

Visible-Light Photoredox Catalysis for the Synthesis of Fluorinated Aromatic Compounds

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
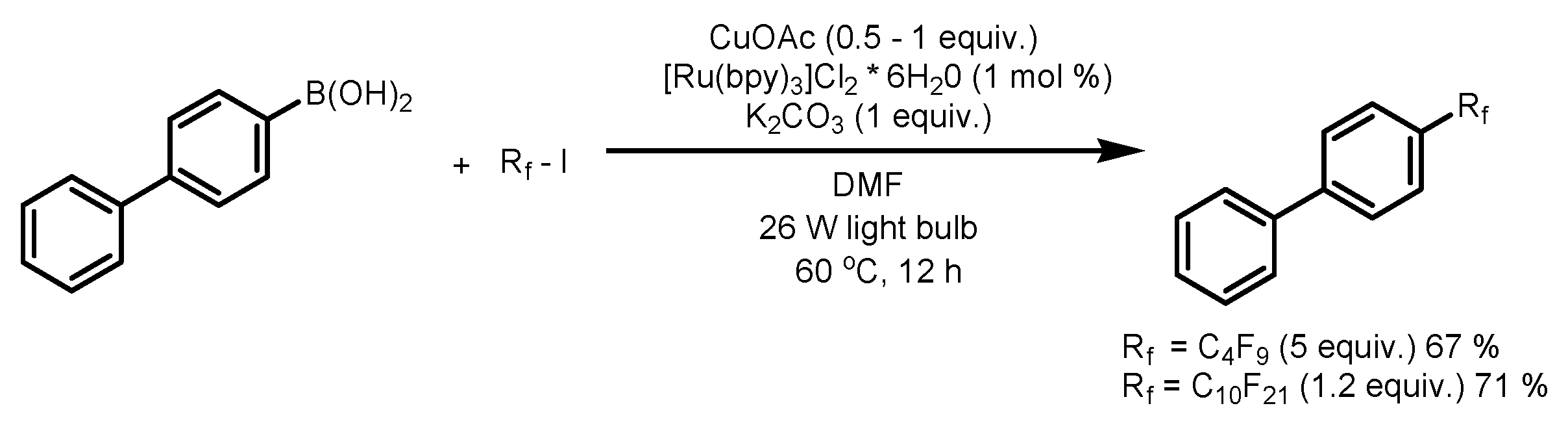{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
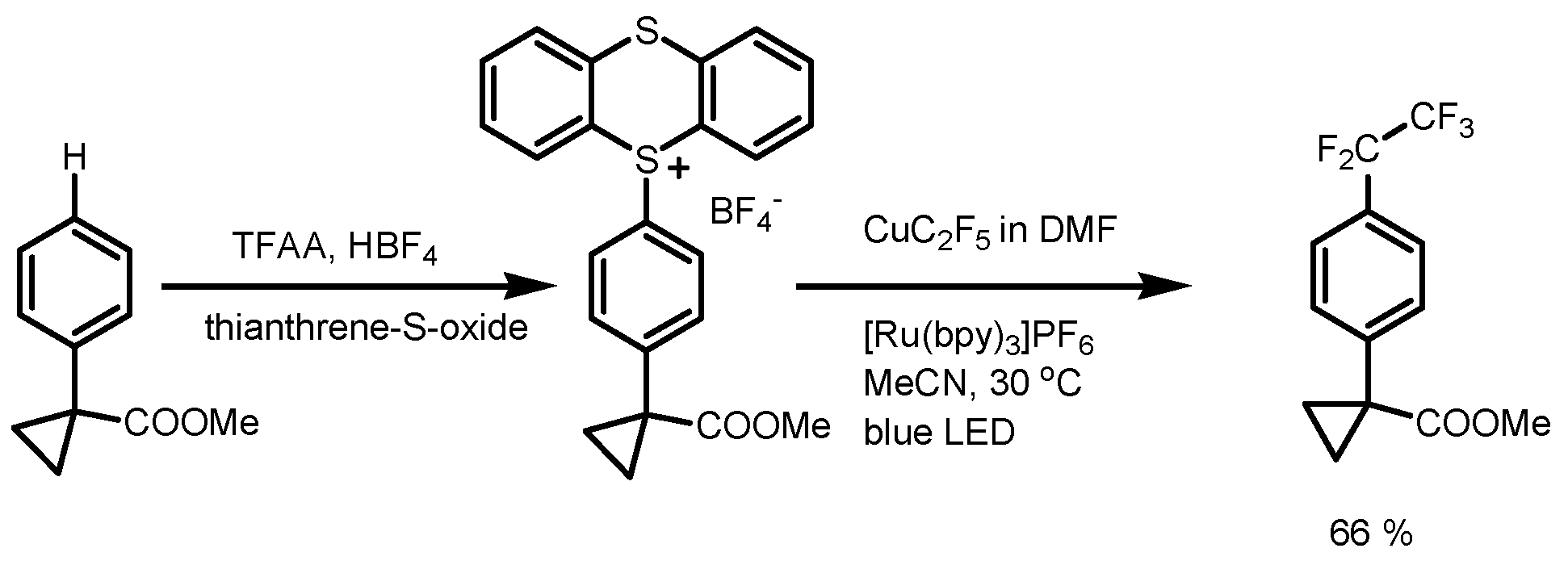
2. Trifluoromethylation of Arenes
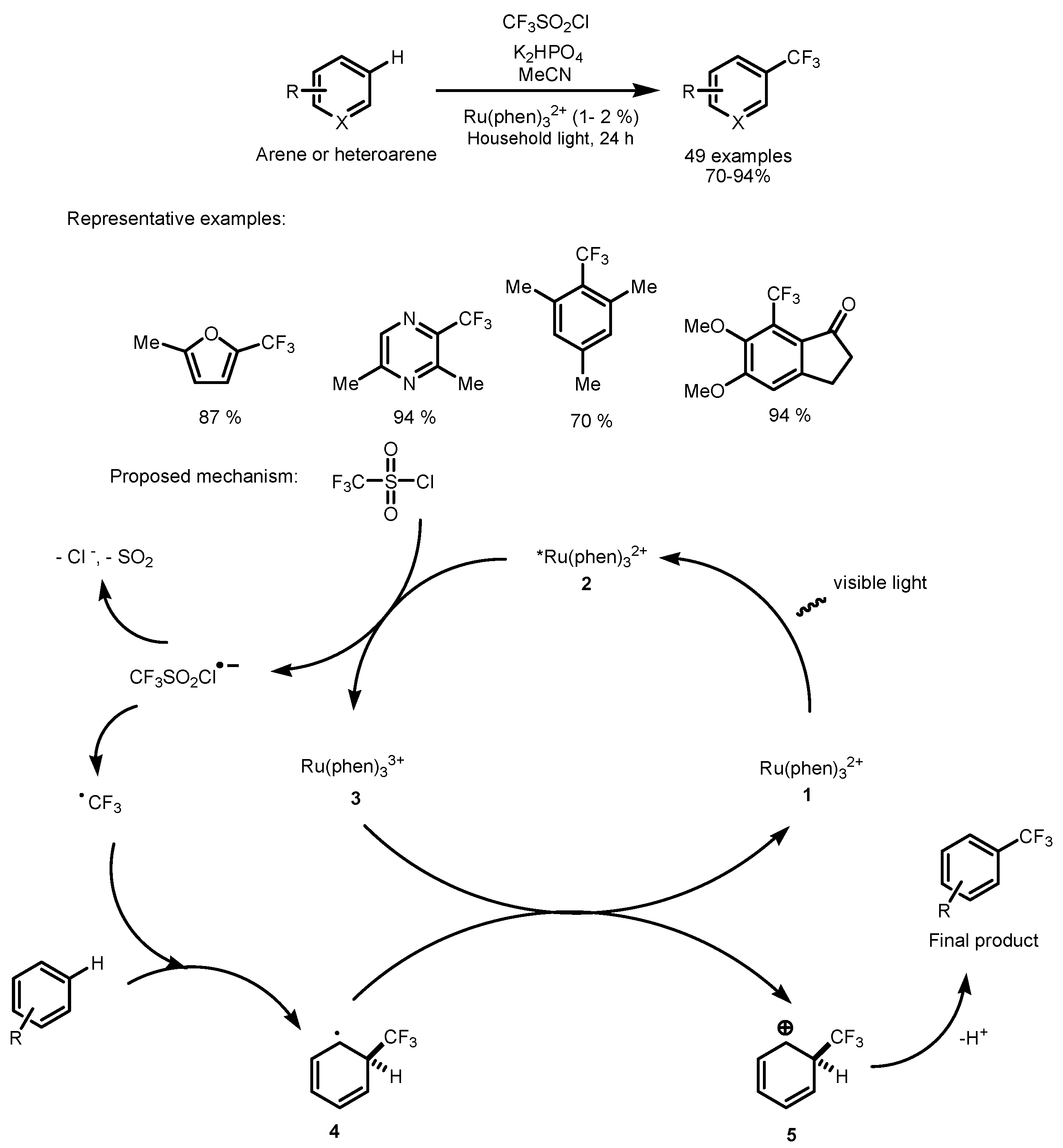
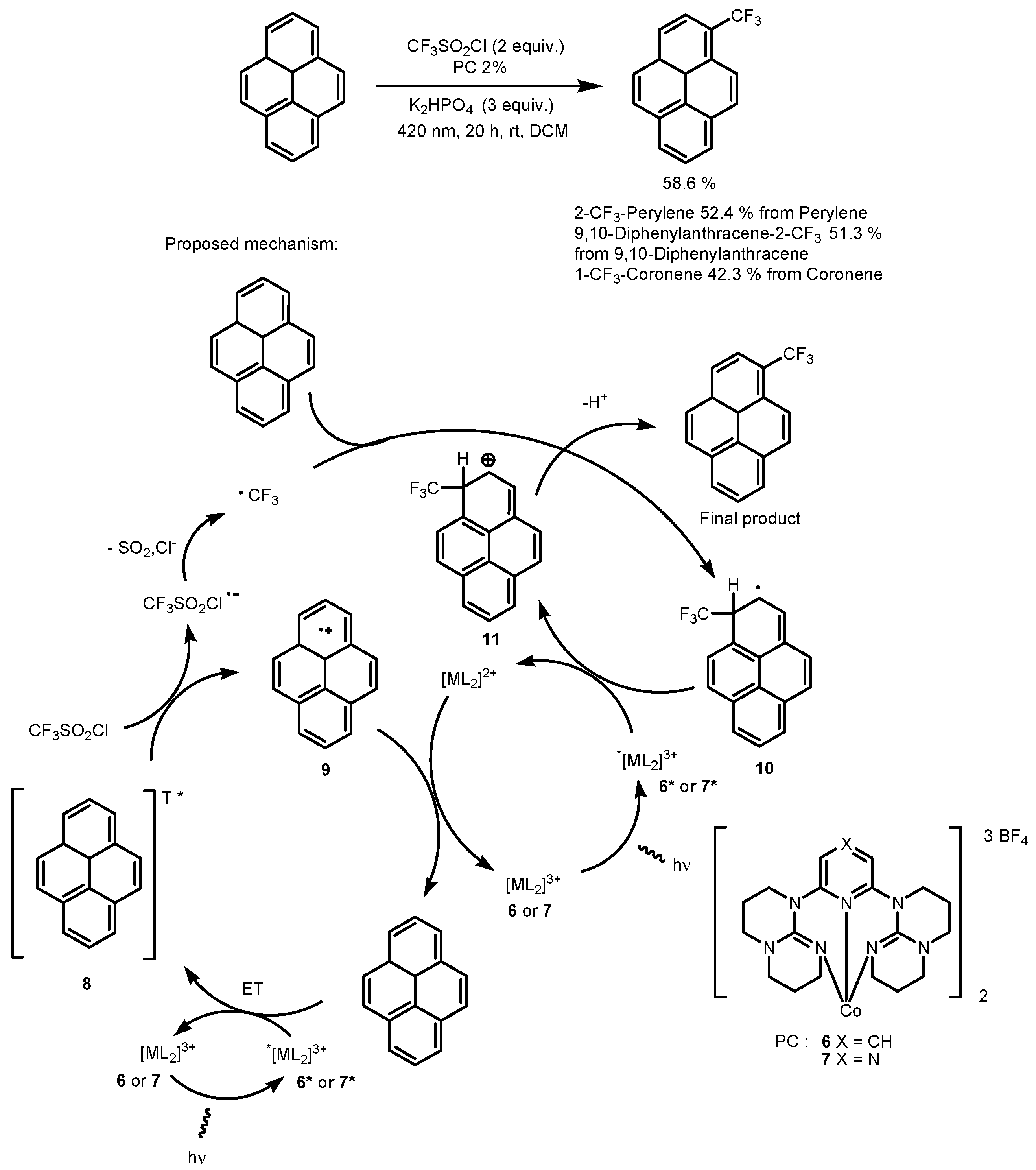
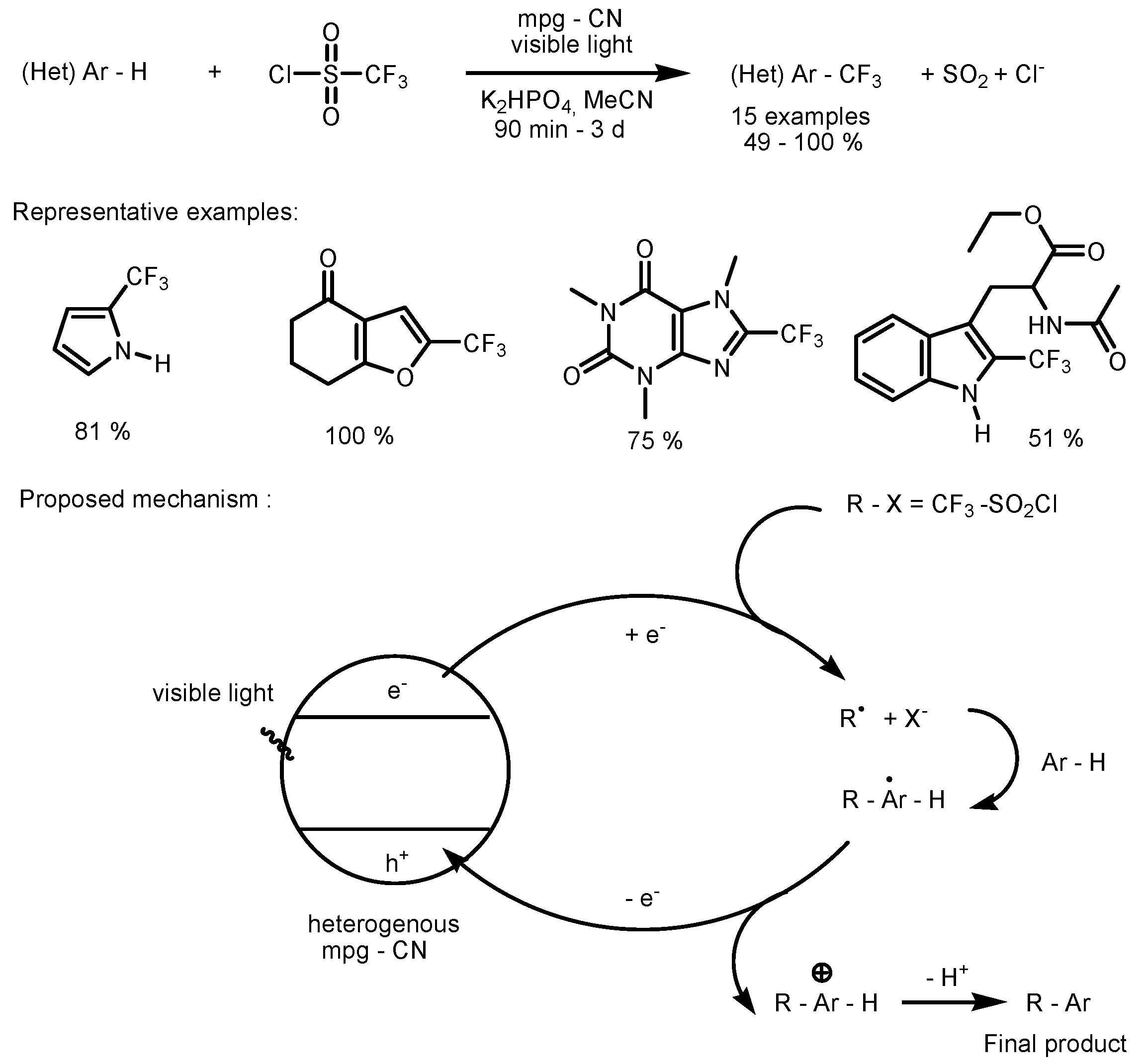
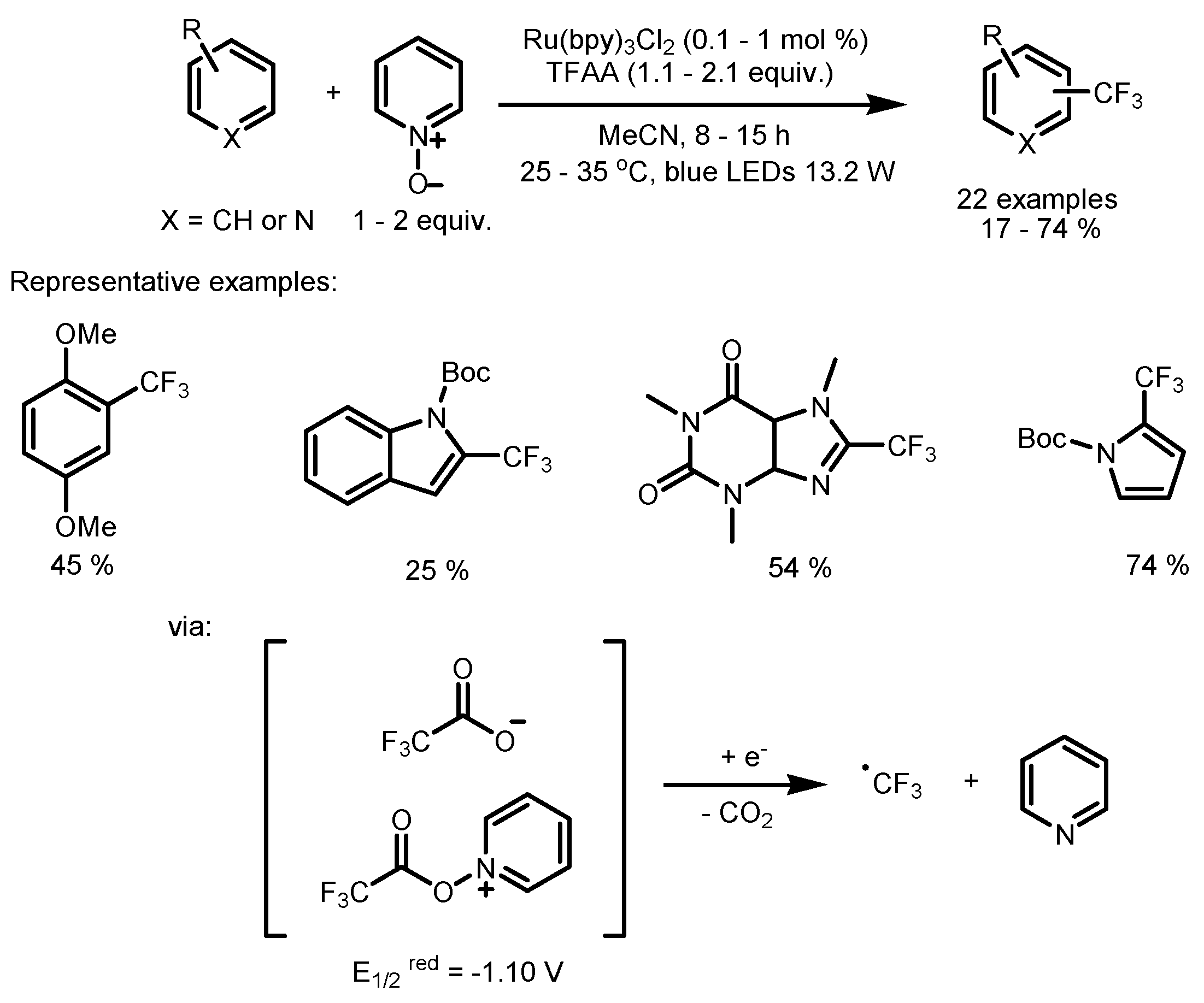
2.1. CF3SO2Cl as the Fluorinating Reactant
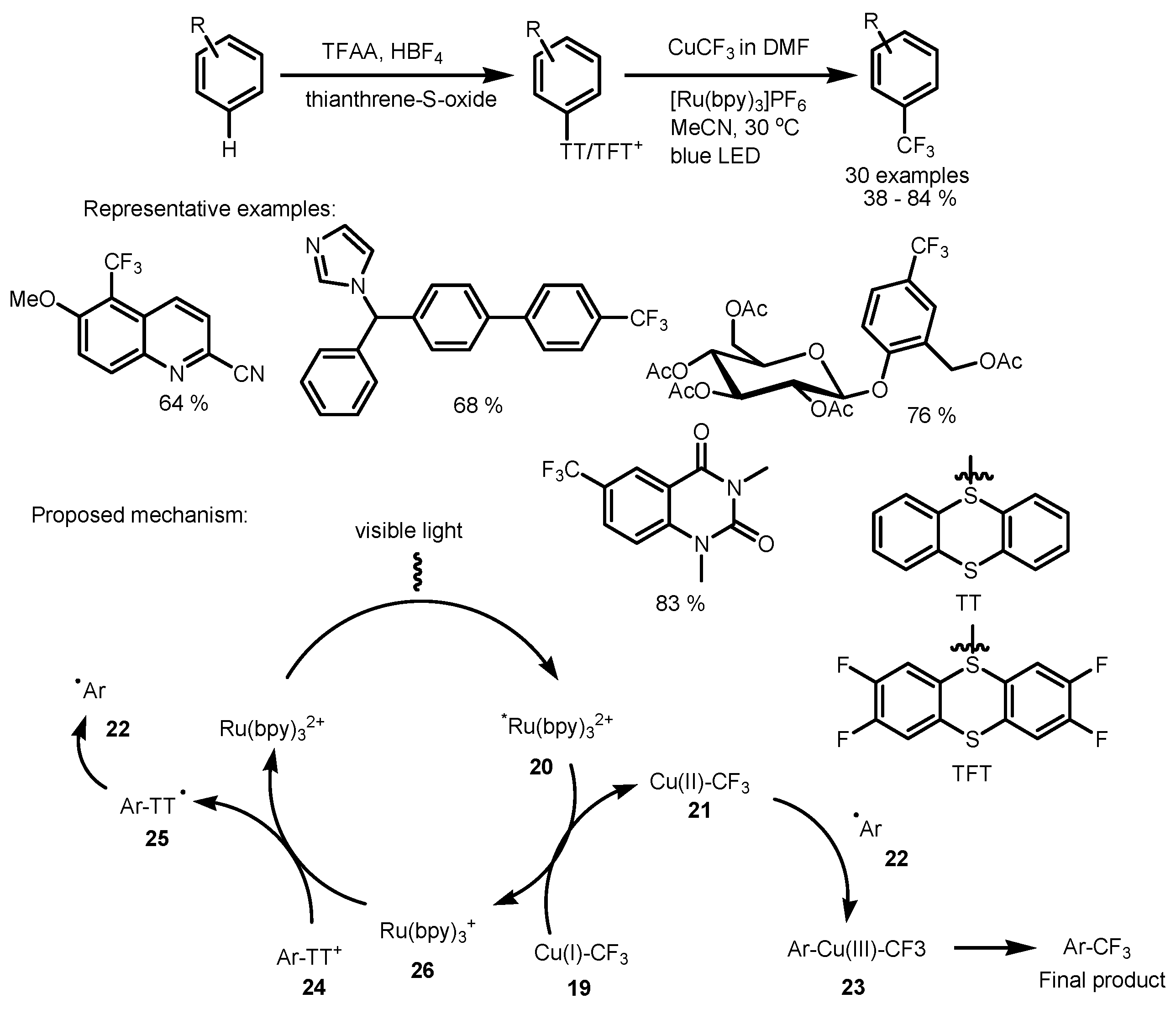
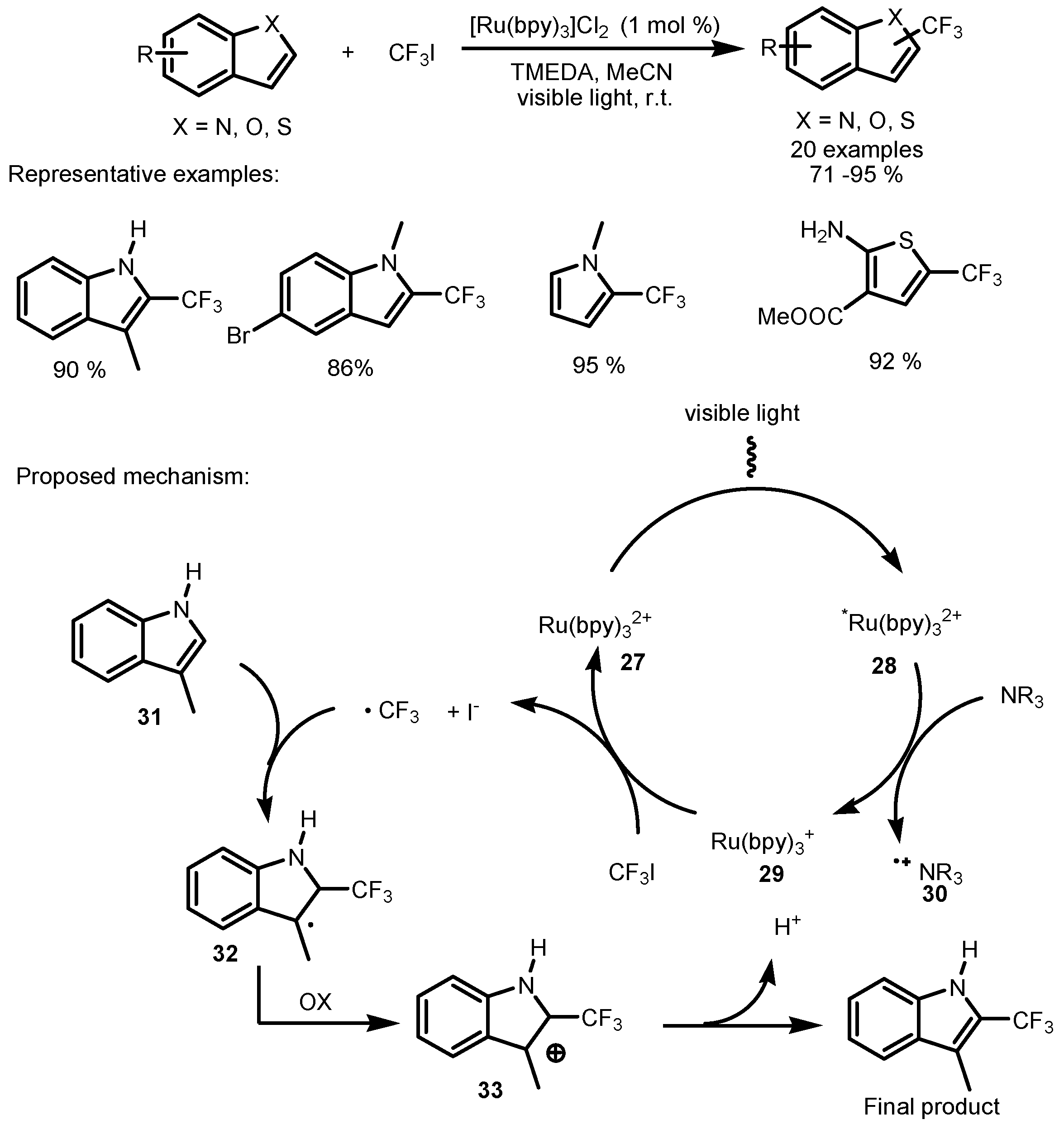
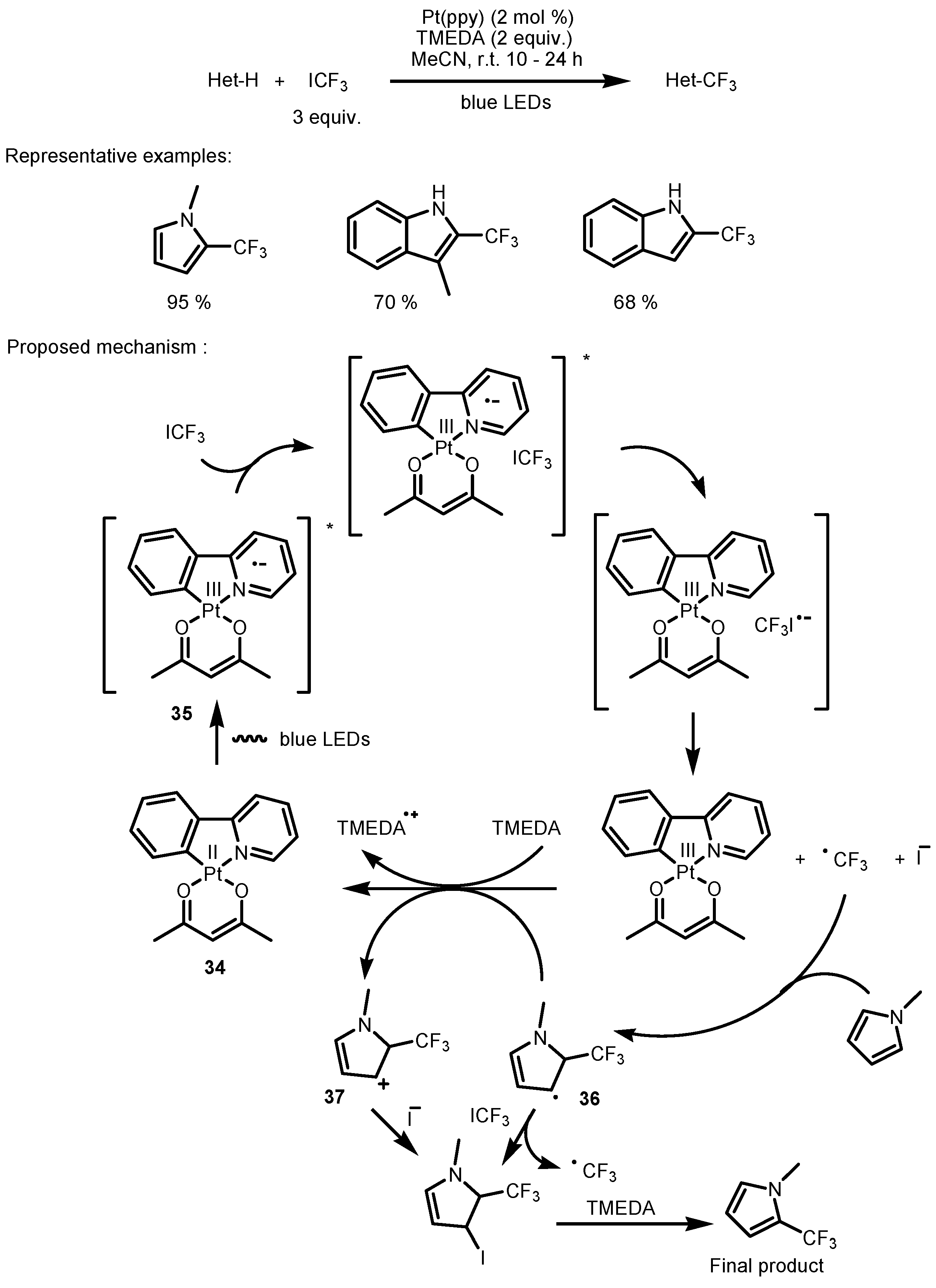
2.2. CF3I as the Fluorinating Reactant
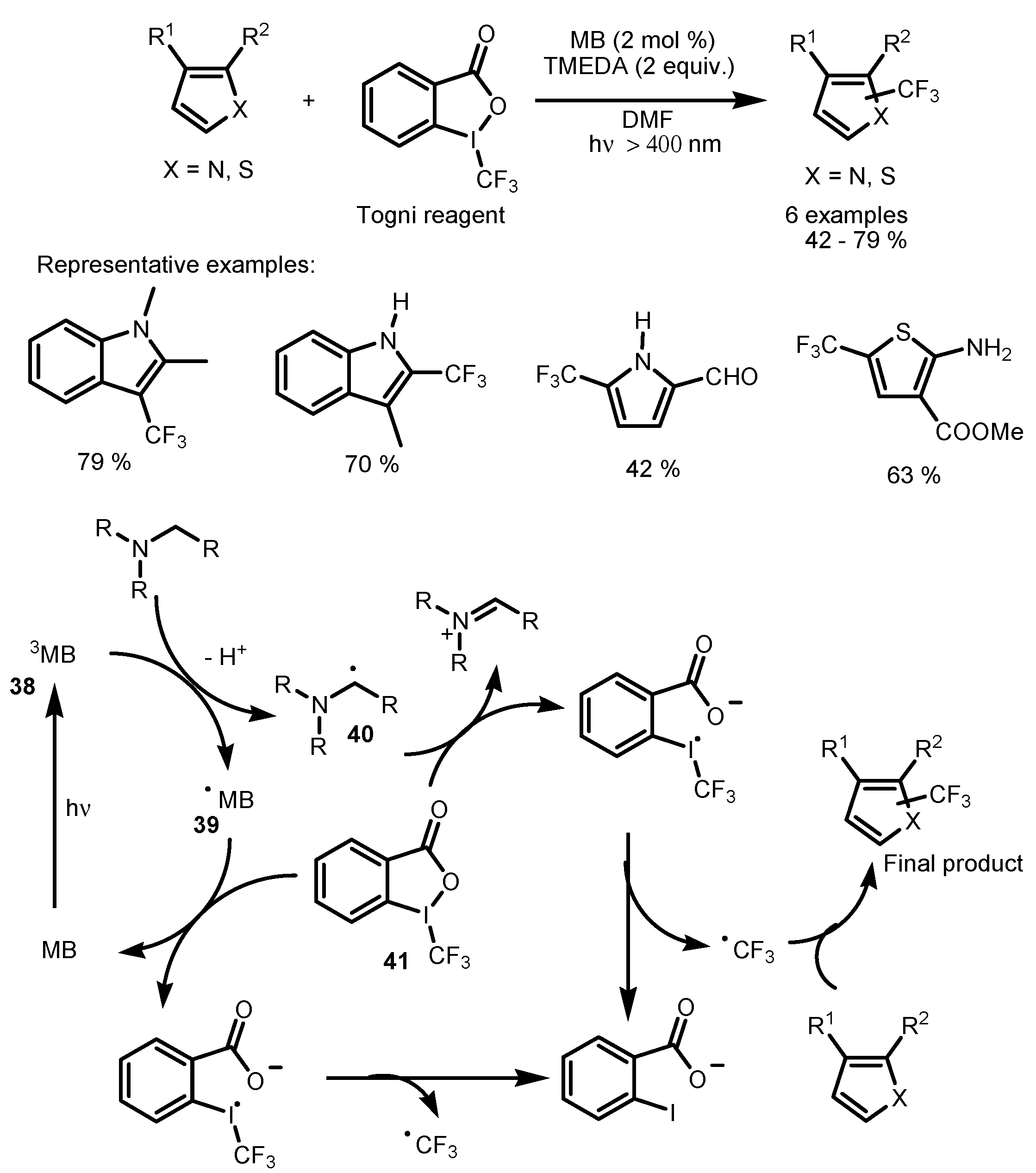
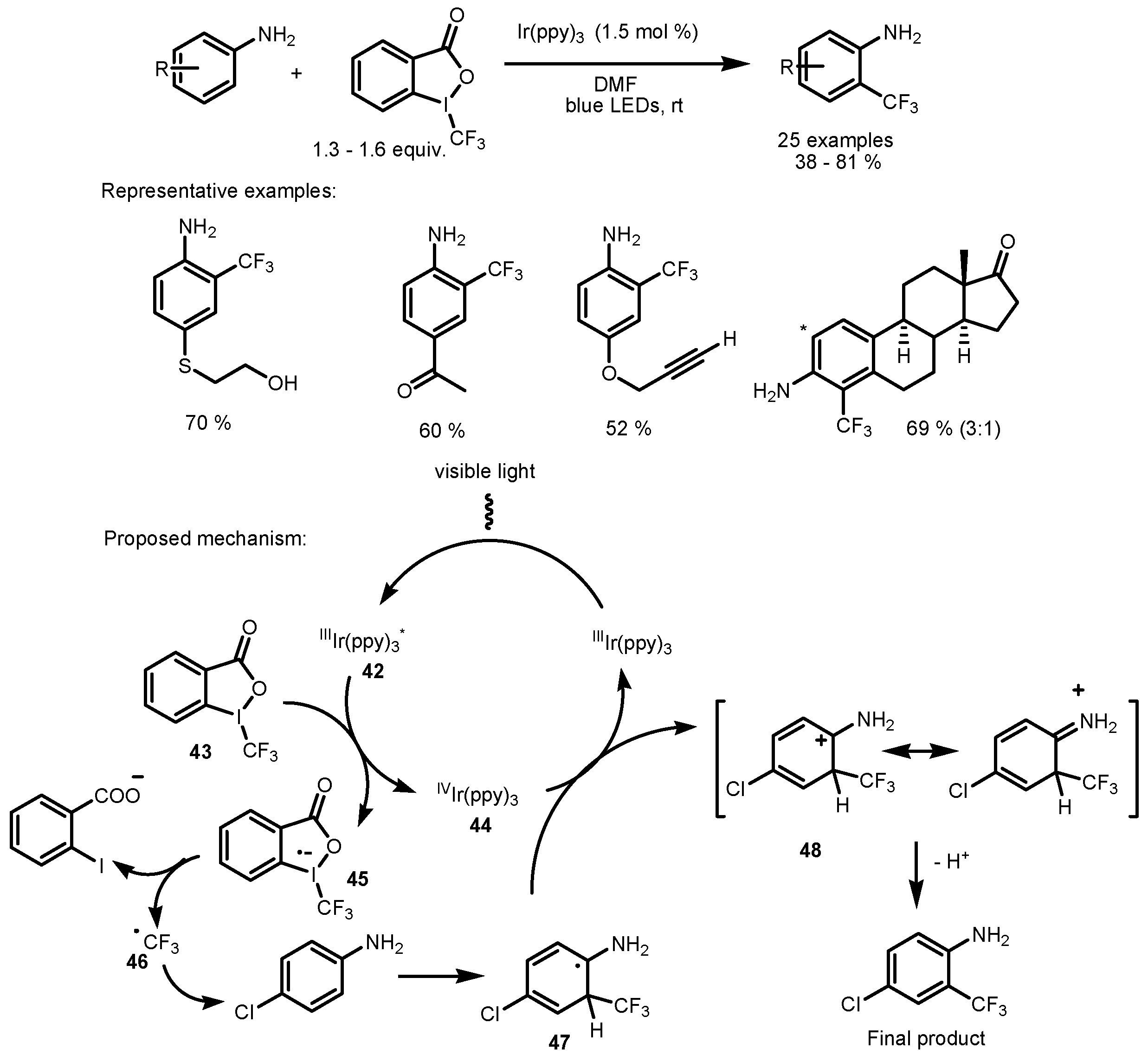
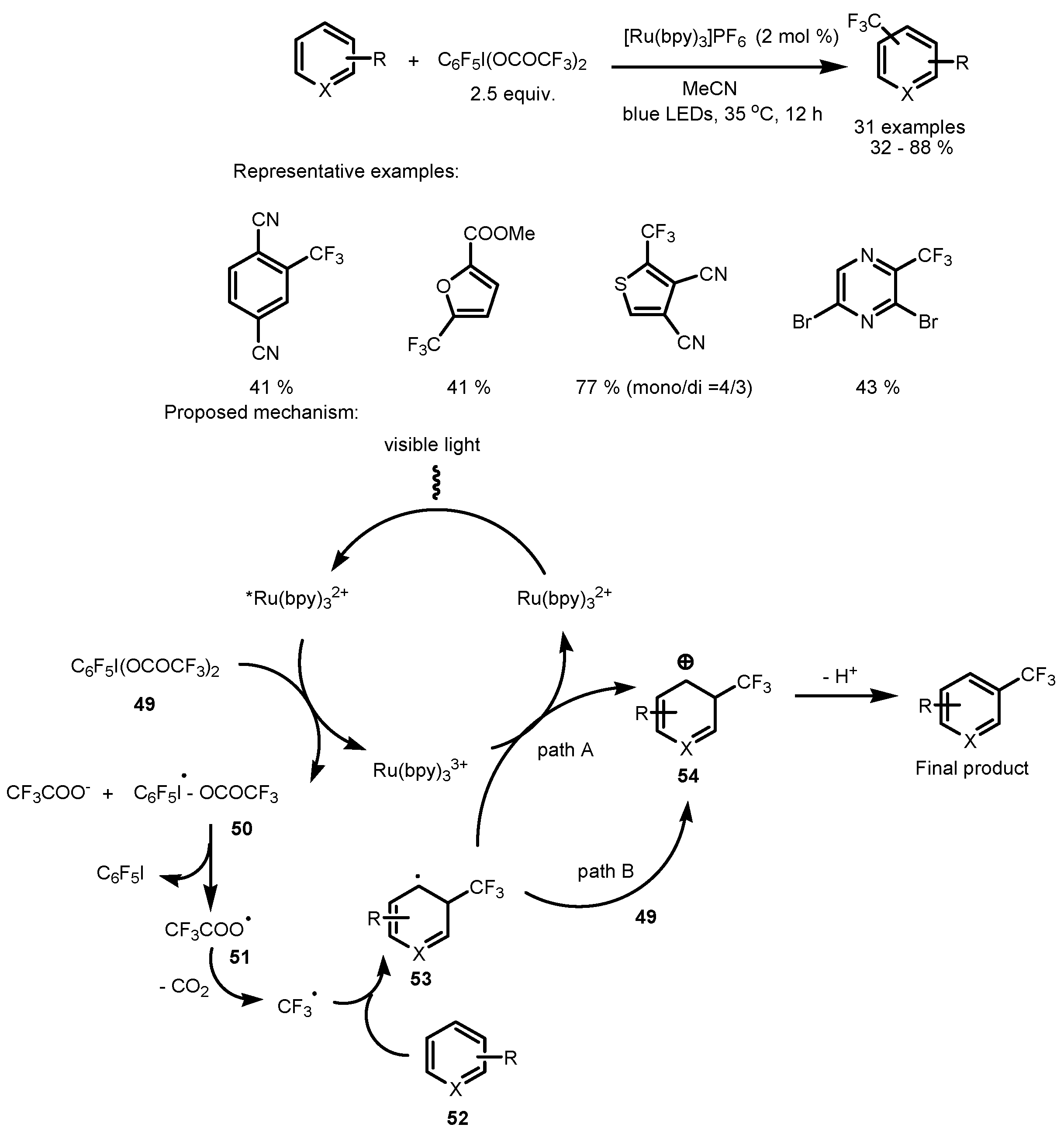
2.3. Hypervalent Iodine and Sulfur Compounds as the Fluorinating Reactants
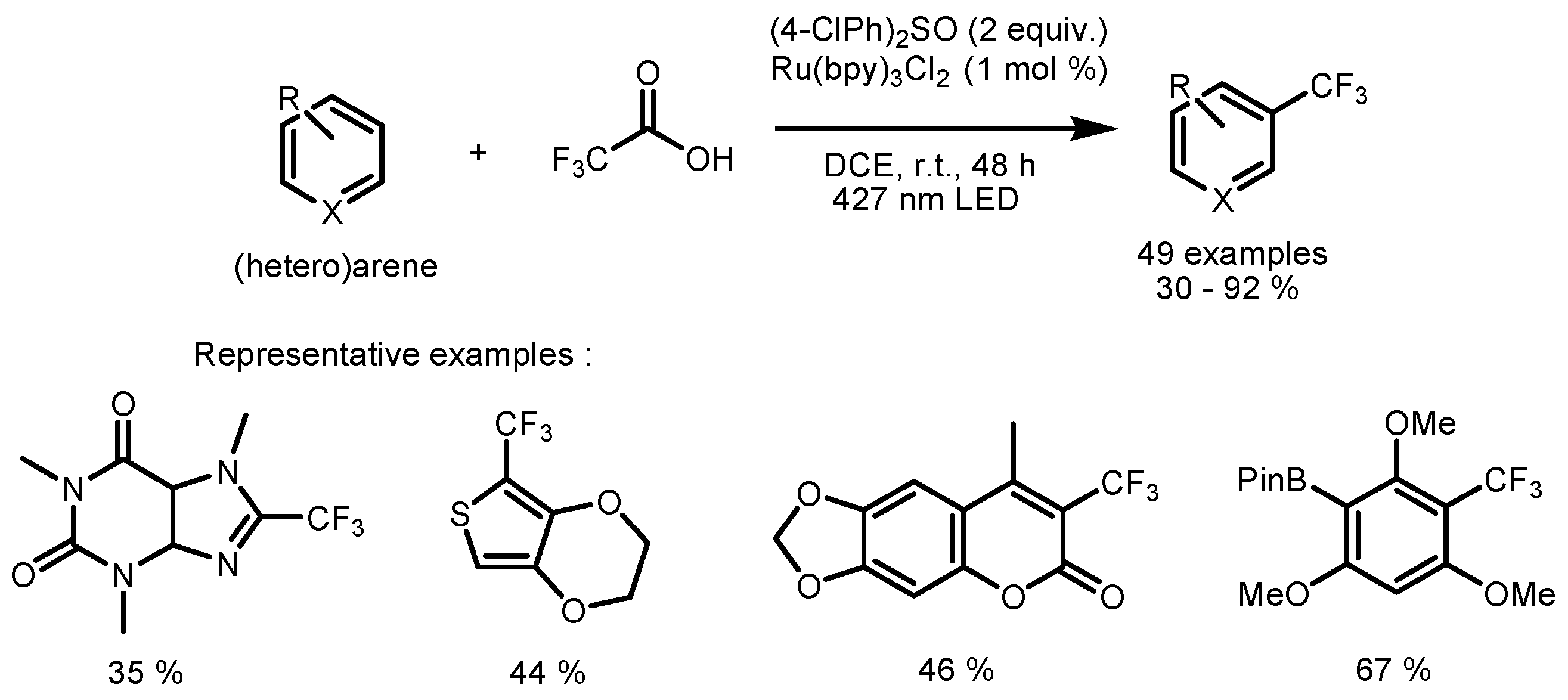
2.4. Trifluoromethyl Acid and Anhydride as the Fluorinating Reactants
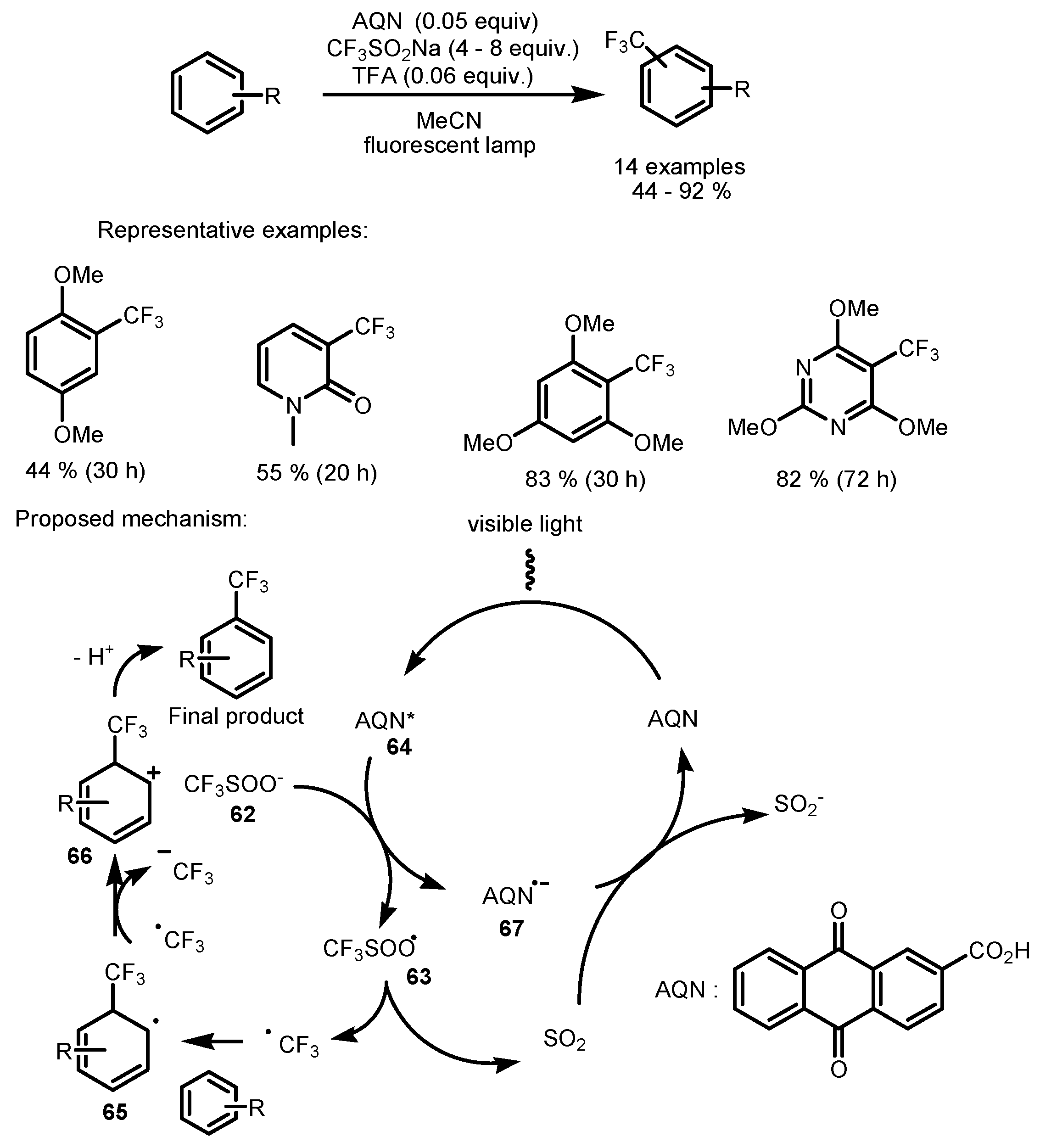
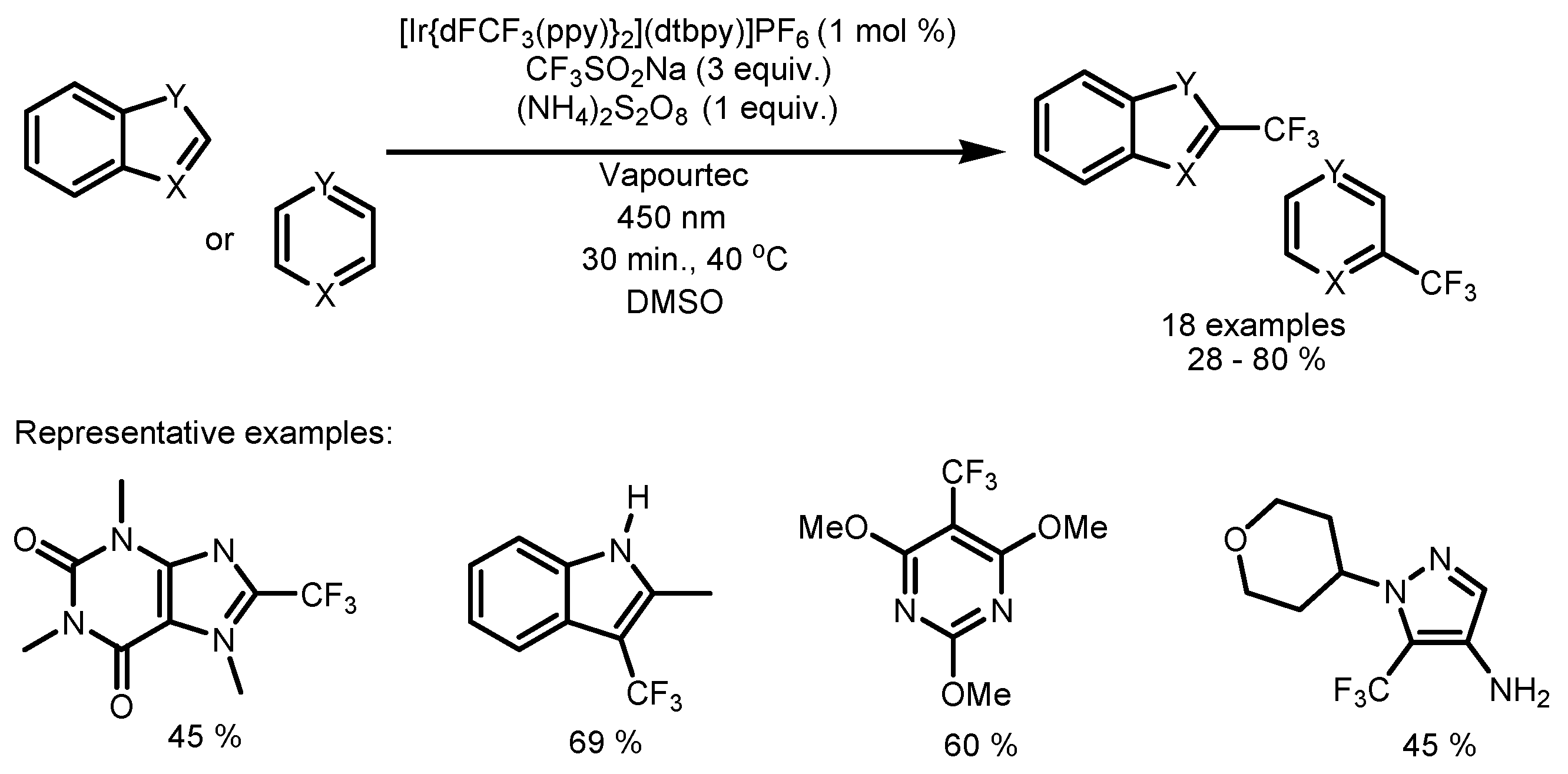
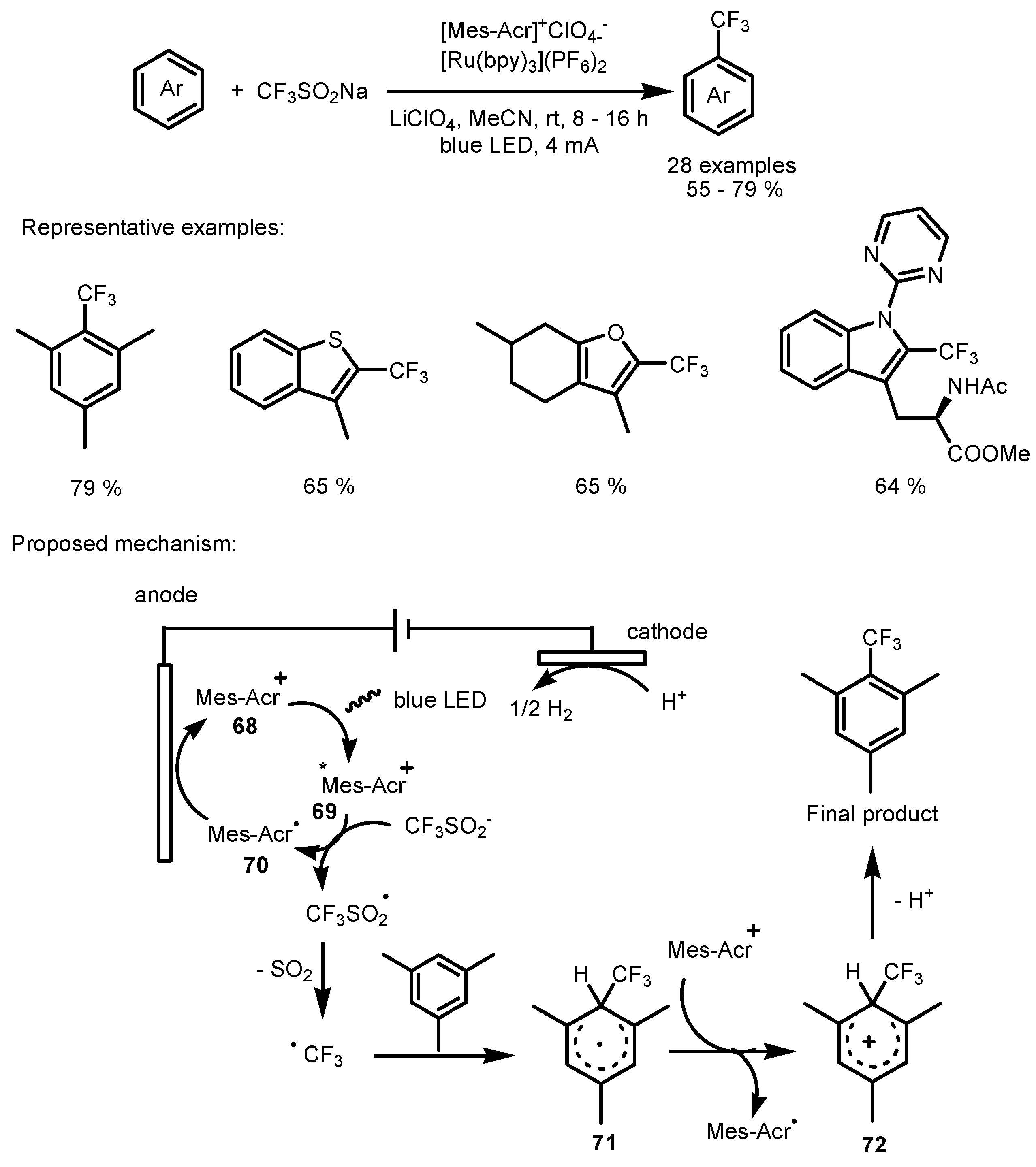
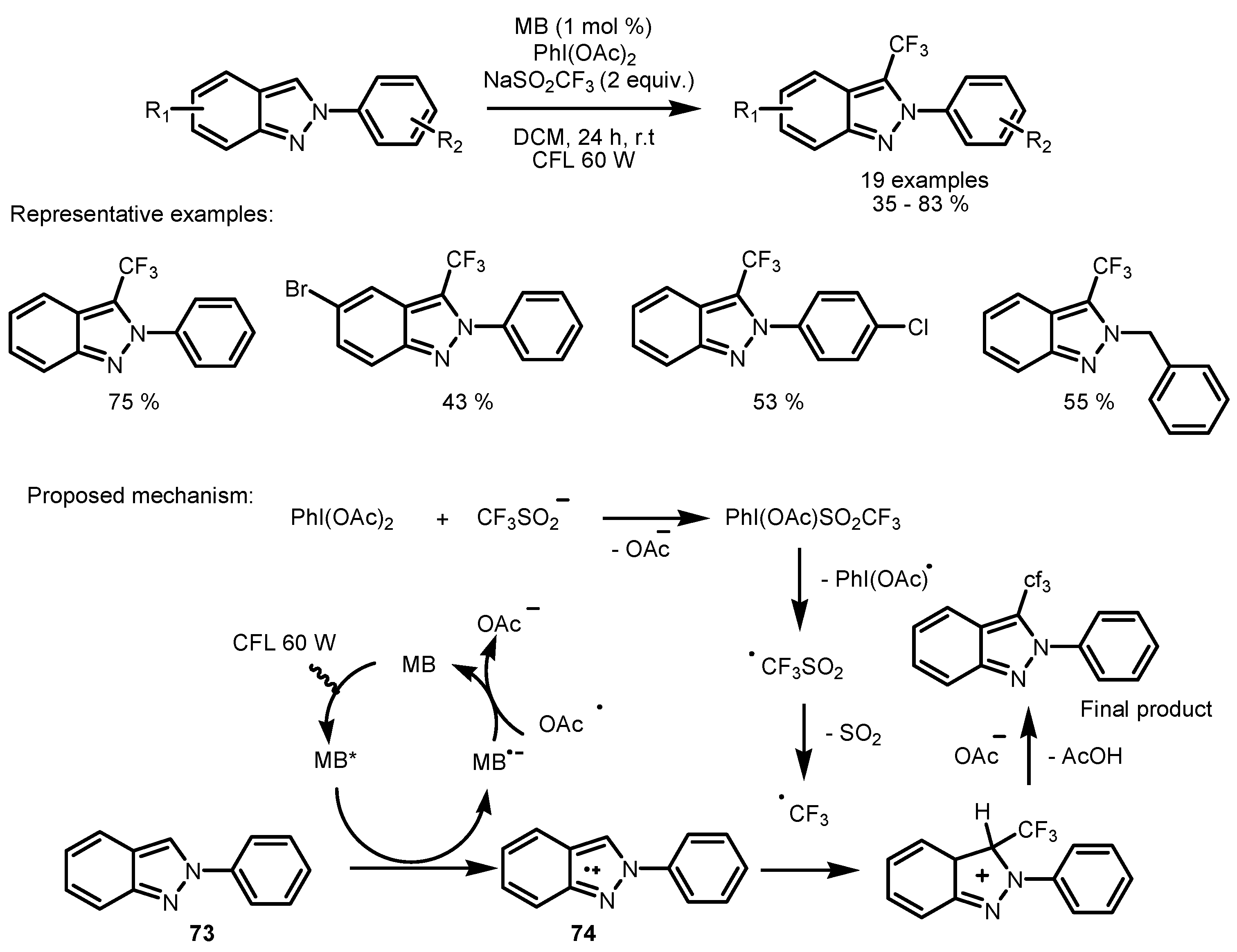
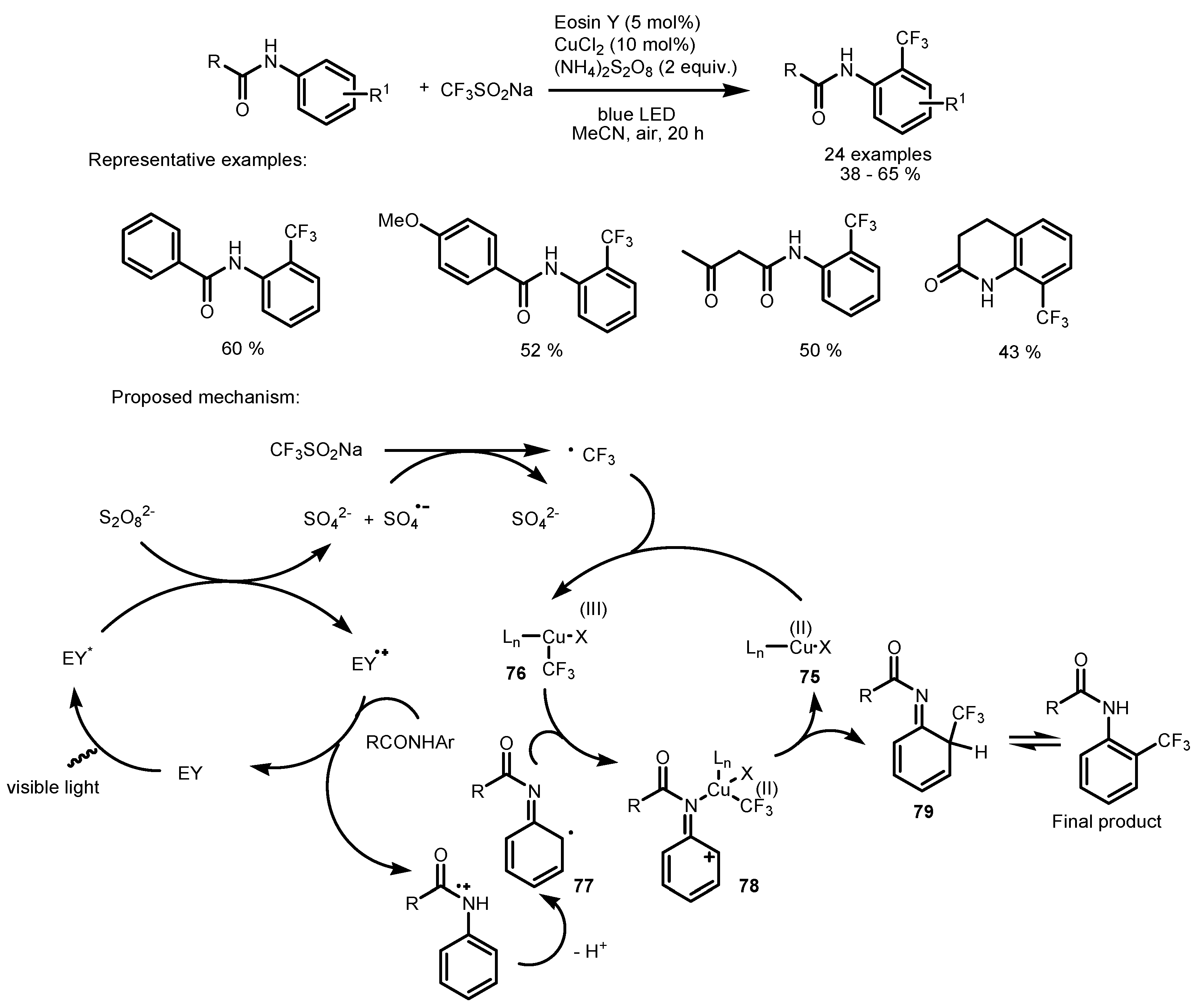
2.5. Sodium Triflinate as the Fluorinating Reactant
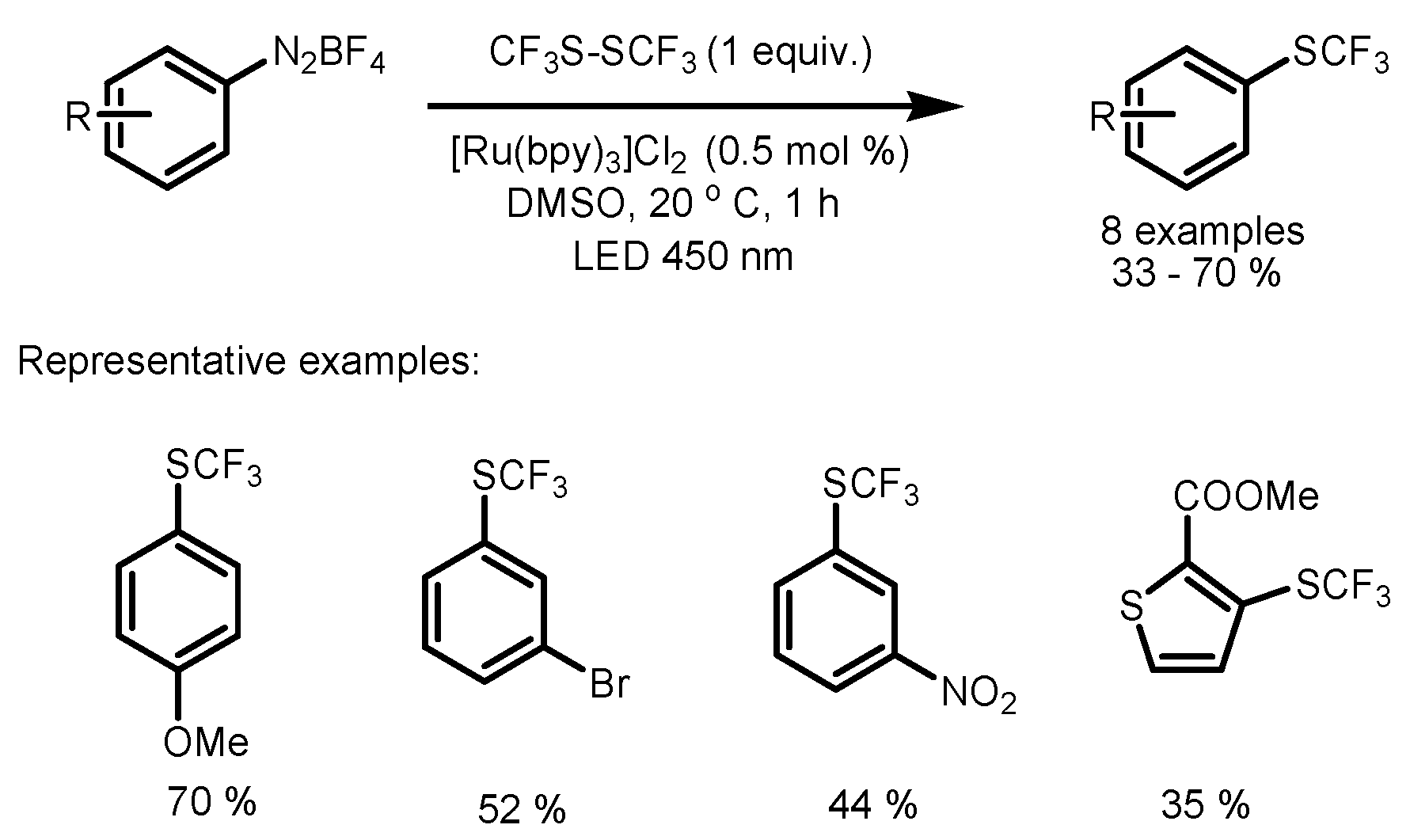
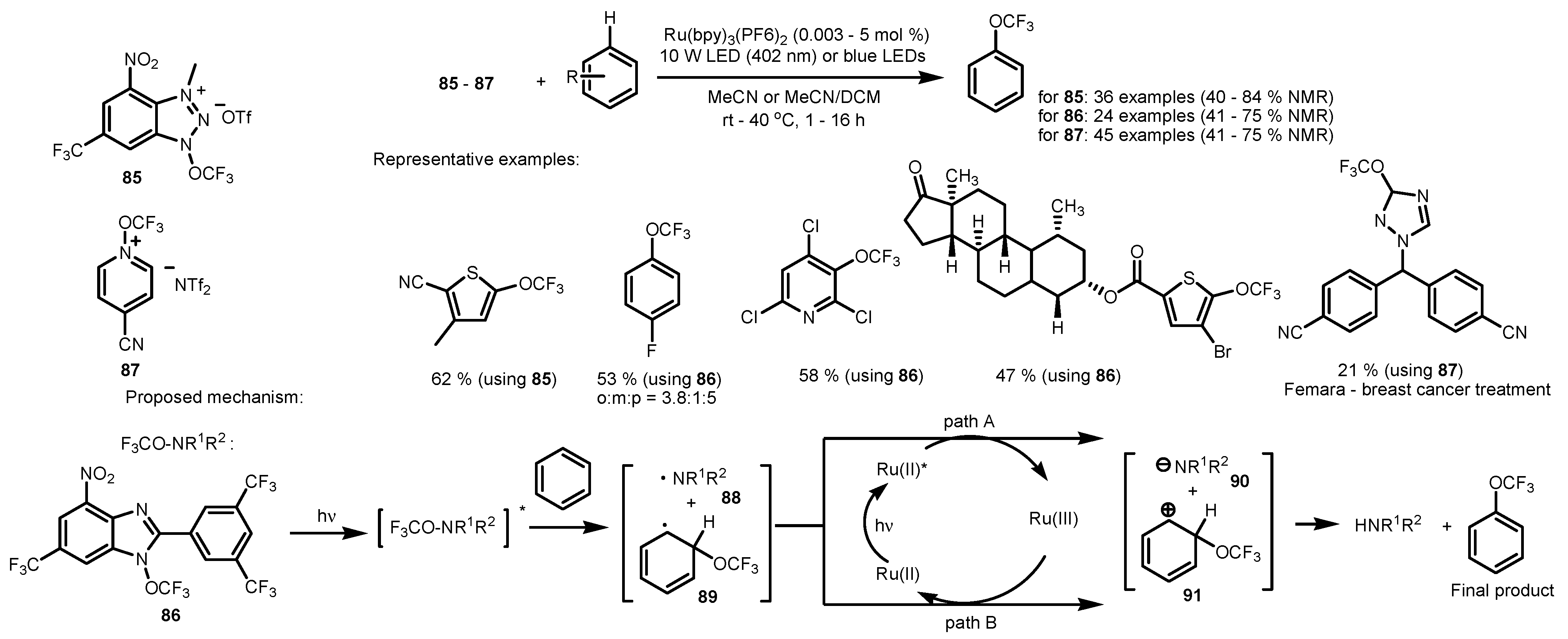
3. Trifluoromethylthiolation and Trifluoromethoxylation of Arenes
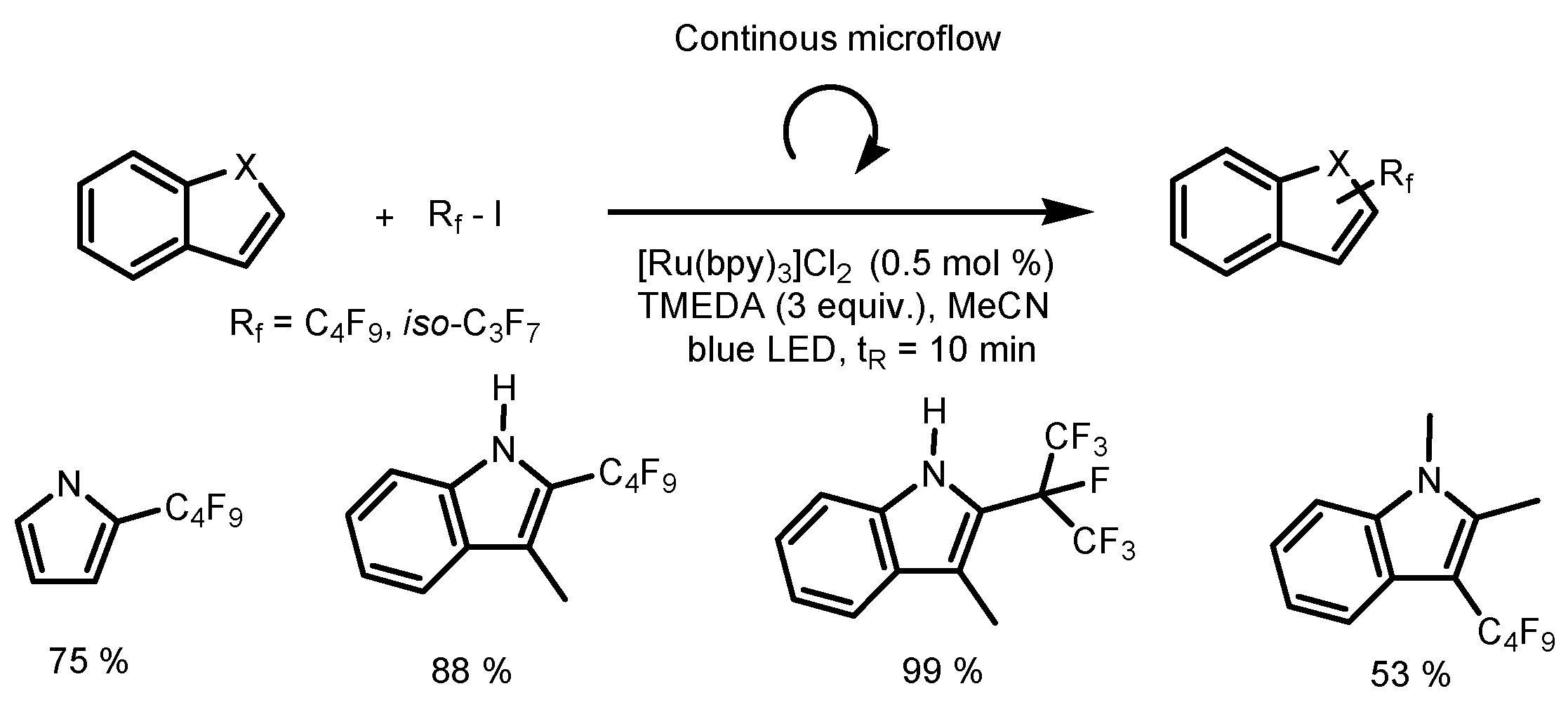
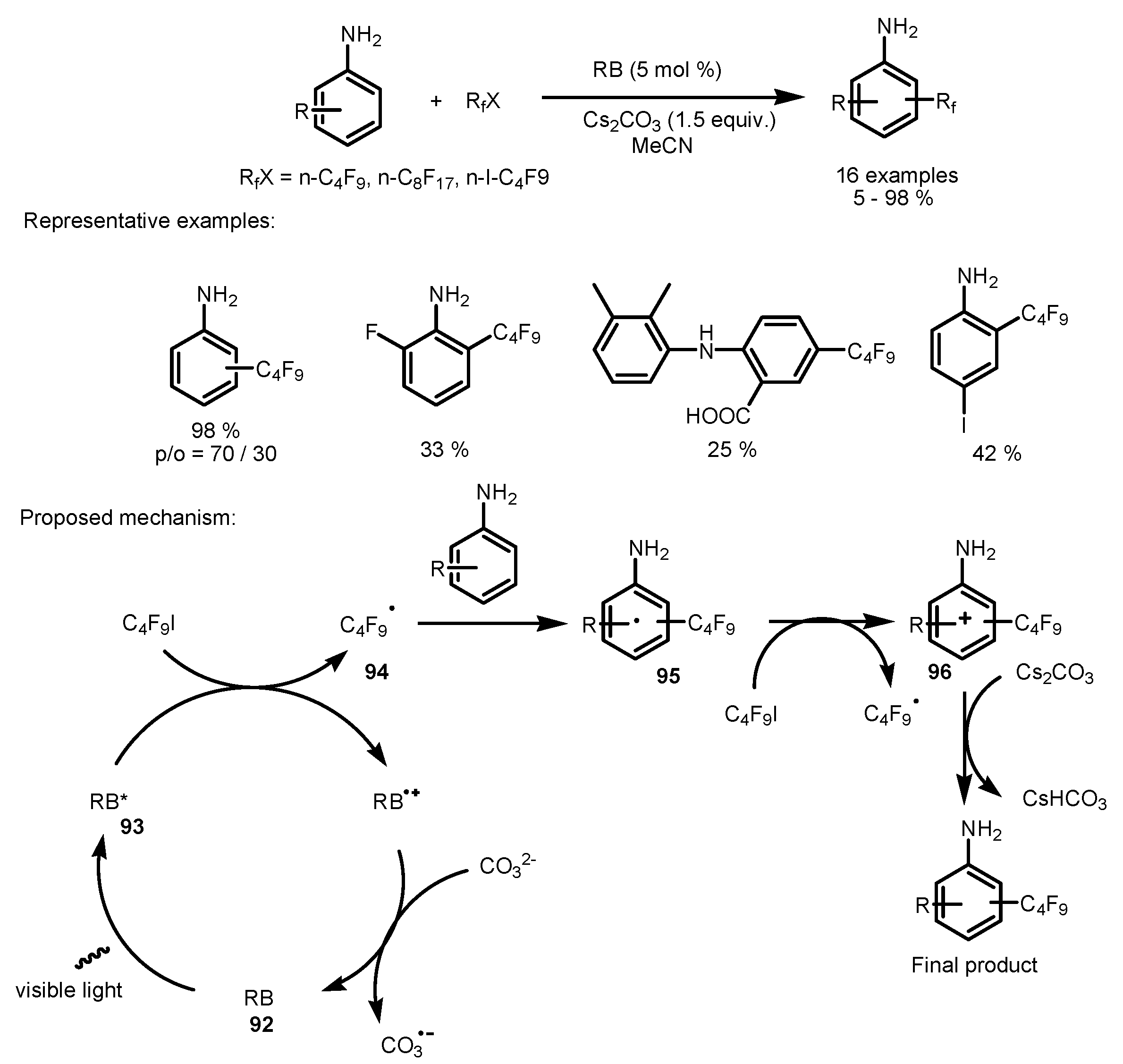
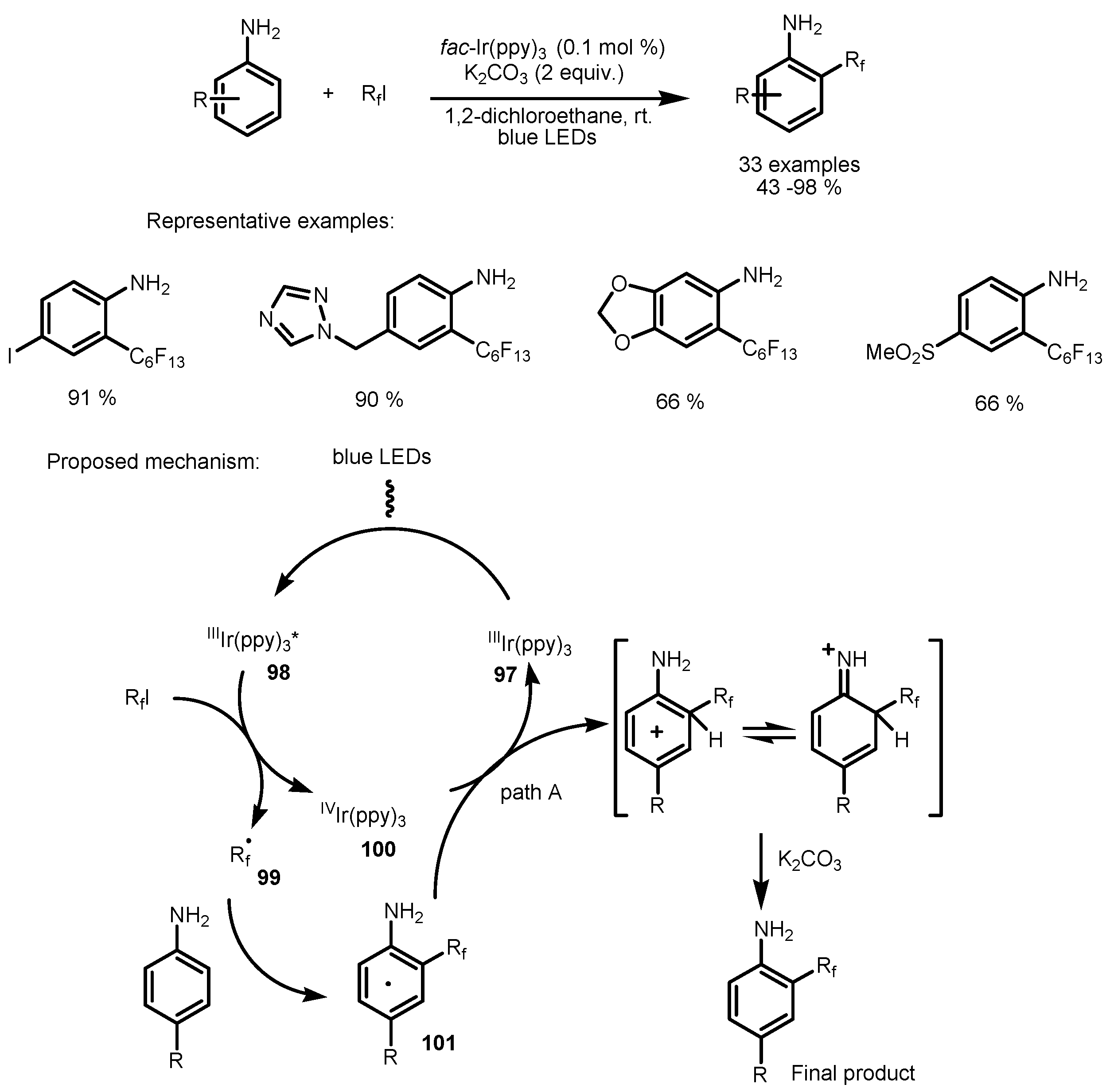
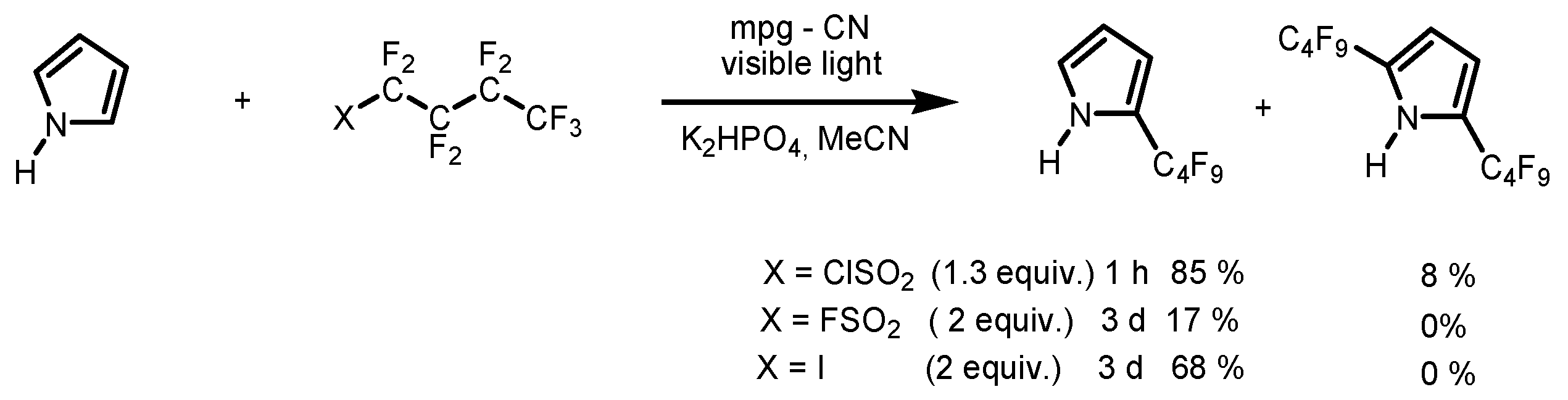
4. Perfluoroalkylation of Arenes
5. Monofluorination of Arenes
6. Fluorosulfonation of Arenes
7. Tandem Photocatalytic Fluorination–Aromatization Processes
8. Conclusions
Funding
Conflicts of Interest
References
- Muller, K.; Faeh, C.; Diederich, F. Fluorine in pharmaceuticals: Looking beyond intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef]
- Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Hird, M. Fluorinated liquid crystals—Properties and applications. Chem. Soc. Rev. 2007, 36, 2070–2095. [Google Scholar] [CrossRef] [PubMed]
- Kirk, K.L. Fluorination in medicinal chemistry: Methods, strategies, and recent developments. Org. Process Res. Dev. 2008, 12, 305–321. [Google Scholar] [CrossRef]
- O’Hagan, D. Understanding organofluorine chemistry. An introduction to the C–F bond. Chem. Soc. Rev. 2008, 37, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chen, Z.; Wu, W.; Mo, Y. How the generalized anomeric effect influences the conformational preference. Chem. Eur. J. 2013, 19, 1436–1444. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Liu, L.Q.; Wang, G.M.; Ouyang, H.; Li, Y.J. Catalyst-free room-temperature decarboxylative tri- or tetrafunctionalization of alkynyl carboxylic acids with N-fluorobenzenesulfonimide (NFSI) and diselenides. Green Chem. 2018, 20, 604–608. [Google Scholar] [CrossRef]
- Wang, X.; Lei, J.; Liu, Y.J.; Ye, Y.; Li, J.Z.; Sun, K. Fluorination and fluoroalkylation of alkenes/alkynes to construct fluoro-containing heterocycles. Org. Chem. Front. 2021, 8, 2079–2109. [Google Scholar] [CrossRef]
- Sun, K.; Wang, S.N.; Feng, R.R.; Zhang, Y.X.; Wang, X.; Zhang, Z.G.; Zhang, B. Copper-catalyzed radical selenodifluoromethylation of alkenes: Access to CF2-containing γ-lactams. Org. Lett. 2019, 21, 2052–2055. [Google Scholar] [CrossRef]
- Li, Y.; Tan, L.; Wang, Z.H.; Qian, H.L.; Shi, Y.B.; Hu, W.P. Air-stable n-type semiconductor: core-perfluoroalkylated perylene bisimides. Org. Lett. 2008, 10, 529–532. [Google Scholar] [CrossRef]
- Matsugi, M.; Curran, D.P. Synthesis, reaction, and recycle of light fluorous Grubbs−Hoveyda catalysts for alkene metathesis. J. Org. Chem. 2005, 70, 1636–1642. [Google Scholar] [CrossRef]
- Tattersall, F.D.; Rycroft, W.; Cumberbatch, M.; Mason, G.; Tye, S.; Williamson, D.J.; Hale, J.J.; Mills, S.G.; Finke, P.E.; MacCoss, M.; et al. The novel NK1 receptor antagonist MK-0869 (L-754,030) and its water soluble phosphoryl prodrug, L-758,298, inhibit acute and delayed cisplatin-induced emesis in ferrets. Neuropharmacology 2000, 39, 652–663. [Google Scholar] [CrossRef]
- Chauret, N.; Guay, D.; Li, C.; Day, S.; Silva, J.; Blouin, M.; Ducharme, Y.; Yergey, J.A.; Nicoll-Griffith, D.A. Improving metabolic stability of phosphodiesterase-4 inhibitors containing a substituted catechol: Prevention of reactive intermediate formation and covalent binding. Bioorganic Med. Chem. Lett. 2002, 12, 2149–2152. [Google Scholar] [CrossRef]
- Robertson, J.F.R.; Come, S.E.; Jones, S.E.; Beex, F.; Kaufmann, M.; Makris, A.; Nortier, J.W.R.; Possinger, K.; Rutqvist, L.E. Endocrine treatment options for advanced breast cancer—The role of fulvestrant. Eur. J. Cancer 2005, 41, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible light photoredox catalysis with transition metal complexes: Applications in organic synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef] [PubMed]
- Xuan, J.; Xiao, W.J. Visible light photoredox catalysis. Angew. Chem. Int. Ed. 2012, 51, 6828–6838. [Google Scholar] [CrossRef] [PubMed]
- Tóth, B.L.; Tischler, O.; Novák, Z. Recent advances in dual transition metal–visible light photoredox catalysis. Tetrahedron Lett. 2016, 57, 4505–4513. [Google Scholar] [CrossRef]
- Shaw, M.H.; Twilton, J.; MacMillan, D.W.C. Photoredox catalysis in organic chemistry. J. Org. Chem. 2016, 81, 6898–6926. [Google Scholar] [CrossRef] [PubMed]
- Kancherla, R.; Muralirajan, K.; Sagadevan, A.; Rueping, M. Visible light-induced excited-state transition-metal catalysis. Trends Chem. 2019, 1, 510–523. [Google Scholar] [CrossRef]
- McAtee, R.C.; McClain, E.J.; Stephenson, C.R.J. Illuminating photoredox catalysis. Trends Chem. 2019, 1, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.J.; Hu, A.; Zuo, Z. Photocatalytic alkoxy radical-mediated transformations. Tetrahedron Lett. 2018, 59, 2103–2111. [Google Scholar] [CrossRef]
- Courant, T.; Masson, G. Recent progress in visible-light photoredox-catalyzed intermolecular 1,2-difunctionalization of double bonds via an ATRA-type mechanism. J. Org. Chem. 2016, 81, 6945–6952. [Google Scholar] [CrossRef]
- Lantaño, B.; Torviso, M.R.; Bonesi, S.M.; Barata-Vallejoa, S.; Postigo, A. Advances in metal-assisted non-electrophilic fluoroalkylation reactions of organic compounds. Coord. Chem. Rev. 2015, 285, 76–108. [Google Scholar] [CrossRef]
- Lantaño, B.; Postigo, A. Radical fluorination reactions by thermal and photoinduced methods. Org. Biomol. Chem. 2017, 15, 9954–9973. [Google Scholar] [CrossRef] [PubMed]
- Bui, T.T.; Hong, W.P.; Kim, H.-K. Recent Advances in Visible Light-mediated Fluorination. J. Fluor. Chem. 2021, 247, 109794. [Google Scholar] [CrossRef]
- Yakubov, S.; Barham, J.P. Photosensitized direct C–H fluorination and trifluoromethylation in organic synthesis. Beilstein J. Org. Chem. 2020, 16, 2151–2192. [Google Scholar] [CrossRef] [PubMed]
- Akita, M.; Koike, T. Principles and applications of photoredox catalysis: Trifluoromethylation and beyond. J. Synth. Org. Chem. Jpn. 2016, 74, 1036–1046. [Google Scholar] [CrossRef]
- Lantano, B.; Torviso, M.R.; Bonesi, S.M.; Barata-Vallejo, S.; Postigo, A. Late-stage electron-catalyzed perfluoroalkylation of coumarine derivatives—thermal fluoroalkyl radical production from sodium perfluoroalkyl sulfinate salts. J. Fluor. Chem. 2017, 197, 42–48. [Google Scholar]
- Lee, K.N.; Lee, J.W.; Ngai, M.Y. Recent development of catalytic trifluoromethoxylation reactions. Tetrahedron 2018, 74, 7127–7135. [Google Scholar] [CrossRef]
- Barata-Vallejo, S.; Postigo, A. New visible-light-triggered photocatalytic trifluoromethylation reactions of carbon–carbon multiple bonds and (hetero)aromatic compounds. Chem. Eur. J. 2020, 26, 11065–11084. [Google Scholar] [CrossRef]
- Baguia, H.; Evano, G. Direct perfluoroalkylation of C-H bonds in (hetero)arenes. Chem. Eur. J. 2022, 28, e202200975. [Google Scholar] [CrossRef]
- Liu, T.; Liu, J.; He, J.; Hong, Y.; Zhou, H.; Liu, Y.-L.; Tang, S. Recent advances in photoinduced perfluoroalkylation using perfluoroalkyl halides as the radical precursors. Synthesis 2022, 54, 1919–1938. [Google Scholar]
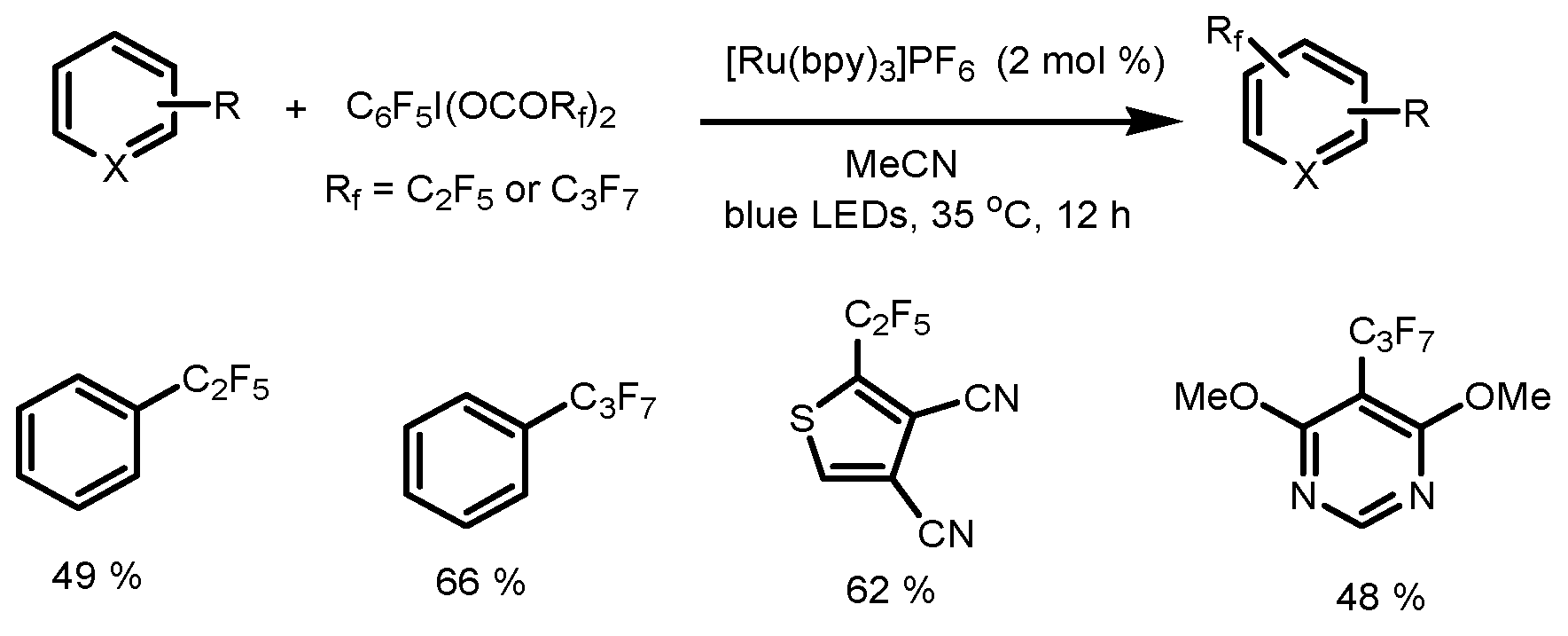
- Barata-Vallejo, S.; Bonesi, S.M.; Postigo, A. Perfluoroalkylation reactions of (hetero)arenes. RSC Adv. 2015, 5, 62498–62518. [Google Scholar] [CrossRef]
- Lemos, A.; Lemaire, C.; Luxena, A. Progress in difluoroalkylation of organic substrates by visible light photoredox catalysis. Adv. Synth. Catal. 2019, 361, 1500–1537. [Google Scholar] [CrossRef] [PubMed]
- Ojima, I. Fluorine in Medical Chemistry and Chemical Biology; Wiley-Blackwell: Chichester, UK, 2009. [Google Scholar]
- Jeschke, P. The unique role of fluorine in the design of active ingredients for modern crop protection. ChemBioChem 2004, 5, 570–589. [Google Scholar] [CrossRef]
- Nagib, D.A.; MacMillan, D.W.C. Trifluoromethylation of arenes and heteroarenes by means of photoredox catalysis. Nature 2011, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.K.; Li, C.; Hanan, G.S.; Zysman-Colman, E. Blue-emissive cobalt (III) complexes and their use in the photocatalytic trifluoromethylation of polycyclic aromatic hydrocarbons. Angew. Chem. Int. Ed. 2018, 57, 8027–8031. [Google Scholar] [CrossRef] [PubMed]
- Baar, M.; Blechert, S. Graphitic carbon nitride polymer as a recyclable photoredox catalyst for fluoroalkylation of arenes. Chem. Eur. J. 2015, 21, 526–530. [Google Scholar] [CrossRef]
- Maiti, B.; Abramov, A.; Pérez-Ruiz, R.; Díaz Díaz, D. The prospect of photochemical reactions in confined gel media. Acc. Chem. Res. 2019, 52, 1865–1876. [Google Scholar] [CrossRef]
- Strekowski, L.; Hojjat, M.; Patterson, S.E.; Kiselyov, A.S. Experimental and computational studies of trifluoromethylation of aromatic amines by the system trifluoroiodomethane-zinc-sulfur dioxide. J. Heterocycl. Chem. 1994, 31, 1413–1416. [Google Scholar] [CrossRef]
- Sato, K.; Omote, M.; Ando, A.; Kumadaki, I. Rhodium-catalyzed novel trifluoromethylation at the α-position of α,β-unsaturated ketones. Org. Lett. 2004, 6, 4359–4361. [Google Scholar] [CrossRef]
- Mikami, K.; Tomita, Y.; Ichikawa, Y.; Amikura, K.; Itoh, Y. Radical trifluoromethylation of ketone silyl enol ethers by activation with dialkylzinc. Org. Lett. 2006, 8, 4671–4673. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Houk, K.N.; Mikami, K. Experimental and theoretical studies on radical trifluoromethylation of titanium ate and lithium enolates. J. Org. Chem. 2006, 71, 8918–8925. [Google Scholar] [CrossRef] [PubMed]
- Kino, T.; Nagase, Y.; Ohtsuka, Y.; Yamamoto, K.; Uraguchi, D.; Tokuhisa, K.; Yamakawa, T. Trifluoromethylation of various aromatic compounds by CF3I in the presence of Fe(II) compound, H2O2 and dimethylsulfoxide. J. Fluor. Chem. 2010, 131, 98–105. [Google Scholar] [CrossRef]
- Ye, Y.; Lee, S.H.; Sanford, M.S. Silver-mediated trifluoromethylation of arenes using TMSCF3. Org. Lett. 2011, 13, 5464–5467. [Google Scholar] [CrossRef] [PubMed]
- Andrieux, C.P.; Gelis, L.; Medebielle, M.; Pinson, J.; Saveant, J.M. Outer-sphere dissociative electron transfer to organic molecules: A source of radicals or carbanions? Direct and indirect electrochemistry of perfluoroalkyl bromides and iodides. J. Am. Chem. Soc. 1990, 112, 3509–3520. [Google Scholar] [CrossRef]
- Ye, Y.; Sanford, M.S. Merging visible-light photocatalysis and transition-metal catalysis in the copper-catalyzed trifluoromethylation of boronic acids with CF3I. J. Am. Chem. Soc. 2012, 134, 9034–9037. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Berger, F.; Jia, H.; Ford, J.; Wortman, A.; Börgel, J.; Genicot, C.; Ritter, T. Aryl sulfonium salts for site-selective late-stage trifluoromethylation. Angew. Chem. Int. Ed. 2019, 58, 14615–14619. [Google Scholar] [CrossRef]
- Iqbal, N.; Choi, S.; Ko, E.; Cho, E.J. Trifluoromethylation of heterocycles via visible light photoredox catalysis. Tetrahedron Lett. 2012, 53, 2005–2008. [Google Scholar] [CrossRef]
- Choi, W.J.; Choi, S.; Ohkubo, K.; Fukuzumi, S.; Cho, E.J.; You, Y. Mechanisms and applications of cyclometalated Pt(II) complexes in photoredox catalytic trifluoromethylation. Chem. Sci. 2015, 6, 1454–1464. [Google Scholar] [CrossRef]
- Zhdankin, V.V. Hypervalent Iodine Chemistry: Preparation, Structure and Synthetic Applications of Polyvalent Iodine Compounds; Wiley: Chichester, UK, 2013. [Google Scholar]
- Varvoglis, A. Aryliodine(III) dicarboxylates. Chem. Soc. Rev. 1981, 10, 377–407. [Google Scholar] [CrossRef]
- Frohn, H.-J.; Hirschberg, M.E.; Wenda, A.; Bardin, V.V. Explored routes to unknown polyfluoroorganyliodine hexafluorides, RFIF6. J. Fluor. Chem. 2008, 129, 459–473. [Google Scholar] [CrossRef]
- Yoshimura, A.; Zhdankin, V.V. Advances in synthetic applications of hypervalent iodine compounds. Chem. Rev. 2016, 116, 3328–3435. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Studer, A. Iodine(III) reagents in radical chemistry. Acc. Chem. Res. 2017, 50, 1712–1724. [Google Scholar] [CrossRef] [PubMed]
- Pitre, S.P.; McTiernan, C.D.; Ismaili, H.; Scaiano, J.C. Metal-free photocatalytic radical trifluoromethylation utilizing methylene blue and visible light irradiation. ACS Catal. 2014, 4, 2530–2535. [Google Scholar] [CrossRef]
- Xie, J.; Yuan, X.; Abdukader, A.; Zhu, C.; Ma, J. Visible-light-promoted radical C−H trifluoromethylation of free anilines. Org. Lett. 2014, 16, 1768–1771. [Google Scholar] [CrossRef]
- Yang, B.; Yu, D.; Xu, X.-H.; Qing, F.-L. Visible-light photoredox decarboxylation of perfluoroarene iodine(III) trifluoroacetates for C−H trifluoromethylation of (hetero)arenes. ACS Catal. 2018, 8, 2839–2843. [Google Scholar] [CrossRef]
- Le, C.; Chen, T.Q.; Liang, T.; Zhang, P.; MacMillan, D.W.C. A radical approach to the copper oxidative addition problem: Trifluoromethylation of bromoarenes. Science 2018, 360, 1010–1014. [Google Scholar] [CrossRef]
- Beatty, J.W.; Douglas, J.J.; Cole, K.P.; Stephenson, C.R.J. A scalable and operati onally simple radical trifluoromethylation. Nat. Commun. 2015, 6, 7919–7925. [Google Scholar] [CrossRef]
- Beatty, J.W.; Douglas, J.J.; Miller, R.; McAtee, R.C.; Cole, K.P.; Stephenson, C.J.P. Photochemical perfluoroalkylation with pyridine N-Oxides: Mechanistic insights and performance on a kilogram scale. Chem 2016, 1, 456–472. [Google Scholar] [CrossRef]
- Yin, D.; Su, D.; Jin, J. Photoredox catalytic trifluoromethylation and perfluoroalkylation of arenes using trifluoroacetic and related carboxylic acids. Cell Rep. Phys. Sci. 2020, 1, 100141. [Google Scholar] [CrossRef]
- Lefebvre, Q. Toward sustainable trifluoromethylation reactions: Sodium triflinate under the spotlight. Synlett 2016, 28, 19–23. [Google Scholar] [CrossRef]
- Zhang, C. Application of Langlois’ reagent in trifluoromethylation reactions. Adv. Synth. Catal. 2014, 356, 2895–2906. [Google Scholar] [CrossRef]
- Cui, L.; Matusaki, Y.; Tada, N.; Miura, T.; Uno, B.; Itoh, A. Metal-free direct CH perfluoroalkylation of arenes and heteroarenes using a photoredox organocatalyst. Adv. Synth. Catal. 2013, 355, 2203–2207. [Google Scholar] [CrossRef]
- Abdiaj, I.; Bottecchia, C.; Alcazar, J.; Noёl, T. Visible light-induced trifluoromethylation of highly functionalized arenes and heteroarenes in continuous flow. Synthesis 2017, 49, 4978–4985. [Google Scholar]
- Qiu, Y.; Scheremetjew, A.; Finger, L.H.; Ackermann, L. Electrophotocatalytic undirected C-H trifluoromethylations of (het)arenes. Chem. Eur. J. 2020, 26, 3241–3246. [Google Scholar] [CrossRef] [PubMed]
- Murugan, A.; Babu, V.N.; Polu, A.; Sabarinathan, N.; Bakthadoss, M.; Sharada, D.S. Regioselective C3−H trifluoromethylation of 2H-indazole under transition-metal-free photoredox catalysis. J. Org. Chem. 2019, 84, 7796–7803. [Google Scholar] [CrossRef]
- Tian, C.; Wang, Q.; Wang, X.; An, G.; Li, G. Visible-light mediated ortho-trifluoromethylation of aniline derivatives. J. Org. Chem. 2019, 84, 14241–14247. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, B.; Ma, N.; Shu, S.; Chen, Y.; Yang, S.; Huang, Z.; Shi, D.; Zhao, Y. Photoredox-catalyst-enabled para-selective trifluoromethylation of tert-butyl arylcarbamates. Angew. Chem. Int. Ed. 2021, 60, 19030–19034. [Google Scholar] [CrossRef]
- Zou, L.; Li, P.; Wanga, B.; Wang, L. Visible-light-induced Pd-catalyzed orthotrifluoromethylation of acetanilides with CF3SO2Na under ambient conditions in the absence of an external photocatalyst. Chem. Commun. 2019, 55, 3737–3740. [Google Scholar] [CrossRef]
- Koziakov, D.; Majek, M.; Von Wangelin, A.J. Radical aromatic trifluoromethylthiolation: Photoredox catalysis vs. base mediation. Eur. J. Org. Chem. 2017, 45, 6722–6725. [Google Scholar] [CrossRef]
- Dear, R.E.A.; Gilbert, E.E. Preparation of bis-[trifluoromethyl] disulfide. Synthesis 1972, 6, 310. [Google Scholar] [CrossRef]
- Barata-Vallejo, S.; Bonesi, S.M.; Postigo, A. Trifluoromethoxylation reactions of (hetero) arenes, olefinic systems and aliphatic saturated substrates. Chem. Eur. J. 2022, 28, e202201776. [Google Scholar]
- Zheng, W.; Morales-Rivera, C.A.; Lee, J.W.; Liu, P.; Ngai, M.Y. Catalytic C−H trifluoromethoxylation of arenes and heteroarenes. Angew. Chem. Int. Ed. 2018, 57, 9645–9649. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Lee, J.W.; Morales-Rivera, C.A.; Liu, P.; Ngai, M.Y. Redox-active reagents for photocatalytic generation of the OCF3 radical and (hetero)aryl C−H trifluoromethoxylation. Angew. Chem. Int. Ed. 2018, 57, 13795–13799. [Google Scholar] [CrossRef] [PubMed]
- Jelier, B.J.; Tripet, P.F.; Pietrasiak, E.; Franzoni, I.; Jeschke, G.; Togni, A. Radical trifluoromethoxylation of arenes triggered by a visible-light-mediated N−O bond redox fragmentation. Angew. Chem. Int. Ed. 2018, 57, 13784–13789. [Google Scholar] [CrossRef] [PubMed]
- Filler, R.; Kobayashi, Y. Biomedical Aspects of Fluorine Chemistry; Kodansha Ltd.: Tokyo, Japan, 1982. [Google Scholar]
- Resnati, G. Synthesis of chiral and bioactive fluoroorganic compounds. Tetrahedron 1993, 49, 9385–9445. [Google Scholar] [CrossRef]
- Kirsh, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications; John Wiley & Sons: New York, NY, USA, 2004. [Google Scholar]
- Uneyama, K. Organofluorine Chemistry; Blackwell Publishing Ltd.: Ames, IA, USA, 2006. [Google Scholar]
- Straathof, N.J.W.; Gemoets, H.P.L.; Wang, X.; Schouten, J.C.; Hessel, V.; Nol, T. Rapid trifluoromethylation and perfluoroalkylation of five-membered heterocycles by photoredox catalysis in continuous flow. ChemSusChem 2014, 7, 1612–1617. [Google Scholar] [CrossRef]
- Straathof, N.J.W.; Van Osch, D.J.G.P.; Schouten, A.; Wang, X.; Schouten, J.C.; Hessel, V.; Noël, T. Visible light photocatalytic metal-free perfluoroalkylation of heteroarenes in continuous flow. J. Flow Chem. 2014, 4, 12–17. [Google Scholar] [CrossRef]
- Barata-Vallejo, S.; Yerien, D.E.; Postigo, A. Benign perfluoroalkylation of aniline derivatives through photoredox organocatalysis under visible-light irradiation. Eur. J. Org. Chem. 2015, 36, 7869–7875. [Google Scholar] [CrossRef]
- He, C.Y.; Gu, J.W.; Zhang, X. Visible-light-mediated direct perfluoroalkylation and trifluoromethylation of free anilines. Tetrahedron Lett. 2017, 58, 3939–3941. [Google Scholar] [CrossRef]
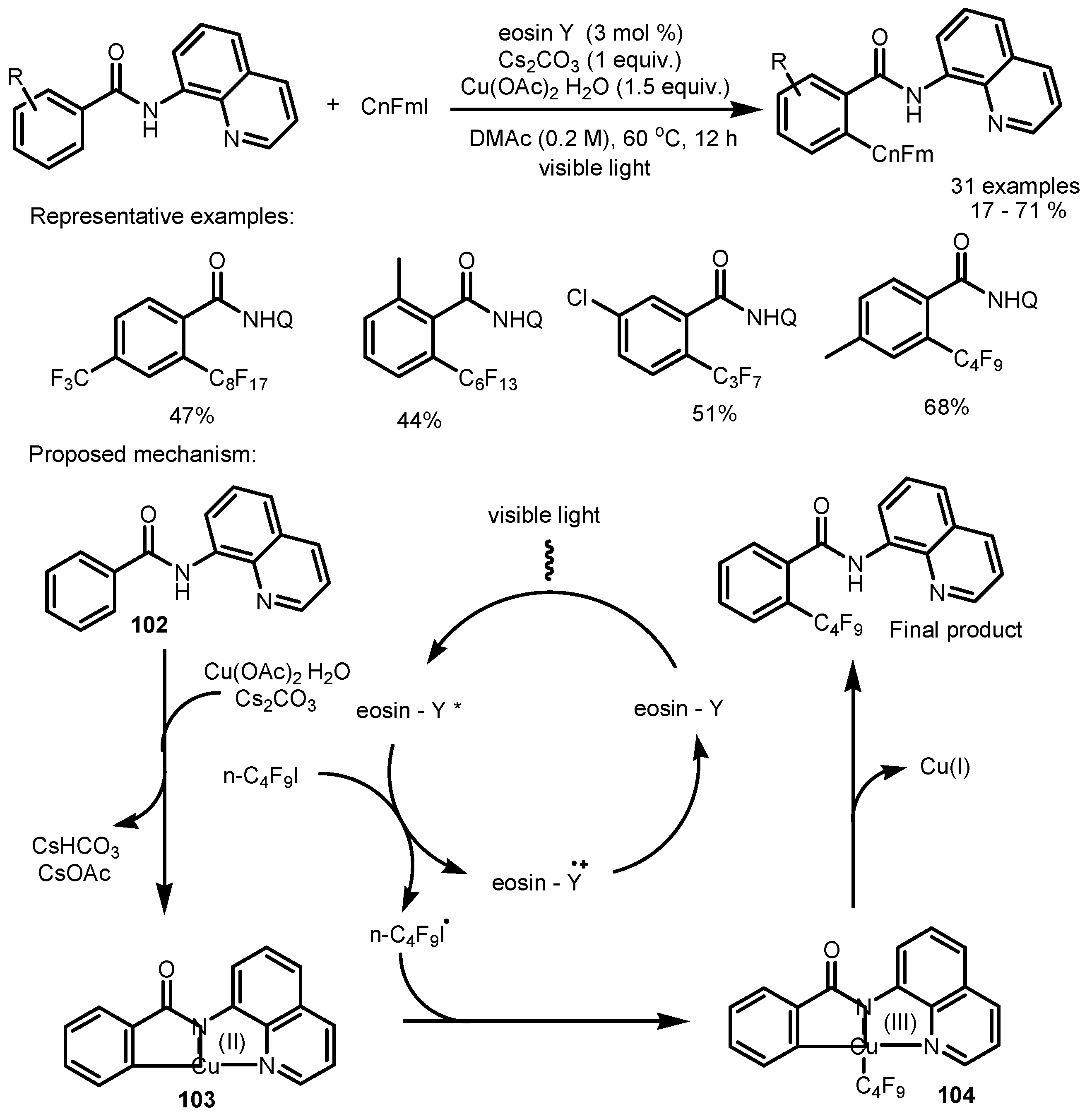
- Chen, X.; Tan, Z.; Gui, Q.; Hu, L.; Liu, J.; Wu, J.; Wang, G. Photocatalytic/Cu-promoted C-H activations: Visible-light-induced ortho-selective perfluoroalkylation of benzamides. Chem. Eur. J. 2016, 22, 6218–6222. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, M.; Yoshida, M. Redox system for perfluoroalkylation of arenes and a-methylstyrene derivatives using titanium oxide as photocatalyst. J. Fluor. Chem. 2009, 130, 926–932. [Google Scholar] [CrossRef]
- Filippini, G.; Longobardo, F.; Forster, L.; Criado, A.; Di Carmine, G.; Nasi, L.; D’Agostino, C.; Melchionna, M.; Fornasiero, P.; Prato, M. Light-driven, heterogeneous organocatalysts for novel C-C-bond formation towards valuable perfluoroalkylated intermediates. Sci. Adv. 2020, 6, eabc9923. [Google Scholar] [CrossRef] [PubMed]
- Tasnim, T.; Ryan, C.; Christensen, M.L.; Fennell, C.J.; Pitre, S.P. Radical perfluoroalkylation enabled by a catalytically generated halogen bonding complex and visible light irradiation. Org. Lett. 2022, 24, 446–450. [Google Scholar] [CrossRef]
- Liang, T.; Neumann, C.N.; Ritter, T. Introduction of fluorine and fluorine-containing functional groups. Angew. Chem. Int. Ed. 2013, 52, 8214–8264. [Google Scholar]
- Yang, X.Y.; Wu, T.; Phipps, R.J.; Toste, F.D. Advances in catalytic enantioselective fluorination, mono-, di-, and trifluoromethylation, and trifluoromethylthiolation reactions. Chem. Rev. 2015, 115, 826–870. [Google Scholar] [CrossRef]
- Kollonitsch, J.; Barash, L.; Doldouras, G.A. Photofluorination with fluoroxytrifluoromethane, a general method for the synthesis of organic fluorine compounds. Direct fluorination of bioactive molecules. J. Am. Chem. Soc. 1970, 92, 7494–7495. [Google Scholar] [CrossRef]
- Simons, J.H. The seven ages of fluorine chemistry. J. Fluor. Chem. 1986, 32, 7–24. [Google Scholar] [CrossRef]
- Yang, L.; Dong, D.; Revankar, H.M.; Zhang, C.P. Recent progress on fluorination in aqueous media. Green Chem. 2017, 19, 3951–3992. [Google Scholar] [CrossRef]
- Chatalova-Sazepin, C.; Hemelaere, R.; Paquin, J.F.; Sammis, G.M. Recent advances in radical fluorination. Synthesis 2015, 47, 2554–2569. [Google Scholar]
- Muñoz-Molina, J.M.; Belderrain, T.R.; Pérez, P. J Recent advances in copper-catalyzed radical C–H bond activation using N–F reagents. Synthesis 2021, 53, 51–64. [Google Scholar] [CrossRef]
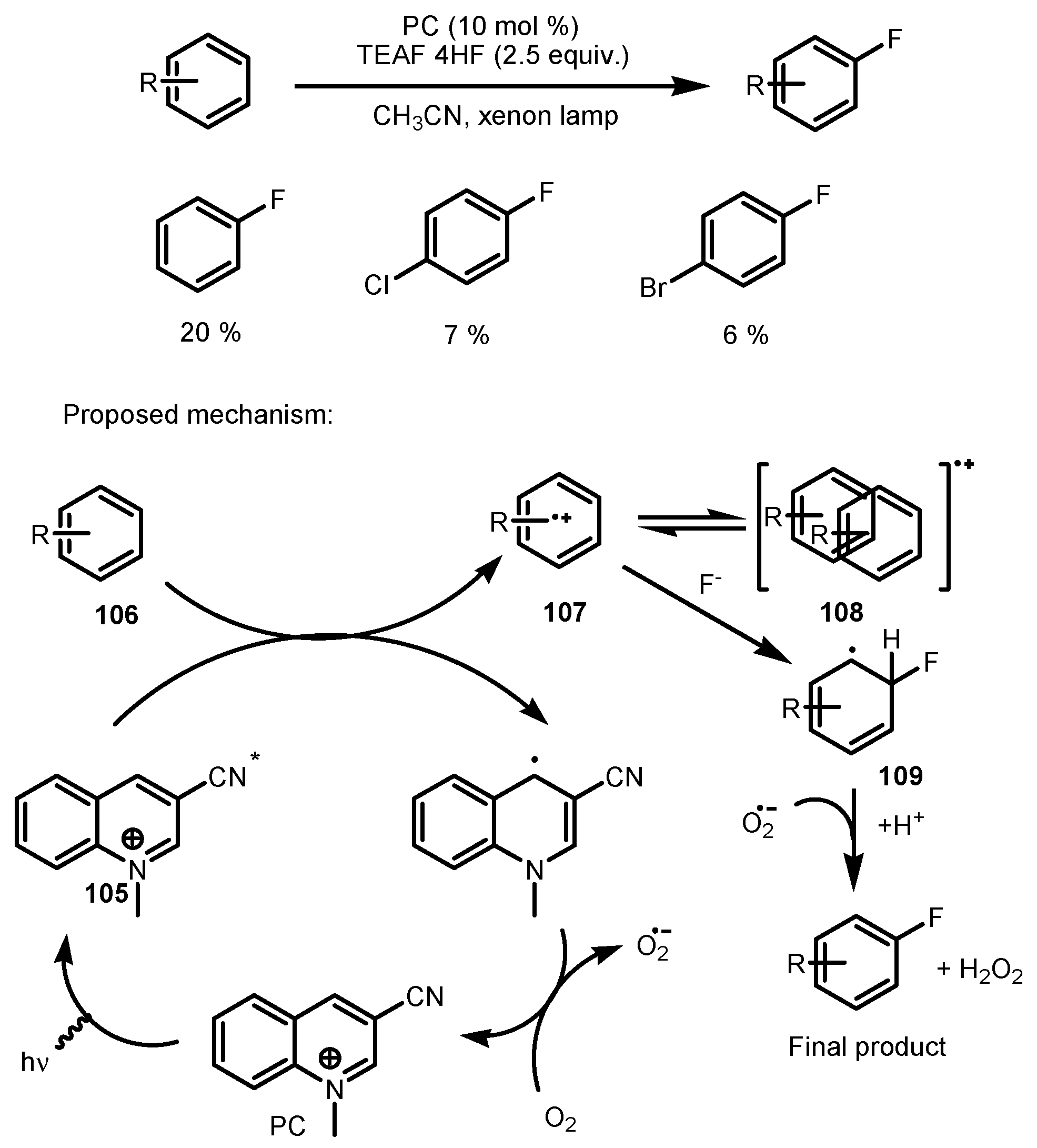
- Ohkubo, K.; Fujimoto, A.; Fukuzumi, S. Photocatalytic monofluorination of benzene by fluoride via photoinduced electron transfer with 3-cyano-1-methylquinolinium. J. Phys. Chem. 2013, 117, 10719–10725. [Google Scholar] [CrossRef] [PubMed]
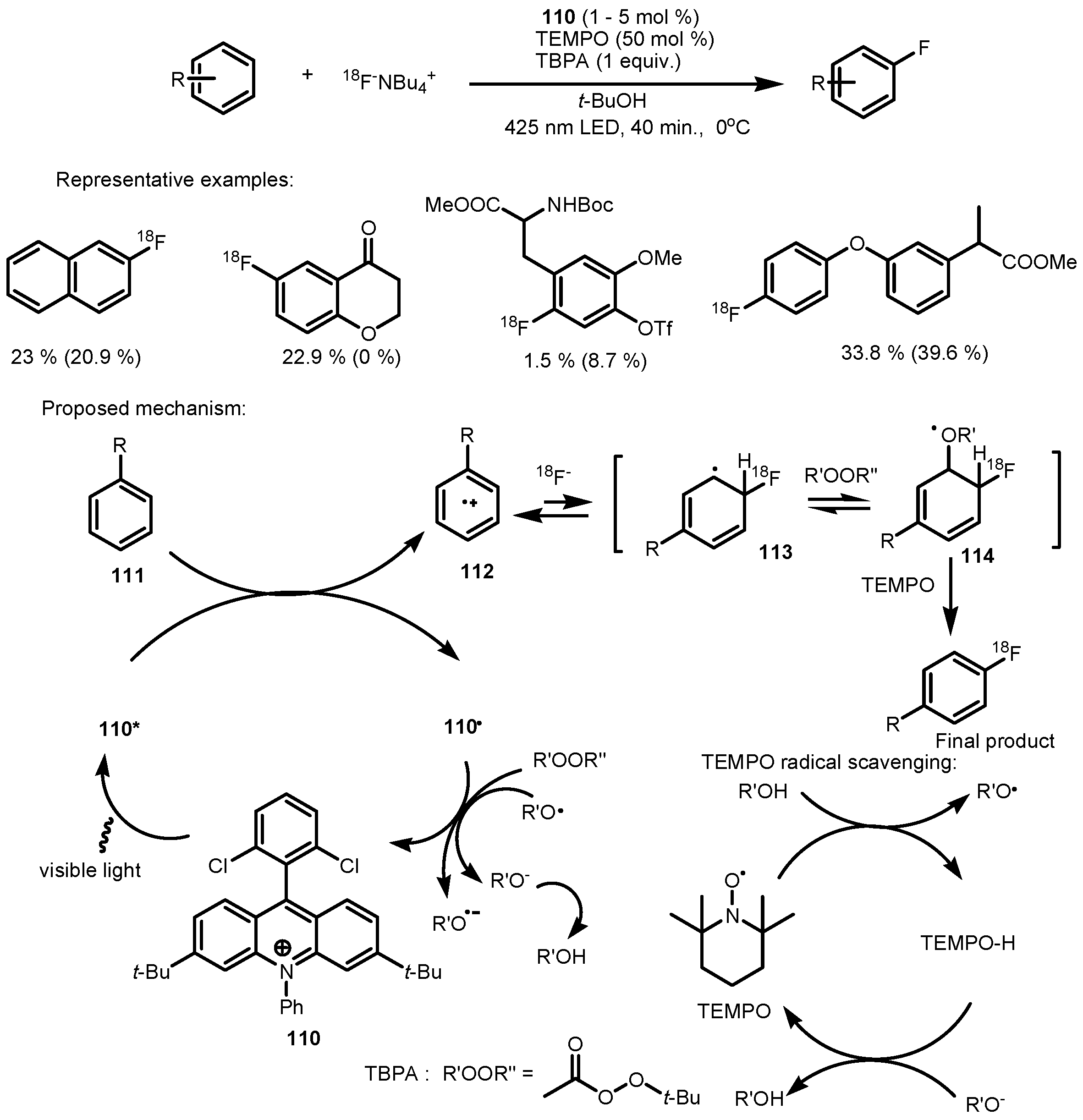
- Chen, W.; Huang, Z.; Tay, N.E.S.; Giglio, B.; Wang, M.; Wang, H.; Wu, Z.; Nicewicz, D.A.; Li, Z. Direct arene C−H fluorination with 18F− via organic photoredox catalysis. Science 2019, 364, 1170–1174. [Google Scholar] [CrossRef]
- Wang, L.; White, A.R.; Chen, W.; Wu, Z.; Nicewicz, D.A.; Li, Z. Direct radiofluorination of arene C−H bonds via photoredox catalysis using a peroxide as the terminal oxidant. Org. Lett. 2020, 22, 7971–7975. [Google Scholar] [CrossRef]
- Li, J.; Chen, J.; Sang, R.; Ham, W.S.; Plutschack, M.B.; Berger, F.; Chabbra, S.; Schnegg, A.; Genicot, C.; Ritter, T. Photoredox catalysis with aryl sulfonium salts enables site-selective late-stage fluorination. Nat. Chem. 2020, 12, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Louvel, D.; Chelagha, A.; Rouillon, J.; Payard, P.A.; Khrouz, L.; Monnereau, C.; Tlili, A. Metal-free visible-light synthesis of arylsulfonyl fluorides: Scope and mechanism. Chem. Eur. J. 2021, 27, 8704–8708. [Google Scholar] [CrossRef]
- Xiao, Q.; Lu, M.; Deng, Y.; Jian, J.-X.; Tong, Q.-X.; Zhong, J.-J. Photoinduced radical cascade cyclization: A metal-free approach to access difluoroalkylated dioxodibenzothiazepines. Org. Lett. 2021, 23, 9303–9308. [Google Scholar] [CrossRef]
- Qi, X.-K.; Zhang, H.; Pan, Z.-T.; Liang, R.-B.; Zhu, C.-M.; Li, J.-H.; Tong, Q.-X.; Gao, X.-W.; Wu, L.Z.; Zhong, J.-J. Photoinduced synthesis of fluorinated dibenz [b,e] azepines via radical triggered cyclization. Chem. Commun. 2019, 55, 10848–10851. [Google Scholar] [CrossRef]
- Jiang, H.; Cheng, Y.; Wang, R.; Zheng, M.; Zhang, Y.; Yu, S. Synthesis of 6-alkylated phenanthridine derivatives using photoredox neutral somophilic isocyanide insertion. Angew. Chem. Int. Ed. 2013, 52, 13289–13292. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Yuan, X.; Jiang, H.; Wang, R.; Ma, J.; Zhang, Y.; Yu, S. Regiospecific synthesis of 1-trifluoromethylisoquinolines enabled by photoredox somophilic vinyl isocyanide insertion. Adv. Synth. Catal. 2014, 356, 2859–2866. [Google Scholar] [CrossRef]
- Mao, S.; Wang, H.; Liu, L.; Wang, X.; Zhou, M.D.; Lia, L. Trifluoromethylation/difluoromethylation-initiated radical cyclization of o-alkenyl aromatic isocyanides for direct construction of 4-cyano-2-trifluoromethyl/difluoromethyl-containing quinolines. Adv. Synth. Catal. 2020, 362, 2274–2279. [Google Scholar] [CrossRef]
- Chu, X.Q.; Xie, T.; Li, L.; Ge, D.; Shen, Z.L.; Loh, T.P. Combining fluoroalkylation and defluorination to enable formal [3 + 2 + 1] heteroannulation by using visible-light photoredox organocatalysis. Org. Lett. 2018, 20, 2749–2752. [Google Scholar] [CrossRef] [PubMed]






































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kliś, T. Visible-Light Photoredox Catalysis for the Synthesis of Fluorinated Aromatic Compounds. Catalysts 2023, 13, 94. https://doi.org/10.3390/catal13010094
Kliś T. Visible-Light Photoredox Catalysis for the Synthesis of Fluorinated Aromatic Compounds. Catalysts. 2023; 13(1):94. https://doi.org/10.3390/catal13010094
Chicago/Turabian StyleKliś, Tomasz. 2023. "Visible-Light Photoredox Catalysis for the Synthesis of Fluorinated Aromatic Compounds" Catalysts 13, no. 1: 94. https://doi.org/10.3390/catal13010094
APA StyleKliś, T. (2023). Visible-Light Photoredox Catalysis for the Synthesis of Fluorinated Aromatic Compounds. Catalysts, 13(1), 94. https://doi.org/10.3390/catal13010094