
Improving Catalytic Activity towards the Direct Synthesis of H2O2 through Cu Incorporation into AuPd Catalysts
 ,
,
Abstract
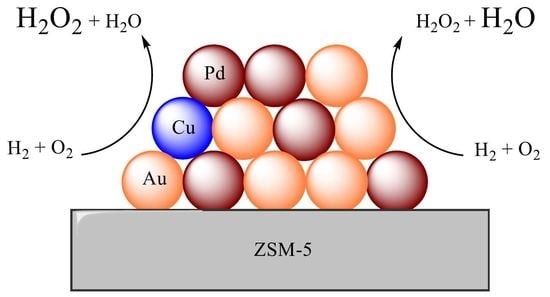
1. Introduction
2. Experimental Section
2.1. Catalyst Preparation
2.2. Catalyst Testing
2.3. Direct Synthesis of H2O2
2.4. Gas Replacement Experiments for the Direct Synthesis of H2O2
2.5. Catalyst Reusability in the Direct Synthesis and Degradation of H2O2
2.6. Degradation of H2O2
2.7. Characterisation
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lewis, R.J.; Hutchings, G.J. Recent Advances in the Direct Synthesis of H2O2. ChemCatChem 2019, 11, 298–308. [Google Scholar] [CrossRef]
- Campos-Martin, J.M.; Blanco-Brieva, G.; Fierro, J.L. Hydrogen peroxide synthesis: An outlook beyond the anthraquinone process. Angew. Chem. Int. Ed. 2006, 45, 6962–6984. [Google Scholar] [CrossRef]
- Blaser, B.; Karl-Heinz, W.; Schiefer, J. (Henkel AG and Co KGaA), Stabilizing Agent for Peroxy-Compounds and Their Solutions. U.S. Patent 3122417A, 3 June 1959. [Google Scholar]
- Gao, G.; Tian, Y.; Gong, X.; Pan, Z.; Yong, K.; Zong, B. Advances in the production technology of hydrogen peroxide. Chin. J. Catal. 2020, 41, 1039–1047. [Google Scholar] [CrossRef]
- Lewis, R.J.; Ueura, K.; Liu, X.; Fukuta, Y.; Davies, T.E.; Morgan, D.J.; Chen, L.; Qi, J.; Singleton, J.; Edwards, J.K.; et al. Highly efficient catalytic production of oximes from ketones using in situ generated H2O2. Science 2022, 376, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Crombie, C.M.; Lewis, R.J.; Kovačič, D.; Morgan, D.J.; Slater, T.J.A.; Davies, T.E.; Edwards, J.K.; Skjøth-Rasmussen, M.S.; Hutchings, G.J. The Influence of Reaction Conditions on the Oxidation of Cyclohexane via the in-situ Production of H2O2. Catal. Lett. 2021, 151, 164–171. [Google Scholar] [CrossRef]
- Jin, Z.; Wang, L.; Zuidema, E.; Mondal, K.; Zhang, M.; Zhang, J.; Wang, C.; Meng, X.; Yang, H.; Mesters, C.; et al. Hydrophobic zeolite modification for in situ peroxide formation in methane oxidation to methanol. Science 2020, 367, 193–197. [Google Scholar] [CrossRef]
- Ford, D.C.; Nilekar, A.U.; Xu, Y.; Mavrikakis, M. Partial and complete reduction of O2 by hydrogen on transition metal surfaces. Surf. Sci. 2010, 604, 1565–1575. [Google Scholar] [CrossRef]
- Voloshin, Y.; Halder, R.; Lawal, A. Kinetics of hydrogen peroxide synthesis by direct combination of H2 and O2 in a microreactor. Catal. Today 2007, 125, 40–47. [Google Scholar] [CrossRef]
- Wilson, N.M.; Flaherty, D.W. Mechanism for the Direct Synthesis of H2O2 on Pd Clusters: Heterolytic Reaction Pathways at the Liquid–Solid Interface. J. Am. Chem. Soc. 2016, 138, 574. [Google Scholar] [CrossRef]
- Adams, J.S.; Chemburkar, A.; Priyadarshini, P.; Ricciardulli, T.; Lu, Y.; Maliekkal, V.; Sampath, A.; Winikoff, S.; Karim, A.M.; Neurock, M.; et al. Solvent molecules form surface redox mediators in situ and cocatalyze O2 reduction on Pd. Science 2021, 371, 626–632. [Google Scholar] [CrossRef]
- Ricciardulli, T.; Gorthy, S.; Adams, J.S.; Thompson, C.; Karim, A.M.; Neurock, M.; Flaherty, D.W. Effect of Pd Coordination and Isolation on the Catalytic Reduction of O2 to H2O2 over PdAu Bimetallic Nanoparticles. J. Am. Chem. Soc. 2021, 143, 5445–5464. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Ntainjua, E.N.; Morgan, D.J.; Davies, T.E.; Carley, A.F.; Freakley, S.J.; Hutchings, G.J. Improving the performance of Pd based catalysts for the direct synthesis of hydrogen peroxide via acid incorporation during catalyst synthesis. Catal. Commun. 2021, 161, 106358. [Google Scholar] [CrossRef]
- Flaherty, D.W. Direct Synthesis of H2O2 from H2 and O2 on Pd Catalysts: Current Understanding, Outstanding Questions, and Research Needs. ACS Catal. 2018, 8, 1520–1527. [Google Scholar] [CrossRef]
- Selinsek, M.; Deschner, B.J.; Doronkin, D.E.; Sheppard, T.L.; Grunwaldt, J.; Dittmeyer, R. Revealing the Structure and Mechanism of Palladium during Direct Synthesis of Hydrogen Peroxide in Continuous Flow Using Operando Spectroscopy. ACS Catal. 2018, 8, 2546–2557. [Google Scholar] [CrossRef]
- Henkel, H.; Weber, W. (Henkel AG and Co KGaA), Manufacture of Hydrogen Peroxid. U.S. Patent 1,108,752, 25 August 1914. [Google Scholar]
- Priyadarshini, P.; Ricciardulli, T.; Adams, J.S.; Yun, Y.S.; Flaherty, D.W. Effects of bromide adsorption on the direct synthesis of H2O2 on Pd nanoparticles: Formation rates, selectivities, and apparent barriers at steady-state. J. Catal. 2021, 399, 24–40. [Google Scholar] [CrossRef]
- Pospelova, T.A.; Kobozev, N. Russ. J. Phys. Chem. 1961, 35, 1192–1197.
- Wilson, N.M.; Priyadarshini, P.; Kunz, S.; Flaherty, D.W. Direct synthesis of H2O2 on Pd and AuxPd1 clusters: Understanding the effects of alloying Pd with Au. J. Catal. 2018, 357, 163–175. [Google Scholar] [CrossRef]
- Richards, T.; Lewis, R.J.; Morgan, D.J.; Hutchings, G.J. The Direct Synthesis of Hydrogen Peroxide Over Supported Pd-Based Catalysts: An Investigation into the Role of the Support and Secondary Metal Modifiers. Catal. Lett. 2022, 1–9. [Google Scholar] [CrossRef]
- Brehm, J.; Lewis, R.J.; Morgan, D.J.; Davies, T.E.; Hutchings, G.J. The Direct Synthesis of Hydrogen Peroxide over AuPd Nanoparticles: An Investigation into Metal Loading. Catal. Lett. 2022, 152, 254–262. [Google Scholar] [CrossRef]
- Freakley, S.J.; He, Q.; Harrhy, J.H.; Lu, L.; Crole, D.A.; Morgan, D.J.; Ntainjua, E.N.; Edwards, J.K.; Carley, A.F.; Borisevich, A.Y.; et al. Palladium-tin catalysts for the direct synthesis of H2O2 with high selectivity. Science 2016, 351, 965–968. [Google Scholar] [CrossRef]
- Wang, S.; Lewis, R.J.; Doronkin, D.E.; Morgan, D.J.; Grunwaldt, J.; Hutchings, G.J.; Behrens, S. The direct synthesis of hydrogen peroxide from H2 and O2 using Pd-Ga and Pd-In catalysts. Catal. Sci. Technol. 2020, 10, 1925–1935. [Google Scholar] [CrossRef]
- Crole, D.A.; Underhill, R.; Edwards, J.K.; Shaw, G.; Freakley, S.J.; Hutchings, G.J.; Lewis, R.J. The direct synthesis of hydrogen peroxide from H2 and O2 using PdNi/TiO2 catalysts. Philos. Trans. R. Soc. A 2020, 378, 20200062. [Google Scholar] [CrossRef] [PubMed]
- Crombie, C.M.; Lewis, R.J.; Taylor, R.L.; Morgan, D.J.; Davies, T.E.; Folli, A.; Murphy, D.M.; Edwards, J.K.; Qi, J.; Jiang, H.; et al. Enhanced Selective Oxidation of Benzyl Alcohol via In Situ H2O2 Production over Supported Pd-Based Catalysts. ACS Catal. 2021, 11, 2701–2714. [Google Scholar] [CrossRef]
- Cao, K.; Yang, H.; Bai, S.; Xu, Y.; Yang, C.; Wu, Y.; Xie, M.; Cheng, T.; Shao, Q.; Huang, X. Efficient Direct H2O2 Synthesis Enabled by PdPb Nanorings via Inhibiting the O–O Bond Cleavage in O2 and H2O2. ACS Catal. 2021, 11, 1106–1118. [Google Scholar] [CrossRef]
- Tian, P.; Xuan, F.; Ding, D.; Sun, Y.; Xu, X.; Li, W.; Si, R.; Xu, J.; Han, Y. Revealing the role of tellurium in palladium-tellurium catalysts for the direct synthesis of hydrogen peroxide. J. Catal. 2020, 385, 21–29. [Google Scholar] [CrossRef]
- Bernardotto, G.; Menegazzo, F.; Pinna, F.; Signoretto, M.; Cruciani, G.; Strukul, G. New Pd–Pt and Pd–Au catalysts for an efficient synthesis of H2O2 from H2 and O2 under very mild conditions. Appl. Catal. A 2009, 358, 129–135. [Google Scholar] [CrossRef]
- Quon, S.; Jo, D.Y.; Han, G.; Han, S.S.; Seo, M.; Lee, K. Role of Pt atoms on Pd(1 1 1) surface in the direct synthesis of hydrogen peroxide: Nano-catalytic experiments and DFT calculations. J. Catal. 2018, 368, 237–247. [Google Scholar] [CrossRef]
- Deguchi, T.; Yamano, H.; Takenouchi, S.; Iwamoto, M. Enhancement of catalytic activity of Pd-PVP colloid for direct H2O2 synthesis from H2 and O2 in water with addition of 0.5 atom% Pt or Ir. Catal. Sci. Technol. 2018, 8, 1002–1015. [Google Scholar] [CrossRef]
- Kim, M.; Han, G.; Xiao, X.; Song, J.; Hong, J.; Jung, E.; Kim, H.; Ahn, J.; Han, S.S.; Lee, K.; et al. Anisotropic growth of Pt on Pd nanocube promotes direct synthesis of hydrogen peroxide. Appl. Surf. Sci. 2021, 562, 150031. [Google Scholar] [CrossRef]
- Xu, J.; Ouyang, L.; Da, G.; Song, Q.; Yang, X.; Han, Y. Pt promotional effects on Pd–Pt alloy catalysts for hydrogen peroxide synthesis directly from hydrogen and oxygen. J. Catal. 2012, 285, 74–82. [Google Scholar] [CrossRef]
- Chen, Q.; Beckman, E.J. Direct synthesis of H2O2 from O2 and H2 over precious metal loaded TS-1 in CO2. Green Chem. 2007, 9, 802–808. [Google Scholar] [CrossRef]
- Gong, X.; Lewis, R.J.; Zhou, S.; Morgan, D.J.; Davies, T.E.; Liu, X.; Kiely, C.J.; Zong, B.; Hutchings, G.J. Enhanced catalyst selectivity in the direct synthesis of H2O2 through Pt incorporation into TiO2 supported AuPd catalysts. Catal. Sci. Technol. 2020, 10, 4635–4644. [Google Scholar] [CrossRef]
- Lewis, R.J.; Ueura, K.; Fukuta, Y.; Davies, T.E.; Morgan, D.J.; Paris, C.B.; Singleton, J.; Edwards, J.K.; Freakley, S.J.; Yamamoto, Y.; et al. Cyclohexanone ammoximation via in situ H2O2 production using TS-1 supported catalysts. Green Chem. 2022; Advance Article. [Google Scholar] [CrossRef]
- Lewis, R.J.; Ueura, K.; Fukuta, Y.; Freakley, S.J.; Kang, L.; Wang, R.; He, Q.; Edwards, J.K.; Morgan, D.J.; Yamamoto, Y.; et al. The Direct Synthesis of H2O2 Using TS-1 Supported Catalysts. ChemCatChem 2019, 11, 1673–1680. [Google Scholar] [CrossRef]
- Nguyen, H.V.; Kim, K.Y.; Nam, H.; Lee, S.Y.; Yu, T.; Seo, T.S. Centrifugal microfluidic device for the high-throughput synthesis of Pd@AuPt core–shell nanoparticles to evaluate the performance of hydrogen peroxide generation. Lab Chip 2020, 20, 3293–3301. [Google Scholar] [CrossRef]
- Ham, H.C.; Hwang, G.S.; Han, J.; Nam, S.W.; Lim, T.H. On the Role of Pd Ensembles in Selective H2O2 Formation on PdAu Alloys. J. Phys. Chem. C 2009, 113, 12943–12945. [Google Scholar] [CrossRef]
- Li, J.; Ishihara, T.; Yoshizawa, K. Theoretical Revisit of the Direct Synthesis of H2O2 on Pd and Au@Pd Surfaces: A Comprehensive Mechanistic Study. J. Phys. Chem. C 2011, 115, 25359–25367. [Google Scholar] [CrossRef]
- Richards, T.; Harrhy, J.H.; Lewis, R.J.; Howe, A.G.R.; Suldecki, G.M.; Folli, A.; Morgan, D.J.; Davies, T.E.; Loveridge, E.J.; Crole, D.A.; et al. A residue-free approach to water disinfection using catalytic in situ generation of reactive oxygen species. Nat. Catal. 2021, 4, 575–585. [Google Scholar] [CrossRef]
- Lewis, R.J.; Bara-Estaun, A.; Agarwal, N.; Freakley, S.J.; Morgan, D.J.; Hutchings, G.J. The Direct Synthesis of H2O2 and Selective Oxidation of Methane to Methanol Using HZSM-5 Supported AuPd Catalysts. Catal. Lett. 2019, 149, 3066–3075. [Google Scholar] [CrossRef]
- Kang, J.; Puthiaraj, P.; Ahn, W.; Park, E.D. Direct synthesis of oxygenates via partial oxidation of methane in the presence of O2 and H2 over a combination of Fe-ZSM-5 and Pd supported on an acid-functionalized porous polymer. Appl. Catal. A 2020, 602, 117711. [Google Scholar] [CrossRef]
- Lyu, J.; Wei, J.; Niu, L.; Lu, C.; Hu, Y.; Xiang, Y.; Zhang, G.; Zhang, Q.; Ding, C.; Li, X. Highly efficient hydrogen peroxide direct synthesis over a hierarchical TS-1 encapsulated subnano Pd/PdO hybrid. RSC Adv. 2019, 9, 13398–13402. [Google Scholar] [CrossRef] [PubMed]
- Barnes, A.; Lewis, R.J.; Morgan, D.J.; Davies, T.E.; Hutchings, G.J. Enhancing catalytic performance of AuPd catalysts towards the direct synthesis of H2O2 through incorporation of base metals. Catal. Sci. Technol. 2022, 12, 1986–1995. [Google Scholar] [CrossRef]
- Santos, A.; Lewis, R.J.; Malta, G.; Howe, A.G.R.; Morgan, D.J.; Hampton, E.; Gaskin, P.; Hutchings, G.J. Direct Synthesis of Hydrogen Peroxide over Au–Pd Supported Nanoparticles under Ambient Conditions. Int. Eng. Chem. Res. 2019, 58, 12623–12631. [Google Scholar] [CrossRef]
- Edwards, J.K.; Thomas, A.; Carley, A.F.; Herzing, A.A.; Kiely, C.J.; Hutchings, G.J. Au–Pd supported nanocrystals as catalysts for the direct synthesis of hydrogen peroxide from H2 and O2. Green Chem. 2008, 10, 388–394. [Google Scholar] [CrossRef]
- Xu, F.; Zhao, L.; Zhao, F.; Deng, L.; Hu, L.; Zeng, B. Electrodeposition of AuPdCu Alloy Nanoparticles on a Multi-Walled Carbon Nanotube Coated Glassy Carbon Electrode for the Electrocatalytic Oxidation and Determination of Hydrazine. Int. J. Electrochem. Sci. 2014, 9, 2832–2847. [Google Scholar]
- Ab Rahim, M.H.; Armstrong, R.D.; Hammond, C.; Dimitratos, N.; Freakley, S.J.; Forde, M.M.; Morgan, D.J.; Lalev, G.; Jenkins, R.L.; Lopez-Sanchez, J.A.; et al. Low temperature selective oxidation of methane to methanol using titania supported gold palladium copper catalysts. Catal. Sci. Technol. 2016, 6, 3410–3418. [Google Scholar] [CrossRef]
- Brehm, J.; Lewis, R.J.; Richards, T.; Qin, T.; Morgan, D.J.; Davies, T.E.; Chen, L.; Liu, X.; Hutchings, G.J. Enhancing the Chemo-Enzymatic One-Pot Oxidation of Cyclohexane via In Situ H2O2 Production over Supported Pd-Based Catalysts. ACS Catal. 2022, 12, 11776–11789. [Google Scholar] [CrossRef]
- Joshi, A.M.; Delgass, W.N.; Thomson, K.T. Investigation of Gold−Silver, Gold−Copper, and Gold−Palladium Dimers and Trimers for Hydrogen Peroxide Formation from H2 and O2. J. Phys. Chem. C 2007, 111, 7384–7395. [Google Scholar] [CrossRef]
- Wilson, A.R.; Sun, K.; Chi, M.; White, R.M.; LeBeau, J.M.; Lamb, H.H.; Wiley, B.J. From Core–Shell to Alloys: The Preparation and Characterization of Solution-Synthesized AuPd Nanoparticle Catalysts. J. Phys. Chem. C 2013, 117, 17557–17566. [Google Scholar] [CrossRef]
- Zhu, B.; Thrimurthulu, G.; Delannoy, L.; Louis, C.; Mottet, C.; Creuze, J.; Legrand, B.; Guesmi, H. Evidence of Pd segregation and stabilization at edges of AuPd nano-clusters in the presence of CO: A combined DFT and DRIFTS study. J. Catal. 2013, 308, 272–281. [Google Scholar] [CrossRef]
- Bollinger, M.A.; Vannice, M.A. A kinetic and DRIFTS study of low-temperature carbon monoxide oxidation over Au—TiO2 catalysts. Appl. Catal. B 1996, 8, 417–443. [Google Scholar] [CrossRef]
- Marx, S.; Krumeich, F.; Baiker, A. Surface Properties of Supported, Colloid-Derived Gold/Palladium Mono- and Bimetallic Nanoparticles. J. Phys. Chem. C 2011, 115, 8195. [Google Scholar] [CrossRef]
- Carter, J.H.; Althahban, S.; Nowicka, E.; Freakley, S.J.; Morgan, D.J.; Shah, P.M.; Golunski, S.; Kiely, C.J.; Hutchings, G.J. Synergy and Anti-Synergy between Palladium and Gold in Nanoparticles Dispersed on a Reducible Support. ACS Catal. 2016, 6, 6623–6633. [Google Scholar] [CrossRef] [PubMed]
- Tian, P.; Ding, D.; Sun, Y.; Xuan, F.; Xu, X.; Xu, J.; Han, Y. Theoretical study of size effects on the direct synthesis of hydrogen peroxide over palladium catalysts. J. Catal. 2019, 369, 95. [Google Scholar] [CrossRef]
- Dissanayake, D.P.; Lunsford, J.H. Evidence for the Role of Colloidal Palladium in the Catalytic Formation of H2O2 from H2 and O2. J. Catal. 2002, 206, 173. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
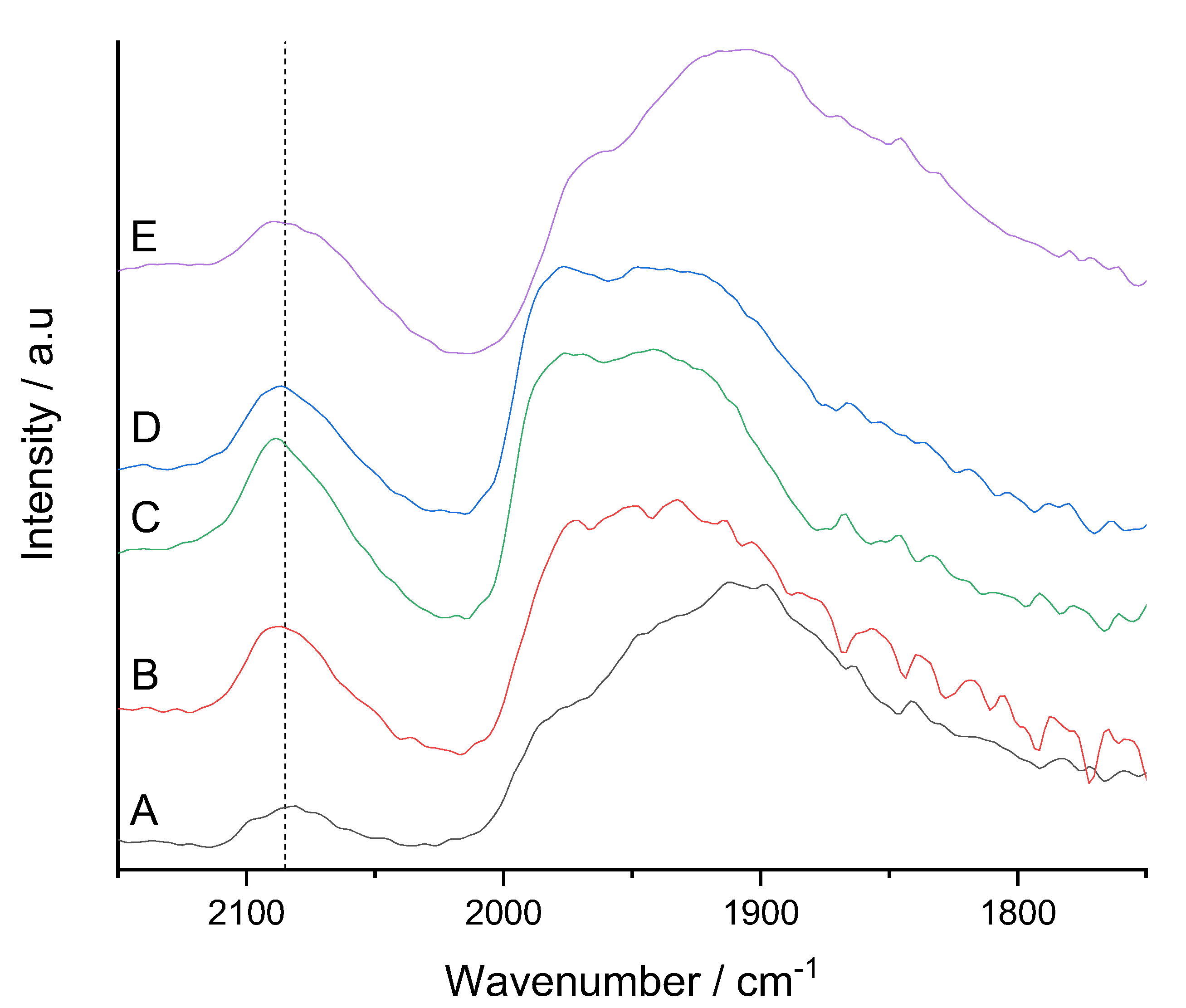{kind=link}
{kind=link}
{kind=link}
| Catalyst | Mean Particle Size/nm (Standard Deviation) | Productivity/molH2O2kgcat−1h−1 (H2O2 Conc./wt.%) |
|---|---|---|
| 1%AuPd(1.00)/ZSM-5 | 3.7 (1.43) | 69 (0.14) |
| 1%AuPd(0.975)Cu(0.025)/ZSM-5 | 4.4 (1.93) | 115 (0.23) |
| Catalyst | Productivity/molH2O2kgcat−1h−1 | Initial Reaction Rate/mmolH2O2mmolmetal−1h−1 | Degradation/molH2O2kgcat−1h−1 | Metal Leached/% | |||||
|---|---|---|---|---|---|---|---|---|---|
| Fresh | Used | Fresh | Used | Fresh | Used | Au | Pd | Cu | |
| 1%AuPd(1.00))/ZSM-5 | 69 | 57 | 37.6 | 27.0 | 320 | 397 | 0 | 0.17 | - |
| 1%AuPd(0.975)Cu(0.025)/ZSM-5 | 115 | 64 | 48.5 | 27.1 | 529 | 231 | 0 | 0.13 | BDL |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barnes, A.; Lewis, R.J.; Morgan, D.J.; Davies, T.E.; Hutchings, G.J. Improving Catalytic Activity towards the Direct Synthesis of H2O2 through Cu Incorporation into AuPd Catalysts. Catalysts 2022, 12, 1396. https://doi.org/10.3390/catal12111396
Barnes A, Lewis RJ, Morgan DJ, Davies TE, Hutchings GJ. Improving Catalytic Activity towards the Direct Synthesis of H2O2 through Cu Incorporation into AuPd Catalysts. Catalysts. 2022; 12(11):1396. https://doi.org/10.3390/catal12111396
Chicago/Turabian StyleBarnes, Alexandra, Richard J. Lewis, David J. Morgan, Thomas E. Davies, and Graham J. Hutchings. 2022. "Improving Catalytic Activity towards the Direct Synthesis of H2O2 through Cu Incorporation into AuPd Catalysts" Catalysts 12, no. 11: 1396. https://doi.org/10.3390/catal12111396
APA StyleBarnes, A., Lewis, R. J., Morgan, D. J., Davies, T. E., & Hutchings, G. J. (2022). Improving Catalytic Activity towards the Direct Synthesis of H2O2 through Cu Incorporation into AuPd Catalysts. Catalysts, 12(11), 1396. https://doi.org/10.3390/catal12111396