
Nickel and Cobalt Ilmenites-Based Catalysts for Upgrading Pyrolytic Oil during Pyrolysis of Waste Tires

 , , ,
, , ,  ,
,  and
and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Characterization of Feedstock and Catalysts
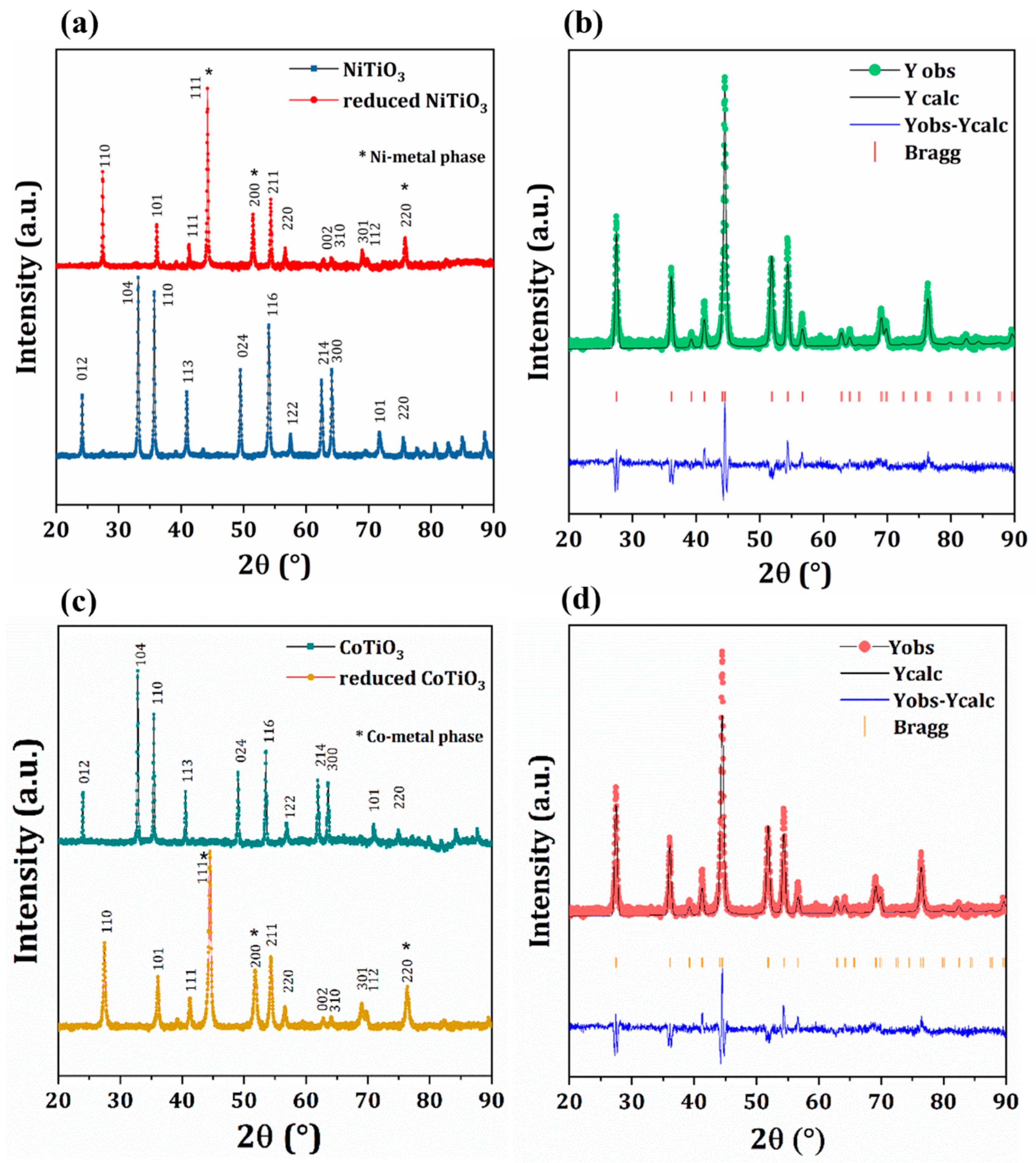
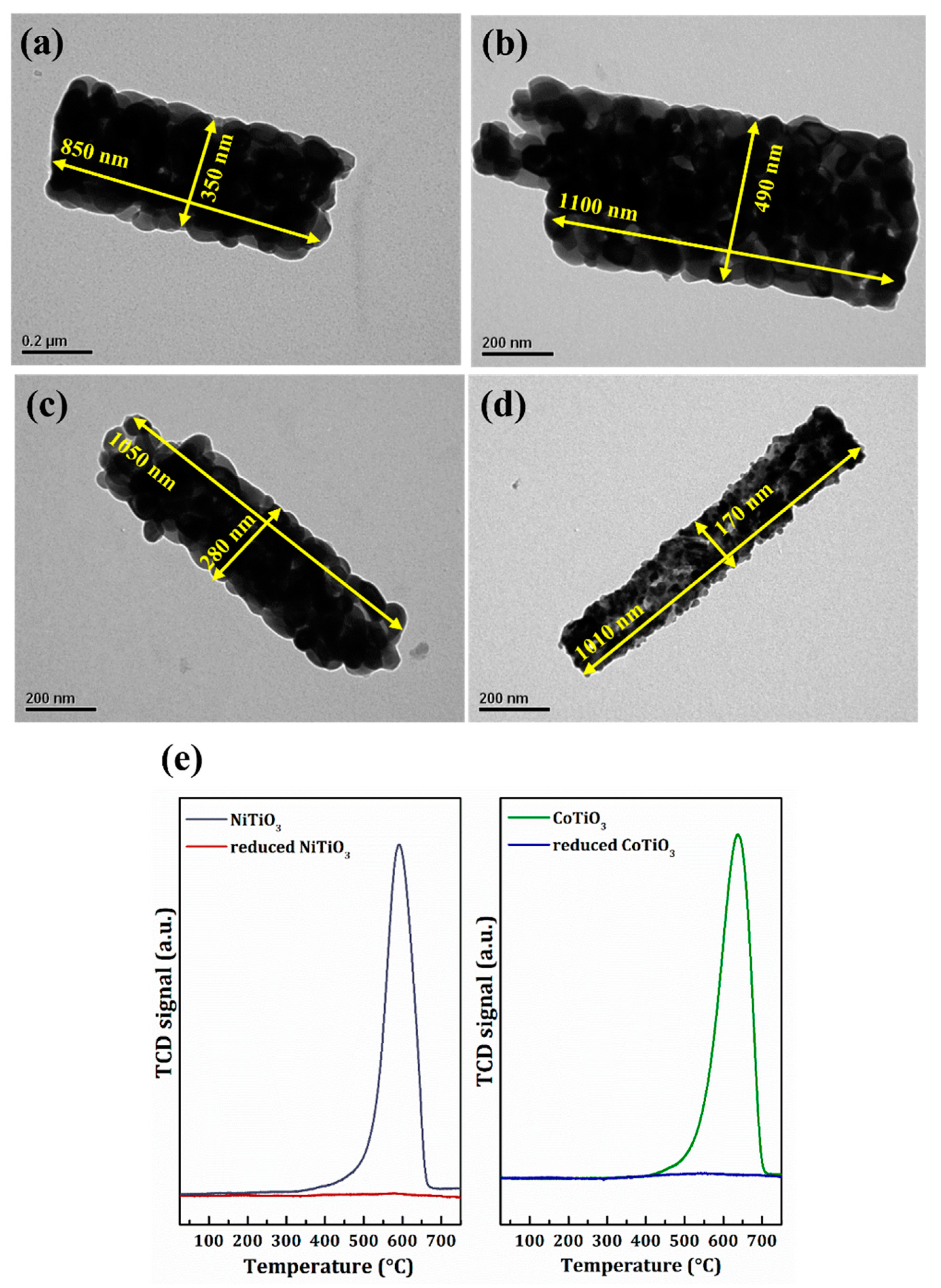
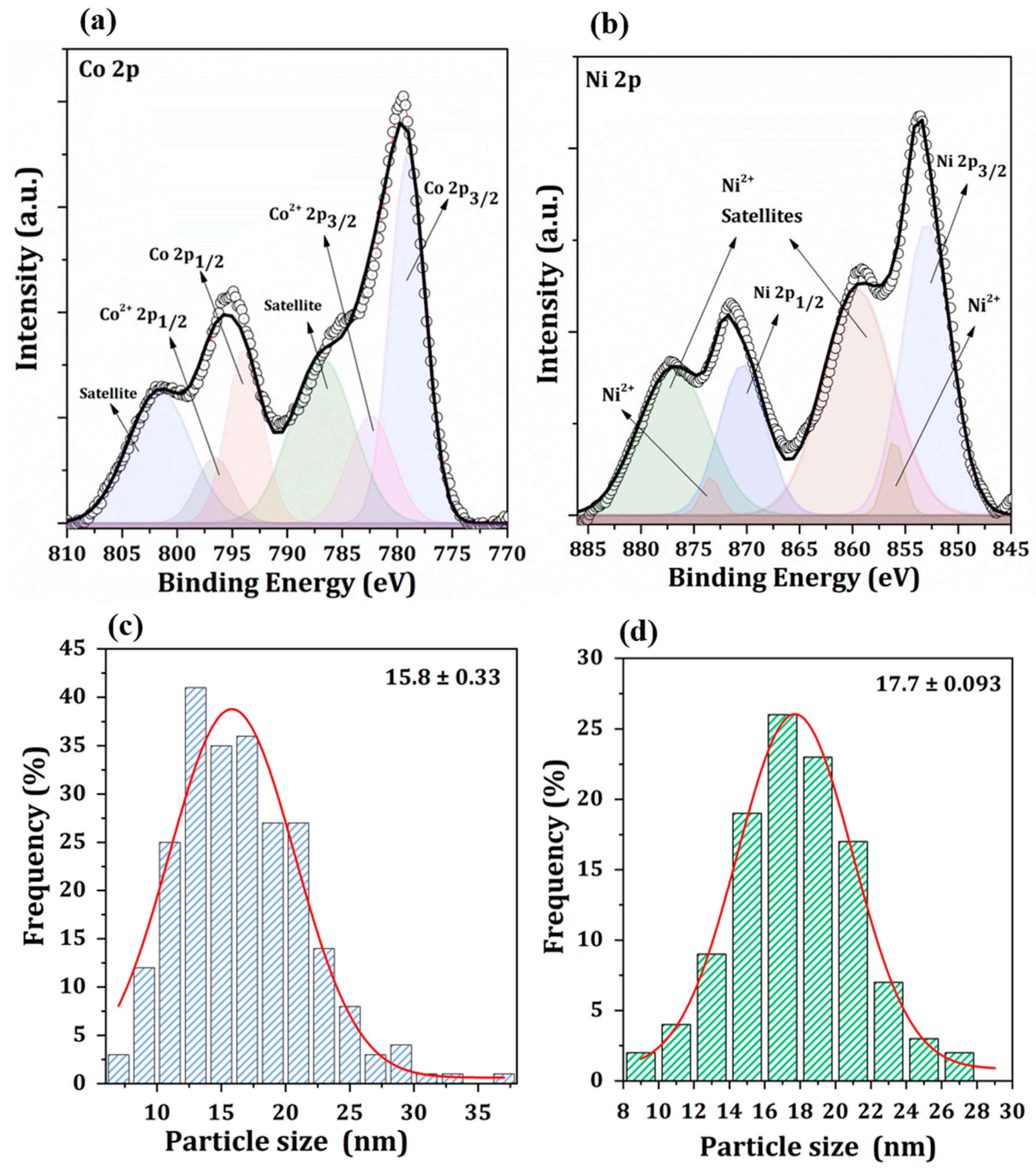
2.2. Catalyst Characterization
2.3. Pyrolysis Reaction of Waste Tires Carried out in a Py-GC/MS System
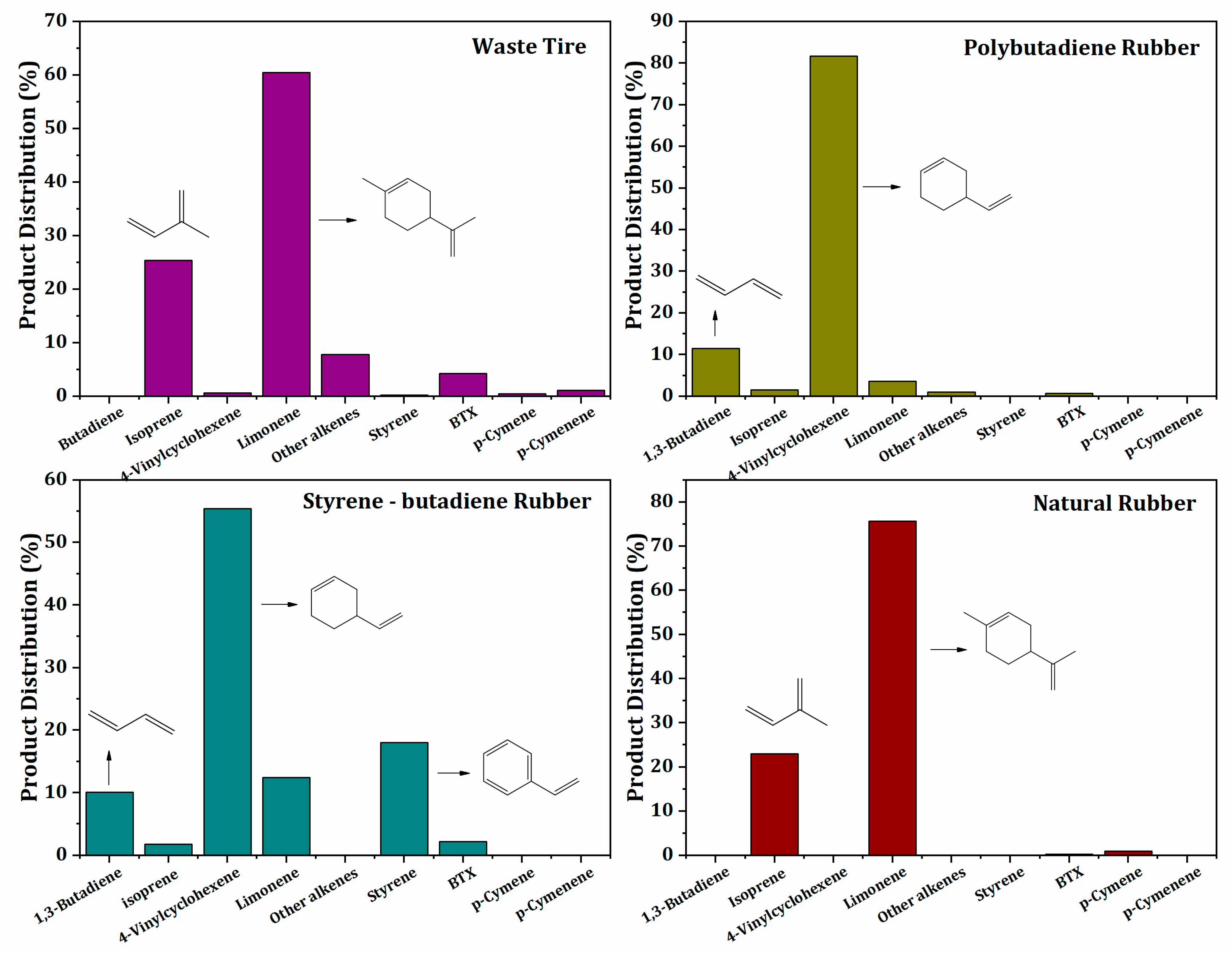
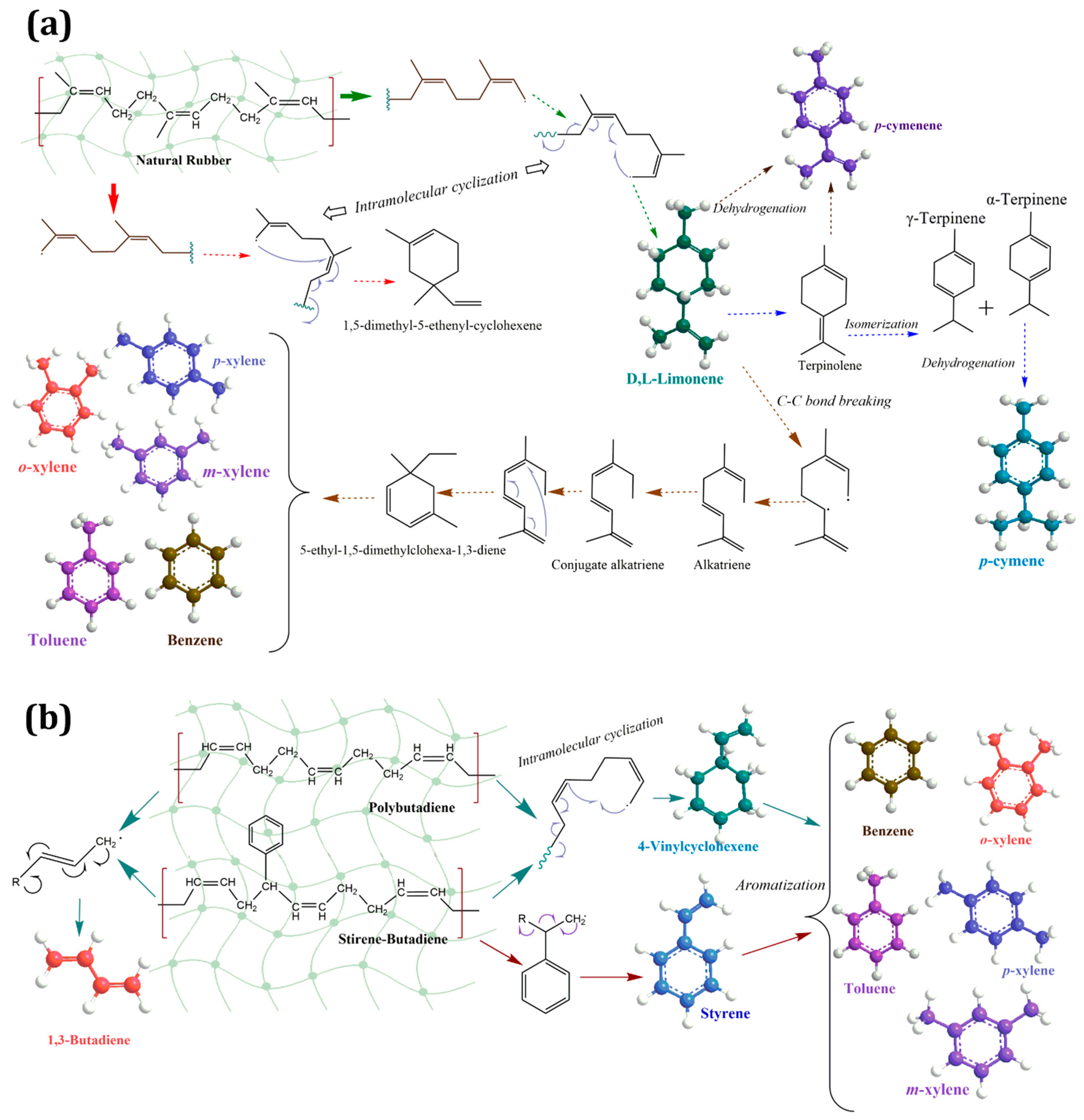
2.3.1. Main Products and Reaction Pathways during Uncatalyzed Pyrolysis Reaction of Waste Tires and Their Components (NR, BR, SBR)
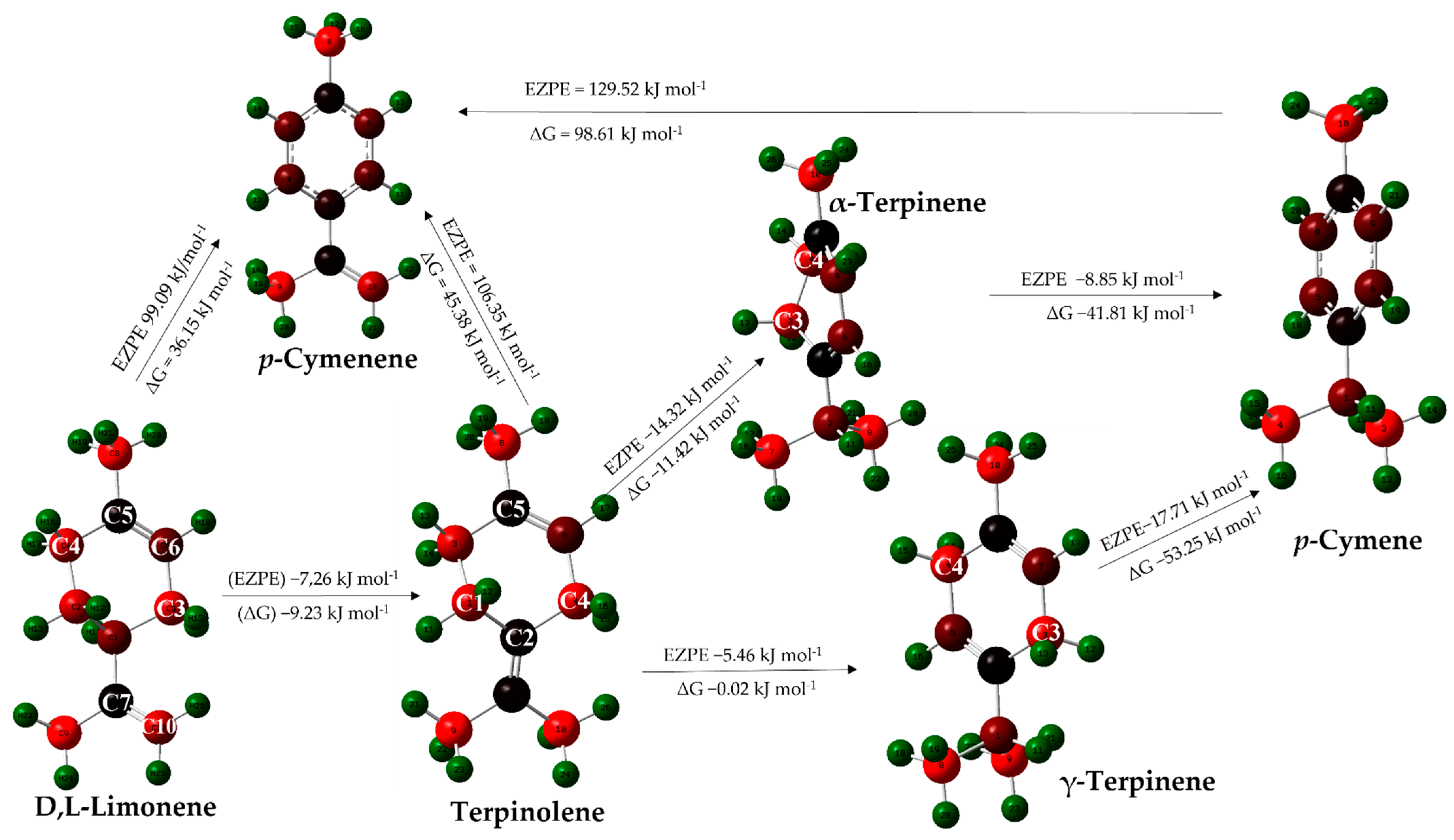
2.3.2. Computational Study of Secondary Reactions Occurring on D,L-Limonene for the Formation of Aromatic Terpenes (p-Cymene and p-Cymenene)
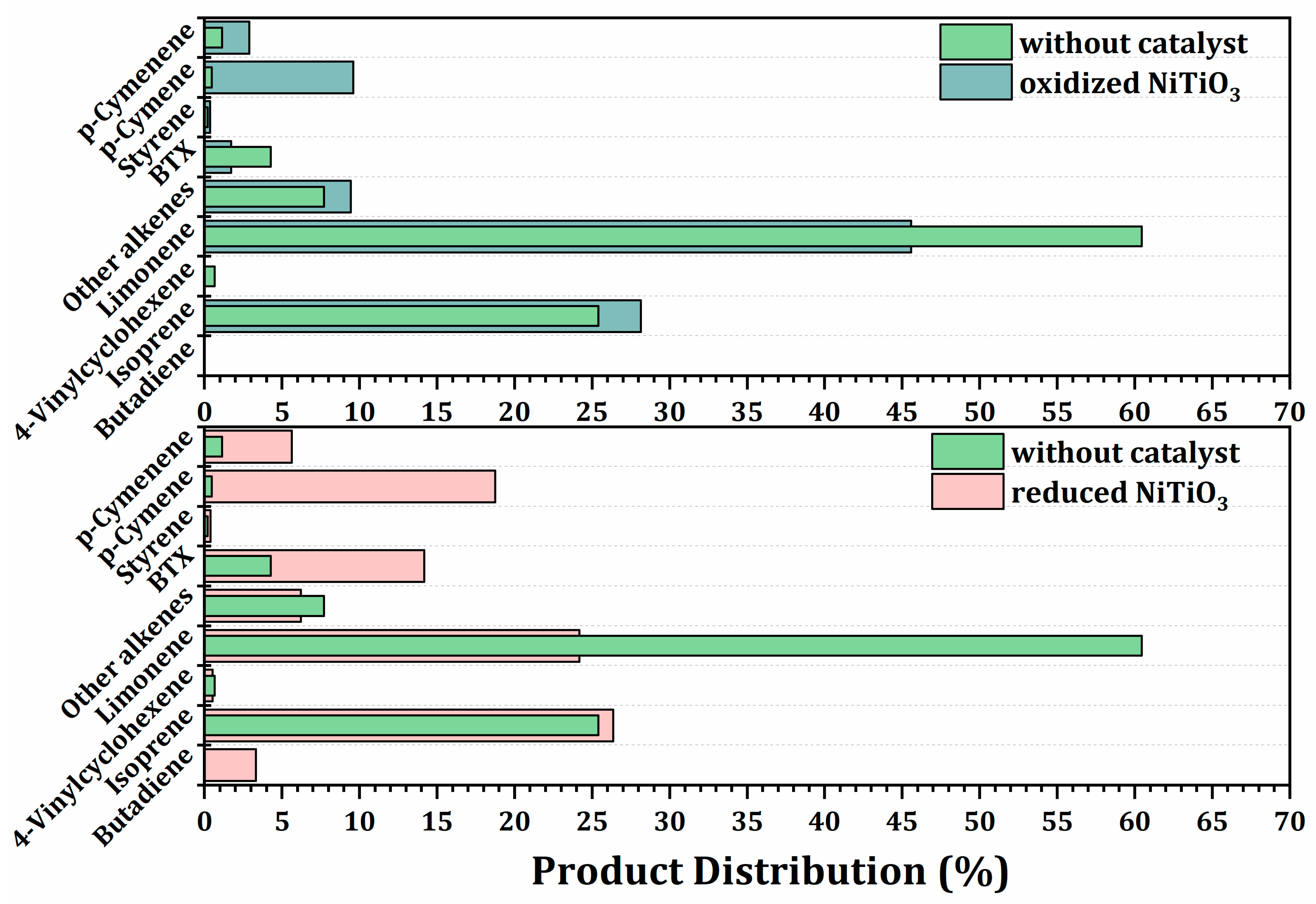
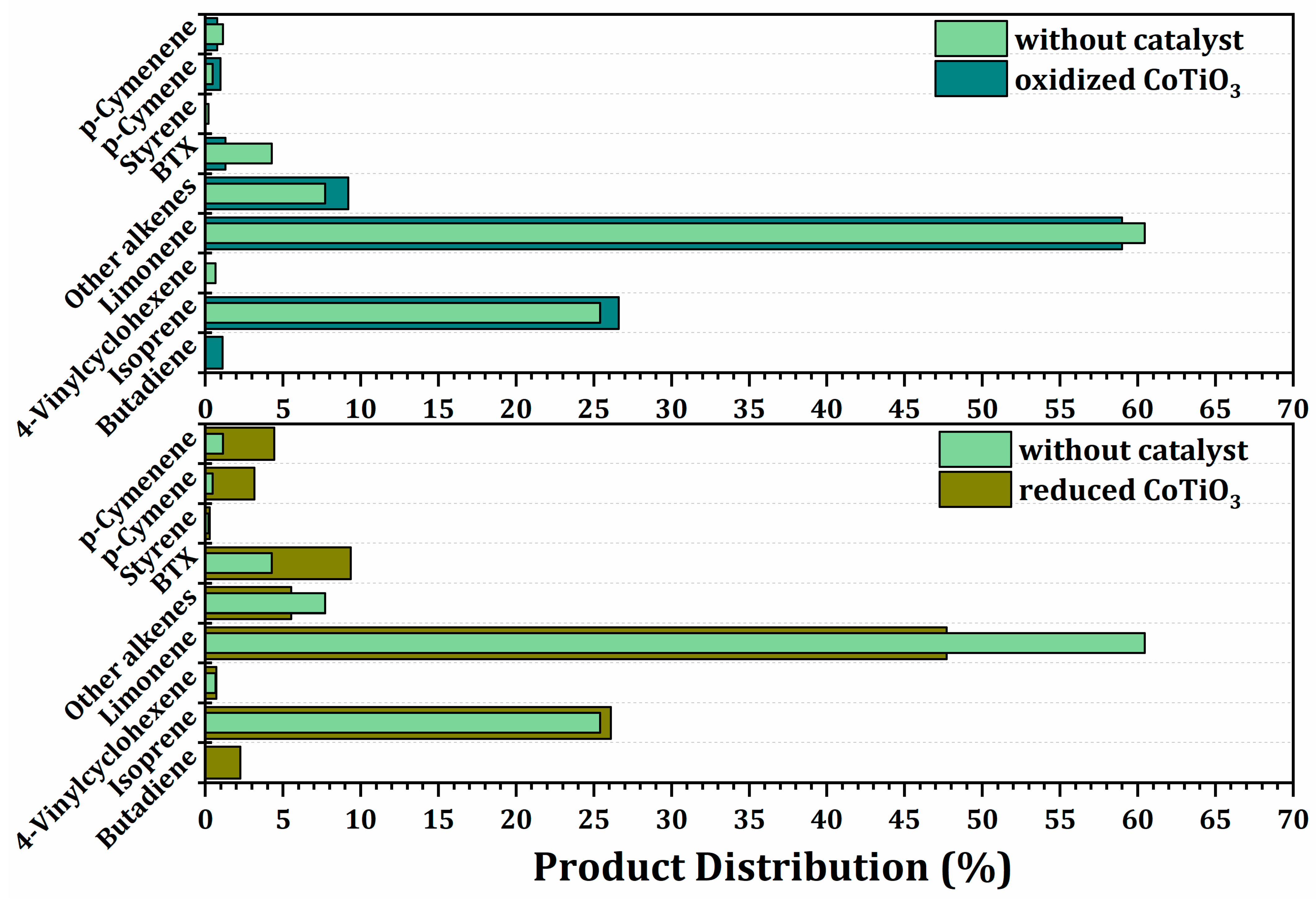
2.3.3. Effect of the Oxidized and Reduced Ilmenites on Secondary Reactions during Pyrolysis of Waste Tires
3. Materials and Methods
3.1. Materials and Chemical Reagents
3.2. Characterization of Waste Tires
3.3. Catalyst Synthesis and Characterization
3.3.1. Preparation of the Catalysts
3.3.2. Characterization Techniques
3.4. Computational Methods
3.5. Pyrolysis Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tire Industry Project, End-of-Life Tire (ELT) Management Toolkit. Available online: https://www.wbcsd.org/Sector-Projects/Tire-Industry-Project/End-of-Life-Tires-ELTs (accessed on 15 July 2022).
- WBCSD & The Tire Industry Project, Global ELT Management-A Global State of Knowledge on Regulation, Management Systems, Impacts of Recovery and Technologies. Available online: https://www.wbcsd.org/Sector-Projects/Tire-Industry-Project/End-of-Life-Tires-ELTs (accessed on 15 July 2022).
- Gao, N.; Wang, F.; Quan, C.; Santamaria, L.; Lopez, G.; Williams, P.T. Tire pyrolysis char: Processes, properties, upgrading and applications. Prog. Energy Combust. Sci. 2022, 93, 101022–101059. [Google Scholar] [CrossRef]
- Arabiourrutia, M.; Lopez, G.; Artetxe, M.; Alvarez, J.; Bilbao, J.; Olazar, M. Waste tyre valorization by catalytic pyrolysis—A review. Renew. Sustain. Energy Rev. 2020, 129, 109932–109956. [Google Scholar] [CrossRef]
- Wang, Z.C.; Duan, P.G.; Wang, K. From waste tire to high value-added chemicals: An analytical Py-GC/TOF–MS study. Environ. Sci. Pollut. Res. 2022, 29, 72117–72125. [Google Scholar] [CrossRef]
- Khalil, U.; Vongsvivut, J.; Shahabuddin, M.; Samudrala, S.P.; Srivatsa, S.C.; Bhattacharya, S. A study on the performance of coke resistive cerium modified zeolite Y catalyst for the pyrolysis of scrap tyres in a two-stage fixed bed reactor. Waste Manag. 2020, 102, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Muenpol, S.; Jitkarnka, S. Effects of Fe supported on zeolites on structures of hydrocarbon compounds and petrochemicals in waste tire-derived pyrolysis oils. J. Anal. Appl. Pyrolysis 2016, 117, 147–156. [Google Scholar] [CrossRef]
- Olazar, M.; Aguado, R.; Arabiourrutia, M.; Lopez, G.; Barona, A.; Bilbao, J. Catalyst effect on the composition of tire pyrolysis products. Energy Fuels 2008, 22, 2909–2916. [Google Scholar] [CrossRef]
- Li, W.; Huang, C.; Li, D.; Huo, P.; Wang, M.; Han, L.; Chen, G.; Li, H.; Li, X.; Wang, Y.; et al. Derived oil production by catalytic pyrolysis of scrap tires, Cuihua Xuebao/Chinese. J. Catal. 2016, 37, 526–532. [Google Scholar] [CrossRef]
- Tavera Ruiz, C.P.; Gauthier-Maradei, P.; Capron, M.; Pirez, C.; Gardoll, O.; Katryniok, B.; Dumeignil, F. Transformation of dl Limonene into Aromatic Compounds Using Supported Heteropolyacid Catalysts. Catal. Letters. 2019, 149, 328–337. [Google Scholar] [CrossRef]
- Dũng, N.A.; Tanglumlert, W.; Wongkasemjit, S.; Jitkarnka, S. Roles of ruthenium on catalytic pyrolysis of waste tire and the changes of its activity upon the rate of calcination. J. Anal. Appl. Pyrolysis 2010, 87, 256–262. [Google Scholar] [CrossRef]
- Yuwapornpanit, R.; Jitkarnka, S. Cu-doped catalysts and their impacts on tire-derived oil and sulfur removal. J. Anal. Appl. Pyrolysis 2015, 111, 200–208. [Google Scholar] [CrossRef]
- Osorio-Vargas, P.; Lick, I.D.; Pizzio, L.R.; Alejandro-Martín, S.; Casas-Ledón, Y.; Poblete, J.; Casella, M.L.; Arteaga-Pérez, L.E. Using tungstophosphoric acid-modified CeO2, TiO2, and SiO2 catalysts to promote secondary reactions leading to aromatics during waste tire pyrolysis. Mol. Catal. 2022, 531, 112682. [Google Scholar] [CrossRef]
- Osorio-Vargas, P.; Shanmugaraj, K.; Herrera, C.; Campos, C.H.; Torres, C.C.; Castillo-Puchi, F.; Arteaga-Pérez, L.E. Valorization of Waste Tires via Catalytic Fast Pyrolysis Using Palladium Supported on Natural Halloysite. Ind. Eng. Chem. Res. 2021, 51, 18806–18816. [Google Scholar] [CrossRef]
- Osorio-Vargas, P.; Menares, T.; Lick, D.; Casella, M.L.; Romero, R.; Jiménez, R.; Arteaga-Pérez, L.E. Tuning the product distribution during the catalytic pyrolysis of waste tires: The effect of the nature of metals and the reaction temperature. Catal. Today 2021, 372, 164–174. [Google Scholar] [CrossRef]
- Osorio-Vargas, P.; Campos, C.H.; Torres, C.C.; Herrera, C.; Shanmugaraj, K.; Bustamante, T.M.; Diaz de Leon, J.; Medina, F.; Arteaga-Pérez, L.E. Catalytic pyrolysis of used tires on noble-metal-based catalysts to obtain high-value chemicals: Reaction pathways. Catal. Today 2021, 394–396, 475–485. [Google Scholar] [CrossRef]
- Williams, P.T.; Brindle, A.J. Catalytic pyrolysis of tyres: Influence of catalyst temperature. Fuel 2002, 81, 2425–2434. [Google Scholar] [CrossRef]
- Muenpol, S.; Yuwapornpanit, R.; Jitkarnka, S. Valuable petrochemicals, petroleum fractions, and sulfur compounds in oils derived from waste tyre pyrolysis using five commercial zeolites as catalysts: Impact of zeolite properties. Clean Technol. Environ. Policy 2015, 17, 1149–1159. [Google Scholar] [CrossRef]
- Williams, P.T.; Brindle, A.J. Aromatic chemicals from the catalytic pyrolysis of scrap tyres. J. Anal. Appl. Pyrolysis 2003, 67, 143–164. [Google Scholar] [CrossRef]
- Buhl, D.; Weyrich, P.A.; Hölderich, W.F. Preparation of p-cymene from mixtures of terpenes as renewable feedstock. Stud. Surf. Sci. Catal. 1999, 121, 191–196. [Google Scholar] [CrossRef]
- Tibbetts, J.D.; Bull, S.D. p-Menthadienes as Biorenewable Feedstocks for a Monoterpene-Based Biorefinery. Adv. Sustain. Syst. 2021, 5, 2000292. [Google Scholar] [CrossRef]
- Tavera-Ruiz, C.; Gauthier-Maradei, P.; Capron, M.; Ferreira-Beltran, D.; Palencia-Blanco, C.; Morin, J.C.; Dumeignil, F. An Alternative to the Cymenes Production from Scrap Tire Rubber Using Heteropolyacid Catalysts. Waste Biomass Valorization 2019, 10, 3057–3069. [Google Scholar] [CrossRef]
- Sun, H.; Xu, X.; Kim, H.; Jung, W.; Zhou, W.; Shao, Z. Electrochemical Water Splitting: Bridging the Gaps between Fundamental Research and Industrial Applications. Energy Environ. Mater. 2022, e12441. [Google Scholar] [CrossRef]
- Du, J.; Xiang, D.; Zhou, K.; Wang, L.; Yu, J.; Xia, H.; Zhao, L.; Liu, H.; Zhou, W. Electrochemical hydrogen production coupled with oxygen evolution, organic synthesis, and waste reforming. Nano Energy 2022, 104, 107875. [Google Scholar] [CrossRef]
- Morales, R.; Campos, C.H.; Fierro, J.L.G.; Fraga, M.A.; Pecchi, G. Enhancing xylose aqueous-phase hydrogenation catalytic performance of A-site Ce substituted and B-site Rh doped reduced perovskites. Mol. Catal. 2017, 436, 182–189. [Google Scholar] [CrossRef]
- Pereñiguez, R.; Gonzalez-delaCruz, V.M.; Caballero, A.; Holgado, J.P. LaNiO3 as a precursor of Ni/La2O3 for CO2 reforming of CH 4: Effect of the presence of an amorphous NiO phase. Appl. Catal. B Environ. 2012, 123–124, 324–332. [Google Scholar] [CrossRef]
- Horrillo-Martínez, P.; Virolleaud, M.A.; Jaekel, C. Selective Palladium-Catalyzed Dehydrogenation of Limonene to Dimethylstyrene. ChemCatChem 2010, 2, 175–181. [Google Scholar] [CrossRef]
- Sanchez-Vazquez, S.A.; Sheppard, T.D.; Evans, J.R.G.; Hailes, H.C. The selective conversion of D-limonene to p,α-dimethylstyrene. RSC Adv. 2014, 4, 61652–61655. [Google Scholar] [CrossRef]
- Williams, P.T.; Besler, S. Pyrolysis- thermogravimetric analysis of tires. Fuel 1995, 74, 1277–1283. [Google Scholar] [CrossRef]
- Miguel, G.S.; Aguado, J.; Serrano, D.P.; Escola, J.M. Thermal and catalytic conversion of used tyre rubber and its polymeric constituents using Py-GC/MS. Appl. Catal. B Environ. 2006, 64, 209–219. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- González-Vera, D.; Bustamante, T.M.; de León, J.N.D.; Dinamarca, R.; Morales, R.; Osorio-Vargas, P.A.; Torres, C.C.; Campos, C.H. Chemoselective nitroarene hydrogenation over Ni-Pd alloy supported on TiO2 prepared from ilmenite-type PdxNi1−xTiO3. Mater. Today Commun. 2020, 24, 101091. [Google Scholar] [CrossRef]
- Osorio-Vargas, P.; Flores-González, N.A.; Navarro, R.M.; Fierro, J.L.G.; Campos, C.H.; Reyes, P. Improved stability of Ni/Al2O3 catalysts by effect of promoters (La2O3, CeO2) for ethanol steam-reforming reaction. Catal. Today 2015, 259, 27–38. [Google Scholar] [CrossRef]
- Madhavi, J. Comparison of average crystallite size by X-ray peak broadening and Williamson–Hall and size–strain plots for VO2+ doped ZnS/CdS composite nanopowder. SN Appl. Sci. 2019, 1, 1509. [Google Scholar] [CrossRef]
- Luo, Y.-R. Handbook of Bond Dissociation Energies in Organic Compounds; CRC Press: Boca Raton, FL, USA, 2002; Volume 126. [Google Scholar] [CrossRef]
- Golub, M. Thermal rearrangements of unsaturated polymers. Rubber Chem. Technol. 1978, 51, 677–685. [Google Scholar] [CrossRef]
- Chien, J.C.W.; Kiang, J.K. Polymer reactions-x Thermal pyrolysis of poly(isoprene) *. Eur. Polym. J. 1979, 15, 1059–1065. [Google Scholar] [CrossRef]
- Kobe, K.A.; Romans, R.T. Dehydrogenation of p-Cymene. Ind. Eng. Chem. 1951, 43, 1755–1758. [Google Scholar] [CrossRef]
- Corma Canos, A.; Iborra, S.; Velty, A. Chemical routes for the transformation of biomass into chemicals. Chem. Rev. 2007, 107, 2411–2502. [Google Scholar] [CrossRef]
- Busca, G. Metal catalysts for hydrogenations and dehydrogenations. In Heterogeneous Catalytic Materials; Elsevier: Amsterdam, The Netherlands, 2014; pp. 297–343. [Google Scholar] [CrossRef]
- Osorio-Vargas, P.; Lick, I.D.; Sobrevía, F.; Correa-Muriel, D.; Menares, T.; Manrique, R.; Casella, M.L.; Arteaga-Pérez, L.E. Thermal Behavior, Reaction Pathways and Kinetic Implications of Using a Ni/SiO2 Catalyst for Waste Tire Pyrolysis. Waste Biomass Valorization 2021, 12, 6465–6479. [Google Scholar] [CrossRef]
- Qu, Y.; Zhou, W.; Fu, H. Porous cobalt titanate nanorod: A new candidate for visible light-driven photocatalytic water oxidation. ChemCatChem 2014, 6, 265–270. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision, C.01, (n.d.); Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proximate Analysis (wt%) | Elemental Analysis | ||||
|---|---|---|---|---|---|
| (wt%) | (mg kg−1) | ||||
| Moisture content | 1.2 | C | 79.54 | Al | 955 |
| Volatile matter | 58.8 | H | 7.33 | Ca | 2687 |
| Fixed carbon | 30.2 | N | 0.47 | Fe | 4164 |
| Ash | 9.89 | S | 1.48 | K | 706 |
| O 1 | 2.77 | Zn | 1121 | ||
| Si | 883 | Na | 396 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Correa-Muriel, D.; Valencia-Sánchez, H.; Cortes-Hernández, H.; González-Vera, D.; Herrera, J.; Campos, C.H.; Casella, M.L.; Arteaga-Perez, L.E.; Osorio-Vargas, P. Nickel and Cobalt Ilmenites-Based Catalysts for Upgrading Pyrolytic Oil during Pyrolysis of Waste Tires. Catalysts 2022, 12, 1437. https://doi.org/10.3390/catal12111437
Correa-Muriel D, Valencia-Sánchez H, Cortes-Hernández H, González-Vera D, Herrera J, Campos CH, Casella ML, Arteaga-Perez LE, Osorio-Vargas P. Nickel and Cobalt Ilmenites-Based Catalysts for Upgrading Pyrolytic Oil during Pyrolysis of Waste Tires. Catalysts. 2022; 12(11):1437. https://doi.org/10.3390/catal12111437
Chicago/Turabian StyleCorrea-Muriel, Daniela, Hoover Valencia-Sánchez, Héctor Cortes-Hernández, Daniela González-Vera, Javiera Herrera, Cristian H. Campos, Mónica L. Casella, Luis E. Arteaga-Perez, and Paula Osorio-Vargas. 2022. "Nickel and Cobalt Ilmenites-Based Catalysts for Upgrading Pyrolytic Oil during Pyrolysis of Waste Tires" Catalysts 12, no. 11: 1437. https://doi.org/10.3390/catal12111437
APA StyleCorrea-Muriel, D., Valencia-Sánchez, H., Cortes-Hernández, H., González-Vera, D., Herrera, J., Campos, C. H., Casella, M. L., Arteaga-Perez, L. E., & Osorio-Vargas, P. (2022). Nickel and Cobalt Ilmenites-Based Catalysts for Upgrading Pyrolytic Oil during Pyrolysis of Waste Tires. Catalysts, 12(11), 1437. https://doi.org/10.3390/catal12111437