Abstract
There is currently considerable interest in the intensification of biocatalytic processes to reduce the cost of goods for biocatalytically produced chemicals, including pharmaceuticals and advanced pharmaceutical intermediates. Continuous-flow biocatalysis shows considerable promise as a method for process intensification; however, the reliance of some reactions on the use of diffusible cofactors (such as the nicotinamide cofactors) has proven to be a technical barrier for key enzyme classes. This minireview covers attempts to overcome this limitation, including the cofactor recapture and recycling retention of chemically modified cofactors. For the latter, we also consider the state of science for cofactor modification, a field reinvigorated by the current interest in continuous-flow biocatalysis.
1. Introduction
Modern biocatalysis has been adopted for a wide variety of chemical syntheses in the pharmaceutical and fine chemicals industry [1,2,3,4]. The rise of biocatalysis has been fueled by the intrinsic properties of enzymes, including their exquisite chemo-, regio- and stereoselectivities (which removes the need for protecting groups and reduces the formation of side products), biodegradability, compatibility with each other and their intrinsic ‘engineerability’ [5,6,7,8]. Many important enzymes used in biocatalysis require a cofactor to effectively execute their catalytic activity. In some cases, the cofactor is naturally regenerated as part of the catalytic cycle, and no further intervention is required [9]. Unfortunately, this is not true for all cofactor-dependent enzymes, especially for reduction/oxidation enzymes such as dehydrogenases and oxidases. In these cases, the mediator of the hydride transfer is usually a diffusible nicotinamide cofactor, which is stoichiometrically consumed and needs to be constantly supplied in the system in the correct oxidation state to drive the reaction [9].
Biotransformation involving nicotinamide-dependent enzymes is an essential tool for the asymmetric synthesis of active pharmaceutical intermediates (APIs). Enantiopure amines, for example, can be accessed via nicotinamide-dependent monoamine oxidases, amine dehydrogenases, imine reductases and reductive aminases [10]. Other nicotinamide-fueled reactions include ene-reductases that reduce prochiral sp2 carbons in activated C=C bonds [11] and alcohol dehydrogenases and ketoreductases [12] that reduce ketones to chiral alcohols [13]. Nicotinamide cofactors represent the most common low-potential redox cofactor used in biocatalysis. With a redox midpoint potential of −320 mV, NAD(P)/NAD(P)H can transfer hydrides directly to the substrate or via the oxidation or reduction of other cofactors, such as flavins. While there are other potentially complementary cofactors with a low redox potential, such as cofactor F420, they are still underexplored and insufficiently developed for wide adoption in biocatalysis [14].
The cost of naturally sourced nicotinamide cofactors would represent an obstacle to the upscaling and implementation of nicotinamide-dependent biocatalysts [15,16,17,18,19,20] had strategies for recycling the cofactor not been developed [15,17,19,20,21]. Comprehensive reviews of cofactor recycling strategies for batch mode reactions, including enzymatic and electro- and photochemical systems, are available [22,23,24,25]. Enzymatic recycling is by far the most commonly employed method, and there are two broad approaches: enzyme-coupled and substrate-coupled regeneration (Figure 1) [17,18].
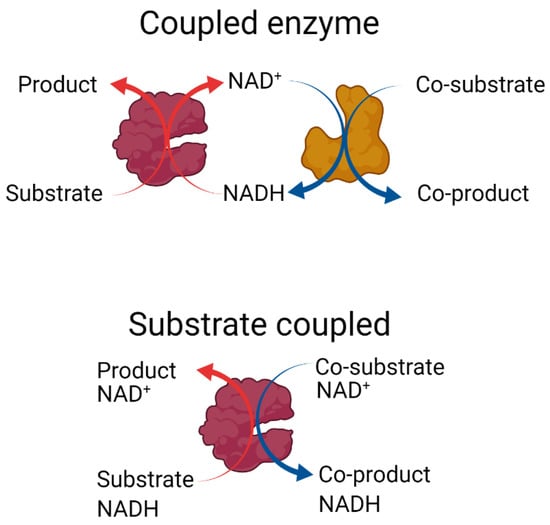
Figure 1.
Enzymatic recycling of nicotinamide cofactors in batch reactors. Top: The coupled enzyme approach to cofactor recycling. An NADH-dependent enzyme (maroon) converts a substrate to the desired product. A second enzyme (mustard) is included to convert a co-substrate to a co-product while regenerating the nicotinamide cofactor. Bottom: The substrate-coupled recycling approach uses a single enzyme (maroon) that both converts the substrate to the desired product at the expense of NADH and then recycles the nicotinamide cofactor via a second reaction, in which a sacrificial co-substrate is converted to a co-product [17]. Created with https://biorender.com (accessed on 4 November 2022).
The current research trend towards the intensification of processes, particularly those conducted through continuous-flow operation, promises to deliver biocatalytic processes with a reduced cost and environmental impact. A wide range of design approaches are currently available for continuous-flow biocatalysis on scales from nanofluidic reactors to liter-scale reactors, based on chip, coil, packed/fluidized bed and membrane designs [26,27,28]. Enzymes can be unmodified or immobilized onto the surface of the reactor using a carrier, such as agarose, silica or other beads, monoliths or carrier-free immobilization via all enzyme hydrogels [26,27,29]. There are excellent reviews of reactor design by Tamborini et al. and de Santis et al. [26,27]. However, the focus of this review is the provision of cofactors in continuous-flow reactors (rather than reactor design, per se). In this review, we will discuss the emerging methods under investigation for NAD(P)+/NAD(P)H use in continuous-flow reactors, including the current methods for the synthesis of cofactors modified to enable their tethering in reactors.
2. NAD(P)H Use in Continuous-Flow Biocatalysis
Continuous-flow biocatalysis systems use compact modular reactors that often contain heterogeneous biocatalysts for the manufacturing of pharmaceuticals, APIs, and fine chemicals [30] and can deliver a better process control, reduced production and substrate inhibition, fewer process steps, simplified product purification and safer operation than would be the case if one were to run the same reactions in sequential batch reactors [27,31,32,33]. However, the use of nicotinamide-dependent biocatalytic processes in a continuous flow can be challenging, and there are two general aspects of cofactor-dependent processes that require solutions to enable their implementation: (i) cofactor recycling—a recycling system must be introduced to the process to regenerate a reduced or oxidized cofactor, as required; and (ii) cofactor recovery—the regenerated cofactor must be retained in the reactor or captured for reuse. With respect to cofactor retention, there are several reports of the chemical modification of NAD(P)+/NAD(P)H designed to enable their covalent attachment to the reactor or enzymes in the reactor [34,35]. The chemical modification of cofactors is still a major challenge for this approach and, thus, will be covered in detail in Section 3.
Cofactor recycling has been achieved by co-immobilizing enzymes with their cofactor-recycling counterparts to enable NAD(P)+/NAD(P)H recycling in continuous-flow reactors, which has been widely reported, principally that of the packed bed and microfluidic varieties. Thompson et al. (2019) [36] described the flow properties of five different immobilized nicotinamide-dependent biocatalytic systems in EziG™ beads. These are controlled-porosity glass beads functionalized with an organic polymer and Fe2+ cations that exhibit a high affinity for His-tagged proteins. In this work, enzymes including the amine dehydrogenase from Caldalkalibacillus termarium (ChiAmDH) and formate dehydrogenase from Candida boidinii (CbFDH) were co-immobilized in packed bed reactors. The immobilized biocatalysts were subsequently fed with a mixture of 4-fluorophenyl acetone, ammonium formate and NAD+. For more than 3 h of continuous operation, no loss in productivity was recorded, and a 68% rate of conversion was obtained, with an STY (space time yield) of more than 300 g L−1 day−1. The authors identified the instability of CbFDH as the major limitation affecting the operation of this system, since the addition of exogenous NADH to the system facilitated the ChiAmDH activity after the reactor lost CbFDH activity. Mattey et al. (2021) [37] used several combinations of co-immobilized NADP+-dependent imine reductase (IR) and glucose dehydrogenase (GDH) in EziG™ beads in continuous-flow cascades to obtain different asymmetric amines with concomitant cofactor regeneration. An imine reductive reactor (IR80-GDH@EziG™) was coupled in a three-step cascade in flow to produce the API 4-O-methylnorbelladine from isovanilyl alcohol with a reported STY of 2.26 g L−1 h−1. Padrosa et al. [38] immobilized biocatalysts for the regeneration of the amine donor L-alanine for the production of vanillyl amine via a transaminase. The system consisted of co-immobilized NADH-dependent alanine dehydrogenase from Halomonas elongata (HeAlaDH), which was used to regenerate L-alanine from pyruvate, and CbFDH, used to regenerate NADH from NAD+. When HeAlaDH and CbFDH were simultaneously co-immobilized, higher rates of vanillin amination were observed, suggesting that, in this system, NADH is more widely available for the HeAlaDH. Peschke et al. [39] used a streptavidin-binding peptide/streptavidin immobilization platform in a microfluidic continuous-flow biocatalysis reactor. For the proof-of-concept, the authors studied the multistep synthesis of a diol with two NADP/NADPH-dependent ketoreductases (KREDs): an (R)-selective alcohol dehydrogenase from Lactobacillus brevis, ATCC 14869 (LbADH), and a (S)-selective methylglyoxal reductase Gre2p from Saccharomyces cerevisae, YJM293. A coupled enzyme approach was employed in the system for nicotinamide regeneration via glucose dehydrogenase from Bacillus subtilis. The enzymes were fused to a streptavidin-binding peptide tag and immobilized onto streptavidin-functionalized beads. The ketoreductase-functionalized beads were subsequently placed in microchannels with immobilized GDH, and the system was fed with 5-nitrononane-2,8-dione. The authors reported conversions of 73%, with an excellent enantiopurity.
There are two broad strategies that have been adopted to enable cofactor recovery: cofactor retention and cofactor recapture (Figure 2). Cofactor recapture involves the introduction of a unit operation to the process so as to isolate the cofactor from the substrate and co-products and recycle it before it is then reintroduced at the start of the process. Cofactor retention, on the other hand, confines and regenerates the cofactor in the same micro-environment as the enzyme via ionic or covalent molecular interactions or a size-dependent permeable membrane. Often, the chemical modification of the cofactor is necessary to enable cofactor retention (Figure 2). For cofactor recapture, some investigators have suggested the inclusion of an in-line unit operation in their processes so as to separate the cofactor from the product and reinclude it into the system. Contente and Paradasi [40] explored this strategy in a continuous two-step cascade towards high value alcohols. The reductive packed bed bioreactor consisted of co-immobilized ketoreductase and glucose dehydrogenase from Bacillus megaterium (BmGDH). The latter enzyme regenerated NADPH from NADP+, which was extracted in-line in the aqueous phase from the collected fractions and reintroduced into the system. NADPH was retained and recycled by this system after more than five days of continuous recirculation. Padrosa et al. [41] implemented a similar nicotinamide cofactor recovery process in the continuous-flow production of L-pipecolic acid from L-lysine. In this work, the oxidized nicotinamide cofactor in the product phase was isolated with a yield of more than 87% in a column packed with Amberlyst® A26 and recirculated with L-lysine into a packed bed reactor containing immobilized lysine dehydrogenase from Geobacillus stearothermophilus (Gs-Lys6DH) and pyrroline-5-carboxylate reductase from Halomonas elongata (He-P5C). Baumer et al. [42] also reported a closed-loop strategy using a substrate-coupled approach to regenerate nicotinamide in bioreductive packed bed reactors. A series of ketones were reduced via immobilized halo-tagged alcohol dehydrogenase from Lactobacillus brevis (LbADH), and isopropanol was employed as the hydride donor to regenerate NADH. Using this design, no loss in performance was recorded over 32 h of operation, and products with an excellent enantiomeric purity and yields greater than 70% were recorded.
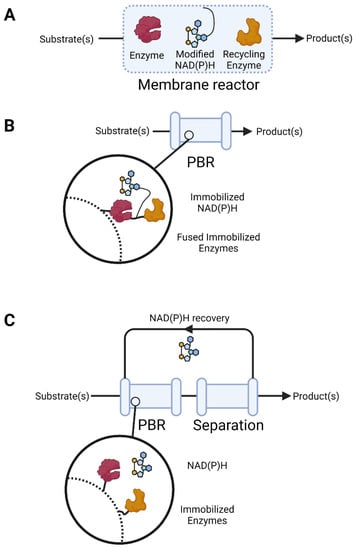
Figure 2.
Successful approaches to supplying NAD(P)H to biocatalytic continuous-flow reactors. (A) Increasing the mass of NAD(P)+/NAD(P)H to enable its retention in membrane reactors; (B) tethering of both enzymes and cofactors to allow for the use, retention, and recycling of NAD(P)+/NAD(P)H in a packed bed reactor (PBR); (C) tether enzymes for the use and recycling of the cofactor, with the in-line separation and recovery of the cofactor for reuse. The enzymes (maroon and mustard) and cofactor (blue) are shown, the direction of flow is indicated by the black arrows, magnified sections showing the molecular arrangement of the enzymes and cofactors are shown in black circles, and the dashed lines represent the solid surfaces of the reactors. This figure was produced using https://biorender.com (Accessed 4 November 2022).
In addition to the closed-loop recapture and reuse of the cofactor, engineering approaches that allow for the physical retention of modified semi-synthetic nicotinamide cofactors in membrane reactors have been explored, beginning with foundational studies in the 1980s. These foundational studies explored the attachment of different soluble polymers, such as poly(ethylene glycol) (PEG) and poly(ethyleneimine) (PEI), to NAD(P)H to increase the molecular weight of the cofactor, enabling the retention of the water-soluble modified cofactor in a membrane reactor [43,44,45,46]. Wichmann et al. [47,48] described the production of L-leucine from α-keto-isocaproate in a membrane reactor employing modified PEG-1000-NADH and L-leucine dehydrogenase, along with an FDH-coupled recycling system used to regenerate the consumed PEG-1000-NADH. The enzymes and modified cofactor were contained in an ultrafiltration membrane, whose pore size was suitable for retaining the PEG-1000-NADH. Substrate conversions higher than 90% were reported over the first 25 days of the reactor’s continuous operation (with 99.7% as the maximum conversion, recorded around the twentieth day) and an STY of 42.5 g L−1 d−1 were determined.
The chemical modification of nicotinamide cofactors has also been used as a route for the immobilization/co-immobilization of enzymes and cofactors via flexible swinging arms. The immobilization of cofactors on enzymes or surfaces has the advantage of enabling their retention in a variety of reactor designs, including packed bed reactors. The immobilization of nicotinamide cofactors via mobile swinging arms has been exemplified in several studies. Katz et al. [49] tethered functionalized oxidized nicotinamide cofactors to a gold electrode via pyrroloquinoline quinone to build a self-fueled lactate biosensor. Fu et al. [50] demonstrated the utility of an artificial flexible oligonucleotide arm (poly(T)20) for channeling the nicotinamide cofactor in a multienzyme cascade complex containing glucose-6-phosphate dehydrogenase (G6pDH) and malic dehydrogenase (MDH). In this study, poly(T)20-NAD+ was equidistantly co-immobilized between G6pDH and MDH on a DNA double-crossover structure to improve the enzyme activity and specificity. Velasco-Lozano et al. [35] investigated the retention of a nicotinamide cofactor via ionic adsorption onto porous agarose microbeads functionalized with polyethyleneimine (Ag-GPEI). The authors demonstrated that, in formulations of co-immobilized dehydrogenase CbFDH and NAD+, the apparent concentration of the cofactor available for catalysis was 30 times higher compared to those where a soluble enzyme was used.
Kim et al. [51] demonstrated the utility of the surface conjugation of NAD+ to macromolecular alginate (AlgNAD+) as an efficient strategy for preventing cofactor leaching from reactors. In this study, the authors reported on a nanopolymeric biocatalyst that was able to retain and regenerate the nicotinamide cofactor, producing D-mannitol from D-fructose via MDH with parallel NADH regeneration via FDH and formate. The oxidized nicotinamide was covalently conjugated to alginate and subsequently co-encapsulated in pluronic nanocarriers (PNCs) with the dehydrogenases FDH and MDH. Repetitive batch experiments with the polymeric nanoreactors showed that the reusability of the biocatalyst did not affect the D-mannitol production, and supplementation with NAD+ was not required. Hartley et al. [34] used a multicomponent engineering approach to design ‘integrated’ biocatalysts, specifically enzyme fusions that could use and recycle NADH, which were tethered to a semisynthetic modified nicotinamide cofactor. The authors used this approach to engineer a chemo-enzymatic process so as to produce the chiral sugar analogue D-fagomine from glycerol. In this multi-enzymatic cascade, the intermediate product, glycerol-3-phosphate, was oxidized to DHAP with concomitant nicotinamide regeneration via an enzyme fusion containing NAD+-dependent glycerol-3-phosphate dehydrogenase from E. coli (G3PDEc) and water-forming NADH-dependent oxidase from Clostridium aminovalericum (NOXCa). A modified nicotinamide cofactor fused with a PEG24 linker was conjugated to the enzyme fusion. The resulting system had high numbers of turnovers for NAD+ (higher than 10,000 turnovers per cofactor molecule).
Although a promising approach, cofactor immobilization depends on the chemical modification of the cofactor to enable the formation of a covalent bond between the cofactor and its flexible linker. The chemical modification of NAD(P)+/NAD(P)H remains challenging, and the major routes used to achieve it are reviewed in the next section.
3. Engineering Nicotinamide Cofactors for Continuous-Flow Biocatalysis
The synthetic structural modification of nicotinamide cofactors is a key technology that enables immobilization. Investigations on the chemical modification of NAD(P)+/NAD(P)H first came to prominence in the 1970s for the purpose of immobilization onto polymeric materials and macromolecular modifiers, such as beads, gels, (nano) fibers and microcapsules [52,53,54]. This approach was used to attach cofactors to surfaces, enabling their incorporation into membrane reactors and affinity chromatographic systems. This was arguably the predecessor of flow biocatalysis. These early systems were further developed to enable the co-immobilization of both the enzyme and cofactor to promote cofactor retention, colocalization and regeneration [55,56].
The modification of nicotinamide nucleosides (and their reduced counterparts) is challenging because the cofactors have likely evolved to achieve chemical stability so as to avoid undesirable biological outcomes. The redox mechanism of the nicotinamide cofactors involves what is often referred to as an electrochemical-chemical-electrochemical (ECE) mechanism, where the oxidation of NAD(P)H to NAD(P)+ involves two single-electron transfers and one proton transfer [57]. Crystal structure analyses of enzyme-cofactor complexes have revealed that the adenine portion of the cofactor NAD+ is generally solvent-accessible when complexed with an enzyme. Modification via chemical tethering at the adenine ring system, therefore, tends to be most the favorable, leading to the least impact upon the cofactor activity [58]. Derivatization at the nicotinamide moiety has been largely avoided to avoid compromising the redox activity of the cofactor.
The specific structural modifications of nicotinamide cofactors are tailored to their intended applications. Despite redox and structural considerations, derivatization has been reported at almost all the available reactive sites on the molecule: the N6, N(1), C8 (adenine), C7 (adenine), 2′/3′ hydroxy (adenine moiety), 2″/3″ hydroxy (nicotinamide moiety) and the pyrophosphate groups (Figure 3). For immobilization, the modification of NAD(P)+ is most often directed towards the primary amine located at the adenine C6 position (often referred to as the N6 position).
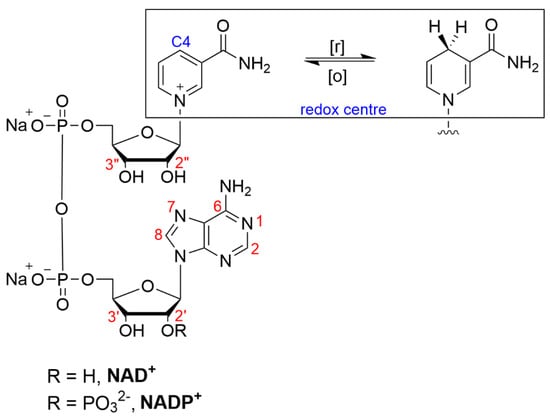
Figure 3.
Structure of β-nicotinamide adenine dinucleotide (phosphate), NAD(P)+, indicating sites of modification (red) and redox chemistry (blue).
Structure/activity relationship studies of the N(1) and N6 NAD+ analogues have shown that the adenine moiety protrudes out of the active enzyme pocket, which is why substitutions are preferred in the case of this moiety [59]. Focusing on adenine derivatization, generally, N(1)-substituted NAD+ derivatives are seldom used as potential candidates in biocatalytic processes, as they have been found to substantially reduce the enzymatic activity compared with the unmodified cofactors, in addition to being less stable molecules than their N6-substituted counterparts [59,60,61]. However, generalizations about which positions can be functionalized with the greatest retention of the activity are not always appropriate. For example, it was found that the N(1)-2-hydroxy-carboxypropyl NADP+ was more active than N6-2-hydroxy-carboxypropyl with glucose-6-phosphate dehydrogenase, glutamate dehydrogenase and aldehyde dehydrogenase [62].
There are several synthetic approaches to modification at the N6 position of NAD(P)+. A common method involves an initial substitution at the N(1) position on the purine ring, followed by a base-catalyzed Dimroth rearrangement and a shift of the substituent between the N(1) endocyclic nitrogen and the exocyclic amine (N6) [63]. A second approach involves the phosphate activation of a potentially substituted mononucleotide, followed by the asymmetric condensation of another mononucleotide via imidazolide, morpholidate, or other intermediates. Substitutions used to enable immobilization at other positions, such as the C2, C8 and 2′-OH positions, are less frequently reported.
3.1. The Dimroth Rearrangement Route
The Dimroth rearrangement route may be favored because the N(1) position is open to facile alkylation by electrophilic reagents under acidic conditions. Iodoacetic acid was used by Mosbach and co-workers to install a carboxymethyl (or, carbamoyl methyl) group at the N(1) position of NAD+ [52] and NADP+, [64] which was followed by the base-mediated translocation of the substituent to the N6 position [55,65]. The rearrangement step was complicated by the need for the prior reduction of the cofactor due to the instability of the oxidized form under basic conditions. Once the reduction and rearrangement of the cofactor were complete, it was re-oxidized enzymatically.
The Dimroth process was later used with propiolactone to form N6-carboxyethyl-NAD+ as well as an epoxide-containing monomer to produce an N6-substituted 2-hydroxyalkylether-NAD+ derivative for the purpose of immobilization (Figure 4) [66,67,68]. NAD+ has also been modified via the same Dimroth rearrangement route, but with ionic end groups that have the potential to interact with membranes charged with the opposite polarity. Here, the cofactor was derivatized with the strongly anionic sulfonatopropyl group using 1,3-propanesultone as a reagent [61,69]. Similarly, strongly cationic N6-2-hydroxy-3-trimethylammonium propyl-NAD+ was prepared in a low yield but showed a good activity [61].
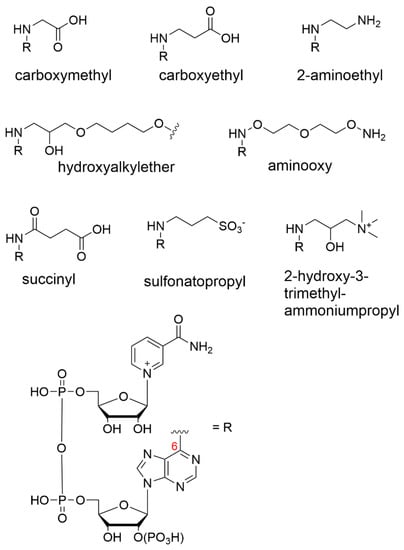
Figure 4.
Various N6 nicotinamide cofactor substituents.
The Bückmann group was the first to install an alkylamine at the N6-position using aziridine as the reactive agent, giving rise to 2-aminoethyl substituents (Figure 4). The group suggested that the cofactor reduction/reoxidation step is unnecessary using this route, as the rearrangement reaction from the N(1) to the N6 position can proceed under mildly basic conditions, thus preventing cofactor degradation.
The presence of the 2′-phosphate in NADP+ can complicate its derivatization via the Dimroth rearrangement method. The Okada group reported that the reaction between NADP+ and propiolactone under slightly acidic conditions, followed by alkaline rearrangement, which results in carboxymethylation at the N6 and 2′-phosphate giving a mixture of products [70,71]. This observation however was not reported by Lowe and Mosbach using similar reaction conditions and reagents [64].
3.2. Phosphate Coupling
Phosphate coupling reactions between two nucleotides are well-known and are often performed between the canonical nucleotides AMP, ADP or ATP and a modified mononucleotide. Typically, anhydrous aprotic solvents are used to prevent the hydrolysis of the activated phosphate back to the native cofactor, and this results in the formation of a symmetrical product containing either purine or pyrimidine bases [72,73]. In terms of the formation of modified pyridine cofactors based on the NAD(P)+ structure, the diphosphate product is not symmetrical, and its synthesis is consequently more nuanced [74,75,76].
The phosphate coupling route permits the modification of a mononucleotide before coupling, such that the derivatization of the adenine can be undertaken on mononucleotides/nucleosides that are less sensitive to base and other harsh reaction conditions. An example of direct derivatization at the N6 position of adenine was reported, [77] where the modified adenine was subsequently coupled to a β-nicotinamide mononucleotide (β-NMN) by a carbodiimide-mediated route to give rise to N6-aminooxy-AMP (Figure 4). The engineered cofactor was later immobilized onto streptavidin-decorated agarose beads. However, the dehydrogenase activity was not measured, as the research was focused on sirtuin-catalyzed deacetylation mechanisms rather than the biocatalytic function.
Various examples of phosphoimidazole activation/coupling routes for the production of NAD(P)+ analogues with alkyl azide/alkyne installation at the N6 position [76] and the purine C2 position have been reported [74,76,78,79]. One approach to this method utilizes 1,1′-carbonyldiimidazole (CDI) to activate β-nicotinamide mononucleotide (β-NMN), forming a phosphoimidazole intermediate. The activated phosphate is then coupled with a 5′-monophosphate—either native adenosine monophosphate (AMP) or an AMP analogue—to achieve the dinucleotide product [80,81]. Morpholidate intermediates can also be used to promote phosphate coupling [72,82,83,84,85].
4. Nicotinamide Cofactor Biomimetics
The use of nicotinamide cofactor biomimetics (NCBs), or artificial redox coenzymes, as substrates for NAD(P)H-dependent oxidoreductases gained attention at the turn of the last century as an approach that can be used to drive reactions with a reduced cost and improve stability compared with those that use natural nicotinamide cofactors (Figure 3). For NCBs, the redox-active nicotinamide moiety is contained within a non-natural, simplified molecular structure [86]. Several recent and comprehensive reviews cover this emerging topic [87,88,89]. Typically, for NCB structures, the ribose phosphate of β-NMN+ is replaced at the pyridine nitrogen position with an aryl, alkyl or alkyl-alcohol (as their triflate or halogenated salt). In some cases, variation occurs at the C5 position, replacing the amide with a carboxylic acid (BAA+) or methyl ester (BAP+) (Figure 5). Although NCBs have the potential to provide chemically facile and cost-effective routes for the attachment of tethers and long linkers, this approach to the provision of NAD(P)+/NAD(P)H surrogates in a continuous flow has not been investigated yet.
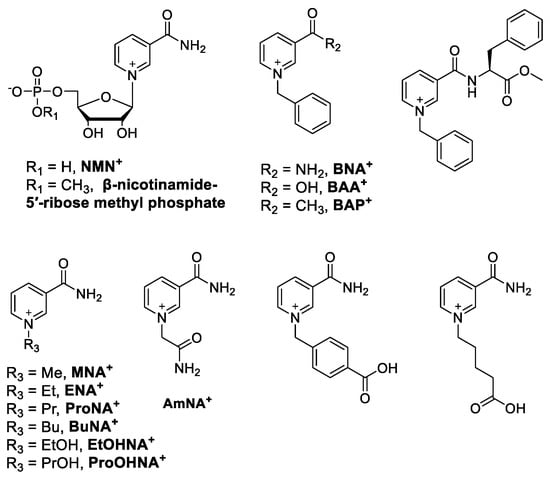
Figure 5.
Selected examples of oxidized NCBs and their abbreviations (where available).
Thus far, NCBs have performed best as substrates for flavin-dependent oxidoreductases, such as enoate reductases (EREDs/OYEs). High activities, optical purity, and satisfactory yields have been obtained for EREDs when supplied with 1-benzyl nicotinamide, BNA+ (Figure 5), as a substrate used in stoichiometric quantities. In these cases, the catalytic efficiency is superior to that of the enzyme with native NAD(P)H [89,90,91,92,93].
The enzymatic conversion of NCBs with wild-type purified alcohol dehydrogenases has been less successful, displaying higher Km values and lower catalytic efficiencies than the natural cofactor [88,89,94,95,96]. The Zhang group improved the activity of 6-phosphogluconate dehydrogenase by 30-fold for NMN+ and 9-fold for BNA+ through directed evolution [97]. The Fraaije group used rational enzyme engineering to improve the activity of an F420:NADPH oxidoreductase by over 100-fold for the purpose of BNA+ recycling. Notably, many NCBs have low redox potentials relative to NAD(P)+, and the low redox potential of cofactor F420 (−360 mV) makes it an excellent candidate for use in efficient NCB recycling cascades [98].
Reduced cofactor biomimetics, 1-methyl-1,4-dihydropyridine-3-carboxamide (MNAH) and 1-benzyl-1,4-dihydropyridine-3-carboxamide (BNAH), have also been shown to be substrates for some flavoenzyme NADH oxidases (NOx) [99,100]. With enzymes that are able to oxidize and reduce NCBs, it may be possible to construct simple NCB regenerating biocatalytic processes. Nowak and co-workers reported the first examples of the enzyme-mediated regeneration of oxidised BNA+ and MNA+ NCBs with an NADH oxidase from Lactobacillus pentosus (LpNox), showing faster reaction rates (kcat) and higher KM values than NAD+ [99,101].
5. Conclusions
Over the course of the development of biocatalysis as a field, cofactor recycling has become a pivotal technology, initially in batch reactions, where diffusion was used to ensure cofactor availability and rapid recycling. As biocatalytic reactor design has become more sophisticated (e.g., compartmentalized continuous flow), it has become clear that additional interventions are required to enable efficient cofactor use and recycling. New unit processes for cofactor recapture have been shown to work well, and chemical modifications of NAD(P)+/NAD(P)H have been demonstrated to be effective routes to cofactor containment.
Methods for the chemical modification of NAD(P)+/NAD(P)H remain difficult and low-yielding, and it is likely that the modified semisynthetic NAD(P)+/NAD(P)H will require improved synthetic routes to become cost-competitive. The use of fully synthetic nicotinamide analogues (NCBs) to replace modified NAD(P)+/NAD(P)H may prove to be a more tractable route for low-cost modified cofactors but will likely rely on enzyme engineering to provide a set of compatible NCBs, biocatalysts and recycling enzymes.
The modification of NAD+/NADH may also have a future role in reactors that employ the in-line recapture and reuse of the cofactor by introducing properties that enable the facile separation of the cofactor from the reaction solvent and products. It is possible to imagine a selection of modified nicotinamide analogues that can be used in a range of solvents, allowing the in-line recapture and re-use strategy to be broadened to include reactions in which the separation of the natural NAD(P)+/NAD(P)H from the reaction would prove difficult.
NCBs have other potential applications. Low-redox NCBs may be able to catalyze reductions that are currently unobtainable through natural nicotinamides, noting that other low-redox cofactors (such as cofactor F420) are available too, pending the further development of cost-effective production and recycling systems [14,102,103,104]. There is also an opportunity to consider the potential of using orthogonal components in complex biocatalytic cascades. This concept was exemplified by the Elk group in a one-pot cascade for the stereo-inversion of chiral sec-alcohols using NAD+- and NADP+-dependent alcohol dehydrogenases of opposing stereo-specificities (i.e., orthogonal oxidation and reduction steps) [105]. By isolating the cofactor used in the oxidation and reduction steps of the cascade, it was possible to drive the reaction in the desired direction. If a range of orthogonal pairings (biocatalyst, recycling partner and NCB) were to be made available, it may become possible to extend this concept to more complex cascades.
Research on cofactor engineering originally peaked in the 1970s and 80s at a time when the applications of chemically modified NAD(P)+/NAD(P)H seemed limited. It is possible that this area of study may see renewed interest as compartmentalized, modular, continuous-flow bioreactors become more widely adopted by the industry.
Author Contributions
Writing—original draft preparation, R.A.R., A.J.N., R.E.S., C.C.W. and C.S.; writing—review and editing, R.A.R., A.J.N., R.E.S., C.C.W. and C.S. All authors have read and agreed to the published version of the manuscript.
Funding
R.A.R. was supported by a QUT South American scholarship from Queensland University of Technology and a top-up grant from the CSIRO Synthetic Biology Future Science Platform. A.J.N. was supported by the CSIRO Synthetic Biology Future Science Platform.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Adams, J.P.; Brown, M.J.B.; Diaz-Rodriguez, A.; Lloyd, R.C.; Roiban, G.D. Biocatalysis: A pharma perspective. Adv. Synth. Catal. 2019, 361, 2421–2432. [Google Scholar] [CrossRef]
- Bornscheuer, U.T.; Huisman, G.W.; Kazlauskas, R.J.; Lutz, S.; Moore, J.C.; Robins, K. Engineering the third wave of biocatalysis. Nature 2012, 485, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Faber, K.; Fessner, W.D.; Turner, N.J. Biocatalysis: Ready to master increasing complexity. Adv. Synth. Catal. 2019, 361, 2373–2376. [Google Scholar] [CrossRef]
- Winkler, C.K.; Schrittwieser, J.H.; Kroutil, W. Power of biocatalysis for organic synthesis. ACS Cent. Sci. 2021, 7, 55–71. [Google Scholar] [CrossRef] [PubMed]
- García-Junceda, E.; Lavandera, I.; Rother, D.; Schrittwieser, J.H. (Chemo) enzymatic cascades—Nature’s synthetic strategy transferred to the laboratory. J. Mol. Catal. B Enzym. 2015, 114, 1–6. [Google Scholar] [CrossRef]
- Reetz, M.T. Biocatalysis in organic chemistry and biotechnology: Past, present, and future. J. Am. Chem. Soc. 2013, 135, 12480–12496. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Brady, D. The limits to biocatalysis: Pushing the envelope. Chem. Commun. 2018, 54, 6088–6104. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Pereira, P.C. Biocatalysis engineering: The big picture. Chem. Soc. Rev. 2017, 46, 2678–2691. [Google Scholar] [CrossRef]
- Faber, K. Biocatalytic applications. In Biotransformations in Organic Chemistry; Springer: Cham, Switzerland, 2011; pp. 31–313. [Google Scholar]
- Grogan, G. Synthesis of chiral amines using redox biocatalysis. Curr. Opin. Chem. Biol. 2018, 43, 15–22. [Google Scholar] [CrossRef]
- Toogood, H.S.; Scrutton, N.S. Discovery, characterization, engineering, and applications of ene-reductases for industrial biocatalysis. ACS Catal. 2018, 8, 3532–3549. [Google Scholar] [CrossRef]
- Rodriguez-Abetxuko, A.; Reifs, A.; Sanchez-deAlcazar, D.; Beloqui, A. A Versatile Chemoenzymatic Nanoreactor that Mimics NAD(P)H Oxidase for the In Situ Regeneration of Cofactors. Angew. Chem. Int. Ed. 2022, 61, e202206926. [Google Scholar]
- Kroutil, W.; Mang, H.; Edegger, K.; Faber, K. Recent advances in the biocatalytic reduction of ketones and oxidation of sec-alcohols. Curr. Opin. Chem. Biol. 2004, 8, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Scott, C.; Grogan, G. F420-dependent enzymes-potential for applications in biotechnology. Trends Biotechnol. 2013, 31, 63–64. [Google Scholar] [CrossRef] [PubMed]
- Chenault, H.K.; Whitesides, G.M. Regeneration of nicotinamide cofactors for use in organic synthesis. Appl. Biochem. Biotechnol. 1987, 14, 147–197. [Google Scholar] [CrossRef]
- Fukuzumi, S.; Lee, Y.-M.; Nam, W. Catalytic recycling of NAD(P)H. J. Inorg. Biochem. 2019, 199, 110777. [Google Scholar] [CrossRef]
- Hummel, W.; Gröger, H. Strategies for regeneration of nicotinamide coenzymes emphasizing self-sufficient closed-loop recycling systems. J. Biotechnol. 2014, 191, 22–31. [Google Scholar] [CrossRef]
- Weckbecker, A.; Gröger, H.; Hummel, W. Regeneration of nicotinamide coenzymes: Principles and applications for the synthesis of chiral compounds. In Biosystems Engineering I; Springer: Berlin/Heidelberg, Germany, 2010; pp. 195–242. [Google Scholar]
- Wu, H.; Tian, C.; Song, X.; Liu, C.; Yang, D.; Jiang, Z. Methods for the regeneration of nicotinamide coenzymes. Green Chem. 2013, 15, 1773–1789. [Google Scholar] [CrossRef]
- Zhao, H.; Van Der Donk, W.A. Regeneration of cofactors for use in biocatalysis. Curr. Opin. Biotechnol. 2003, 14, 583–589. [Google Scholar] [CrossRef]
- Rodriguez, C.; Lavandera, I.; Gotor, V. Recent advances in cofactor regeneration systems applied to biocatalyzed oxidative processes. Current Org. Chem. 2012, 16, 2525–2541. [Google Scholar] [CrossRef]
- Wang, X.; Saba, T.; Yiu, H.H.P.; Howe, R.F.; Anderson, J.A.; Shi, J. Cofactor NAD(P)H regeneration inspired by heterogeneous pathways. Chem 2017, 2, 621–654. [Google Scholar] [CrossRef]
- Mordhorst, S.; Andexer, J.N. Round, round we go–strategies for enzymatic cofactor regeneration. Nat. Prod. Rep. 2020, 37, 1316–1333. [Google Scholar] [CrossRef] [PubMed]
- Tassano, E.; Hall, M. Enzymatic self-sufficient hydride transfer processes. Chem. Soc. Rev. 2019, 48, 5596–5615. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.Q.; Feng, J.Q.; Cui, J.W.; Jiang, G.D.; Harrison, W.; Zang, X.; Zhou, J.H.; Wang, B.J.; Zhao, H.M. Photoinduced chemomimetic biocatalysis for enantioselective intermolecular radical conjugate addition. Nat. Catal. 2022, 5, 586–593. [Google Scholar] [CrossRef]
- De Santis, P.; Meyer, L.E.; Kara, S. The rise of continuous flow biocatalysis—Fundamentals, very recent developments and future perspectives. React. Chem. Eng. 2020, 5, 2155–2184. [Google Scholar] [CrossRef]
- Tamborini, L.; Fernandes, P.; Paradisi, F.; Molinari, F. Flow bioreactors as complementary tools for biocatalytic process intensification. Trends Biotechnol. 2018, 36, 73–88. [Google Scholar] [CrossRef]
- Woltinger, J.; Karau, A.; Leuchtenberger, W.; Drauz, K. Membrane reactors at Degussa. In Technology Transfer in Biotechnology: From Lab to Industry to Production; Springer: Berlin, Germany, 2005; Volume 92, pp. 289–316. [Google Scholar]
- Bitterwolf, P.; Ott, F.; Rabe, K.S.; Niemeyer, C.M. Imine Reductase Based All-Enzyme Hydrogel with Intrinsic Cofactor Regeneration for Flow Biocatalysis. Micromachines 2019, 10, 783. [Google Scholar] [CrossRef]
- Gutmann, B.; Cantillo, D.; Kappe, C.O. Continuous-flow technology—A tool for the safe manufacturing of active pharmaceutical ingredients. Angew. Chem. Int. Ed. 2015, 54, 6688–6728. [Google Scholar] [CrossRef]
- Hessel, V.; Kralisch, D.; Kockmann, N.; Noël, T.; Wang, Q. Novel process windows for enabling, accelerating, and uplifting flow chemistry. ChemSusChem 2013, 6, 746–789. [Google Scholar] [CrossRef]
- Newman, S.G.; Jensen, K.F. The role of flow in green chemistry and engineering. Green Chem. 2013, 15, 1456–1472. [Google Scholar] [CrossRef]
- Thompson, M.P.; Peñafiel, I.; Cosgrove, S.C.; Turner, N.J. Biocatalysis using immobilized enzymes in continuous flow for the synthesis of fine chemicals. Org. Process Res. Dev. 2018, 23, 9–18. [Google Scholar] [CrossRef]
- Hartley, C.J.; Williams, C.C.; Scoble, J.A.; Churches, Q.I.; North, A.; French, N.G.; Nebl, T.; Coia, G.; Warden, A.C.; Simpson, G. Engineered enzymes that retain and regenerate their cofactors enable continuous-flow biocatalysis. Nat. Catal. 2019, 2, 1006–1015. [Google Scholar] [CrossRef]
- Velasco-Lozano, S.; Benítez-Mateos, A.I.; López-Gallego, F. Co-immobilized phosphorylated cofactors and enzymes as self-sufficient heterogeneous biocatalysts for chemical processes. Angew. Chem. Int. Ed. 2017, 56, 771–775. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.P.; Derrington, S.R.; Heath, R.S.; Porter, J.L.; Mangas-Sanchez, J.; Devine, P.N.; Truppo, M.D.; Turner, N.J. A generic platform for the immobilisation of engineered biocatalysts. Tetrahedron 2019, 75, 327–334. [Google Scholar] [CrossRef]
- Mattey, A.P.; Ford, G.J.; Citoler, J.; Baldwin, C.; Marshall, J.R.; Palmer, R.B.; Thompson, M.; Turner, N.J.; Cosgrove, S.C.; Flitsch, S.L. Development of Continuous Flow Systems to Access Secondary Amines Through Previously Incompatible Biocatalytic Cascades. Angew. Chem. Int. Ed. 2021, 60, 18660–18665. [Google Scholar] [CrossRef]
- Roura Padrosa, D.; Nisar, Z.; Paradisi, F. Efficient amino donor recycling in amination reactions: Development of a new alanine dehydrogenase in continuous flow and dialysis membrane reactors. Catalysts 2021, 11, 520. [Google Scholar] [CrossRef]
- Peschke, T.; Skoupi, M.; Burgahn, T.; Gallus, S.; Ahmed, I.; Rabe, K.S.; Niemeyer, C.M. Self-immobilizing fusion enzymes for compartmentalized biocatalysis. ACS Catal. 2017, 7, 7866–7872. [Google Scholar] [CrossRef]
- Contente, M.L.; Paradisi, F. Self-sustaining closed-loop multienzyme-mediated conversion of amines into alcohols in continuous reactions. Nat. Catal. 2018, 1, 452–459. [Google Scholar] [CrossRef]
- Padrosa, D.R.; Benítez-Mateos, A.I.; Calvey, L.; Paradisi, F. Cell-free biocatalytic syntheses of L-pipecolic acid: A dual strategy approach and process intensification in flow. Green Chem. 2020, 22, 5310–5316. [Google Scholar] [CrossRef]
- Baumer, B.; Classen, T.; Pohl, M.; Pietruszka, J. Efficient Nicotinamide Adenine Dinucleotide Phosphate [NADP (H)] Recycling in Closed-Loop Continuous Flow Biocatalysis. Adv. Synth. Catal. 2020, 362, 2894–2901. [Google Scholar] [CrossRef]
- Kula, M.-R.; Wandrey, C. [2] Continuous enzymatic transformation in an enzyme-membrane reactor with simultaneous NADH regeneration. Methods Enzymol. 1987, 136, 9–21. [Google Scholar]
- Månsson, M.-O.; Mosbach, K. Immobilized active coenzymes. Meth. Enzymol. 1987, 136, 3–9. [Google Scholar]
- Mosbach, K.; Larsson, P.-O.; Lowe, C. Immobilized coenzymes. Methods Enzymol. 1976, 44, 859–887. [Google Scholar] [PubMed]
- Wichmann, R.; Vasic-Racki, D. Cofactor regeneration at the lab scale. Technol. Transf. Biotechnol. 2005, 92, 225–260. [Google Scholar]
- Wichmann, R.; Wandrey, C.; Bückmann, A.F.; Kula, M.R. Continuous enzymatic transformation in an enzyme membrane reactor with simultaneous NAD(H) regeneration. Biotechnol. Bioeng. 1981, 23, 2789–2802. [Google Scholar] [CrossRef]
- Wandrey, C.; Wichmann, R. Production of l-amino acids in the membrane reactor. Biotech. 1987, 1, 85–92. [Google Scholar]
- Katz, E.; Bückmann, A.F.; Willner, I. Self-powered enzyme-based biosensors. J. Am. Chem. Soc. 2001, 123, 10752–10753. [Google Scholar] [CrossRef]
- Fu, J.; Yang, Y.R.; Johnson-Buck, A.; Liu, M.; Liu, Y.; Walter, N.G.; Woodbury, N.W.; Yan, H. Multi-enzyme complexes on DNA scaffolds capable of substrate channelling with an artificial swinging arm. Nat. Nanotechnol. 2014, 9, 531–536. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, K.; Tae, G.; Kwon, I. Nano-Entrapping Multiple Oxidoreductases and Cofactor for All-In-One Nanoreactors. ACS Sustain. Chem. Eng. 2021, 9, 6741–6747. [Google Scholar] [CrossRef]
- Lindberg, M.; Larsson, P.O.; Mosbach, K. A new immobilized NAD+ analogue, its application in affinity chromatography and as a functioning coenzyme. Eur. J. Biochem. 1973, 40, 187–193. [Google Scholar] [CrossRef]
- Schmidt, H.L.; Grenner, G. Coenzyme Properties of NAD+ Bound to Different Matrices through the Amino Group in the 6-Position. Eur. J. Biochem. 1976, 67, 295–302. [Google Scholar] [CrossRef]
- Wykes, J.R.; Dunnill, P.; Lilly, M.D. The preparation of soluble high molecular weight NAD derivative active as a cofactor. Biochem. Biophys. Acta Gen. Subj. 1972, 286, 260–268. [Google Scholar] [CrossRef]
- Mosbach, K. Immobilised enzymes. FEBS Lett. 1976, 62, E80–E95. [Google Scholar] [CrossRef]
- Zhang, M.; Mullens, C.; Gorski, W. Coimmobilization of dehydrogenases and their cofactors in electrochemical biosensors. Anal. Chem. 2007, 79, 2446–2450. [Google Scholar] [CrossRef] [PubMed]
- Radoi, A.; Compagnone, D. Recent advances in NADH electrochemical sensing design. Bioelectrochemistry 2009, 76, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.J.; Buehner, M.; Chandrasekhar, K.; Ford, G.C.; Hackert, M.L.; Liljas, A.; Rossmann, M.G.; Smiley, I.E.; Allison, W.S.; Everse, J. Structure-function relationships in lactate dehydrogenase. Proc. Natl. Acad. Sci. USA 1973, 70, 1968–1972. [Google Scholar] [CrossRef]
- Hendle, J.; BÜCkmann, A.F.; Aehle, W.; Schomburg, D.; Schmid, R.D. Structure/activity relationship of adenine-modified NAD derivatives with respect to porcine heart lactate dehydrogenase isozyme H4 simulated with molecular mechanics. Eur. J. Biochem. 1993, 213, 947–956. [Google Scholar] [CrossRef]
- Grenner, G.; Schmidt, H.L.; Voelkl, W. Coenzym-Eigenschaften einiger in l- und N6-stellung des Adeninringes substituierterN AD+-Derivate. Z. Physiol. Chem. 1976, 357, 887–902. [Google Scholar]
- Ottolina, G.; Carrea, G.; Riva, S.; Bückmann, A.F. Coenzymatic properties of low molecular-weight and macromolecular N6-derivatives of NAD+ and NADP+ with dehydrogenases of interest for organic synthesis. Enzym. Microb. Technol. 1990, 12, 596–602. [Google Scholar] [CrossRef]
- Zappelli, P.; Pappa, R.; Rossodivita, A.; Re, L. Preparation and Coenzymic Activity of Soluble Polyethyleneimine-Bound NADP+ Derivatives. Eur. J. Biochem. 1977, 72, 309–315. [Google Scholar] [CrossRef]
- Engel, J.D. Mechanism of the Dimroth rearrangement in adenosine. Biochem. Biophys. Res. Commun. 1975, 64, 581–586. [Google Scholar] [CrossRef]
- Lowe, C.R.; Mosbach, K. The synthesis of adenine-substituted derivatives of NADP+ and their potential as active coenzymes and affinity adsorbents. Eur. J. Biochem. 1974, 49, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, J.; Vieille, C. Activity of select dehydrogenases with Sepharose-immobilized N6-carboxymethyl-NAD. Bioengineered 2015, 6, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, T.; Itami, M.; Yomo, T.; Urabe, I.; Yamada, Y.; Okada, H. Improved methods for the preparation of N6-(2-carboxyethyl)-NAD and poly (ethylene glycol)-bound NAD (H). J. Ferment. Bioeng. 1991, 71, 447–449. [Google Scholar] [CrossRef]
- Muramatsu, M.; Urabe, I.; Yamada, Y.; Okada, H. Synthesis and kinetic properties of a new NAD+ derivative carrying a vinyl group. Eur. J. Biochem. 1977, 80, 111–117. [Google Scholar] [CrossRef]
- Fuller, C.W.; Rubin, J.R.; Bright, H.J. A simple procedure for covalent immobilization of NADH in a soluble and enzymically active form. Eur. J. Biochem. 1980, 103, 421–430. [Google Scholar] [CrossRef]
- Carrea, G.; Ottolina, G.; Riva, S.; Danieli, B.; Lesma, G.; Palmisano, G. Alkylation of Adenine, Adenosine, and NAD+ with 1, 3-Propanesultone. Synthesis of N6-(3-sulfonatopropyl)-NAD+, a new NAD+ derivative with substantial coenzyme activity. Helv. Chim. Acta 1988, 71, 762–772. [Google Scholar] [CrossRef]
- Okuda, K.; Suntinanalerts, P.; Miyoshi, S.; Urabe, I.; Yamada, Y.; Okada, H. Preparation and characterization of NADP derivatives alkylated at 2′-phosphate and 6-amino groups. Eur. J. Biochem. 1985, 147, 241–247. [Google Scholar] [CrossRef]
- Sogin, D.C. 2′, 3′-Cyclic NADP as a substrate for 2′, 3′-cyclic nucleotide 3′-phosphohydrolase. J. Neurochem. 1976, 27, 1333–1337. [Google Scholar] [CrossRef]
- Reiss, J.R.; Moffatt, J.G. Dismutation Reactions of Nucleoside Polyphosphates. III. The Synthesis of α, ι-Dinucleoside 5′-Polyphosphates1. J. Org. Chem. 1965, 30, 3381–3387. [Google Scholar] [CrossRef]
- Appy, L.; Chardet, C.; Peyrottes, S.; Roy, B. Synthetic Strategies for Dinucleotides Synthesis. Molecules 2019, 24, 4334. [Google Scholar] [CrossRef]
- Buntz, A.; Wallrodt, S.; Gwosch, E.; Schmalz, M.; Beneke, S.; Ferrando-May, E.; Marx, A.; Zumbusch, A. Real-time cellular imaging of protein poly (ADP-ribos) ylation. Angew. Chem. Int. Ed. 2016, 55, 11256–11260. [Google Scholar] [CrossRef] [PubMed]
- Hughes, N.A.; Kenner, G.W.; Todd, A. Codehydrogenases. Part III. A synthesis of diphosphopyridine nucleotide (cozymase), and some observations on the synthesis of triphosphopyridine nucleotide. J. Chem. Soc. 1957, 3733–3738. [Google Scholar] [CrossRef]
- Wallrodt, S.; Buntz, A.; Wang, Y.; Zumbusch, A.; Marx, A. Bioorthogonally Functionalized NAD+ Analogues for In-Cell Visualization of Poly (ADP-Ribose) Formation. Angew. Chem. Int. Ed. 2016, 55, 7660–7664. [Google Scholar] [CrossRef] [PubMed]
- Cen, Y.; Falco, J.N.; Xu, P.; Youn, D.Y.; Sauve, A.A. Mechanism-based affinity capture of sirtuins. Org. Biomol. Chem. 2011, 9, 987–993. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.T.; Zhang, X.-N.; Courouble, V.V.; Strutzenberg, T.S.; Pei, H.; Stiles, B.L.; Louie, S.G.; Griffin, P.R.; Zhang, Y. A Bifunctional NAD+ for Profiling Poly-ADP-Ribosylation-Dependent Interacting Proteins. ACS Chem. Biol. 2021, 16, 389–396. [Google Scholar] [CrossRef]
- Wang, Y.; Rösner, D.; Grzywa, M.; Marx, A. Chain-Terminating and Clickable NAD+ Analogues for Labeling the Target Proteins of ADP-Ribosyltransferases. Angew. Chem. Int. Ed. 2014, 53, 8159–8162. [Google Scholar] [CrossRef]
- Maeda, M.; Patel, A.D.; Hampton, A. Formation of ribonucleotide 2′, 3′-cyclic carbonates during conversion of ribonucleoside 5′-phosphates to diphosphates and triphosphates by the phosphorimidazolidate procedure. Nucleic Acids Res. 1977, 4, 2843–2853. [Google Scholar] [CrossRef]
- Zatorski, A.; Goldstein, B.M.; Colby, T.D.; Jones, J.P.; Pankiewicz, K.W. Potent inhibitors of human inosine monophosphate dehydrogenase type II. fluorine-substituted analogs of thiazole-4-carboxamide adenine dinucleotide. J. Med. Chem. 1995, 38, 1098–1105. [Google Scholar] [CrossRef]
- Lynch, J.; áP Volante, R.; Reider, P. A chemical synthesis of nicotinamide adenine dinucleotide (NAD+). Chem. Commun. 1999, 729–730. [Google Scholar] [CrossRef]
- Pergolizzi, G.; Butt, J.N.; Bowater, R.P.; Wagner, G.K. A novel fluorescent probe for NAD-consuming enzymes. Chem. Commun. 2011, 47, 12655–12657. [Google Scholar] [CrossRef]
- Pergolizzi, G.; Cominetti, M.M.D.; Butt, J.N.; Field, R.A.; Bowater, R.P.; Wagner, G.K. Base-modified NAD and AMP derivatives and their activity against bacterial DNA ligases. Org. Biomol. Chem. 2015, 13, 6380–6398. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wagner, G.K.; Weber, K.; Garnham, C.; Morgan, A.J.; Galione, A.; Guse, A.H.; Potter, B.V.L. 2′-Deoxy cyclic adenosine 5′-diphosphate ribose derivatives: Importance of the 2′-hydroxyl motif for the antagonistic activity of 8-substituted cADPR derivatives. J. Med. Chem. 2008, 51, 1623–1636. [Google Scholar] [CrossRef] [PubMed]
- Ansell, R.J.; Lowe, C.R. Artificial redox coenzymes: Biomimetic analogues of NAD+. Appl. Microbiol. Biotechnol. 1999, 51, 703–710. [Google Scholar] [CrossRef]
- Paul, C.E.; Arends, I.W.C.E.; Hollmann, F. Is simpler better? Synthetic nicotinamide cofactor analogues for redox chemistry. ACS Catal. 2014, 4, 788–797. [Google Scholar] [CrossRef]
- Paul, C.E.; Hollmann, F. A survey of synthetic nicotinamide cofactors in enzymatic processes. Appl. Microbiol. Biotechnol. 2016, 100, 4773–4778. [Google Scholar] [CrossRef]
- Zachos, I.; Nowak, C.; Sieber, V. Biomimetic cofactors and methods for their recycling. Curr. Opin. Chem. Biol. 2019, 49, 59–66. [Google Scholar] [CrossRef]
- Geddes, A.; Paul, C.E.; Hay, S.; Hollmann, F.; Scrutton, N.S. Donor–acceptor distance sampling enhances the performance of “better than nature” nicotinamide coenzyme biomimetics. J. Am. Chem. Soc. 2016, 138, 11089–11092. [Google Scholar] [CrossRef]
- Knaus, T.; Paul, C.E.; Levy, C.W.; De Vries, S.; Mutti, F.G.; Hollmann, F.; Scrutton, N.S. Better than nature: Nicotinamide biomimetics that outperform natural coenzymes. J. Am. Chem. Soc. 2016, 138, 1033–1039. [Google Scholar] [CrossRef]
- Lutz, J.; Hollmann, F.; Ho, T.V.; Schnyder, A.; Fish, R.H.; Schmid, A. Bioorganometallic chemistry: Biocatalytic oxidation reactions with biomimetic NAD+/NADH co-factors and [Cp* Rh (bpy) H]+ for selective organic synthesis. J. Organomet. Chem. 2004, 689, 4783–4790. [Google Scholar] [CrossRef]
- Paul, C.E.; Gargiulo, S.; Opperman, D.J.; Lavandera, I.; Gotor-Fernández, V.; Gotor, V.; Taglieber, A.; Arends, I.W.C.E.; Hollmann, F. Mimicking Nature: Synthetic Nicotinamide Cofactors for C=C Bioreduction Using Enoate Reductases. Org. Lett. 2013, 15, 180–183. [Google Scholar] [CrossRef]
- Campbell, E.; Meredith, M.; Minteer, S.D.; Banta, S. Enzymatic biofuel cells utilizing a biomimetic cofactor. Chem. Commun. 2012, 48, 1898–1900. [Google Scholar] [CrossRef] [PubMed]
- Kara, S.; Schrittwieser, J.H.; Hollmann, F.; Ansorge-Schumacher, M.B. Recent trends and novel concepts in cofactor-dependent biotransformations. Appl. Microbiol. Biotechnol. 2014, 98, 1517–1529. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Danishefsky, S.J. The total synthesis of proteasome inhibitors TMC-95A and TMC-95B: Discovery of a new method to generate cis-propenyl amides. Angew. Chem. Int. Ed. 2002, 114, 530–533. [Google Scholar] [CrossRef]
- Huang, R.; Chen, H.; Upp, D.M.; Lewis, J.C.; Zhang, Y.-H.P.J. A high-throughput method for directed evolution of NAD(P)+-dependent dehydrogenases for the reduction of biomimetic nicotinamide analogues. ACS Catal. 2019, 9, 11709–11719. [Google Scholar] [CrossRef] [PubMed]
- Drenth, J.; Yang, G.; Paul, C.E.; Fraaije, M.W. A Tailor-Made Deazaflavin-Mediated Recycling System for Artificial Nicotinamide Cofactor Biomimetics. ACS Catal. 2021, 11, 11561–11569. [Google Scholar] [CrossRef]
- Nowak, C.; Beer, B.C.; Pick, A.; Roth, T.; Lommes, P.; Sieber, V. A water-forming NADH oxidase from Lactobacillus pentosus suitable for the regeneration of synthetic biomimetic cofactors. Front. Microbiol. 2015, 6, 957. [Google Scholar] [CrossRef]
- Nowak, C.; Pick, A.; Csepei, L.I.; Sieber, V. Characterization of biomimetic cofactors according to stability, redox potentials, and enzymatic conversion by NADH oxidase from Lactobacillus pentosus. ChemBioChem 2017, 18, 1944–1949. [Google Scholar] [CrossRef]
- Nowak, C.; Pick, A.; Lommes, P.; Sieber, V. Enzymatic reduction of nicotinamide biomimetic cofactors using an engineered glucose dehydrogenase: Providing a regeneration system for artificial cofactors. ACS Catal. 2017, 7, 5202–5208. [Google Scholar] [CrossRef]
- Bashiri, G.; Rehan, A.M.; Greenwood, D.R.; Dickson, J.M.J.; Baker, E.N. Metabolic engineering of cofactor F420 production in Mycobacterium smegmatis. PLoS ONE 2010, 5, e15803. [Google Scholar] [CrossRef]
- Shah, M.V.; Antoney, J.; Kang, S.W.; Warden, A.C.; Hartley, C.J.; Nazem-Bokaee, H.; Jackson, C.J.; Scott, C. Cofactor F420-dependent enzymes: An under-explored resource for asymmetric redox biocatalysis. Catalysts 2019, 9, 868. [Google Scholar] [CrossRef]
- Shah, M.V.; Nazem-Bokaee, H.; Antoney, J.; Kang, S.W.; Jackson, C.J.; Scott, C. Improved production of the non-native cofactor F420 in Escherichia coli. Sci. Rep. 2021, 11, 21774. [Google Scholar] [CrossRef] [PubMed]
- Voss, C.V.; Gruber, C.C.; Faber, K.; Knaus, T.; Macheroux, P.; Kroutil, W. Orchestration of concurrent oxidation and reduction cycles for stereoinversion and deracemisation of sec-alcohols. J. Am. Chem. Soc. 2008, 130, 13969–13972. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).