
Coupling Reactions on Secondary Allylic, Propargylic, and Alkyl Carbons Using Organoborates/Ni and RMgX/Cu Reagents


Abstract
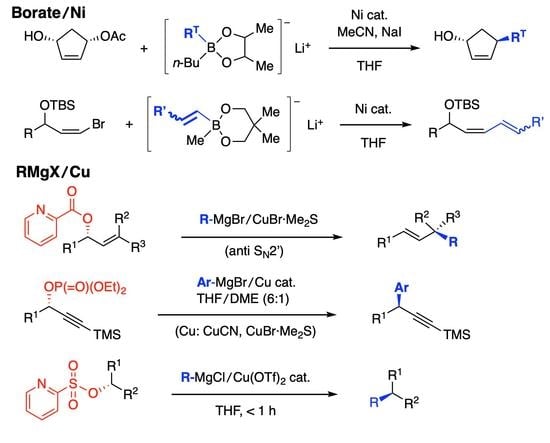
1. Introduction
2. Design of Reactive Borates
3. Preparation of Borates
4. Allylic Coupling
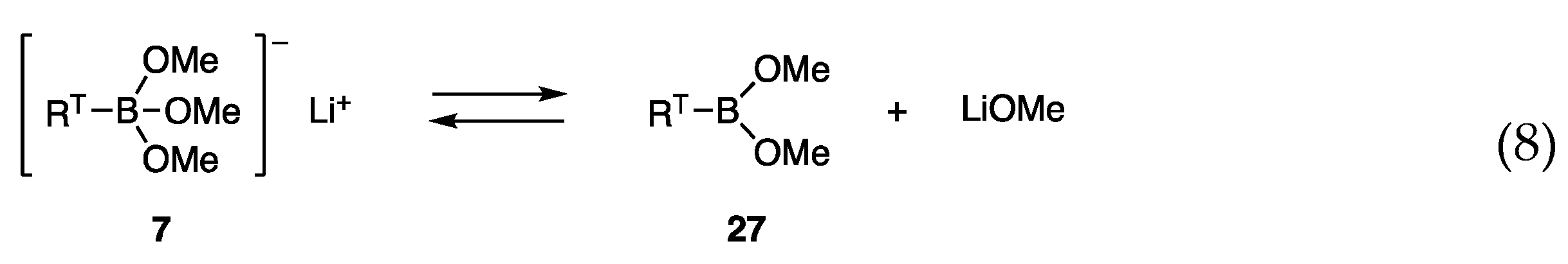
4.1. Trimethoxyborates
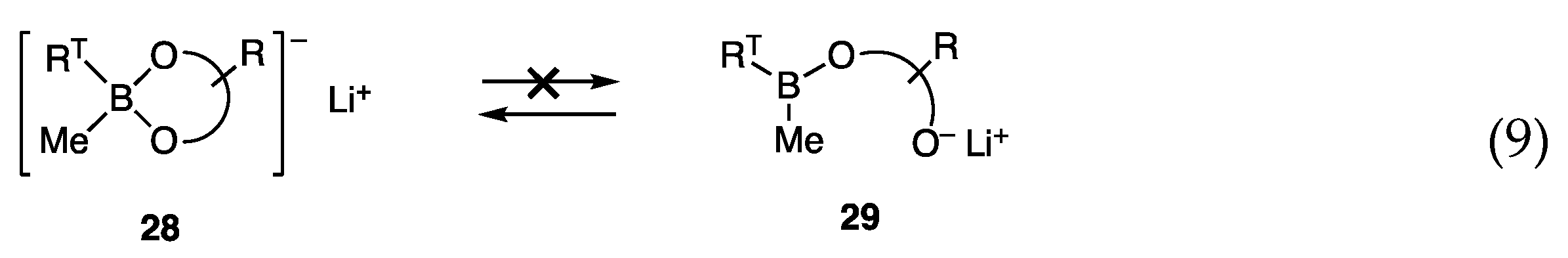
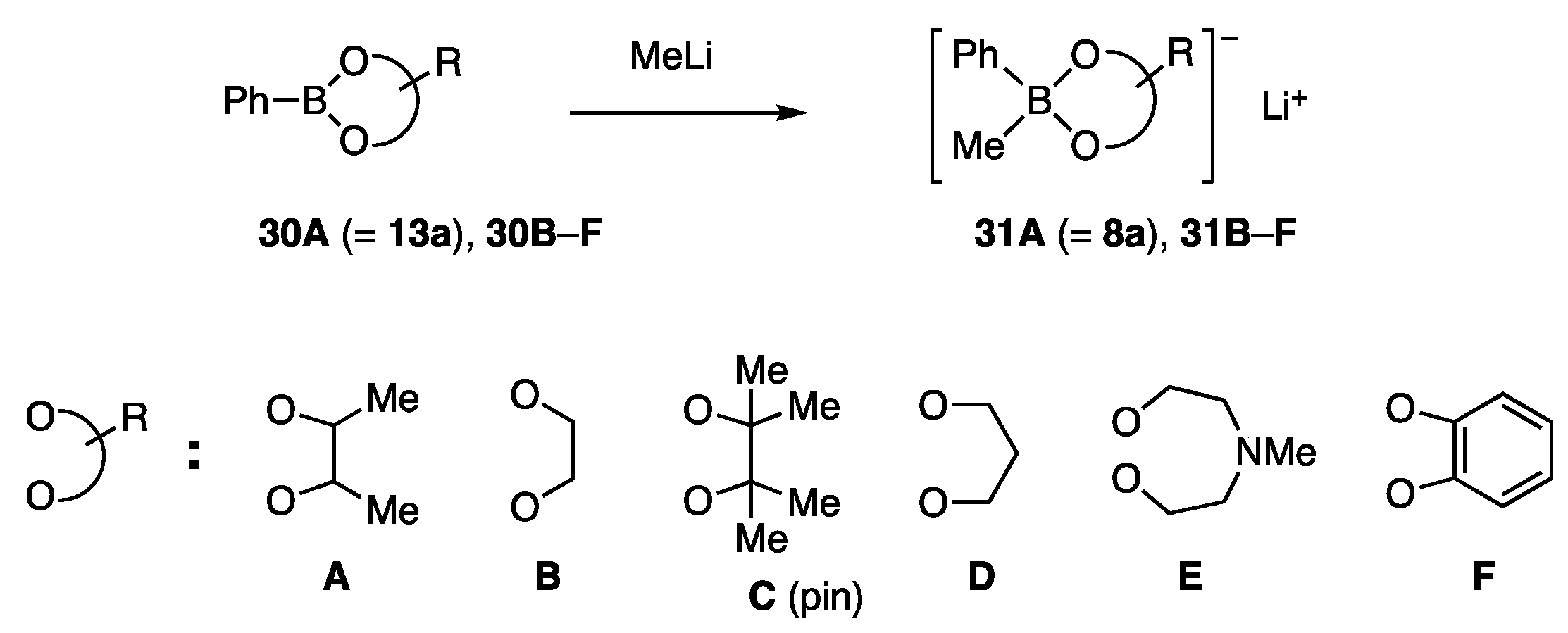
4.2. Borates with Alkanediol Ligands
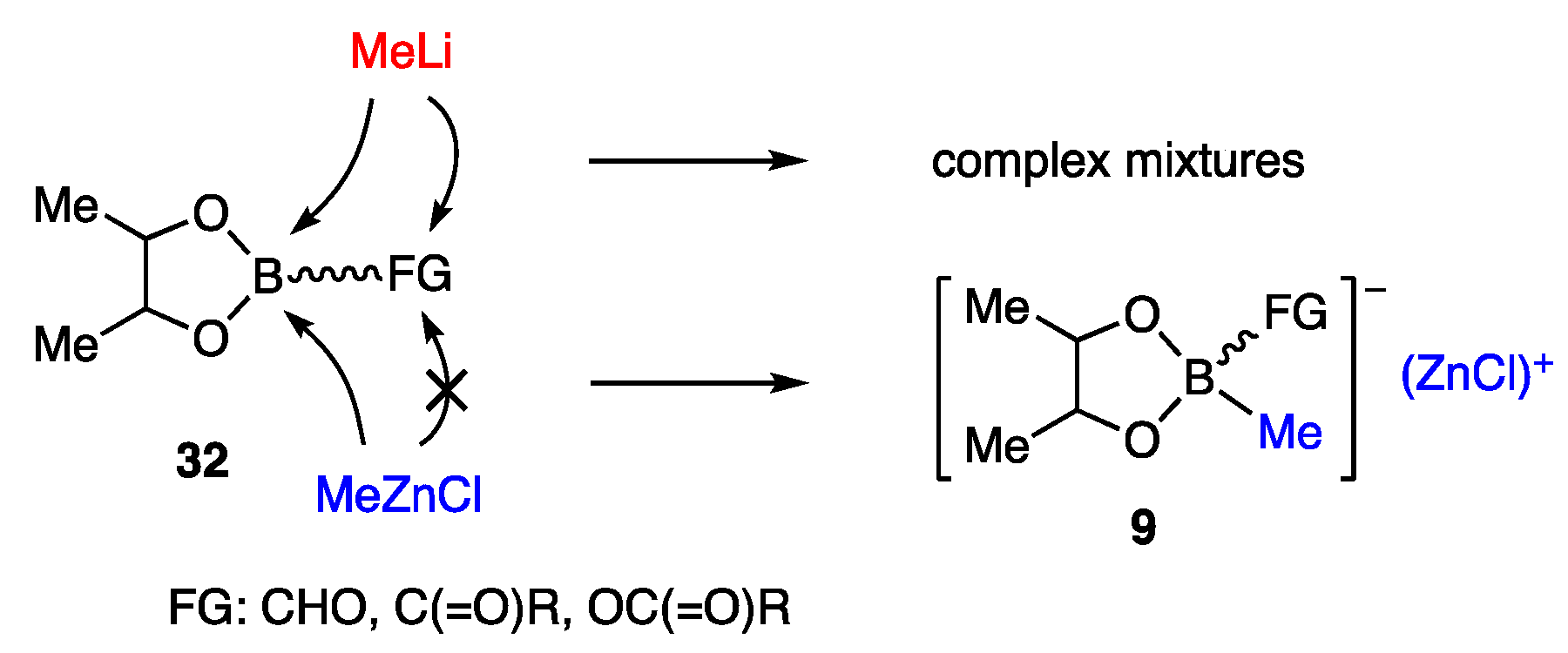
4.3. Carbonyll Group-Friendly Zinc Borates
4.4. Allylic Coupling on Cyclopentenediol Monoacetate
4.5. Determination of the Stereochemistry between RT and OH in the Major Products 4
4.6. Synthetic Application of Allylic Coupling
4.6.1. Prostaglandin A2
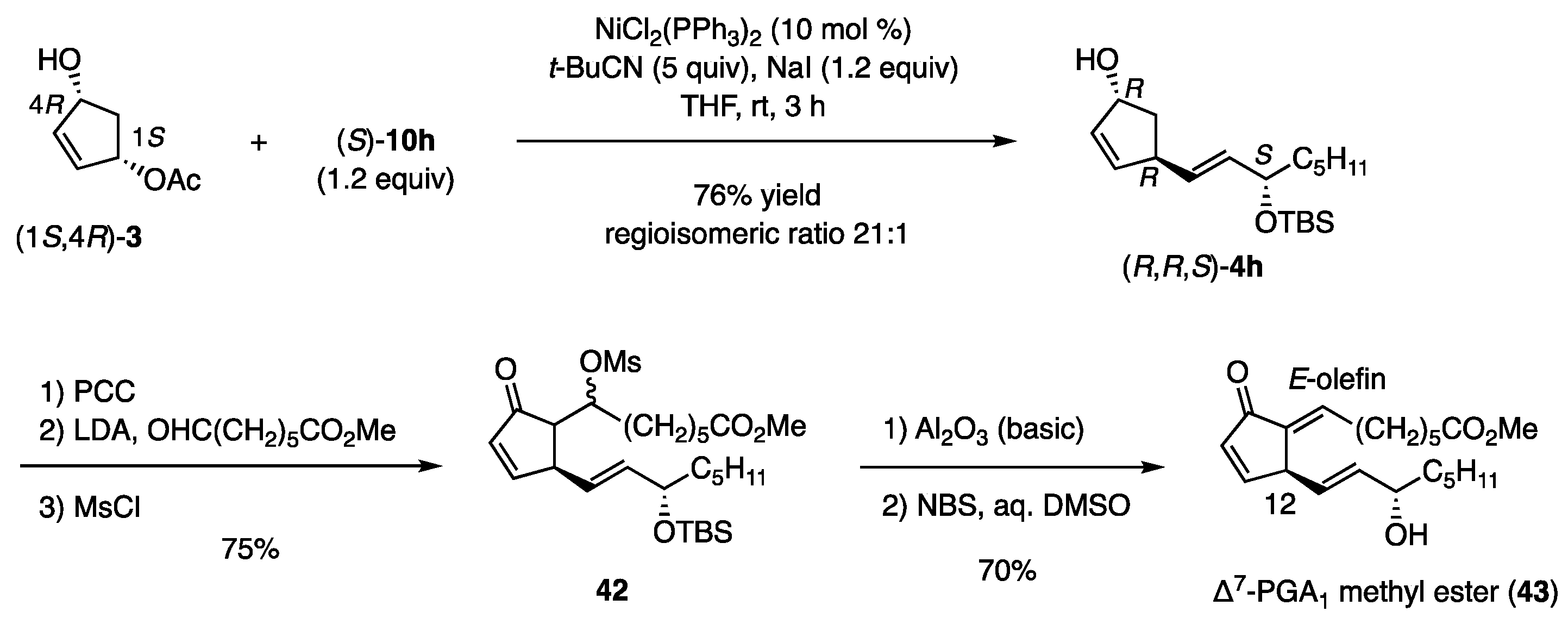
4.6.2. Δ7-PGA1 Methyl Ester
4.6.3. Aristeromycin
5. Alkenyl-Alkenyl Coupling Reaction
5.1. Optimal Alkanediol Ligand for Alkenyl Borates
5.2. Synthetic Application of the Alkenyl-Alkenyl Coupling
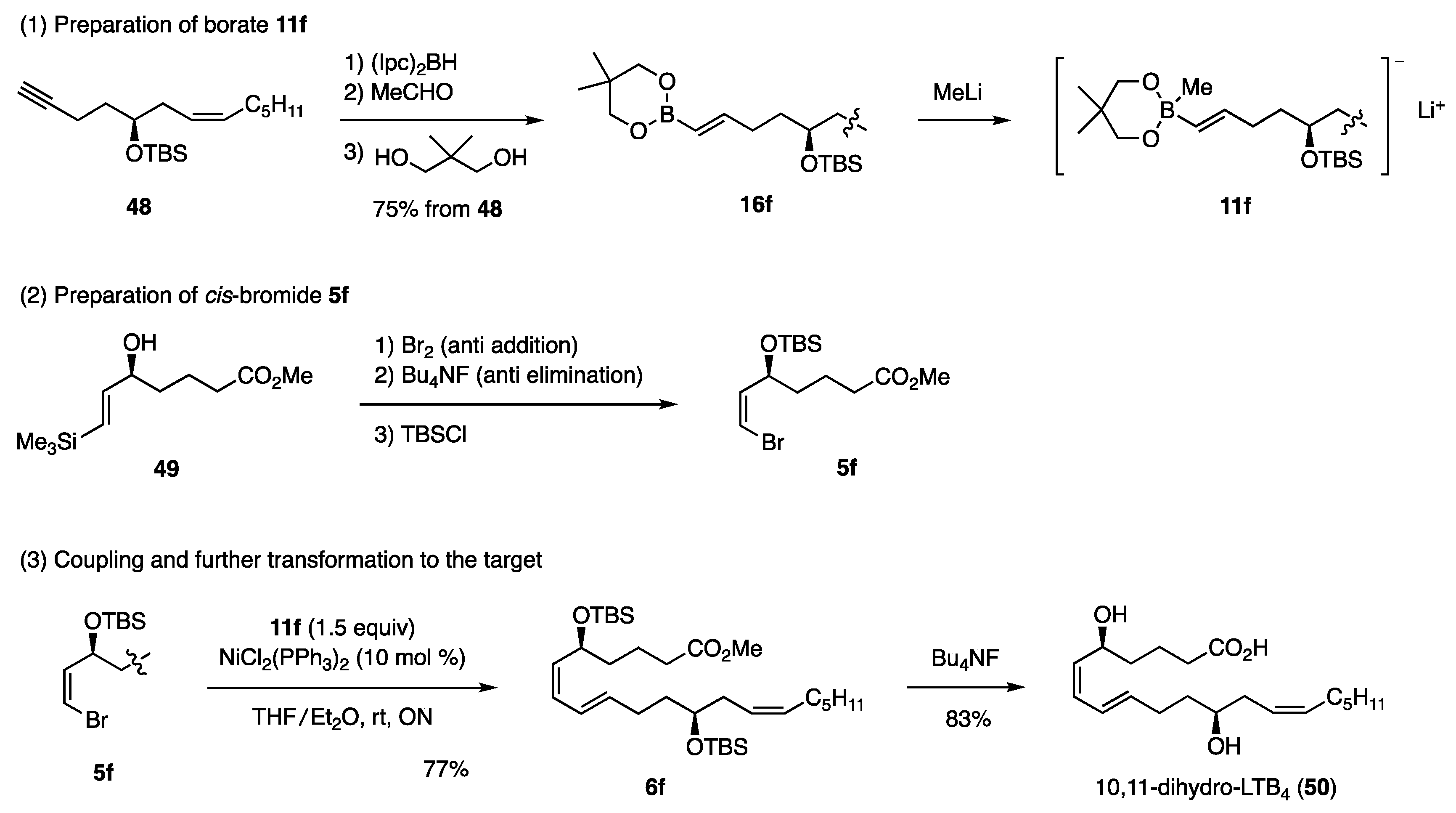
5.2.1. Synthesis of 10,11-Dihydro-LTB4
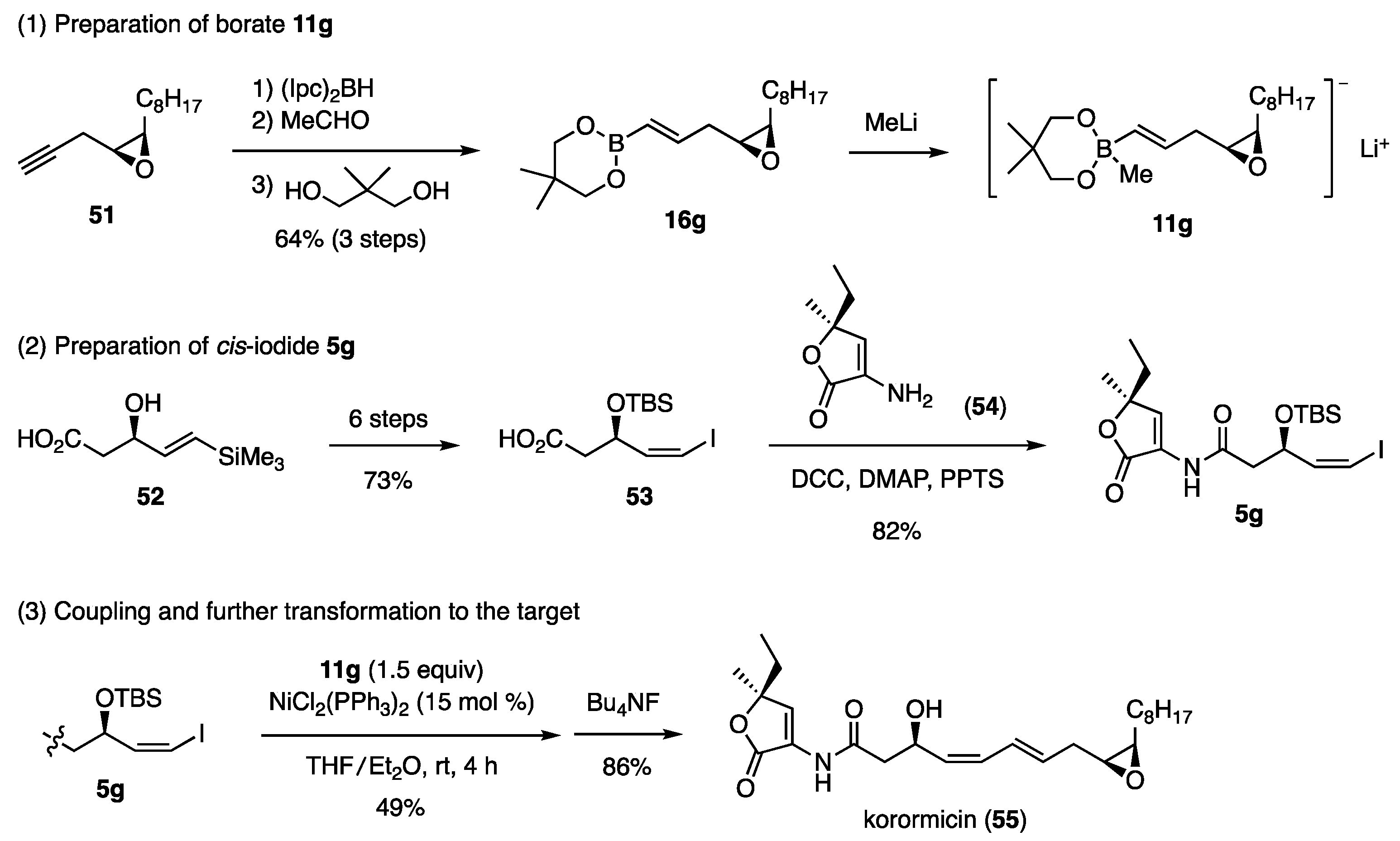
5.2.2. Synthesis of Korormicin
5.3. Functionalization of the Alkenyl-Alkenyl Coupling Products
5.4. C(sp2)–C(sp2) Coupling
6. Beyond Borates
6.1. Limitation of Borates
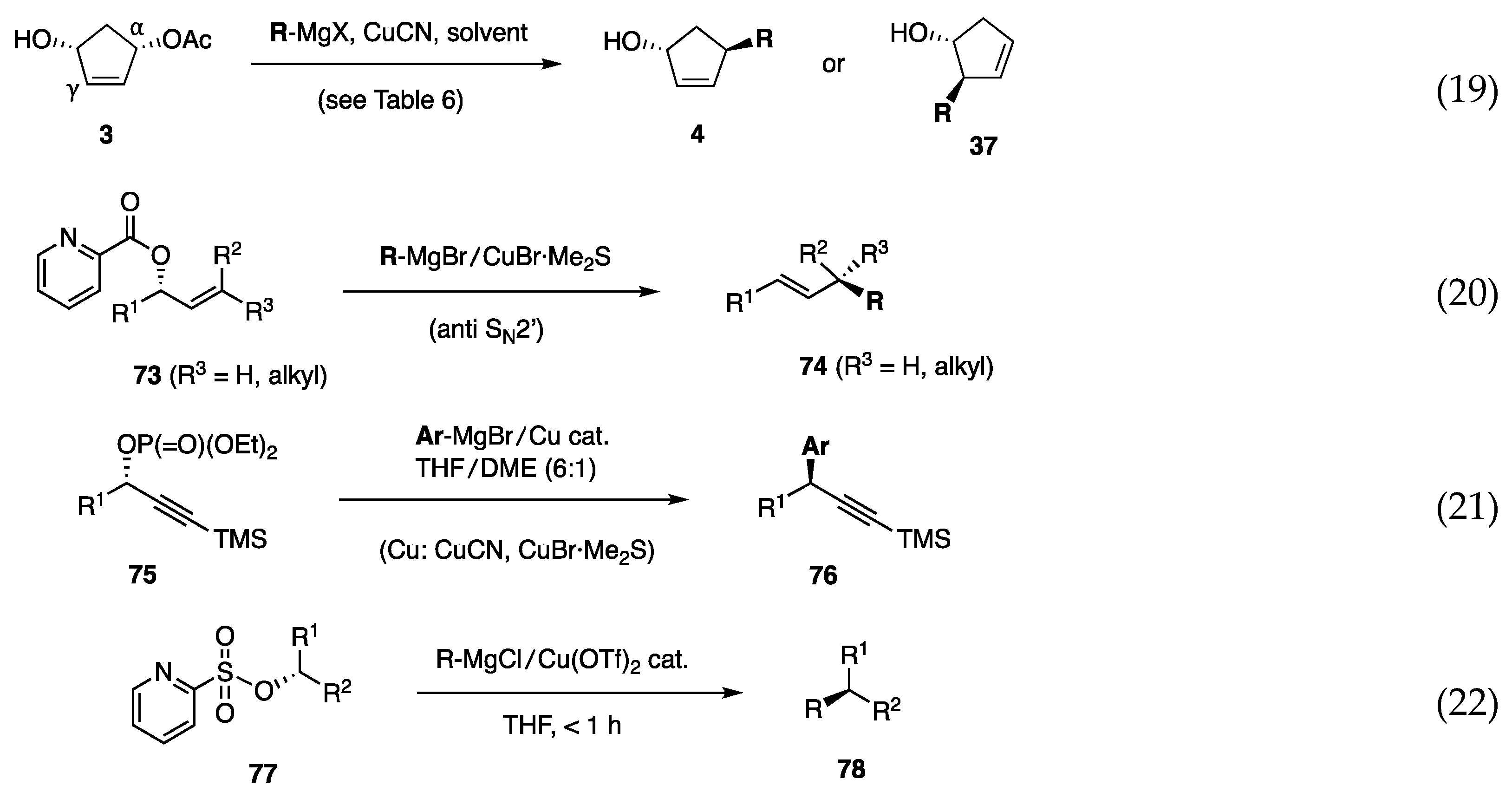
6.2. Substitution of Cyclopentenediol Monoacetate 3
6.3. Substitution of Secondary Allylic Picolinates

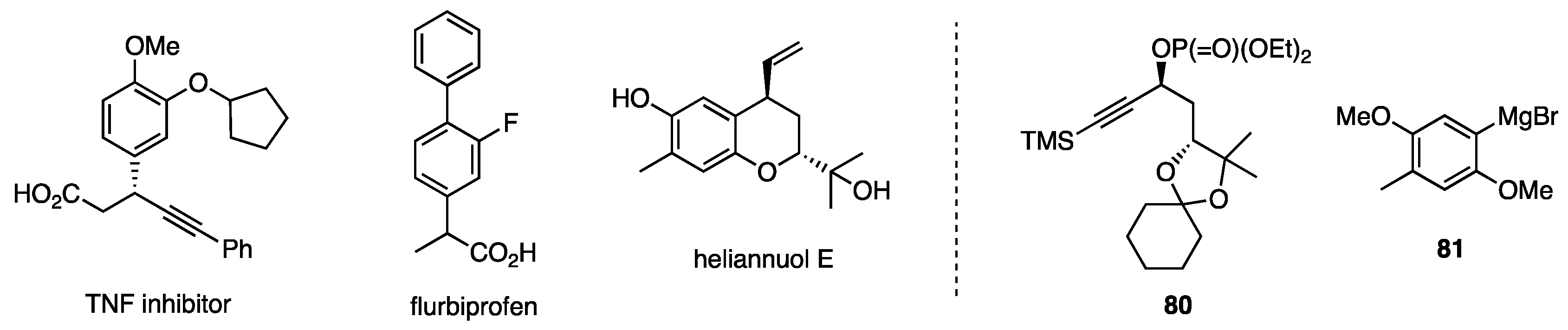
6.4. Substitution of Secondary Propargylic Phosphates

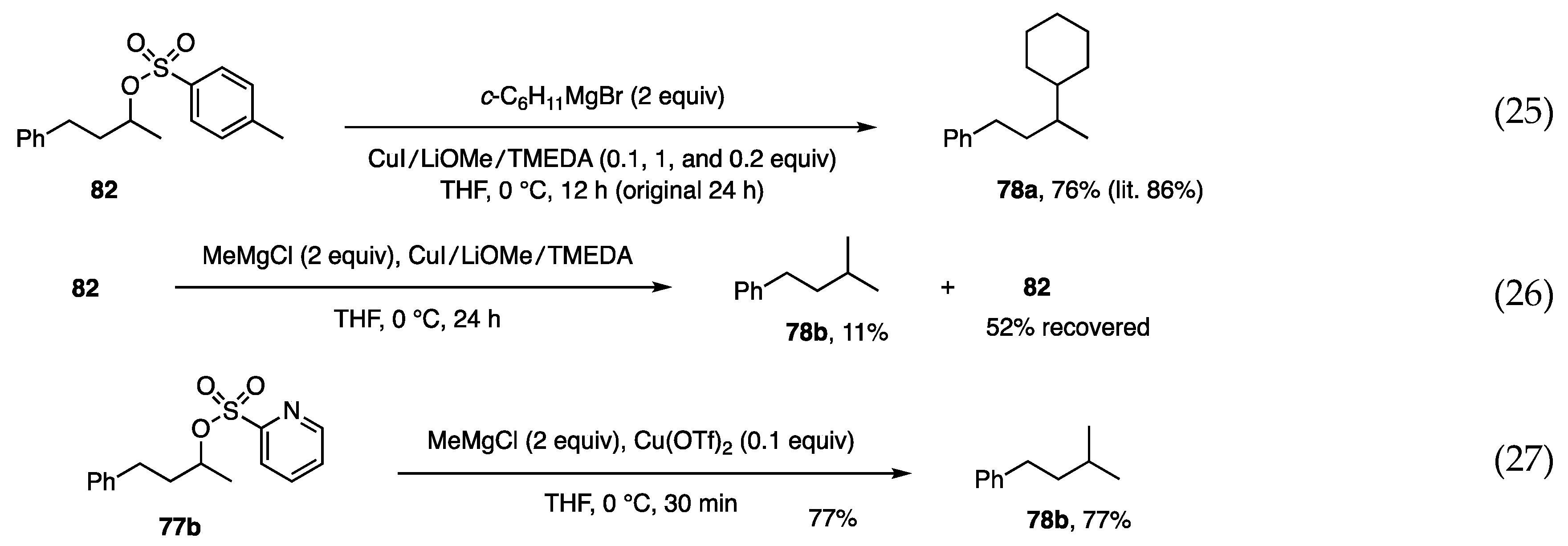
6.5. Substitution at Secondary Alkyl Carbons
7. Summary
Funding
Data Availability Statement
Conflicts of Interest
References
- Tsuji, J. Palladium Reagents and Catalysts: Innovations in Organic Synthesis; Wiley: Chichester, UK, 1995; pp. 1–574. ISBN 978-0-471-97202-0. [Google Scholar]
- Tsuji, J. Palladium Reagents and Catalysts: New Perspectives for the 21st Century; Wiley: Chichester, UK, 2004; pp. 1–670. ISBN 978-0-470-85032-9. [Google Scholar]
- Stork, G.; Tsuji, J. Lithium-Ammonia Reduction of α,β-Unsaturated Ketones. II. Formation and Alkylation of A β-Carbanion Intermediate. J. Am. Chem. Soc. 1961, 83, 2783–2784. [Google Scholar] [CrossRef]
- Stork, G.; Rosen, P.; Goldman, N.; Coombs, R.V.; Tsuji, J. Alkylation and Carbonation of Ketones by Trapping the Enolates from the Reduction of α,β-Unsaturated Ketones. J. Am. Chem. Soc. 1965, 87, 275–286. [Google Scholar] [CrossRef]
- Miyaura, N.; Yamada, K.; Suginome, H.; Suzuki, A. Novel and convenient method for the stereo-and regiospecific synthesis of conjugated alkadienes and alkenynes via the palladium-catalyzed cross-coupling reaction of 1-alkenylboranes with bromoalkenes and bromoalkynes. J. Am. Chem. Soc. 1985, 107, 972–980. [Google Scholar] [CrossRef]
- Pagett, A.B.; Lloyd-Jones, G.C. Suzuki–Miyaura Cross-Coupling. Org. React. 2019, 100, 547–620. [Google Scholar] [CrossRef]
- Lennox, A.J.J.; Lloyd-Jones, G.C. Selection of boron reagents for Suzuki–Miyaura coupling. Chem. Soc. Rev. 2013, 43, 412–443. [Google Scholar] [CrossRef]
- Ghorai, D.; Cristòfol, À.; Kleij, A.W. Nickel-Catalyzed Allylic Substitution Reactions: An Evolving Alternative. Eur. J. Inorg. Chem. 2021, 2022, e202100820. [Google Scholar] [CrossRef]
- Kobayashi, Y. Modern Organonickel Chemistry; Tamaru., Y., Ed.; Wiley: Chichester, UK, 2005; Chapter 3; pp. 56–101. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Ikeda, E. Nickel-catalysed substitution reactions of allylic carbonates with aryl- and alkenyl-borates. J. Chem. Soc. Chem. Commun. 1994, 1789–1790. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Mizojiri, R.; Ikeda, E. Nickel-Catalyzed Coupling Reaction of 1,3-Disubstituted Secondary Allylic Carbonates and Lithium Aryl- and Alkenylborates. J. Org. Chem. 1996, 61, 5391–5399. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Tokoro, Y.; Watatani, K. Zinc borates: Functionalized hard nucleophiles for coupling reaction with secondary allylic acetates. Eur. J. Org. Chem. 2000, 2000, 3825–3834. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Murugesh, M.G.; Nakano, M.; Takahisa, E.; Usmani, S.B.; Ainai, T. A New Method for Installation of Aryl and Alkenyl Groups onto a Cyclopentene Ring and Synthesis of Prostaglandins. J. Org. Chem. 2002, 67, 7110–7123. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Nakayama, Y.; Mizojiri, R. Nickel-catalyzed coupling reaction of sterically congested cis bromides and lithium alkenylborates. Tetrahedron 1998, 54, 1053–1062. [Google Scholar] [CrossRef]
- Kobayashi, Y.; William, A.D.; Mizojiri, R. Scope and limitation of the nickel-catalyzed coupling reaction between lithium borates and mesylate. J. Organometal. Chem. 2002, 653, 91–97. [Google Scholar] [CrossRef]
- Suzuki, M.; Oda, Y.; Noyori, R. Palladium(0) catalyzed reaction of 1,3-diene epoxides. A useful method for the site-specific oxygenation of 1,3-dienes. J. Am. Chem. Soc. 1979, 101, 1623–1625. [Google Scholar] [CrossRef]
- Grisso, B.A.; Johnson, J.R.; Mackenzie, P.B. Nickel-catalyzed, chlorotrialkylsilane-assisted conjugate addition of alkenyltributyltin reagents to α,β-unsaturated aldehydes. Evidence for a [1-[(trialkylsilyl)oxy]allyl]nickel(II) mechanism. J. Am. Chem. Soc. 1992, 114, 5160–5165. [Google Scholar] [CrossRef]
- Trost, B.M.; Verhoeven, T.R. Allylic alkylation. Palladium-catalyzed substitutions of allylic carboxylates. Stereo- and regiochemistry. J. Am. Chem. Soc. 1980, 102, 4730–4743. [Google Scholar] [CrossRef]
- Newton, R.F.; Reynolds, D.P.; Davies, J.; Kay, P.B.; Roberts, S.M.; Wallace, T.W. Enantiocomplementary total asymmetric syntheses of prostaglandin A2. J. Chem. Soc. Perkin Trans. 1983, 1, 683–685. [Google Scholar] [CrossRef]
- Noyori, R.; Suzuki, M. Organic Synthesis of Prostaglandins: Advancing Biology. Science 1993, 259, 44–45. [Google Scholar] [CrossRef]
- Tokoro, Y.; Kobayashi, Y. Realisation of highly stereoselective dihydroxylation of the cyclopentene in the synthesis of (−)-aristeromycin. Chem. Commun. 1999, 807–809. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Morita, M. Cutting-Edge of Organic Synthesis and Chemical Biology of Bioactive Molecules; Kobayashi, Y., Ed.; Springer Nature: Singapore, 2019; Chapter 9; pp. 193–231. ISBN 978-981-13-6243-9. Available online: https://link.springer.com/chapter/10.1007/978-981-13-6244-6_9 (accessed on 22 November 2022).
- Nakayama, Y.; Kumar, G.B.; Kobayashi, Y. Synthesis of 10,11-Dihydroleukotriene B4 Metabolites via a Nickel-Catalyzed Coupling Reaction of cis-Bromides and trans-Alkenyl Borates. J. Org. Chem. 2000, 65, 707–715. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Shimazaki, T.; Taguchi, H.; Sato, F. Highly stereocontrolled total synthesis of leukotriene B4, 20-hydroxyleukotriene B4, leukotriene B3, and their analogs. J. Org. Chem. 1990, 55, 5324–5335. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Yoshida, S.; Nakayama, Y. Total synthesis of korormicin. Eur. J. Org. Chem. 2001, 2001, 1873–1881. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Yoshida, S.; Asano, M.; Takeuchi, A.; Acharya, H.P. Nickel-Catalyzed Coupling Producing (2Z)-2,4-Alkadien-1-ols, Conversion to (E)-3-Alkene-1,2,5-triol Derivatives, and Synthesis of Decarestrictine D. J. Org. Chem. 2007, 72, 1707–1716. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Asano, M.; Yoshida, S.; Takeuchi, A. Stereoselective Synthesis of Decarestrictine D from a Previously Inaccessible (2Z,4E)-Alkadienyl Alcohol Precursor. Org. Lett. 2005, 7, 1533–1536. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; William, A.D. Coupling Reactions of α-Bromoalkenyl Phosphonates with Aryl Boronic Acids and Alkenyl Borates. Org. Lett. 2002, 4, 4241–4244. [Google Scholar] [CrossRef]
- Kobayashi, Y.; William, A.D. Palladium- and Nickel-Catalyzed Coupling Reactions of α-Bromoalkenylphosphonates with Arylboronic Acids and Lithium Alkenylborates. Adv. Synth. Catal. 2004, 346, 1749–1757. [Google Scholar] [CrossRef]
- Ito, M.; Matsuumi, M.; Murugesh, M.G.; Kobayashi, Y. Scope and Limitation of Organocuprates, and Copper or Nickel Catalyst-Modified Grignard Reagents for Installation of an Alkyl Group onto cis-4-Cyclopentene-1,3-diol Monoacetate. J. Org. Chem. 2001, 66, 5881–5889. [Google Scholar] [CrossRef] [PubMed]
- Kiyotsuka, Y.; Acharya, H.P.; Katayama, Y.; Hyodo, T.; Kobayashi, Y. Picolinoxy Group, a New Leaving Group for anti SN2′ Selective Allylic Substitution with Aryl Anions Based on Grignard Reagents. Org. Lett. 2008, 10, 1719–1722. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Takashima, Y.; Motoyama, Y.; Isogawa, Y.; Katagiri, K.; Tsuboi, A.; Ogawa, N. α- and γ-Regiocontrol and Enantiospecificity in the Copper-Catalyzed Substitution Reaction of Propargylic Phosphates with Grignard Reagents. Chem. Eur. J. 2020, 27, 3779–3785. [Google Scholar] [CrossRef]
- Shinohara, R.; Morita, M.; Ogawa, N.; Kobayashi, Y. Use of the 2-Pyridinesulfonyloxy Leaving Group for the Fast Copper-Catalyzed Coupling Reaction at Secondary Alkyl Carbons with Grignard Reagents. Org. Lett. 2019, 21, 3247–3251. [Google Scholar] [CrossRef]
- Ainai, T.; Matsuumi, M.; Kobayashi, Y. Efficient Total Synthesis of 12-oxo-PDA and OPC-8:0. J. Org. Chem. 2003, 68, 7825–7832. [Google Scholar] [CrossRef]
- Nonaka, H.; Ogawa, N.; Maeda, N.; Wang, Y.-G.; Kobayashi, Y. Stereoselective synthesis of epi-jasmonic acid, tuberonic acid, and 12-oxo-PDA. Org. Biomol. Chem. 2010, 8, 5212–5223. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, N.; Kobayashi, Y. Synthesis of the amino acid conjugates of epi-jasmonic acid. Amino Acids 2011, 42, 1955–1966. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Nakata, K.; Ainai, T. New Reagent System for Attaining High Regio-and Stereoselectivities in Allylic Displacement of 4-Cyclopentene-1,3-diol Monoacetate with Aryl- and Alkenylmagnesium Bromides. Org. Lett. 2004, 7, 183–186. [Google Scholar] [CrossRef]
- Nakata, K.; Kiyotsuka, Y.; Kitazume, T.; Kobayashi, Y. Realization of Anti-SN2′ Selective Allylation of 4-Cyclopentene-1,3-diol Monoester with Aryl- and Alkenyl-Zinc Reagents. Org. Lett. 2008, 10, 1345–1348. [Google Scholar] [CrossRef] [PubMed]
- Nakata, K.; Kobayashi, Y. Aryl-and Alkenyllithium Preparations and Copper-Catalyzed Reaction between the Derived Magnesium Reagents and the Monoacetate of 4-Cyclopentene-1,3-diol. Org. Lett. 2005, 7, 1319–1322. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y. Alkyl Pyridinesulfonates and Allylic Pyridinecarboxylates, New Boosters for the Substitution at Secondary Carbons. Heterocycles 2020, 100, 499. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Shimoda, M. Cutting-Edge of Organic Synthesis and Chemical Biology of Bioactive Molecules; Kobayashi, Y., Ed.; Springer Nature: Singapore, 2019; Chapter 7; pp. 145–169. ISBN 978-981-13-6243-9. Available online: https://link.springer.com/chapter/10.1007/978-981-13-6244-6_7 (accessed on 22 November 2022).
- Kiyotsuka, Y.; Kobayashi, Y. Formation of Chiral C(sp3)−C(sp) Bond by Allylic Substitution of Secondary Allylic Picolinates and Alkynyl Copper Reagents. J. Org. Chem. 2009, 74, 7489–7495. [Google Scholar] [CrossRef]
- Wang, Q.; Kobayashi, Y. Allylic Substitution on Cyclopentene and -hexene Rings with Alkynylcopper Reagents. Org. Lett. 2011, 13, 6252–6255. [Google Scholar] [CrossRef]
- Kiyotsuka, Y.; Katayama, Y.; Acharya, H.P.; Hyodo, T.; Kobayashi, Y. New General Method for Regio- and Stereoselective Allylic Substitution with Aryl and Alkenyl Coppers Derived from Grignard Reagents. J. Org. Chem. 2009, 74, 1939–1951. [Google Scholar] [CrossRef]
- Ozaki, T.; Kobayashi, Y. Synthesis of (−)-mesembrine using the quaternary carbon-constructing allylic substitution. Org. Chem. Front. 2015, 2, 328–335. [Google Scholar] [CrossRef]
- Kawashima, H.; Kaneko, Y.; Sakai, M.; Kobayashi, Y. Synthesis of Cyclobakuchiols A, B, and C by Using Conformation-Controlled Stereoselective Reactions. Chem. Eur. J. 2013, 20, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Ozaki, T. Concise Synthesis of (–)-Axenol by Using Stereocontrolled Allylic Substitution. Synlett 2015, 26, 1085–1088. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Wada, K.; Sakai, M.; Kawashima, H.; Ogawa, N. Efficient Synthesis of Anastrephin via the Allylic Substitution for Quaternary Carbon Construction. Synlett 2016, 27, 1428–1432. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Saeki, R.; Nanba, Y.; Suganuma, Y.; Morita, M.; Nishimura, K. Synthesis of the Verapamil Intermediate through the Quaternary Carbon-Constructing Allylic Substitution. Synlett 2017, 28, 2655–2659. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Yamaguchi, K.; Morita, M. Regio-and stereoselective SN2′ reaction of an allylic picolinate in the synthesis of LY426965. Tetrahedron 2018, 74, 1826–1831. [Google Scholar] [CrossRef]
- Hyodo, T.; Kiyotsuka, Y.; Kobayashi, Y. Synthesis of the Active Form of Loxoprofen by Using Allylic Substitutions in Two Steps. Org. Lett. 2009, 11, 1103–1106. [Google Scholar] [CrossRef]
- Takashima, Y.; Isogawa, Y.; Tsuboi, A.; Ogawa, N.; Kobayashi, Y. Synthesis of a TNF inhibitor, flurbiprofen and an i-Pr analogue in enantioenriched forms by copper-catalyzed propargylic substitution with Grignard reagents. Org. Biomol. Chem. 2021, 19, 9906–9909. [Google Scholar] [CrossRef]
- Ogawa, N.; Uematsu, C.; Kobayashi, Y. Stereoselective Synthesis of (–)-Heliannuol E by α-Selective Propargyl Substitution. Synlett 2021, 32, 2071–2074. [Google Scholar] [CrossRef]
- Studte, C.; Breit, B. Zinc-Catalyzed Enantiospecific sp3–sp3 Cross-Coupling of α-Hydroxy Ester Triflates with Grignard Reagents. Angew. Chem. Int. Ed. 2008, 47, 5451–5455. [Google Scholar] [CrossRef]
- Brand, G.J.; Studte, C.; Breit, B. Iterative Synthesis of (Oligo) deoxypropionates via Zinc-Catalyzed Enantiospecific sp3−sp3 Cross-Coupling. Org. Lett. 2009, 11, 4668–4670. [Google Scholar] [CrossRef]
- Yang, C.-T.; Zhang, Z.-Q.; Liang, J.; Liu, J.-H.; Lu, X.-Y.; Chen, H.-H.; Liu, L. Copper-Catalyzed Cross-Coupling of Nonactivated Secondary Alkyl Halides and Tosylates with Secondary Alkyl Grignard Reagents. J. Am. Chem. Soc. 2012, 134, 11124–11127. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Oda, A.; Bobinski, T.; Li, H.; Matsueda, Y.; Negishi, E.-I. Highly Efficient, Convergent, and Enantioselective Synthesis of Phthioceranic Acid. Angew. Chem. Int. Ed. 2015, 54, 9319–9322. [Google Scholar] [CrossRef] [PubMed]

































{kind=link}
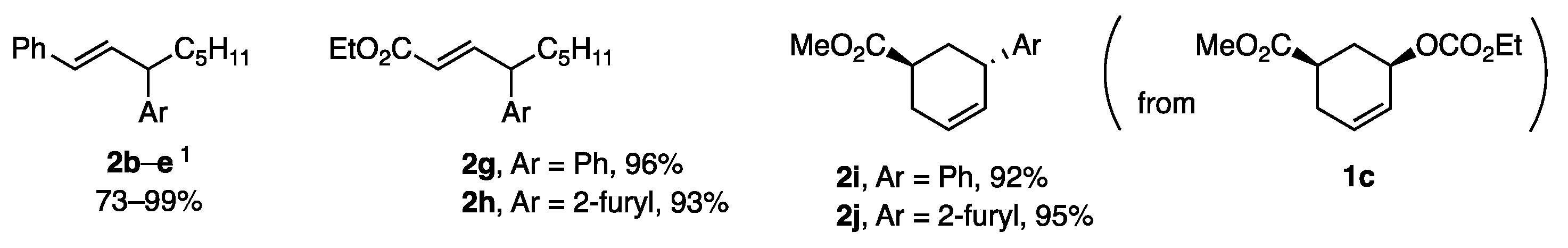{kind=link}
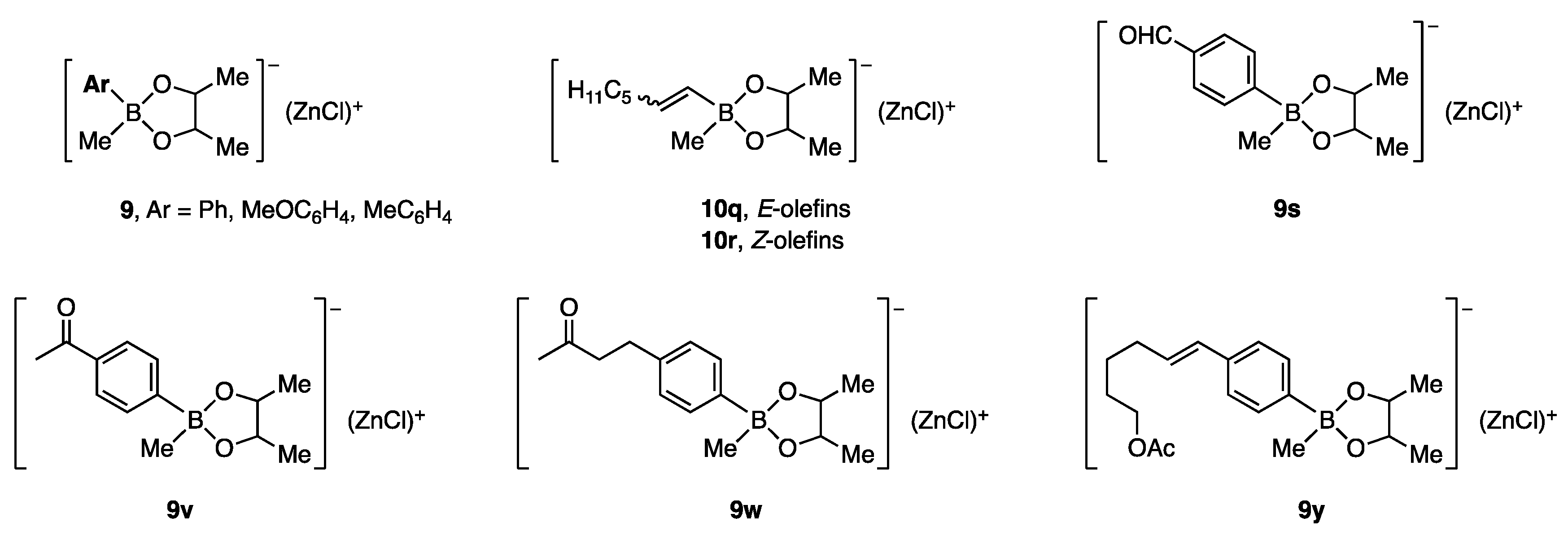{kind=link}
{kind=link}
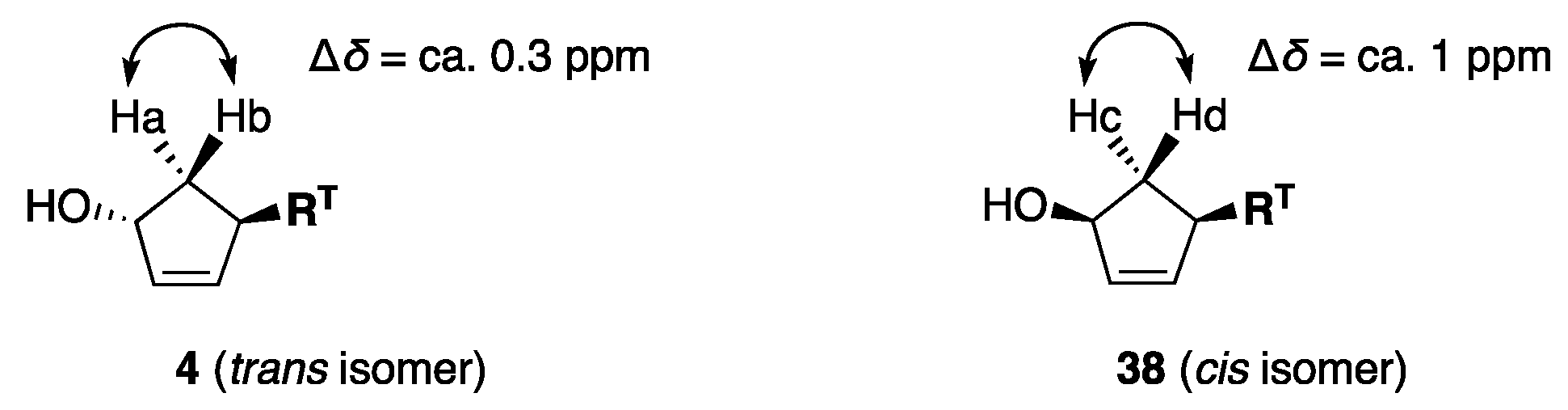{kind=link}
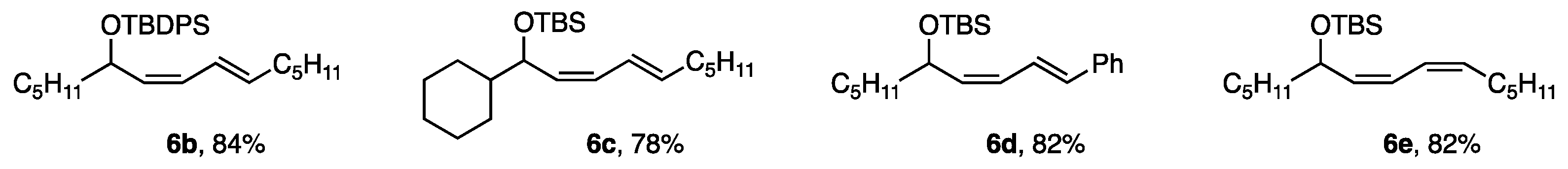{kind=link}
{kind=link}
{kind=link}
{kind=link}
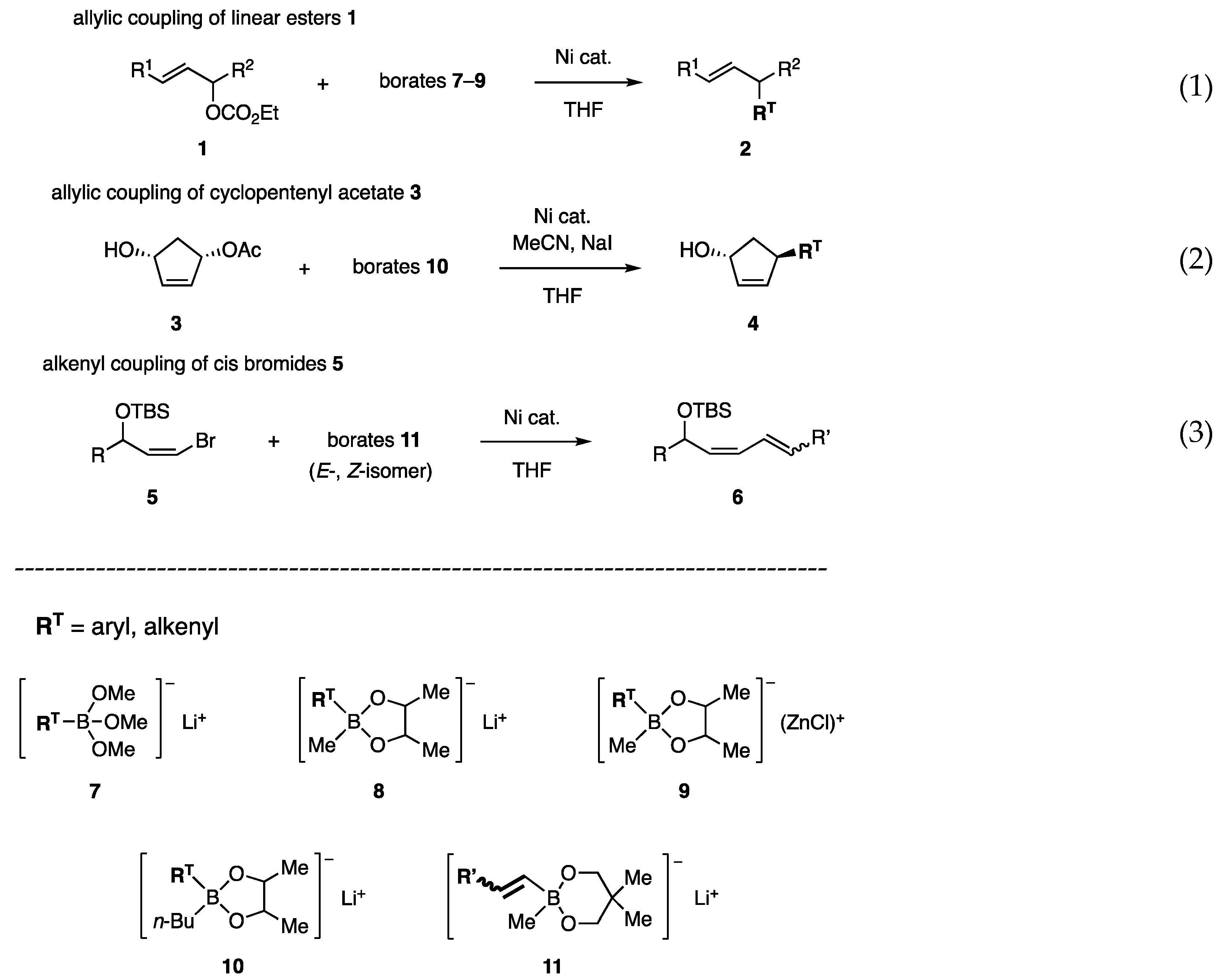{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Cat. | 2, Ar | 2 (%) | 19 (%) |
|---|---|---|---|---|
| 1 | Pd(PPh3)2 | 2a, Ph | <10 | 66 |
| 2 | Pd(dppf) | 2a, Ph | 0 | 54 |
| 3 | NiCl2(PPh3)2 | 2a, Ph | 97 | 0 |
| 4 | NiCl2(dppf) | 2a, Ph | 98 | 0 |
| 5 | NiCl2(dppf) | 2b, 2-MeOC6H4 | 86 | 0 |
| 6 | NiCl2(dppf) | 2c, 3-MeOC6H4 | 81 | 0 |
| 7 | NiCl2(dppf) | 2d, 4-MeOC6H4 | 89 | 0 |
| 8 | NiCl2(dppf) | 2e, 2-furyl | 88 | 0 |

| Entry | Zinc Reagent | Additive | 2a, Yield (%) | Byproducts 1 (%) |
|---|---|---|---|---|
| 1 | MeZnCl | - | 88 | 9 |
| 2 | MeZnBr | - | 85 | 6 |
| 3 | MeZnCl | DMI | 87 | 0 |
| 4 | MeZnCl | DMF | 80 | 0 |

| Entry | Suffix for RT | RT | Yield | 4/37 | |
|---|---|---|---|---|---|
| Additive | No Additive | ||||
 | |||||
| 1 1 | a | R = H | 84 | 13:1 | 0.9:1 |
| 2 | b | R = 3-Me | 80 | 7:1 | 1.2:1 |
| 3 | c | R = 4-Me | 81 | 12:1 | 1.5:1 |
| 4 | d | R = 3-MeO | 84 | 6:1 | 0.7:1 |
| 5 | e | R = 4-MeO | 81 | 9:1 | – |
| 6 | f |  | 89 | 6:1 | 3.0:1 |
| 7 | g |  | 85 | 5:1 | 1.3:1 |
 | |||||
| 8 | h | R = n-C5H11 | 67 | 15:1 | 5.4:1 |
| 9 | i | R = c-C6H11 | 85 | 15:1 | 5.5:1 |
| 10 | j | R = CH2OPh | 80 | 17:1 | 8.0:1 |
| 11 | k |  | 85 | 6:1 | 2.5:1 |

| Entry | Borate | Diol Ligand | Temp. | 6a, Yield (%) | Conversion (%) |
|---|---|---|---|---|---|
| 1 | 47A | A | 40–45 °C 1 | 76 | 100 |
| 2 | 47C | C | rt | - | 0 |
| 3 | 47D | D | rt | - | 68 |
| 4 | 47G (=11a) | G | rt | 90 | 100 |
| 5 | 47H | H | rt | - | 79 |
| 6 | 47I | I | rt | - | 0 |

| Entry | Substrate | Borate | Cat. | 67a, Yield (%) | 67a/68 1 |
|---|---|---|---|---|---|
| 1 | 65a | 8a | NiCl2(PPh3)2 | 24 | 57:43 |
| 2 | 65a | 8a | NiCl2(dppf) | 32 | 55:45 |
| 3 | 65a | 10a | NiCl2(PPh3)2 | 95 | >95:5 |
| 4 | 66 | 10a | NiCl2(PPh3)2 | 83 | >95:5 |

| Entry | n-BuMgCl/CuCN | Solvent | 4m/37m | Yield (%) |
|---|---|---|---|---|
| 1 | 1:1 | Et2O | 14:86 | 37 |
| 2 | 2:1 | Et2O | 7:93 | 85 |
| 3 | 10:1 | Et2O | 8:92 | 82 |
| 4 | 1:1 | THF | 7:93 | 97 |
| 5 | 2:1 | THF | 93:7 | 94 |
| 6 | 10:1 | THF | 94:6 | 100 |
| 7 | 1:1 | Et2O | 4:96 | 77 |
| 8 | 2:1 | Et2O | 6:94 | 88 |
| 9 | 10:1 | Et2O | 5:95 | 94 |
| 10 | 1:1 | THF | 10:90 | 89 |
| 11 | 2:1 | THF | 71:29 | 98 |
| 12 | 10:1 | THF | 73:27 | 96 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kobayashi, Y. Coupling Reactions on Secondary Allylic, Propargylic, and Alkyl Carbons Using Organoborates/Ni and RMgX/Cu Reagents. Catalysts 2023, 13, 132. https://doi.org/10.3390/catal13010132
Kobayashi Y. Coupling Reactions on Secondary Allylic, Propargylic, and Alkyl Carbons Using Organoborates/Ni and RMgX/Cu Reagents. Catalysts. 2023; 13(1):132. https://doi.org/10.3390/catal13010132
Chicago/Turabian StyleKobayashi, Yuichi. 2023. "Coupling Reactions on Secondary Allylic, Propargylic, and Alkyl Carbons Using Organoborates/Ni and RMgX/Cu Reagents" Catalysts 13, no. 1: 132. https://doi.org/10.3390/catal13010132
APA StyleKobayashi, Y. (2023). Coupling Reactions on Secondary Allylic, Propargylic, and Alkyl Carbons Using Organoborates/Ni and RMgX/Cu Reagents. Catalysts, 13(1), 132. https://doi.org/10.3390/catal13010132