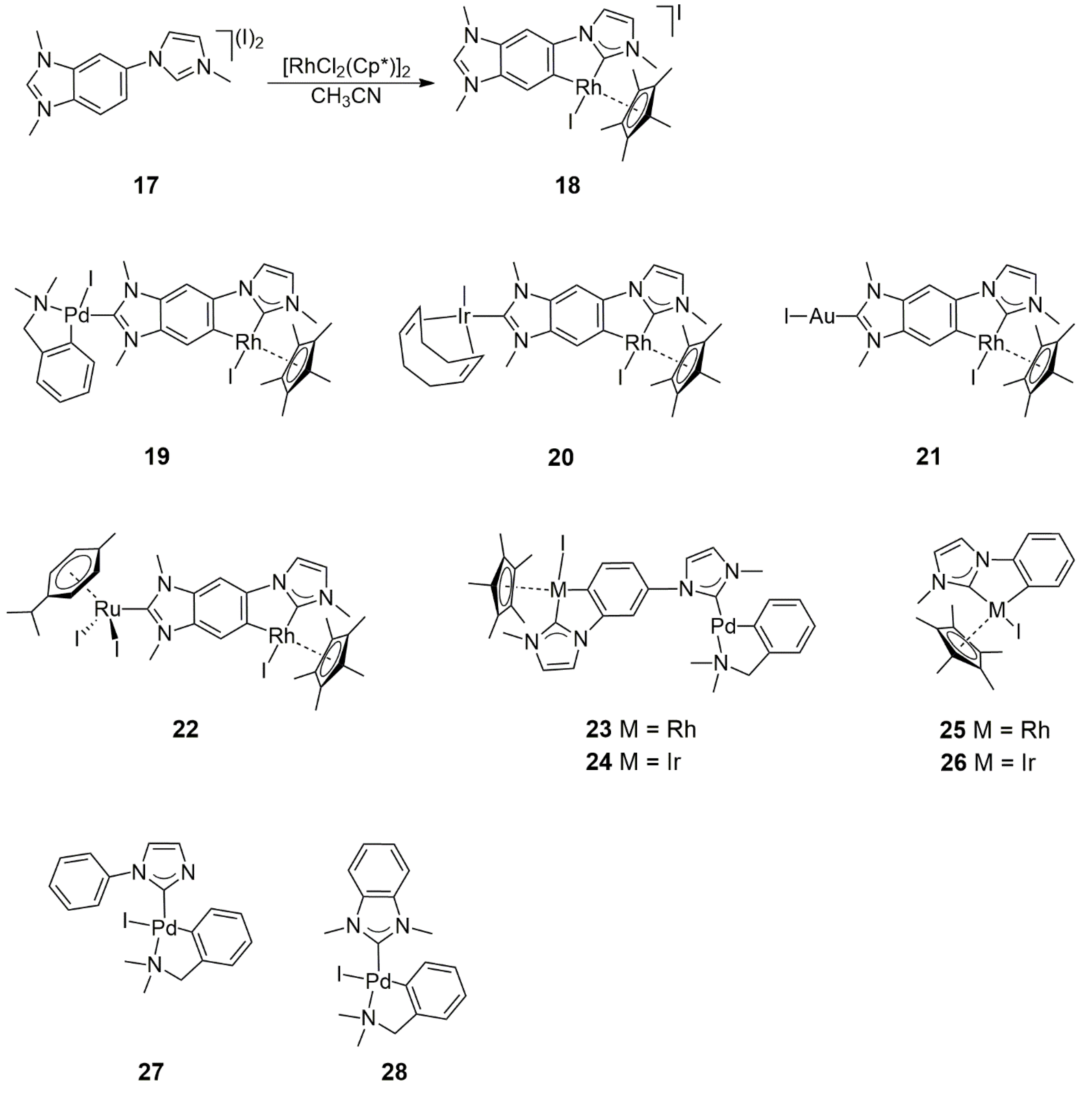
A mixed imidazolium/benzimidazolium bis(carbene) precursor, 17 (
Scheme 5), was synthesized by Hahn, Peris, and co-workers [
26]. The reaction of this compound with 1 equiv [IrCl
2(Cp*)]
2 (Cp* = pentamethylcyclopentadienyl anion) in the presence of NaOAc as a base led to the formation of a mixture of monometallic iridium complexes. However, under similar conditions, [RhCl
2(Cp*)]
2 selectively yielded
18, in which Rh binds to the C2 carbenic carbon of the imidazole moiety and simultaneously orthometalates the phenyl ring of the benzimidazolium unit (
Scheme 5). The formation of such a metallacycle contributes to the selective formation of the imidazol-2-ylidene type complex, while the benzimidazolium part remains available for further reactions. Indeed, the benzimidazole-ylidene complexes Rh/Pd (
19), Rh/Ir (
20), Rh/Au (
21), and Rh/Ru (
22) were prepared from
18 with the use of the Ag
2O-based method of synthesis. For a comparison of catalytic purposes, several other heterobimetallic complexes and their monometallic fragments (
23–
28) were synthesized (
Scheme 5).
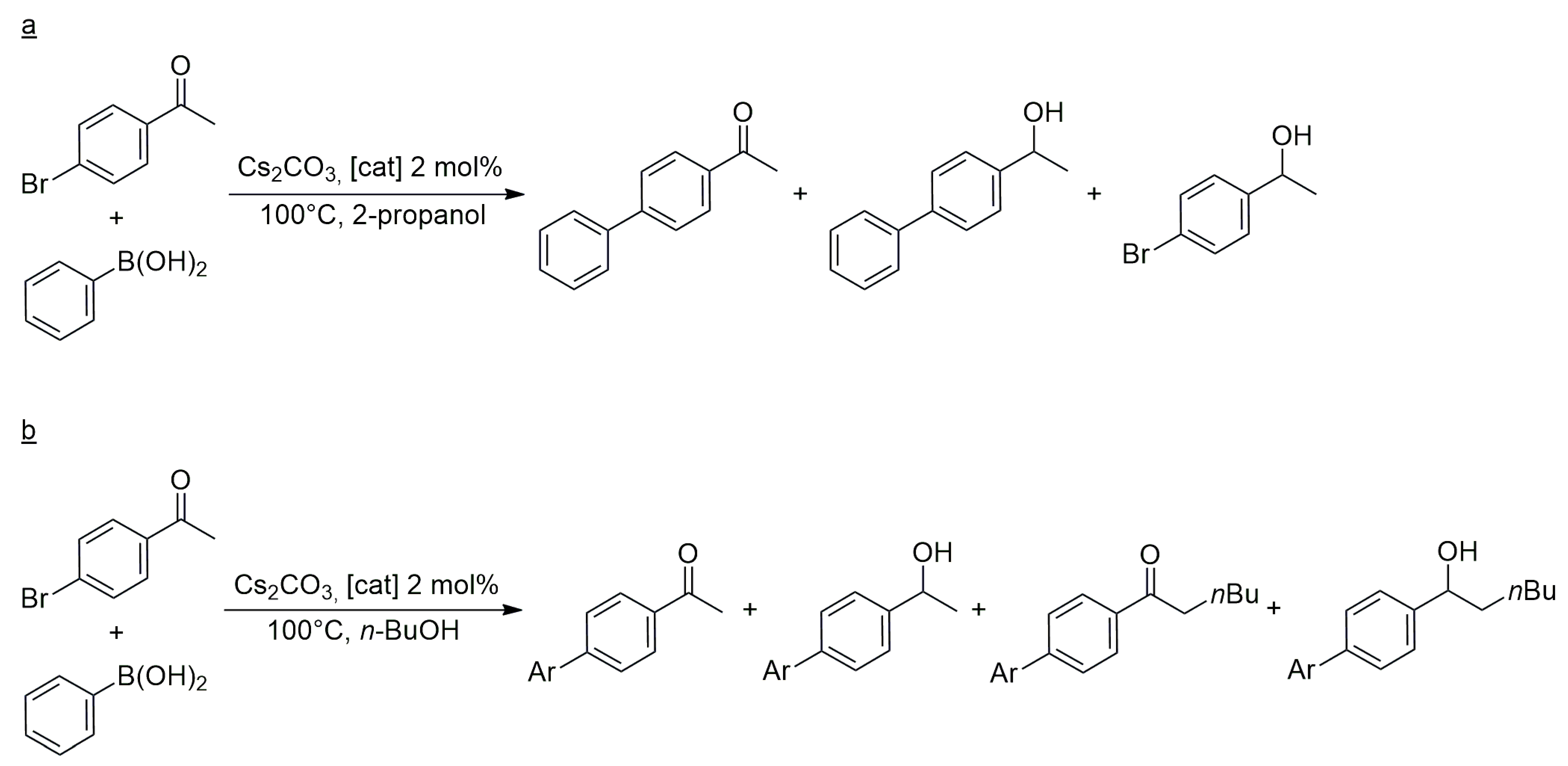
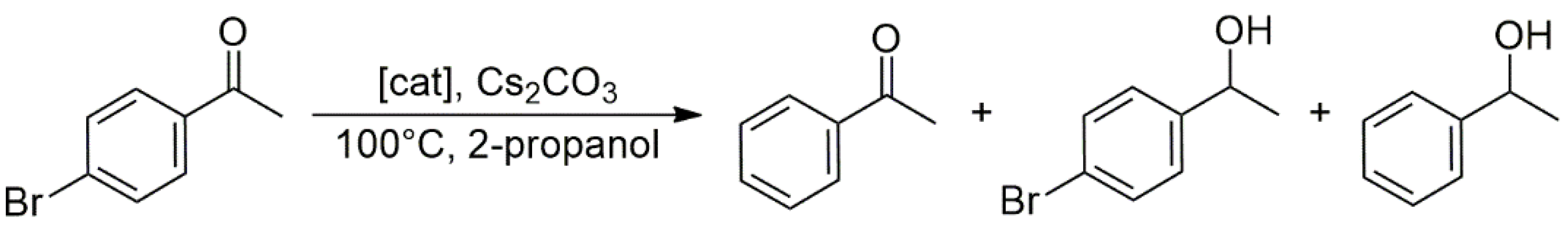
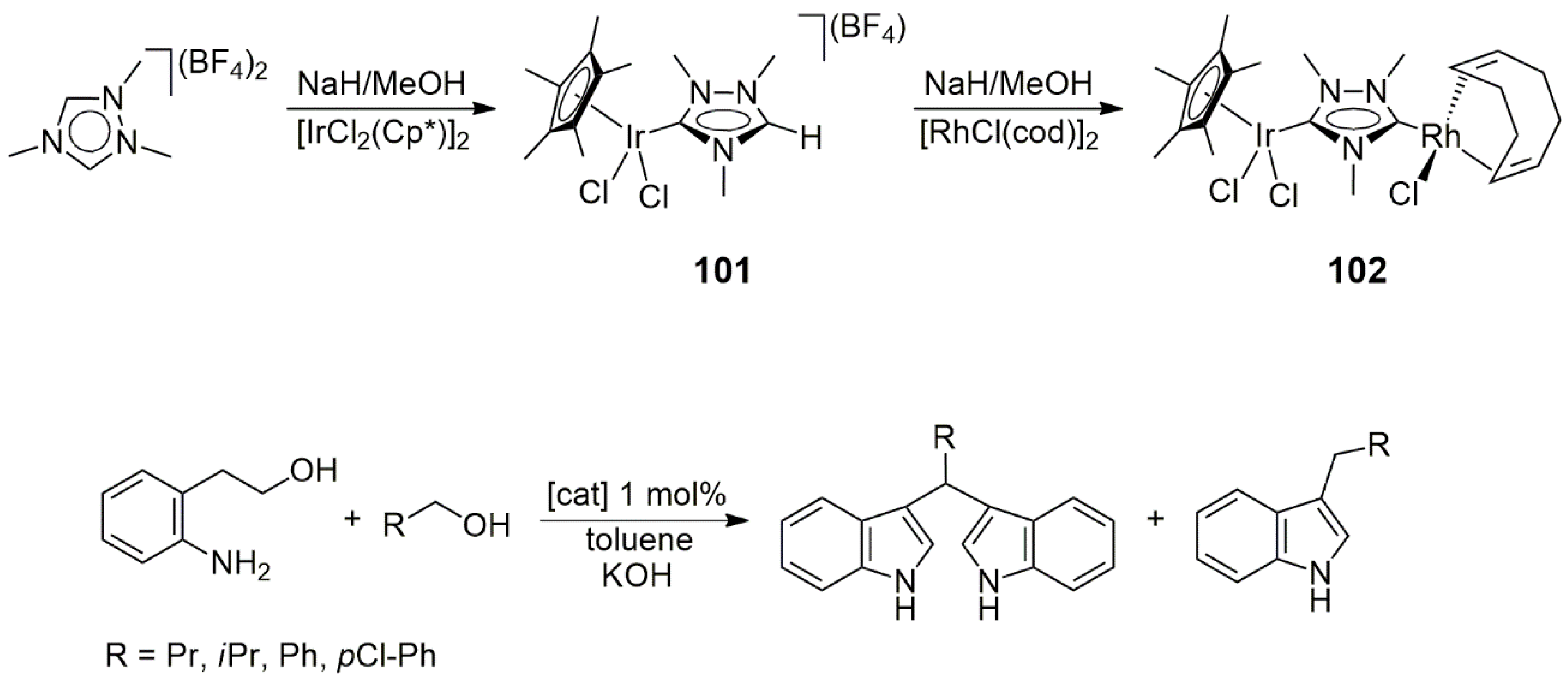
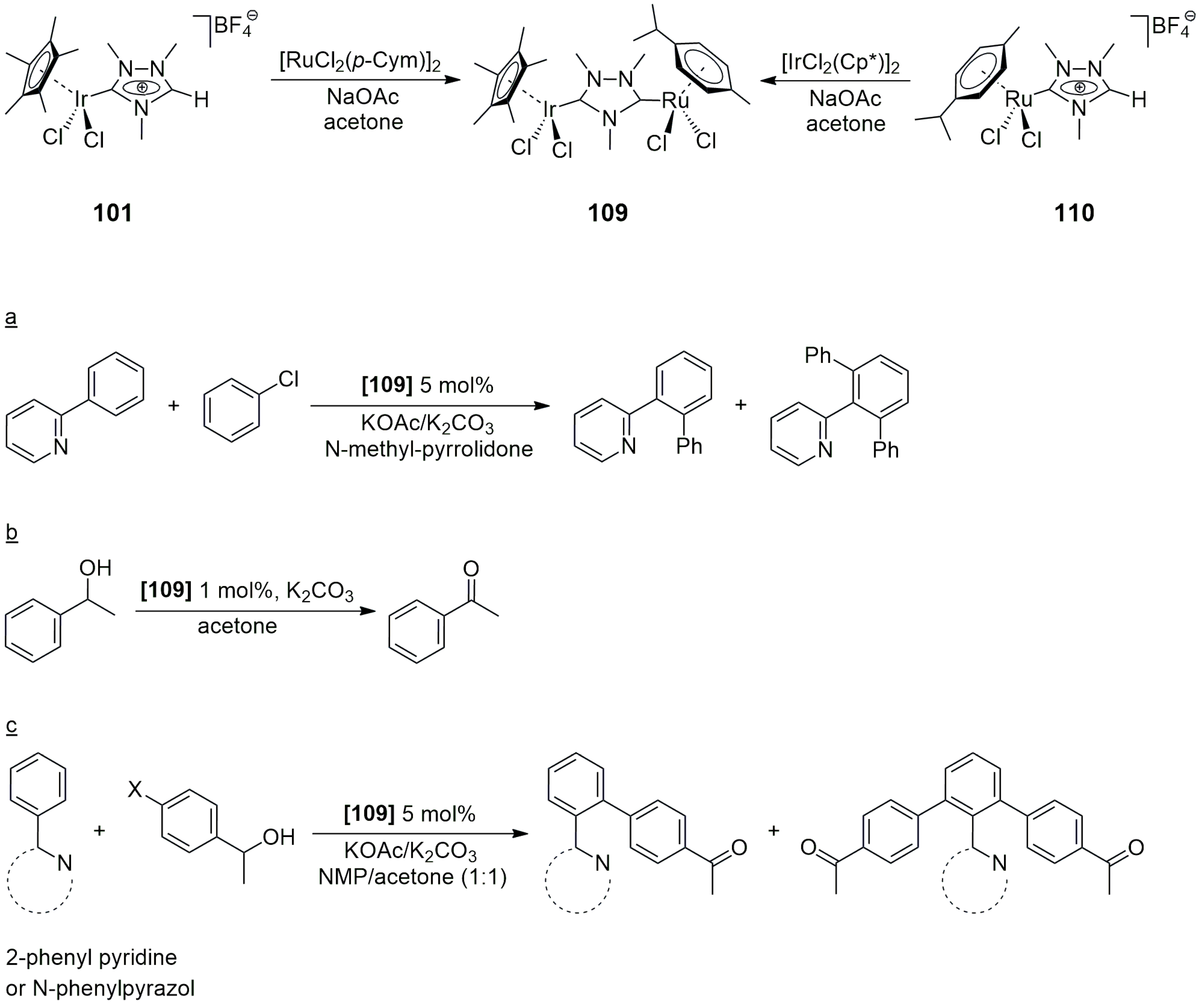
The catalytic properties of the new heterobimetallic NHC complexes were examined in two reactions. First, the tandem Suzuki–Miyaura
C–
C coupling/transfer hydrogenation of 4-bromoacetophenone was studied with phenylboronic acid as a coupling partner and 2-propanol as a hydrogen donor and solvent (
Scheme 6). At a temperature of 100 °C, and with Cs
2CO
3 as a base, the catalytic reactions proceeded with 99% conversion and afforded only the coupled product (4-phenylacetophenone) and its hydrogenated derivative 1-(4-biphenyl)ethanol. The reaction in the absence of a metal catalyst yielded 54% 4-Br-phenylethanol, but no debromination of the latter to 1-phenylethanol was observed. Concerning the relative activities of heterobimetallic NHC complexes,
19 (Rh/Pd) actively catalyzed the
C–
C coupling of 4-bromoacetophenone with phenylboronic acid, but its activity in the further transfer hydrogenation of the resulting biphenyl-methylketone was lower than that of
23 and
24. The latter two catalysts provided 82% and 84% yields of 1-(4-biphenyl)ethanol, respectively, in contrast to
19, which afforded only 53% (all yields at 99% total conversion of 4-bromoacetophenone). On the basis of kinetic measurements, the reaction could be regarded as a conventional consecutive reaction in which the first intermediate, the product of
C–
C coupling, accumulated up to 85% before further hydrogenation by H-transfer from 2-propanol. This behavior shows that the Pd-catalyzed
C–
C coupling proceeds faster than the Rh-catalyzed transfer hydrogenation. Nevertheless, cooperation between the two metals in the heterobimetallic complexes cannot be excluded since, in certain cases, the reactions proceeded faster with heterobimetallic catalysts, e.g.,
23 and
24, than with equimolar mixtures of the monomeric ‘fragments’, such as (
25 +
28), and (
26 +
28).
With the same catalyst, 4-bromoacetophenone was coupled to phenylboronic acid in
n-butyl alcohol, in which case, the product mixture contained α-
n-butylated products, too (
Scheme 6) [
26]. Catalysts
23 and
24 were found more effective for α-
n-butylation than mixtures of (
25 +
27) and (
26 +
27). Again, the time-dependent reaction profile showed a typical consecutive reaction. The higher activity of heterobimetallic complexes relative to the monomeric analogs suggested some additivity or cooperativity between the two metal ions in the catalyst complexes; however, the mechanism of such cooperations was not established.
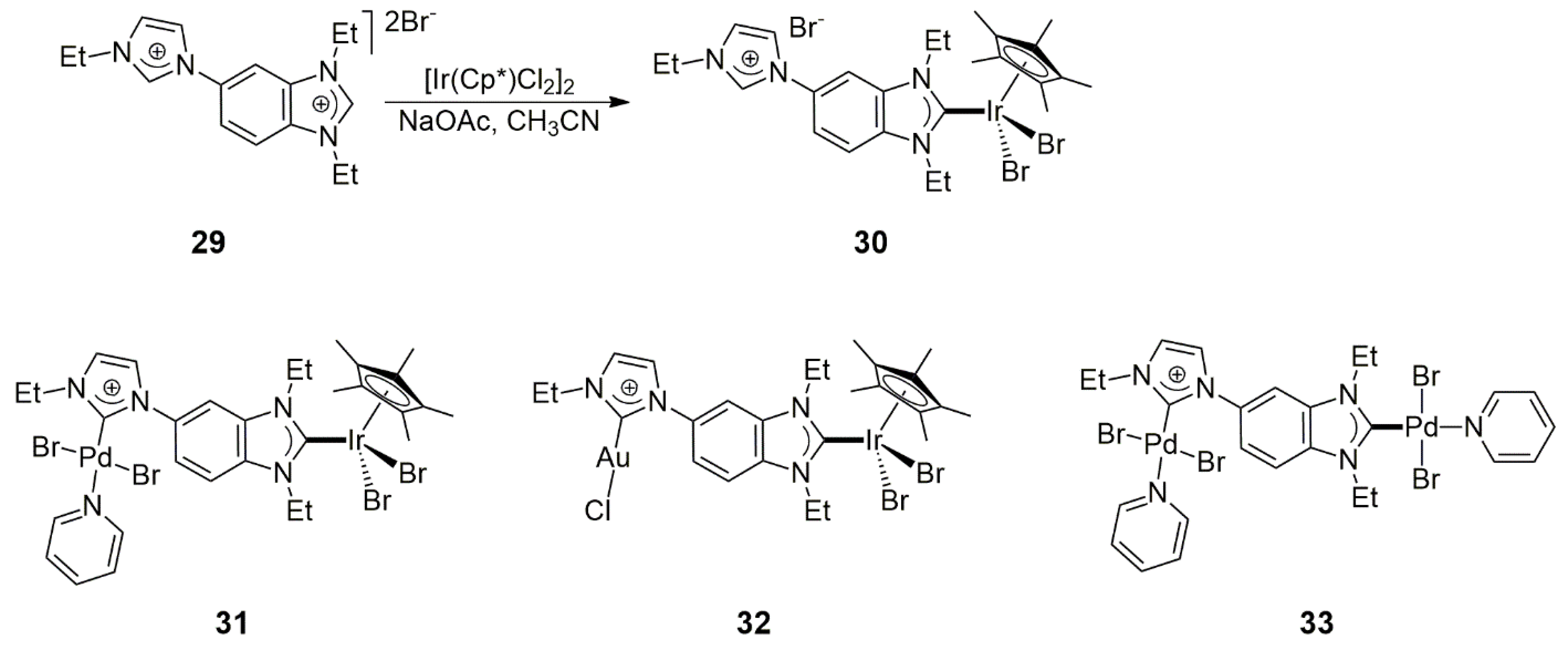
In a related study, Nishad, Kumar, and Rit [
27] found that reaction of
29 and 0.5 equiv [IrCl
2(Cp*)]
2 afforded selectively the monometallic Ir-carbene complex
30, with a pendent N-ethylimidazolium unit. Further metalations with Pd(OAc)
2 or Ag
2O/Au(THT) yielded the bimetallic complexes
31 (Ir/Pd) and
32 (Ir/Au), respectively. For comparison, the analogous Pd/Pd homobimetallic complex
33 was also prepared (
Scheme 7).
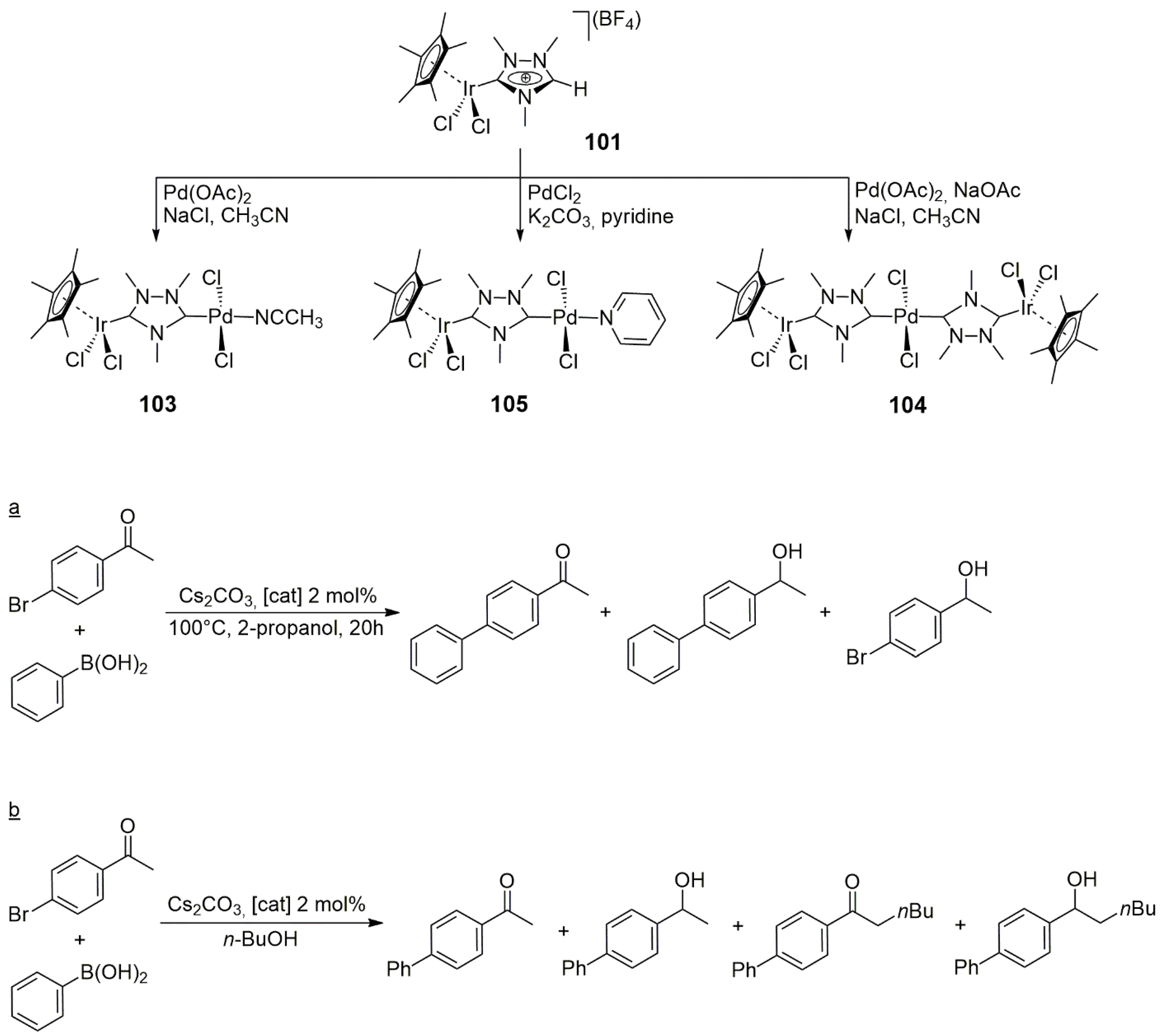
Complexes
31 (Ir/Pd) and
33 (Pd/Pd) were used as catalysts in the tandem Suzuki–Miyaura coupling of 4-bromoacetophenone with phenylboronic acid, followed by transfer hydrogenation with the use of 2-propanol and KO
tBu (O
tBu = tert-butylate) (
Scheme 6a). Under optimal conditions (1 mol% catalyst, 100 °C, 12 h, 2-propanol as solvent) the Ir/Pd catalyst (
31) led to the formation of the
C–
C-coupled and hydrogenated product with an 87% yield. Under the same conditions, catalysis by
33 (Pd/Pd) yielded only 61% 1-(4-biphenyl)ethanol, accompanied by 27% 1-phenylethanol arising from the debromination and transfer hydrogenation of 4-bromoacetophenone. These results suggested that there was some cooperation between the metals in the heterobimetallic catalyst, leading to higher activity in the case of the Ir/Pd catalyst over the Pd/Pd complex. Interestingly, under similar experimental conditions to the above,
31 also showed high catalytic activity in the tandem defluorination/transfer hydrogenation of mixed (4-fluorophenyl)-alkyl- or arylketones, in which it led to the formation of the defluorinated products in 80%, 97%, and 96% (alkyl = methyl, ethyl; aryl = phenyl, respectively). In these reactions, too, 2-propanol served both as a hydrogen donor and solvent.
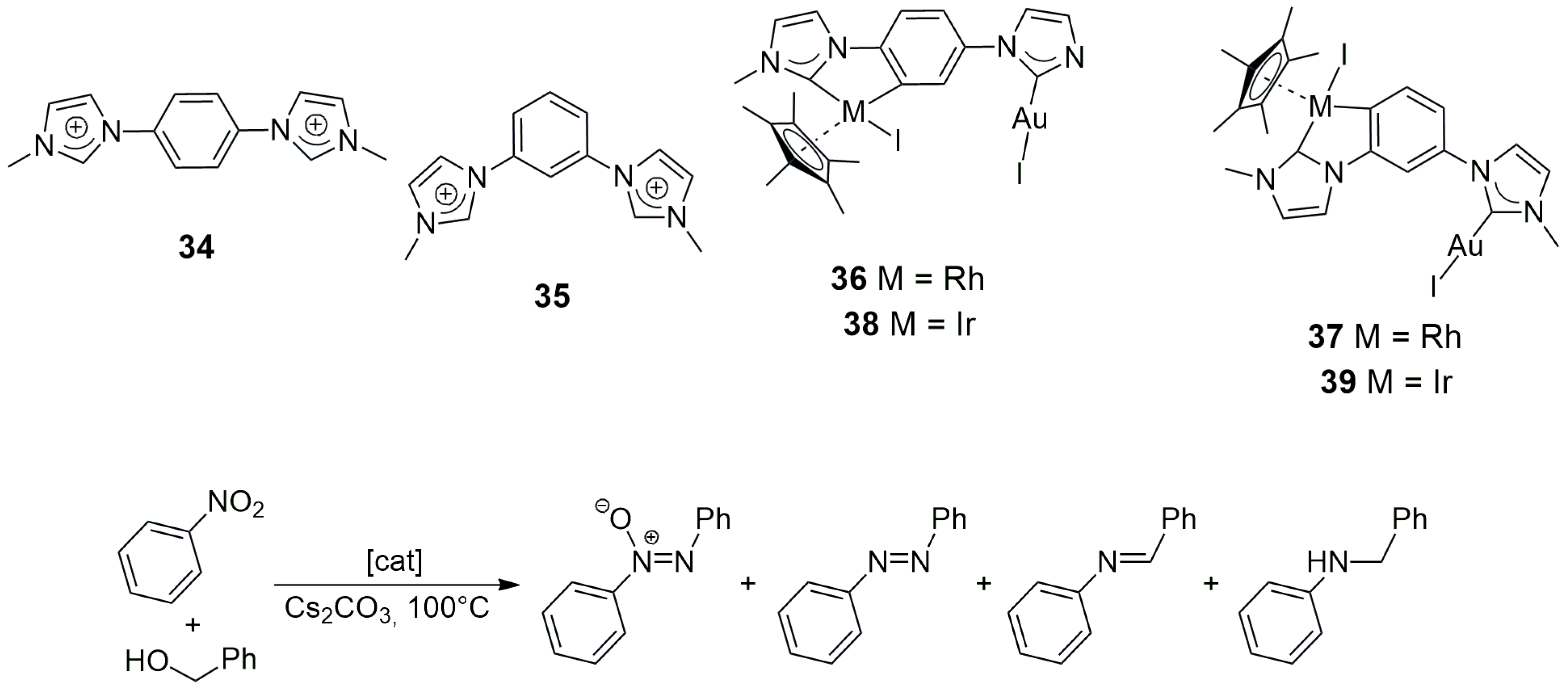
Hahn, Peris, and co-workers applied the 1,4-phenylene-bridged (
34) and the 1,3-phenylene-bridged (
35) N-methylimidazolium iodides to the synthesis of heterobimetallic Rh/Au (
36,
37) and Ir/Au (
38,
39) complexes (
Scheme 8) [
28]. In the first step of sequential metalation,
34 and
35 reacted with either [RhCl
2(Cp*)]
2 or [IrCl
2(Cp*)]
2, yielding the corresponding cyclometalated Rh/NHC or Ir/NHC intermediates, respectively. Next, these intermediates were treated with Ag
2O/Au(THT), affording the desired heterobimetallic Rh/Au (
36,
38) and Ir/Au (
37,
39) products. Overall yields were in the 21–56% range.
The catalysts were tested in the tandem condensation of nitrobenzene with benzyl alcohol and imine reduction (
Scheme 8) with a 0.5% catalyst loading and Cs
2CO
3 as a base at 100 °C for 22 h in benzyl alcohol as the solvent. With the exception of
38, all other new heterobimetallic NHC-complex catalysts led to 100% conversion, which was higher than the conversions generally obtained with equimolar mixtures of analogous monometallic ‘fragments’. The highest imine yield (62%) was obtained with catalyst
36 (Rh/Au) with only 13% secondary amine (the hydrogenated product). It could be concluded that the cooperation of the two metal ions in the heterobimetallic complexes with
meta-bis-NHC ligands was stronger than in those with
para-bis-NHC ligands, which was demonstrated by the highest activity of
39 for amine formation (82%).
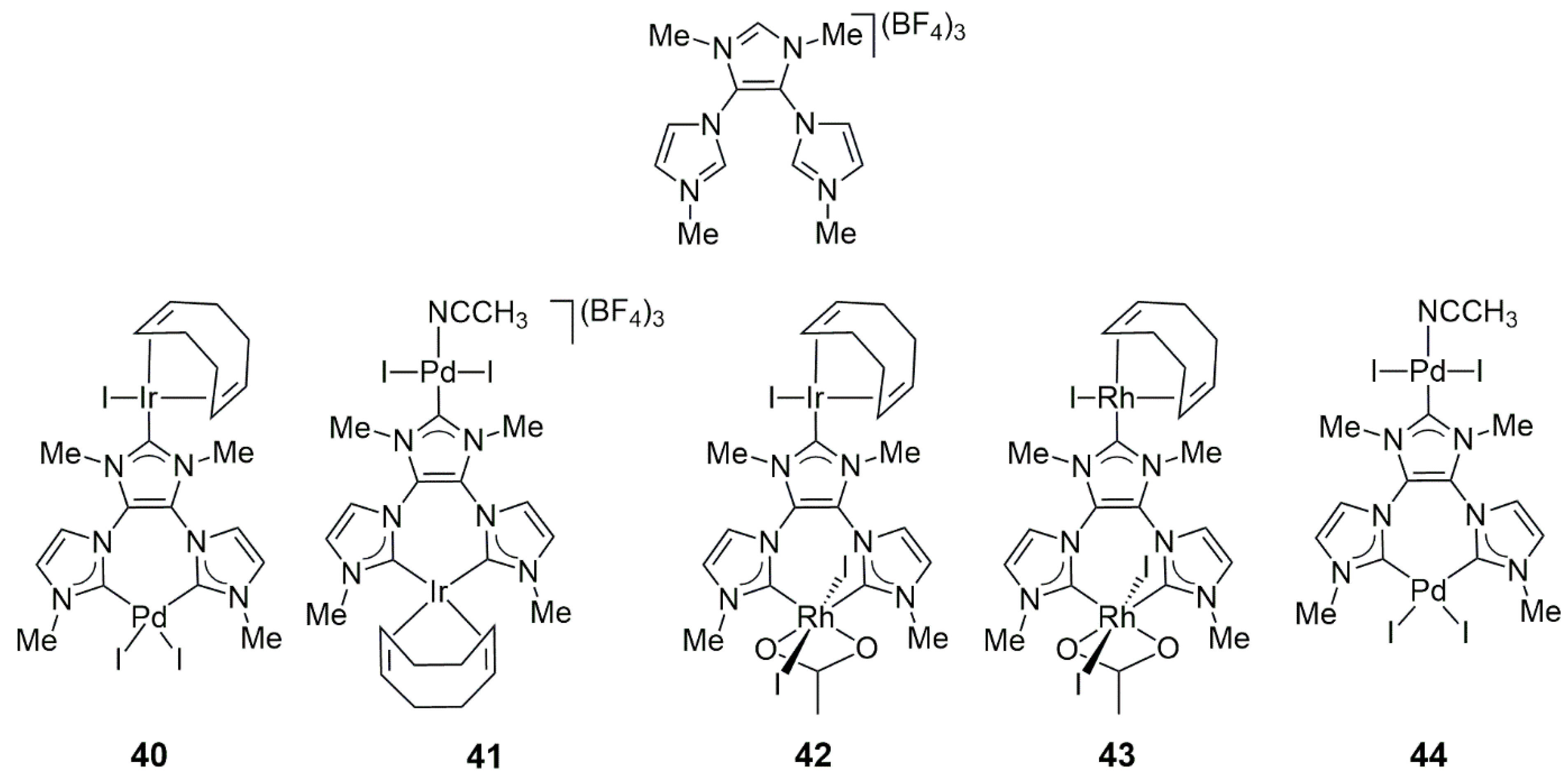
Gonell, Poyatos, Mata, and Peris synthesized a Y-shaped tris-N-heterocyclic carbene ligand precursor and applied it to the synthesis of Rh-, Ir-, and Pd-based homo- and heterobimetallic complexes
40,
41,
42, and
43 by sequential metalation with the use of [RhCl(cod)]
2, [IrCl(cod)]
2, or Pd(OAc)
2 (
Scheme 9) [
29]. It was found that this Y-shaped ligand reacted with the first equivalent of the metal compounds with the formation of chelate bis-NHC complexes, which—upon the addition of the next equivalent of metal compounds—could bind another metal ion on the C2 carbene atom of their third imidazolium group. The order of addition of the metal precursor to the ligand determined which metal ion was coordinated in a chelate or monodentate fashion (see
40 vs.
41). Interestingly, despite the use of [RhCl(cod)]
2 for the preparation of
43 in the presence of NaOAc, the chelated rhodium was always in the Rh(III) oxidation state. The solid-state structures of
40 and
43 were determined by X-ray diffraction, which showed a through-space Ir-Pd distance of 6.76 Å in
40 and a Rh(I)-Rh(III) distance of 7.003 Å in
43. For comparison purposes, the homobimetallic compound
44 (Pd/Pd) was also prepared.
In the tandem dehalogenation/transfer hydrogenation of 4-bromoacetophenone (
Scheme 10), the Ir(chelate)/Pd(monodentate) catalyst
41 yielded 1-phenylethanol in close to quantitative yield (1–2% catalyst loading, 100 °C, 20–48 h, 2-propanol as a solvent and H-donor). In contrast, Ir(monodentate)/Pd(chelate)
40 was much less active in transfer hydrogenation and, under the same conditions, led to the formation of acetophenone in 52–89% yields. A 1:1 mixture of related (albeit not strictly analogous) binuclear Ir/Ir and Pd/Pd complexes catalyzed the dehalogenation step efficiently; however, the transfer hydrogenation proceeded only to 25%. As expected,
41 was also an effective catalyst for the tandem Suzuki–Miyaura coupling/transfer hydrogenation of 4-bromoacetophenone with phenylboronic acid (
Scheme 6), in which the ratio of the biphenyl methyl ketone and 1-(4-biphenyl)ethanol products strongly varied with the reaction time (from 82% to 16% and from 0% to 69%, respectively, in the 0.5–20 h range of reaction time). It could be concluded that there was substantial catalytic cooperation between the two metal ions; still, Pd was mainly responsible for dehalogenation, while Ir was necessary for the effective catalysis of transfer hydrogenation.
It was also demonstrated that 40 and 41 were very active catalysts for the cyclization of 2-(ortho-)aminophenyl ethyl alcohol to indole (GC yields > 99%) in toluene at 110 °C for 2 h. Similarly, the same complexes catalyzed the acylation of bromobenzene with n-hexanal, resulting in its full conversion to butyl phenyl ketone at 115 °C in DMF for 16 h. However, the details of these reactions were not reported.
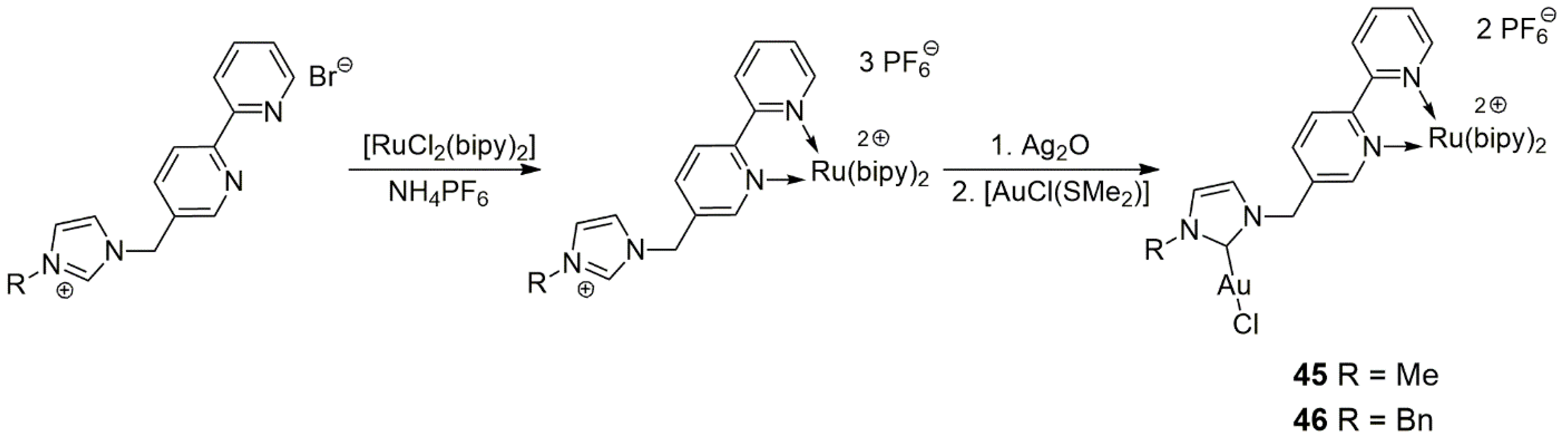
Hemmert, Gornitzka, and co-workers were interested in the effect of a Au-NHC unit on the photophysical properties of [Ru(bipy)
3]
2+ (bipy = 2,2′-bipyridine) within the same heterobimetallic complex [
30]. Conversely, they also wished to reveal the effect of the [Ru(bipy)
3]
2+ part of the molecule on the cytotoxic, antileishmanial, and antimalarial activities of the Au-NHC moiety. For this purpose, a 2,2′-bipyridine unit was attached via a methylene linker to methyl-,
n-butyl-, and benzylimidazole in the reaction of the azole with 5-(bromomethyl)-2,2′-bipyridine. In the reaction of [RuCl
2(bipy)
2] with the functionalized azolium salt, an octahedral Ru(II) complex was formed with three bipy ligands, one of which carried a pendant azolium group. With the treatment of this intermediate with Ag
2O and then with AuCl(SMe
2), heterobimetallic Ru-bipy/Au-NHC complexes
45 and
46 were obtained, differing only in the substituent of the NHC-carbene part (
Scheme 11). The main conclusion of this study was that the photophysical properties of the Au/Ru heterobimetallic complexes were very similar to those of [Ru(bipy)
3]
2+ and that without Ru(II) coordination, the compounds showed low biological activity in comparison to the Au(I) complex. In line with the original purpose of the studies, no catalysis by compounds
45 and
46 was reported.
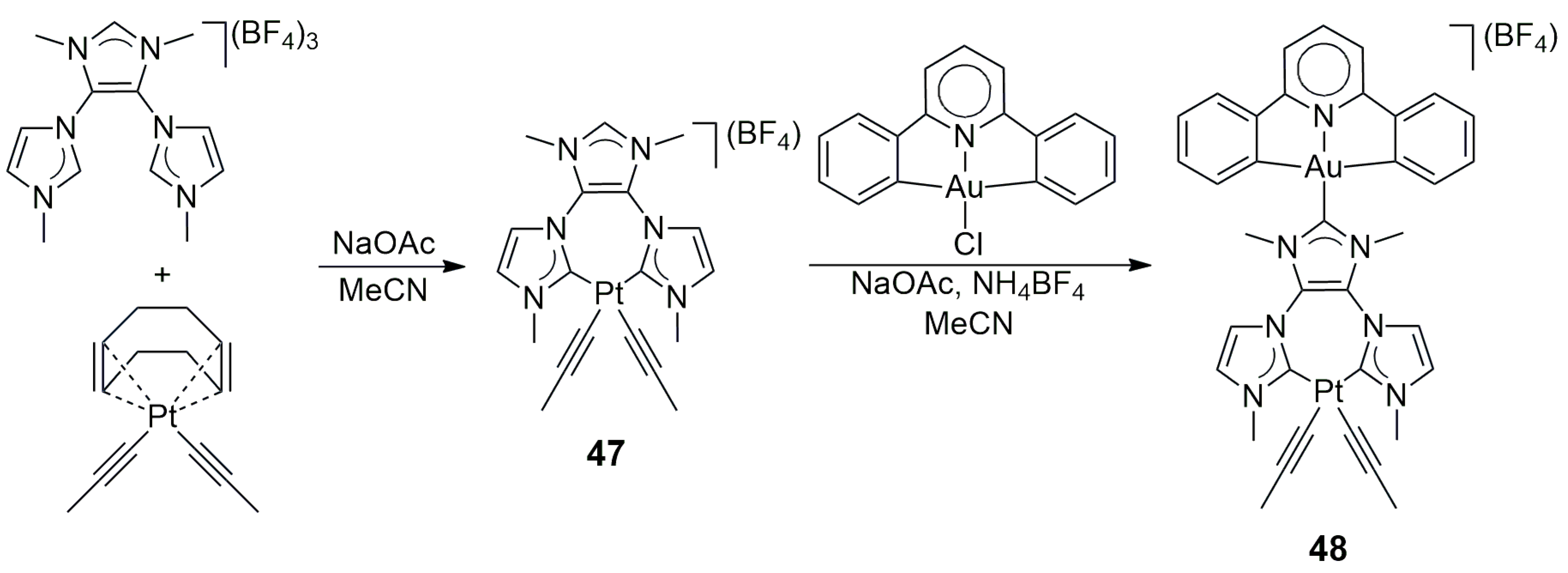
In a related study [
31], Gonell, Poyatos, and Peris synthesized an Au/Pt heterobimetallic NHC complex based on the Y-shaped tris-NHC ligand precursor developed by them earlier [
29]. In the first step, the ligand precursor tris-azolium salt was reacted with the bis(alkynyl) complex [Pt(CCPh
2)(cod)]. This reaction resulted in complex
47, which contained the Pt(CCPh)
2 moiety in chelate coordination. The reaction of this compound with a pincer-type (CNC)Au(I)Cl complex in the presence of NaOAc in acetonitrile led to the desired heterobimetallic Au/Pt product
48 (
Scheme 12). In addition to NMR spectroscopy and HR mass spectrometry,
48 was characterized by UV-Vis absorption and emission spectroscopy, as well as by cyclic voltammetry and differential pulse voltammetry. Both
47 and
48 were found emissive in the solid state but non-emissive in solution. It could also be concluded that the emission was centered on the Pt(II) fragment.
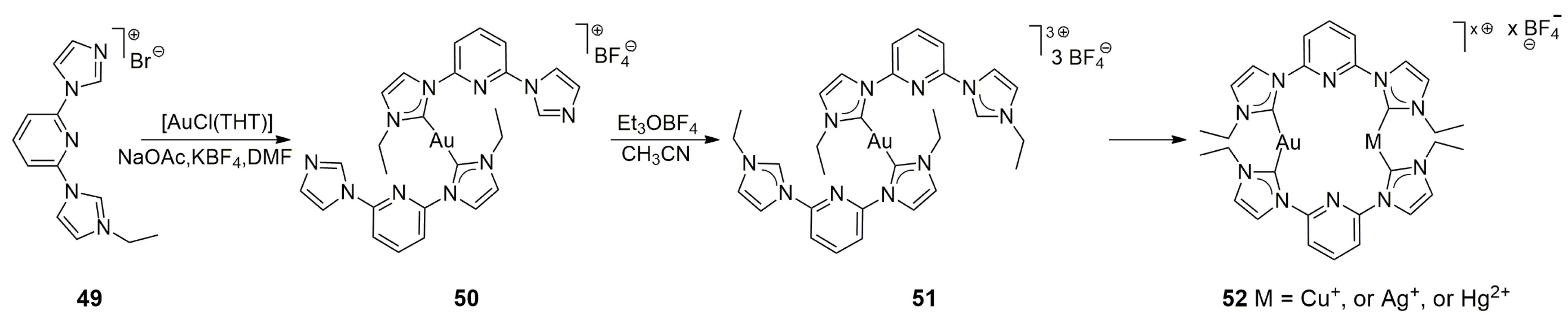
Barnard and co-workers planned to prepare heterobimetallic complexes based on the pro-ligand
49 [
32]. However, treatment of this ligand with 0.5 equiv of AuCl(THT) in the presence of a weak base such as NaOAc produced only a homobimetallic Au(I) complex, together with unreacted
49 and other unidentified products. A postmetalation ligand synthesis was developed, according to which the second imidazolium moiety was synthesized only after the initial metalation step with AuCl(THT). The ethylation of
50 with Et
3OBF
4 in CH
3CN led to complex
51, in which the second imidazolium was metalated to yield the corresponding Au/M complexes (
52), where M = Cu
+, Ag
+, or Hg
2+ (
Scheme 13). Although the complexes
52 were emissive in the solid state, they were not luminescent in solutions. Nevertheless, according to the solid-state structures (X-ray), all heterobimetallic complexes support heterometallophilic interactions, and their structures may give further insight into the luminescent properties of such heterobimetallic compounds.
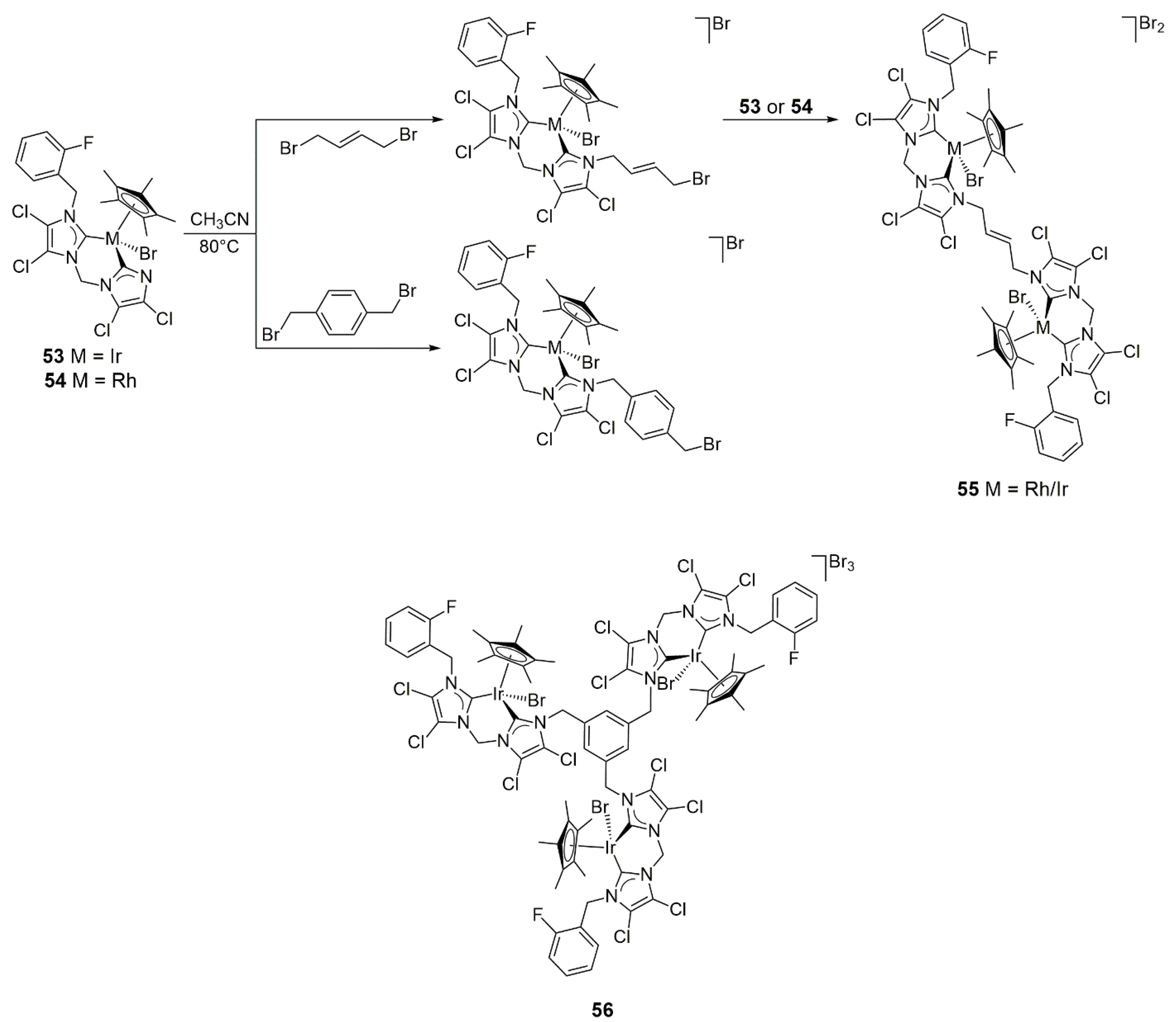
A different postmetalation synthetic method was developed by Hahn, Jin, and co-workers for the preparation of multidentate NHC complexes [
33]. The Ir- or Rh-containing bis-NHC complexes
53 and
54, which contained a protic NHC ligand unit, were reacted with trans-1,4-dibromo-2-butene or with 1,4-bis(dibromomethyl)benzene, leading to the N-alkylated (benzylated) derivatives with pendant bromomethyl groups. The further reaction of these intermediates with
53 or
54 gave tetrakis-NHC complexes, of which
55 is an Ir/Rh heterobimetallic NHC compound (
Scheme 14). In addition to characterization by NMR and HRMS, the solid-state structure of
55 was determined by X-ray crystallography. However, no catalysis was reported for this new heterobimetallic complex. When
53 reacted with 1,3,5-tris(bromomethyl)benzene instead of 1,4-bis(bromomethyl)benzene, the same synthetic strategy led to the homometallic hexakis-NHC iridium complex
56 (
Scheme 14).
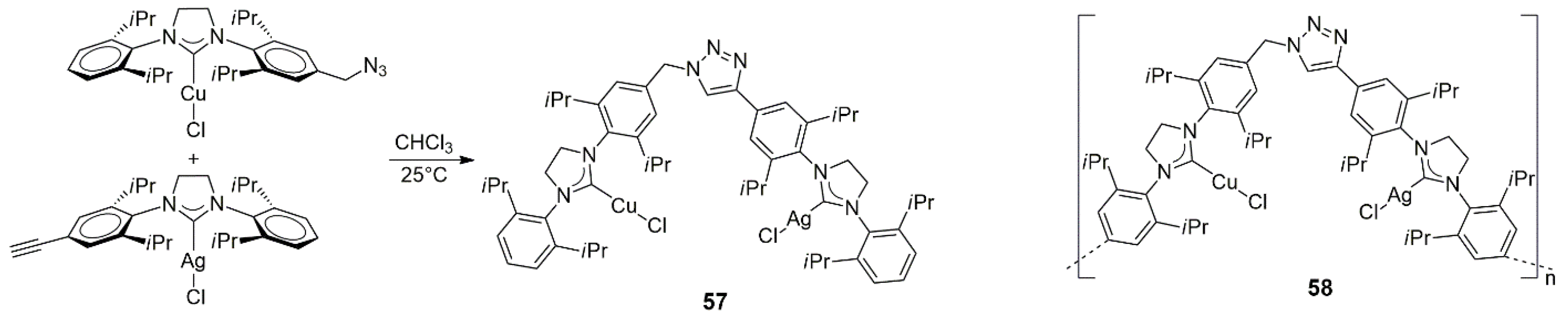
Hahn, Li, and co-workers reported a general strategy for the synthesis of oligomeric or polymeric NHC complexes having strictly pre-determined positions of the NHC units and metal ions [
34]. The strategy is based on the click reaction of suitable monomeric NHC–metal complexes (secondary building units or SBUs) having
p-azidophenyl and
p-ethynylphenyl NHC wingtips (
Scheme 15). The azide group could be directly attached to the aromatic ring or via a flexible methylene linker. The reaction of the azide-containing SBUs with phenylethynyl-substituted NHC complexes resulted in the formation of polymers with alternating metal–NHC units joined together by 1,2,3-triazole (or the more flexible methylene-triazole) linkers along the polymeric chain. In cases when one of the SBUs was an azide-modified copper complex, the click reaction proceeded rapidly without an external catalyst and, depending on the combination and molar ratios of the SBUs, resulted in heterodimetallic dimers, trimers, or polymers (only the Cu/Ag bis-NHC complex
57 and the −[Cu~Ag]
n− polymer
58 are shown in
Scheme 15). The Cu/Ag derivatives could be further transmetalated in reaction with, e.g., allylpalladium(II) chloride dimer [PdCl(allyl)]
2 to yield analogous Cu/Pd polymers.
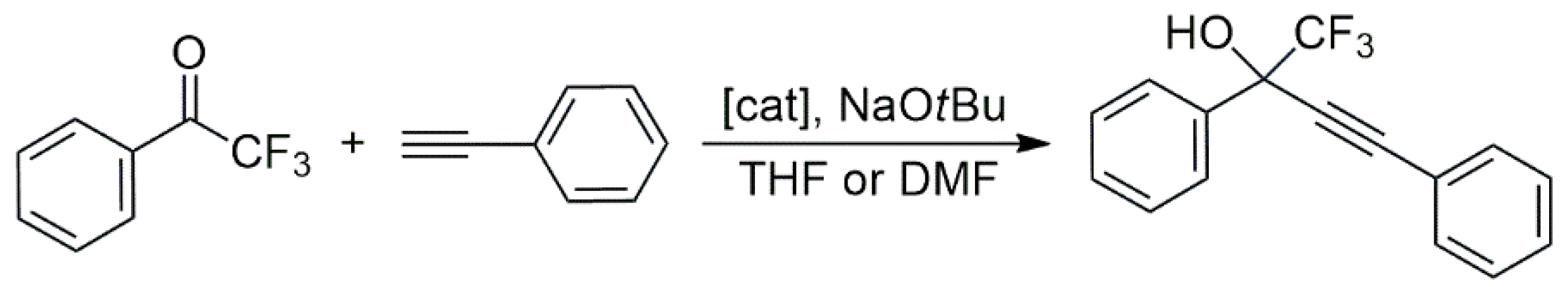
The Cu/Ag heterobimetallic bis-NHC complex
57 and the corresponding polymer
58 were studied in the catalytic alkynylation of 2,2,2-trifluoro-1-phenylethanone with various terminal alkynes (
Scheme 16). Detailed measurements with phenylacetylene as a model substrate showed that both
57 and
58 exhibited high catalytic activity, leading to 96 and 95% yields of the fluorinated propargylic alcohol at a catalyst loading as low as 0.5 mol%. (For solubility reasons,
57 was used in THF, while
58 was in DMF.) Control experiments with a Cu/Ag polymer with a rigid triazol linker showed diminished catalytic activity (67% yield) compared to that of
58 (99% yield), pointing to the importance of the flexibility of the polymer chain. Importantly, a 1:1 mixture of [CuCl(SI
iPr)] (SI
iPr = 1,3-bis(2,6-isopropylphenyl)imidazoline-2-ylidene) and [AgCl(SI
iPr)] (mononuclear analogs of the NHC complex units in
57 and
58) also showed much inferior activity (65% in THF and 39% in DMF). The latter observation strongly supports the assumption that there is cooperativity between Cu(I) and Ag(I) in the catalysis of this reaction, which also requires the flexibility of the polymer. Other terminal alkynes also reacted smoothly with
57 and
58 as the catalysts.
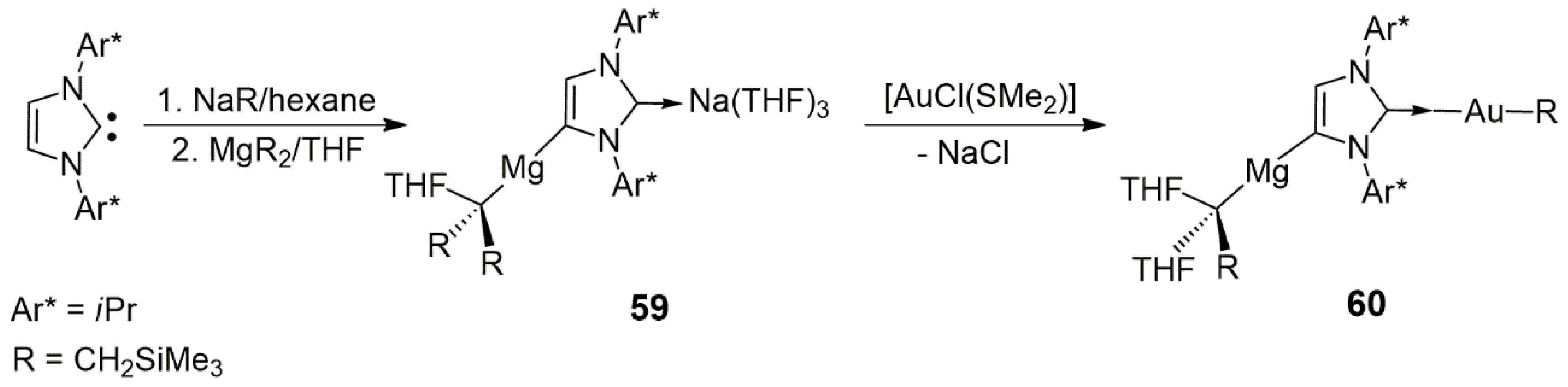
Anionic N-heterocyclic dicarbenes are mostly used as bridging ligands between two main-group elements. In a recent paper, Hevia et al. described the magnesiation of the common NHC-carbene IPr (1,3-bis(2,6-isopropylphenyl)imidazole-2-ylidene) [
35]. The reaction of IPr and NaR (R = CH
2SiMe
3) in hexane yielded a polymeric anionic sodium–NHC intermediate, which could be further metalated with the use of MgR
2 in THF to yield the heterobimetallic product
59. In this complex, magnesium was coordinated to the abnormal C4 carbene position of IPr (
Scheme 17). The reaction of
59 and AuCl(SMe
2) in toluene at −70 °C resulted in the novel Mg/Au heterobimetallic complex
60. In this compound, magnesium retained its C4 coordination; however, one of the CH
2SiMe
3 groups was transferred from Mg to Au, while the coordination sphere of Mg was filled with an additional THF molecule originally bound to Na in
59. The reaction is accompanied by the formation of NaCl, the toluene-insolubility of which also drives the process toward the formation of
60 by creating a coordination vacancy at C2 (
Scheme 17). This study opened new pathways for the synthesis of anionic and abnormal Mg-NHC complexes and their application in transmetalation reactions, leading to new heterobimetallic Mg/Au complexes. No catalysis by the new compounds was reported.
2.2.2. Cyclometalation-Assisted Synthesis of Heterobimetallic NHC Complexes and Their Catalytic Properties
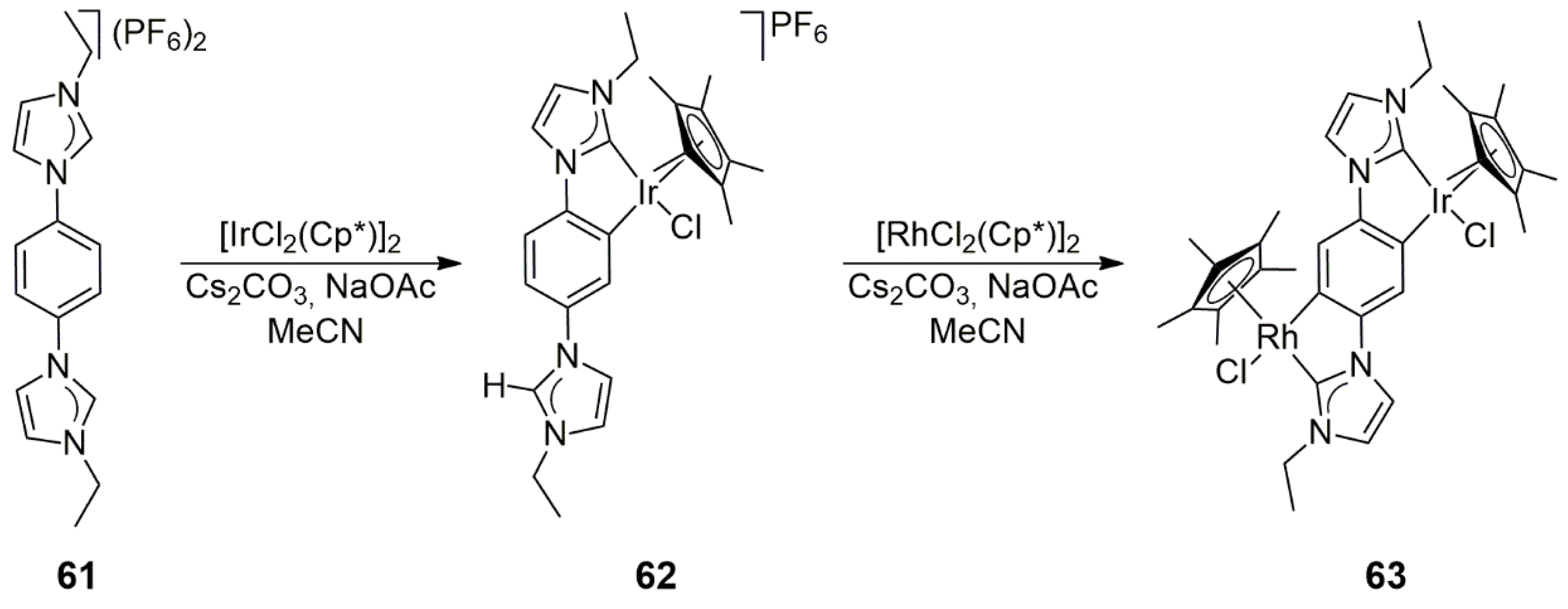
In addition to the formation of NHC complexes through metal–carbene coordination, product stability is often increased by the orthometalation of a suitable aryl substituent attached to an imidazolium nitrogen. Such a case was revealed by Maity, Hahn, and co-workers [
36] in the reaction of 1,4-bis(3-ethyl-1-imidazolium)benzene
61, originally synthesized by You et al. [
37] and later by the Hahn group via a modified procedure [
38]. As shown in
Scheme 18, the reaction of
61 with [IrCl
2(Cp*)]
2 yielded
62, which afforded the heterobimetallic
63 upon further metalation with [RhCl
2(Cp*)]
2 (both reactions took place in acetonitrile solution in the presence of Cs
2CO
3 and NaOAc). Both Ir(III) and Rh(III) cyclometalated the 1,4-phenylene linker between the NHC units. Orthometalation in
62 and double orthometalation in
63 were established by 2D NMR spectroscopy. Furthermore, the determination of the solid-state structure of
62 unambiguously confirmed this structural feature. The catalytic properties of
62 and
63 were not investigated.
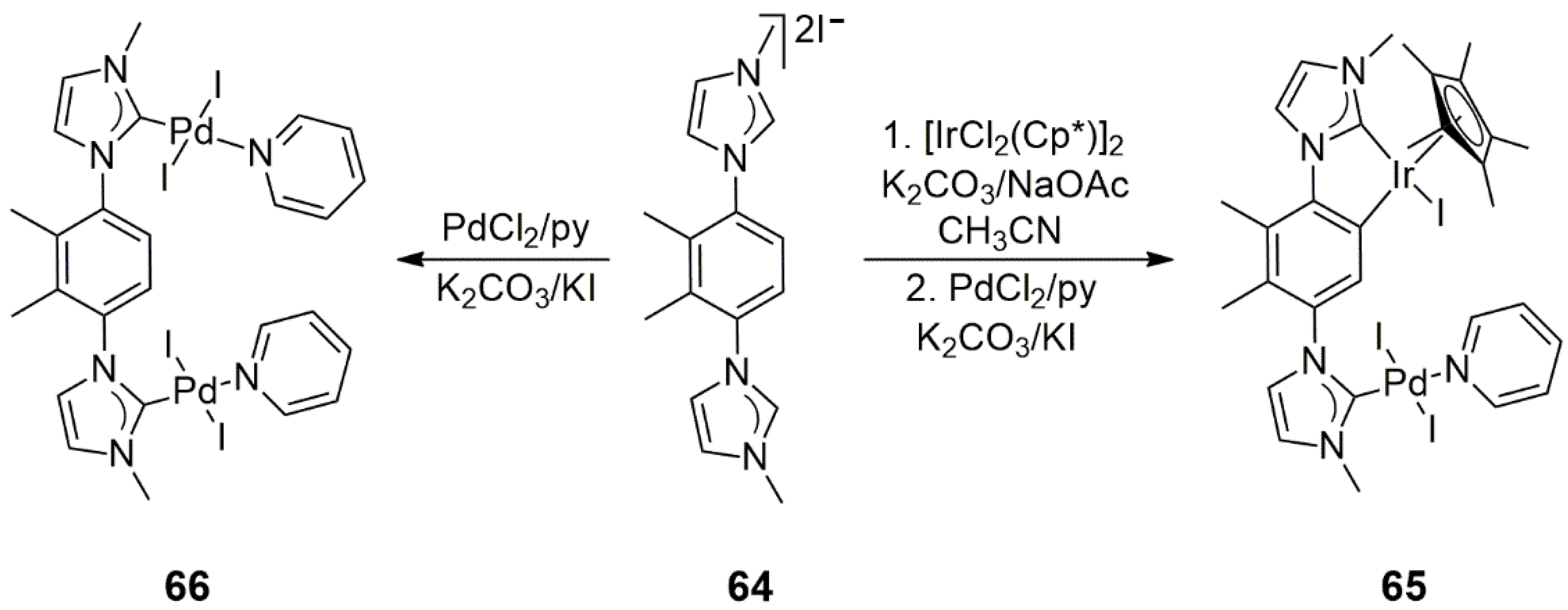
In a related study, Maity and co-workers synthesized 1,4-bis(3-methyl-1-imidazolium)-2,3-dimethyl-benzene
64 (
Scheme 19) [
39]. Although
64 is very similar to the previously mentioned ligand
61, the two methyl substituents on the 1,4-phenylene linker prevented the double orthometalation of the bridging phenylene moiety. In contrast, the reaction of
64 with [IrCl
2(Cp*)]
2 in acetonitrile afforded a monometallic cyclometalated Ir(III) NHC complex, which could be further metalated on the second NHC carbene with PdCl
2 in pyridine. The overall reaction yielded the PEPPSI-type Ir/Pd bimetallic bis-NHC complex
65. An X-ray diffraction study of
65 revealed a 6.14 Å distance between the two metal centers in the solid-state structure. The similar reaction of
64 with PdCl
2/pyridine led to the formation of the analogous Pd/Pd complex
66 (no cyclometalation in this case).
The catalytic activity of the Ir/Pd heterobimetallic bis-NHC complex
65 was studied in the α-arylation of oxindole in toluene solutions (
Scheme 20). In comparison to the homometallic counterpart
66, the heterobimetallic complex
65 showed slightly higher activities both with bromobenzene (32% vs. 26% isolated yields) and with 2-bromotoluene (53% vs. 41% isolated yields). Similarly, in the Suzuki–Miyaura cross-coupling of 4-bromobenzaldehyde and phenylboronic acid,
65 and
66 showed comparable activities (99% vs. 83% conversions in 5 h). Under the same conditions, with 4-toluylboronic acid, the yield of the coupled product was 99% with both catalysts. Note that these reactions were carried out with water as a solvent. Conversely, when the solvent was changed for 2-propanol/THF and KO
tBu was added as a base, tandem Suzuki–Miyaura coupling/transfer hydrogenation took place both with 4-bromobenzaldehyde and 4-bromoacetophenone as substrates. In these reactions,
65 was consistently more efficient, with yields up to 85%, than a 1:1 mixture of its mononuclear Pd(II) and Ir(III) counterparts (highest yield of 41%). It was concluded that the outstanding activity of the Ir/Pd heterobimetallic bis-NHC complex in the tandem
C–
C cross-coupling/transfer hydrogenation reaction was most probably due to the interaction between the Ir and Pd centers in the molecule.
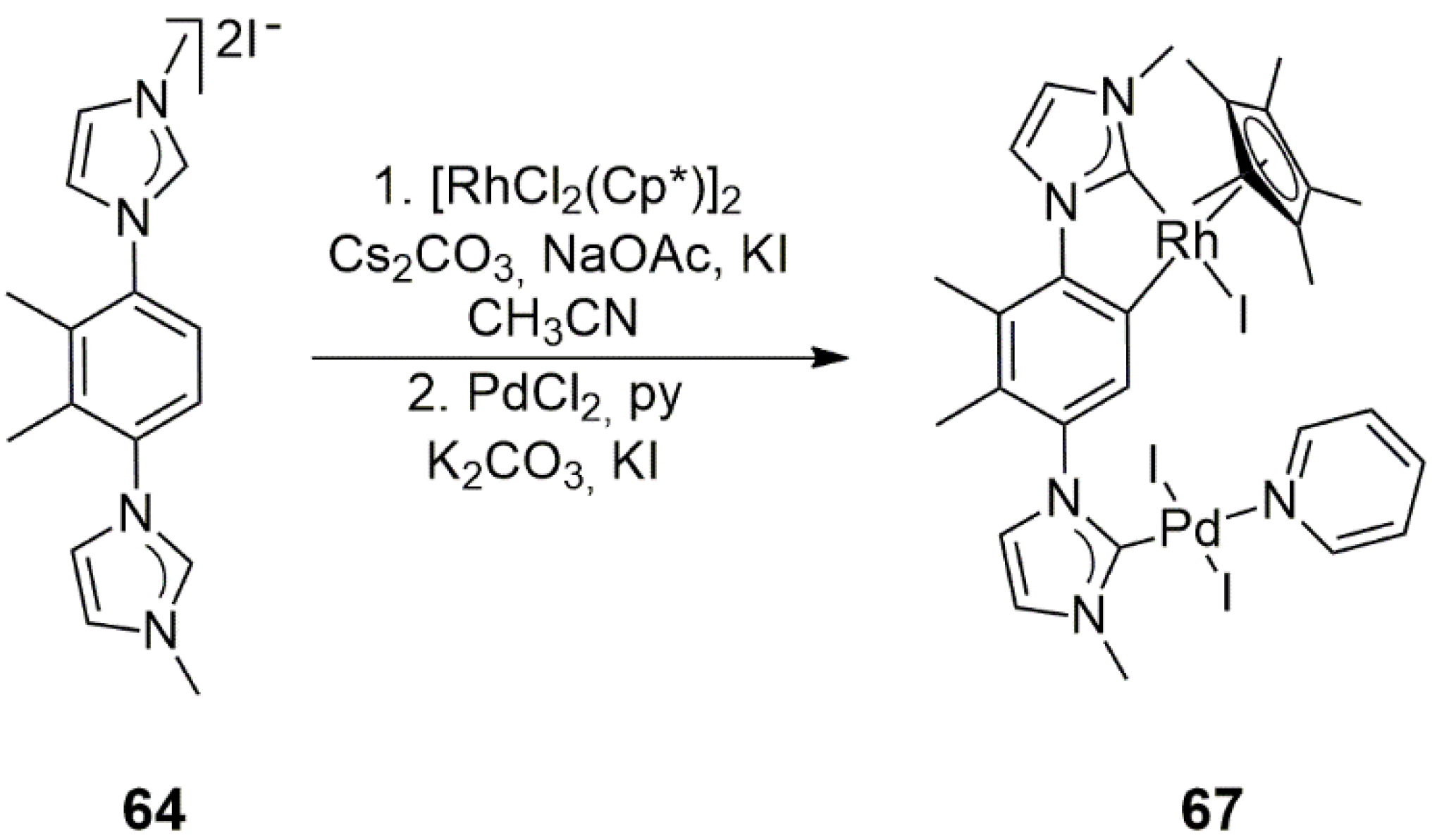
In continuation of their work described in [
39], following the same synthetic strategy, Majumder, Maity, et al. applied ligand
64 to the synthesis of the Rh/Pd analog
67 of the Ir/Pd complex
65 [
40]. Similar to
65,
67 also showed orthometalation (this time with the formation of a rhodacycle), contributing to the complex’s stability (
Scheme 21). Analogous complexes with 4-methoxypyridine and 4-chloropyridine were also obtained and thoroughly characterized by NMR and HRMS methods and, in several cases, by X-ray diffraction measurements. The catalysis of the tandem Suzuki–Miyaura cross-coupling/transfer hydrogenation of 4-bromobenzaldehyde with phenylboronic acid in 2-propanol/THF with KO
tBu as a base was attempted (
Scheme 6). With the Rh/Pd heterobimetallic bis-NHC complex,
67 showed only negligible activity (6% isolated yield of 1-(4-biphenyl)ethanol at 90 °C, 2 h).
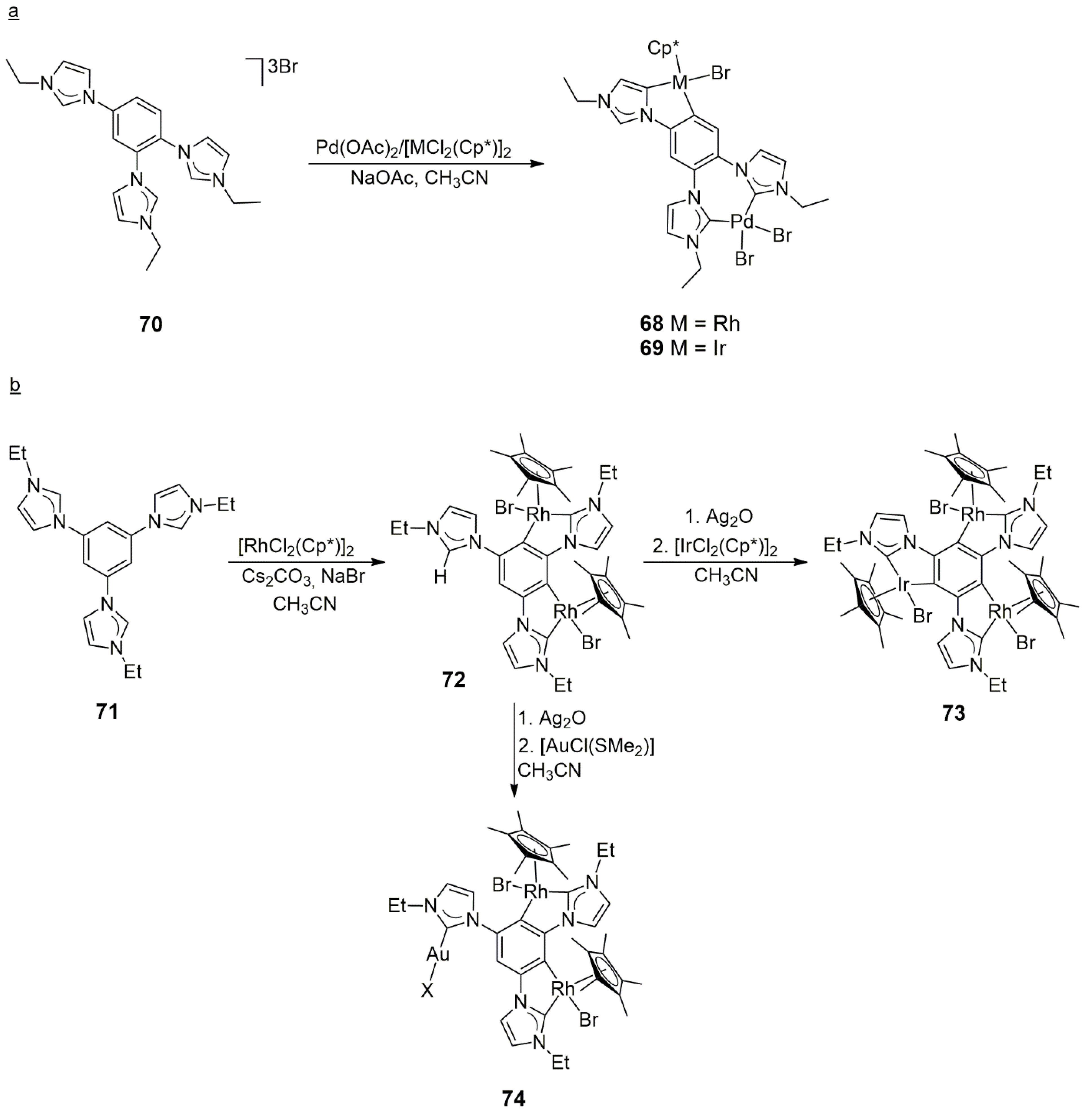
The heterobimetallic tris-NHC complexes
68 (Rh/Pd) and
69 (IrPd) were obtained by the Hahn group with the selective stepwise metalation of the non-symmetric 1,2,4-tris(imidazolium)-substituted tris-NHC ligand precursor
70 [
41]. In the reaction of
70 and Pd(OAc)
2 in DMF, a mononuclear chelate bis-NHC compound was formed, which could be further metalated (CH
3CN, Cs
2CO
3) with the use of [RhCl
2(Cp*)]
2 or [IrCl
2(Cp*)]
2 to yield
68 or
69, respectively. It was also discovered that the same heterobimetallic complexes could be obtained in a one-step reaction of
70 by employing Pd(OAc)
2 together with [RhCl
2(Cp*)]
2 or [IrCl
2(Cp*)]
2, this time in the presence of NaOAc instead of Cs
2CO
3 (
Scheme 22). Pd was always coordinated in a chelate fashion, while Rh or Ir was coordinated by a single NHC donor and orthometalated the phenyl ring. Thorough NMR and HRMS characterizations were supplemented by X-ray diffraction measurements, which showed an Ir-Pd through-space distance of 7.240 Å in
69. Although this metal–metal distance is rather large for cooperative catalysis, this work exemplifies the simple access to heterobimetallic complexes, which may be useful in organic transformations. However, no attempt was made to reveal the catalytic properties of
68 and
69.
Maity, Schulte to Brinke, and Hahn investigated the complexation properties of a symmetric tris-NHC ligand, which was obtained from the 1,3,5-trisimidazolium salt
71 (
Scheme 22) [
42]. The reaction of this precursor with [RhCl
2(Cp*)]
2 in MeCN at 65 °C in the presence of Cs
2CO
3 and NaBr resulted in complex
72, which contained two rhodacycles and one uncomplexed imidazolium unit. The further metalation of
72 was achieved with [IrCl
2(Cp*)]
2 or [AuCl(SMe)
2] (in both cases via the Ag
2O route) to yield Rh/Ir (
73) or Rh/Au (
74) heterobimetallic tris-NHC complexes, respectively. It is of interest that
73 is a triply orthometalated heterotrimetallic complex with three five-membered metalacycles fused to the central phenyl ring. From detailed NMR measurements, it was concluded that the metalation of the third imidazolium moiety did not affect the other two metals and that all metals behaved as independent electronic centers. No catalytic studies with the new heterobimetallic NHC complexes were reported.
2.2.3. Oxidative Addition Reactions in the Synthesis of Heterobimetallic NHC Complexes
The oxidative addition of a carbon–halogen bond of a suitable halogen-substituted N-heterocyclic compound onto a low-valent metal complex may yield N-heterocyclic carbene complexes. This possibility was extensively studied by the Hahn research group with the aim of developing versatile methods for the synthesis of heterobimetallic NHC complexes. Accordingly, the research described below did not include catalytic applications of the heterobimetallic products.
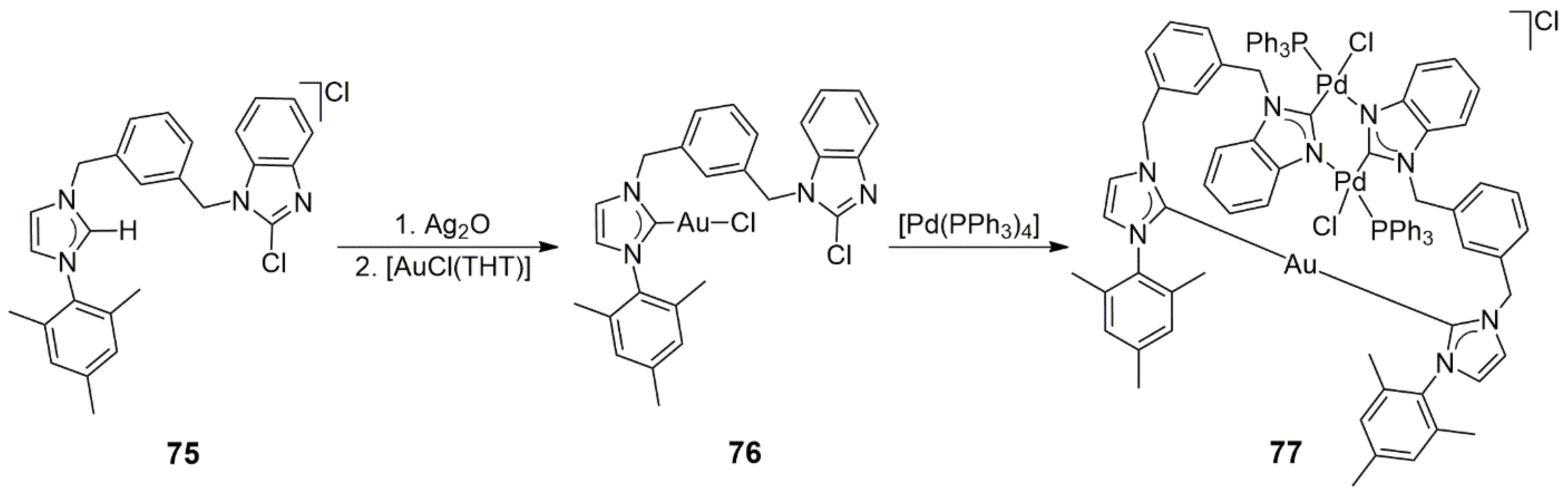
The
m-xylyl-bridged imidazolium/chlorobenzimidazole ligand precursor
75 gave the imidazolylidene–Au complex
76, in which the chlorobenzimidazole moiety remained available for metalation by oxidative addition to [Pd(PPh
3)
4] [
43]. The Pd/Au heterobimetallic product
77 could be isolated both as the chloride (29%) and as the PF
6 salt (56%). An alternative pathway for the synthesis of
77 included the formation of the gold complex of imidazolium-2-ylidene in the first step, followed by the oxidative addition of the C-Cl bond to [Pd(PPh
3)
4]. This example clearly shows the possibility of sequential site-selective metalation due to the different reactivities of the N-CH-N and N-C(Cl)-N groups (
Scheme 23).
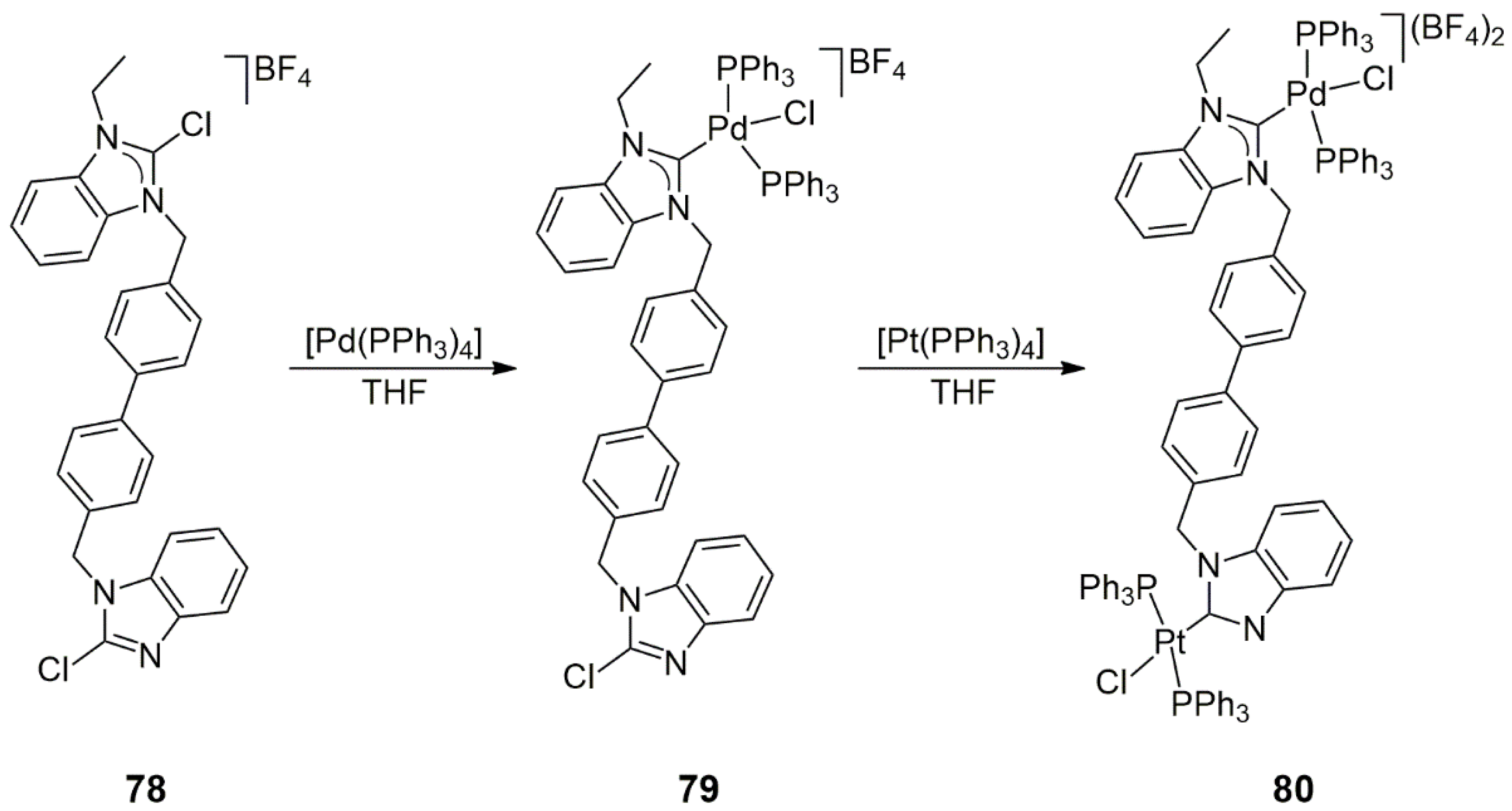
Regioselectivity in double oxidative addition reactions of bis-NHC precursors also resulted in heterobimetallic bis-NHC complexes [
44]. In the reaction of the 4,4′-
p-biphenylene-bridged chlorobenzimidazole/ethylchlorobenzimidazolium compound
78 with one equivalent of [Pd(PPh
3)
4], the monopalladium NHC complex
79 was formed selectively in the oxidative addition reaction of the 1,3-dialkyl-2-chlorobenzimidazolium moiety (3 h, 75 °C, THF). In a slower reaction (18 h, 100 °C), [Pt(PPh
3)
4] underwent oxidative addition at the non-alkylated benzimidazole part, yielding the bimetallic Pd/Pt complex
80 in a 24% yield (
Scheme 24). The analogous Pd/Pd bis-NHC complex could be obtained in the reaction of
78 with [Pd(PPh
3)
4] under somewhat milder conditions (18 h, 75 °C) in a 65% yield, showing the higher reactivity of Pd(0) compared to Pt(0) in oxidative additions.
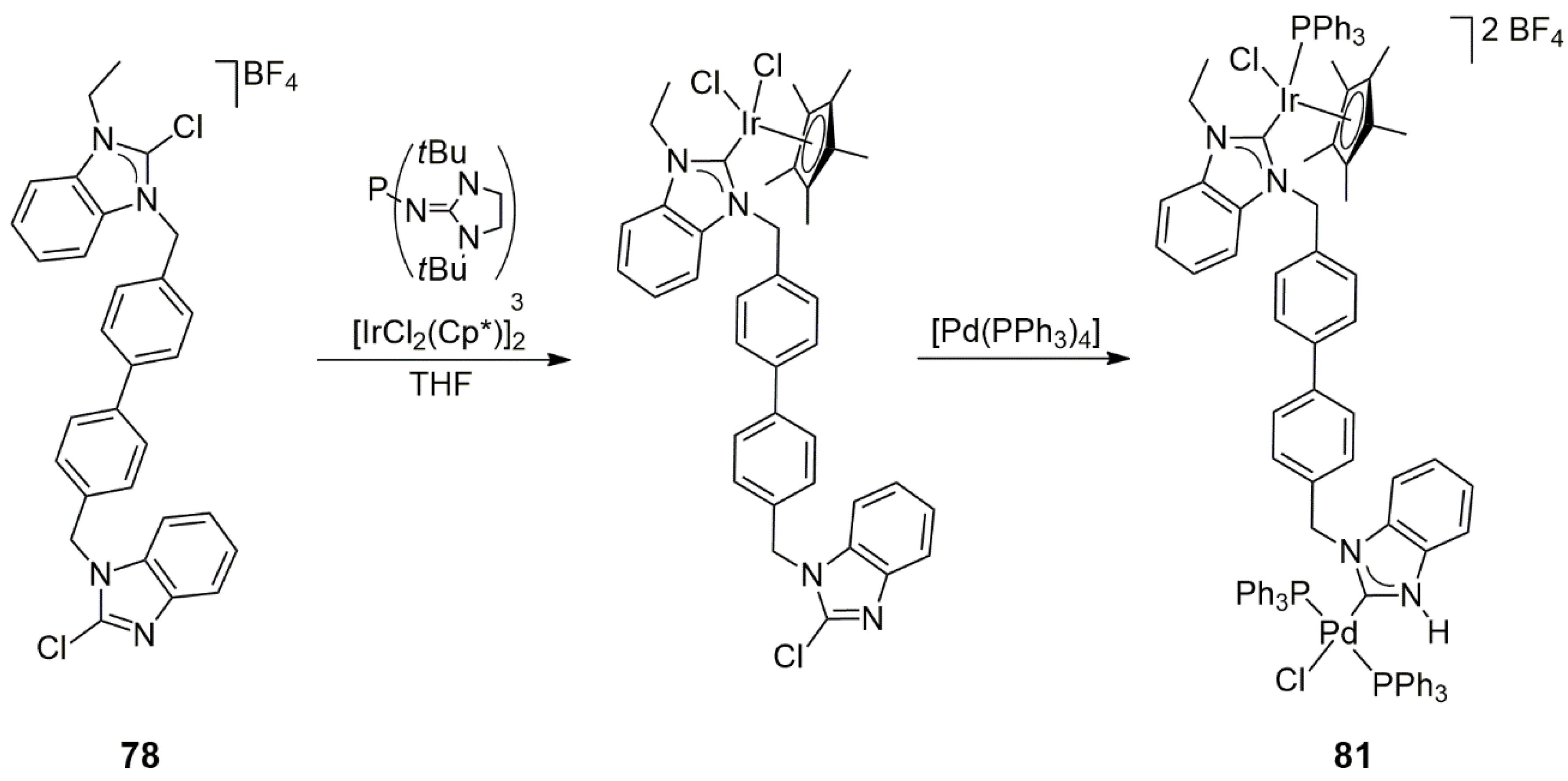
The doubly chlorosubstituted bis-NHC precursor
78 served as the starting material for mono- and heterobimetallic NHC complexes via another synthetic route, too [
45]. Although, in general, electron-rich tertiary phosphines are known to dehalogenate chloroimidazolium salts, the sterically encumbered tris(1,3-
tert-butylimidazolidin-2-ylidenamino)phosphine (
Scheme 25) was found to be particularly useful for the selective removal of chloride from 2-chloroazolium salts. Consequently, it was possible to react the linked 2-chlorobenzimidazole/2-chloro-3-ethylbenzimidazolium salt first with the mentioned phosphine, resulting in the exclusive metalation of the N-ethyl-benzimidazolium part, and then to perform oxidative addition at the chlorobenzimidazole end (
Scheme 25). This strategy led to the isolation of an Ir/Pd heterobimetallic complex,
81, but it has more general utility for obtaining mixed-metal NHC complexes.
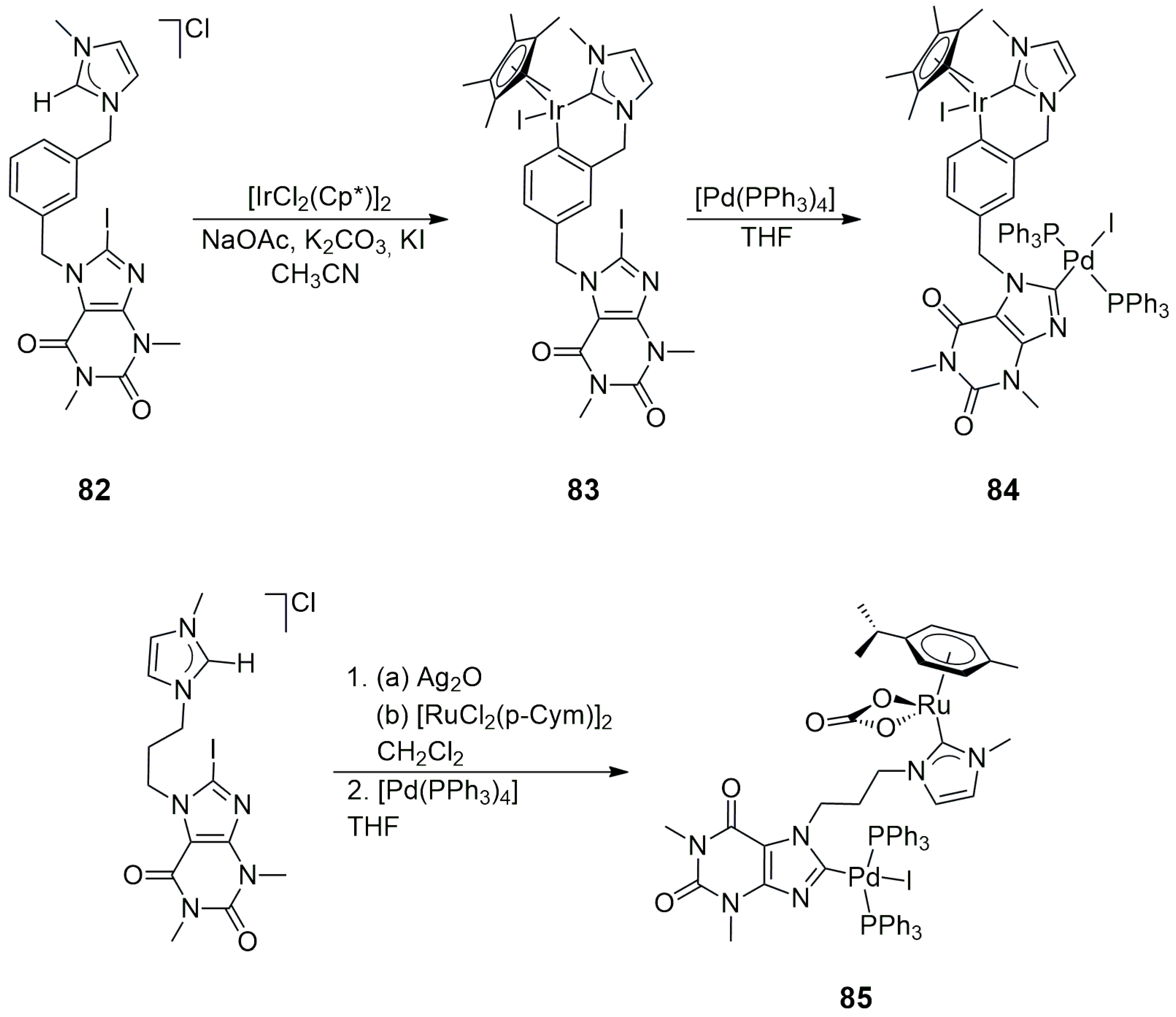
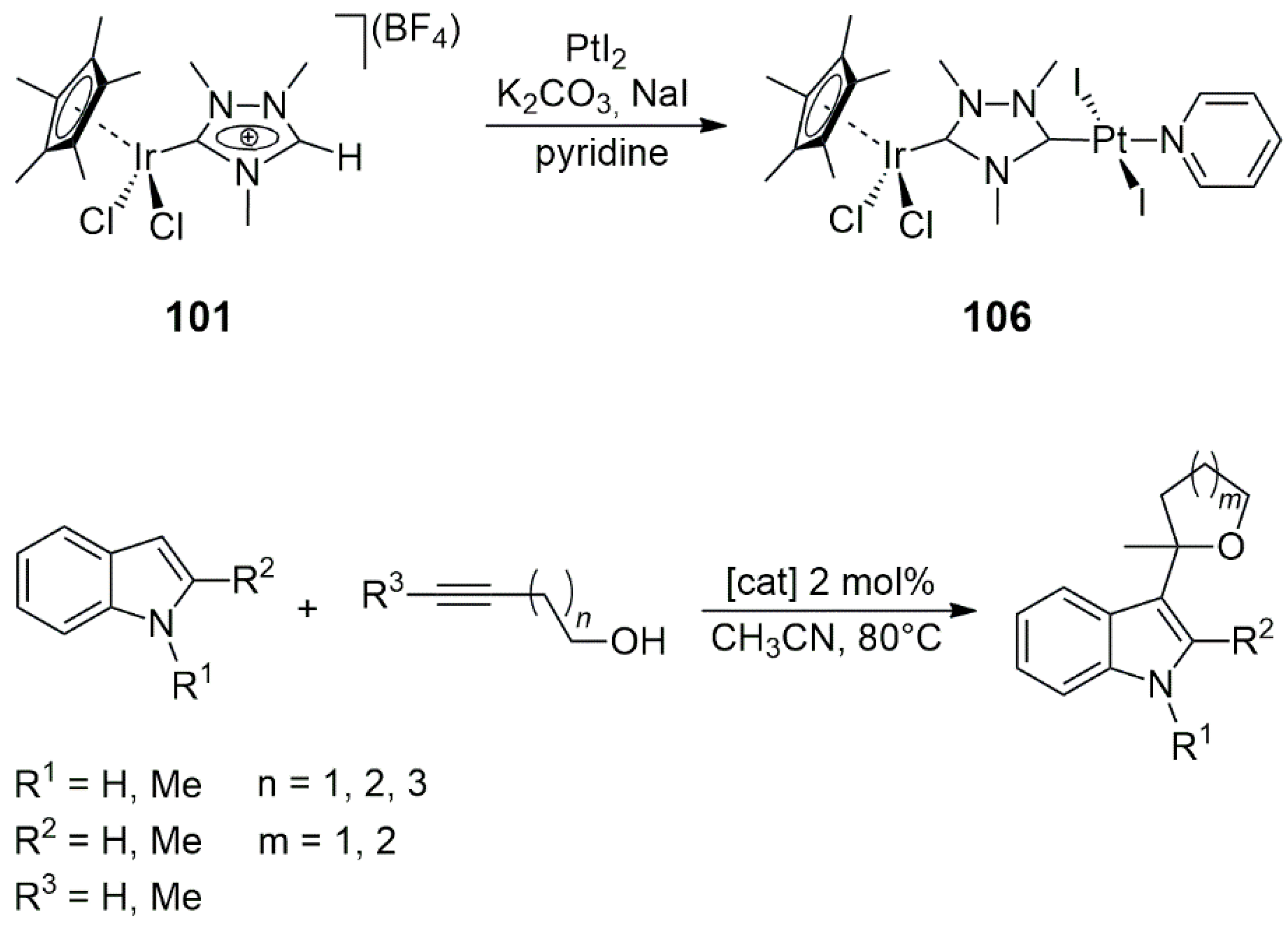
In the bis-NHC precursor
82, an N-ethylimidazolium group and an 8-iodotheophylline unit were joined by an
m-xylyl linker. This NHC precursor was cleanly monometalated at the imidazolium part with [IrCl
2(Cp*)]
2, and the resulting cyclometalated Ir(III) complex
83 was transformed into the Ir/Pd heterobimetallic bis-NHC product
84 under mild conditions (25 °C, in 1 day) (
Scheme 26). The Ru/Pd heterobimetallic compound
85 was similarly obtained by the metalation of the imidazolium part of the propyl-bridged precursor with Ag
2O/[RuCl
2(η
6-
p-cymene)]
2 (
p-cymene = 4-isopropyltoluene), followed by the oxidative addition of C(8)-I in the 8-iodotheophylline unit to [Pd(PPh
3)
4] (
Scheme 26) [
46]. The coordinated carbonato ligand was formed by the oxidation of the CH
2Cl
2 solvent in the presence of silver salts.
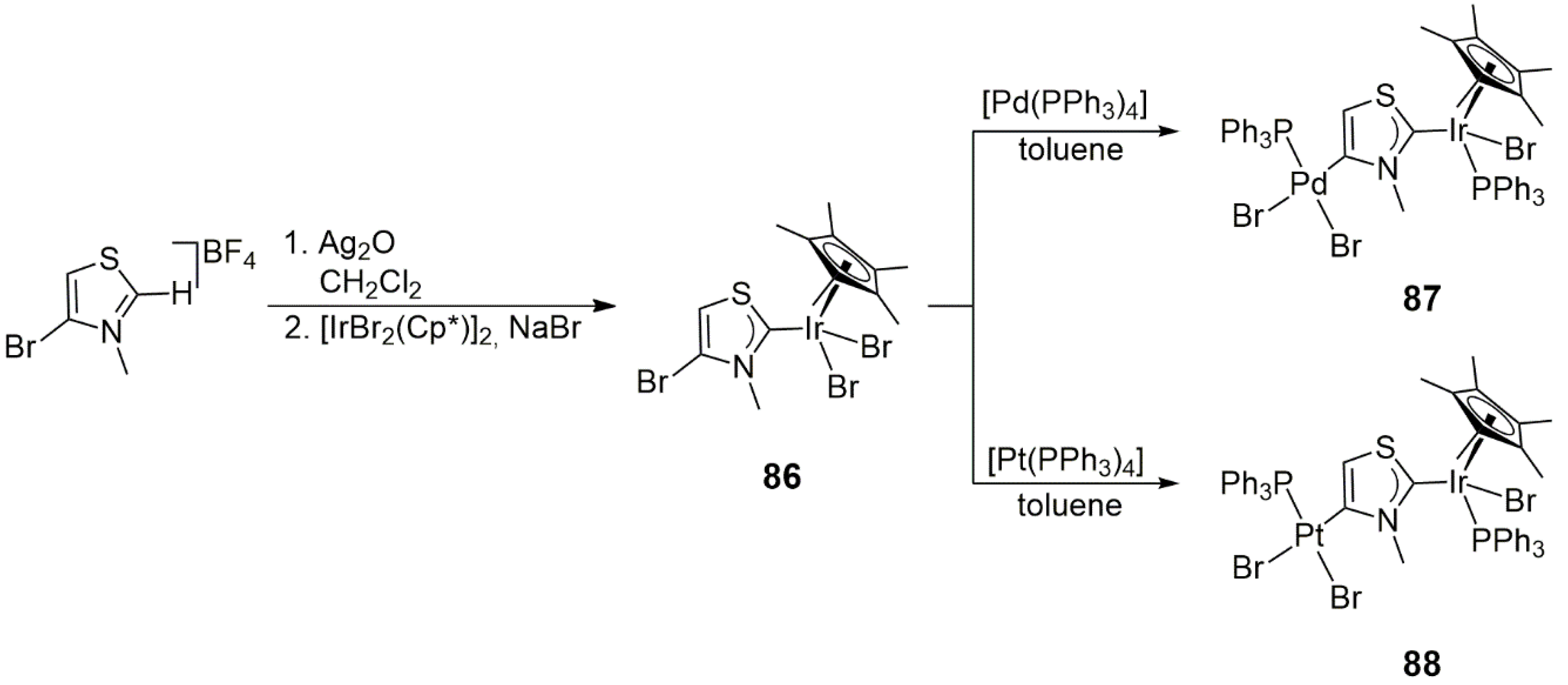
In another study [
47], 4-bromo-3-methylthiazolium tetrafluoroborate was first metalated with [IrBr
2(Cp*)]
2 with the use of the Ag
2O method. This reaction yielded a mononuclear Ir-NHC complex,
86, in which Ir was bound to the carbenic C2 atom of the heterocycle (
Scheme 27). The subsequent oxidative addition of the C4-Br bond to Pd(0) in [Pd(PPh
3)
4] at 90 °C in 24 h resulted in the heterobimetallic Ir/Pd complex
87. Note that the overall reaction included a Br
−/PPh
3 exchange between the Ir and Pd centers. Under similar conditions, but at a temperature of 110 °C, the reaction of [Pt(PPh
3)
4] afforded the analogous Ir/Pt heterobimetallic complex
88, also with a Br
−/PPh
3 exchange between the two metal centers. The reaction of the substitutionally inert [Pt(PPh
3)
4] for only 4 h allowed the isolation of the direct product of oxidative addition, an intermediate en route to
88, still featuring a C4-carbene-bound {PtBr(PPh
3)
2} moiety.
2.2.4. Pendant Group Coordination in the Synthesis of Heterobimetallic NHC Complexes and Their Catalytic Properties
N-heterocyclic carbenes containing a pendant donor group may show altered coordination (and catalytic) properties compared to their counterparts with no such substituents. Such pendant groups may serve as an anchor to facilitate the coordination of the N-heterocyclic carbene ligand, stabilize certain steric coordination geometries, and alter the electronic properties of the metal center in the carbene complex compared to complexes with exclusively NHC coordination. A case in point is the formation and catalytic properties of complexes obtained with the use of the NHC precursor
89 (
Scheme 28), which was studied in much detail by Kühn, Baratta, and co-workers.
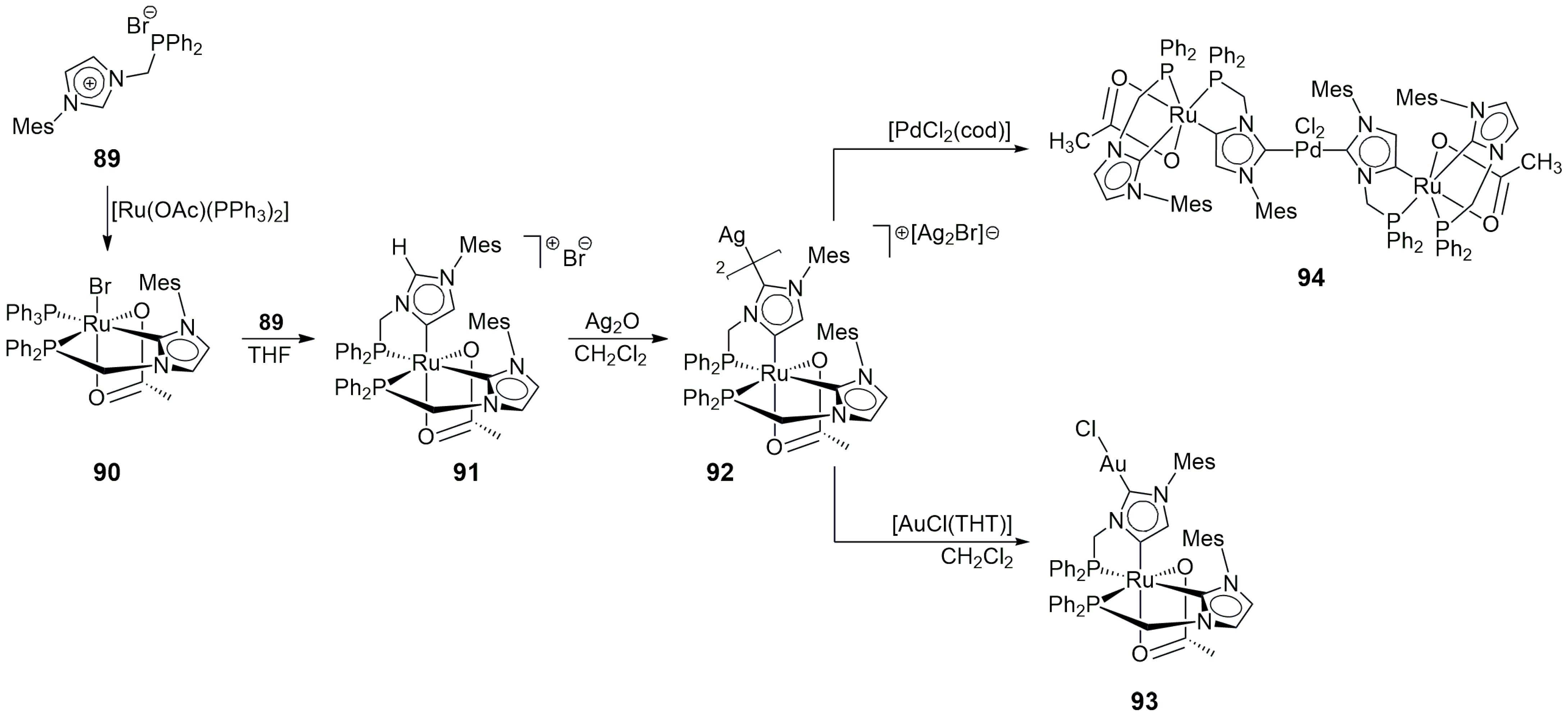
The reaction of [Ru(OAc)
2(PPh
3)
3] with one equivalent of
89.HBr in the presence of NaOAc in
tert-BuOH led to the clean formation of the mononuclear normal Ru(II)-NHC complex
90. However, a second equivalent of
89 yielded a cationic Ru(II)-NHC product
91, in which the second NHC ligand is coordinated on its C4-position (abnormal NHC) [
48]. The formation of such a normal/abnormal NHC-coordinated metal complex may be due to the steric requirements of the bulky mesityl groups in
89, together with the strong coordination of the substituent –PPh
2 group to Ru(II). The treatment of
91 with Ag
2O afforded the cationic dimer
92 ([AgBr
2]
– as a counterion), with the two
91 units connected by the bis-NHC coordination of Ag(I), which—at the same time—transformed the originally abnormal NHC moieties into N-heterocyclic dicarbene (NHDC) ligands (
Scheme 28). Compound
92 was the first example of an NHDC complex containing two different d-block elements.
Compound
92 proved to be a versatile starting material for the preparation of heterobimetallic NHDC complexes. The reaction of this dimer with [AuCl(THT)] gave the monomeric neutral Ru/Au compound
93, while with [PdCl
2(cod)], the neutral Pd/Ru dimer
94 was formed, with a structure strictly analogous to that of
92 [
49].
The monometallic 91 displayed very high catalytic activity in the transfer hydrogenation of various ketones, such as acetophenone, benzophenone, and cyclohexanone, to the respective alcohols. The reactions took place with a 0.1% catalyst loading in refluxing 2-propanol with NaOiPr as a base; turnover frequencies up to 49,000 h−1 were achieved.
Catalyst
94 was studied in the tandem Suzuki–Miyaura
C–
C cross-coupling of various bromoacetophenones and boronic acids/transfer hydrogenation of the resulting ketones (
Scheme 6a). In highly basic 2-propanol solutions, Suzuki–Miyaura cross-coupling was accompanied by extensive dehalogenation. However, under optimized conditions in toluene/2-propanol as a solvent and KOH as a base, yields of the respective alcohols in the range of 93–98% were obtained (according to GC or NMR analysis), showing the high functional group tolerance of the catalyst. In the reaction of 4-bromoacetophenone and phenylboronic acid, the time course of the reaction displayed the fast formation of the
C–
C-coupled product (acetylbiphenyl), followed by a slower transfer hydrogenation to 1-(4-biphenyl)ethanol. Under the same conditions, an equimolar mixture of
91 + [PdCl
2(IMes)
2] produced a lower yield of 1-(4-biphenyl)ethanol (76% vs. 95 with
94) and the formation of several unidentified byproducts, thereby demonstrating the superior selectivity of the heterobimetallic catalyst
94 in this tandem Suzuki–Miyaura coupling/transfer hydrogenation.
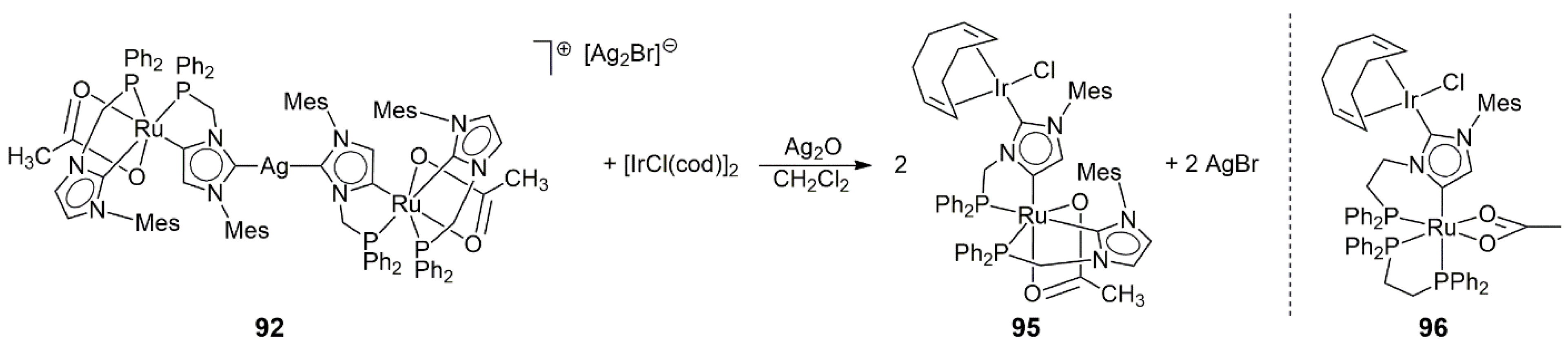
The Ag(I)-linked cationic dimer
92 was applied to the synthesis of the neutral Ru/Ir heterobimetallic complex
95, too, through its reaction with [IrCl(cod)]
2 via the Ag
2O route in CH
2Cl
2 (
Scheme 29) [
50]. With the use of cyclic voltammetry measurements, it was found that the Ru(II)/Ru(III) redox process occurred at a 70 mV lower potential in
95 than in
92, suggesting an interaction between the Ir and Ru centers. For comparison, the normal/abnormal (NHDC) dimetalated (Ir/Ru) doubly phosphine-coordinated complex
96 was also prepared (
Scheme 29). The solid-state structures of all new compounds were determined by X-ray crystallography.
The catalytic activities of
91,
95, and
96 in the transfer hydrogenation of acetophenone were compared and are displayed in
Table 1.
It could be concluded from the data in
Table 1 that the coordination of a second metal (Ir) decreased the catalytic activity of the mononuclear normal/abnormal Ru(II) NHDC complex to a large extent. Similar to the results of cyclic voltammetry, this also indicates a strong metal–metal interaction; however, in this case, the interaction manifests itself in a large drop in catalytic activity. These data indirectly suggest that the N-C
H-N imidazolium proton in
91 could play an important role in catalysis.
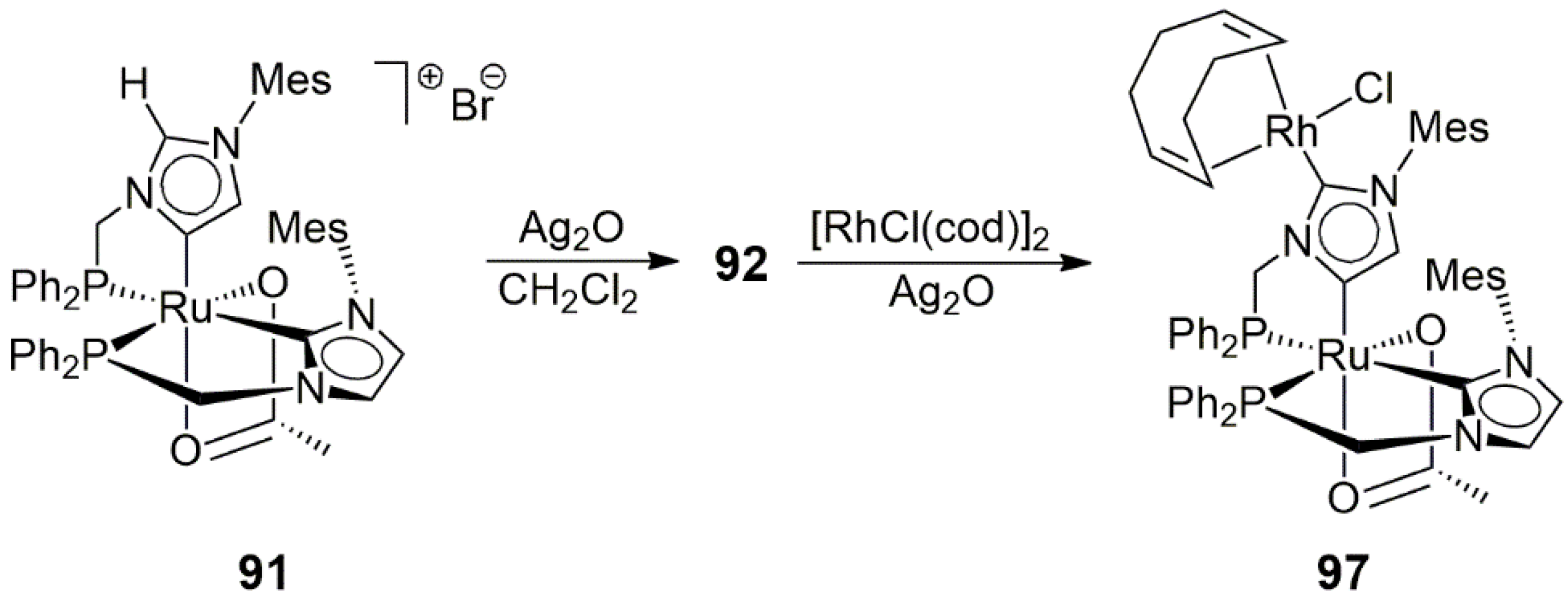
Kühn and co-workers metalated
91 with the use of Ag
2O and then [RhCl(cod)
2] to afford the Ru/Rh heterobimetallic NHDC complex
97 (
Scheme 30) (solid-state structure determined by X-ray diffraction) with the intermediate formation of the Ag-linked dimer
92 [51]. The catalytic efficiency of
97 was tested in the transfer hydrogenation of acetophenone with basic 2-propanol as a H-donor (0.1 mol% catalyst, 2 mol% NaO
íPr, 80 °C). In 25 min, the reaction reached equilibrium, which allowed the calculation of a TOF = 5700 h
−1. Under comparable conditions, the activity of the Ru/Ir complex
95 could be characterized by TOF = 6700 h
−1 (
Table 1). A comparison of these data shows that, similar to the case of
95, a significant drop in catalytic activity occurred upon the coordination of a second metal (Rh) to the mononuclear normal/abnormal bis(imidazolylidene)Ru(II) complex. However, the loss of catalytic activity was more or less the same in the case of the two complexes
95 and
97, which may indicate the decisive role of the Ru(II) center in the studied complexes in the catalysis of acetophenone transfer hydrogenation.



















































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
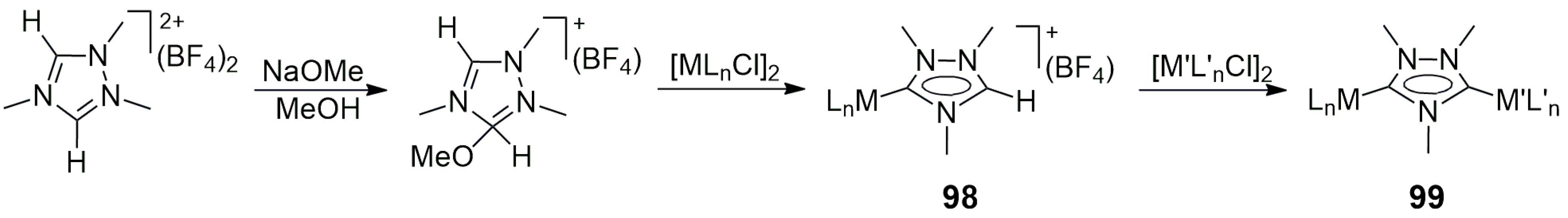{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
