
Carboxylate-Assisted Carboxylation of Thiophene with CO2 in the Solvent-Free Carbonate Medium

Abstract
:1. Introduction
2. Results and Discussions
2.1. Carboxylation Reaction in the Carbonate/Carboxylate Mixed Salts
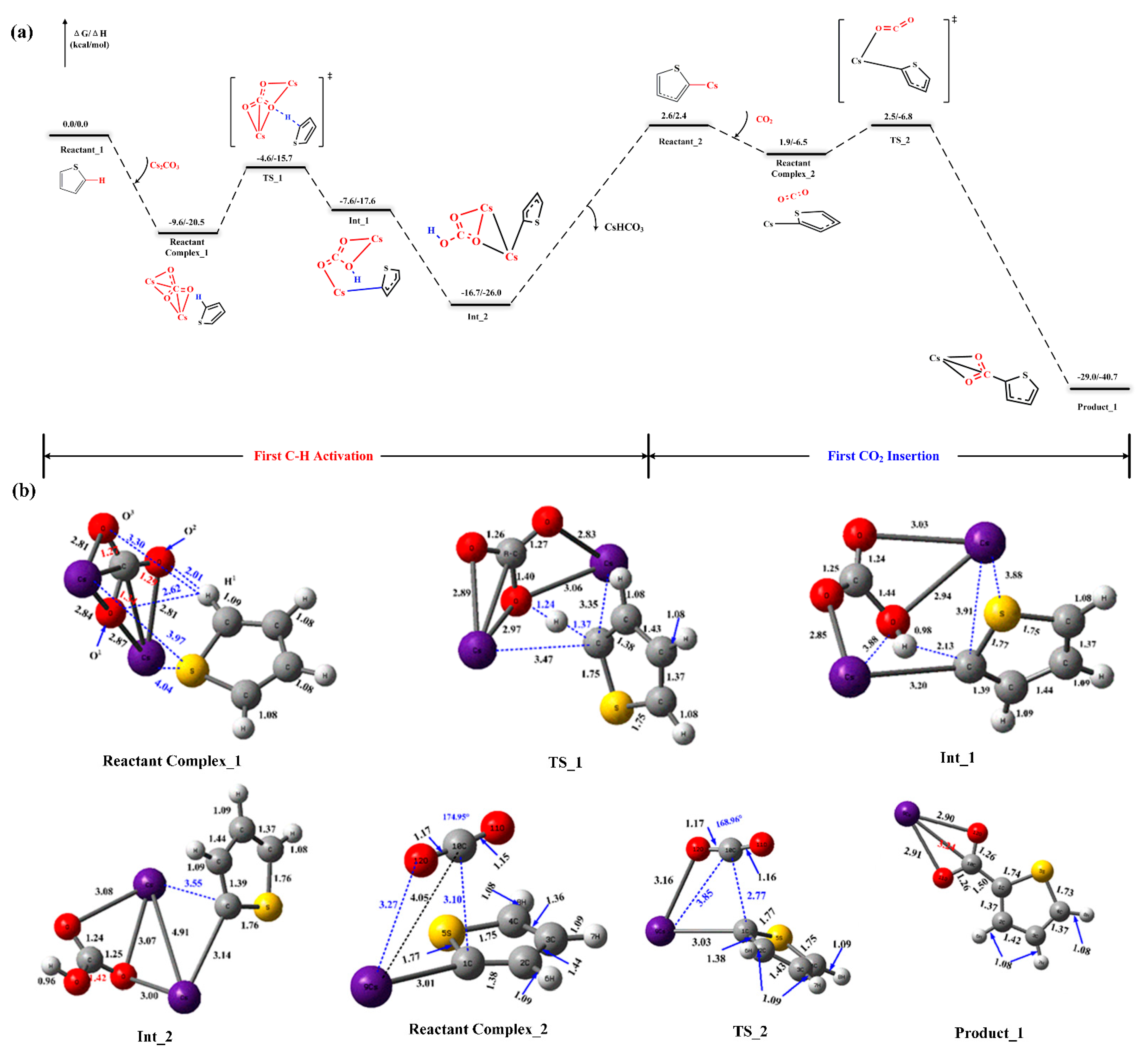
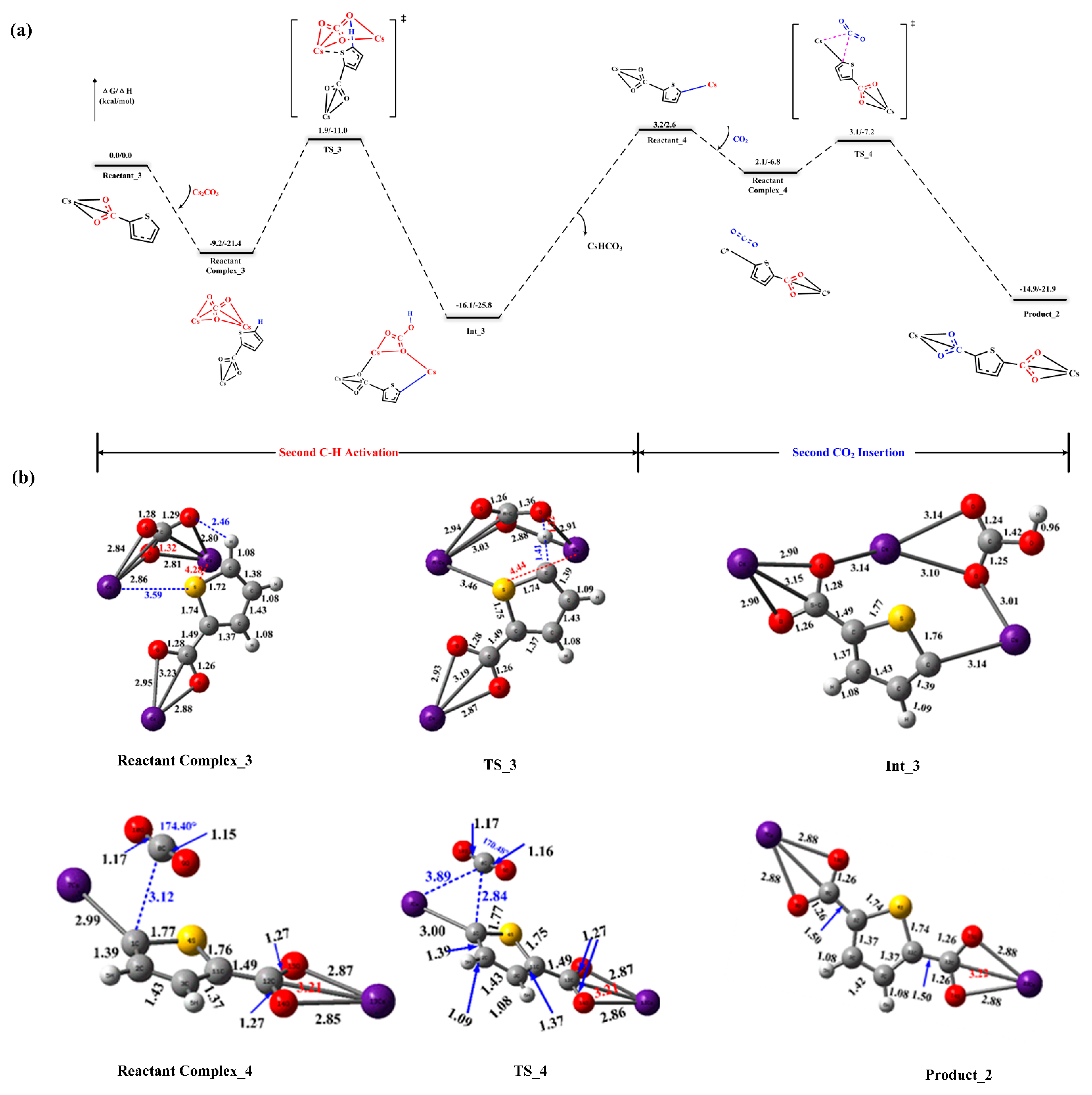
2.2. Reaction Mechanism of Cs2CO3-Promoted C–H Carboxylation with CO2
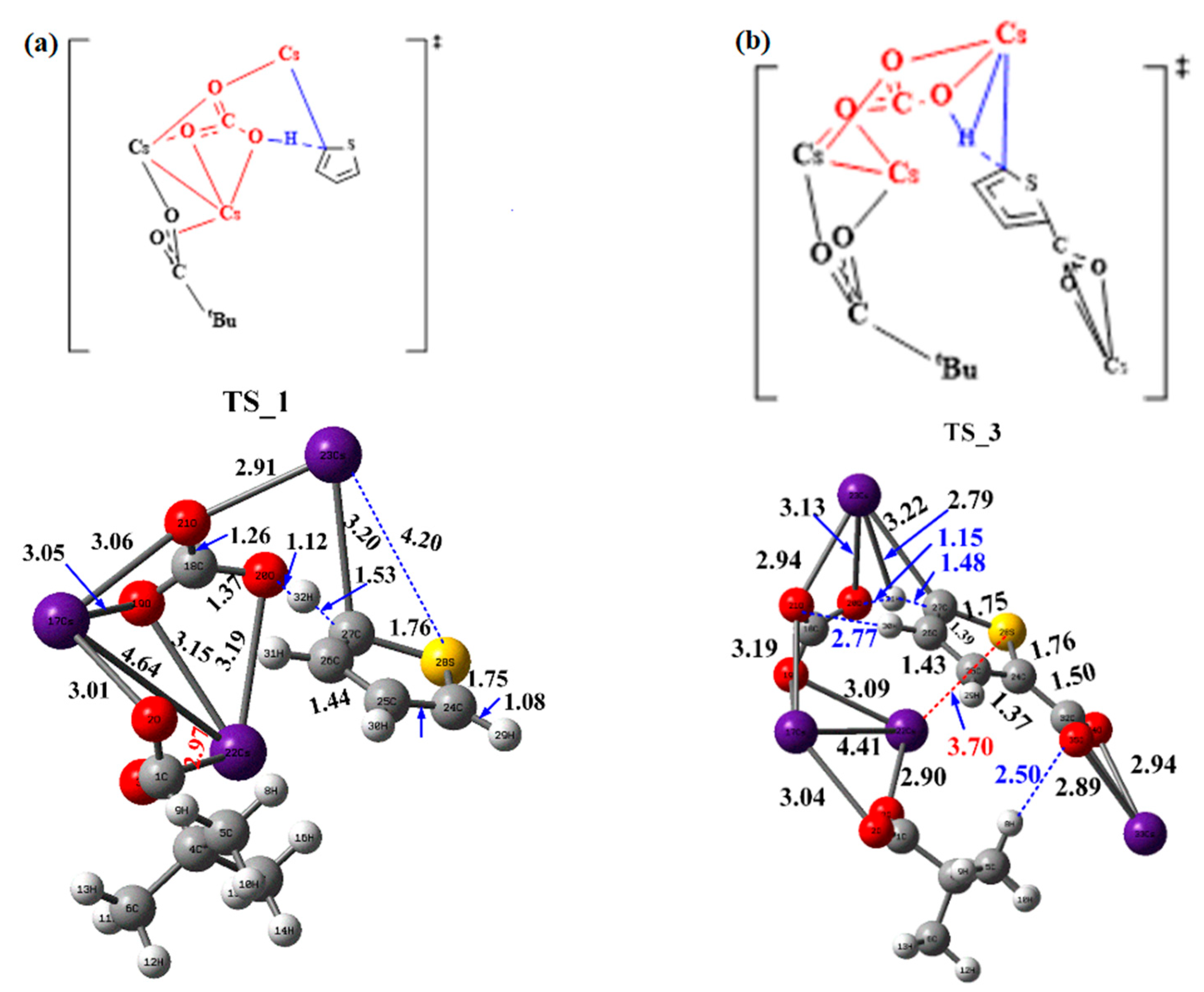
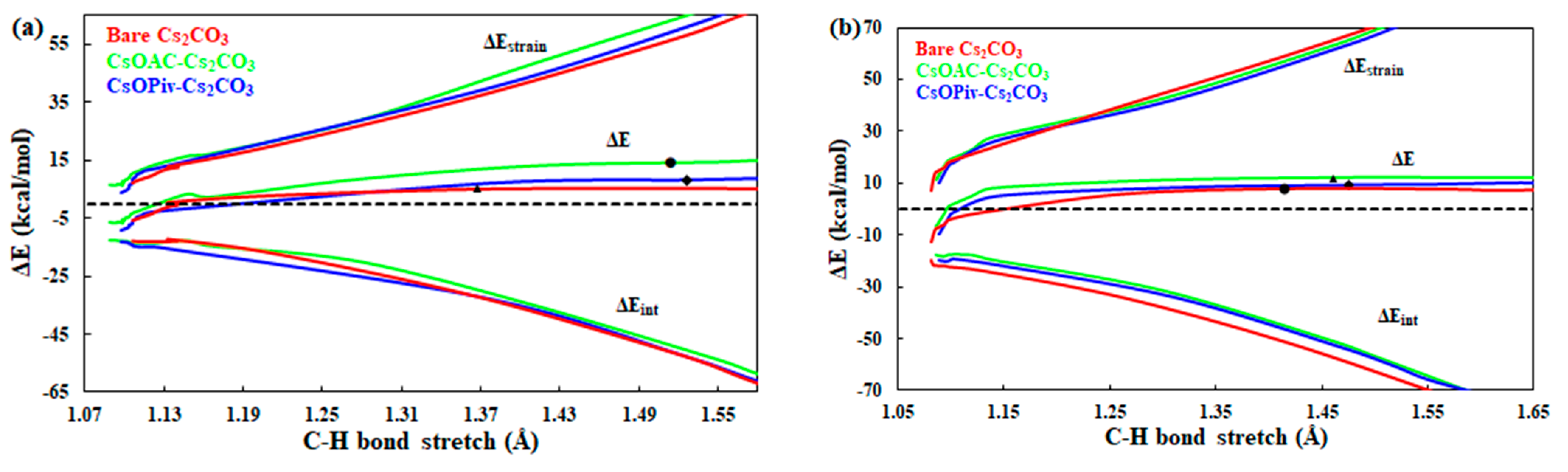
2.3. Carboxylate-Assisted Cs2CO3-Mediated C–H Carboxylation with CO2
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sakakura, T.; Choi, J.C.; Yasuda, H. Transformation of carbon dioxide. Chem. Rev. 2007, 107, 2365–2387. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Larrosa, I. C–H carboxylation of aromatic compounds through CO2 fixation. ChemSusChem 2017, 10, 3317–3332. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.; Li, M.; Zhang, J.; Sun, B.; Mo, F. C-H bond carboxylation with carbon dioxide. ChemSusChem 2019, 12, 6–39. [Google Scholar] [CrossRef]
- Liu, A.H.; Yu, B.; He, L.N. Catalytic conversion of carbon dioxide to carboxylic acid derivatives. Greenh. Gases 2015, 5, 17–33. [Google Scholar] [CrossRef]
- Olah, G.A.; Torok, B.; Joschek, J.P.; Bucsi, I.; Esteves, P.M.; Rasul, G.; Prakash, G.K.S. Efficient chemoselective carboxylation of aromatics to arylcarboxylic acids with a superelectrophilically activated carbon dioxide-Al2Cl6/Al system. J. Am. Chem. Soc. 2002, 124, 11379–11391. [Google Scholar] [CrossRef] [PubMed]
- Munshi, P.; Beckman, E.J. Effect of incubation of CO2 and Lewis acid on the generation of toluic acid from toluene and CO2. Ind. Eng. Chem. Res. 2009, 48, 1059–1062. [Google Scholar] [CrossRef]
- Munshi, P.; Beckman, E.J.; Padmanabhan, S. Combined influence of fluorinated solvent and base in Friedel-Crafts reaction of toluene and CO2. Ind. Eng. Chem. Res. 2010, 49, 6678–6682. [Google Scholar] [CrossRef]
- Nemoto, K.; Yoshida, H.; Egusa, N.; Morohashi, N.; Hattori, T. Direct carboxylation of arenes and halobenzenes with CO2 by the combined use of AlBr3 and R3SiCl. J. Org. Chem. 2010, 75, 7855–7862. [Google Scholar] [CrossRef]
- Nemoto, K.; Onozawa, S.; Egusa, N.; Morohashi, N.; Hattori, T. Carboxylation of indoles and pyrroles with CO2 in the presence of dialkylaluminum halides. Tetrahedron Lett. 2009, 50, 4512–4514. [Google Scholar] [CrossRef]
- Nemoto, K.; Onozawa, S.; Konno, M.; Morohashi, N.; Hattori, T. Direct carboxylation of thiophenes and benzothiophenes with the aid of EtAlCl2. Bull. Chem. Soc. Jpn. 2012, 85, 369–371. [Google Scholar] [CrossRef]
- Tanaka, S.; Watanabe, K.; Tanaka, Y.; Hattori, T. EtAlCl2/2,6-disubstituted pyridine-mediated carboxylation of alkenes with carbon dioxide. Org. Lett. 2016, 18, 2576–2579. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cheng, Z. Performance of combined use of chlorosilanes and AlCl3 in the carboxylation of toluene with CO2. AIChE J. 2017, 63, 185–191. [Google Scholar] [CrossRef]
- Boogaerts, I.I.F.; Nolan, S.P. Carboxylation of C-H Bonds using N-heterocyclic carbene gold(I) complexes. J. Am. Chem. Soc. 2010, 132, 8858–8859. [Google Scholar] [CrossRef] [PubMed]
- Boogaerts, I.I.F.; Fortman, G.C.; Furst, M.R.L.; Cazin, C.S.J.; Nolan, S.P. Carboxylation of N-H/C-H bonds using N-heterocyclic carbene copper(I) complexes. Angew. Chem. Int. Edit. 2010, 49, 8674–8677. [Google Scholar] [CrossRef] [PubMed]
- Yoo, W.J.; Capdevila, M.G.; Du, X.; Kobayashi, S. Base-mediated carboxylation of unprotected indole derivatives with carbon dioxide. Org. Lett. 2012, 14, 5326–5329. [Google Scholar] [CrossRef] [PubMed]
- Fenner, S.; Ackermann, L. C-H carboxylation of heteroarenes with ambient CO2. Green Chem. 2016, 18, 3804–3807. [Google Scholar] [CrossRef] [Green Version]
- Shigeno, M.; Hanasaka, K.; Sasaki, K.; Kumada, N.K.; Kondo, Y. Direct carboxylation of electron-rich heteroarenes promoted by LiO-tBu with CsF and [18]crown-6. Chem. Eur. J. 2019, 25, 3235–3239. [Google Scholar]
- Shigeno, M.; Sasaki, K.; Kumada, N.K.; Kondo, Y. Double-carboxylation of two C-H bonds in 2-alkylheteroarenes using LiO-t-Bu/CsF. Org. Lett. 2019, 21, 4515–4519. [Google Scholar] [CrossRef]
- Shigeno, M.; Tohara, I.; Kumada, N.K.; Kondo, Y. Direct C-2 carboxylation of 3-substituted indoles using a combined Brønsted base consisting of LiO-tBu/CsF/18-crown-6. Eur. J. Org. Chem. 2020, 2020, 1987–1991. [Google Scholar] [CrossRef]
- Lee, M.; Hwang, Y.K.; Kwak, J. Ag(I)-catalyzed C-H carboxylation of thiophene derivatives. Organometallics 2021, 40, 3136–3144. [Google Scholar] [CrossRef]
- Sugimoto, H.; Kawata, I.; Taniguchi, H.; Fujiwara, Y. Palladium-catalyzed carboxylation of aromatic compounds with carbon dioxide. J. Organomet. Chem. 1984, 266, C44–C46. [Google Scholar] [CrossRef]
- Vechorkin, O.; Hirt, N.; Hu, X. Carbon dioxide as the C1 source for direct C-H functionalization of aromatic heterocycles. Org. Lett. 2010, 12, 3567–3569. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Dick, G.R.; Yoshino, T.; Kanan, M.W. Carbon dioxide utilization via carbonate-promoted C-H carboxylation. Nature 2016, 531, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Dick, G.R.; Frankhouser, A.D.; Banerjee, A.; Kanan, M.W. A scalable carboxylation route to furan-2,5-dicarboxylic acid. Green Chem. 2017, 19, 2966–2972. [Google Scholar] [CrossRef]
- Lankenau, A.W.; Kanan, M.W. Polyamide monomers via carbonate-promoted C-H carboxylation of furfurylamine. Chem. Sci. 2020, 11, 248–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, K.; Fu, Y.; Li, J.N.; Liu, L.; Guo, Q.X. What are the pKa values of C-H bonds in aromatic heterocyclic compounds in DMSO? Tetrahedron 2007, 63, 1568–1576. [Google Scholar] [CrossRef]
- Porter, T.M.; Kanan, M.W. Carbonate-promoted C-H carboxylation of electron-rich heteroarenes. Chem. Sci. 2020, 11, 11936–11944. [Google Scholar] [CrossRef]
- Banerjee, A.; Kanan, M.W. Carbonate-promoted hydrogenation of carbon dioxide to multicarbon carboxylates. ACS Cent. Sci. 2018, 4, 606–613. [Google Scholar] [CrossRef]
- Lafrance, M.; Fagnou, K. Palladium-Catalyzed Benzene Arylation: Incorporation of catalytic pivalic acid as a proton shuttle and a key element in catalyst design. J. Am. Chem. Soc. 2006, 128, 16496–16497. [Google Scholar] [CrossRef]
- Zhao, D.; Wang, W.; Lian, S.; Yang, F.; Lan, J.; You, J. Phosphine-free, palladium-catalyzed arylation of heterocycles through C-H bond activation with pivalic acid as a cocatalyst. Chem. Eur. J. 2009, 15, 1337–1340. [Google Scholar] [CrossRef]
- Xu, H.; Muto, K.; Yamaguchi, J.; Zhao, C.; Itami, K.; Musaev, D.G. Key mechanistic features of Ni-catalyzed C−H/C−O biaryl coupling of azoles and naphthalen-2-yl pivalates. J. Am. Chem. Soc. 2014, 136, 14834–14844. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013; Available online: https://www.scienceopen.com/document?vid=839f33cc-9114-4a55-8f1a-3f1520324ef5 (accessed on 5 February 2022).
- Wang, Y.; Guo, C.; Shen, J.; Sun, Y.; Niu, Y.; Li, P.; Liu, G.; Wei, X. A sustainable and green route to futan-2,5-dicarboxylaic acid by direct carboxylation of 2-furoic acid and CO2. J. CO2 Util. 2021, 48, 101524. [Google Scholar] [CrossRef]
- Anslyn, E.V.; Dougherty, D.A. Modern Physical Organic Chemistry; University Science Book: Sausalito, CA, USA, 2006; pp. 87–95. [Google Scholar]
- Xu, L.; Haines, B.; Ajitha, M.; Murakami, K.; Itami, K.; Musaev, D. Roles of base in the Pd-catalyzed annulative chlorophenylene dimerization. ACS Catal. 2020, 10, 3059–3073. [Google Scholar] [CrossRef]
- Wolters, L.P.; Bickelhaupt, F.M. The activation strain model and molecular orbital theory. WIREs Comput. Mol. Sci. 2015, 5, 324–343. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Interaction region indicator: A simple real space function clearly revealing both chemical bonds and weak interactions. Chem.-Methods 2021, 1, 231–239. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]

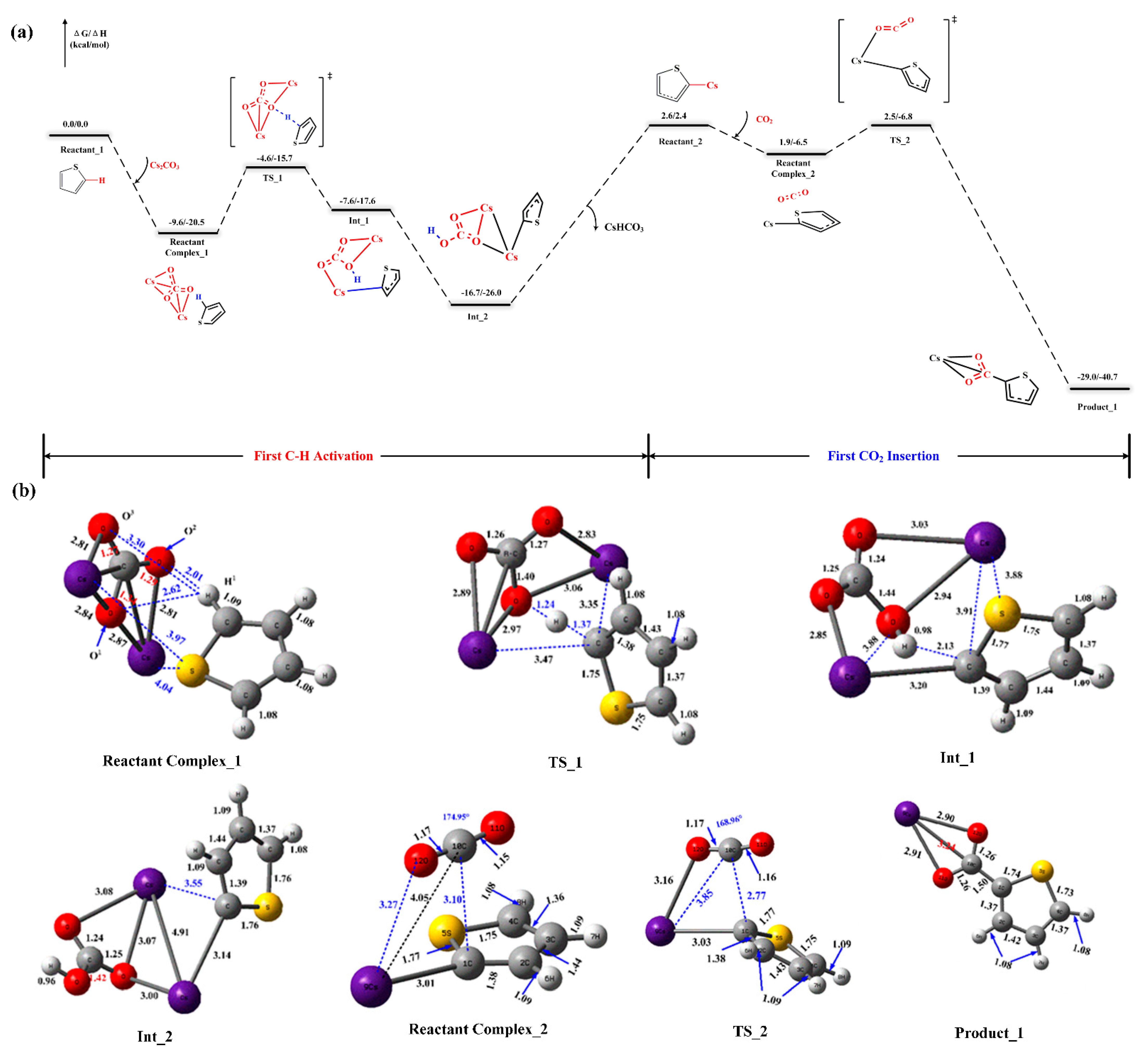{kind=link}
{kind=link}
{kind=link}
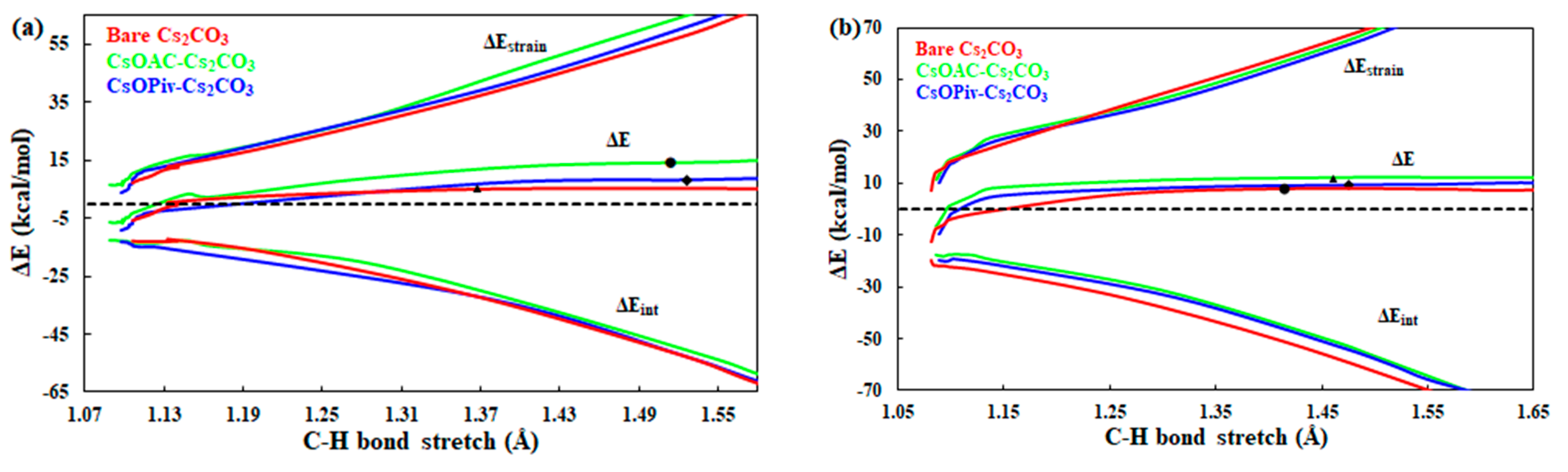{kind=link}
| T (°C) | PCO2 (bar) | nthio (mmol) | nsalts (mmol) | Mixed Salt | 2-Carboxylate (μmol/g) | 2,5-Dicarboxylate (μmol/g) | Yield (%) |
|---|---|---|---|---|---|---|---|
| 200 | 8 | 2 | 3 | Cs2CO3/cesium acetate | 2.38 | 0.00 | 0.08 |
| 220 | 8 | 2 | 3 | Cs2CO3/cesium acetate | 4.35 | 19.56 | 0.78 |
| 280 | 8 | 2 | 3 | Cs2CO3/cesium acetate | 13.34 | 82.26 | 3.11 |
| 300 | 8 | 2 | 3 | Cs2CO3/cesium acetate | 33.99 | 118.97 | 4.98 |
| 320 | 8 | 2 | 3 | Cs2CO3/cesium acetate | 33.74 | 111.75 | 4.74 |
| 300 | 8 | 2 | 3 | Cs2CO3/cesium formate | 4.38 | 21.91 | 0.86 |
| 300 | 8 | 2 | 3 | Cs2CO3/cesium oxalate | 0.00 | 7.17 | 0.23 |
| 280 | 8 | 2 | 3 | Cs2CO3/cesium pivalate | 131.49 | 124.38 | 8.33 |
| 300 | 8 | 2 | 3 | Cs2CO3/cesium pivalate | 102.46 | 109.78 | 6.91 |
| 280 | / | 2 | 3 | Cs2CO3/cesium pivalate | 0 | 0 | 0 |
| 300 | 8 | 2 | 3 | Cs2CO3/potassium acetate | 25.40 | 80.43 | 3.44 |
| 280 | 8 | 2 | 3 | Cs2CO3/potassium acetate | 20.71 | 53.85 | 2.43 |
| 280 | 8 | 2 | 3 | Cs2CO3/potassium isobutyrate | 47.49 | 32.22 | 2.59 |
| 280 | 8 | 2 | 3 | Cs2CO3/potassium pivalate | 42.03 | 22.63 | 2.10 |
| 280 | 8 | 2 | 3 | K2CO3/cesium pivalate | 26.08 | 7.24 | 1.08 |
| 280 | 8 | 2 | 3 | K2CO3/potassium pivalate | 0 | 0 | 0 |
| Entry | T (°C) | PCO2 (bar) | nthio (mmol) | CO32− Ratio (%) | 2-Carboxylate (μmol/g) | 2,5-Dicarboxylate (μmol/g) | Yield a (%) |
|---|---|---|---|---|---|---|---|
| 1 | 280 | 2 | 2 | 40 | 49.75 | 31.51 | 2.65 |
| 2 | 280 | 4 | 2 | 40 | 56.58 | 26.72 | 2.71 |
| 3 | 280 | 6 | 2 | 40 | 102.78 | 95.70 | 6.46 |
| 4 | 280 | 8 | 2 | 40 | 131.49 | 124.38 | 8.33 |
| 5 | 280 | 10 | 2 | 40 | 104.20 | 88.57 | 6.27 |
| 6 | 280 | 8 | 4 | 40 | 189.81 | 436.16 | 20.37 |
| 7 | 280 | 8 | 6 | 40 | 246.61 | 760.74 | 32.79 |
| 8 | 280 | 8 | 8 | 40 | 291.66 | 1010.26 | 42.38 |
| 9 | 280 | 8 | 10 | 40 | 352.46 | 852.45 | 39.22 |
| 10 | 280 | 8 | 2 | 20 | 114.36 | 110.78 | 7.33 |
| 11 | 280 | 8 | 2 | 60 | 60.52 | 85.74 | 4.76 |
| 12 | 280 | 8 | 2 | 80 | 18.85 | 26.39 | 1.47 |
| Carboxylate | Activated H-Position | (kcal/mol) | TS C-H Length (Å) | TS O-H Length (Å) | TS Cs-C Length (Å) |
|---|---|---|---|---|---|
| bare | α-H | 11.90 | 1.367 | 1.238 | 3.346 |
| 5-H | 18.29 | 1.414 | 1.220 | 3.489 | |
| formate | α-H | 20.21 | 1.532 | 1.127 | 3.473 |
| 5-H | 22.38 | 1.528 | 1.125 | 3.636 | |
| acetate | α-H | 20.99 | 1.515 | 1.130 | 3.190 |
| 5-H | 19.74 | 1.461 | 1.161 | 3.212 | |
| pivalate | α-H | 16.86 | 1.527 | 1.234 | 3.198 |
| 5-H | 19.62 | 1.476 | 1.150 | 3.219 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.; Shi, P.; Zeng, A. Carboxylate-Assisted Carboxylation of Thiophene with CO2 in the Solvent-Free Carbonate Medium. Catalysts 2022, 12, 369. https://doi.org/10.3390/catal12040369
Zhang Q, Shi P, Zeng A. Carboxylate-Assisted Carboxylation of Thiophene with CO2 in the Solvent-Free Carbonate Medium. Catalysts. 2022; 12(4):369. https://doi.org/10.3390/catal12040369
Chicago/Turabian StyleZhang, Qingjun, Pengyuan Shi, and Aiwu Zeng. 2022. "Carboxylate-Assisted Carboxylation of Thiophene with CO2 in the Solvent-Free Carbonate Medium" Catalysts 12, no. 4: 369. https://doi.org/10.3390/catal12040369
APA StyleZhang, Q., Shi, P., & Zeng, A. (2022). Carboxylate-Assisted Carboxylation of Thiophene with CO2 in the Solvent-Free Carbonate Medium. Catalysts, 12(4), 369. https://doi.org/10.3390/catal12040369