
Improved the Methanol Electro-Oxidation and Carbon Monoxide Tolerance for Direct Methanol Fuel Cells Using Strontium Molybdate



Abstract
:1. Introduction
2. Results and Discussion
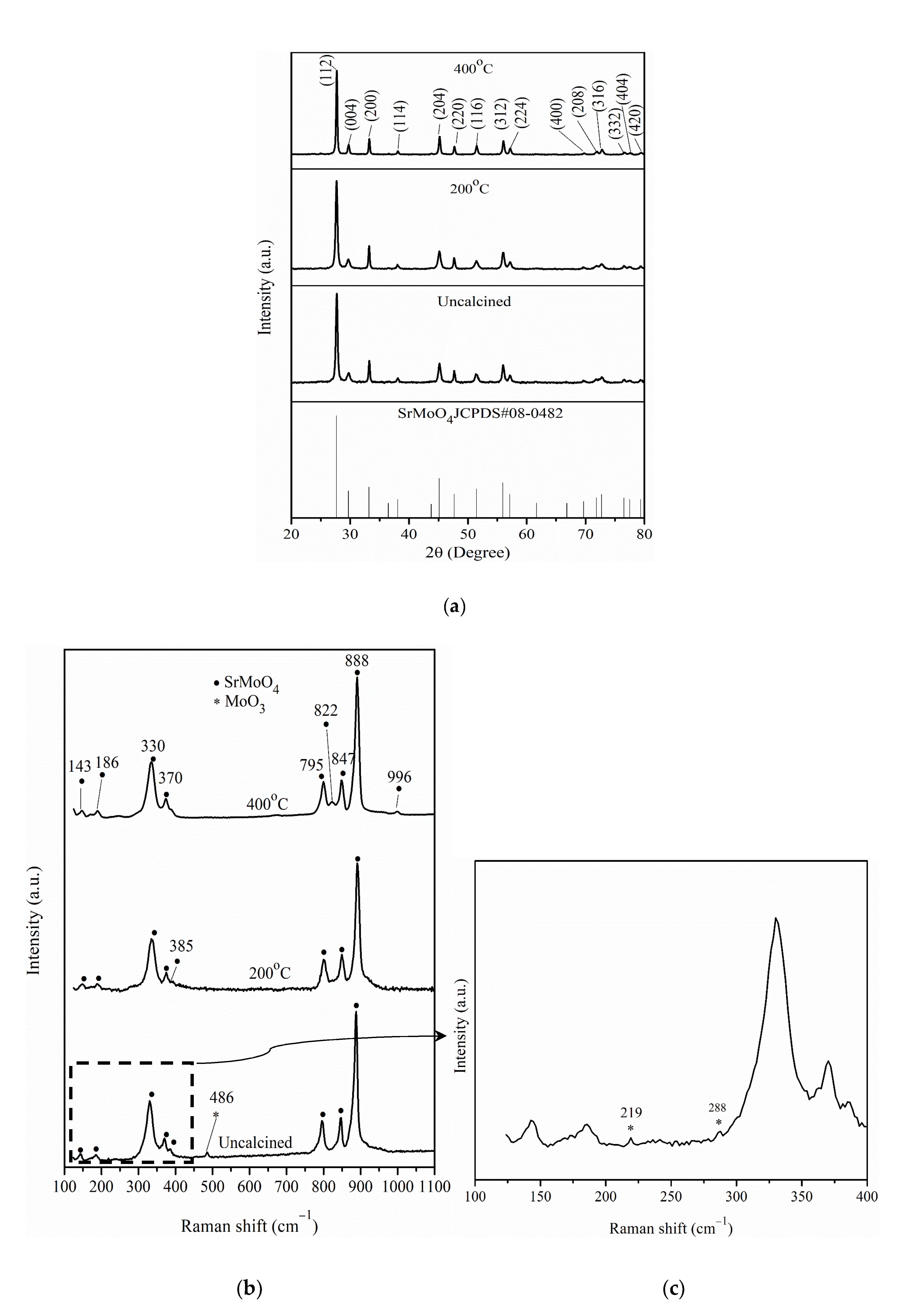
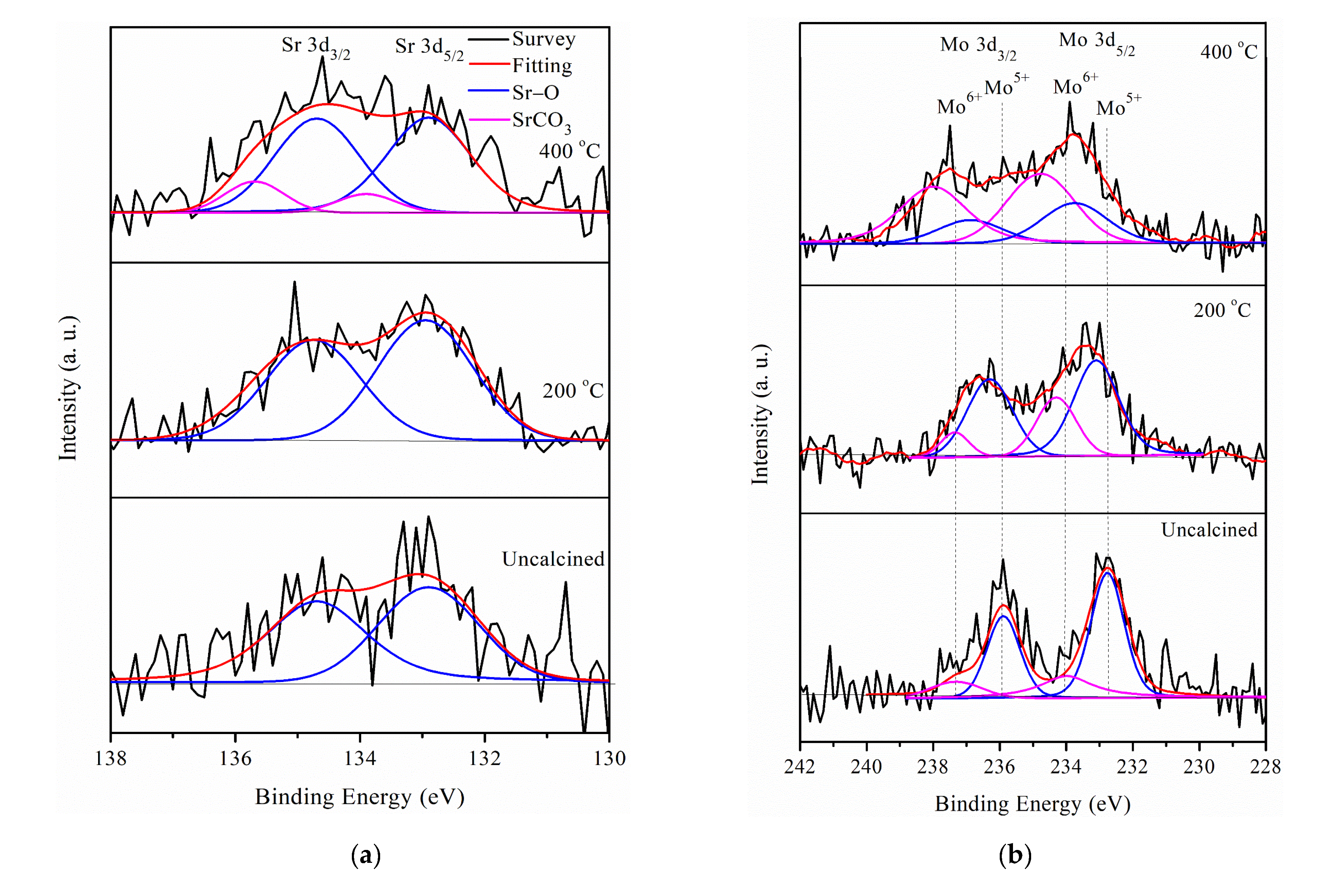
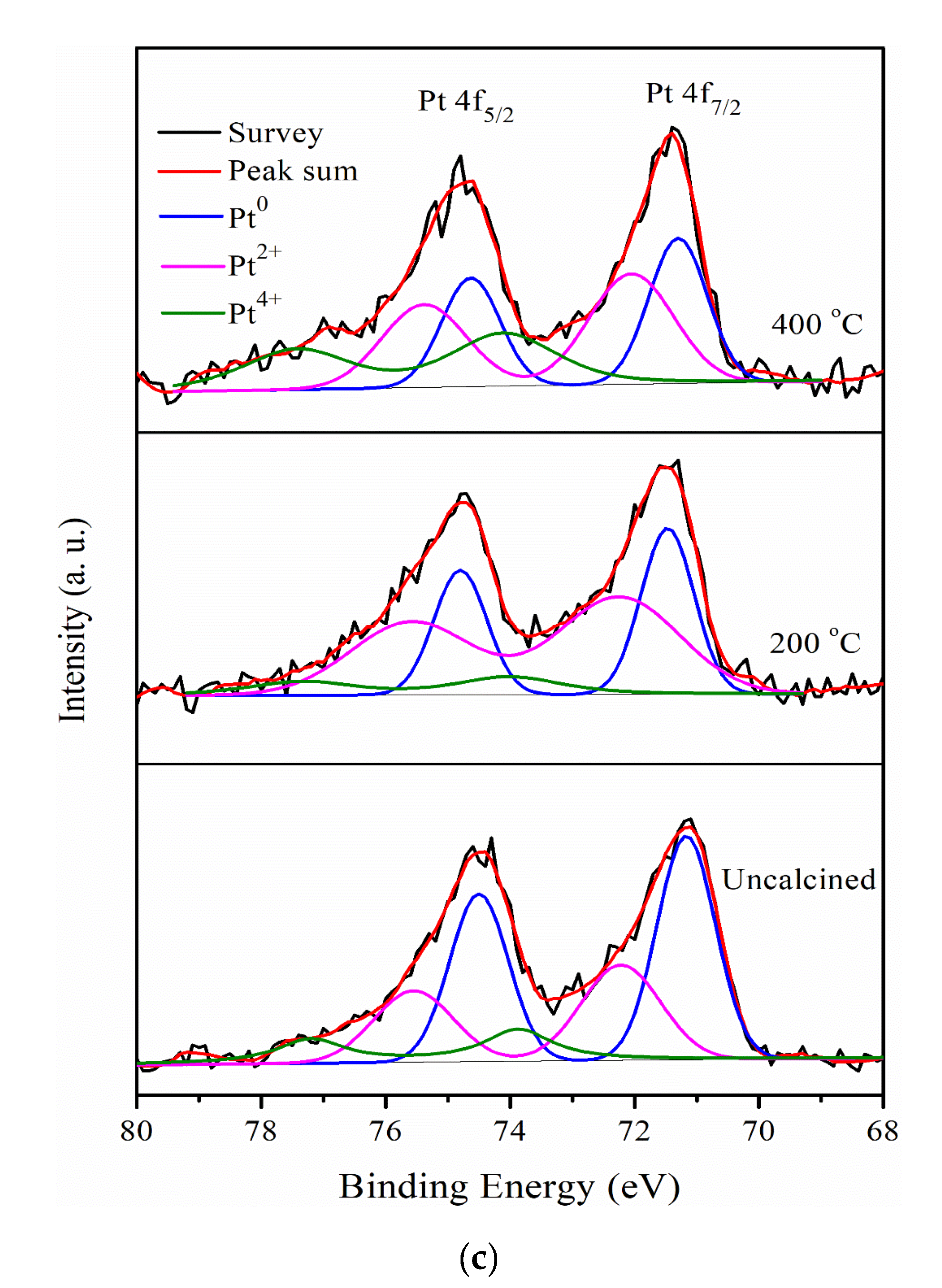
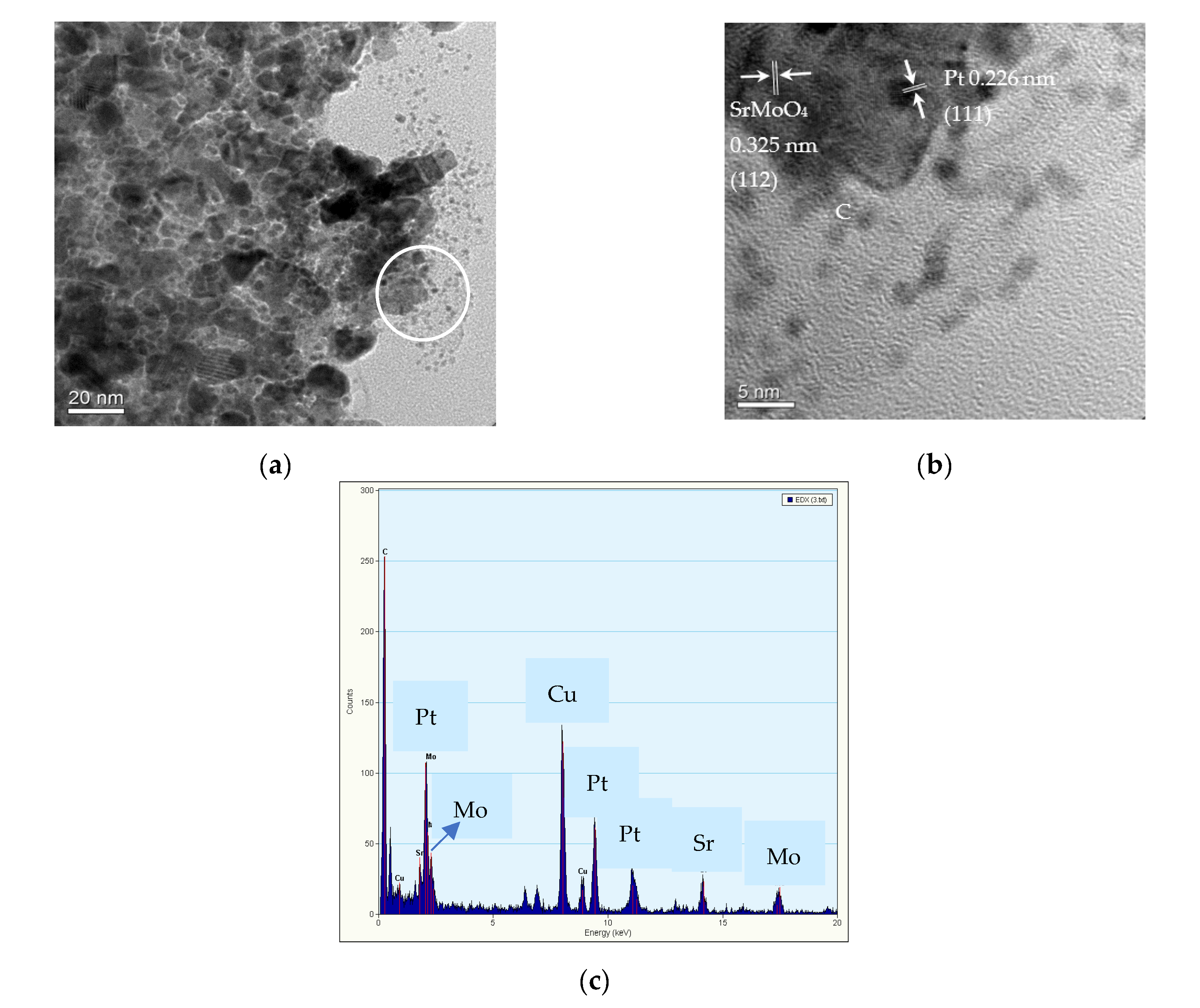
2.1. Characterization of Pt/different Calcination Temperature Sr0.5Mo0.5O4−δ-C Electrocatalysts
2.2. MOR and CO Tolerance for Strontium Molybdate with Different Calcination Temperatures
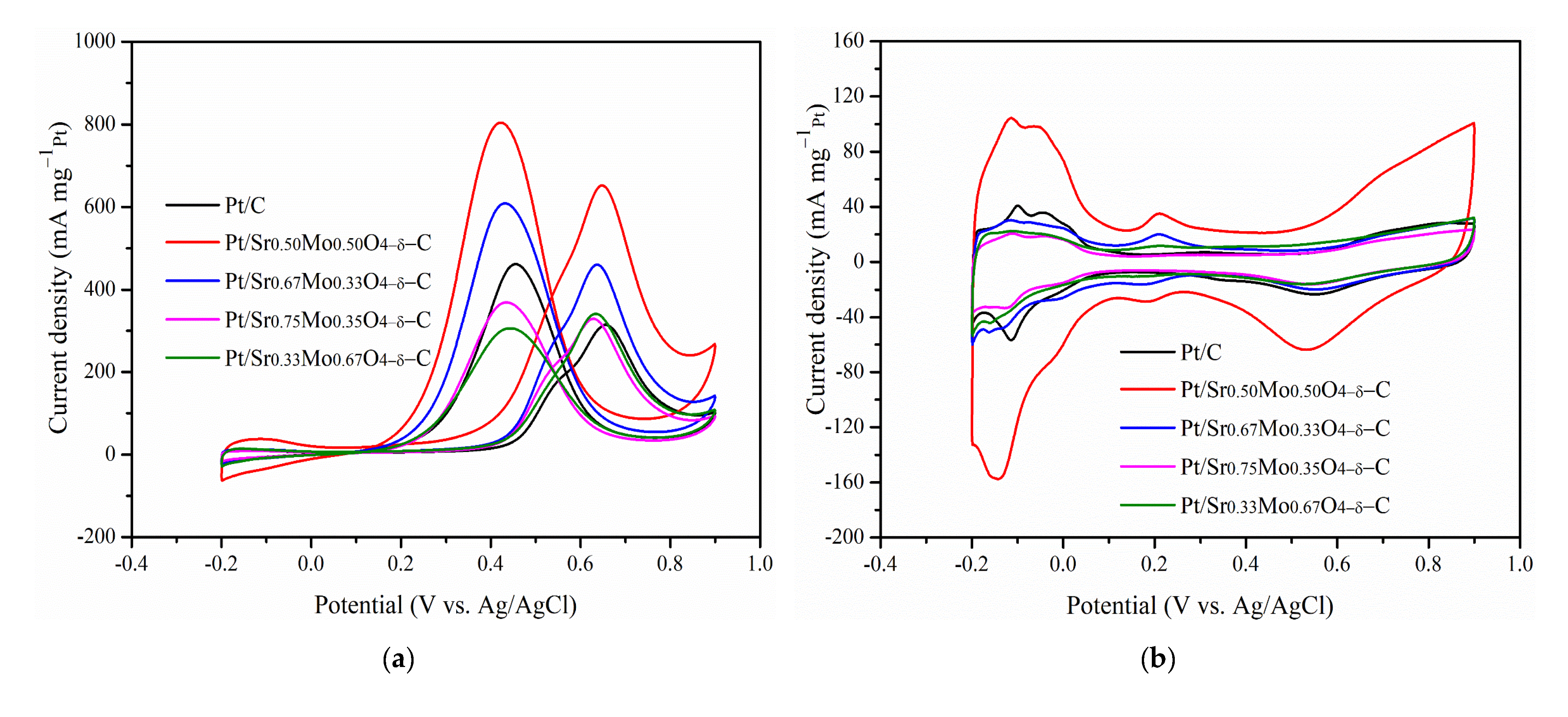
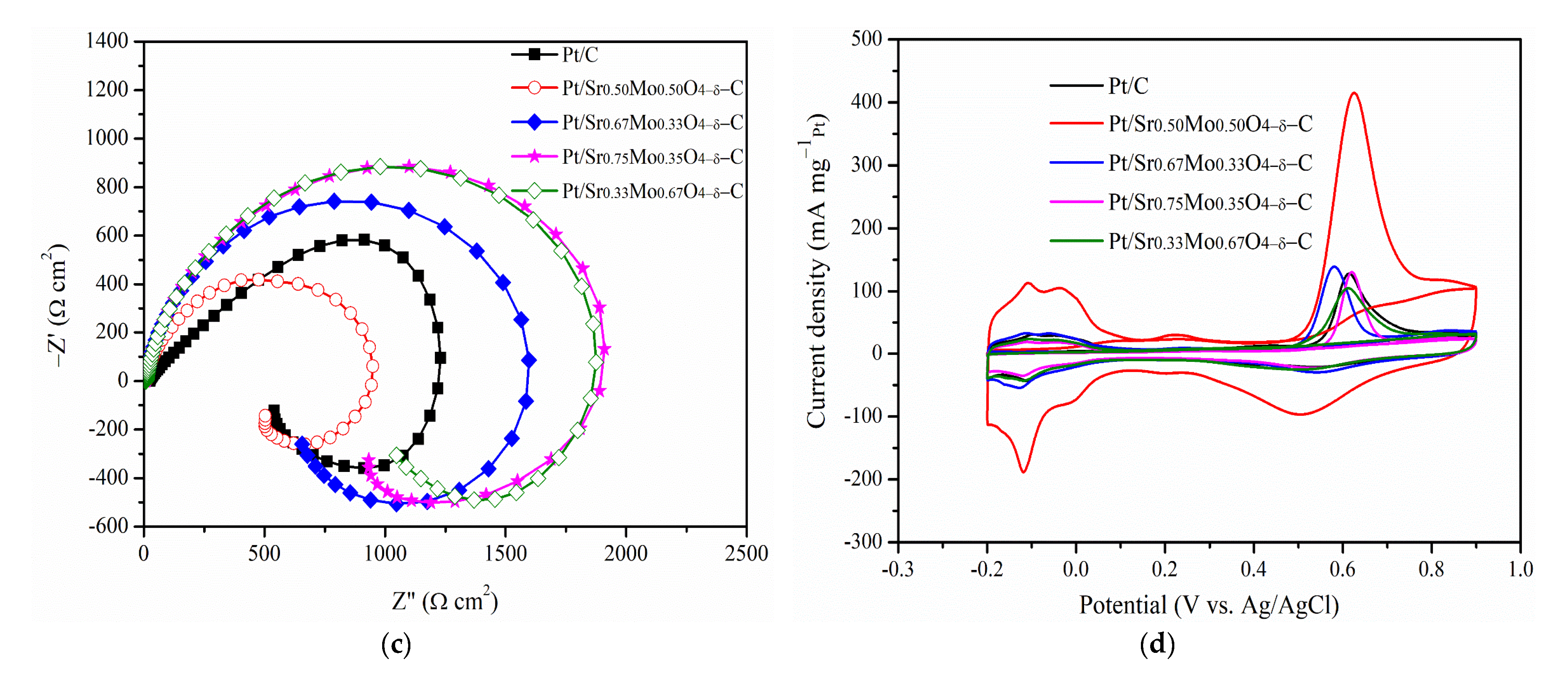
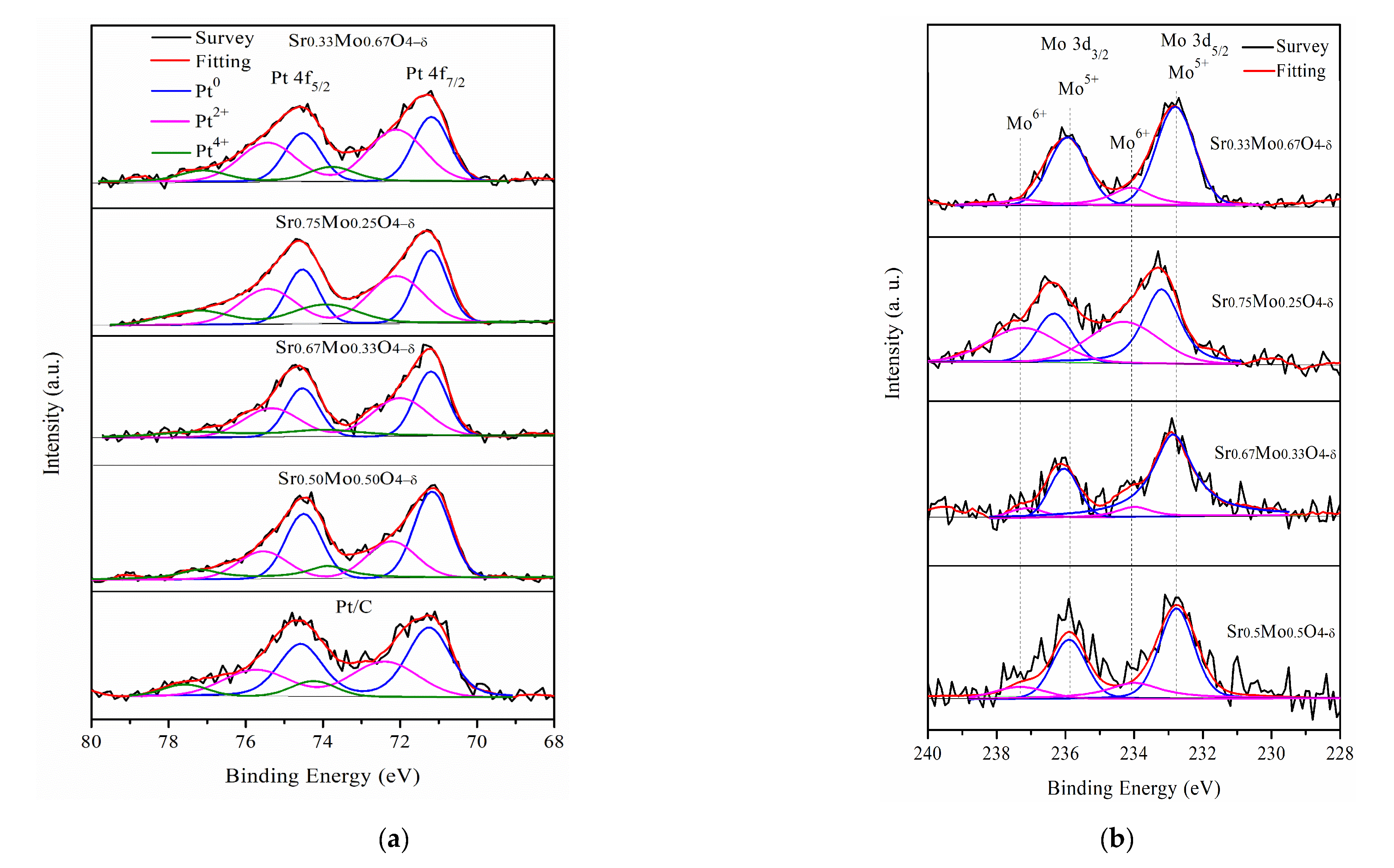
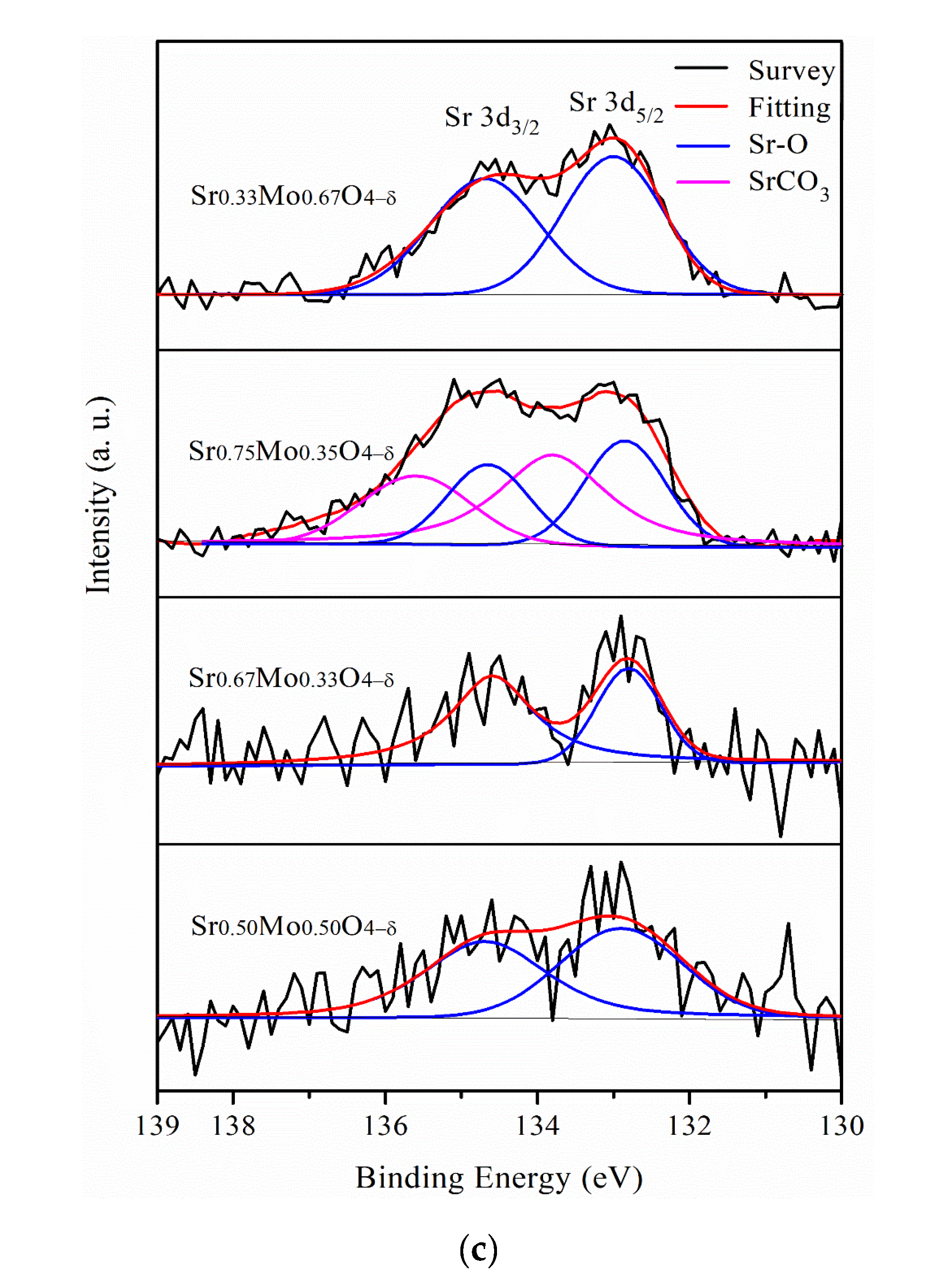
2.3. MOR and CO Tolerance of Strontium Molybdate Oxide Prepared with Different Sr and Mo Ratio Contacts
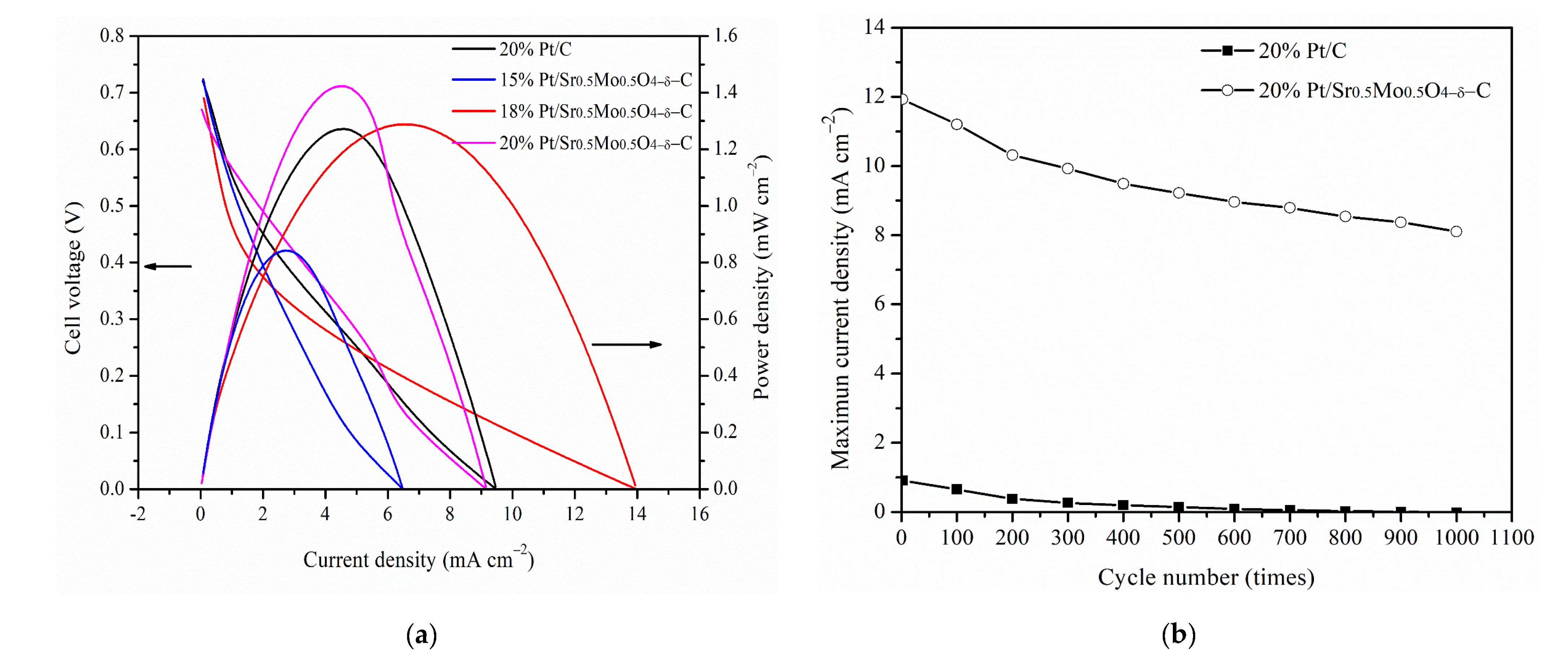
2.4. DMFCs Performance and Cycling Performance Tests
3. Materials and Methods
3.1. Preparation of SrMoO4
3.2. Pt/SrMoO4-C Preparation
3.3. Material Characterization
3.4. Electrochemical Measurements
3.5. Membrane Electrode Assembly Fabrication and Single-Cell Performance Testing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Simoes, S.; Nijs, W.; Ruiz, P.; Sgobbi, A.; Thiel, C. Comparing policy routes for low-carbon power technology deployment in EU-an energy system analysis. Energy Policy 2017, 101, 353–365. [Google Scholar] [CrossRef]
- Samimi, F.; Hamedi, N.; Rahimpour, M.R. Green methanol production process from indirect CO2 conversion: RWGS reactor versus RWGS membrane reactor. J. Environ. Chem. Eng. 2019, 7, 102813. [Google Scholar] [CrossRef]
- Rifal, M.; Sinaga, N. Impact of methanol-gasoline fuel blend on the fuel consumption and exhaust emission of a SI engine. In Proceedings of the 3rd International Conference on Advanced Materials Science and Technology (ICAMST 2015), Semarang, Indonesia, 6–7 October 2015. [Google Scholar]
- Du, C.Y.; Zhao, T.S.; Yang, W.W. Effect of methanol crossover on the cathode behavior of a DMFC: A half-cell investigation. Electrochim. Acta 2007, 52, 5266–5271. [Google Scholar] [CrossRef]
- Yajima, T.; Uchida, H.; Watanabe, M. In-Situ ATR-FTIR spectroscopic study of electro-oxidation of methanol and adsorbed CO at Pt-Ru alloy. J. Phys. Chem. B 2004, 108, 2654–2659. [Google Scholar] [CrossRef]
- Seiler, T.; Savinova, E.R.; Friedrich, K.A.; Stimming, U. Poisoning of PtRu/C catalysts in the anode of a direct methanol fuel cell: A DEMS study. Electrochim. Acta 2004, 49, 3927–3936. [Google Scholar] [CrossRef]
- Kaur, A.; Kaur, G.; Singh, P.P.; Kaushal, S. Supported bimetallic nanoparticles as anode catalysts for direct methanol fuel cells: A review. Int. J. Hydrogen Energy 2021, 46, 15820–15849. [Google Scholar] [CrossRef]
- Cheng, X.; Shi, Z.; Glass, N.; Zhang, L.; Zhang, J.; Songa, D.; Liu, Z.S.; Wang, H.; Shen, J. A review of PEM hydrogen fuel cell contamination: Impacts, mechanisms, and mitigation. J. Power Sources 2007, 16, 739–756. [Google Scholar] [CrossRef]
- Yamanaka, T.; Takeguchi, T.; Wang, G.; Muhamad, E.N.; Ueda, W. Particle size dependence of CO tolerance of anode PtRu catalysts for polymer electrolyte fuel cells. J. Power Sources 2010, 195, 6398–6404. [Google Scholar] [CrossRef]
- Wang, J.; Xi, J.; Bai, Y.; Shen, Y.; Sunc, J.; Chen, L.; Zhu, W.; Qiu, X. Structural designing of Pt-CeO2/CNTs for methanol electro-oxidation. J. Power Sources 2007, 164, 555–560. [Google Scholar] [CrossRef]
- Xu, C.; Shen, P.K. Electrochamical oxidation of ethanol on Pt-CeO2/C catalysts. J. Power Sources 2005, 142, 27–29. [Google Scholar] [CrossRef]
- Zhao, H.; Zheng, Z.; Li, J.; Jia, H.; Wong, K.W.; Zhang, Y.; Lau, W.M. Substitute of expensive Pt with improved electrocatalytic performance and higher resistance to CO poisoning for methanol oxidation: The case of synergistic Pt-Co3O4 nanocomposite. Nano Micro Lett. 2013, 5, 296–302. [Google Scholar] [CrossRef]
- Amin, R.S.; Hameed, R.M.A.; El-Khatib, K.M.; Youssef, M.E.; Elzatahry, A.A. Pt–NiO/C anode electrocatalysts for direct methanol fuel cells. Electrochim. Acta 2012, 59, 499–508. [Google Scholar] [CrossRef]
- Sun, X.; Gao, K.; Pang, X.; Yang, H.; Volinsky, A.A. Electrochemical oxidation of methanol on Pt-SnOx/C catalysts characterized by electrochemistry methods. J. Electrochem. Soc. 2015, 162, F1540–F1548. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Kundu, P.P. Pt–Ru/Al2O3–C nanocomposites as direct methanol fuel cell catalysts for electrooxidation of methanol in acidic medium. RSC Adv. 2015, 5, 93539–93546. [Google Scholar] [CrossRef]
- Jayaraman, S.; Jaramillo, T.F.; Baeck, S.H.; McFarland, E.W. Synthesis and characterization of Pt-WO3 as methanol oxidation catalysts for fuel cells. J. Phys. Chem. B 2005, 109, 22958–22966. [Google Scholar] [CrossRef]
- Li, Y.; Liu, C.; Liu, Y.; Feng, B.; Li, L.; Pan, H.; Kellogg, W.; Higgins, D.; Wu, G. Sn-doped TiO2 modified carbon to support Pt anode catalysts for direct methanol fuel cells. J. Power Sources 2015, 286, 354–361. [Google Scholar] [CrossRef]
- Tan, Q.; Du, C.; Sun, Y.; Du, L.; Yin, G.; Gao, Y. Nickel-doped ceria nanoparticles for promoting catalytic activity of Pt/C for ethanol electrooxidation. J. Power Sources 2014, 263, 310–314. [Google Scholar] [CrossRef]
- Ordóñez, L.C.; Roquero, P.; Ramírez, J.; Sebastian, P.J. Methanol electro-oxidation on bimetallic PtMo/C catalysts and Pt/C-Mo/C mechanical mixtures. Int. J. Electrochem. Sci. 2016, 11, 5364–5379. [Google Scholar] [CrossRef]
- Justin, P.; Rao, G.R. Methanol oxidation on MoO3 promoted Pt/C electrocatalyst. Int. J. Hydrogen Energy 2011, 36, 5875–5884. [Google Scholar] [CrossRef]
- Ioroi, T.; Yasuda, K.; Siroma, Z.; Fujiwara, N.; Miyazaki, Y. Enhanced CO-Tolerance of carbon-supported platinum and molybdenum oxide anode catalyst. J. Electrochem. Soc. 2003, 150, A1225–A1230. [Google Scholar] [CrossRef]
- Jothi, P.R.; Kannan, S.; Velayutham, G. Enhanced methanol electro-oxidation over in-situ carbon and graphene supported one dimensional NiMoO4 nanorods. J. Power Sources 2015, 277, 350–359. [Google Scholar] [CrossRef]
- Zhang, G.; Yanga, Z.; Zhanga, W.; Wanga, Y. Nanosized Mo-doped CeO2 enhances the electrocatalytic property of the Pt anode catalyst in direct methanol fuel cells. J. Mater. Chem. A 2017, 5, 1481–1487. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, C.; Yu, X.; Osgood, H.; Wu, G. CeO2-modified α-MoO3 nanorods as a synergistic support for Pt nanoparticles with enhanced COads tolerance during methanol oxidation. Phys. Chem. Chem. Phys. 2017, 19, 330. [Google Scholar] [CrossRef] [PubMed]
- Diczhazi, D.; Borbath, I.; Bakos, I.; Szijjarto, G.P.; Tompos, A.; Paszti, Z. Design of Mo-doped mixed oxide–carbon composite supports for Pt-based electrocatalysts: The nature of the Mo-Pt interaction. Catal. Today 2021, 366, 31–40. [Google Scholar] [CrossRef]
- Lu, G.P.; Ma, X.B.; Yang, H.F.; Kong, D.S.; Feng, Y.Y. Highly active Pt catalysts promoted by molybdenum-doped SnO2 for methanol electrooxidation. Int. J. Hydrogen Energy 2015, 40, 5889–5896. [Google Scholar] [CrossRef]
- Mikhaylovskaya, Z.A.; Buyanova, E.S.; Petrova, S.A.; Nikitina, А.А. Sheelite-related strontium molybdates: Synthesis and characterization. Chim. Techno Acta 2018, 5, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Aruna, K.K.; Manoharan, R. Electrochemical hydrogen evolution catalyzed by SrMoO4 spindle particles in acid water. Int. J. Hydrogen Energy 2013, 38, 12695–12703. [Google Scholar] [CrossRef]
- Karthik, R.; Karikalan, N.; Chen, S.M.; Kumar, J.V.; Karuppiah, C.; Muthuraj, V. Assessment of divergent functional properties of seed-like strontium molybdate for the photocatalysis and electrocatalysis of the postharvest scald inhibitor diphenylamine. J. Catal. 2017, 352, 606–616. [Google Scholar] [CrossRef]
- Huerta-Flores, A.M.; Juárez-Ramírez, I.; Torres-Martínez, L.M.; Carrera-Crespo, J.E.; Gómez-Bustamante, T.; Sarabia-Ramos, O. Synthesis of AMoO4 (A = Ca, Sr, Ba) photocatalysts and their potential application for hydrogen evolution and the degradation of tetracycline in water. J. Photochem. Photobiol. A 2018, 356, 29–37. [Google Scholar] [CrossRef]
- Thrane, J.; Lundegaard, L.F.; Beato, P.; Mentzel, U.V.; Thorhauge, M.; Jensen, A.D.; Høj, M. Alkali earth metal molybdates as catalysts for the selective oxidation of methanol to formaldehyde—Selectivity, activity, and stability. Catalysts 2020, 10, 82. [Google Scholar] [CrossRef] [Green Version]
- Wagner, N.; Schulze, M. Change of electrochemical impedance spectra during CO poisoning of the Pt and Pt–Ru anodes in a membrane fuel cell (PEFC). Electrochim. Acta 2003, 48, 3899–3907. [Google Scholar] [CrossRef]
- Almeida, G.R.O.; Sussuchi, E.M.; Meneses, C.T.D.; Salazar-Banda, G.R.; Eguiluz, K.I.B. Influence of the metallic load of Pt/C and Pt0.6-Ru0.4/C nanowires on the electrochemical oxidation of methanol in acid medium. Int. J. Electrochem. Sci. 2017, 12, 7502–7517. [Google Scholar] [CrossRef]
- Vidya, S.; John, A.; Solomon, S.; Thomas, J.K. Optical and dielectric properties of SrMoO4 powders prepared by the combustion synthesis method. Adv. Mater. Res. 2012, 1, 191–204. [Google Scholar] [CrossRef]
- Sczancoski, J.C.; Cavalcante, L.S.; Joya, M.R.; Varela, J.A.; Pizani, P.S.; Longo, E. SrMoO4 powders processed in microwave-hydrothermal: Synthesis, characterization and optical properties. Chem. Eng. J. 2008, 140, 632–637. [Google Scholar] [CrossRef]
- Dieterle, M.; Weinberg, G.; Mestl, G. Raman spectroscopy of molybdenum oxides Part I. Structural characterization of oxygen defects in MoO3-x by DR UV/VIS, Raman spectroscopy and X-ray diffraction. Phys. Chem. Chem. Phys. 2002, 4, 812–821. [Google Scholar] [CrossRef]
- Song, J.; Sun, Y.; Ba, R.; Huang, Z.Y.; Zhang, J.; Sun, Y.; Zhu, Y. Monodisperse Sr–La2O3 hybrid nanofibers for oxidative coupling of methane to synthesize C2 hydrocarbons. Nanoscale 2015, 7, 2260–2264. [Google Scholar] [CrossRef] [PubMed]
- Bagherisereshki, E.; Tran, J.; Lei, F.; Yeung, N.A. Investigation into SrO/SrCO3 for high temperature thermochemical energy Storage. Sol. Energy 2018, 160, 85–93. [Google Scholar] [CrossRef]
- Çiftyürek, E.; Sabolsky, K.; Sabolsky, E.M. Molybdenum and tungsten oxide based gas sensors for high temperature detection of environmentally hazardous sulfur species. Sens. Actuators B Chem. 2016, 237, 262–274. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.S.; Winograd, N.; Davis, R.E. Electron spectroscopy of platinum-oxygen surfaces and application to electrochemical studies. J. Am. Chem. Soc. 1971, 93, 6296–6297. [Google Scholar] [CrossRef]
- Fu, Q.; Saltsburg, H.; Flytzani-Stephanopoulos, M. Active nonmetallic Au and Pt species on ceria-based water-gas shift catalysts. Science 2003, 301, 935–938. [Google Scholar] [CrossRef]
- Václavu, M.; Matolinová, I.; Mysliveček, J.; Fiala, R.; Matolin, V. Anode material for hydrogen polymer membrane fuel cell: Pt – CeO2 RF-sputtered thin films. J. Electrochem. Soc. 2009, 156, B938–B942. [Google Scholar] [CrossRef]
- Wannapop, S.; Thongtem, T.; Thongtem, S. Characterization of donut-like SrMoO4 produced by microwave-hydrothermal process. J. Nanomater. 2013, 2013, 474576. [Google Scholar] [CrossRef] [Green Version]
- Binninger, T.; Fabbri, E.; Kötz, R.; Schmidt, T.J. Determination of the electrochemically active surface area of metal-oxide supported platinum catalyst. J. Electrochem. Soc. 2014, 161, H121–H128. [Google Scholar] [CrossRef]
- Baronia, R.; Goel, J.; Kaswan, J.; Shukla, A.; Singhal, S.K.; Singh, S.P. PtCo/rGO nano-anode catalyst: Enhanced power density with reduced methanol crossover in direct methanol fuel cell. Mater. Renew. Sustain. Energy 2018, 7, 27. [Google Scholar] [CrossRef] [Green Version]
- Hou, Z.; Yi, B.; Yu, H.; Lin, Z.; Zhang, H. CO tolerance electrocatalyst of PtRu-HxMeO3/C (Me = W, Mo) made by composite support method. J. Power Sources 2003, 123, 116–125. [Google Scholar] [CrossRef]
- Li, W.S.; Tian, L.P.; Huan, Q.M.; Li, H.; Chen, H.Y.; Lian, X.P. Catalystic oxidation of methanol on molybdate-modified platinum electrode in sulfuric acid solution. J. Power Sources 2002, 104, 281–288. [Google Scholar] [CrossRef]
- Li, W.; Lu, J.; Du, J.; Lu, D.; Chen, H.; Li, H.; Wu, Y. Electrocatalytic oxidation of methanol on polyaniline-stabilized Pt–HxMoO3 in sulfuric acid solution. Electrochem. Commun. 2005, 7, 406–410. [Google Scholar]
- Silva, C.D.; Morais, L.H.; Gonçalves, R.; Matos, R.; Souza, G.L.C.; Freitas, R.G.; Pereira, E.C. The methanol and CO electro-oxidation onto Ptpc/Co/Pt metallic multilayer nanostructured electrodes: An experimental and theoretical approach. Electrochim. Acta 2018, 280, 197–205. [Google Scholar] [CrossRef]
- Tsukagoshi, Y.; Ishitobi, H.; Nakagawa, N. Improved performance of direct methanol fuel cells with the porous catalyst layer using highly-active nanofiber catalyst. Carbon Resour. Convers. 2018, 1, 61–72. [Google Scholar] [CrossRef]
- Chun, H.J.; Kim, D.B.; Lim, D.H.; Lee, W.D.; Lee, H.I. A synthesisof CO-tolerant Nb2O5-promoted Pt/C catalyst for direct methanol fuel cell; its physical and electrochemical characterization. Int. J. Hydrogen Energy 2010, 35, 6399–6408. [Google Scholar] [CrossRef]
- Wang, Y.; Fachini, E.R.; Cruz, G.; Zhu, Y.; Ishikawa, Y.; Colucci, J.A.; Cabrera, C.R. Effect of surface composition of electrochemically codeposited platinum molybdenum oxide on methanol oxidation. J. Electrochem. Soc. 2001, 148, C222. [Google Scholar] [CrossRef]
- Radhakrishnan, R.; Reed, C.; Oyama, S.T. Variability in the structure of supported MoO3 catalysts: Studies using Raman and X-ray absorption spectroscopy with ab initio calculations. J. Phys. Chem. B 2001, 105, 8519–8530. [Google Scholar] [CrossRef]
- Wu, Q.L.; Zhao, S.X.; Yu, L.; Zheng, X.X.; Wang, Y.F.; Yu, L.Q.; Nan, C.W.; Cao, G. Oxygen vacancy-enriched MoO3−x nanobelts for asymmetric supercapacitors with excellent room/low temperature performance. J. Mater. Chem. A 2019, 7, 13205–13214. [Google Scholar] [CrossRef]
- Tsai, M.C.; Nguyen, T.T.; Akalework, N.G.; Pan, C.J.; Rick, J.; Liao, Y.F.; Su, W.N.; Hwang, B.J. Interplay between molybdenum dopant and oxygen vacancies in a TiO2 support enhances the oxygen reduction reaction. ACS Catal. 2016, 6, 6551–6559. [Google Scholar] [CrossRef]
- Chiang, T.H.; Yeh, H.C. A novel synthesis of α-MoO3 nanobelts and the characterization. J. Alloys Compd. 2014, 585, 535–541. [Google Scholar] [CrossRef]
- Huang, H.; Sun, D.; Wang, X. Low-defect MWNT-Pt nanocomposite as a high performance electrocatalyst for direct methanol fuel cells. J. Phys. Chem. C 2011, 115, 19405–19412. [Google Scholar] [CrossRef]
- Rodríguez, J.M.D.; Melián, J.A.H.; Peña, J.P. Determination of the real surface area of Pt electrodes by hydrogen adsorption using cyclic voltammetry. J. Chem. Educ. 2000, 77, 1195. [Google Scholar] [CrossRef]
- Shepperd, S.A.; Campbell, S.A.; Smith, J.R.; Lloyd, G.W.; Ralph, T.R.; Walsh, F.C. Electrochemical and microscopic characterization of platinum-coated perfluorosulfonic acid (Nafion 117). Analyst 1998, 123, 1923. [Google Scholar] [CrossRef]
- Pan, C.J.; Sarma, L.S.; Hwang, B.J. Formation and Characterization of Bimetallic Nanoparticles in Electrochemistry. In Handbook of Nanoelectrochemistry; Aliofkhazraei, M., Makhlouf, A., Eds.; Springer: New York, NY, USA, 2015; pp. 169–239. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrocatalysts | Mo5+ (%) | Mo6+ (%) | Pt0 (%) | Pt2+ (%) | Pt4+ (%) |
|---|---|---|---|---|---|
| Pt/Uncalcined Sr0.5Mo0.5O4−δ–C | 74.0 | 36.0 | 54.6 | 31.6 | 13.8 |
| Pt/200 Sr0.5Mo0.5O4−δ–C | 72.6 | 27.4 | 38.2 | 51.6 | 10.4 |
| Pt/400 Sr0.5Mo0.5O4−δ–C | 29.5 | 70.5 | 34.6 | 38.2 | 27.2 |
| Electrocatalysts | Forward Peak Current Density (mA/cm2) | ECSAH (m2/g) | Onset Potential-H (mV) | Rct (Ω cm2) | ECSACO (m2/g) | Onset Potential-co (mV) |
|---|---|---|---|---|---|---|
| Pt/uncalcined MoO3x–C | 9.50 | 45.37 | 399 | 1410 | 62.78 | 535 |
| Pt/uncalcined Sr0.5Mo0.5O4-δ–C | 12.56 | 116.53 | 344 | 940 | 168.14 | 476 |
| Pt/200 Sr0.5Mo0.5O4-δ–C | 4.41 | 25.12 | 411 | 4600 | 23.57 | 430 |
| Pt/400 Sr0.5Mo0.5O4-δ–C | 3.62 | 22.52 | 411 | 5682 | 20.32 | 450 |
| Electrocatalysts | Forward Peak Current Density (mA cm–2) | ECSAH (m2 g–1) | Onset Potential-H (mV) | ECSACO (m2 g–1) | Onset Potential-co (mV) | Rct (Ω cm2) |
|---|---|---|---|---|---|---|
| Pt/C | 8.87 | 63.81 | 405 | 52.61 | 515 | 1220 |
| Pt/Sr0.5Mo0.5O4−δ-C | 12.56 | 116.53 | 344 | 168.14 | 476 | 940 |
| Pt/Sr0.67Mo0.33O4−δ-C | 8.87 | 25.12 | 369 | 44.38 | 465 | 1590 |
| Pt/Sr0.75Mo0.25O4-δ-C | 6.35 | 24.15 | 377 | 31.20 | 533 | 1900 |
| Pt/Sr0.3Mo0.67O4-δ-C | 6.58 | 19.90 | 365 | 37.95 | 489 | 1860 |
| Materials | Pt0 (%) | Pt2+ (%) | Pt4+ (%) |
|---|---|---|---|
| Pt/C | 53.1 | 36.4 | 10.5 |
| Pt/Sr0.50Mo0.50O4−δ-C | 54.6 | 31.6 | 13.8 |
| Pt/Sr0.67Mo0.33O4−δ-C | 42.5 | 42.7 | 14.8 |
| Pt/Sr0.75Mo0.25O4-δ-C | 38.1 | 42 | 19.9 |
| Pt/Sr0.33Mo0.67O4-δ-C | 39.2 | 48.5 | 12.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiang, T.H.; Hsu, J.-W. Improved the Methanol Electro-Oxidation and Carbon Monoxide Tolerance for Direct Methanol Fuel Cells Using Strontium Molybdate. Catalysts 2022, 12, 676. https://doi.org/10.3390/catal12070676
Chiang TH, Hsu J-W. Improved the Methanol Electro-Oxidation and Carbon Monoxide Tolerance for Direct Methanol Fuel Cells Using Strontium Molybdate. Catalysts. 2022; 12(7):676. https://doi.org/10.3390/catal12070676
Chicago/Turabian StyleChiang, Tzu Hsuan, and Jia-Wei Hsu. 2022. "Improved the Methanol Electro-Oxidation and Carbon Monoxide Tolerance for Direct Methanol Fuel Cells Using Strontium Molybdate" Catalysts 12, no. 7: 676. https://doi.org/10.3390/catal12070676
APA StyleChiang, T. H., & Hsu, J.-W. (2022). Improved the Methanol Electro-Oxidation and Carbon Monoxide Tolerance for Direct Methanol Fuel Cells Using Strontium Molybdate. Catalysts, 12(7), 676. https://doi.org/10.3390/catal12070676