
High-Performance of Electrocatalytic CO2 Reduction on Defective Graphene-Supported Cu4S2 Cluster

Abstract
:
1. Introduction
2. Results and Discussion
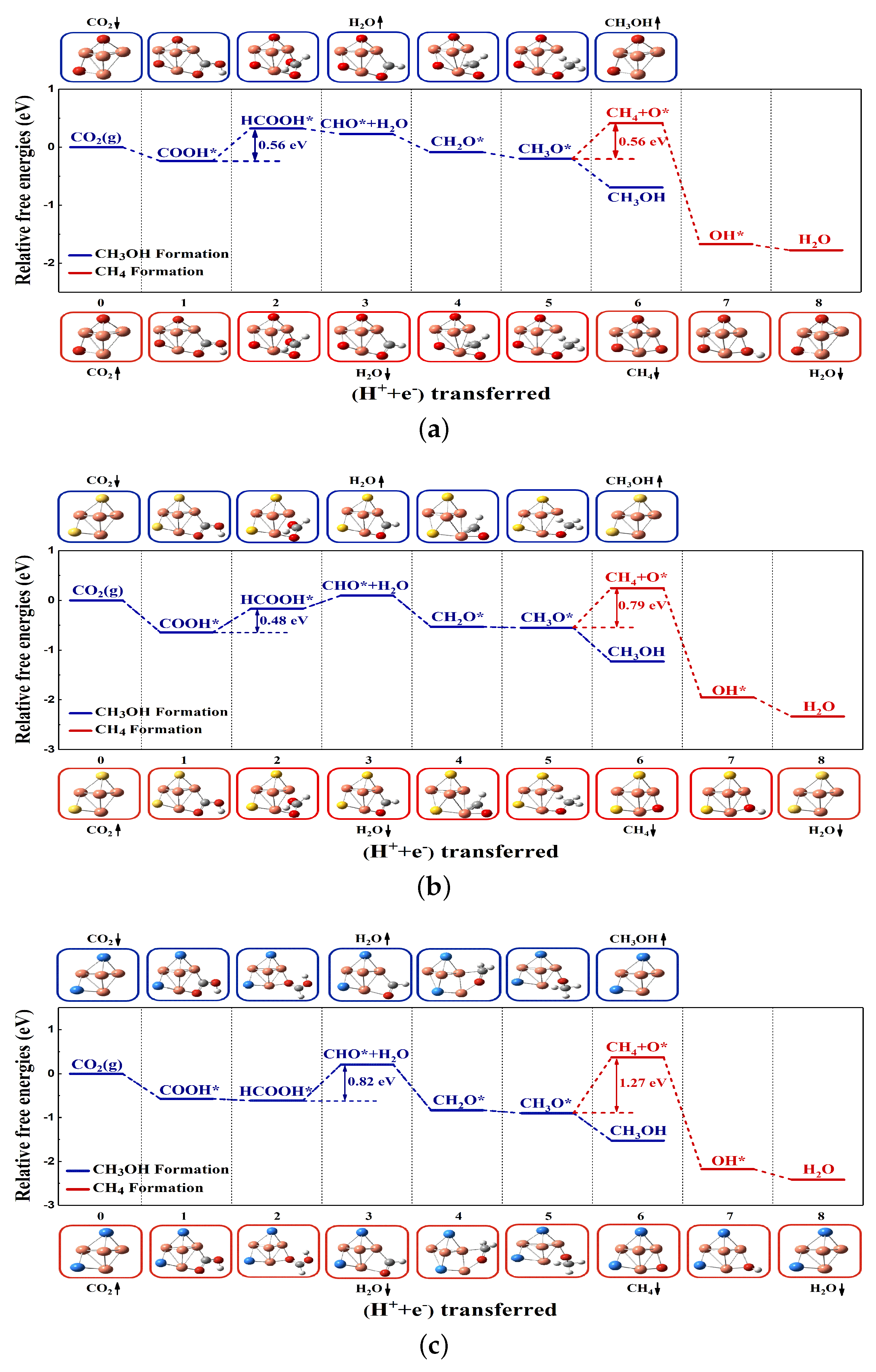
2.1. Mechanism of CO2RR on the Cu4X2 Cluster
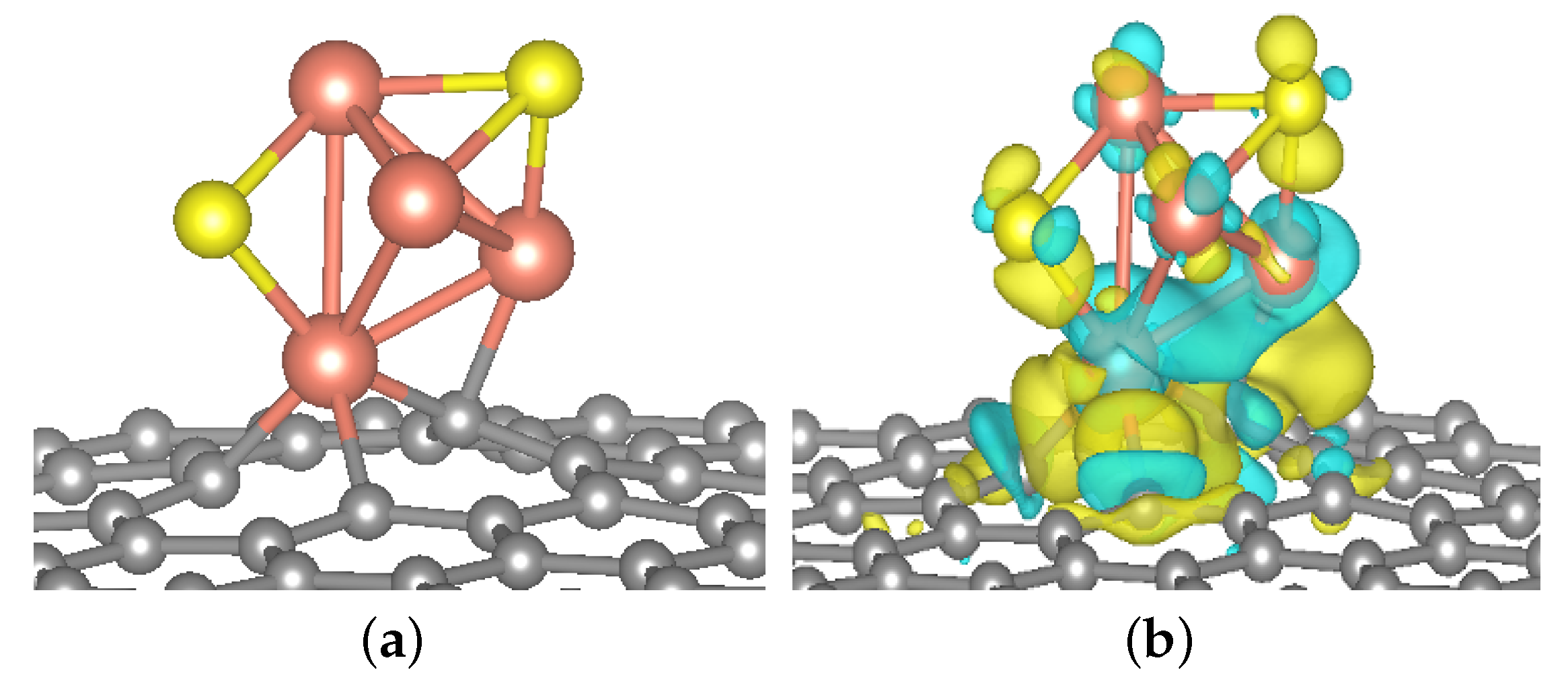
2.1.1. Cu4X2 Cluster and Electrochemical Adsorption of CO2
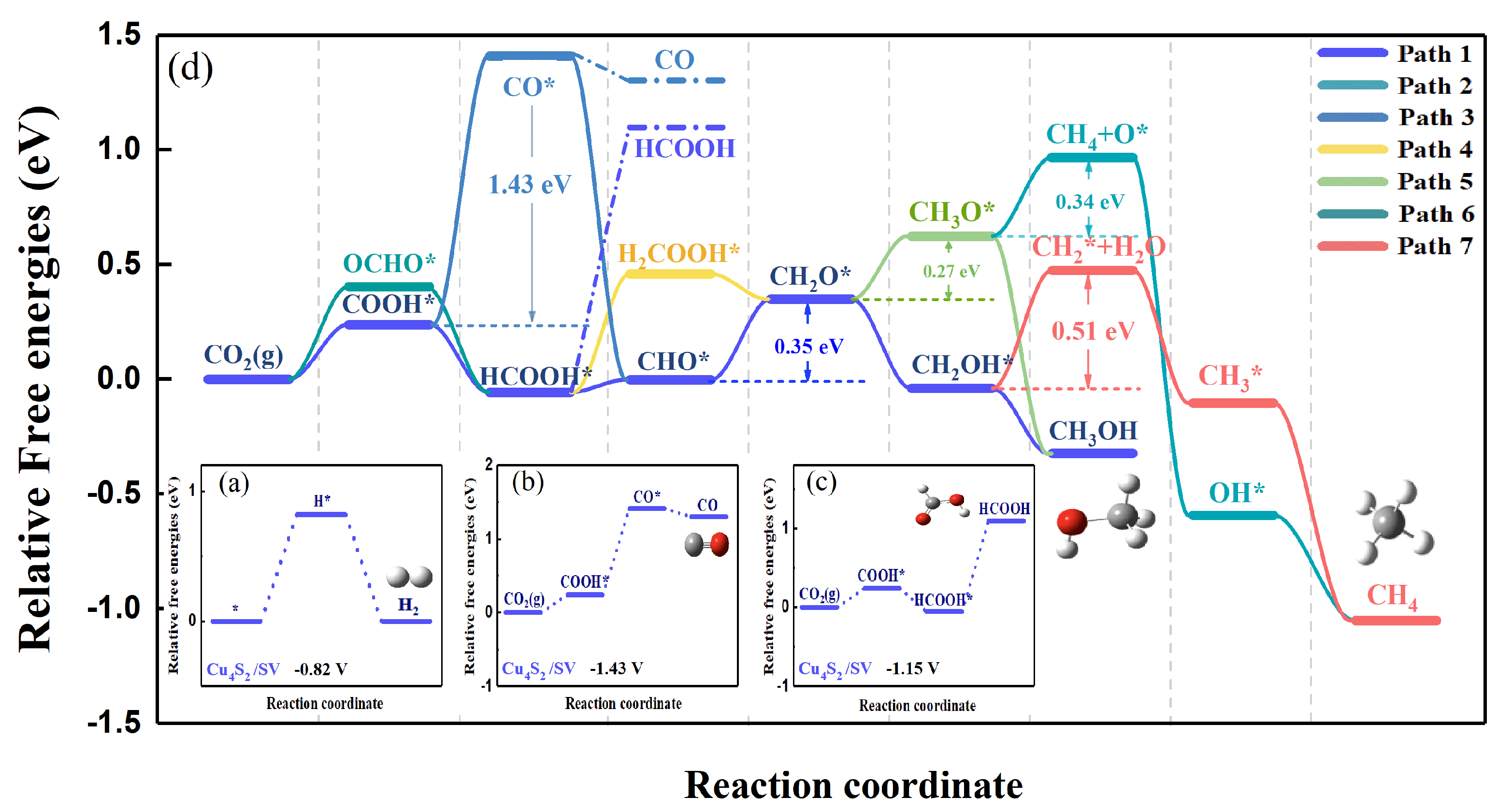
2.1.2. Reaction Pathway of CO2RR on the Cu4X2 Cluster
2.1.3. The Catalytic Performance on Cu4X2 Cluster
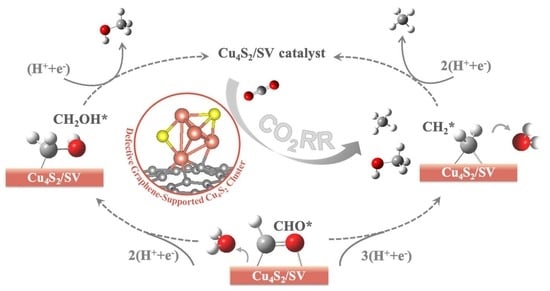
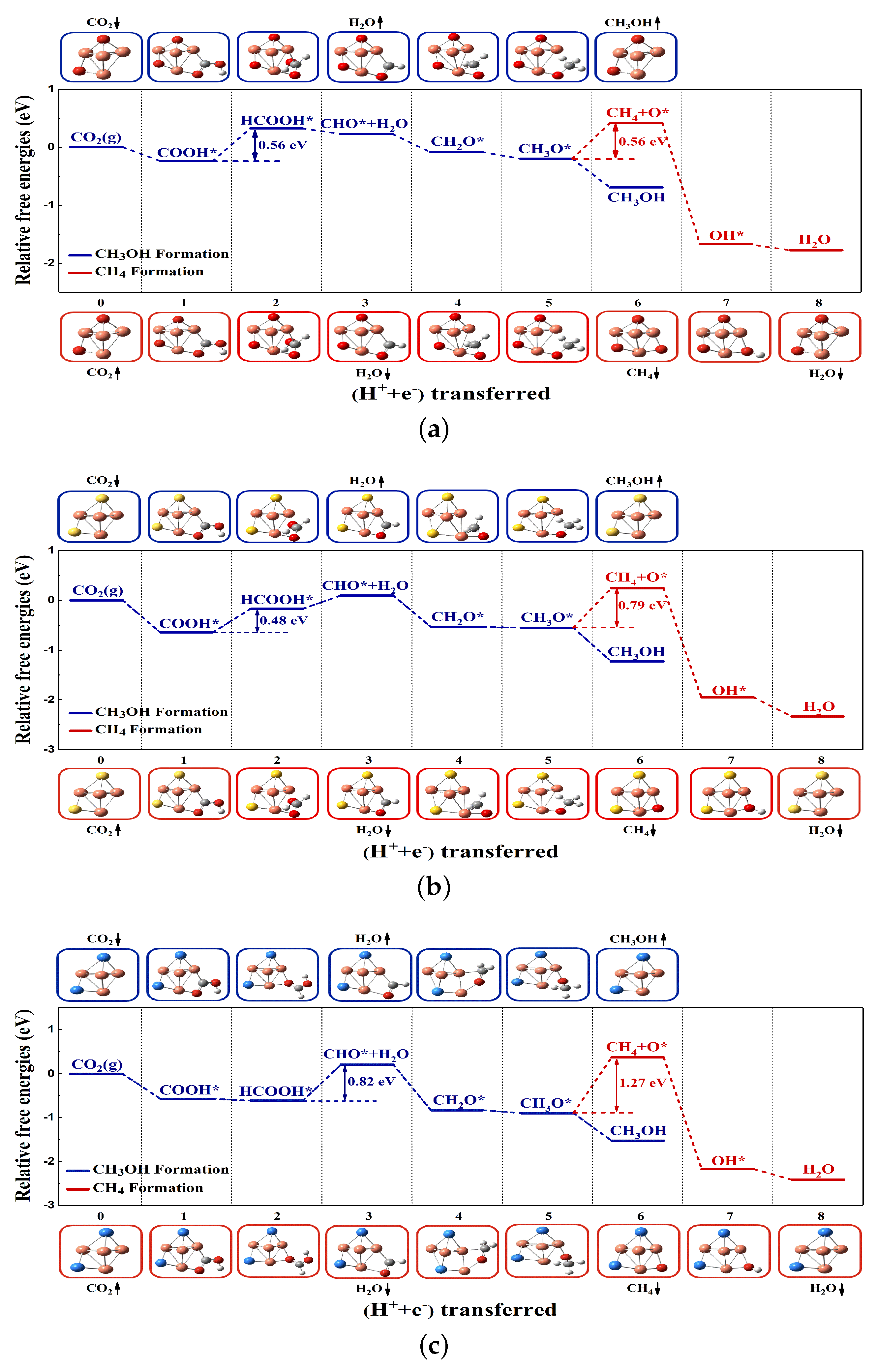
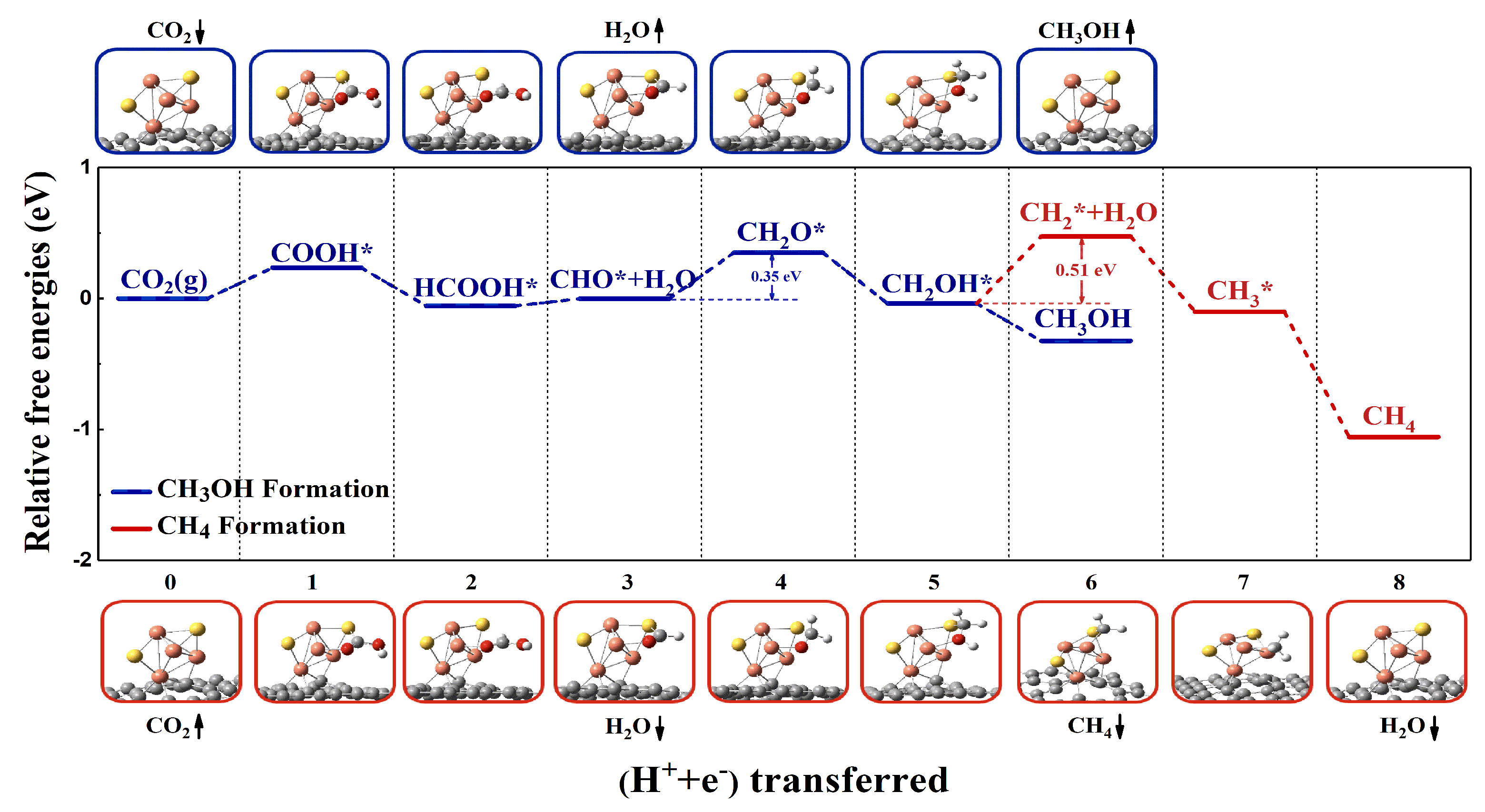
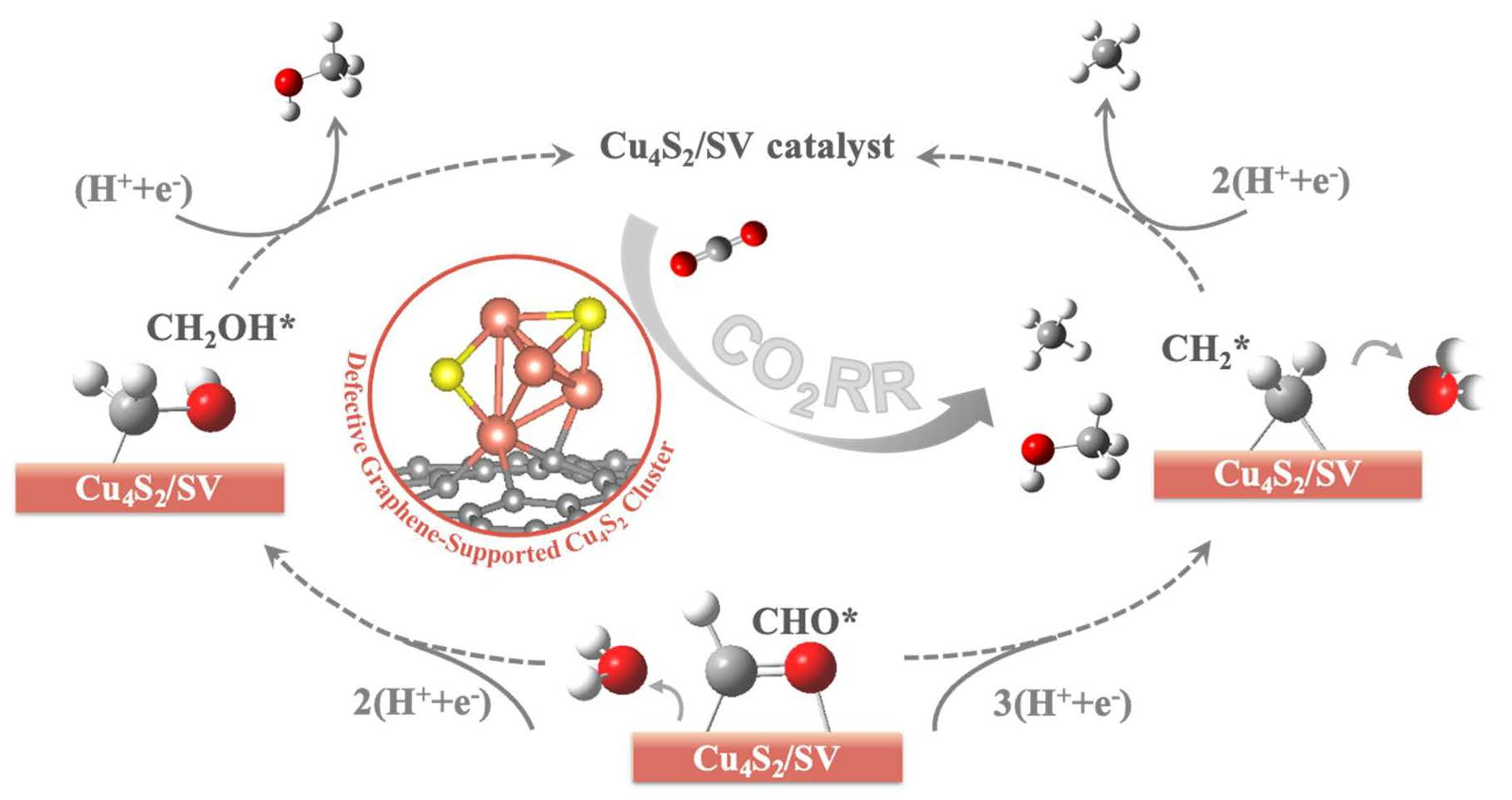
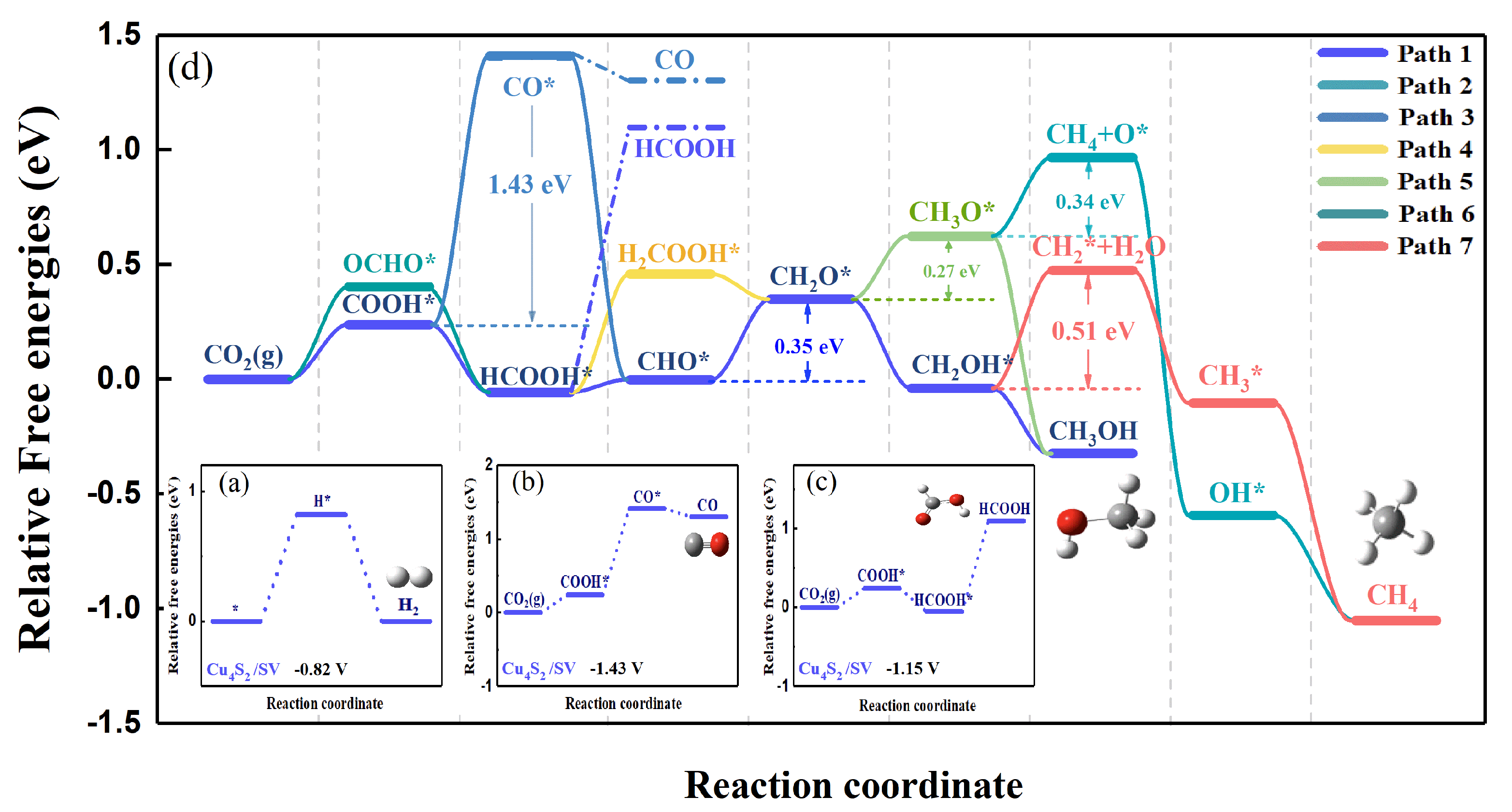
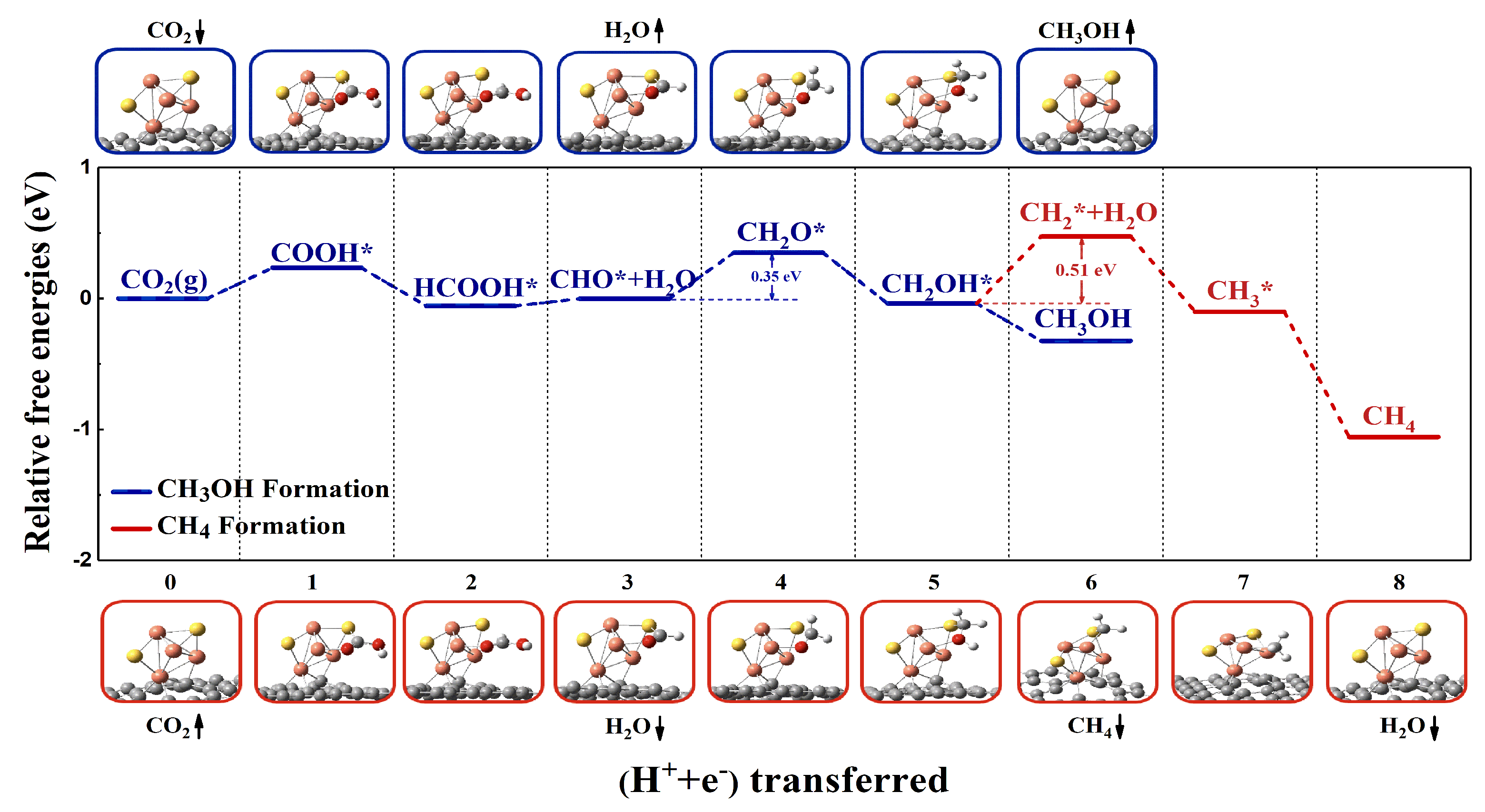
2.2. CO2RR Pathway on the Defective Graphene Supported Cu4S2 Cluster
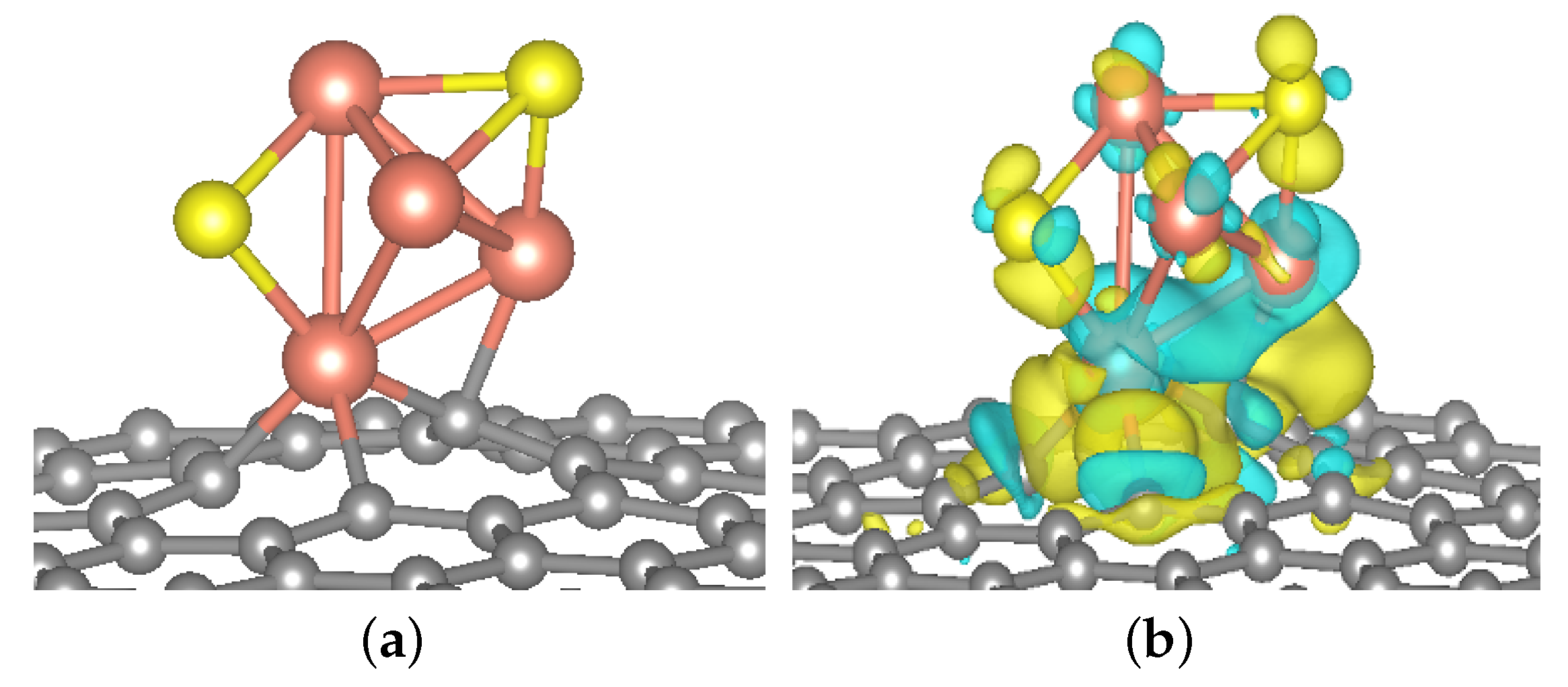
2.2.1. Geometric Structure and Stability of the Catalyst Model
2.2.2. Reaction Pathway of CO2RR on Cu4S2/SV
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dinh, C.; Burdyny, T.; Kibria, M.G.; Seifitokaldani, A.; Gabardo, C.M.; De, A.; Kiani, A.; Edwards, J.P.; De Luna, P.; Bushuyev, O.S.; et al. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 2018, 360, 783–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kortlever, R.; Shen, J.; Schouten, K.J.P.; Calle Vallejo, F.; Koper, M.T. Catalysts and reaction pathways for the electrochemical reduction of carbon dioxide. J. Phys. Chem. Lett. 2015, 6, 4073–4082. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703–3727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Federsel, C.; Jackstell, R.; Beller, M. State-of-the-art catalysts for hydrogenation of carbon dioxide. Angew. Chem. Int. Ed. Engl. 2010, 49, 6254–6257. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y. Electrochemical CO2 Reduction on Metal Electrodes. In Modern Aspects of Electrochemistry; Springer: New York, NY, USA, 2018; Volume 42, pp. 89–189. [Google Scholar] [CrossRef]
- Jiao, J.; Lin, R.; Liu, S.; Cheong, W.C.; Zhang, C.; Chen, Z.; Pan, Y.; Tang, J.; Wu, K.; Hung, S.F.; et al. Copper atom-pair catalyst anchored on alloy nanowires for selective and efficient electrochemical reduction of CO2. Nat. Chem. 2019, 11, 222–228. [Google Scholar] [CrossRef]
- Olah, G.A.; Goeppert, A.; Prakash, G.S. Chemical recycling of carbon dioxide to methanol and dimethyl ether: From greenhouse gas to renewable, environmentally carbon neutral fuels and synthetic hydrocarbons. J. Org. Chem. 2009, 74, 487–498. [Google Scholar] [CrossRef]
- Rawat, K.S.; Mahata, A.; Pathak, B. Thermochemical and electrochemical CO2 reduction on octahedral Cu nanocluster: Role of solvent towards product selectivity. J. Catal. 2017, 349, 118–127. [Google Scholar] [CrossRef]
- Roberts, F.S.; Kuhl, K.P.; Nilsson, A. High selectivity for ethylene from carbon dioxide reduction over copper nanocube electrocatalysts. Angew. Chem. Int. Ed. 2015, 127, 5268–5271. [Google Scholar] [CrossRef]
- Li, J.; Kuang, Y.; Meng, Y.; Tian, X.; Hung, W.H.; Zhang, X.; Li, A.; Xu, M.; Zhou, W.; Ku, C.S.; et al. Electroreduction of CO2 to formate on a copper-based electrocatalyst at high pressures with high energy conversion efficiency. J. Am. Chem. Soc. 2020, 142, 7276–7282. [Google Scholar] [CrossRef]
- Reddy, K.P.; Deshpande, P.A. Density functional theory study of the immobilization and hindered surface migration of Pt3 and Pt4 nanoclusters over defect-ridden graphene: Implications for heterogeneous catalysis. ACS Appl. Nano Mater. 2021, 4, 9068–9079. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, Y.; Shi, L.; Wu, M.; Ouyang, Y.; Wang, J. Dynamic structure change of Cu nanoparticles on carbon supports for CO2 electro-reduction toward multicarbon products. InfoMat 2021, 3, 1285–1294. [Google Scholar] [CrossRef]
- Shi, R.; Zhao, J.; Liu, S.; Sun, W.; Li, H.; Hao, P.; Li, Z.; Ren, J. Nitrogen-doped graphene supported copper catalysts for methanol oxidative carbonylation: Enhancement of catalytic activity and stability by nitrogen species. Carbon 2018, 130, 185–195. [Google Scholar] [CrossRef]
- Carlsson, J.M.; Scheffler, M. Structural, electronic, and chemical properties of nanoporous carbon. Phys. Rev. Lett. 2006, 96, 046806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Liu, M.; Sharma, P.P.; Yadav, R.M.; Ma, L.; Yang, Y.; Zou, X.; Zhou, X.D.; Vajtai, R.; Yakobson, B.I.; et al. Incorporation of nitrogen defects for efficient reduction of CO2 via two-electron pathway on three-dimensional graphene foam. Nano Lett. 2016, 16, 466–470. [Google Scholar] [CrossRef]
- Lim, D.H.; Jo, J.H.; Shin, D.Y.; Wilcox, J.; Ham, H.C.; Nam, S.W. Carbon dioxide conversion into hydrocarbon fuels on defective graphene-supported Cu nanoparticles from first principles. Nanoscale 2014, 6, 5087–5092. [Google Scholar] [CrossRef]
- Liu, C.; He, H.; Zapol, P.; Curtiss, L.A. Computational studies of electrochemical CO2 reduction on subnanometer transition metal clusters. Phys. Chem. Chem. Phys. 2014, 16, 26584–26599. [Google Scholar] [CrossRef]
- Ma, Z.; Tsounis, C.; Kumar, P.V.; Han, Z.; Wong, R.J.; Toe, C.Y.; Zhou, S.; Bedford, N.M.; Thomsen, L.; Ng, Y.H.; et al. Enhanced Electrochemical CO2 Reduction of Cu@CuxO Nanoparticles Decorated on 3D Vertical Graphene with Intrinsic sp3-type Defect. Adv. Funct. 2020, 30, 1910118. [Google Scholar] [CrossRef]
- Hu, Q.; Han, Z.; Wang, X.; Li, G.; Wang, Z.; Huang, X.; Yang, H.; Ren, X.; Zhang, Q.; Liu, J.; et al. Facile synthesis of sub-nanometric copper clusters by double confinement enables selective reduction of carbon dioxide to methane. J. Mater. Chem. A 2020, 59, 19054–19059. [Google Scholar] [CrossRef]
- Perez, G.; Marcandalli, G.; Figueiredo, M.C.; Calle, V.F.; Koper, M.T. Structure-and potential-dependent cation effects on CO reduction at copper single-crystal electrodes. J. Am. Chem. Soc. 2017, 139, 16412–16419. [Google Scholar] [CrossRef]
- Liu, C.; Yang, B.; Tyo, E.; Seifert, S.; DeBartolo, J.; Zapol, P.; Vajda, S.; Curtiss, L.A. Carbon dioxide conversion to methanol over size-selected Cu4 clusters at low pressures. J. Am. Chem. Soc. 2015, 137, 8676–8679. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Liu, C.; Halder, A.; Tyo, E.C.; Martinson, A.B.; Seifert, S.; Zapol, P.; Curtiss, L.A.; Vajda, S. Copper cluster size effect in methanol synthesis from CO2. J. Phys. Chem. C 2017, 121, 10406–10412. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, Y.; Liang, Y.; Li, J.; Zhang, A.; Zheng, H.; Geng, Z.; Li, F.; Zeng, J. Molecular Stabilization of Sub-Nanometer Cu Clusters for Selective CO2 Electromethanation. ChemSusChem 2022, 15, e202102010. [Google Scholar] [CrossRef] [PubMed]
- Passalacqua, R.; Parathoner, S.; Centi, G.; Halder, A.; Tyo, E.C.; Yang, B.; Seifert, S.; Vajda, S. Electrochemical behaviour of naked sub-nanometre sized copper clusters and effect of CO2. Catal. Sci. Technol. 2016, 6, 6977–6985. [Google Scholar] [CrossRef]
- Wei, W.; Lu, Y.; Chen, W.; Chen, S. One-pot synthesis, photoluminescence, and electrocatalytic properties of subnanometer-sized copper clusters. J. Am. Chem. Soc. 2011, 133, 2060–2063. [Google Scholar] [CrossRef] [PubMed]
- Schutz, M.; Muhr, M.; Freitag, K.; Gemel, C.; Kahlal, S.; Saillard, J.Y.; Da Silva, A.C.; Da Silva, J.L.; Fassler, T.F.; Fischer, R.A. Contrasting structure and bonding of a copper-rich and a zinc-rich intermetalloid Cu/Zn cluster. Inorg. Chem. 2020, 59, 9077–9085. [Google Scholar] [CrossRef]
- Su, X.; Jiang, Z.; Zhou, J.; Liu, H.; Zhou, D.; Shang, H.; Ni, X.; Peng, Z.; Yang, F.; Chen, W.; et al. Complementary Operando Spectroscopy identification of in-situ generated metastable charge-asymmetry Cu2-CuN3 clusters for CO2 reduction to ethanol. Nat. Commun. 2022, 13, 1322. [Google Scholar] [CrossRef]
- Uzunova, E.L.; Seriani, N.; Mikosch, H. CO2 conversion to methanol on Cu(I) oxide nanolayers and clusters: An electronic structure insight into the reaction mechanism. Phys. Chem. Chem. Phys. 2015, 17, 11088–11094. [Google Scholar] [CrossRef]
- Zhao, Z.; Peng, X.; Liu, X.; Sun, X.; Shi, J.; Han, L.; Li, G.; Luo, J. Efficient and stable electroreduction of CO2 to CH4 on CuS nanosheet arrays. J. Mater. Chem. A 2017, 5, 20239–20243. [Google Scholar] [CrossRef]
- Lu, Y.F.; Dong, L.Z.; Liu, J.; Yang, R.X.; Liu, J.J.; Zhang, Y.; Zhang, L.; Wang, Y.R.; Li, S.L.; Lan, Y.Q. Predesign of Catalytically Active Sites via Stable Coordination Cluster Model System for Electroreduction of CO2 to Ethylene. Angew. Chem. Int. Ed. 2021, 60, 26210–26217. [Google Scholar] [CrossRef]
- Zhang, J.; Dai, L. Heteroatom-doped graphitic carbon catalysts for efficient electrocatalysis of oxygen reduction reaction. ACS Catal. 2015, 5, 7244–7253. [Google Scholar] [CrossRef]
- Navalon, S.; Dhakshinamoorthy, A.; Alvaro, M.; Antonietti, M.; Garcia, H. Active sites on graphene-based materials as metal-free catalysts. Chem. Soc. Rev. 2017, 46, 4501–4529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Yuan, Y.; Hu, Y.; Zhang, J.; Tang, Y.; Ren, B. Density functional theory study of the structures and electronic properties of copper and sulfur doped copper clusters. Comput. Theor. Chem. 2016, 1080, 47–55. [Google Scholar] [CrossRef]
- Zhang, Q.Y.; Zhao, Q.F.; Liang, X.M.; Wang, X.L.; Ma, F.X.; Suo, B.B.; Zou, W.L.; Han, H.X.; Song, Q.; Wu, Q.; et al. Computational studies of electrochemical CO2 reduction on chalcogen doped Cu4 cluster. Int. J. Hydrogen Energy 2018, 43, 9935–9942. [Google Scholar] [CrossRef]
- Natesakhawat, S.; Lekse, J.W.; Baltrus, J.P.; Ohodnicki Jr, P.R.; Howard, B.H.; Deng, X.; Matranga, C. Active sites and structure–activity relationships of copper-based catalysts for carbon dioxide hydrogenation to methanol. ACS Catal. 2012, 2, 1667–1676. [Google Scholar] [CrossRef]
- Carugno, S.; Chassaing, E.; Rosso, M.; Gonzalez, G.A. Enhanced electrochemical oxidation of methanol on copper electrodes modified by electrocorrosion and electrodeposition. Mater. Chem. Phys. 2014, 143, 1012–1017. [Google Scholar] [CrossRef]
- Periasamy, A.P.; Liu, J.; Lin, H.M.; Chang, H.T. Synthesis of copper nanowire decorated reduced graphene oxide for electro-oxidation of methanol. J. Mater. Chem. A 2013, 1, 5973–5981. [Google Scholar] [CrossRef]
- Peterson, A.A.; Abild Pedersen, F.; Studt, F.; Rossmeisl, J.; Norskov, J.K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 2010, 3, 1311–1315. [Google Scholar] [CrossRef]
- Zhang, J.; Dolg, M. ABCluster: The artificial bee colony algorithm for cluster global optimization. Phys. Chem. Chem. Phys. 2015, 17, 24173–24181. [Google Scholar] [CrossRef]
- Hertwig, R.H.; Koch, W. On the parameterization of the local correlation functional. What is Becke-3-LYP? Chem. Phys. Lett. 1997, 268, 345–351. [Google Scholar] [CrossRef]
- Rassolov, V.A.; Ratner, M.A.; Pople, J.A.; Redfern, P.C.; Curtiss, L.A. 6–31G* basis set for third-row atoms. J. Comput. Chem. 2001, 22, 976–984. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Neese, F.; Wennmohs, F.; Hansen, A. Efficient and accurate local approximations to coupled-electron pair approaches: An attempt to revive the pair natural orbital method. J. Chem. Phys. 2009, 130, 114108. [Google Scholar] [CrossRef] [PubMed]
- Kupka, T.; Stachów, M.; Nieradka, M.; Kaminsky, J.; Pluta, T.; Sauer, S.P. From CCSD(T)/aug-cc-pVTZ-J to CCSD(T) complete basis set limit isotropic nuclear magnetic shieldings via affordable DFT/CBS calculations. Magn. Reson. Chem. 2011, 49, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Takano, Y.; Houk, K. Benchmarking the conductor-like polarizable continuum model (CPCM) for aqueous solvation free energies of neutral and ionic organic molecules. J. Chem. Theory Comput. 2005, 1, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Mathew, K.; Kolluru, V.S.C.; Hennig, R.G. VASPsol: Implicit Solvation and Electrolyte Model for Density-Functional Theory. 2018. Available online: https://github.com/henniggroup/VASPsol (accessed on 1 November 2021).
- Norskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jonsson, H. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Zhang, J.; Glezakou, V.A.; Rousseau, R.; Nguyen, M.T. NWPEsSe: An adaptive-learning global optimization algorithm for nanosized cluster systems. J. Chem. Theory Comput. 2020, 16, 3947–3958. [Google Scholar] [CrossRef]
- Malloum, A.; Fifen, J.J.; Conradie, J. Exploration of the potential energy surface of the ethanol hexamer. J. Chem. Phys. 2019, 150, 124308. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Wang, N.; Zhang, L.; Guo, X.; Huang, S. Analysis and assessment of the structural, electronic properties of (ZrH2)n (n = 5 − 24) clusters: Density function theory calculations. Comput. Theor. Chem. 2020, 1188, 112940. [Google Scholar] [CrossRef]
- Malloum, A.; Conradie, J. Solvent effects on the structures of the neutral ammonia clusters. Comput. Theor. Chem. 2020, 1191, 113042. [Google Scholar] [CrossRef]
- Malloum, A.; Fifen, J.J.; Conradie, J. Large-Sized Ammonia Clusters and Solvation Energies of the Proton in Ammonia. J. Comput. Chem. 2020, 41, 21–30. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster | Electrochemical Adsorption of CO2 | |||||
|---|---|---|---|---|---|---|
| COOH* | δ+ | Geometric Parameters | ||||
| CuO |  | 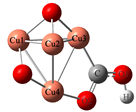 | −0.24 | 0.55 | ||
| 1.91 | 1.89 | |||||
| 1.33 | 1.26 | |||||
| 0.98 | 114.9 | |||||
| CuS | 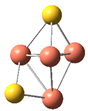 |  | −0.65 | 0.52 | ||
| 1.92 | 1.90 | |||||
| 1.33 | 1.26 | |||||
| 0.98 | 115.3 | |||||
| CuSe | 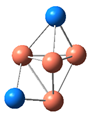 | 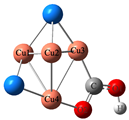 | −0.57 | 0.52 | ||
| 1.93 | 1.91 | |||||
| 1.33 | 1.27 | |||||
| 0.98 | 115.2 | |||||
| Cluster | CH3O* | Bond Length | Bond Energy | Adsorption Energy | ||
|---|---|---|---|---|---|---|
| CuO |  | 1.83 | 1.38 | 53.93 | 95.46 | −1.59 |
| CuO |  | 1.85 | 1.37 | 48.04 | 101.90 | −0.57 |
| CuS |  | 1.89 | 1.41 | 55.35 | 78.30 | −1.53 |
| CuS | 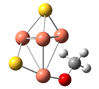 | 1.84 | 1.37 | 41.71 | 96.76 | −0.33 |
| CuSe | 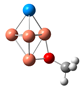 | 1.89 | 1.41 | 53.98 | 77.23 | −2.01 |
| CuSe | 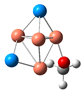 | 1.87 | 1.41 | 49.44 | 98.31 | −0.65 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.; Li, Y.; Zhu, H.; Suo, B. High-Performance of Electrocatalytic CO2 Reduction on Defective Graphene-Supported Cu4S2 Cluster. Catalysts 2022, 12, 454. https://doi.org/10.3390/catal12050454
Zhang Q, Li Y, Zhu H, Suo B. High-Performance of Electrocatalytic CO2 Reduction on Defective Graphene-Supported Cu4S2 Cluster. Catalysts. 2022; 12(5):454. https://doi.org/10.3390/catal12050454
Chicago/Turabian StyleZhang, Qiyan, Yawei Li, Haiyan Zhu, and Bingbing Suo. 2022. "High-Performance of Electrocatalytic CO2 Reduction on Defective Graphene-Supported Cu4S2 Cluster" Catalysts 12, no. 5: 454. https://doi.org/10.3390/catal12050454
APA StyleZhang, Q., Li, Y., Zhu, H., & Suo, B. (2022). High-Performance of Electrocatalytic CO2 Reduction on Defective Graphene-Supported Cu4S2 Cluster. Catalysts, 12(5), 454. https://doi.org/10.3390/catal12050454