
Multicatalytic Hybrid Materials for Biocatalytic and Chemoenzymatic Cascades—Strategies for Multicatalyst (Enzyme) Co-Immobilization

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Co-Immobilization of Enzymes
2.1. Co-Immobilization of Enzymes into Solid Supports
2.1.1. Co-Immobilization of Enzymes through Adsorption
2.1.2. Co-Immobilization of Enzymes through Covalent Bonds
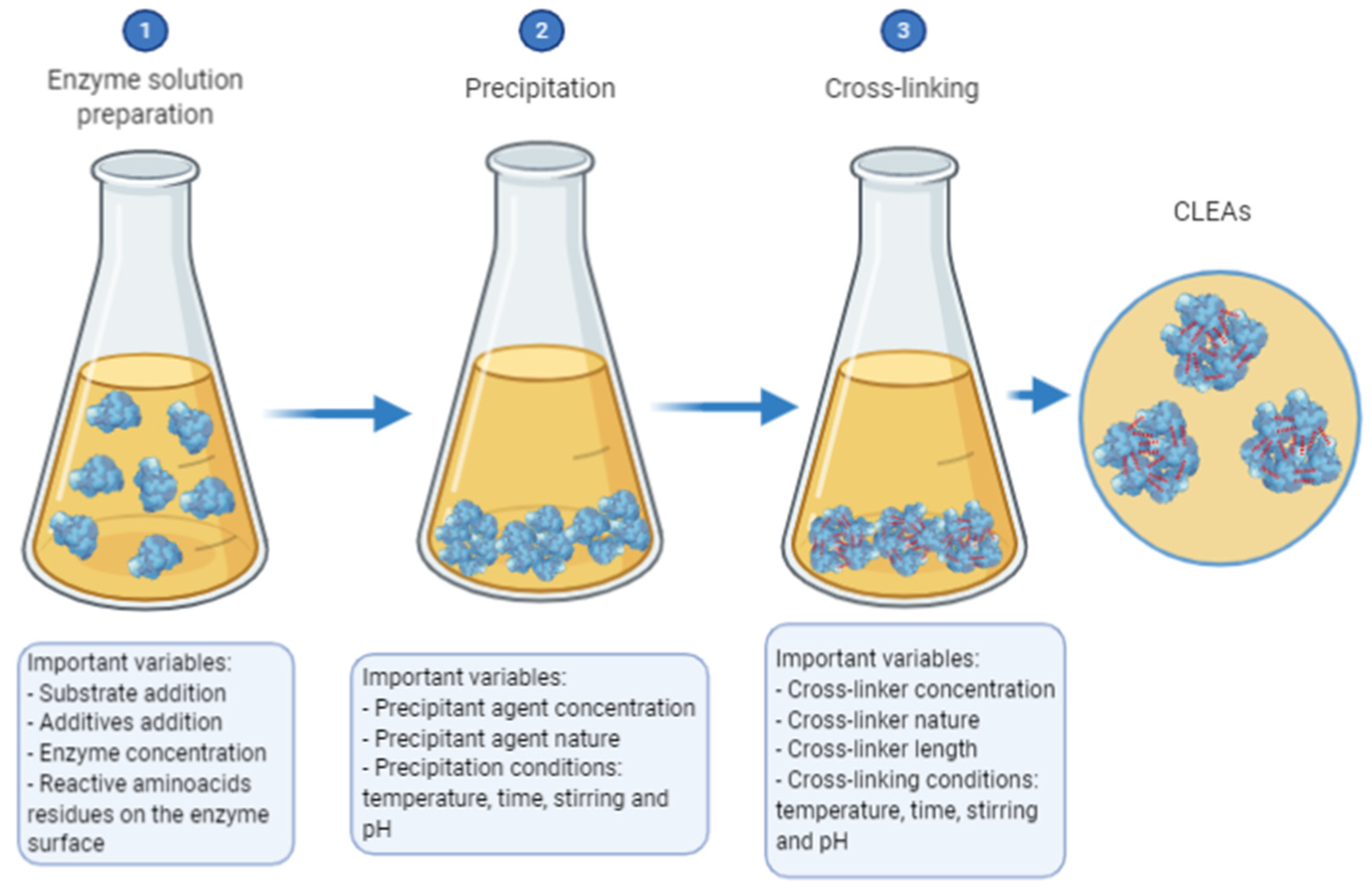
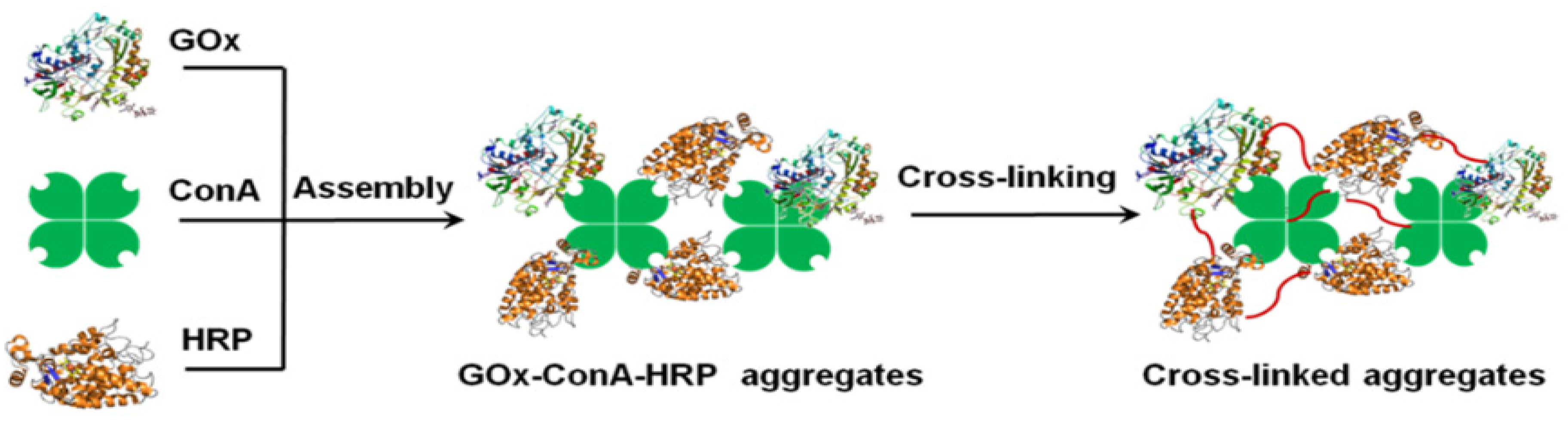
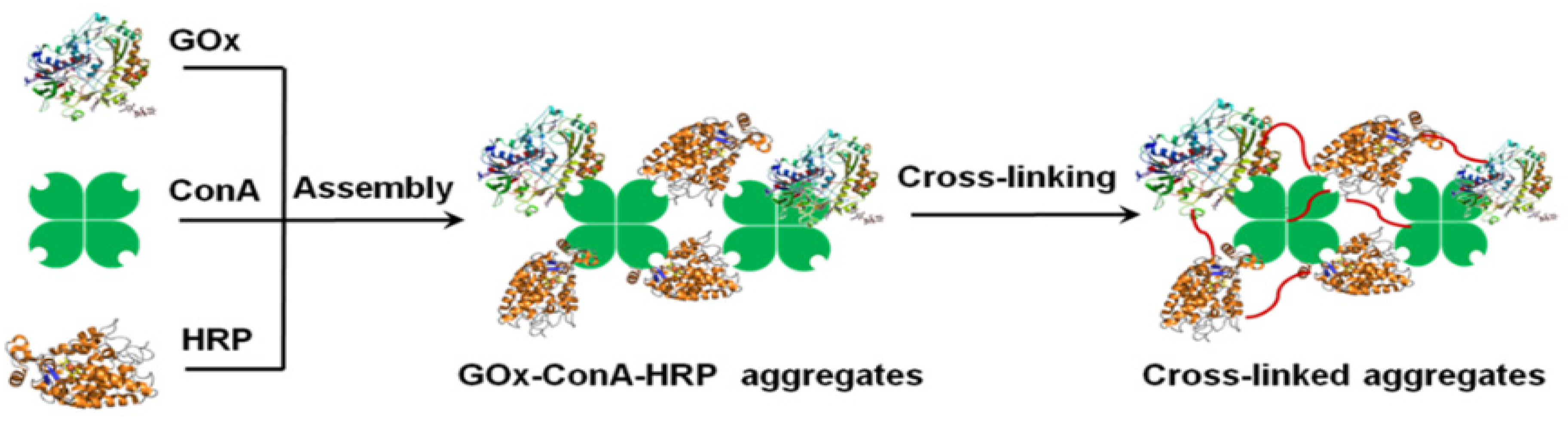
2.2. Cross-Linked Enzymes Aggregates—CLEAs
2.3. Enzyme Co-Immobilization by Encapsulation
2.3.1. Gel Encapsulation
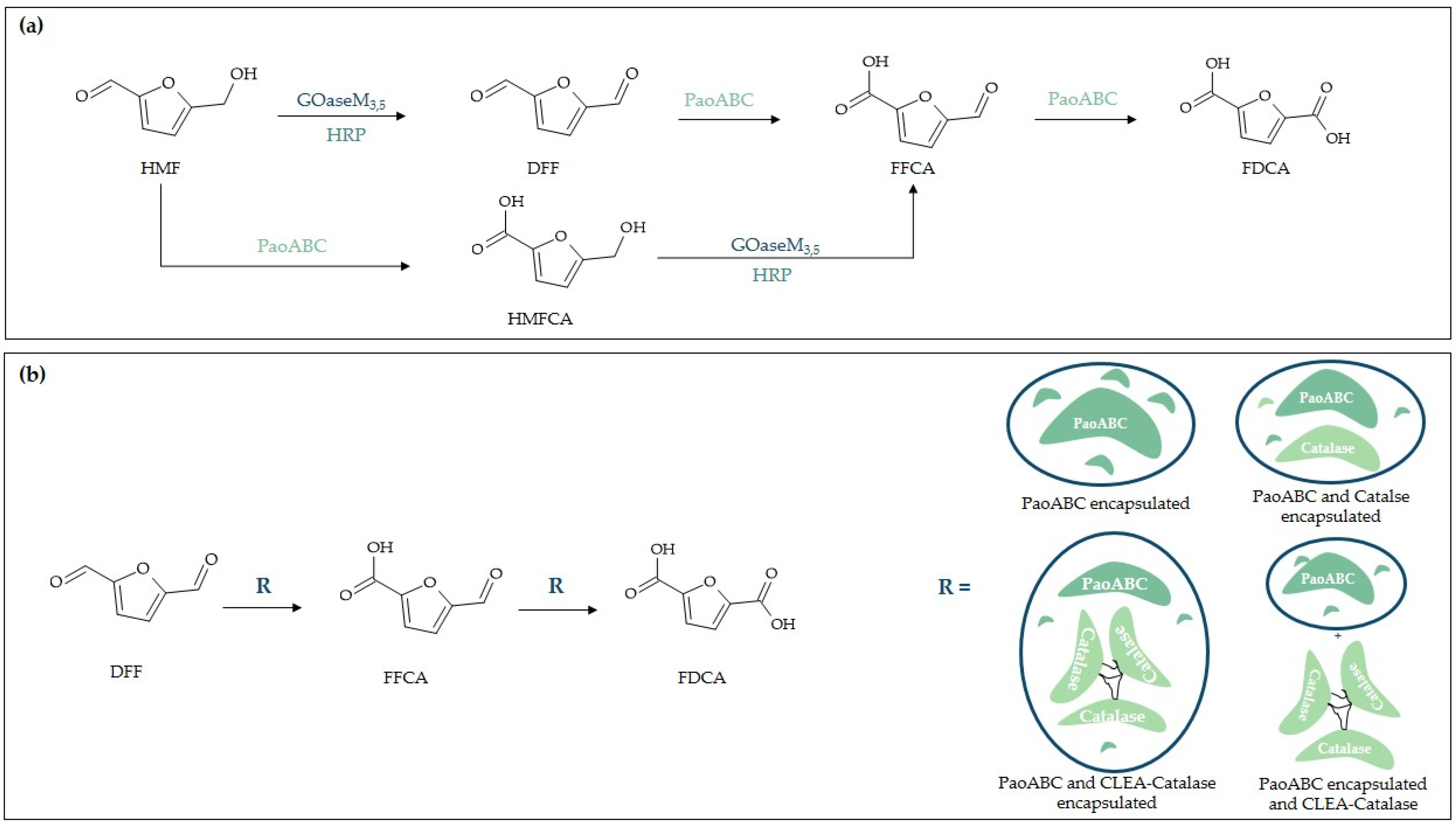
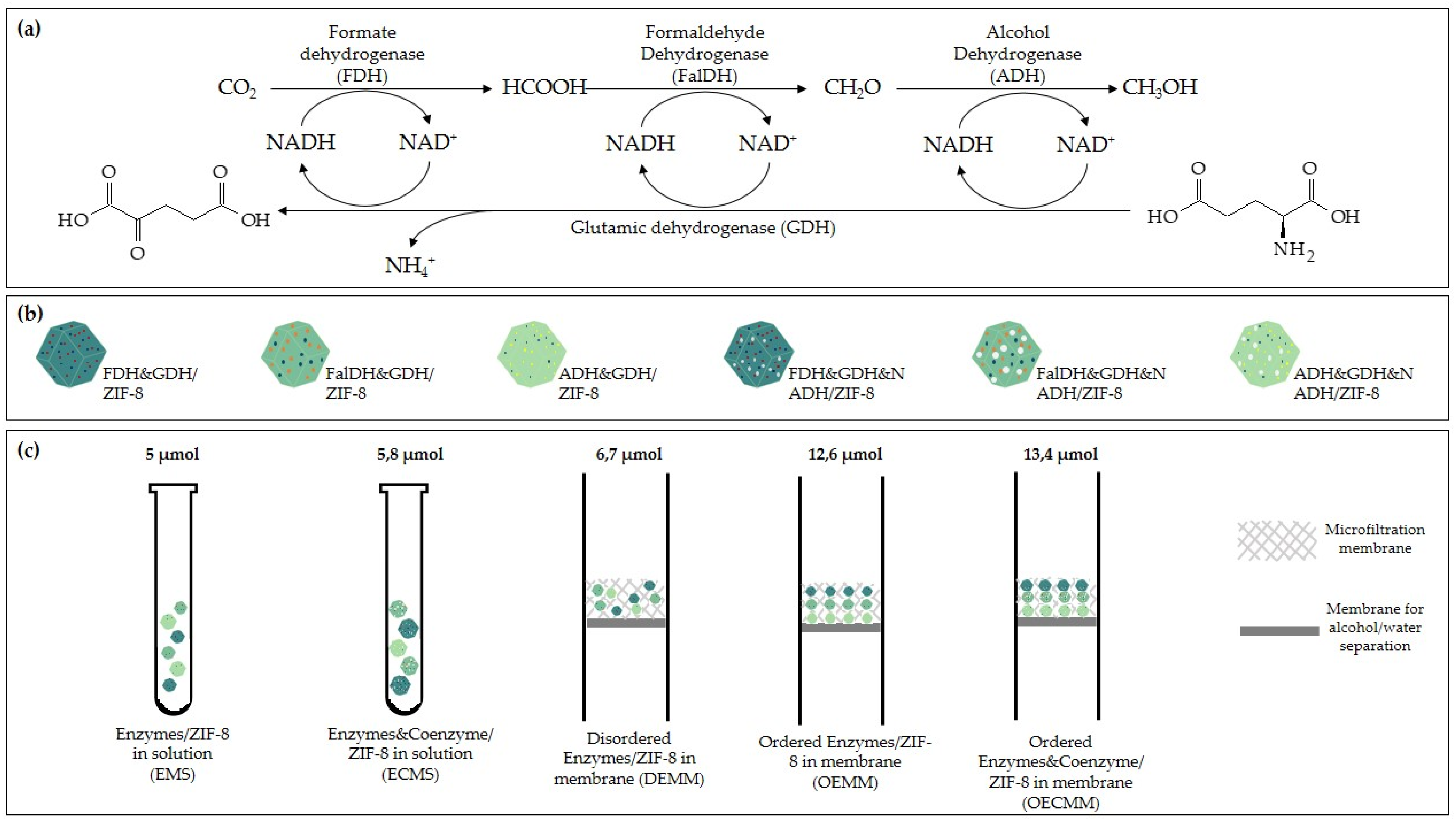
2.3.2. Metal–Organic Framework (MOF) Encapsulation
2.3.3. Nanofibers Encapsulation
2.3.4. Polymersomes
3. Co-Immobilization of Enzymes and Non-Enzymatic Catalysts
3.1. Co-Immobilization of Enzymes and Metals in Siliceous Materials
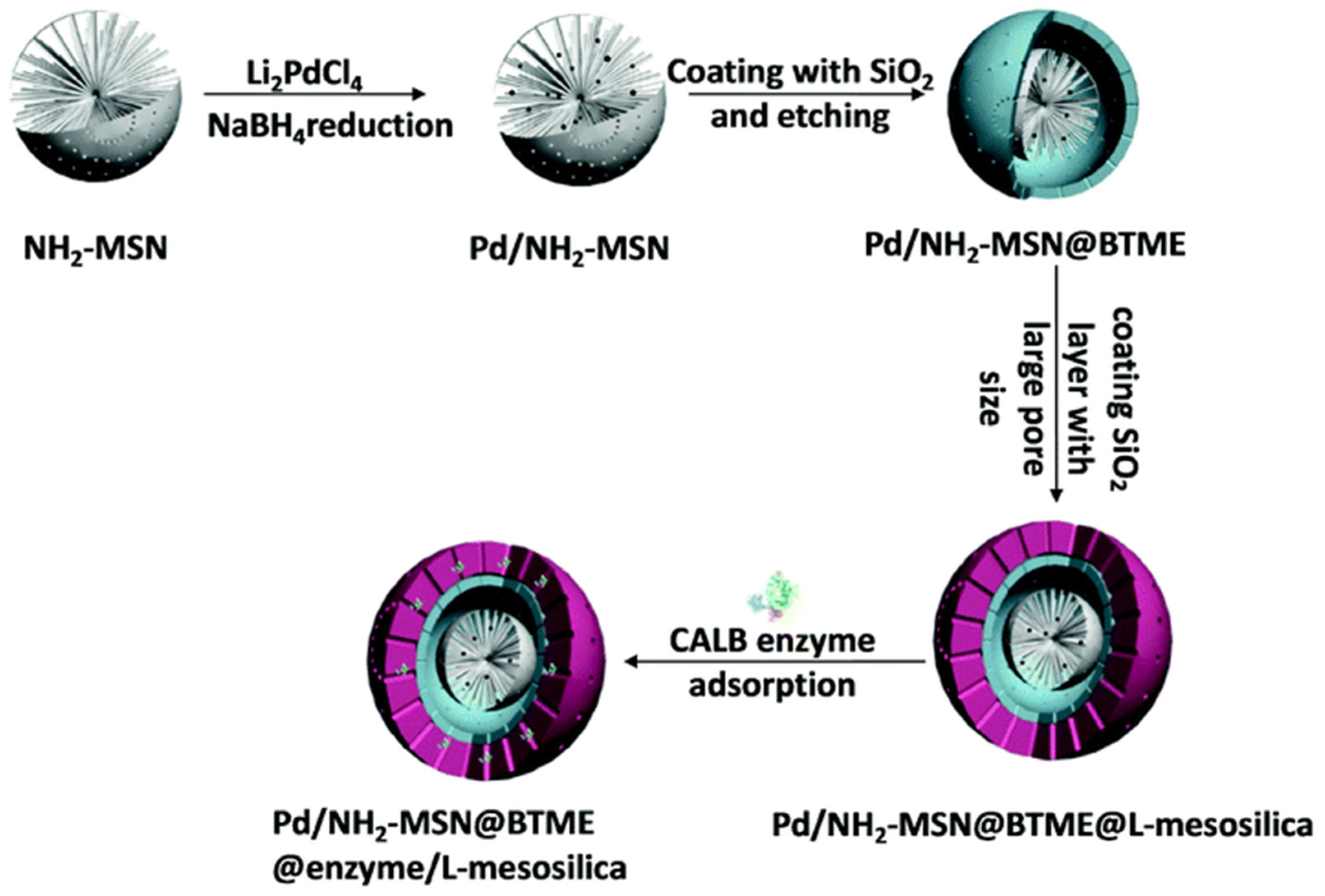
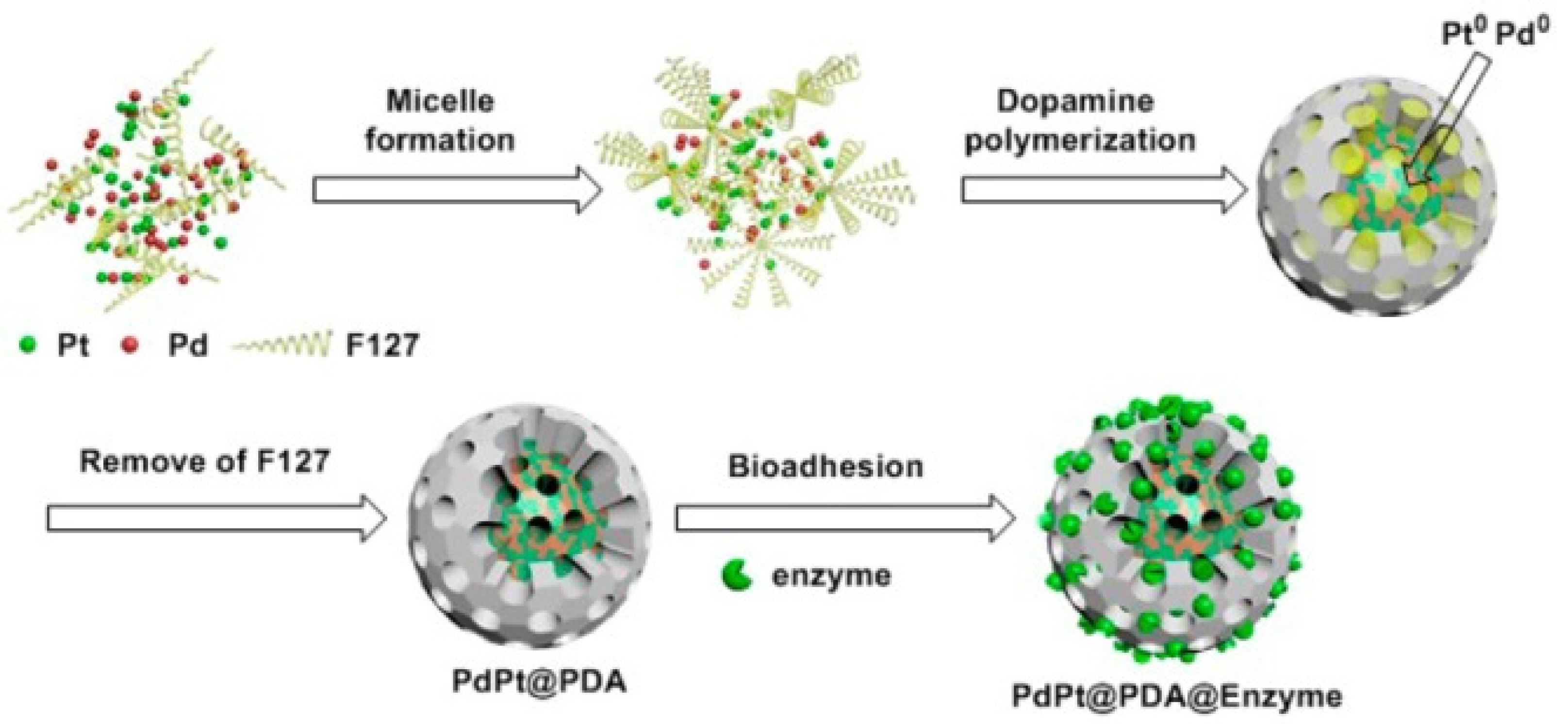
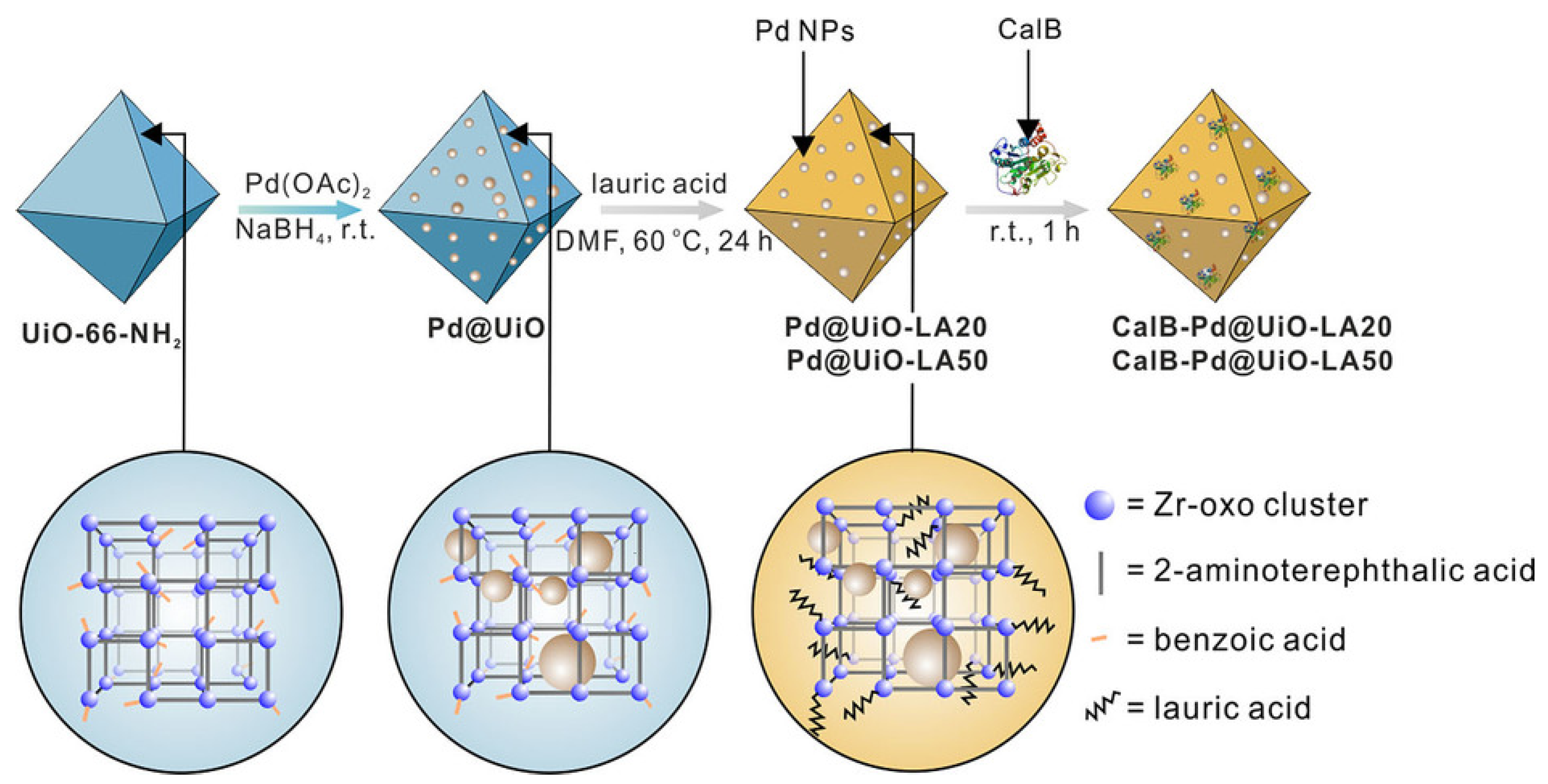
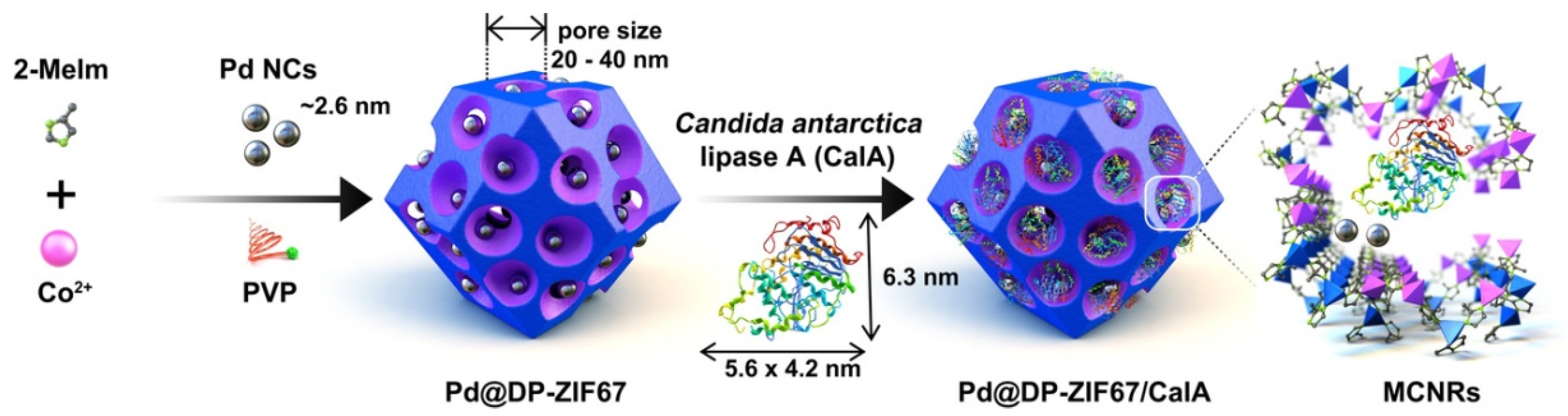
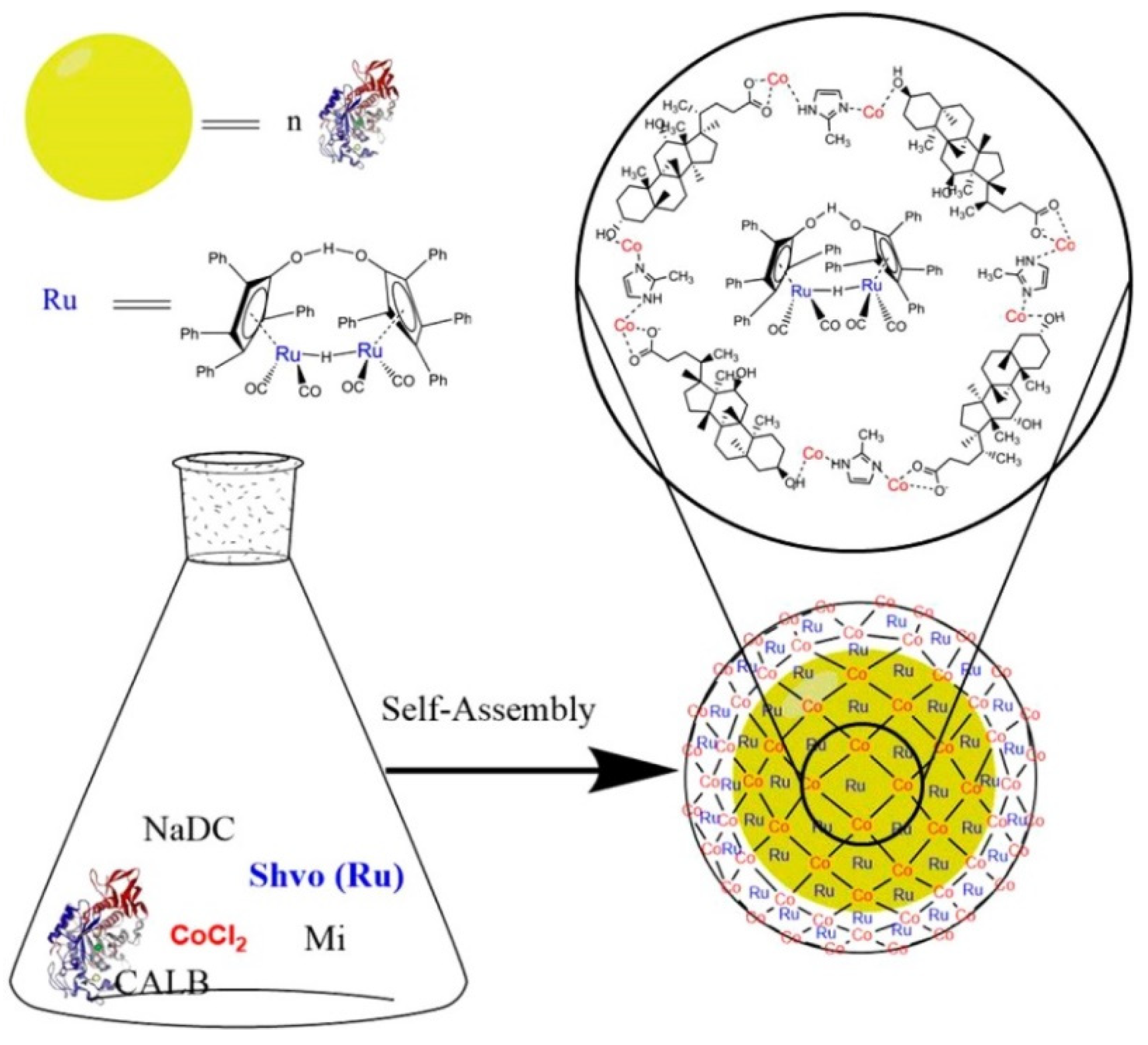
3.2. Co-Immobilization of Enzymes and Metals in Non-Siliceous Materials
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ni, Y.; Holtmann, D.; Hollmann, F. How Green is Biocatalysis? To Calculate is To Know. ChemCatChem 2014, 6, 930–943. [Google Scholar] [CrossRef]
- Turner, N.J.; O’Reilly, E. Biocatalytic retrosynthesis. Nat. Chem. Biol. 2013, 9, 285–288. [Google Scholar] [CrossRef]
- De Souza, R.O.M.A.; Miranda, L.S.M.; Bornscheuer, U.T. A Retrosynthesis Approach for Biocatalysis in Organic Synthesis. Chem. A Eur. J. 2017, 23, 12040–12063. [Google Scholar] [CrossRef] [Green Version]
- Sheldon, R.A.; Brady, D. The limits to biocatalysis: Pushing the envelope. Chem. Commun. 2018, 54, 6088–6104. [Google Scholar] [CrossRef]
- Bornscheuer, U.T.; Huisman, G.W.; Kazlauskas, R.J.; Lutz, S.; Moore, J.C.; Robins, K. Engineering the third wave of biocatalysis. Nature 2012, 485, 185–194. [Google Scholar] [CrossRef]
- Costa, I.C.R.; de Souza, R.O.M.A.; Bornscheuer, U.T. Asymmetric synthesis of serinol-monoesters catalyzed by amine transaminases. Tetrahedron Asymmetry 2017, 28, 1183–1187. [Google Scholar] [CrossRef]
- Woodley, J.M. Accelerating the implementation of biocatalysis in industry. Appl. Microbiol. Biotechnol. 2019, 103, 4733–4739. [Google Scholar] [CrossRef]
- Sheldon, R.A. Enzyme Immobilization: The Quest for Optimum Performance. Adv. Synth. Catal. 2007, 349, 1289–1307. [Google Scholar] [CrossRef]
- Sheldon, R.A.; van Pelt, S. Enzyme immobilisation in biocatalysis: Why, what and how. Chem. Soc. Rev. 2013, 42, 6223–6235. [Google Scholar] [CrossRef] [Green Version]
- Schrittwieser, J.H.; Velikogne, S.; Hall, M.; Kroutil, W. Artificial Biocatalytic Linear Cascades for Preparation of Organic Molecules. Chem. Rev. 2018, 118, 270–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, L.; van Langen, L.; Sheldon, R.A. Immobilised enzymes: Carrier-bound or carrier-free? Curr. Opin. Biotechnol. 2003, 14, 387–394. [Google Scholar] [CrossRef]
- Schoffelen, S.; van Hest, J.C.M. Chemical approaches for the construction of multi-enzyme reaction systems. Curr. Opin. Struct. Biol. 2013, 23, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Losada-Garcia, N.; Cabrera, Z.; Urrutia, P.; Garcia-Sanz, C.; Andreu, A.; Palomo, J.M. Recent Advances in Enzymatic and Chemoenzymatic Cascade Processes. Catalysts 2020, 10, 1258. [Google Scholar] [CrossRef]
- Ricca, E.; Brucher, B.; Schrittwieser, J.H. Multi-enzymatic cascade reactions: Overview and perspectives. Adv. Synth. Catal. 2011, 353, 2239–2262. [Google Scholar] [CrossRef]
- Schmid-Dannert, C.; López-Gallego, F. Advances and opportunities for the design of self-sufficient and spatially organized cell-free biocatalytic systems. Curr. Opin. Chem. Biol. 2019, 49, 97–104. [Google Scholar] [CrossRef]
- Ren, S.; Li, C.; Jiao, X.; Jia, S.; Jiang, Y.; Bilal, M.; Cui, J. Recent progress in multienzymes co-immobilization and multienzyme system applications. Chem. Eng. J. 2019, 373, 1254–1278. [Google Scholar] [CrossRef]
- France, S.P.; Hepworth, L.J.; Turner, N.J.; Flitsch, S.L. Constructing Biocatalytic Cascades: In Vitro and in Vivo Approaches to de Novo Multi-Enzyme Pathways. ACS Catal. 2017, 7, 710–724. [Google Scholar] [CrossRef]
- Zhao, F.; Masci, D.; Tomarelli, E.; Castagnolo, D. Biocatalytic and Chemo-Enzymatic Approaches for the Synthesis of Heterocycles. Synthesis 2020, 52, 2948–2961. [Google Scholar] [CrossRef]
- Denard, C.A.; Hartwig, J.F.; Zhao, H. Multistep One-Pot Reactions Combining Biocatalysts and Chemical Catalysts for Asymmetric Synthesis. ACS Catal. 2013, 3, 2856–2864. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, P.; Gao, S.; Wang, Z.; Luan, P.; González-Sabín, J.; Jiang, Y. Construction of chemoenzymatic cascade reactions for bridging chemocatalysis and Biocatalysis: Principles, strategies and prospective. Chem. Eng. J. 2020, 420, 127659. [Google Scholar] [CrossRef]
- Kourist, R.; González-Sabín, J. Non-Conventional Media as Strategy to Overcome the Solvent Dilemma in Chemoenzymatic Tandem Catalysis. ChemCatChem 2020, 12, 1903–1912. [Google Scholar] [CrossRef]
- Schmidt, S.; Castiglione, K.; Kourist, R. Overcoming the Incompatibility Challenge in Chemoenzymatic and Multi-Catalytic Cascade Reactions. Chem. A Eur. J. 2018, 24, 1755–1768. [Google Scholar] [CrossRef]
- Heuson, E.; Dumeignil, F. The various levels of integration of chemo- and bio-catalysis towards hybrid catalysis. Catal. Sci. Technol. 2020, 10, 7082–7100. [Google Scholar] [CrossRef]
- Li, X.; Cao, X.; Xiong, J.; Ge, J. Enzyme–Metal Hybrid Catalysts for Chemoenzymatic Reactions. Small 2020, 16, 1902751. [Google Scholar] [CrossRef]
- Debecker, D.P.; Smeets, V.; Van der Verren, M.; Meersseman, A.H.; Kinnaer, M.; Devred, F. Hybrid chemoenzymatic heterogeneous catalysts. Curr. Opin. Green Sustain. Chem. 2021, 28, 100437. [Google Scholar] [CrossRef]
- Hwang, E.T.; Lee, S. Multienzymatic Cascade Reactions via Enzyme Complex by Immobilization. ACS Catal. 2019, 9, 4402–4425. [Google Scholar] [CrossRef]
- Hirata, D.B.; Albuquerque, T.L.; Rueda, N.; Virgen-Ortíz, J.J.; Tacias-Pascacio, V.G.; Fernandez-Lafuente, R. Evaluation of different immobilized lipases in transesterification reactions using tributyrin: Advantages of the heterofunctional octyl agarose beads. J. Mol. Catal. B Enzym. 2016, 133, 117–123. [Google Scholar] [CrossRef]
- Kuo, C.-H.; Shieh, C.-J. Biocatalytic Process Optimization. Catalysts 2020, 10, 1303. [Google Scholar] [CrossRef]
- Cantone, S.; Ferrario, V.; Corici, L.; Ebert, C.; Fattor, D.; Spizzo, P.; Gardossi, L. Efficient immobilisation of industrial biocatalysts: Criteria and constraints for the selection of organic polymeric carriers and immobilisation methods. Chem. Soc. Rev. 2013, 42, 6262–6276. [Google Scholar] [CrossRef] [Green Version]
- Gomes-Ruffi, C.R.; da Cunha, R.H.; Almeida, E.L.; Chang, Y.K.; Steel, C.J. Effect of the emulsifier sodium stearoyl lactylate and of the enzyme maltogenic amylase on the quality of pan bread during storage. LWT 2012, 49, 96–101. [Google Scholar] [CrossRef] [Green Version]
- Homaei, A.A.; Sariri, R.; Vianello, F.; Stevanato, R. Enzyme immobilization: An update. J. Chem. Biol. 2013, 6, 185–205. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, M. Ordered Mesoporous Materials for Bioadsorption and Biocatalysis. Chem. Mater. 2005, 17, 4577–4593. [Google Scholar] [CrossRef]
- Kallenberg, A.I.; van Rantwijk, F.; Sheldon, R.A. Immobilization of Penicillin G Acylase: The Key to Optimum Performance. Adv. Synth. Catal. 2005, 347, 905–926. [Google Scholar] [CrossRef]
- Pierre, A.C. The sol-gel encapsulation of enzymes. Biocatal. Biotransform. 2004, 22, 145–170. [Google Scholar] [CrossRef]
- Gutarra, M.L.E.; Miranda, L.S.M.; de Souza, R.O.M.A. Enzyme Immobilization for Organic Synthesis. In Organic Synthesis Using Biocatalysis; Elsevier: Amsterdam, The Netherlands, 2016; pp. 99–126. [Google Scholar]
- Garcia-Galan, C.; Berenguer-Murcia, Á.; Fernandez-Lafuente, R.; Rodrigues, R.C. Potential of Different Enzyme Immobilization Strategies to Improve Enzyme Performance. Adv. Synth. Catal. 2011, 353, 2885–2904. [Google Scholar] [CrossRef]
- Tran, D.N.; Balkus, K.J. Perspective of Recent Progress in Immobilization of Enzymes. ACS Catal. 2011, 1, 956–968. [Google Scholar] [CrossRef]
- Oliveira, F.L.; França, S.A.; Castro, A.M.; Alves de Souza, R.O.M.; Esteves, P.M.; Gonçalves, R.S.B. Enzyme Immobilization in Covalent Organic Frameworks: Strategies and Applications in Biocatalysis. Chempluschem 2020, 85, 2051–2066. [Google Scholar] [CrossRef]
- Scherer, R.P.; Dallago, R.L.; Penna, F.G.; Bertella, F.; de Oliveira, D.; de Oliveira, J.V.; Pergher, S.B.C. Influence of process parameters on the immobilization of commercial porcine pancreatic lipase using three low-cost supports. Biocatal. Agric. Biotechnol. 2012, 1, 290–294. [Google Scholar] [CrossRef]
- Gérardin, C.; Reboul, J.; Bonne, M.; Lebeau, B. Ecodesign of ordered mesoporous silica materials. Chem. Soc. Rev. 2013, 42, 4217–4255. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.F.; Li, Y.T.; Yang, V.C. Biomedical Application of Immobilized Enzymes. J. Pharm. Sci. 2000, 89, 979–990. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, S.F. Immobilization and Stabilization of Biomaterials for Biosensor Applications. Appl. Biochem. Biotechnol. 2001, 96, 225–238. [Google Scholar] [CrossRef]
- Wu, L.-C.; Cheng, C.-M. Flow-injection enzymatic analysis for glycerol and triacylglycerol. Anal. Biochem. 2005, 346, 234–240. [Google Scholar] [CrossRef]
- Arroyo, M.; de la Mata, I.; Acebal, C.; Pilar Castillón, M. Biotechnological applications of penicillin acylases: State-of-the-art. Appl. Microbiol. Biotechnol. 2003, 60, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Girelli, A.M.; Mattei, E. Application of immobilized enzyme reactor in on-line high performance liquid chromatography: A review. J. Chromatogr. B 2005, 819, 3–16. [Google Scholar] [CrossRef]
- Taylor, R.F. Protein Immobilization; Dekker, M., Ed.; Taylor & Francis: New York, NY, USA, 1991. [Google Scholar]
- Rao, S.V.; Anderson, K.W.; Bachas, L.G. Oriented immobilization of proteins. Mikrochim. Acta 1998, 128, 127–143. [Google Scholar] [CrossRef]
- Wilchek, M.; Miron, T. Oriented versus random protein immobilization. J. Biochem. Biophys. Methods 2003, 55, 67–70. [Google Scholar] [CrossRef]
- Turková, J. Oriented immobilization of biologically active proteins as a tool for revealing protein interactions and function. J. Chromatogr. B Biomed. Sci. Appl. 1999, 722, 11–31. [Google Scholar] [CrossRef]
- Wilchek, M.; Miron, T.; Kohn, J.B.T.-M. in E. Affinity chromatography. In Part C: Enzyme Purification and Related Techniques; Academic Press: Cambridge, MA, USA, 1984; Volume 104, pp. 3–55. ISBN 0076-6879. [Google Scholar]
- Carvalho, N.B.; Lima, Á.S.; Soares, C.M.F. Use of Modified Silicas for Lipase Immobilization. Quim. Nova 2014, 38, 399–409. [Google Scholar] [CrossRef]
- Sun, Q.; Fu, C.-W.; Aguila, B.; Perman, J.; Wang, S.; Huang, H.-Y.; Xiao, F.-S.; Ma, S. Pore Environment Control and Enhanced Performance of Enzymes Infiltrated in Covalent Organic Frameworks. J. Am. Chem. Soc. 2018, 140, 984–992. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; He, Q.; Shao, Q.; Zuo, Y.; Wang, F.; Ni, H. Preparation and Characterization of Monodispersed Microfloccules of TiO2 Nanoparticles with Immobilized Multienzymes. ACS Appl. Mater. Interfaces 2011, 3, 3300–3307. [Google Scholar] [CrossRef]
- Le, M.; Means, G.E. NAD+/NADH recycling by coimmobilized lactate dehydrogenase and glutamate dehydrogenase. Enzyme Microb. Technol. 1998, 23, 49–57. [Google Scholar] [CrossRef]
- Liu, X.; Du, X.; Feng, J.; Wu, M.-B.; Lin, J.; Guan, J.; Wang, T.; Zhang, Z. Co-immobilization of Short-Chain Dehydrogenase/Reductase and Glucose Dehydrogenase for the Efficient Production of (±)-Ethyl Mandelate. Catal. Lett. 2019, 149, 1710–1720. [Google Scholar] [CrossRef]
- Bachosz, K.; Synoradzki, K.; Staszak, M.; Pinelo, M.; Meyer, A.S.; Zdarta, J.; Jesionowski, T. Bioconversion of xylose to xylonic acid via co-immobilized dehydrogenases for conjunct cofactor regeneration. Bioorg. Chem. 2019, 93, 102747. [Google Scholar] [CrossRef]
- Bachosz, K.; Zdarta, J.; Marczak, Ł.; Błażewicz, J.; Jesionowski, T. A highly effective approach to cofactor regeneration and subsequent membrane separation of bioconversion products: Kinetic parameters and effect of process conditions. Bioresour. Technol. Rep. 2020, 9, 100365. [Google Scholar] [CrossRef]
- Luo, J.; Meyer, A.S.; Mateiu, R.V.; Pinelo, M. Cascade catalysis in membranes with enzyme immobilization for multi-enzymatic conversion of CO2 to methanol. N. Biotechnol. 2015, 32, 319–327. [Google Scholar] [CrossRef]
- Zhong, C.; Duić, B.; Bolivar, J.M.; Nidetzky, B. Three-Enzyme Phosphorylase Cascade Immobilized on Solid Support for Biocatalytic Synthesis of Cello−oligosaccharides. ChemCatChem 2020, 12, 1350–1358. [Google Scholar] [CrossRef]
- Valikhani, D.; Bolivar, J.M.; Dennig, A.; Nidetzky, B. A tailor-made, self-sufficient and recyclable monooxygenase catalyst based on coimmobilized cytochrome P450 BM3 and glucose dehydrogenase. Biotechnol. Bioeng. 2018, 115, 2416–2425. [Google Scholar] [CrossRef]
- Rocha-Martín, J.; de las Rivas, B.; Muñoz, R.; Guisán, J.M.; López-Gallego, F. Rational Co-Immobilization of Bi-Enzyme Cascades on Porous Supports and their Applications in Bio-Redox Reactions with In Situ Recycling of Soluble Cofactors. ChemCatChem 2012, 4, 1279–1288. [Google Scholar] [CrossRef]
- Velasco-Lozano, S.; Santiago-Arcos, J.; Mayoral, J.A.; López-Gallego, F. Co-immobilization and Colocalization of Multi-Enzyme Systems for the Cell-Free Biosynthesis of Aminoalcohols. ChemCatChem 2020, 12, 3030–3041. [Google Scholar] [CrossRef]
- Peirce, S.; Virgen-Ortíz, J.J.; Tacias-Pascacio, V.G.; Rueda, N.; Bartolome-Cabrero, R.; Fernandez-Lopez, L.; Russo, M.E.; Marzocchella, A.; Fernandez-Lafuente, R. Development of simple protocols to solve the problems of enzyme coimmobilization. Application to coimmobilize a lipase and a β-galactosidase. RSC Adv. 2016, 6, 61707–61715. [Google Scholar] [CrossRef]
- Mallin, H.; Wulf, H.; Bornscheuer, U.T. A self-sufficient Baeyer–Villiger biocatalysis system for the synthesis of ɛ-caprolactone from cyclohexanol. Enzyme Microb. Technol. 2013, 53, 283–287. [Google Scholar] [CrossRef]
- Delgove, M.A.F.; Valencia, D.; Solé, J.; Bernaerts, K.V.; De Wildeman, S.M.A.; Guillén, M.; Álvaro, G. High performing immobilized Baeyer-Villiger monooxygenase and glucose dehydrogenase for the synthesis of ε-caprolactone derivative. Appl. Catal. A Gen. 2019, 572, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Solé, J.; Caminal, G.; Schürmann, M.; Álvaro, G.; Guillén, M. Co-immobilization of P450 BM3 and glucose dehydrogenase on different supports for application as a self-sufficient oxidative biocatalyst. J. Chem. Technol. Biotechnol. 2019, 94, 244–255. [Google Scholar] [CrossRef]
- Giannakopoulou, A.; Patila, M.; Spyrou, K.; Chalmpes, N.; Zarafeta, D.; Skretas, G.; Gournis, D.; Stamatis, H. Development of a Four-Enzyme Magnetic Nanobiocatalyst for Multi-Step Cascade Reactions. Catalysts 2019, 9, 995. [Google Scholar] [CrossRef] [Green Version]
- Torabizadeh, H.; Montazeri, E. Nano co-immobilization of α-amylase and maltogenic amylase by nanomagnetic combi-cross-linked enzyme aggregates method for maltose production from corn starch. Carbohydr. Res. 2020, 488, 107904. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, S.; Landarani–Isfahani, A.; Moghadam, M.; Tangestaninejad, S.; Mirkhani, V.; Mohammadpoor-Baltork, I. Development of a novel bi-enzymatic silver dendritic hierarchical nanostructure cascade catalytic system for efficient conversion of starch into gluconic acid. Chem. Eng. J. 2019, 356, 423–435. [Google Scholar] [CrossRef]
- Chen, R.; Wei, Q.; Wei, X.; Liu, Y.; Zhang, X.; Chen, X.; Yin, X.; Xie, T. Stable and efficient immobilization of bi-enzymatic NADPH cofactor recycling system under consecutive microwave irradiation. PLoS ONE 2020, 15, e0242564. [Google Scholar] [CrossRef]
- Kirupa, S.M.; Ravikumar, R.; Naresh, K.M.; Sivakumar, U. Development of co-immobilized tri-enzyme biocatalytic system for one-pot pretreatment of four different perennial lignocellulosic biomass and evaluation of their bioethanol production potential. Bioresour. Technol. 2018, 269, 227–236. [Google Scholar] [CrossRef]
- El-Zahab, B.; Jia, H.; Wang, P. Enabling multienzyme biocatalysis using nanoporous materials. Biotechnol. Bioeng. 2004, 87, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Küchler, A.; Adamcik, J.; Mezzenga, R.; Schlüter, A.D.; Walde, P. Enzyme immobilization on silicate glass through simple adsorption of dendronized polymer–enzyme conjugates for localized enzymatic cascade reactions. RSC Adv. 2015, 5, 44530–44544. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, Y.; Yuan, Q.; Liang, H. Preparation of multi-enzyme co-immobilized nanoparticles by bis-aryl hydrazone bond conjugation. Biotechnol. Appl. Biochem. 2016, 63, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Ou, X.-Y.; Guo, Z.-W.; Zeng, Y.-J.; Zong, M.-H.; Lou, W.-Y. Co-immobilization of multiple enzymes by self-assembly and chemical crosslinking for cofactor regeneration and robust biocatalysis. Int. J. Biol. Macromol. 2020, 162, 445–453. [Google Scholar] [CrossRef]
- Tan, C.Y.; Hirakawa, H.; Suzuki, R.; Haga, T.; Iwata, F.; Nagamune, T. Immobilization of a Bacterial Cytochrome P450 Monooxygenase System on a Solid Support. Angew. Chemie Int. Ed. 2016, 55, 15002–15006. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yong, Y.; Ge, J.; Liu, Z. Lectin Agglutinated Multienzyme Catalyst with Enhanced Substrate Affinity and Activity. ACS Catal. 2016, 6, 3789–3795. [Google Scholar] [CrossRef]
- Hu, X.; Liu, L.; Chen, D.; Wang, Y.; Zhang, J.; Shao, L. Co-expression of the recombined alcohol dehydrogenase and glucose dehydrogenase and cross-linked enzyme aggregates stabilization. Bioresour. Technol. 2017, 224, 531–535. [Google Scholar] [CrossRef]
- Aranaz, I.; Acosta, N.; Férnandez-Valle, M.E.; Heras, A. Optimization of d-amino acid production catalyzed by immobilized multi-enzyme system in polyelectrolyte complex gel capsules. J. Mol. Catal. B Enzym. 2015, 121, 45–52. [Google Scholar] [CrossRef]
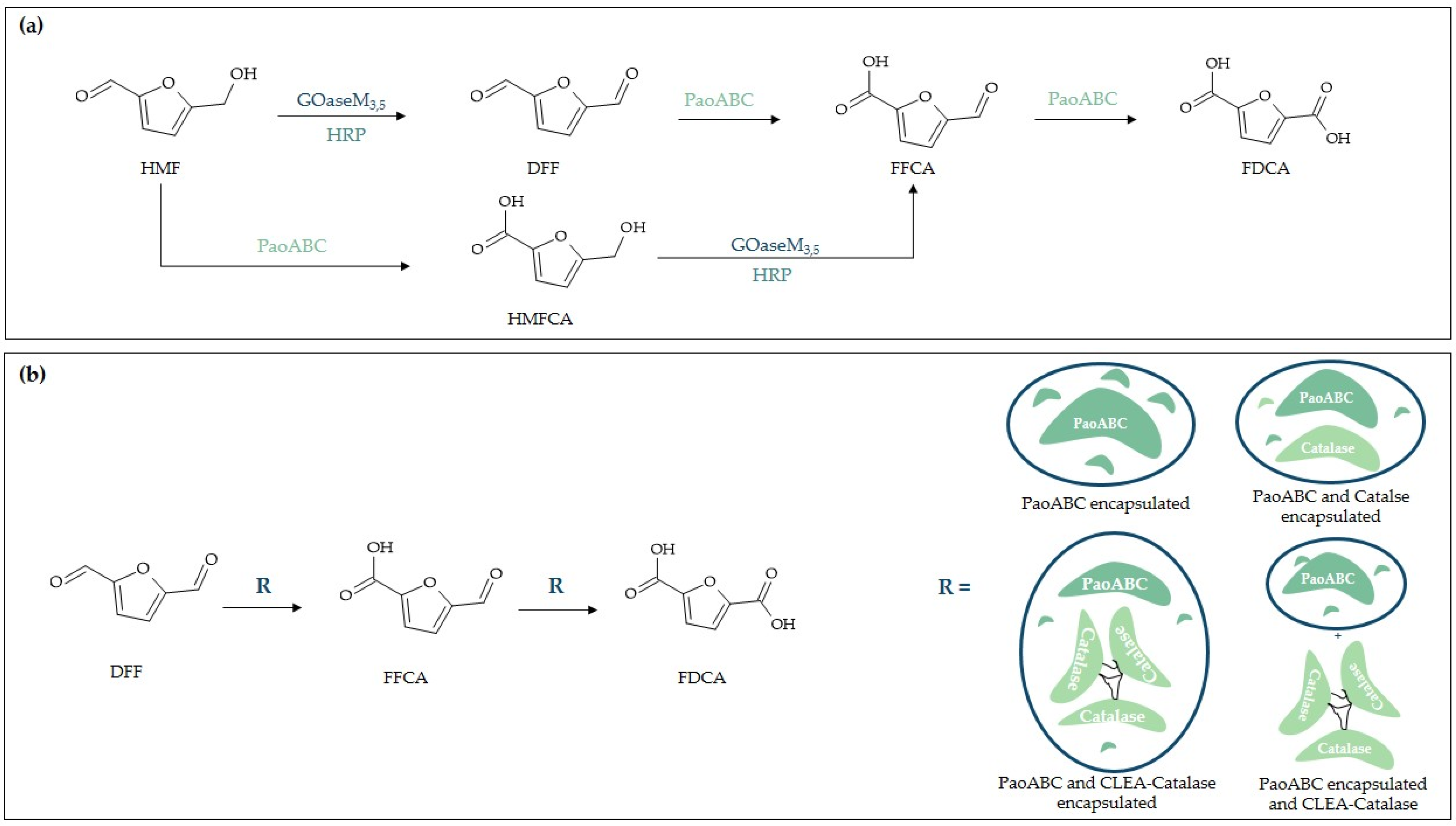
- McKenna, S.M.; Mines, P.; Law, P.; Kovacs-Schreiner, K.; Birmingham, W.R.; Turner, N.J.; Leimkühler, S.; Carnell, A.J. The continuous oxidation of HMF to FDCA and the immobilisation and stabilisation of periplasmic aldehyde oxidase (PaoABC). Green Chem. 2017, 19, 4660–4665. [Google Scholar] [CrossRef]
- Long, J.; Pan, T.; Xie, Z.; Xu, X.; Jin, Z. Effective production of lactosucrose using β-fructofuranosidase and glucose oxidase co-immobilized by sol–gel encapsulation. Food Sci. Nutr. 2019, 7, 3302–3316. [Google Scholar] [CrossRef]
- Long, J.; Pan, T.; Xie, Z.; Xu, X.; Jin, Z. Co-immobilization of β-fructofuranosidase and glucose oxidase improves the stability of Bi-enzymes and the production of lactosucrose. LWT 2020, 128, 109460. [Google Scholar] [CrossRef]
- Chen, W.-H.; Vázquez-González, M.; Zoabi, A.; Abu-Reziq, R.; Willner, I. Biocatalytic cascades driven by enzymes encapsulated in metal–organic framework nanoparticles. Nat. Catal. 2018, 1, 689–695. [Google Scholar] [CrossRef]
- Zhu, D.; Ao, S.; Deng, H.; Wang, M.; Qin, C.; Zhang, J.; Jia, Y.; Ye, P.; Ni, H. Ordered Coimmobilization of a Multienzyme Cascade System with a Metal Organic Framework in a Membrane: Reduction of CO2 to Methanol. ACS Appl. Mater. Interfaces 2019, 11, 33581–33588. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Wang, Z.; Bilal, M.; Feng, Y.; Jiang, Y.; Jia, S.; Cui, J. Co-immobilization multienzyme nanoreactor with co-factor regeneration for conversion of CO2. Int. J. Biol. Macromol. 2020, 155, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Su, Z.; Wang, P.; Ma, G.; Zhang, S. Tethering of Nicotinamide Adenine Dinucleotide Inside Hollow Nanofibers for High-Yield Synthesis of Methanol from Carbon Dioxide Catalyzed by Coencapsulated Multienzymes. ACS Nano 2015, 9, 4600–4610. [Google Scholar] [CrossRef] [PubMed]
- Klermund, L.; Poschenrieder, S.T.; Castiglione, K. Biocatalysis in Polymersomes: Improving Multienzyme Cascades with Incompatible Reaction Steps by Compartmentalization. ACS Catal. 2017, 7, 3900–3904. [Google Scholar] [CrossRef]
- Jesionowski, T.; Zdarta, J.; Krajewska, B. Enzyme immobilization by adsorption: A review. Adsorption 2014, 20, 801–821. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, M.; Kostrov, X. Immobilization of enzymes on porous silicas—Benefits and challenges. Chem. Soc. Rev. 2013, 42, 6277–6289. [Google Scholar] [CrossRef]
- Mokhtar, N.F.; Rahman, R.N.Z.R.A.; Muhd, N.N.D.; Mohd, S.F.; Mohamad, A.M.S. The Immobilization of Lipases on Porous Support by Adsorption and Hydrophobic Interaction Method. Catalysts 2020, 10, 744. [Google Scholar] [CrossRef]
- Amirnejat, S.; Movahedi, F.; Masrouri, H.; Mohadesi, M.; Kassaee, M.Z. Silica nanoparticles immobilized benzoylthiourea ferrous complex as an efficient and reusable catalyst for one-pot synthesis of benzopyranopyrimidines. J. Mol. Catal. A Chem. 2013, 378, 135–141. [Google Scholar] [CrossRef]
- Basri, M.; Kassim, M.A.; Mohamad, R.; Ariff, A.B. Optimization and kinetic study on the synthesis of palm oil ester using Lipozyme TL IM. J. Mol. Catal. B Enzym. 2013, 85–86, 214–219. [Google Scholar] [CrossRef] [Green Version]
- Beck, J.S.; Vartuli, J.C.; Roth, W.J.; Leonowicz, M.E.; Kresge, C.T.; Schmitt, K.D.; Chu, C.T.W.; Olson, D.H.; Sheppard, E.W.; McCullen, S.B.; et al. A new family of mesoporous molecular sieves prepared with liquid crystal templates. J. Am. Chem. Soc. 1992, 114, 10834–10843. [Google Scholar] [CrossRef]
- Jaros, D.; Rohm, H. Enzymes Exogenous to Milk in Dairy Technology | Transglutaminase. Encycl. Dairy Sci. 2011, 297–300. [Google Scholar] [CrossRef]
- Xu, Y.; Zhou, G.; Wu, C.; Li, T.; Song, H. Improving adsorption and activation of the lipase immobilized in amino-functionalized ordered mesoporous SBA-15. Solid State Sci. 2011, 13, 867–874. [Google Scholar] [CrossRef]
- Yang, J.; Hu, Y.; Jiang, L.; Zou, B.; Jia, R.; Huang, H. Enhancing the catalytic properties of porcine pancreatic lipase by immobilization on SBA-15 modified by functionalized ionic liquid. Biochem. Eng. J. 2013, 70, 46–54. [Google Scholar] [CrossRef]
- Zou, B.; Song, C.; Xu, X.; Xia, J.; Huo, S.; Cui, F. Enhancing stabilities of lipase by enzyme aggregate coating immobilized onto ionic liquid modified mesoporous materials. Appl. Surf. Sci. 2014, 311, 62–67. [Google Scholar] [CrossRef]
- Ramani, K.; Karthikeyan, S.; Boopathy, R.; Kennedy, L.J.; Mandal, A.B.; Sekaran, G. Surface functionalized mesoporous activated carbon for the immobilization of acidic lipase and their application to hydrolysis of waste cooked oil: Isotherm and kinetic studies. Process Biochem. 2012, 47, 435–445. [Google Scholar] [CrossRef]
- Fernandez-Lafuente, R. Lipase from Thermomyces lanuginosus: Uses and prospects as an industrial biocatalyst. J. Mol. Catal. B Enzym. 2010, 62, 197–212. [Google Scholar] [CrossRef]
- Mozhaev, V.V.; Melik-nubarov, N.S.; Sergeeva, M.V.; Šikšnis, V.; Martinek, K. Strategy for Stabilizing Enzymes Part One: Increasing Stability of Enzymes via their Multi-Point Interaction with a Support. Biocatalysis 1990, 3, 179–187. [Google Scholar] [CrossRef]
- Agostinelli, E.; Belli, F.; Tempera, G.; Mura, A.; Floris, G.; Toniolo, L.; Vavasori, A.; Fabris, S.; Momo, F.; Stevanato, R. Polyketone polymer: A new support for direct enzyme immobilization. J. Biotechnol. 2007, 127, 670–678. [Google Scholar] [CrossRef]
- Orrego, C.E.; Salgado, N.; Valencia, J.S.; Giraldo, G.I.; Giraldo, O.H.; Cardona, C.A. Novel chitosan membranes as support for lipases immobilization: Characterization aspects. Carbohydr. Polym. 2010, 79, 9–16. [Google Scholar] [CrossRef]
- Thyparambil, A.A.; Wei, Y.; Latour, R.A. Experimental characterization of adsorbed protein orientation, conformation, and bioactivity. Biointerphases 2015, 10, 19002. [Google Scholar] [CrossRef] [Green Version]
- Gayet, J.-C.; Fortier, G. Drug Release from New Bioartificial Hydrogel. Artif. Cells Blood Substit. Biotechnol. 1995, 23, 605–611. [Google Scholar] [CrossRef]
- Cao, S.-L.; Huang, Y.-M.; Li, X.-H.; Xu, P.; Wu, H.; Li, N.; Lou, W.-Y.; Zong, M.-H. Preparation and Characterization of Immobilized Lipase from Pseudomonas Cepacia onto Magnetic Cellulose Nanocrystals. Sci. Rep. 2016, 6, 20420. [Google Scholar] [CrossRef] [Green Version]
- Souza, C.J.F.; Garcia-Rojas, E.E.; Favaro-Trindade, C.S. Lactase (β-galactosidase) immobilization by complex formation: Impact of biopolymers on enzyme activity. Food Hydrocoll. 2018, 83, 88–96. [Google Scholar] [CrossRef]
- Rodrigues, R.C.; Barbosa, O.; Ortiz, C.; Berenguer-Murcia, Á.; Torres, R.; Fernandez-Lafuente, R. Amination of enzymes to improve biocatalyst performance: Coupling genetic modification and physicochemical tools. RSC Adv. 2014, 4, 38350–38374. [Google Scholar] [CrossRef] [Green Version]
- Elias, N.; Chandren, S.; Razak, F.I.A.; Jamalis, J.; Widodo, N.; Wahab, R.A. Characterization, optimization and stability studies on Candida rugosa lipase supported on nanocellulose reinforced chitosan prepared from oil palm biomass. Int. J. Biol. Macromol. 2018, 114, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Pessela, B.C.C.; Fernández-Lafuente, R.; Fuentes, M.; Vián, A.; García, J.L.; Carrascosa, A.V.; Mateo, C.; Guisán, J.M. Reversible immobilization of a thermophilic β-galactosidase via ionic adsorption on PEI-coated Sepabeads. Enzyme Microb. Technol. 2003, 32, 369–374. [Google Scholar] [CrossRef]
- Fuentes, M.; Pessela, B.C.C.; Maquiese, J.V.; Ortiz, C.; Segura, R.L.; Palomo, J.M.; Abian, O.; Torres, R.; Mateo, C.; Fernández-Lafuente, R.; et al. Reversible and Strong Immobilization of Proteins by Ionic Exchange on Supports Coated with Sulfate-Dextran. Biotechnol. Prog. 2004, 20, 1134–1139. [Google Scholar] [CrossRef]
- Orrego, H.A.; Romero-Fernández, M.; Millán-Linares, M.; Yust, M.; Guisán, J.; Rocha-Martin, J. Stabilization of Enzymes by Multipoint Covalent Attachment on Aldehyde-Supports: 2-Picoline Borane as an Alternative Reducing Agent. Catalysts 2018, 8, 333. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, M.; Ashjari, M.; Garmroodi, M.; Yousefi, M.; Karkhane, A.A. The use of isocyanide-based multicomponent reaction for covalent immobilization of Rhizomucor miehei lipase on multiwall carbon nanotubes and graphene nanosheets. RSC Adv. 2016, 6, 72275–72285. [Google Scholar] [CrossRef]
- Li, X.; Wang, X.; Ye, G.; Xia, W.; Wang, X. Polystyrene-based diazonium salt as adhesive: A new approach for enzyme immobilization on polymeric supports. Polymer 2010, 51, 860–867. [Google Scholar] [CrossRef]
- Barbosa, O.; Ortiz, C.; Berenguer-Murcia, Á.; Torres, R.; Rodrigues, R.C.; Fernandez-Lafuente, R. Glutaraldehyde in bio-catalysts design: A useful crosslinker and a versatile tool in enzyme immobilization. RSC Adv. 2014, 4, 1583–1600. [Google Scholar] [CrossRef] [Green Version]
- Veronese, F.M.; Morpurgo, M. Bioconjugation in pharmaceutical chemistry. Farmaco 1999, 54, 497–516. [Google Scholar] [CrossRef]
- Mateo, C.; Abian, O.; Fernandez-Lafuente, R.; Guisan, J.M. Reversible enzyme immobilization via a very strong and nondistorting ionic adsorption on support-polyethylenimine composites. Biotechnol. Bioeng. 2000, 68, 98–105. [Google Scholar] [CrossRef]
- Rubentheren, V.; Ward, T.A.; Chee, C.Y.; Tang, C.K. Processing and analysis of chitosan nanocomposites reinforced with chitin whiskers and tannic acid as a crosslinker. Carbohydr. Polym. 2015, 115, 379–387. [Google Scholar] [CrossRef]
- Manan, F.M.A.; Attan, N.; Zakaria, Z.; Keyon, A.S.A.; Wahab, R.A. Enzymatic esterification of eugenol and benzoic acid by a novel chitosan-chitin nanowhiskers supported Rhizomucor miehei lipase: Process optimization and kinetic assessments. Enzyme Microb. Technol. 2018, 108, 42–52. [Google Scholar] [CrossRef]
- Elias, N.; Chandren, S.; Attan, N.; Mahat, N.A.; Razak, F.I.A.; Jamalis, J.; Wahab, R.A. Structure and properties of oil palm-based nanocellulose reinforced chitosan nanocomposite for efficient synthesis of butyl butyrate. Carbohydr. Polym. 2017, 176, 281–292. [Google Scholar] [CrossRef]
- Van de Velde, F.; Lourenço, N.D.; Pinheiro, H.M.; Bakker, M. Carrageenan: A Food-Grade and Biocompatible Support for Immobilisation Techniques. Adv. Synth. Catal. 2002, 344, 815–835. [Google Scholar] [CrossRef]
- Jackson, E.; Correa, S.; Betancor, L. Cellulose-Based Nanosupports for Enzyme Immobilization. In Polymers and Polymeric Composites: A Reference Series; Springer: Cham, Switzerland, 2019; pp. 1235–1253. [Google Scholar]
- Lin, N.; Dufresne, A. Nanocellulose in biomedicine: Current status and future prospect. Eur. Polym. J. 2014, 59, 302–325. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.A.; Akhtar, S.; Husain, Q. Direct immobilization of polyphenol oxidases on Celite 545 from ammonium sulphate fractionated proteins of potato (Solanum tuberosum). J. Mol. Catal. B Enzym. 2006, 40, 58–63. [Google Scholar] [CrossRef]
- Sulaiman, S.; Mokhtar, M.N.; Naim, M.N.; Baharuddin, A.S.; Salleh, M.A.M.; Sulaiman, A. Development of Cellulose Nano fibre (CNF) Derived from Kenaf Bast Fibre and its Potential in Enzyme Immobilization Support. Malays. J. Anal. Sci. 2016, 20, 309–317. [Google Scholar] [CrossRef]
- Luo, X.; Zhang, L. Immobilization of Penicillin G Acylase in Epoxy-Activated Magnetic Cellulose Microspheres for Improvement of Biocatalytic Stability and Activities. Biomacromolecules 2010, 11, 2896–2903. [Google Scholar] [CrossRef]
- No, H.K.; Meyers, S.P. Preparation and Characterization of Chitin and Chitosan—A Review. J. Aquat. Food Prod. Technol. 1995, 4, 27–52. [Google Scholar] [CrossRef]
- Elias, N.; Wahab, R.A.; Chandren, S.; Abdul Razak, F.I.; Jamalis, J. Effect of operative variables and kinetic study of butyl butyrate synthesis by Candida rugosa lipase activated by chitosan-reinforced nanocellulose derived from raw oil palm leaves. Enzyme Microb. Technol. 2019, 130, 109367. [Google Scholar] [CrossRef]
- Wan Ngah, W.S.; Teong, L.C.; Hanafiah, M.A.K.M. Adsorption of dyes and heavy metal ions by chitosan composites: A review. Carbohydr. Polym. 2011, 83, 1446–1456. [Google Scholar] [CrossRef]
- Abd Manan, F.M.; Attan, N.; Zakaria, Z.; Mahat, N.A.; Abdul, W.R. Insight into the Rhizomucor miehei lipase supported on chitosan-chitin nanowhiskers assisted esterification of eugenol to eugenyl benzoate. J. Biotechnol. 2018, 280, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Lopez, L.; Pedrero, S.G.; Lopez-Carrobles, N.; Virgen-Ortíz, J.J.; Gorines, B.C.; Otero, C.; Fernandez-Lafuente, R. Physical crosslinking of lipase from Rhizomucor miehei immobilized on octyl agarose via coating with ionic polymers. Process Biochem. 2017, 54, 81–88. [Google Scholar] [CrossRef]
- Ji, Q.; Wang, B.; Tan, J.; Zhu, L.; Li, L. Immobilized multienzymatic systems for catalysis of cascade reactions. Process Biochem. 2016, 51, 1193–1203. [Google Scholar] [CrossRef]
- Gkantzou, E.; Chatzikonstantinou, A.V.; Fotiadou, R.; Giannakopoulou, A.; Patila, M.; Stamatis, H. Trends in the development of innovative nanobiocatalysts and their application in biocatalytic transformations. Biotechnol. Adv. 2021, 107738. [Google Scholar] [CrossRef]
- Giannakopoulou, A.; Gkantzou, E.; Polydera, A.; Stamatis, H. Multienzymatic Nanoassemblies: Recent Progress and Applications. Trends Biotechnol. 2020, 38, 202–216. [Google Scholar] [CrossRef]
- Qiu, H.; Li, Y.; Ji, G.; Zhou, G.; Huang, X.; Qu, Y.; Gao, P. Immobilization of lignin peroxidase on nanoporous gold: Enzymatic properties and in situ release of H2O2 by co-immobilized glucose oxidase. Bioresour. Technol. 2009, 100, 3837–3842. [Google Scholar] [CrossRef]
- Mathesh, M.; Liu, J.; Barrow, C.J.; Yang, W. Graphene-Oxide-Based Enzyme Nanoarchitectonics for Substrate Channeling. Chem. A Eur. J. 2017, 23, 304–311. [Google Scholar] [CrossRef]
- Gao, F.; Hu, M.; Li, S.; Zhai, Q.; Jiang, Y. Positional orientating co-immobilization of bienzyme CPO/GOx on mesoporous TiO2 thin film for efficient cascade reaction. Bioprocess Biosyst. Eng. 2019, 42, 1065–1075. [Google Scholar] [CrossRef]
- Pitzalis, F.; Monduzzi, M.; Salis, A. A bienzymatic biocatalyst constituted by glucose oxidase and Horseradish peroxidase immobilized on ordered mesoporous silica. Microporous Mesoporous Mater. 2017, 241, 145–154. [Google Scholar] [CrossRef]
- Rocha-Martin, J.; Velasco-Lozano, S.; Guisán, J.M.; López-Gallego, F. Oxidation of phenolic compounds catalyzed by immobilized multi-enzyme systems with integrated hydrogen peroxide production. Green Chem. 2014, 16, 303–311. [Google Scholar] [CrossRef]
- Guisan, J.M.; Bolivar, J.M.; López-Gallego, F.; Rocha-Martín Editors, J. Immobilization of Enzymes and Cells, 4th ed.; In Methods in Molecular Biology; Springer: New York, NY, USA, 2020; ISBN 9781071602140. [Google Scholar]
- Cao, L.; Van Rantwijk, F.; Sheldon, R.A. Cross-linked enzyme aggregates: A simple and effective method for the immobilization of penicillin acylase. Org. Lett. 2000, 2, 1361–1364. [Google Scholar] [CrossRef]
- Galliani, M.; Santi, M.; Del Grosso, A.; Cecchettini, A.; Santorelli, F.M.; Hofmann, S.L.; Lu, J.Y.; Angella, L.; Cecchini, M.; Signore, G. Cross-Linked Enzyme Aggregates as Versatile Tool for Enzyme Delivery: Application to Polymeric Nanoparticles. Bioconjug. Chem. 2018, 29, 2225–2231. [Google Scholar] [CrossRef]
- Gräslund, S.; Nordlund, P.; Weigelt, J.; Hallberg, B.M.; Bray, J.; Gileadi, O.; Knapp, S.; Oppermann, U.; Arrowsmith, C.; Hui, R.; et al. Protein production and purification. Nat. Methods 2008, 5, 135–146. [Google Scholar] [CrossRef]
- Nallamsetty, S.; Waugh, D.S. A generic protocol for the expression and purification of recombinant proteins in Escherichia coli using a combinatorial His6-maltose binding protein fusion tag. Nat. Protoc. 2007, 2, 383–391. [Google Scholar] [CrossRef] [Green Version]
- Razib, M.S.M.; Rahman, R.N.Z.R.A.; Shariff, F.M.; Ali, M.S.M. Biochemical and structural characterization of cross-linked enzyme aggregates (Cleas) of organic solvent tolerant protease. Catalysts 2020, 10, 55. [Google Scholar] [CrossRef] [Green Version]
- Mafra, A.C.O.; Beltrame, M.B.; Ulrich, L.G.; Giordano, R.D.L.C.; de Arruda Ribeiro, M.P.; Tardioli, P.W. Combined CLEAs of invertase and soy protein for economically feasible conversion of sucrose in a fed-batch reactor. Food Bioprod. Process. 2018, 110, 145–157. [Google Scholar] [CrossRef]
- Vaidya, B.K.; Kuwar, S.S.; Golegaonkar, S.B.; Nene, S.N. Preparation of cross-linked enzyme aggregates of l-aminoacylase via co-aggregation with polyethyleneimine. J. Mol. Catal. B Enzym. 2012, 74, 184–191. [Google Scholar] [CrossRef]
- Arsenault, A.; Cabana, H.; Jones, J.P. Laccase-based CLEAs: Chitosan as a novel cross-linking agent. Enzyme Res. 2011, 2011. [Google Scholar] [CrossRef] [Green Version]
- Li, X.D.; Wu, J.; Jia, D.C.; Wan, Y.H.; Yang, N.; Qiao, M. Preparation of cross-linked glucoamylase aggregates immobilization by using dextrin and xanthan gum as protecting agents. Catalysts 2016, 6, 77. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Dutt, K.; Misra, S.; Raghuwanshi, S.; Saxena, R.K. Characterization of cross-linked immobilized lipase from thermophilic mould Thermomyces lanuginosa using glutaraldehyde. Bioresour. Technol. 2009, 100, 4074–4076. [Google Scholar] [CrossRef] [PubMed]
- Paitaid, P.; H-Kittikun, A. Magnetic Cross-Linked Enzyme Aggregates of Aspergillus oryzae ST11 Lipase Using Polyacrylonitrile Coated Magnetic Nanoparticles for Biodiesel Production. Appl. Biochem. Biotechnol. 2020, 190, 1319–1332. [Google Scholar] [CrossRef]
- Amaral-Fonseca, M.; Kopp, W.; Giordano, R.D.L.C. Fernández-Lafuente, R.; Tardioli, P.W. Preparation of magnetic cross-linked amyloglucosidase aggregates: Solving some activity problems. Catalysts 2018, 8, 496. [Google Scholar] [CrossRef] [Green Version]
- Guimarães, J.R.; de Lima Camargo Giordano, R.; Fernandez-Lafuente, R.; Tardioli, P.W. Evaluation of strategies to produce highly porous cross-linked aggregates of porcine pancreas lipase with magnetic properties. Molecules 2018, 23, 2993. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Vidal, T.; Armenta-Perez, V.P.; Rosales-Rivera, L.C.; Mateos-Díaz, J.C.; Rodríguez, J.A. Cross-linked enzyme aggregates of recombinant Candida antarctica lipase B for the efficient synthesis of olvanil, a nonpungent capsaicin analogue. Biotechnol. Prog. 2019, 35, 1–11. [Google Scholar] [CrossRef]
- Hernández-García, S.; García-García, M.I.; García-Carmona, F. Improving the production, activity, and stability of CLEAs with diepoxides. Biotechnol. Prog. 2017, 33, 1425–1429. [Google Scholar] [CrossRef]
- Velasco-Lozano, S.; López-Gallego, F.; Vázquez-Duhalt, R.; Mateos-Díaz, J.C.; Guisán, J.M.; Favela-Torres, E. Carrier-free immobilization of lipase from candida rugosa with polyethyleneimines by carboxyl-activated cross-linking. Biomacromolecules 2014, 15, 1896–1903. [Google Scholar] [CrossRef]
- Goetze, D.; Foletto, E.F.; da Silva, H.B.; Silveira, V.C.C.; Dal Magro, L.; Rodrigues, R.C. Effect of feather meal as proteic feeder on combi-CLEAs preparation for grape juice clarification. Process Biochem. 2017, 62, 122–127. [Google Scholar] [CrossRef]
- Tudorache, M.; Gheorghe, A.; Viana, A.S.; Parvulescu, V.I. Biocatalytic epoxidation of α-pinene to oxy-derivatives over cross-linked lipase aggregates. J. Mol. Catal. B Enzym. 2016, 134, 9–15. [Google Scholar] [CrossRef]
- Li, H.; Wang, R.; Wang, A.; Zhang, J.; Yin, Y.; Pei, X.; Zhang, P. Rapidly and Precisely Cross-Linked Enzymes Using Bio-Orthogonal Chemistry from Cell Lysate for the Synthesis of (S)-1-(2,6-Dichloro-3-fluorophenyl) Ethanol. ACS Sustain. Chem. Eng. 2020, 8, 6466–6478. [Google Scholar] [CrossRef]
- Jia, J.; Zhang, W.; Yang, Z.; Yang, X.; Wang, N.; Yu, X. Novel magnetic cross-linked cellulase aggregates with a potential application in lignocellulosic biomass bioconversion. Molecules 2017, 22, 269. [Google Scholar] [CrossRef]
- Sheldon, R. CLEAs, Combi-CLEAs and ‘Smart’ Magnetic CLEAs: Biocatalysis in a Bio-Based Economy. Catalysts 2019, 9, 261. [Google Scholar] [CrossRef] [Green Version]
- Dalziel, K. Physical Significance of Michaelis Constants. Nature 1962, 196, 1203–1205. [Google Scholar] [CrossRef]
- Yang, T.; Kim, Y.-J.; Roy, J.K.; Kim, Y.-W. Combined Cross-Linked Enzyme Aggregates of Monoamine Oxidase and Putrescine Oxidase as a Bifunctional Biocatalyst for Determination of Biogenic Amines in Foods. Catalysts 2019, 9, 579. [Google Scholar] [CrossRef] [Green Version]
- Majewski, M.B.; Howarth, A.J.; Li, P.; Wasielewski, M.R.; Hupp, J.T.; Farha, O.K. Enzyme encapsulation in metal–organic frameworks for applications in catalysis. CrystEngComm 2017, 19, 4082–4091. [Google Scholar] [CrossRef]
- McKenna, S.M.; Leimkühler, S.; Herter, S.; Turner, N.J.; Carnell, A.J. Enzyme cascade reactions: Synthesis of furandicarboxylic acid (FDCA) and carboxylic acids using oxidases in tandem. Green Chem. 2015, 17, 3271–3275. [Google Scholar] [CrossRef]
- Wu, X.; Ge, J.; Yang, C.; Hou, M.; Liu, Z. Facile synthesis of multiple enzyme-containing metal–organic frameworks in a biomolecule-friendly environment. Chem. Commun. 2015, 51, 13408–13411. [Google Scholar] [CrossRef]
- Liang, H.; Jiang, S.; Yuan, Q.; Li, G.; Wang, F.; Zhang, Z.; Liu, J. Co-immobilization of multiple enzymes by metal coordinated nucleotide hydrogel nanofibers: Improved stability and an enzyme cascade for glucose detection. Nanoscale 2016, 8, 6071–6078. [Google Scholar] [CrossRef]
- Song, J.; He, W.; Shen, H.; Zhou, Z.; Li, M.; Su, P.; Yang, Y. Exquisitely designed magnetic DNA nanocompartment for enzyme immobilization with adjustable catalytic activity and improved enzymatic assay performance. Chem. Eng. J. 2020, 390, 124488. [Google Scholar] [CrossRef]
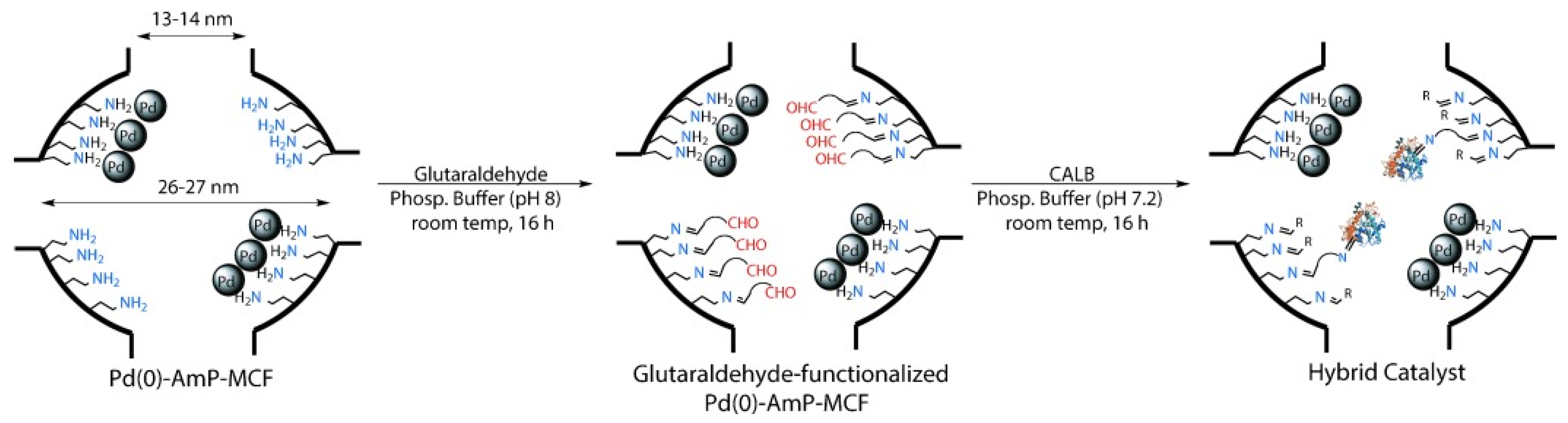
- Heuson, E.; Froidevaux, R.; Itabaiana, I.; Wojcieszak, R.; Capron, M.; Dumeignil, F. Optimisation of catalysts coupling in multi-catalytic hybrid materials: Perspectives for the next revolution in catalysis. Green Chem. 2021, 23, 1942–1954. [Google Scholar] [CrossRef]
- Engström, K.; Johnston, E.V.; Verho, O.; Gustafson, K.P.J.; Shakeri, M.; Tai, C.-W.; Bäckvall, J.-E. Co-immobilization of an enzyme and a metal into the compartments of mesoporous silica for cooperative tandem catalysis: An artificial metalloenzyme. Angew. Chemie Int. Ed. 2013, 52, 14006–14010. [Google Scholar] [CrossRef] [Green Version]
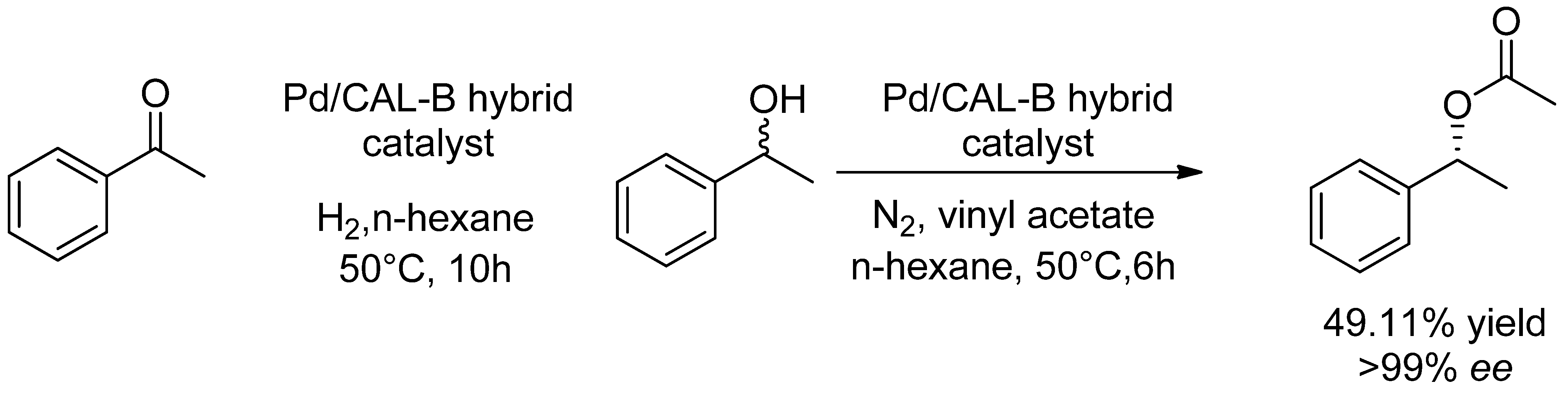
- Magadum, D.B.; Yadav, G.D. Design of tandem catalyst by co-immobilization of metal and enzyme on mesoporous foam for cascaded synthesis of (R)-phenyl ethyl acetate. Biochem. Eng. J. 2018, 129, 96–105. [Google Scholar] [CrossRef]
- Magadum, D.B.; Yadav, G.D. One-pot synthesis of (R)-1-(pyridin-4-yl)ethyl acetate using tandem catalyst prepared by co-immobilization of palladium and lipase on mesoporous foam: Optimization and kinetic modeling. Chirality 2017, 29, 811–823. [Google Scholar] [CrossRef]
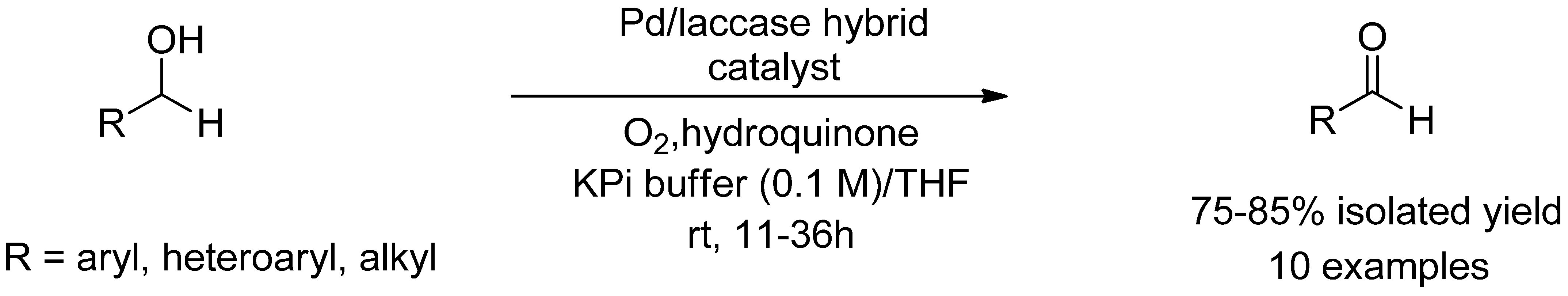
- Moradi, S.; Shokri, Z.; Ghorashi, N.; Navaee, A.; Rostami, A. Design and synthesis of a versatile cooperative catalytic aerobic oxidation system with co-immobilization of palladium nanoparticles and laccase into the cavities of MCF. J. Catal. 2020, 382, 305–319. [Google Scholar] [CrossRef]
- Shokri, Z.; Azimi, N.; Moradi, S.; Rostami, A. A novel magnetically separable laccase-mediator catalyst system for the aerobic oxidation of alcohols and 2-substituted-2,3-dihydroquinazolin-4(1 H)-ones under mild conditions. Appl. Organomet. Chem. 2020, 34, e5899. [Google Scholar] [CrossRef]
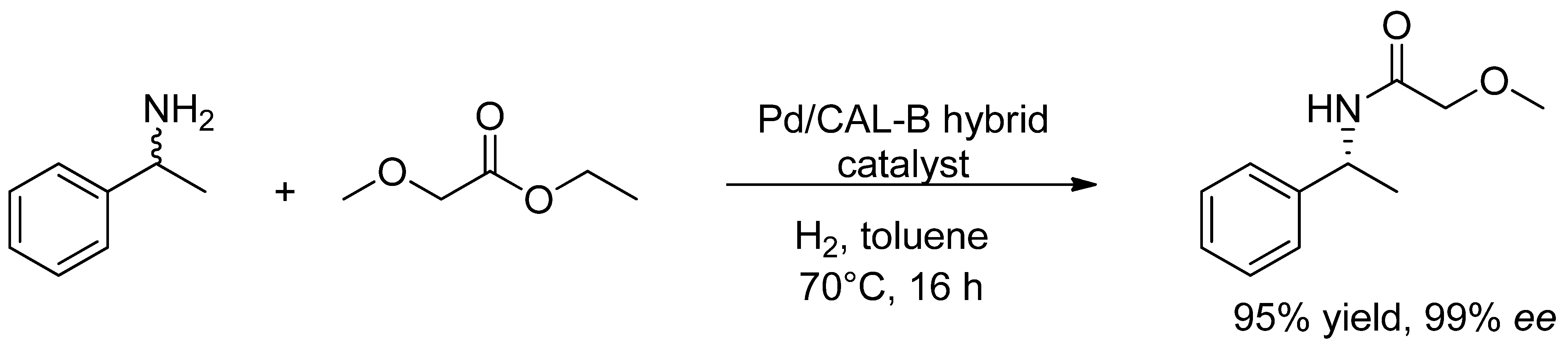
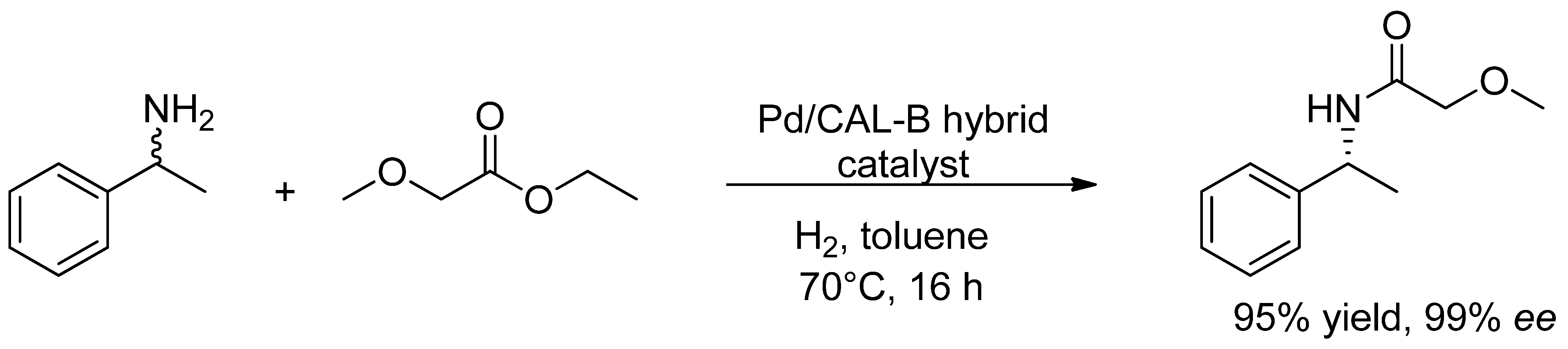
- De Souza, S.P.; Leão, R.A.C.; Bassut, J.F.; Leal, I.C.R.; Wang, S.; Ding, Q.; Li, Y.; Lam, F.L.-Y.; de Souza, R.O.M.A.; Itabaiana, I., Jr. New Biosilified Pd-lipase hybrid biocatalysts for dynamic resolution of amines. Tetrahedron Lett. 2017, 58, 4849–4854. [Google Scholar] [CrossRef]
- Zhang, X.; Jing, L.; Chang, F.; Chen, S.; Yang, H.; Yang, Q. Positional immobilization of Pd nanoparticles and enzymes in hierarchical yolk–shell@shell nanoreactors for tandem catalysis. Chem. Commun. 2017, 53, 7780–7783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Hübner, R.; Wang, Y.; Zhang, E.; Zhou, Y.; Dong, S.; Wu, C. Surface-Functionalized Mesoporous Nanoparticles as Heterogeneous Supports to Transfer Bifunctional Catalysts into Organic Solvents for Tandem Catalysis. ACS Appl. Nano Mater. 2018, 1, 6378–6386. [Google Scholar] [CrossRef] [Green Version]
- Ferraz, C.A.; do Nascimento, M.A.; Almeida, R.F.O.; Sergio, G.G.; Junior, A.A.T.; Dalmônico, G.; Caraballo, R.; Finotelli, P.V.; Leão, R.A.C.; Wojcieszak, R.; et al. Synthesis and characterization of a magnetic hybrid catalyst containing lipase and palladium and its application on the dynamic kinetic resolution of amines. Mol. Catal. 2020, 493, 111106. [Google Scholar] [CrossRef]
- Gao, L.; Wang, Z.; Liu, Y.; Liu, P.; Gao, S.; Gao, J.; Jiang, Y. Co-immobilization of metal and enzyme into hydrophobic nanopores for highly improved chemoenzymatic asymmetric synthesis. Chem. Commun. 2020, 56, 13547–13550. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Wang, Z.; Ma, L.; Liu, Y.; Gao, J.; Jiang, Y. Mesoporous Core–Shell Nanostructures Bridging Metal and Biocatalyst for Highly Efficient Cascade Reactions. ACS Catal. 2020, 10, 1375–1380. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, N.; Zhang, E.; Han, Y.; Qi, Z.; Ansorge-Schumacher, M.B.; Ge, Y.; Wu, C. Heterogeneous Metal-Organic-Framework-Based Biohybrid Catalysts for Cascade Reactions in Organic Solvent. Chem. A Eur. J. 2019, 25, 1716–1721. [Google Scholar] [CrossRef]
- Wu, Y.; Shi, J.; Mei, S.; Katimba, H.A.; Sun, Y.; Wang, X.; Liang, K.; Jiang, Z. Concerted Chemoenzymatic Synthesis of α-Keto Acid through Compartmentalizing and Channeling of Metal–Organic Frameworks. ACS Catal. 2020, 10, 9664–9673. [Google Scholar] [CrossRef]
- Dutta, S.; Kumari, N.; Dubbu, S.; Jang, S.W.; Kumar, A.; Ohtsu, H.; Kim, J.; Cho, S.H.; Kawano, M.; Lee, I.S. Highly Mesoporous Metal-Organic Frameworks as Synergistic Multimodal Catalytic Platforms for Divergent Cascade Reactions. Angew. Chemie 2020, 132, 3444–3450. [Google Scholar] [CrossRef]
- Xia, H.; Li, Z.; Zhong, X.; Li, B.; Jiang, Y.; Jiang, Y. HKUST-1 catalyzed efficient in situ regeneration of NAD+ for dehydrogenase mediated oxidation. Chem. Eng. Sci. 2019, 203, 43–53. [Google Scholar] [CrossRef]
- Li, H.; Qiu, C.; Cao, X.; Lu, Y.; Li, G.; He, X.; Lu, Q.; Chen, K.; Ouyang, P.; Tan, W. Artificial Nanometalloenzymes for Cooperative Tandem Catalysis. ACS Appl. Mater. Interfaces 2019, 11, 15718–15726. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, N.; Hübner, R.; Tan, D.; Löffler, M.; Facsko, S.; Zhang, E.; Ge, Y.; Qi, Z.; Wu, C. Enzymes Immobilized on Carbon Nitride (C 3 N 4) Cooperating with Metal Nanoparticles for Cascade Catalysis. Adv. Mater. Interfaces 2019, 6, 1801664. [Google Scholar] [CrossRef]
- Reetz, M.T.; Schimossek, K. Lipase-catalyzed dynamic kinetic resolution of chiral amines: Use of palladium as the racemization catalyst. Chim. Int. J. Chem. 1996, 50, 668–669. [Google Scholar]
- Shakeri, M.; Tai, C.; Göthelid, E.; Oscarsson, S.; Bäckvall, J.-E. Small Pd nanoparticles supported in large pores of mesocellular foam: An excellent catalyst for racemization of amines. Chem. A Eur. J. 2011, 17, 13269–13273. [Google Scholar] [CrossRef] [PubMed]



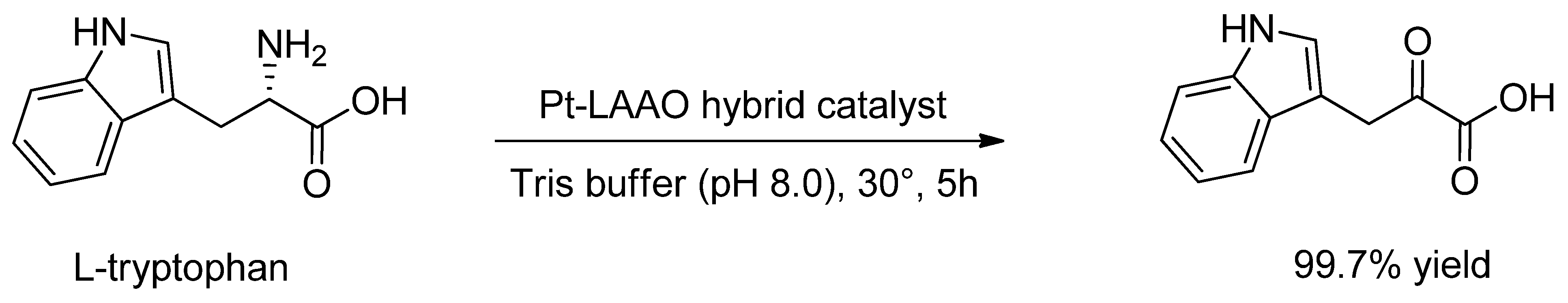

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Matrix | Immobilization Method | Enzymes | Applications |
|---|---|---|---|
| TiO2 nanoparticles | adsorption | GDH/PDOR/GDHt | Conversion of glycerol to 1,3-propanediol [53] |
| Amberlite-XAD7 | adsorption | LDH/GlDH | Reductive amination of α-ketoglutarate [54] |
| Silica gel | adsorption | SDR/G-DH | Reduction of ethyl benzoylformate to ethyl (S)-mandelate [55] |
| Silica-coated magnetite nanoparticle | adsorption | XADH/ADH | Oxidation of xylose to xylonic acid [56,57] |
| Cellulose membrane | adsorption | FDH/FaldDH/ADH | Methanol synthesis from CO2 [58] |
| Anioinic macroporous poly)methyl methacrilate particles (ReliSorb SP400) | adsorption | ScP/CbP/CdP | Synthesis of cello-oligosaccharides [59] |
| Anioinic macroporous poly)methyl methacrilate particles (ReliSorb SP400) | adsorption | P450/G-DH | C-H hydroxylation reactions [60] |
| Ni2+-containing agarose beads | Metal coordination, covalent bond | ADH/GlDH | Alcohol oxidation, ketone reduction [61] |
| Co2+-containing agarose microbeads | Metal coordination | ADH/ωTA/L-AlaDH | Alcohol oxidation/reductive amination cascade [62] |
| Octyl-agarose beads | Adsorption, ion exchange | CAL-B/β-Gal | Ester hydrolysis [63] |
| Amino-functionalized methacrylic polymer | Covalent bond | PDH/CHMO | Alcohol oxidation/Bayer–Villiger reaction cascade [64] |
| Amino-functionalized agarose | Covalent bond | CHMO/G-DH | Bayer–Villiger reaction [65] |
| Epoxy-functionalized agarose | Covalent bond | P450/G-DH | C-H hydroxylation [66] |
| Amino-functionalized iron-based magnetic nanoparticles | Covalent bond | CelA/β-Gl/GOx/HRP | Carboxymethylcellulose hydrolysis [67] |
| Amino-functionalized iron-based magnetic nanoparticles | Covalent bond/Combi-CLEAs | α-AMA/MAM | Starch conversion to maltose [68] |
| Amino-functionalized silver dendrites | Covalent bond | DOx/GA | Starch hydrolysis/glucose oxidation cascade [69] |
| Amino-functionalized MCF | Covalent bond | AKR/G-DH | β-ketoester reduction [70] |
| Alginate | Entrapment | AKR/G-DH | β-ketoester reduction [70] |
| Alginate | Entrapment | LacA/CalA/β-Gl | Conversion of lignocellulosic biomass to saccharides [71] |
| Epoxy-functionalized silica | Covalent bond | LDH/G-DH | Pyruvate reduction to lactate [72] |
| Hydrazide-functionalized methacrylate polymer | Covalent bond | GOx/HRP | Glucose oxidation [73] |
| Amino-functionalized polystyrene nanoparticles | Covalent bond | GOx/HRP | Glucose oxidation [74] |
| Amino-functionalized silica nanoparticles | Covalent bond, protein fusion | BDH/FDH | Ketone reduction [75] |
| NHS-magnetic sepharose beads | Covalent bond, protein fusion | P450/PdR/PdX | Hydroxylation reactions [76] |
| - | Combi-CLEA | GOx/HRP | Glucose oxidation [77] |
| - | Combi-CLEA | ADH/G-DH | Ketone reduction [78] |
| Alginate, chitosan | encapsulation | D-HA/D-CA | Hydantoin DKR and decarboxylation [79] |
| Silicious hydrogel | encapsulation | PaoABC/CAT | Aldehyde oxidation [80] |
| Silicious hydrogel | encapsulation | β-FFA/GOx | Production of lactosucrose from sucrose and lactose [81,82] |
| ZIF-8 (MOF) | encapsulation | ADH/LDH | Pyruvate reduction to lactate [83] |
| ZIF-8 (MOF) | encapsulation | FDH/GDH | Methanol synthesis from CO2 [84] |
| ZIF-8 (MOF) | encapsulation | CA/FDH/GDH | Formate synthesis from CO2 [85] |
| Polyurethane hollow nanofibers | encapsulation | FDH/FaldDH/ADH/GlDH | Methanol synthesis from CO2 [86] |
| Polymersome | encapsulation | AGE/NAL/CSS | Synthesis of CMP-N-acetylneuramic acid [87] |
| Reaction | Support-Enzyme Linkage |
|---|---|
| Diazotization | 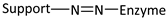 |
| Alkylation and arylation | 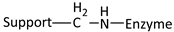 |
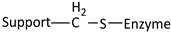 | |
| Schiff’s base formation | 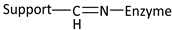 |
| Imidamide formation | 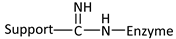 |
| Amidation reaction | 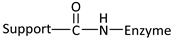 |
| Thiol-Disulphide interchange | 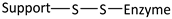 |
| Schiff’s base formation | 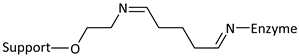 |
| Ugi reaction | 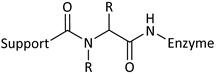 |
| Mercury-sulfide formation |  |
| Gamma-Irradiation induced coupling |  M = matrix radical; E = enzyme radical |
| Matrix | Biocatalyst/Chemocatalyst | Applications |
|---|---|---|
| Siliceous mesocellular foam | CAL-B/Pd | DKR of amines [169]; ketone reduction/KR cascade [170,171] |
| Laccase/Pd | Oxidation of alcohols [172] and 2-substituted-2,3-dihydroquinazolin-4(1H)-ones [173] | |
| Silica nanoparticles | CAL-B/Pd | DKR of amines [174,175]; aldehyde reduction/alcohol esterification cascade [176] |
| Silica-coated iron oxide nanoparticles | CAL-B/Pd | DKR of amines [177] |
| Dendritic organosilica nanoparticles | CAL-B/Pd | DKR of amines [178] |
| Pd/ADH | Pd/Cu-catalyzed Liebeskind–Strogl reaction/bioreduction cascade [178] | |
| PDA nanostructures | Pd, Pt/CAL-B; Pd, Pt/CAL-A | DKR of amines [179] |
| UiO-66 (MOF) | Pd/CAL-B | aldehyde reduction/alcohol esterification cascade [180] |
| Pt/LAAO | Oxidative deamination of L-tryptophan [181] | |
| ZIF-67 (MOF) | Pd/CAL-A | Nitroaldol reaction/DKR of alcohols cascade [182] |
| HKUST-1 (MOF) | Cu/ADH | Benzaldehyde oxidation [183] |
| NaDC/Mi MBN | Shvö’s catalyst/CAL-B | DKR of alcohols and amines [184] |
| Carbon nitride (C3N4) | Pd/CAL-B | aldehyde reduction/alcohol esterification cascade [185] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Júnior, A.A.d.T.; Ladeira, Y.F.X.; França, A.d.S.; Souza, R.O.M.A.d.; Moraes, A.H.; Wojcieszak, R.; Itabaiana, I., Jr.; Miranda, A.S.d. Multicatalytic Hybrid Materials for Biocatalytic and Chemoenzymatic Cascades—Strategies for Multicatalyst (Enzyme) Co-Immobilization. Catalysts 2021, 11, 936. https://doi.org/10.3390/catal11080936
Júnior AAdT, Ladeira YFX, França AdS, Souza ROMAd, Moraes AH, Wojcieszak R, Itabaiana I Jr., Miranda ASd. Multicatalytic Hybrid Materials for Biocatalytic and Chemoenzymatic Cascades—Strategies for Multicatalyst (Enzyme) Co-Immobilization. Catalysts. 2021; 11(8):936. https://doi.org/10.3390/catal11080936
Chicago/Turabian StyleJúnior, Aldo Araújo da Trindade, Yan Ferraz Ximenes Ladeira, Alexandre da Silva França, Rodrigo Octavio Mendonça Alves de Souza, Adolfo Henrique Moraes, Robert Wojcieszak, Ivaldo Itabaiana, Jr., and Amanda Silva de Miranda. 2021. "Multicatalytic Hybrid Materials for Biocatalytic and Chemoenzymatic Cascades—Strategies for Multicatalyst (Enzyme) Co-Immobilization" Catalysts 11, no. 8: 936. https://doi.org/10.3390/catal11080936
APA StyleJúnior, A. A. d. T., Ladeira, Y. F. X., França, A. d. S., Souza, R. O. M. A. d., Moraes, A. H., Wojcieszak, R., Itabaiana, I., Jr., & Miranda, A. S. d. (2021). Multicatalytic Hybrid Materials for Biocatalytic and Chemoenzymatic Cascades—Strategies for Multicatalyst (Enzyme) Co-Immobilization. Catalysts, 11(8), 936. https://doi.org/10.3390/catal11080936