
Theoretical Insights into the Hydrogen Evolution Reaction on the Ni3N Electrocatalyst



Abstract
1. Introduction
2. Results and Discussion
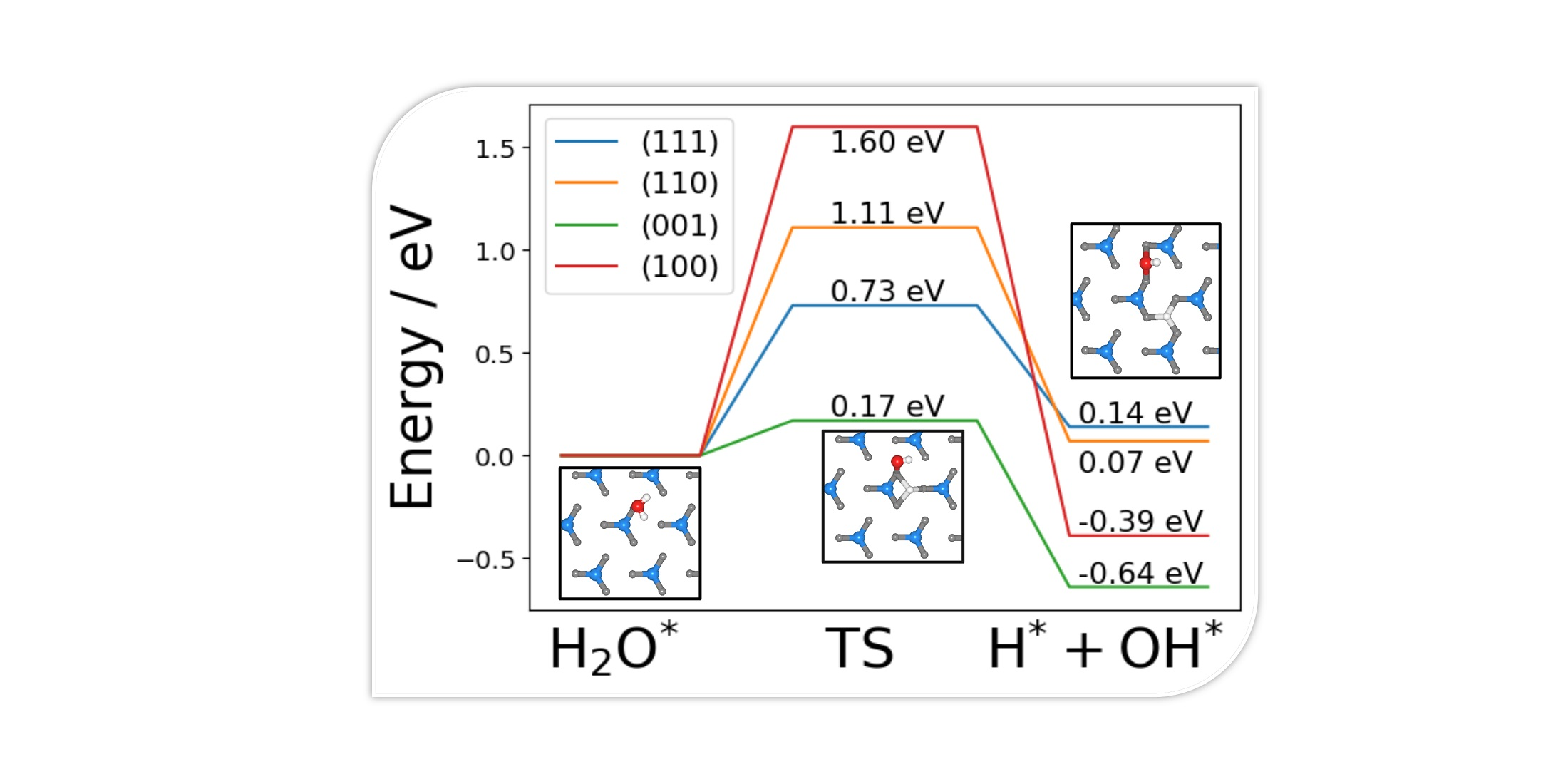
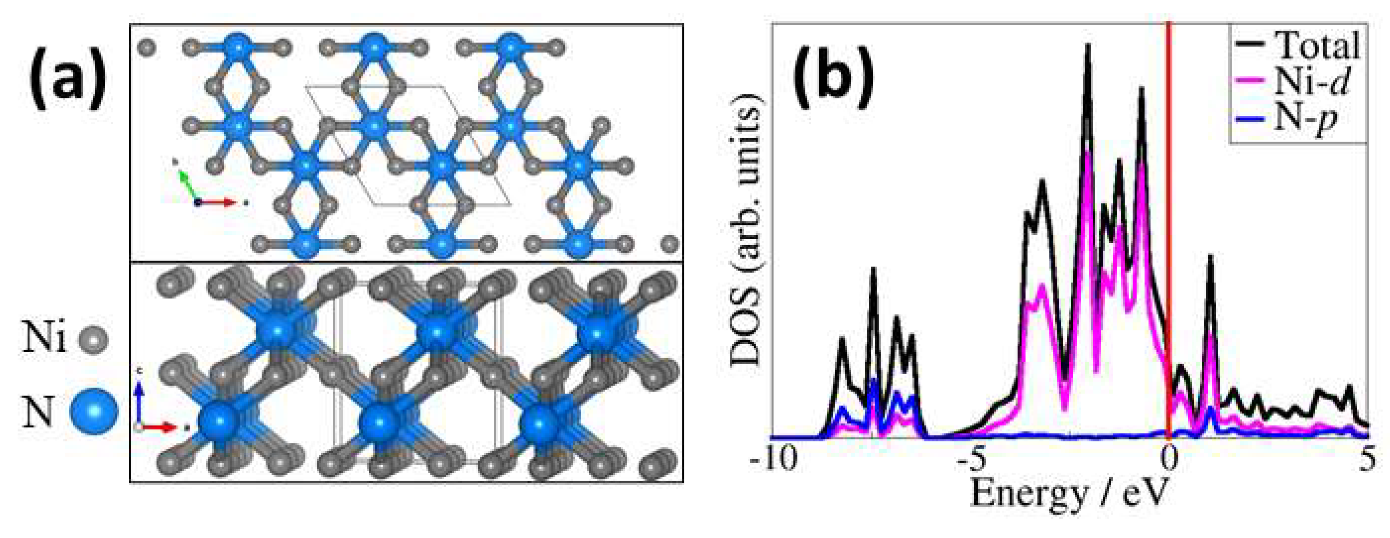
2.1. Bulk and Surface Characterisation of Ni3N
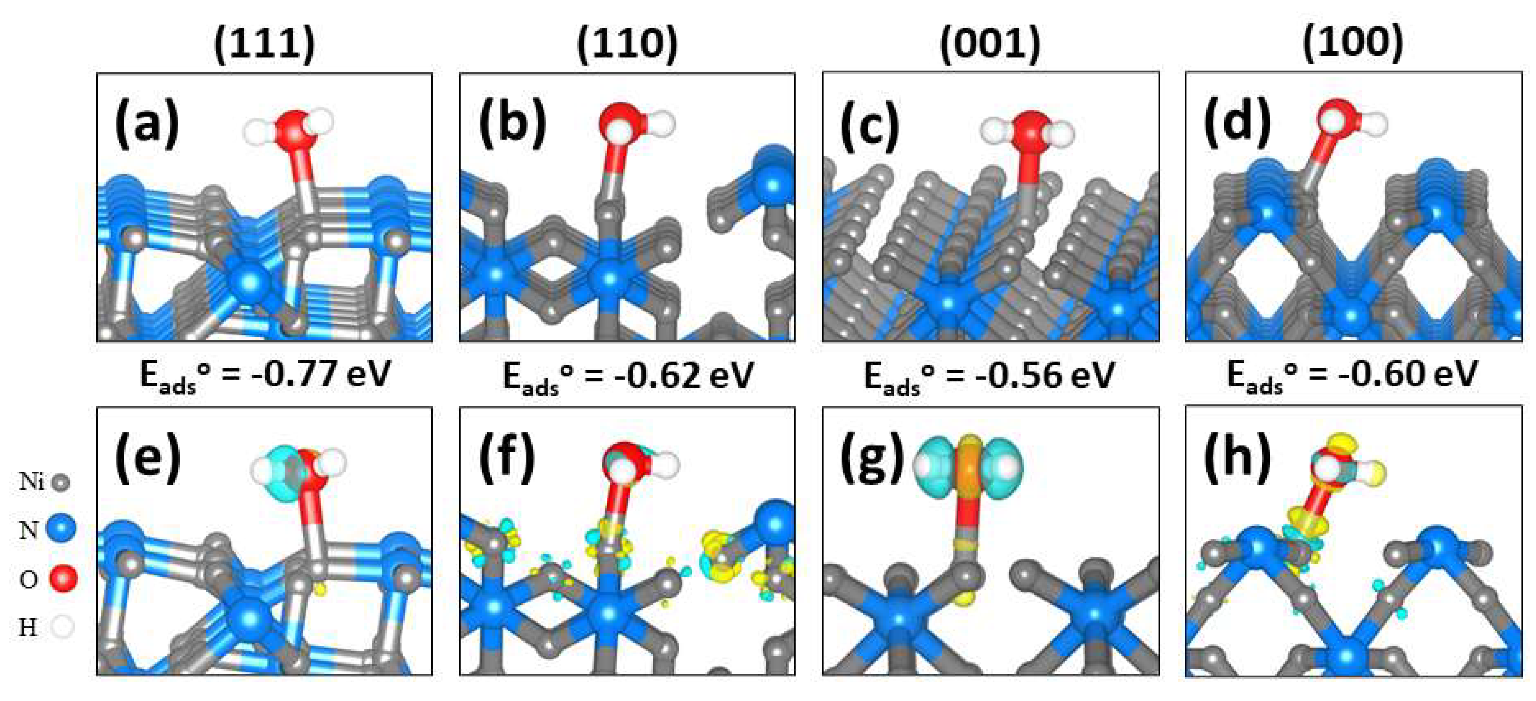
2.2. Molecular Water Adsorption to Ni3N(111), (110), (001), and (100) Surfaces
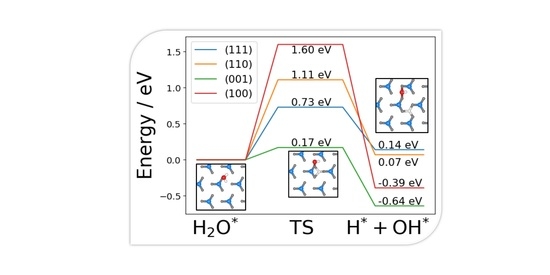
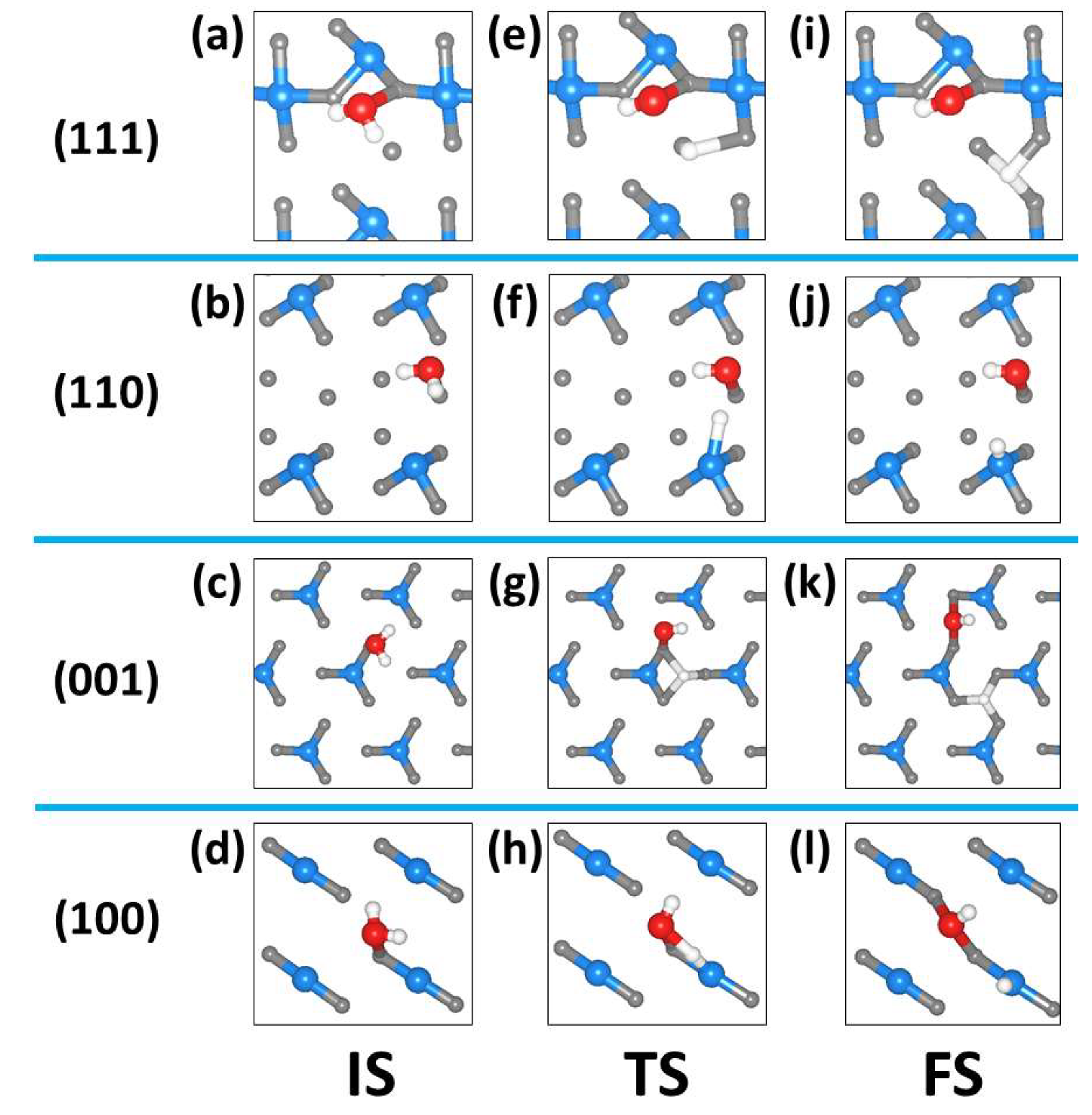
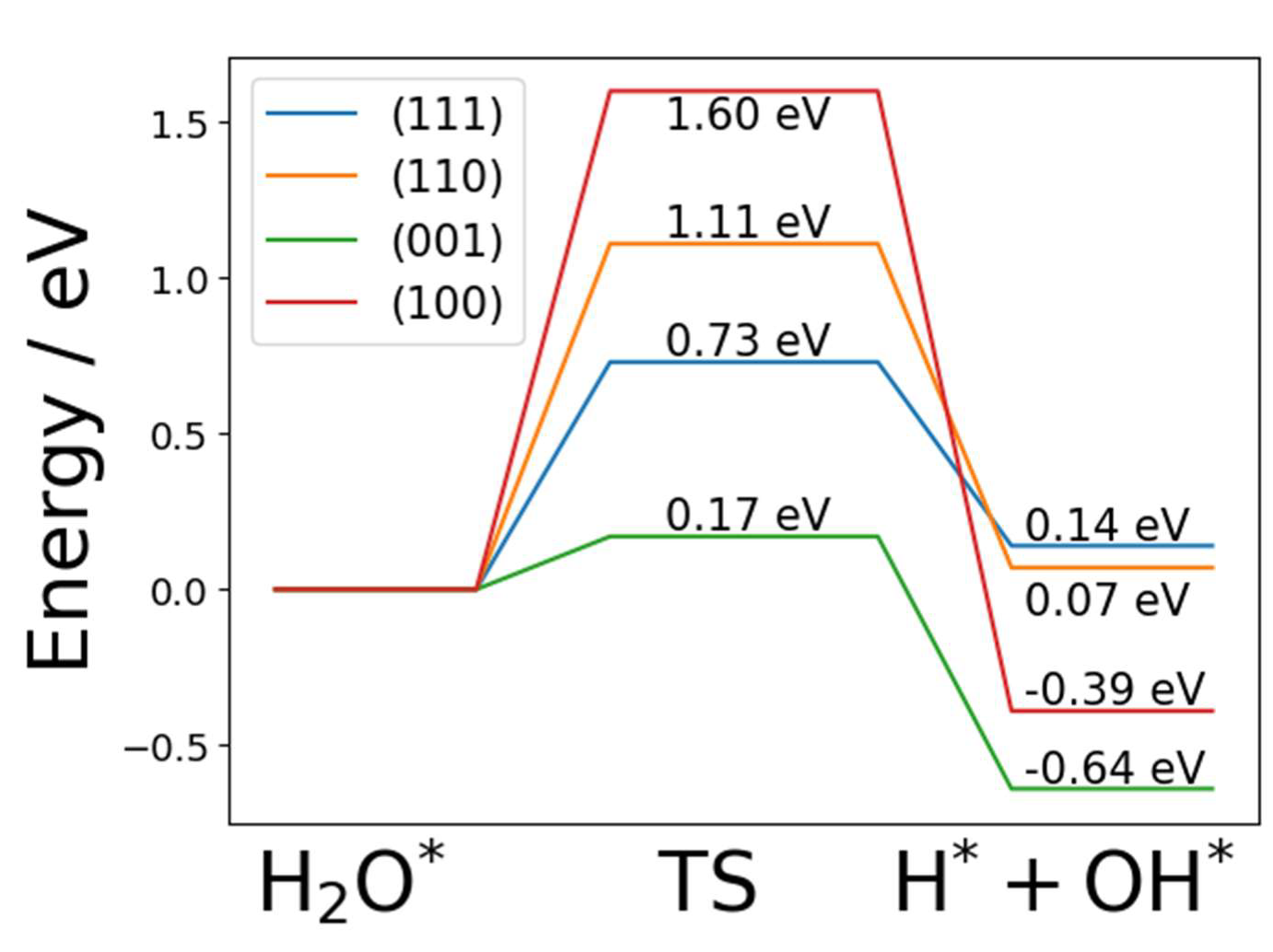
2.3. Dissociated Water Adsorption on Ni3N
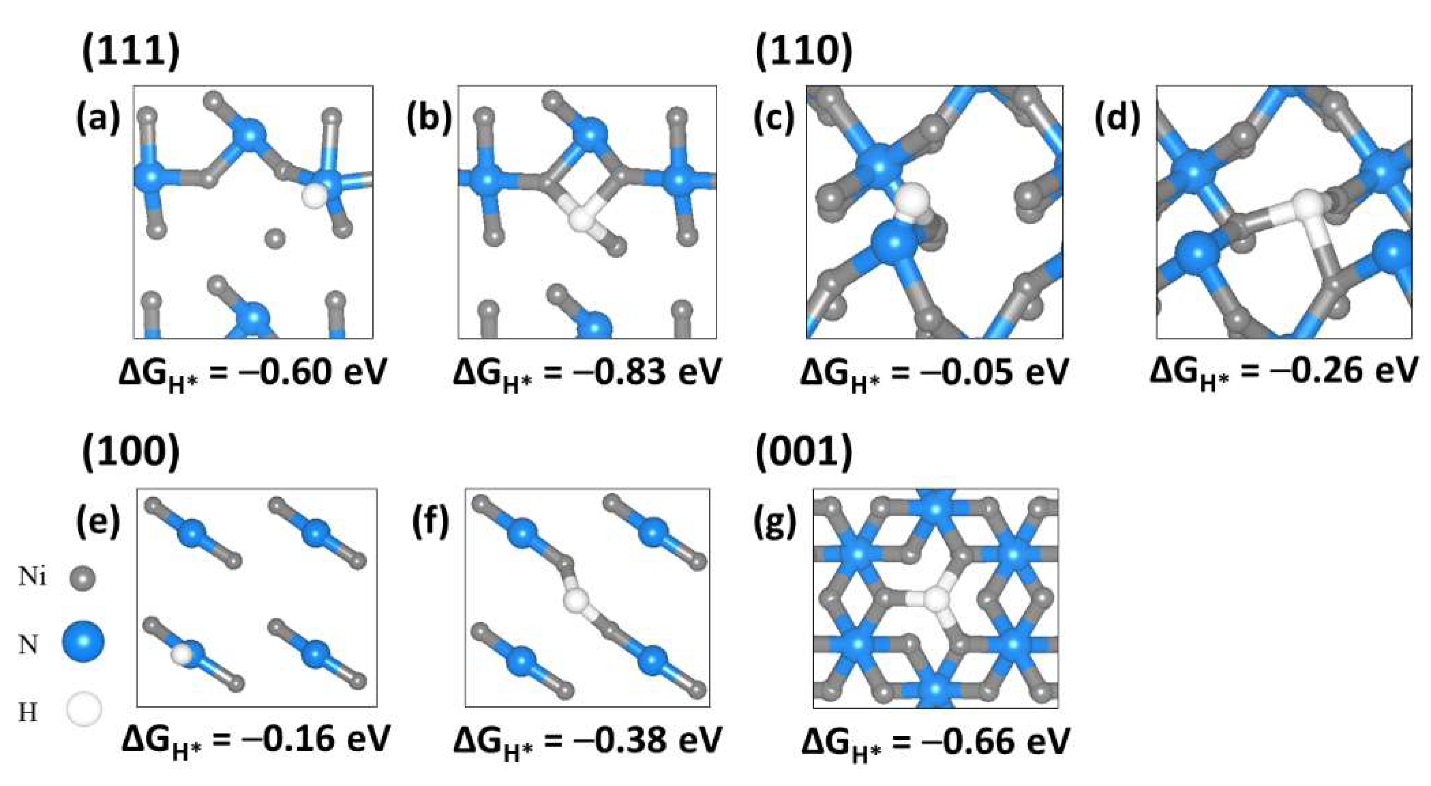
2.4. Hydrogen Adsorption to Ni3N Surfaces
3. Conclusions
4. Computational Details
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Angeli, S.D.; Monteleone, G.; Giaconia, A.; Lemonidou, A.A. State-of-the-art catalysts for CH4 steam reforming at low temperature. Int. J. Hydrogen Energy 2014, 39, 1979–1997. [Google Scholar] [CrossRef]
- Duan, H.; Li, D.; Tang, Y.; He, Y.; Ji, S.; Wang, R.; Lv, H.; Lopes, P.P.; Paulikas, A.P.; Li, H.; et al. High-Performance Rh2P Electrocatalyst for Efficient Water Splitting. J. Am. Chem. Soc. 2017, 139, 5494–5502. [Google Scholar] [CrossRef] [PubMed]
- El-Deab, M.S.; Ohsaka, T. Manganese Oxide Nanoparticles Electrodeposited on Platinum Are Superior to Platinum for Oxygen Reduction. Angew. Chem. Int. Ed. 2006, 45, 5963–5966. [Google Scholar] [CrossRef] [PubMed]
- Noerskov, J.K.; Bligaard, T.; Logadottir, A.; Kitchin, J.R.; Chen, J.G.; Pandelov, S.; Stimming, U.; Nørskov, J.K.; Bligaard, T.; Logadottir, A.; et al. Trends in the Exchange Current for Hydrogen Evolution. J. Electrochem. Soc. 2005, 152, J23–J26. [Google Scholar] [CrossRef]
- McKone, J.R.; Marinescu, S.C.; Brunschwig, B.S.; Winkler, J.R.; Gray, H.B. Earth-abundant hydrogen evolution electrocatalysts. Chem. Sci. 2014, 5, 865–878. [Google Scholar] [CrossRef]
- Li, Y.; Wang, H.; Xie, L.; Liang, Y.; Hong, G.; Dai, H. MoS2 Nanoparticles Grown on Graphene: An Advanced Catalyst for the Hydrogen Evolution Reaction. J. Am. Chem. Soc. 2011, 133, 7296–7299. [Google Scholar] [CrossRef]
- Voiry, D.; Salehi, M.; Silva, R.; Fujita, T.; Chen, M.; Asefa, T.; Shenoy, V.B.; Eda, G.; Chhowalla, M. Conducting MoS2 Nanosheets as Catalysts for Hydrogen Evolution Reaction. Nano Lett. 2013, 13, 6222–6227. [Google Scholar] [CrossRef]
- Chen, T.-Y.; Chang, Y.-H.; Hsu, C.-L.; Wei, K.-H.; Chiang, C.-Y.; Li, L.-J. Comparative study on MoS2 and WS2 for electrocatalytic water splitting. Int. J. Hydrogen Energy 2013, 38, 12302–12309. [Google Scholar] [CrossRef]
- Wu, Z.; Fang, B.; Bonakdarpour, A.; Sun, A.; Wilkinson, D.P.; Wang, D. WS2 nanosheets as a highly efficient electrocatalyst for hydrogen evolution reaction. Appl. Catal. B Environ. 2012, 125, 59–66. [Google Scholar] [CrossRef]
- Schipper, D.E.; Zhao, Z.; Thirumalai, H.; Leitner, A.P.; Donaldson, S.L.; Kumar, A.; Qin, F.; Wang, Z.; Grabow, L.C.; Bao, J.; et al. Effects of Catalyst Phase on the Hydrogen Evolution Reaction of Water Splitting: Preparation of Phase-Pure Films of FeP, Fe2P, and Fe3P and Their Relative Catalytic Activities. Chem. Mater. 2018, 30, 3588–3598. [Google Scholar] [CrossRef]
- Jiao, L.; Zhou, Y.-X.; Jiang, H.-L. Metal–organic framework-based CoP/reduced graphene oxide: High-performance bifunctional electrocatalyst for overall water splitting. Chem. Sci. 2016, 7, 1690–1695. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Rodriguez, J.A. Catalysts for hydrogen evolution from the [NiFe] hydrogenase to the Ni2P(001) surface: The importance of ensemble effect. J. Am. Chem. Soc. 2005, 127, 14871–14878. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Liu, J.; Wang, J.; Ruan, Y.; Ji, X.; Xu, K.; Chen, C.; Wan, H.; Miao, L.; Jiang, J. Interface engineering: The Ni(OH)2/MoS2 heterostructure for highly efficient alkaline hydrogen evolution. Nano Energy 2017, 37, 74–80. [Google Scholar] [CrossRef]
- Danilovic, N.; Subbaraman, R.; Strmcnik, D.; Chang, K.-C.; Paulikas, A.P.; Stamenkovic, V.R.; Markovic, N.M. Enhancing the Alkaline Hydrogen Evolution Reaction Activity through the Bifunctionality of Ni(OH)2/Metal Catalysts. Angew. Chem. Int. Ed. 2012, 51, 12495–12498. [Google Scholar] [CrossRef] [PubMed]
- Subbaraman, R.; Tripkovic, D.; Chang, K.-C.; Strmcnik, D.; Paulikas, A.P.; Hirunsit, P.; Chan, M.; Greeley, J.; Stamenkovic, V.; Markovic, N.M. Trends in activity for the water electrolyser reactions on 3d M(Ni,Co,Fe,Mn) hydr(oxy)oxide catalysts. Nat. Mater. 2012, 11, 550. [Google Scholar] [CrossRef]
- Chaitoglou, S.; Giannakopoulou, T.; Papanastasiou, G.; Tsoutsou, D.; Vavouliotis, A.; Trapalis, C.; Dimoulas, A. Cu vapor-assisted formation of nanostructured Mo2C electrocatalysts via direct chemical conversion of Mo surface for efficient hydrogen evolution reaction applications. Appl. Surf. Sci. 2020, 510, 145516. [Google Scholar] [CrossRef]
- Ma, L.; Ting, L.R.L.; Molinari, V.; Giordano, C.; Yeo, B.S. Efficient hydrogen evolution reaction catalyzed by molybdenum carbide and molybdenum nitride nanocatalysts synthesized via the urea glass route. J. Mater. Chem. A 2015, 3, 8361–8368. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, Y.; Wang, Y.; Al-Enizi, A.M.; Elzatahry, A.A.; Zheng, G. Myriophyllum-like hierarchical TiN@Ni3N nanowire arrays for bifunctional water splitting catalysts. J. Mater. Chem. A 2016, 4, 5713–5718. [Google Scholar] [CrossRef]
- Shalom, M.; Ressnig, D.; Yang, X.; Clavel, G.; Fellinger, T.P.; Antonietti, M. Nickel nitride as an efficient electrocatalyst for water splitting. J. Mater. Chem. A 2015, 3, 8171–8177. [Google Scholar] [CrossRef]
- Pu, Z.; Liu, Q.; Tang, C.; Asiri, A.M.; Sun, X. Ni2P nanoparticle films supported on a Ti plate as an efficient hydrogen evolution cathode. Nanoscale 2014, 6, 11031–11034. [Google Scholar] [CrossRef]
- Tang, C.; Xie, L.; Sun, X.; Asiri, A.M.; He, Y. Highly efficient electrochemical hydrogen evolution based on nickel diselenide nanowall film. Nanotechnology 2016, 27, 20LT02. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, T.; Li, L.; Song, S.; Ding, R. Nickel-based electrodes as catalysts for hydrogen evolution reaction in alkaline media. Ionics 2018, 24, 1121–1127. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, T.; Liu, P.; Liao, Z.; Liu, S.; Zhuang, X.; Chen, M.; Zschech, E.; Feng, X. Efficient hydrogen production on MoNi4 electrocatalysts with fast water dissociation kinetics. Nat. Commun. 2017, 8, 15437. [Google Scholar] [CrossRef]
- Song, F.; Li, W.; Han, G.; Sun, Y. Electropolymerization of Aniline on Nickel-Based Electrocatalysts Substantially Enhances Their Performance for Hydrogen Evolution. ACS Appl. Energy Mater. 2018, 1, 3–8. [Google Scholar] [CrossRef]
- Nong, H.N.; Gan, L.; Willinger, E.; Teschner, D.; Strasser, P. IrOx core-shell nanocatalysts for cost- and energy-efficient electrochemical water splitting. Chem. Sci. 2014, 5, 2955–2963. [Google Scholar] [CrossRef]
- Gong, M.; Zhou, W.; Tsai, M.-C.; Zhou, J.; Guan, M.; Lin, M.-C.; Zhang, B.; Hu, Y.; Wang, D.-Y.; Yang, J.; et al. Nanoscale nickel oxide/nickel heterostructures for active hydrogen evolution electrocatalysis. Nat. Commun. 2014, 5, 4695. [Google Scholar] [CrossRef]
- Zhang, F.-S.; Wang, J.-W.; Luo, J.; Liu, R.-R.; Zhang, Z.-M.; He, C.-T.; Lu, T.-B. Extraction of nickel from NiFe-LDH into Ni2P@NiFe hydroxide as a bifunctional electrocatalyst for efficient overall water splitting. Chem. Sci. 2018, 9, 1375–1384. [Google Scholar] [CrossRef]
- Yan, X.; Tian, L.; Chen, X. Crystalline/amorphous Ni/NiO core/shell nanosheets as highly active electrocatalysts for hydrogen evolution reaction. J. Power Sources 2015, 300, 336–343. [Google Scholar] [CrossRef]
- Popczun, E.J.; McKone, J.R.; Read, C.G.; Biacchi, A.J.; Wiltrout, A.M.; Lewis, N.S.; Schaak, R.E. Nanostructured Nickel Phosphide as an Electrocatalyst for the Hydrogen Evolution Reaction. J. Am. Chem. Soc. 2013, 135, 9267–9270. [Google Scholar] [CrossRef]
- Xing, Z.; Li, Q.; Wang, D.; Yang, X.; Sun, X. Self-supported nickel nitride as an efficient high-performance three-dimensional cathode for the alkaline hydrogen evolution reaction. Electrochim. Acta 2016, 191, 841–845. [Google Scholar] [CrossRef]
- Jiang, N.; Tang, Q.; Sheng, M.; You, B.; Jiang, D.; Sun, Y. Nickel sulfides for electrocatalytic hydrogen evolution under alkaline conditions: A case study of crystalline NiS, NiS2, and Ni3S2 nanoparticles. Catal. Sci. Technol. 2016, 6, 1077–1084. [Google Scholar] [CrossRef]
- Chen, P.; Zhou, T.; Zhang, M.; Tong, Y.; Zhong, C.; Zhang, N.; Zhang, L.; Wu, C.; Xie, Y. 3D Nitrogen-Anion-Decorated Nickel Sulfides for Highly Efficient Overall Water Splitting. Adv. Mater. 2017, 29, 1701584. [Google Scholar] [CrossRef]
- Wang, F.; Li, Y.; Shifa, T.A.; Liu, K.; Wang, F.; Wang, Z.; Xu, P.; Wang, Q.; He, J. Selenium-Enriched Nickel Selenide Nanosheets as a Robust Electrocatalyst for Hydrogen Generation. Angew. Chem. Int. Ed. 2016, 55, 6919–6924. [Google Scholar] [CrossRef]
- Gao, D.; Zhang, J.; Wang, T.; Xiao, W.; Tao, K.; Xue, D.; Ding, J. Metallic Ni3N nanosheets with exposed active surface sites for efficient hydrogen evolution. J. Mater. Chem. A 2016, 4, 17363–17369. [Google Scholar] [CrossRef]
- Wang, Y.; Fu, Z.-W.; Yue, X.-L.; Qin, Q.-Z. Electrochemical Reactivity Mechanism of Ni3N with Lithium. J. Electrochem. Soc. 2004, 151, E162–E167. [Google Scholar] [CrossRef]
- Gillot, F.; Oró-Solé, J.; Palacín, M.R. Nickel nitride as negative electrode material for lithium ion batteries. J. Mater. Chem. 2011, 21, 9997–10002. [Google Scholar] [CrossRef]
- Tang, Q.; Jiang, D. Mechanism of Hydrogen Evolution Reaction on 1T-MoS2 from First Principles. ACS Catal. 2016, 6, 4953–4961. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, T.; Liu, P.; Liu, S.; Dong, R.; Zhuang, X.; Chen, M.; Feng, X. Engineering water dissociation sites in MoS2 nanosheets for accelerated electrocatalytic hydrogen production. Energy Environ. Sci. 2016, 9, 2789–2793. [Google Scholar] [CrossRef]
- Gao, M.; Chen, L.; Zhang, Z.; Sun, X.; Zhang, S. Interface engineering of the Ni(OH)2–Ni3N nanoarray heterostructure for the alkaline hydrogen evolution reaction. J. Mater. Chem. A 2018, 6, 833–836. [Google Scholar] [CrossRef]
- Liu, Q.; Xie, L.; Qu, F.; Liu, Z.; Du, G.; Asiri, A.M.; Sun, X. A porous Ni3N nanosheet array as a high-performance non-noble-metal catalyst for urea-assisted electrochemical hydrogen production. Inorg. Chem. Front. 2017, 4, 1120–1124. [Google Scholar] [CrossRef]
- Leineweber, A.; Jacobs, H.; Hull, S. Ordering of Nitrogen in Nickel Nitride Ni3N Determined by Neutron Diffraction. Inorg. Chem. 2001, 40, 5818–5822. [Google Scholar] [CrossRef]
- Liu, T.; Li, M.; Jiao, C.; Hassan, M.; Bo, X.; Zhou, M.; Wang, H.-L. Design and synthesis of integrally structured Ni3N nanosheets/carbon microfibers/Ni3N nanosheets for efficient full water splitting catalysis. J. Mater. Chem. A 2017, 5, 9377–9390. [Google Scholar] [CrossRef]
- Cross, R.W.; Dzade, N.Y. First-Principles Mechanistic Insights into the Hydrogen Evolution Reaction on Ni2P Electrocatalyst in Alkaline Medium. Catalysts 2020, 10, 307. [Google Scholar] [CrossRef]
- Bai, Y.; Kirvassilis, D.; Xu, L.; Mavrikakis, M. Atomic and molecular adsorption on Ni(111). Surf. Sci. 2019, 679, 240–253. [Google Scholar] [CrossRef]
- Nojima, A.; Yamashita, K. A theoretical study of hydrogen adsorption and diffusion on a W(110) surface. Surf. Sci. 2007, 601, 3003–3011. [Google Scholar] [CrossRef]
- Hansen, M.H.; Stern, L.-A.; Feng, L.; Rossmeisl, J.; Hu, X. Widely available active sites on Ni2P for electrochemical hydrogen evolution – insights from first principles calculations. Phys. Chem. Chem. Phys. 2015, 17, 10823–10829. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zuo, X.; Wang, S.-P.; Yin, J.-W.; Zhang, Y.-N.; Chen, J. Theoretical and experimental design of Pt-Co(OH)2 electrocatalyst for efficient HER performance in alkaline solution. Prog. Nat. Sci. Mater. Int. 2019, 29, 356–361. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, J.; Liu, J.; Zhang, L.; Wan, H.; Miao, L.; Jiang, J. Dual-Descriptor Tailoring: The Hydroxyl Adsorption Energy-Dependent Hydrogen Evolution Kinetics of High-Valance State Doped Ni3N in Alkaline Media. ACS Catal. 2019, 9, 9332–9338. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Y.; Li, J.; Jin, X.; Li, Y.; Qian, Q.; Wang, Y.; El-Harairy, A.; Li, Z.; Zhu, Y.; et al. Vanadium Substitution Steering Reaction Kinetics Acceleration for Ni3N Nanosheets Endows Exceptionally Energy-Saving Hydrogen Evolution Coupled with Hydrazine Oxidation. ACS Appl. Mater. Interfaces 2021, 13, 3881–3890. [Google Scholar] [CrossRef]
- Li, R.-Q.; Liu, Q.; Zhou, Y.; Lu, M.; Hou, J.; Qu, K.; Zhu, Y.; Fontaine, O. 3D self-supported porous vanadium-doped nickel nitride nanosheet arrays as efficient bifunctional electrocatalysts for urea electrolysis. J. Mater. Chem. A 2021, 9, 4159–4166. [Google Scholar] [CrossRef]
- Wang, M.; Ma, W.; Lv, Z.; Liu, D.; Jian, K.; Dang, J. Co-Doped Ni3N Nanosheets with Electron Redistribution as Bifunctional Electrocatalysts for Efficient Water Splitting. J. Phys. Chem. Lett. 2021, 12, 1581–1587. [Google Scholar] [CrossRef]
- Liu, X.; Guo, Y.; Wang, P.; Wu, Q.; Zhang, Q.; Rozhkova, E.A.; Wang, Z.; Liu, Y.; Zheng, Z.; Dai, Y.; et al. Synthesis of Synergistic Nitrogen-Doped NiMoO4/Ni3N Heterostructure for Implementation of an Efficient Alkaline Electrocatalytic Hydrogen Evolution Reaction. ACS Appl. Energy Mater. 2020, 3, 2440–2449. [Google Scholar] [CrossRef]
- Niu, S.; Fang, Y.; Zhou, J.; Cai, J.; Zang, Y.; Wu, Y.; Ye, J.; Xie, Y.; Liu, Y.; Zheng, X.; et al. Manipulating the water dissociation kinetics of Ni3N nanosheets via in situ interfacial engineering. J. Mater. Chem. A 2019, 7, 10924–10929. [Google Scholar] [CrossRef]
- Huang, H.; Yu, C.; Han, X.; Li, S.; Cui, S.; Zhao, C.; Huang, H.; Qiu, J. Interface Engineering of Ni3N@Fe3N Heterostructure Supported on Carbon Fiber for Enhanced Water Oxidation. Ind. Eng. Chem. Res. 2017, 56, 14245–14251. [Google Scholar] [CrossRef]
- Hua, W.; Sun, H.; Liu, H.; Li, Y.; Wang, J.-G. Interface engineered NiMoN/Ni3N heterostructures for enhanced alkaline hydrogen evolution reaction. Appl. Surf. Sci. 2021, 540, 148407. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Watson, G.W.; Kelsey, E.T.; de Leeuw, N.H.; Harris, D.J.; Parker, S.C. Atomistic simulation of dislocations, surfaces and interfaces in MgO. J. Chem. Soc. Faraday Trans. 1996, 92, 433–438. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Surface | d(Ni–O) (Å) | d(O–H) (Å) | φ(H–O–H) (°) | ν (cm−1) | q(Ni) (e−) | q(H2O) (e−) | ||
|---|---|---|---|---|---|---|---|---|
| Clean | H2Oads | |||||||
| (111) | −0.77 | 2.145 | 0.973, 0.983 | 105.6 | 3818, 3687 | 0.450 | 0.548 | 0.031 |
| (110) | −0.62 | 2.047 | 0.981, 0.987 | 103.8 | 3744, 3640 | 0.214 | 0.354 | 0.042 |
| (100) | −0.60 | 2.122 | 0.980, 0.982 | 104.1 | 3811, 3699 | 0.241 | 0.396 | 0.045 |
| (001) | −0.56 | 2.085 | 0.983, 0.986 | 104.5 | 3660, 3532 | 0.184 | 0.340 | 0.027 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cross, R.W.; Rondiya, S.R.; Dzade, N.Y. Theoretical Insights into the Hydrogen Evolution Reaction on the Ni3N Electrocatalyst. Catalysts 2021, 11, 716. https://doi.org/10.3390/catal11060716
Cross RW, Rondiya SR, Dzade NY. Theoretical Insights into the Hydrogen Evolution Reaction on the Ni3N Electrocatalyst. Catalysts. 2021; 11(6):716. https://doi.org/10.3390/catal11060716
Chicago/Turabian StyleCross, Russell W., Sachin R. Rondiya, and Nelson Y. Dzade. 2021. "Theoretical Insights into the Hydrogen Evolution Reaction on the Ni3N Electrocatalyst" Catalysts 11, no. 6: 716. https://doi.org/10.3390/catal11060716
APA StyleCross, R. W., Rondiya, S. R., & Dzade, N. Y. (2021). Theoretical Insights into the Hydrogen Evolution Reaction on the Ni3N Electrocatalyst. Catalysts, 11(6), 716. https://doi.org/10.3390/catal11060716