
Highly Efficient and Selective Carbon-Doped BN Photocatalyst Derived from a Homogeneous Precursor Reconfiguration


 ,
,
Abstract
:
1. Introduction
2. Results
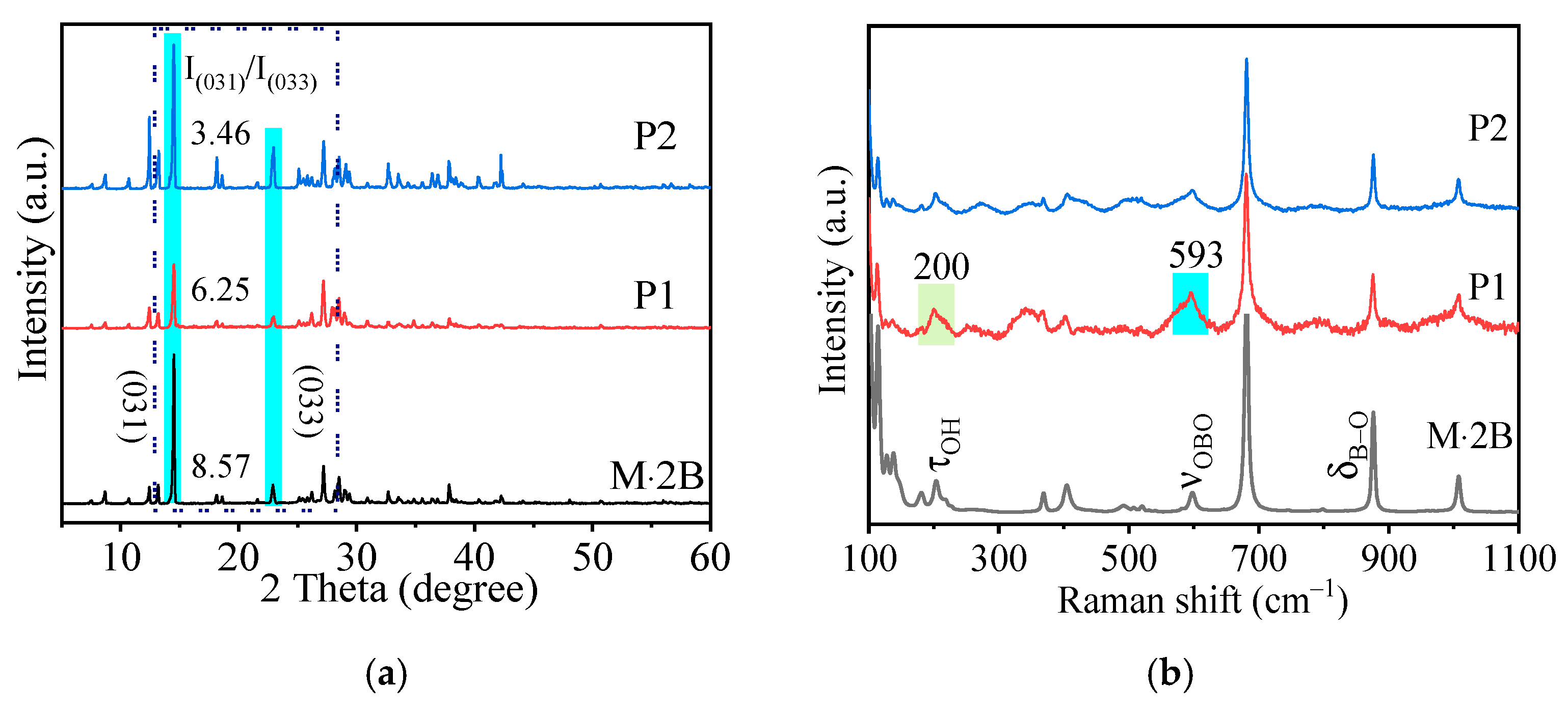
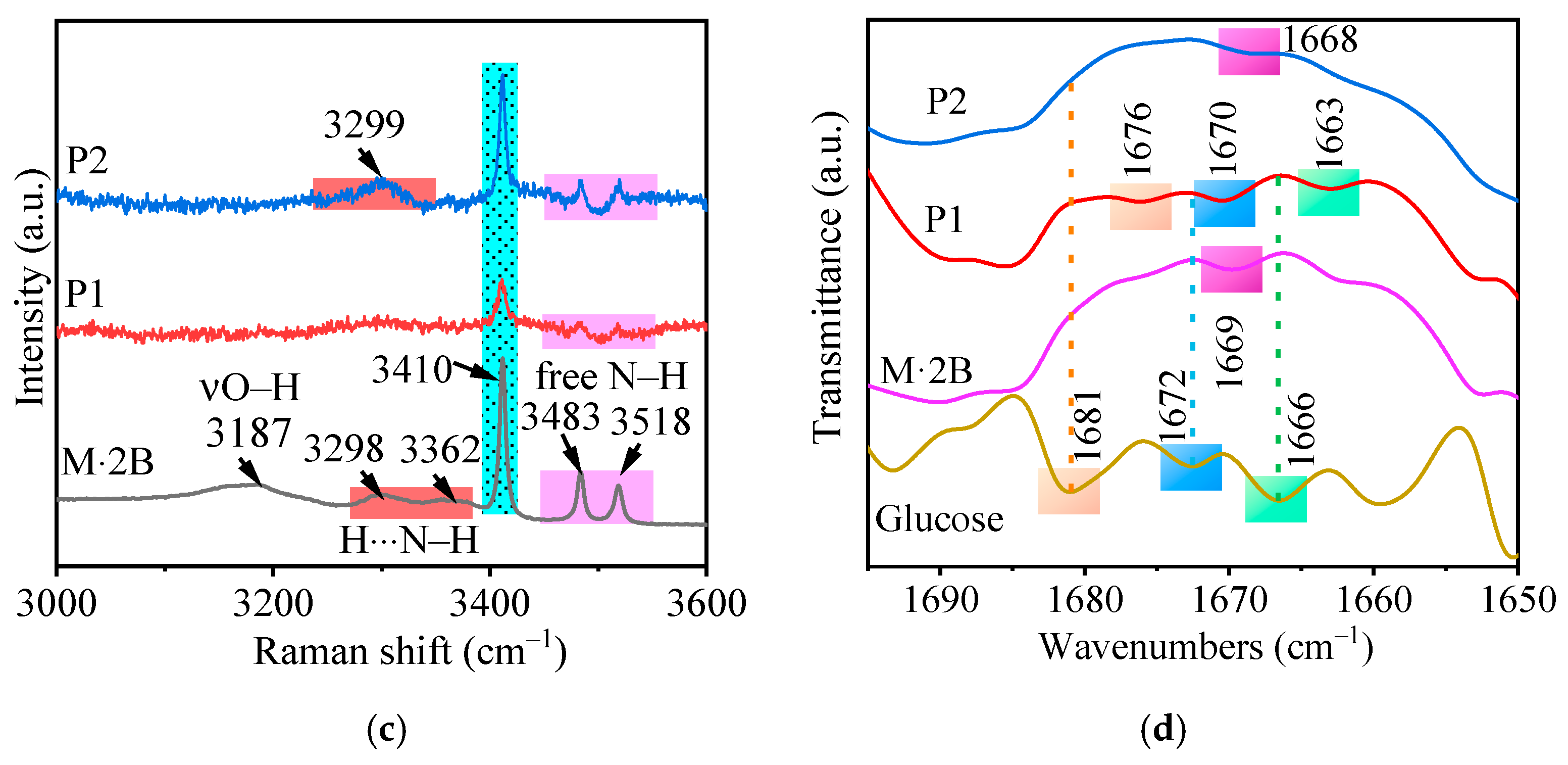
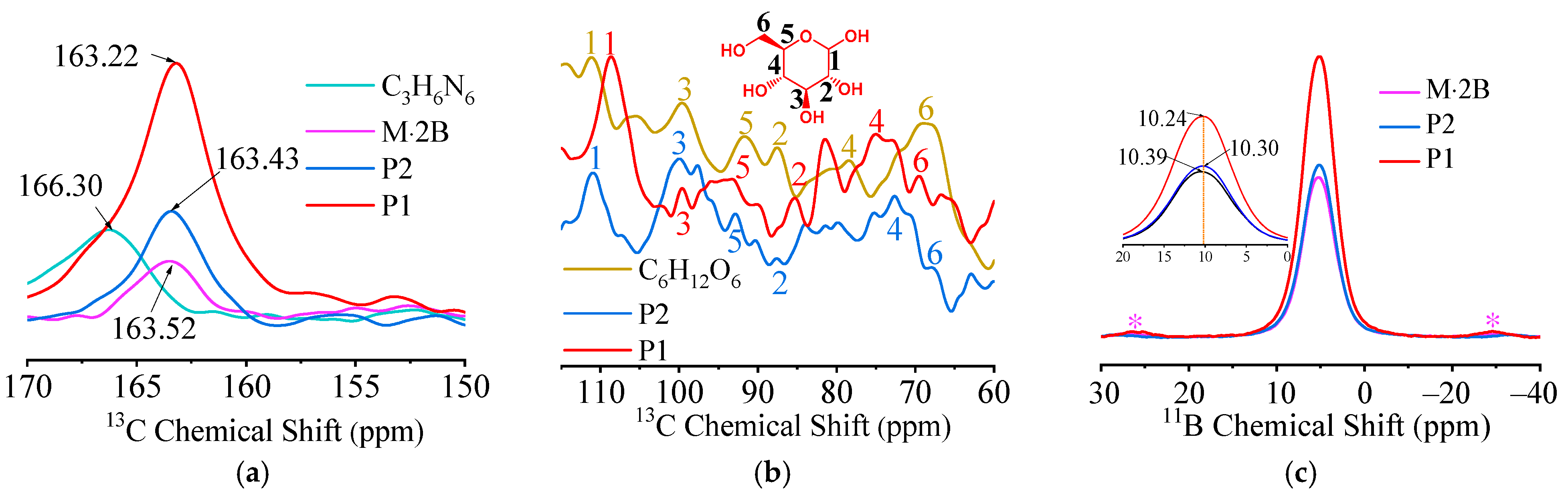
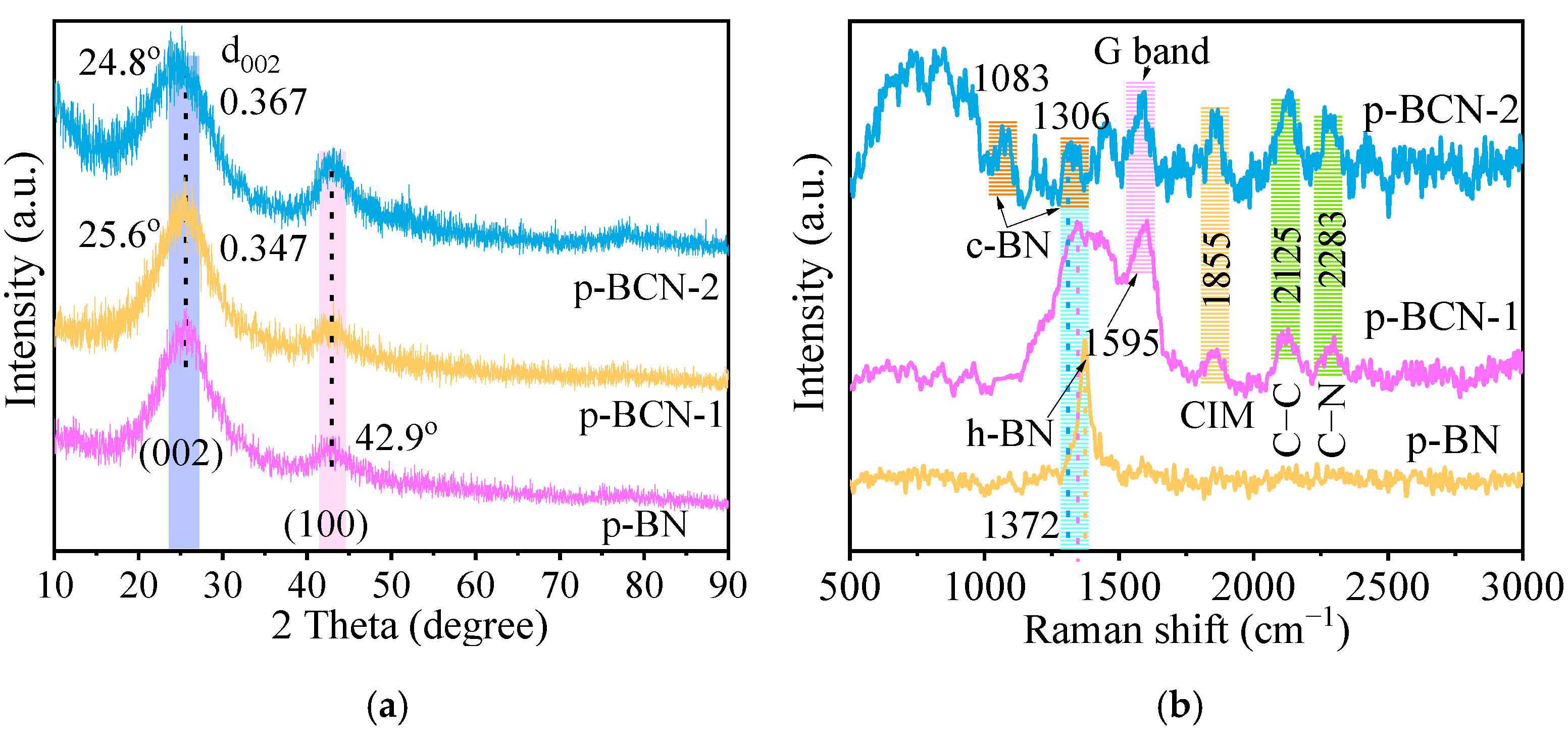
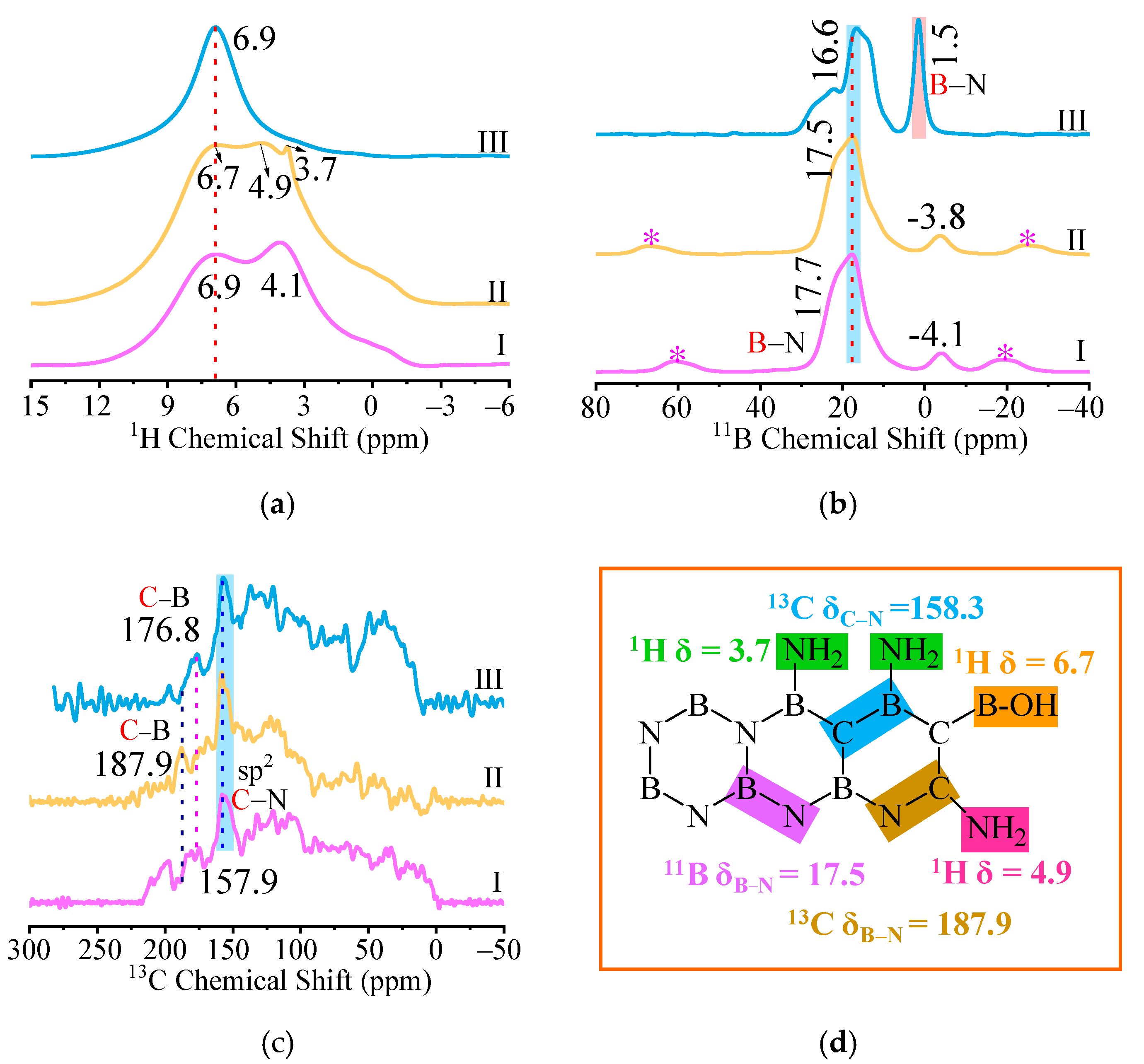
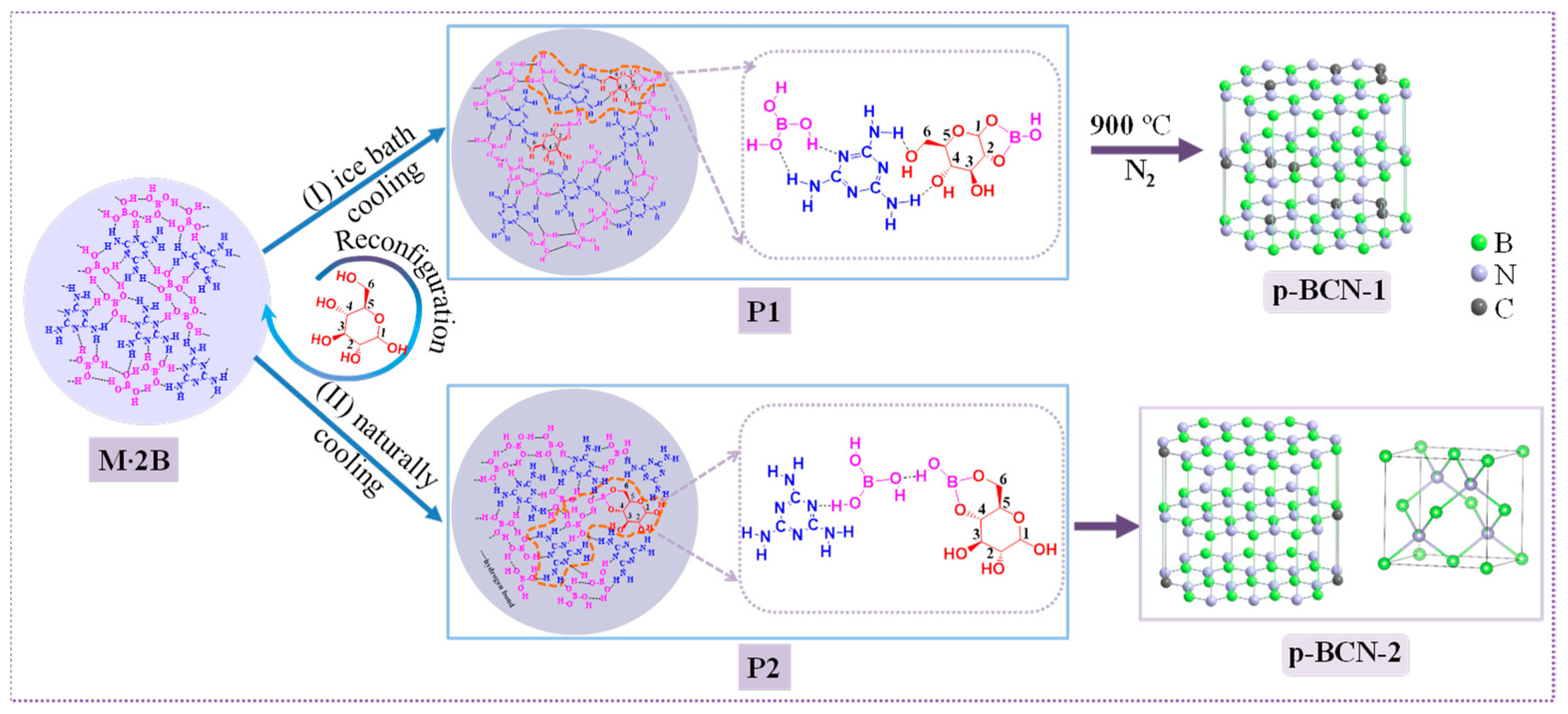
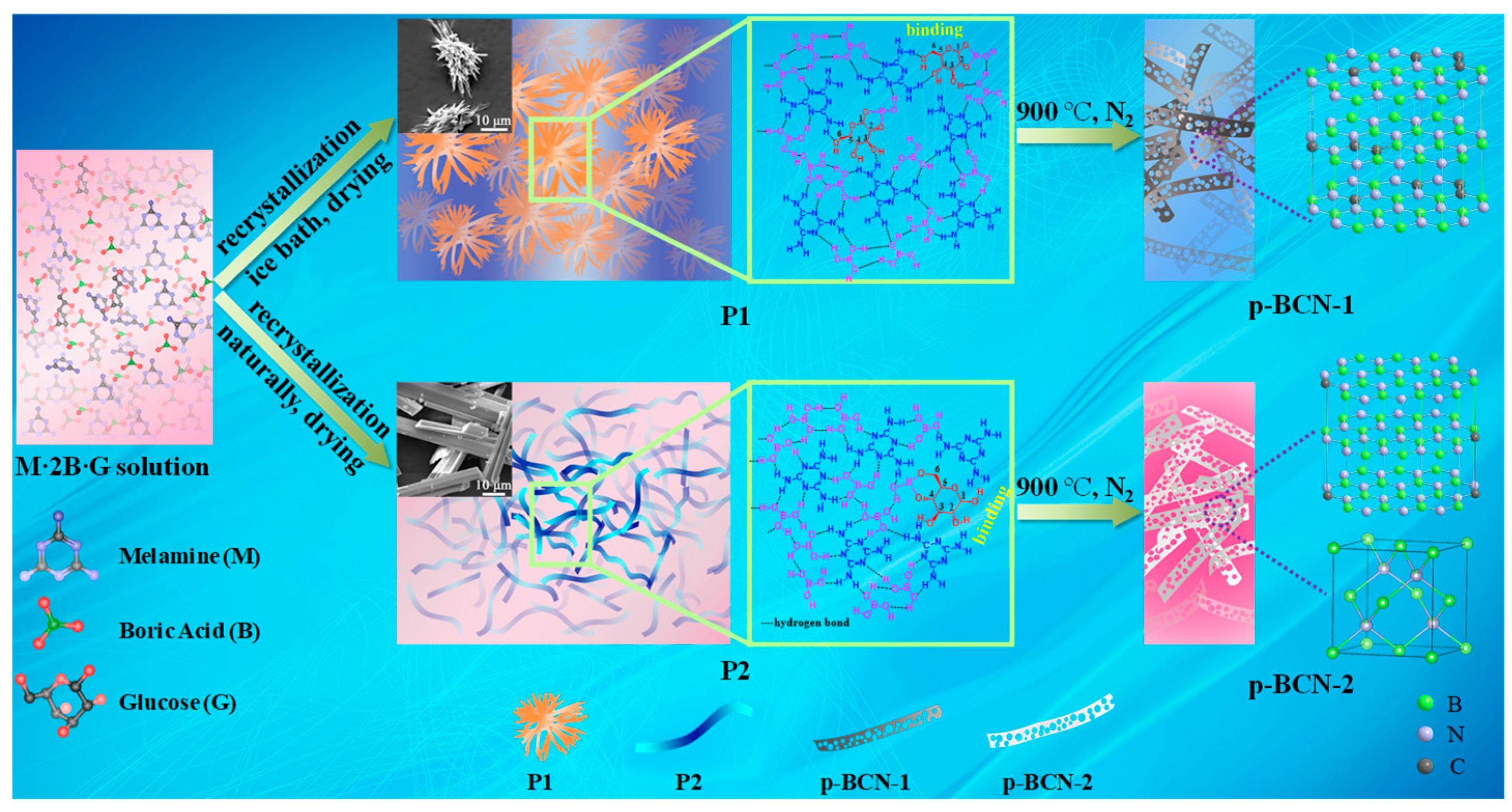
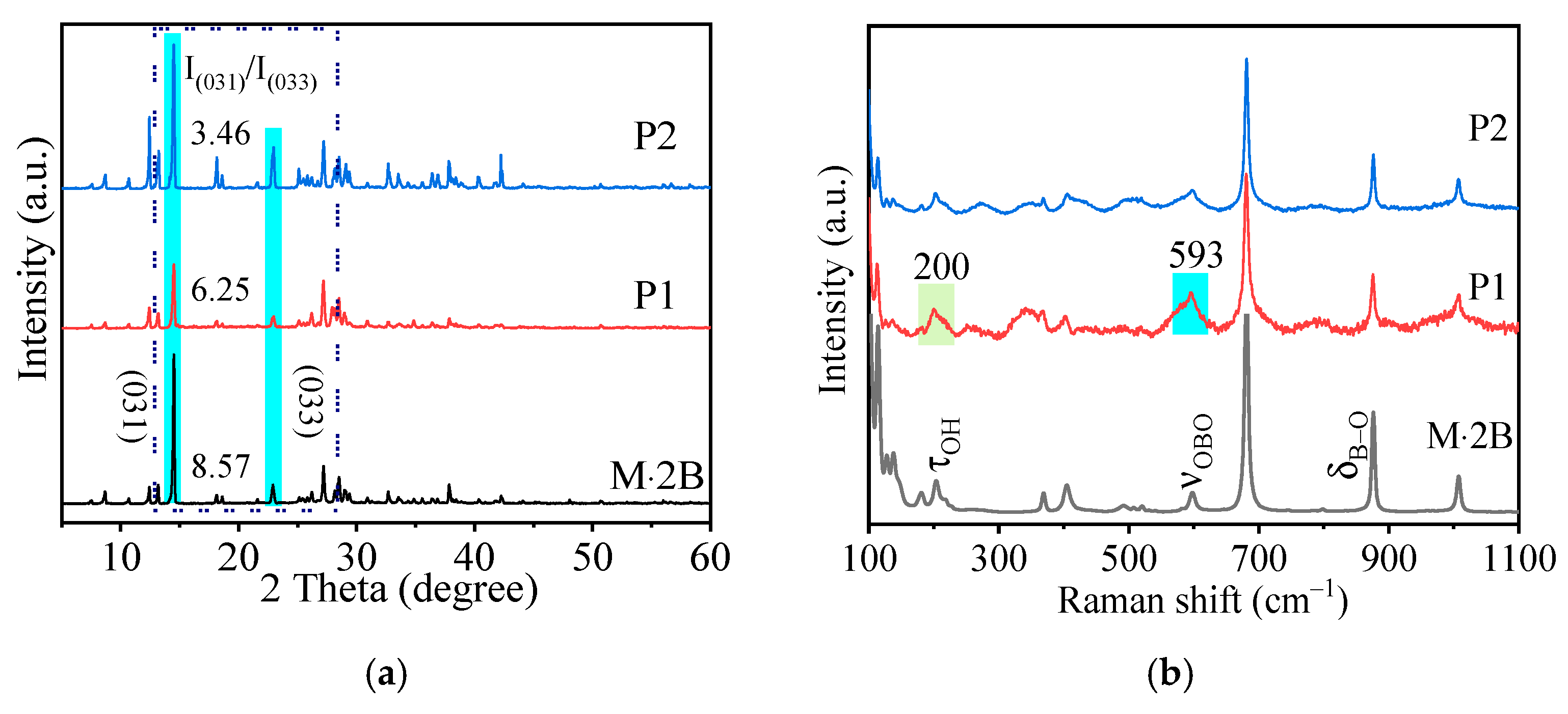
2.1. Characterization of Precursors
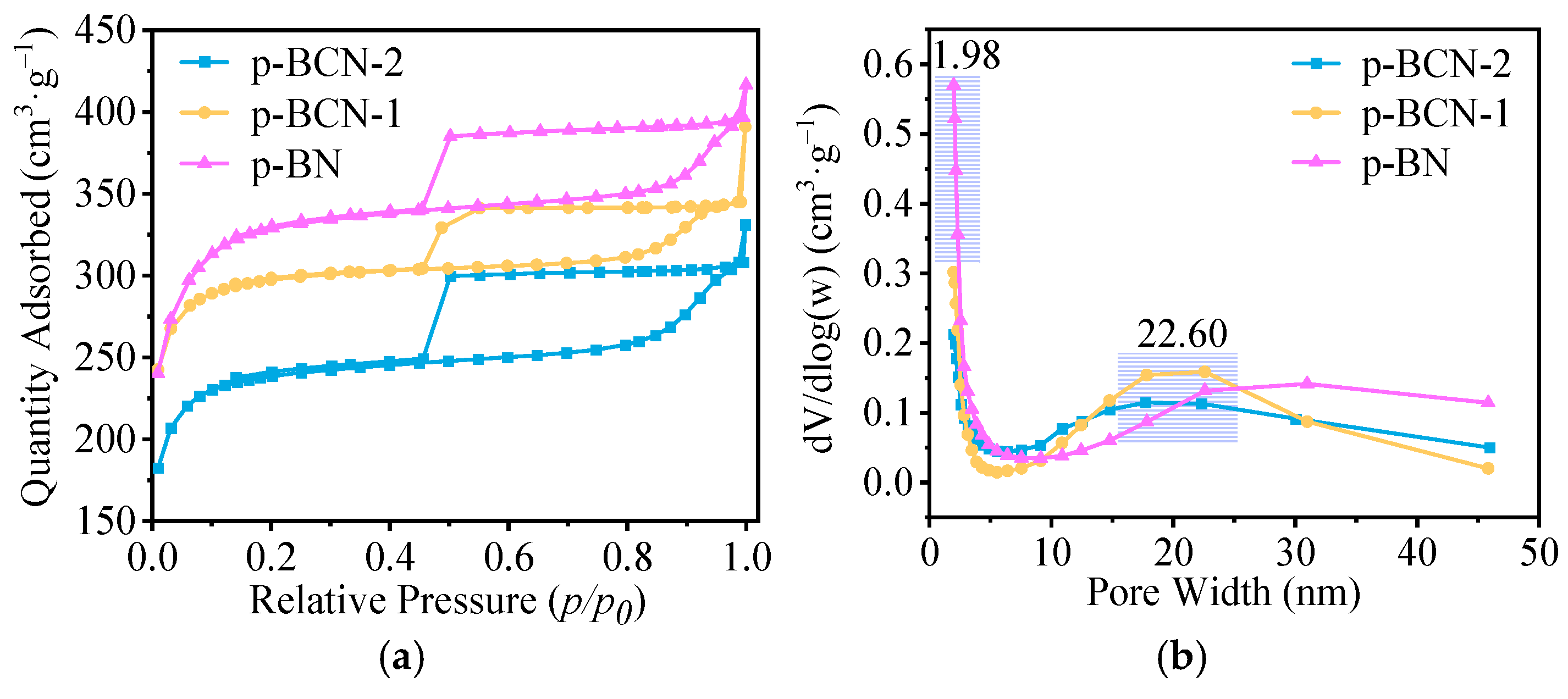
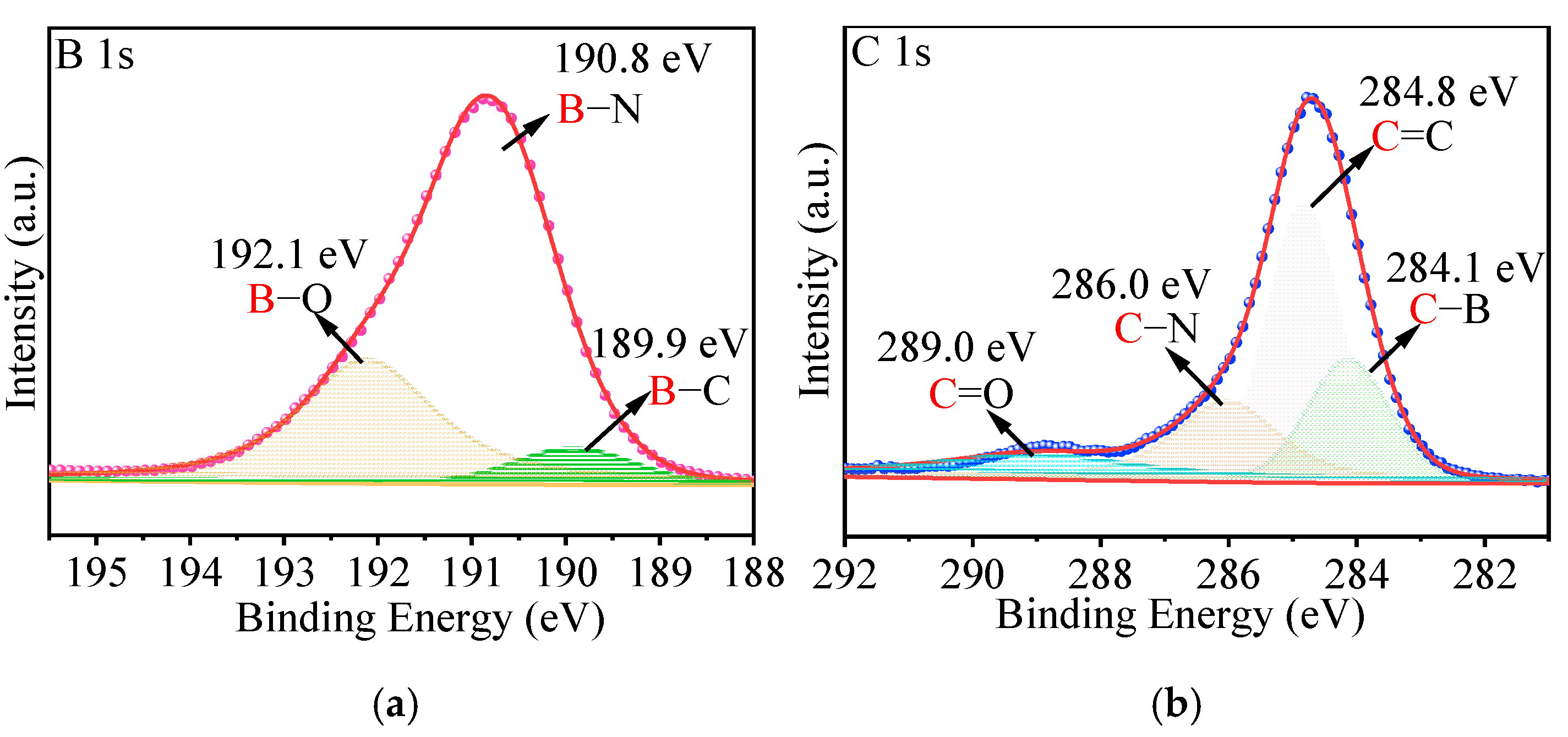
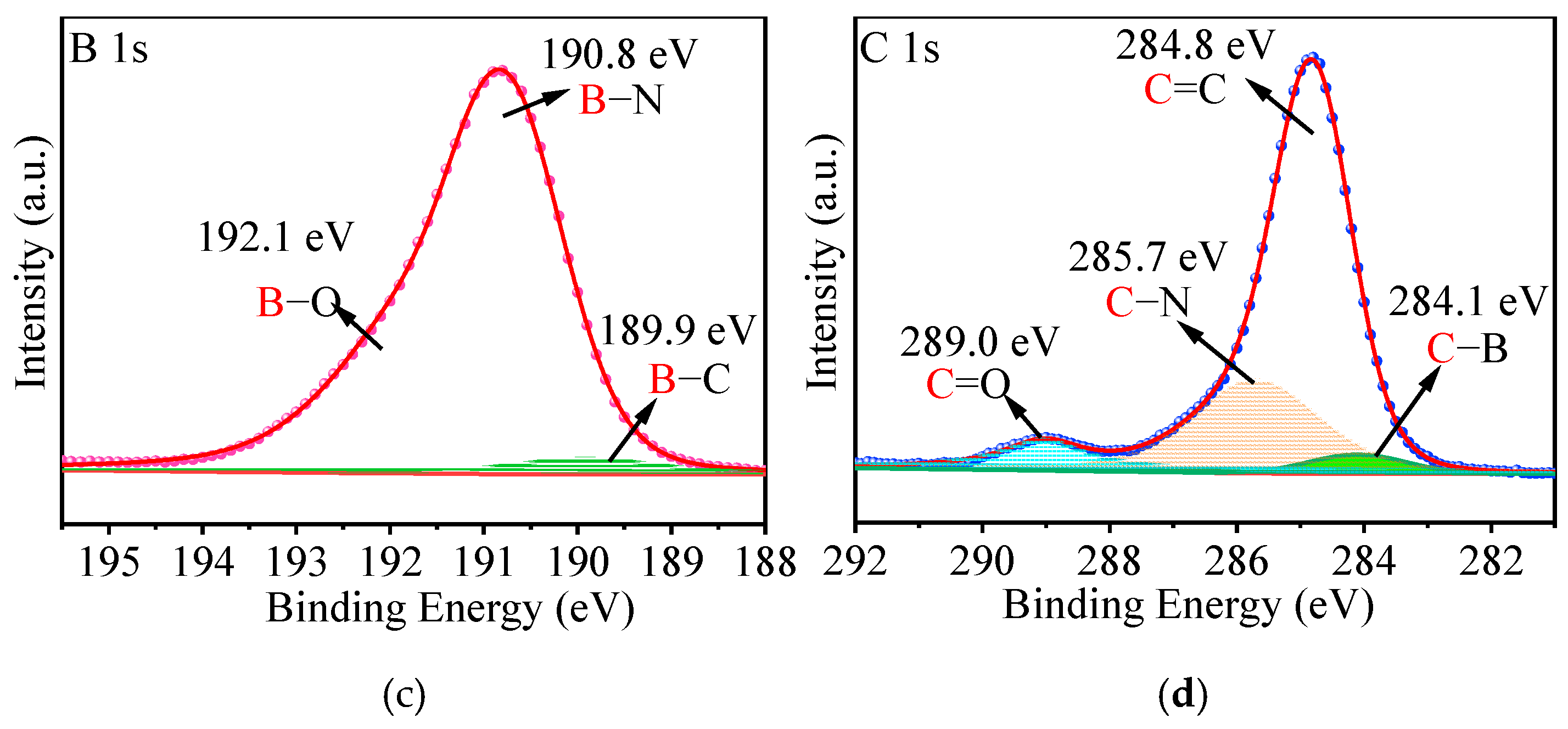
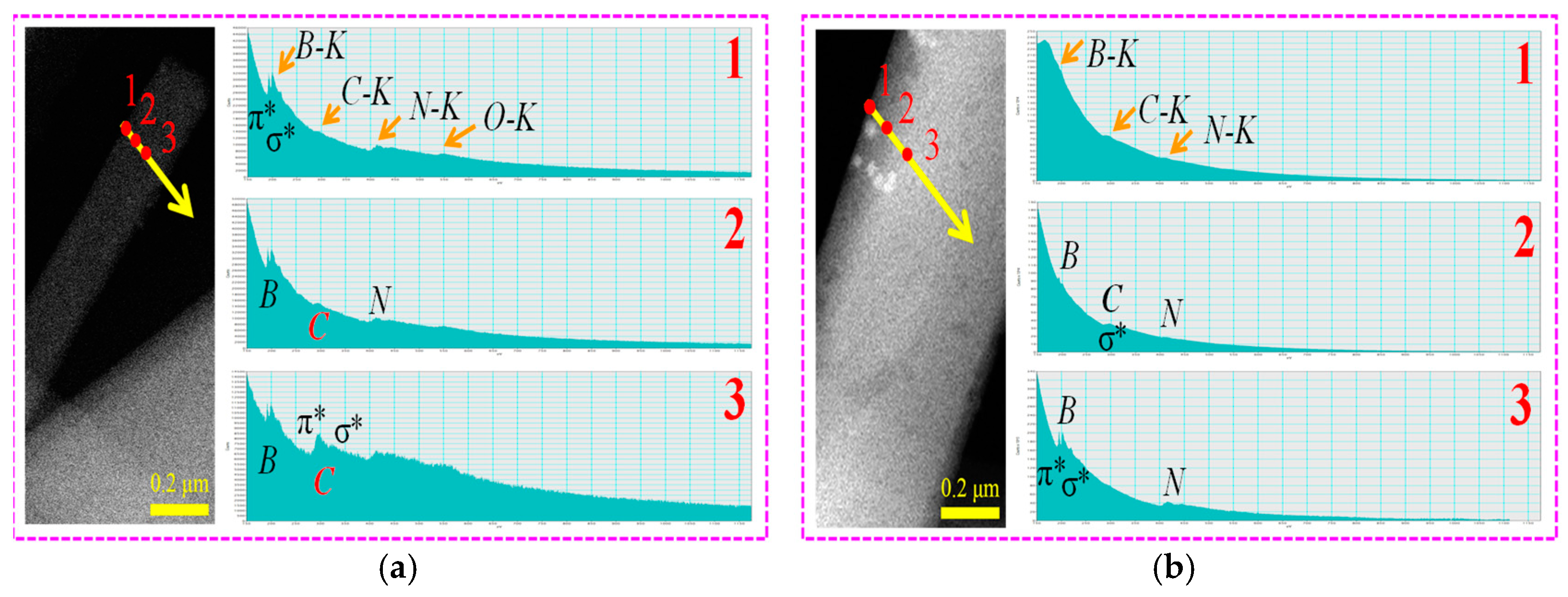
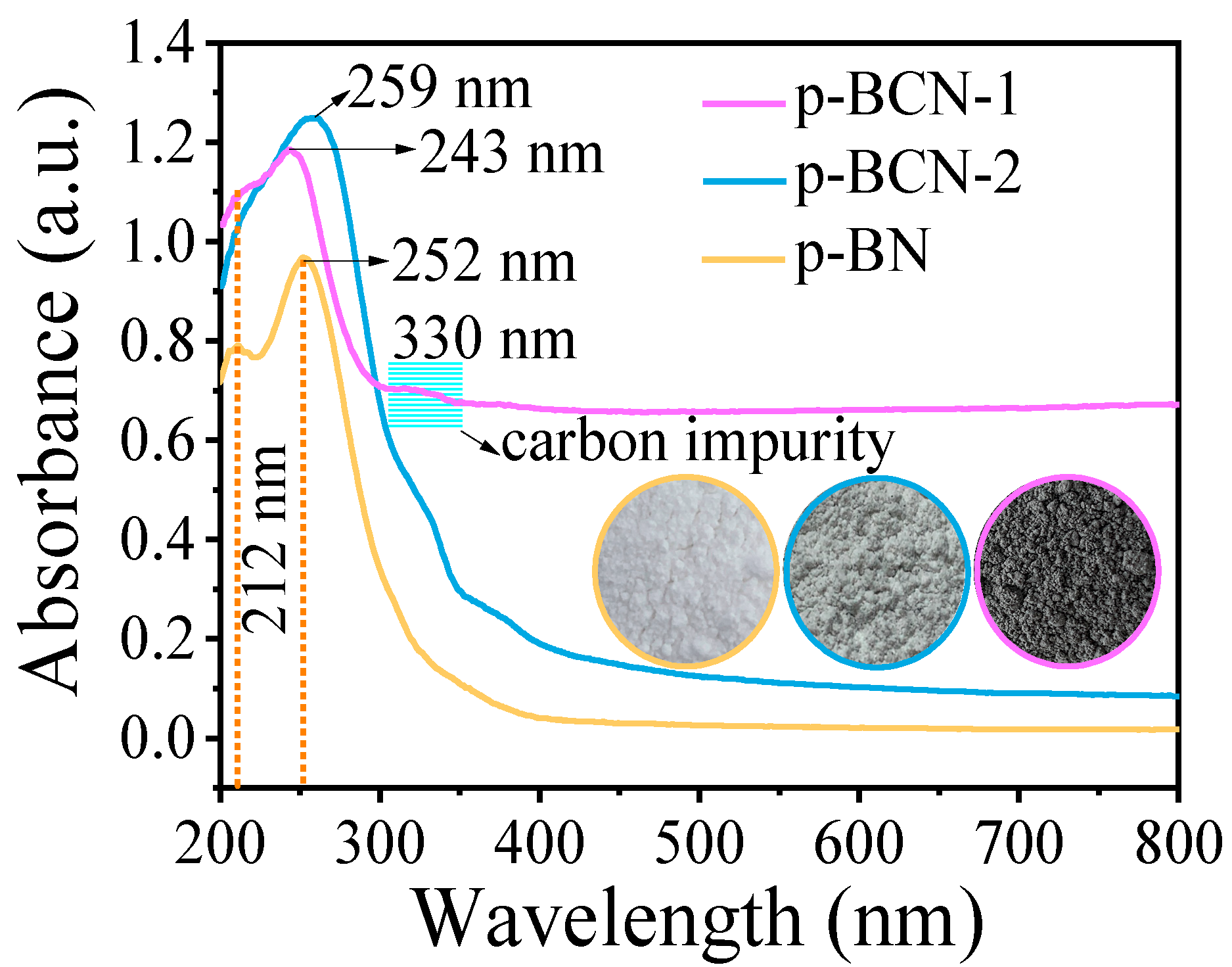
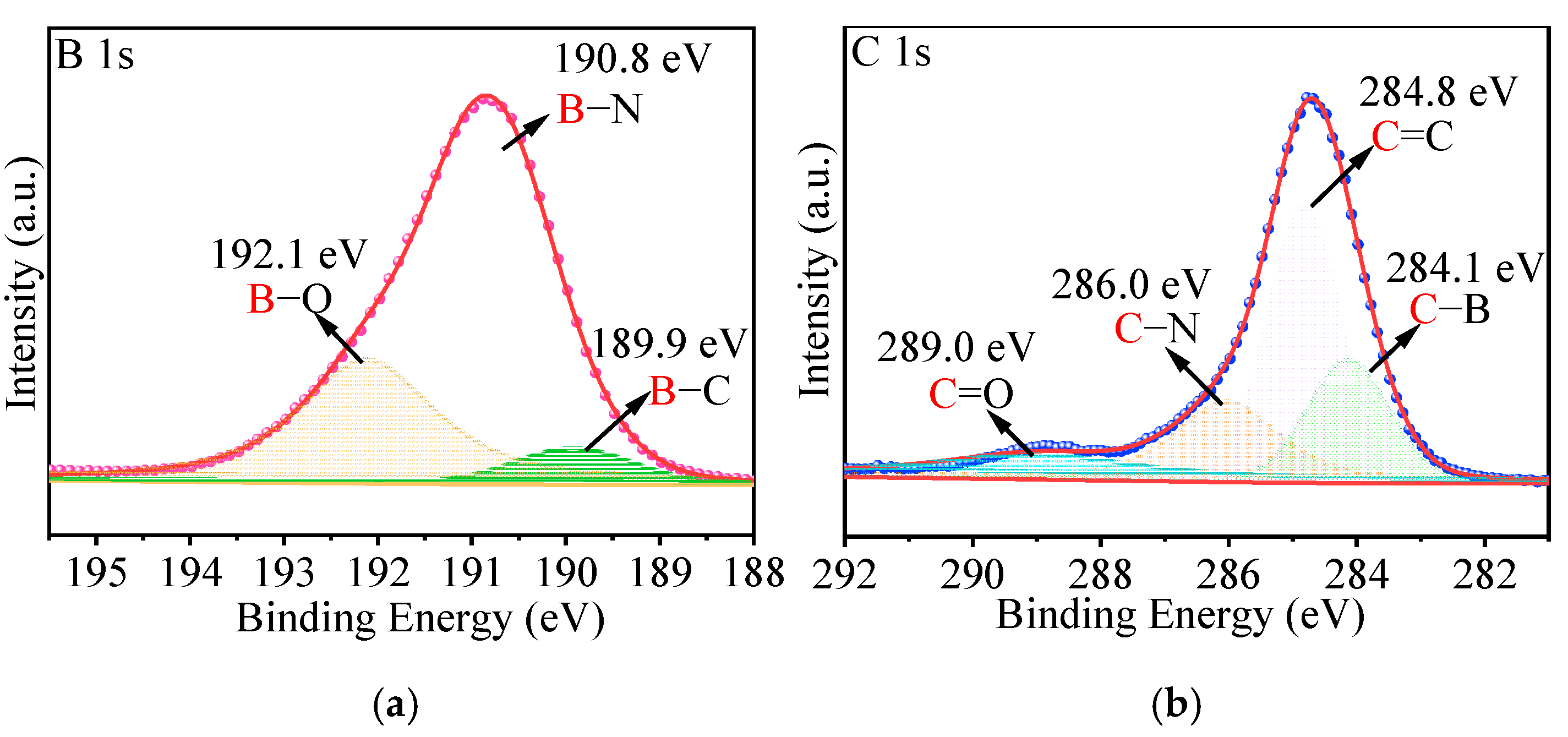
2.2. Characterization of p-BCN Photocatalysts
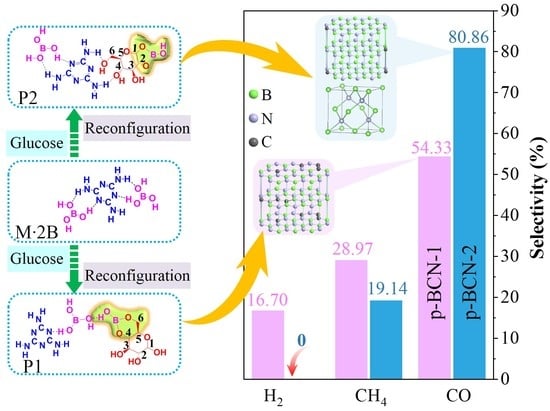
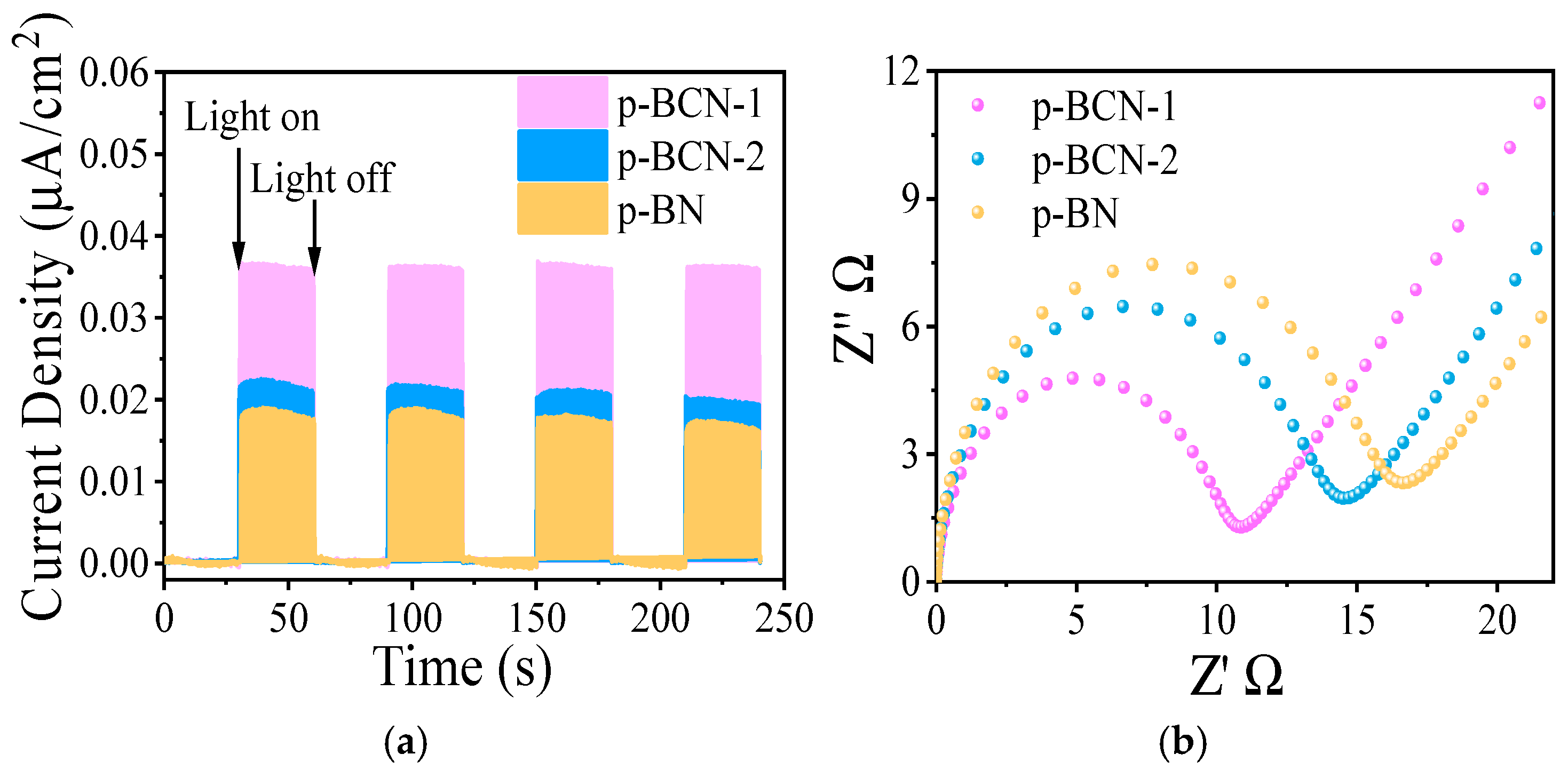
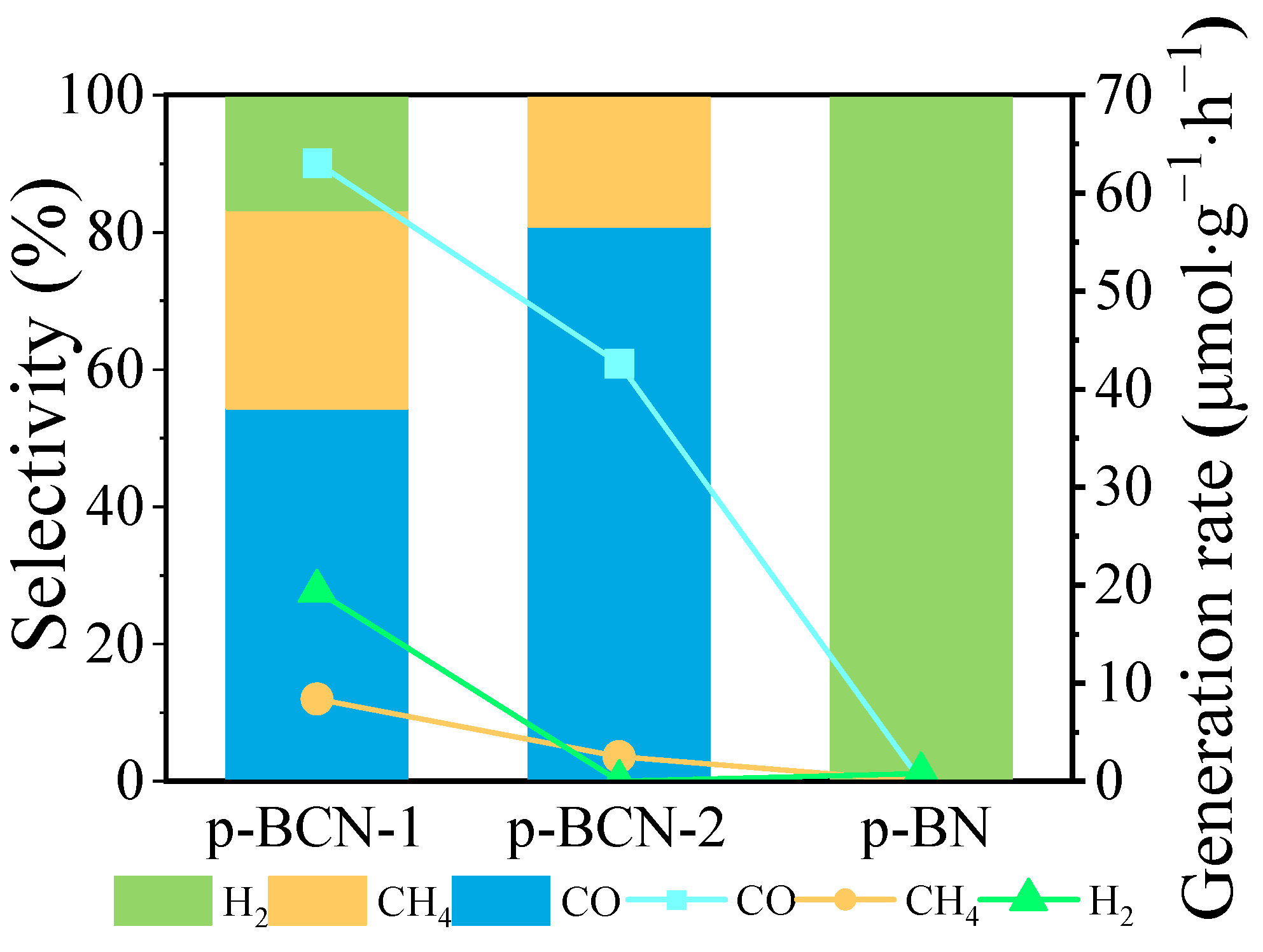
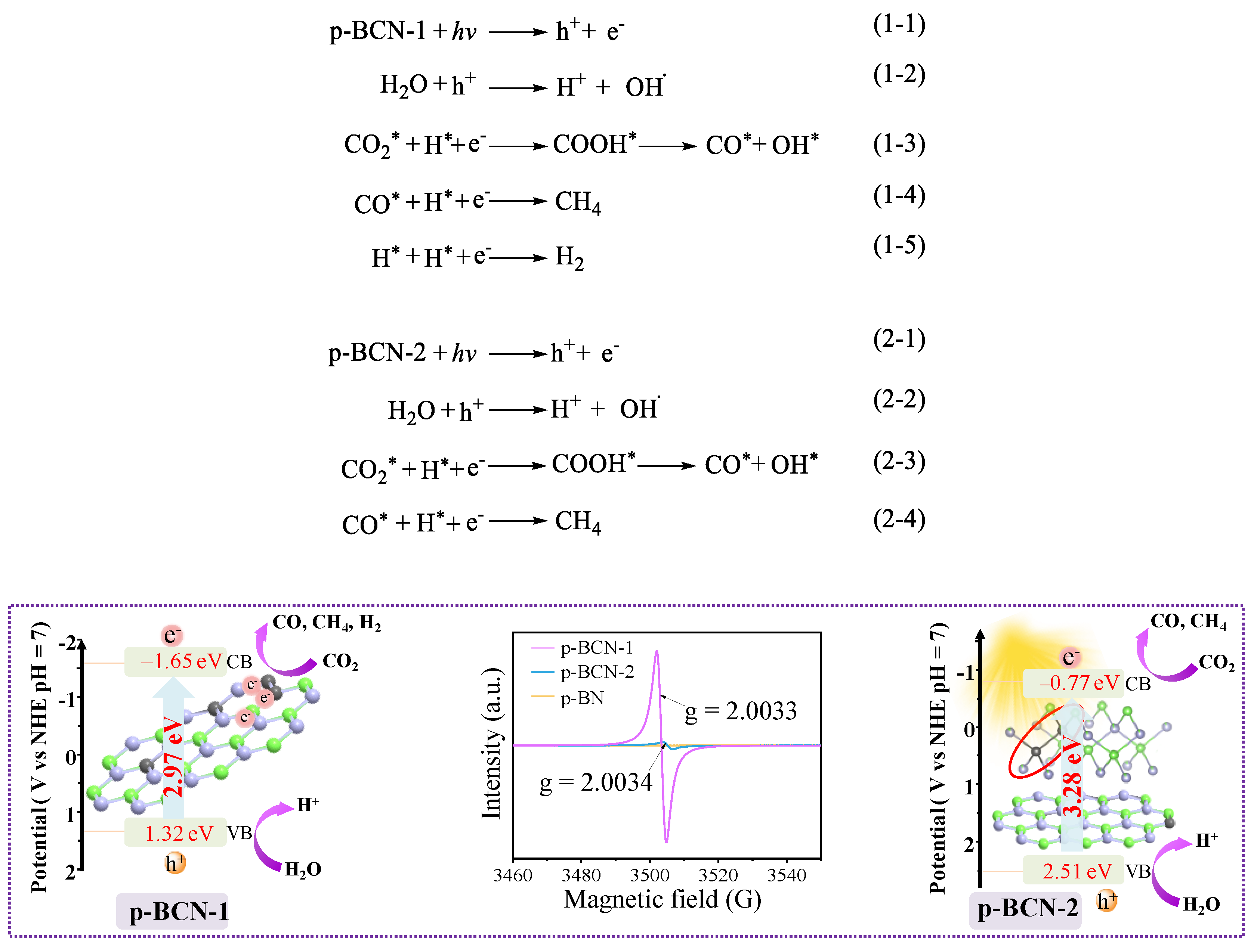
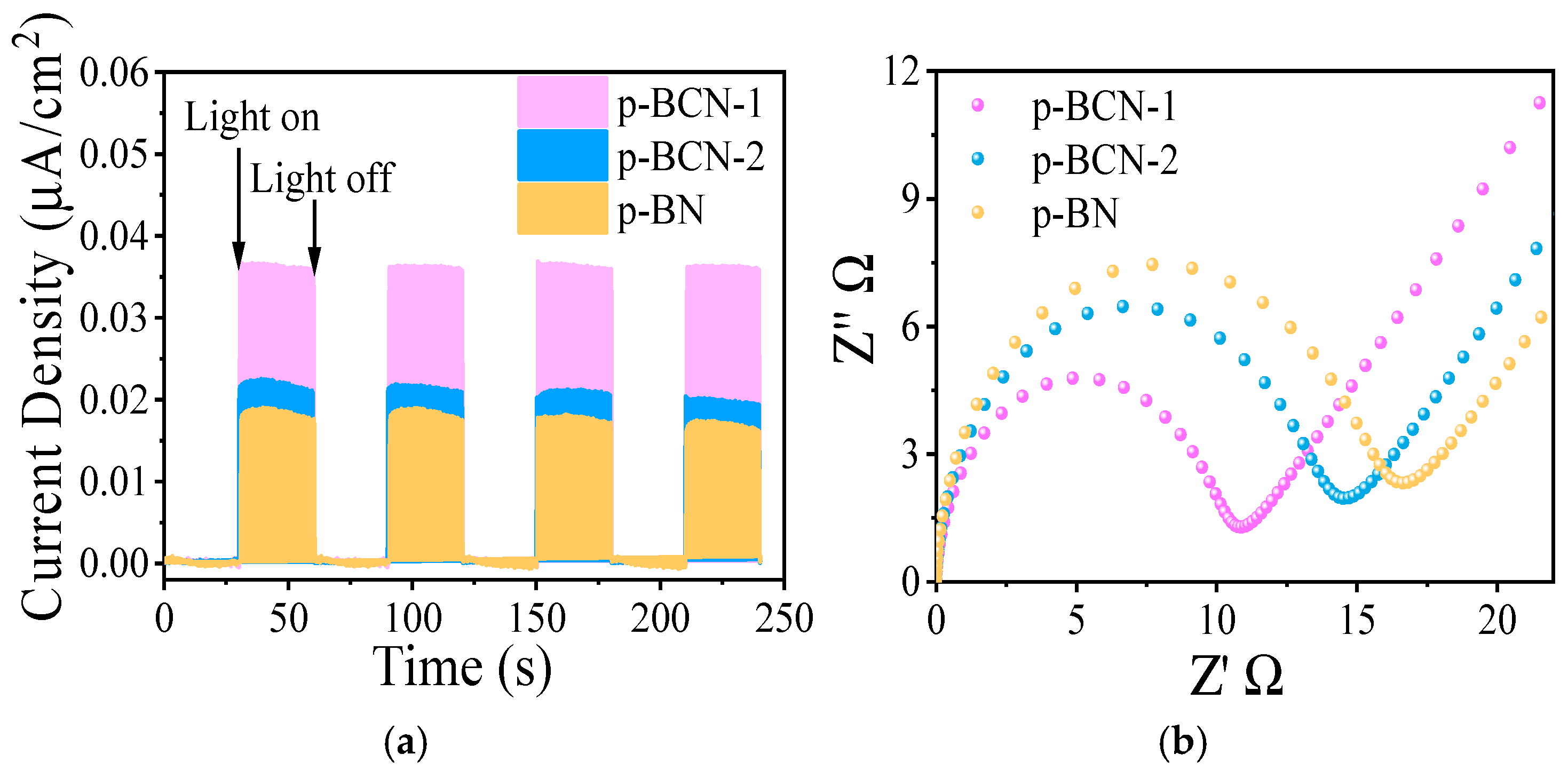
2.3. Photocatalytic CO2 Reduction Performance of p-BCN
3. Materials and Methods
3.1. Materials
3.2. Synthesis of C-Doped BN (p-BCN)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gong, Y.; Xu, Z.-Q.; Li, D.; Zhang, J.; Aharonovich, I.; Zhang, Y. Two-dimensional hexagonal boron nitride for building Next-generation energy-efficient devices. ACS Energy Lett. 2021, 6, 985–996. [Google Scholar] [CrossRef]
- Kim, K.K.; Lee, H.S.; Lee, Y.H. Synthesis of hexagonal boron nitride heterostructures for 2D van der Waals electronics. Chem. Soc. Rev. 2018, 47, 6342–6369. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tan, B.; Zhang, X.; Gao, F.; Hu, Y.; Wang, L.; Duan, X.; Yang, Z.; Hu, P. Atomically thin hexagonal boron nitride and its heterostructures. Adv. Mater. 2020, 33, 2000769. [Google Scholar] [CrossRef] [PubMed]
- Weng, Q.; Wang, X.; Wang, X.; Bando, Y.; Golberg, D. Functionalized hexagonal boron nitride nanomaterials: Emerging properties and applications. Chem. Soc. Rev. 2016, 45, 3989–4012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, C.; Lai, C.; Zhang, C.; Zeng, G.; Huang, D.; Cheng, M.; Hu, L.; Xiong, W.; Chen, M.; Wang, J.; et al. Semiconductor/boron nitride composites: Synthesis, properties, and photocatalysis applications. Appl. Catal. B 2018, 238, 6–18. [Google Scholar] [CrossRef]
- Mendelson, N.; Chugh, D.; Reimers, J.R.; Cheng, T.S.; Gottscholl, A.; Long, H.; Mellor, C.J.; Zettl, A.; Dyakonov, V.; Beton, P.H.; et al. Identifying carbon as the source of visible single-photon emission from hexagonal boron nitride. Nat. Mater. 2020, 20, 321–328. [Google Scholar] [CrossRef]
- Chen, L.; Zhou, M.; Luo, Z.; Wakeel, M.; Asiri, A.M.; Wang, X. Template-free synthesis of carbon-doped boron nitride nanosheets for enhanced photocatalytic hydrogen evolution. Appl. Catal. B 2019, 241, 246–255. [Google Scholar] [CrossRef]
- Huang, C.; Chen, C.; Zhang, M.; Lin, L.; Ye, X.; Lin, S.; Antonietti, M.; Wang, X. Carbon-doped BN nanosheets for metal-free photoredox catalysis. Nat. Commun. 2015, 6, 7698. [Google Scholar] [CrossRef] [Green Version]
- Zheng, M.; Shi, J.; Yuan, T.; Wang, X. Metal-free dehydrogenation of N-heterocycles by ternary h-BCN nanosheets with visible light. Angew. Chem. Int. Ed. 2018, 57, 5487–5491. [Google Scholar] [CrossRef]
- Shi, J.; Yuan, T.; Zheng, M.; Wang, X. Metal-free heterogeneous semiconductor for visible-light photocatalytic decarboxylation of carboxylic acids. ACS Catal. 2021, 11, 3040–3047. [Google Scholar] [CrossRef]
- Guo, F.; Yang, P.; Pan, Z.; Cao, X.-N.; Xie, Z.; Wang, X. Carbon-doped BN nanosheets for the oxidative dehydrogenation of ethylbenzene. Angew. Chem. Int. Ed. 2017, 56, 8231–8235. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Yuan, T.; Shi, J.; Cai, W.; Wang, X. Photocatalytic oxygenation and deoxygenation transformations over BCN nanosheets. ACS Catal. 2019, 9, 8068–8072. [Google Scholar] [CrossRef]
- Wang, B.; Anpo, M.; Lin, J.; Yang, C.; Zhang, Y.; Wang, X. Direct hydroxylation of benzene to phenol on h-BCN nanosheets in the presence of FeCl3 and H2O2 under visible light. Catal. Today 2019, 324, 73–82. [Google Scholar] [CrossRef]
- Zhou, M.; Wang, S.; Yang, P.; Huang, C.; Wang, X. Boron carbon nitride semiconductors decorated with CdS nanoparticles for photocatalytic reduction of CO2. ACS Catal. 2018, 8, 4928–4936. [Google Scholar] [CrossRef]
- Garay, A.L.; Pichon, A.; James, S.L. Solvent-free synthesis of metal complexes. Chem. Soc. Rev. 2007, 36, 846–855. [Google Scholar] [CrossRef]
- Ambika, S.; Devasena, M.; Nambi, I.M. Synthesis, characterization and performance of high energy ball milled meso-scale zero valent iron in Fenton reaction. J. Environ. Manag. 2016, 181, 847–855. [Google Scholar] [CrossRef]
- Blanco, M.C.; Cámara, J.; Gimeno, M.C.; Laguna, A.; James, S.L.; Lagunas, M.C.; Villacampa, M.D. Synthesis of gold-silver luminescent honeycomb aggregates by both solvent-based and solvent-free methods. Angew. Chem. Int. Ed. 2012, 51, 9777–9779. [Google Scholar] [CrossRef]
- Wang, K.; Duan, D.; Wang, R.; Lin, A.; Cui, Q.; Liu, B.; Cui, T.; Zou, B.; Zhang, X.; Hu, J.; et al. Stability of hydrogen-bonded supramolecular architecture under high pressure conditions: Pressure-induced amorphization in melamine-boric acid adduct. Langmuir 2009, 25, 4787–4791. [Google Scholar] [CrossRef]
- Kawsaki, T.; Kuroda, Y.; Nishikawa, H. The crystal structure of melamine diborate. J. Ceram. Soc. Jpn. 1996, 104, 935–938. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Choudhury, A.C.; Rao, N.R. Supramolecular hydrogen-bonded structure of a 1:2 adduct of melamine with boric acid. J. Mol. Struct. 2002, 613, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Atalay, Y.; Avcı, D.; Başoğlu, A.; Okur, İ. Molecular structure and vibrational spectra of melamine diborate by density functional theory and ab initio Hartree-Fock calculations. J. Mol. Struc. THEOCHEM 2005, 713, 21–26. [Google Scholar] [CrossRef]
- Norrild, J.C.; Eggert, H. Evidence for mono-and bisdentate boronate complexes of glucose in the furanose form. Application of 1JC-C coupling constants as a structural probe. J. Am. Chem. Soc. 1995, 117, 1479–1484. [Google Scholar] [CrossRef]
- Franco, A.; Ascenso, J.R.; Ilharco, L.; Diogo, H.P.; André, V.; da Silva, J.A.L. Ribose-borate esters as potential components for prebiological evolution. J. Mol. Struct. 2019, 1184, 281–288. [Google Scholar] [CrossRef]
- Cordes, D.B.; Gamsey, S.; Singaram, B. Fluorescent quantum dots with boronic acid substituted viologens to sense glucose in aqueous solution. Angew. Chem. Int. Ed. 2006, 45, 3829–3832. [Google Scholar] [CrossRef]
- Lu, Q.; An, J.; Duan, Y.; Luo, Q.; Yin, R.; Li, X.; Tang, C.; Wang, D. A strategy for preparing efficient Ag/p-BNNS nanocatalyst with a synergistic effect between Ag and p-BNNS. J. Catal. 2021, 395, 457–466. [Google Scholar] [CrossRef]
- Panicker, C.Y.; Varghese, H.T.; John, A.; Philip, D.; Nogueira, H.I.S. Vibrational spectra of melamine diborate, C3N6H62H3BO3. Spectrochim. Acta A 2002, 58, 1545–1551. [Google Scholar] [CrossRef]
- Bagno, A.; Rastrelli, F.; Saielli, G. Prediction of the 1H and 13C NMR spectra of α-D-glucose in water by DFT methods and MD simulations. J. Org. Chem. 2007, 72, 7373–7381. [Google Scholar] [CrossRef]
- Henderson, W.G.; How, M.J.; Kennedy, G.R.; Mooney, E.F. The interconversion of aqueous boron species and the interaction of borate with diols: A 11B NMR study. Carbohydr. Res. 1973, 28, 1–12. [Google Scholar] [CrossRef]
- Zhang, S.; Mandai, T.; Ueno, K.; Dokko, K.; Watanabe, M. Hydrogen-bonding supramolecular protic salt as an “all-in-one” precursor for nitrogen-doped mesoporous carbons for CO2 adsorption. Nano Energy 2015, 13, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Pascallon, J.; Stambouli, V.; Ilias, S.; Bouchier, D.; Nouet, G.; Silva, F.; Gicquel, A. Deposition of c-BN films on diamond: Influence of the diamond roughness. Mater. Sci. Eng. B 1999, 59, 239–243. [Google Scholar] [CrossRef]
- Taguchi, A.; Schüth, F. Ordered mesoporous materials in catalysis. Microporous Mesoporous Mater. 2005, 77, 1–45. [Google Scholar] [CrossRef]
- Song, Q.; Fang, Y.; Liu, Z.; Li, L.; Wang, Y.; Liang, J.; Huang, Y.; Lin, J.; Hu, L.; Zhang, J.; et al. The performance of porous hexagonal BN in high adsorption capacity towards antibiotics pollutants from aqueous solution. Chem. Eng. J. 2017, 325, 71–79. [Google Scholar] [CrossRef]
- Wang, J.; Hao, J.; Liu, D.; Qin, S.; Portehault, D.; Li, Y.; Chen, Y.; Lei, W. Porous boron carbon nitride nanosheets as efficient metal-free catalysts for the oxygen reduction reaction in both alkaline and acidic solutions. ACS Energy Lett. 2017, 2, 306–312. [Google Scholar] [CrossRef] [Green Version]
- Weng, Q.H.; Wang, X.B.; Zhi, C.Y.; Bando, Y.; Golberg, D. Boron nitride porous microbelts for hydrogen storage. ACS Nano 2013, 7, 1558–1565. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wu, B.; Liu, H.; Huang, L.; Li, Y.; Guo, W.; Chen, X.; Peng, P.; Fu, L.; Yang, Y.; et al. Water-assisted growth of large-sized single crystal hexagonal boron nitride grains. Mater. Chem. Front. 2017, 1, 1836–1840. [Google Scholar] [CrossRef]
- Liu, A.C.Y.; Arenal, R.; Montagnac, G. In situ transmission electron microscopy observation of keV-ion irradiation of single-walled carbon and boron nitride nanotubes. Carbon 2013, 62, 248–255. [Google Scholar] [CrossRef]
- Qin, L.; Yu, J.; Kuang, S.; Xiao, C.; Bai, X. Few-atomic-layered boron carbonitride nanosheets prepared by chemical vapor deposition. Nanoscale 2012, 4, 120–123. [Google Scholar] [CrossRef]
- Endo, M.; Kim, Y.A.; Hayashi, T.; Muramatsu, H.; Terrones, M.; Saito, R.; Villalpando-Paez, F.; Chou, S.G.; Dresselhaus, M.S. Nanotube coalescence-inducing mode: A novel vibrational mode in carbon systems. Small 2006, 2, 1031–1036. [Google Scholar] [CrossRef]
- Dargelos, A.; Karamanis, P.; Pouchan, C. Ab-initio calculations of the IR spectra of dicyanodiacetylene (C6N2) beyond the harmonic approximation. Chem. Phys. Lett. 2019, 723, 155–159. [Google Scholar] [CrossRef]
- Yu, L.L.; Gao, B.; Chen, Z.; Sun, C.T.; Cui, D.L.; Wang, C.J.; Wang, Q.L.; Jiang, M.H. In situ FTIR investigation on phase transformations in BN nanoparticles. Chin. Sci. Bull. 2005, 50, 2827–2831. [Google Scholar]
- Zeng, X.; Chen, H.; He, X.; Zhang, H.; Fang, W.; Du, X.; Li, W.; Huang, Z.; Zhao, L. In-situ synthesis of non-phase-separated boron carbon nitride for photocatalytic reduction of CO2. Environ. Res. 2022, 207, 112178. [Google Scholar] [CrossRef] [PubMed]
- López-Salas, N.; Ferrer, M.L.; Gutiérrez, M.C.; Fierro, J.L.G.; Cuadrado-Collados, C.; Gandara-Loe, J.; Silvestre-Albero, J.; del Monte, F. Hydrogen-bond supramolecular hydrogels as efficient precursors in the preparation of freestanding 3D carbonaceous architectures containing BCNO nanocrystals and exhibiting a high CO2/CH4 adsorption ratio. Carbon 2018, 134, 470–479. [Google Scholar] [CrossRef] [Green Version]
- Lei, W.; Portehault, D.; Liu, D.; Qin, S.; Chen, Y. Porous boron nitride nanosheets for effective water cleaning. Nat. Commun. 2013, 4, 1777. [Google Scholar] [CrossRef] [PubMed]
- Giusto, P.; Cruz, D.; Heil, T.; Tarakina, N.; Patrini, M.; Antonietti, M. Chemical vapor deposition of highly conjugated, transparent boron carbon nitride thin films. Adv. Sci. 2021, 8, 2101602. [Google Scholar] [CrossRef] [PubMed]
- Weng, Q.; Zeng, L.; Chen, Z.; Han, Y.; Jiang, K.; Bando, Y.; Golberg, D. Hydrogen storage in carbon and oxygen Co-doped porous boron nitrides. Adv. Funct. Mater. 2020, 31, 2007381. [Google Scholar] [CrossRef]
- Torii, S.; Jimura, K.; Hayashi, S.; Kikuchi, R.; Takagaki, A. Utilization of hexagonal boron nitride as a solid acid-base bifunctional catalyst. J. Catal. 2017, 355, 176–184. [Google Scholar] [CrossRef]
- Marchetti, P.S.; Kwon, D.; Schmidt, W.R.; Interrante, L.V.; Maciel, G.E. High-field boron-11 magic-angle spinning NMR characterization of boron nitrides. Chem. Mater. 2002, 3, 482–486. [Google Scholar] [CrossRef]
- Bawari, S.; Sharma, K.; Kalita, G.; Madhu, P.K.; Narayanan, T.N.; Mondal, J. Structural evolution of BCN systems from graphene oxide towards electrocatalytically active atomic layers. Mater. Chem. Front. 2020, 4, 2330–2338. [Google Scholar] [CrossRef]
- Portehault, D.; Giordano, C.; Gervais, C.; Senkovska, I.; Kaskel, S.; Sanchez, C.; Antonietti, M. High-surface-area nanoporous boron carbon nitrides for hydrogen storage. Adv. Funct. Mater. 2010, 20, 1827–1833. [Google Scholar] [CrossRef]
- Chen, S.; Li, P.; Xu, S.; Pan, X.; Fu, Q.; Bao, X. Carbon doping of hexagonal boron nitride porous materials toward CO2 capture. J. Mater. Chem. A 2018, 6, 1832–1839. [Google Scholar] [CrossRef]
- Wrackmeyer, B. Carbon-13 NMR spectroscopy of boron compounds. Prog. Nucl. Magn. Reson. Spectrosc. 1979, 12, 227–259. [Google Scholar] [CrossRef]
- Tay, R.Y.; Li, H.; Tsang, S.H.; Zhu, M.; Loeblein, M.; Jing, L.; Leong, F.N.; Teo, E.H.T. Trimethylamine borane: A new single-source precursor for monolayer h-BN single crystals and h-BCN thin films. Chem. Mater. 2016, 28, 2180–2190. [Google Scholar] [CrossRef]
- Liu, F.; Yu, J.; Ji, X.; Qian, M. Nanosheet-structured boron nitride spheres with a versatile adsorption capacity for water cleaning. ACS Appl. Mater. Inter. 2015, 7, 1824–1832. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Chen, C.; Du, M.; Wu, Y.; Ren, C.; Ding, K.; Song, M.; Huang, C. Porous hexagonal boron nitride sheets: Effect of hydroxyl and secondary amino groups on photocatalytic hydrogen evolution. ACS Appl. Nano Mater. 2018, 1, 4566–4575. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Bhattacharya, S.; Das, G.P. Band gap engineering by functionalization of BN sheet. Phys. Rev. B 2012, 85, 035415. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Zhang, R.; Zhou, T.; Jin, S.; Huang, J.; Ye, L.; Huang, Z.; Wang, F.; Zhou, Y. B–O bonds in ultrathin boron nitride nanosheets to promote photocatalytic carbon dioxide conversion. ACS Appl. Mater. Interfaces 2020, 12, 9935–9943. [Google Scholar] [CrossRef]
- Zhao, L.; Ye, F.; Wang, D.; Cai, X.; Meng, C.; Xie, H.; Zhang, J.; Bai, S. Lattice engineering on metal cocatalysts for enhanced photocatalytic reduction of CO2 into CH4. ChemSusChem 2018, 11, 3524–3533. [Google Scholar] [CrossRef]
- Xie, S.; Wang, Y.; Zhang, Q.; Deng, W.; Wang, Y. MgO- and Pt-promoted TiO2 as an efficient photocatalyst for the preferential reduction of carbon dioxide in the presence of water. ACS Catal. 2014, 4, 3644–3653. [Google Scholar] [CrossRef]
- Shankar, R.; Sachs, M.; Francàs, L.; Lubert-Perquel, D.; Kerherve, G.; Regoutz, A.; Petit, C. Porous boron nitride for combined CO2 capture and photoreduction. J. Mater. Chem. A 2019, 7, 23931–23940. [Google Scholar] [CrossRef] [Green Version]
- Weng, Q.; Kvashnin, D.G.; Wang, X.; Cretu, O.; Yang, Y.; Zhou, M.; Zhang, C.; Tang, D.-M.; Sorokin, P.B.; Bando, Y.; et al. Tuning of the optical, electronic, and magnetic properties of boron nitride nanosheets with oxygen doping and functionalization. Adv. Mater. 2017, 29, 1700695. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Jiang, K.; Qiu, X.; Yu, J.; Liu, M. Product selectivity of photocatalytic CO2 reduction reactions. Mater. Today 2020, 32, 222–243. [Google Scholar] [CrossRef]
- Park, H.-a.; Choi, J.H.; Choi, K.M.; Lee, D.K.; Kang, J.K. Highly porous gallium oxide with a high CO2 affinity for the photocatalytic conversion of carbon dioxide into methane. J. Mater. Chem. 2012, 22, 5304. [Google Scholar] [CrossRef]
- White, J.L.; Baruch, M.F.; Pander, J.E., III; Hu, Y.; Fortmeyer, I.C.; Park, J.E.; Zhang, T.; Liao, K.; Gu, J.; Yan, Y.; et al. Light-driven heterogeneous reduction of carbon dioxide: Photocatalysts and photoelectrodes. Chem. Rev. 2015, 115, 12888–12935. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Wang, B.; Wu, N.; Han, C.; Zhang, X.; Shen, S.; Tian, Q.; Qin, C.; Li, P.; Wang, Y. Molecular-scale understanding on the structure evolution from melamine diborate supramolecule to boron nitride fibers. Ceram. Int. 2020, 46, 1083–1090. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Photocatalysts | Formation Rate (μmol·g−1·h−1) | ||
|---|---|---|---|
| CO | CH4 | H2 | |
| BN [56] | 0 | - | 0.7 |
| O/BN [56] | 12.5 | - | 3.3 |
| g-C3N4 [57] | 0.1 | 0.07 | 1.0 |
| TiO2 [58] | 1.2 | 0.38 | 2.1 |
| Pt-TiO2 [58] | 1.1 | 5.2 | 33 |
| Pd-TiO2 [58] | 1.1 | 4.3 | 25 |
| Rh-TiO2 [58] | 0.62 | 3.5 | 18 |
| Au-TiO2 [58] | 1.5 | 3.1 | 20 |
| Ag-TiO2 [58] | 1.7 | 2.1 | 16 |
| Porous BN [59] | 1.17 | - | - |
| p-BN a | 0 | 0 | 0.78 |
| p-BCN-1 a | 63.1 | 8.4 | 19.4 |
| p-BCN-2 a | 42.6 | 2.52 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Q.; An, J.; Duan, Y.; Luo, Q.; Shang, Y.; Liu, Q.; Tang, Y.; Huang, J.; Tang, C.; Yin, R.; et al. Highly Efficient and Selective Carbon-Doped BN Photocatalyst Derived from a Homogeneous Precursor Reconfiguration. Catalysts 2022, 12, 555. https://doi.org/10.3390/catal12050555
Lu Q, An J, Duan Y, Luo Q, Shang Y, Liu Q, Tang Y, Huang J, Tang C, Yin R, et al. Highly Efficient and Selective Carbon-Doped BN Photocatalyst Derived from a Homogeneous Precursor Reconfiguration. Catalysts. 2022; 12(5):555. https://doi.org/10.3390/catal12050555
Chicago/Turabian StyleLu, Qiong, Jing An, Yandong Duan, Qingzhi Luo, Yunyun Shang, Qiunan Liu, Yongfu Tang, Jianyu Huang, Chengchun Tang, Rong Yin, and et al. 2022. "Highly Efficient and Selective Carbon-Doped BN Photocatalyst Derived from a Homogeneous Precursor Reconfiguration" Catalysts 12, no. 5: 555. https://doi.org/10.3390/catal12050555
APA StyleLu, Q., An, J., Duan, Y., Luo, Q., Shang, Y., Liu, Q., Tang, Y., Huang, J., Tang, C., Yin, R., & Wang, D. (2022). Highly Efficient and Selective Carbon-Doped BN Photocatalyst Derived from a Homogeneous Precursor Reconfiguration. Catalysts, 12(5), 555. https://doi.org/10.3390/catal12050555