
Isolation Strategy towards Earth-Abundant Single-Site Co-Catalysts for Photocatalytic Hydrogen Evolution Reaction

 , , ,
, , , 
Abstract

1. Introduction
2. Results and Discussion
2.1. The Site-Isolation Strategy
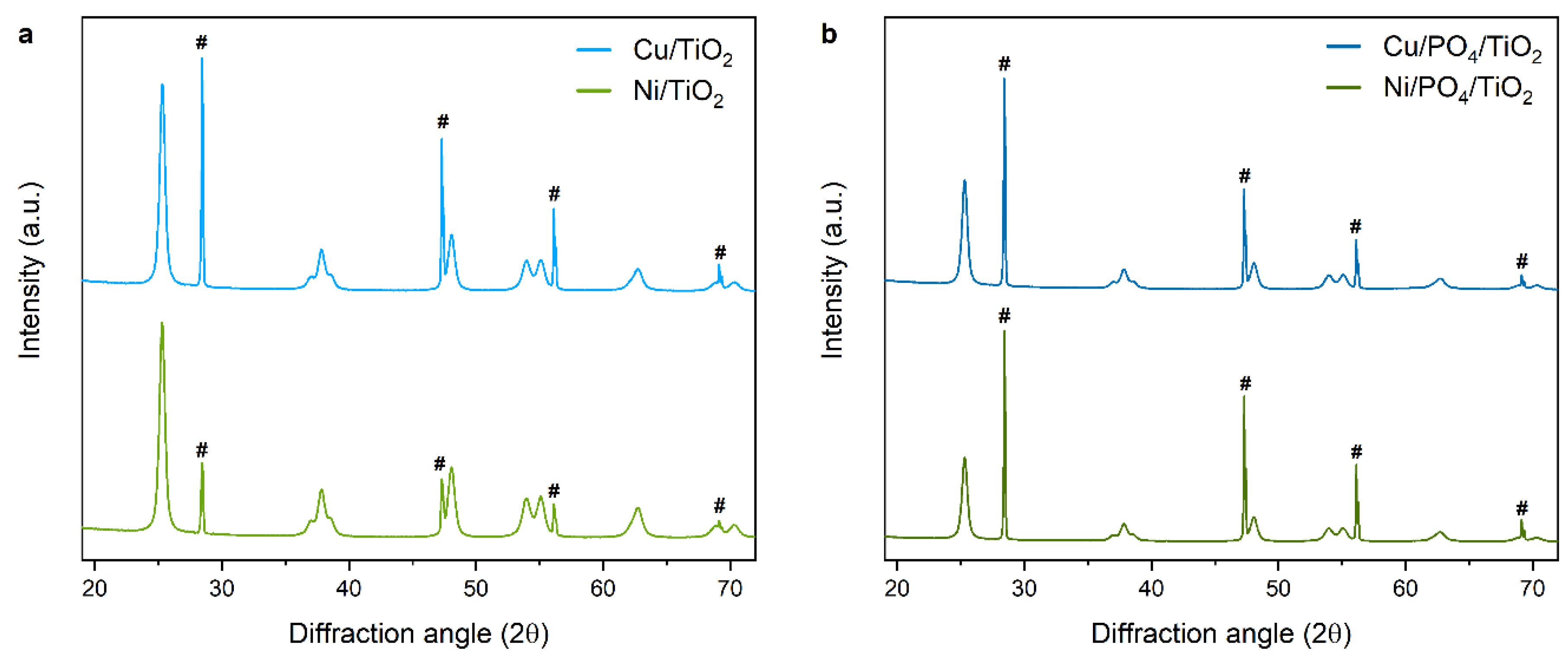
2.2. Characterization
2.2.1. Real Co-Catalyst Loadings
2.2.2. Adsorption and State of Cu and Ni
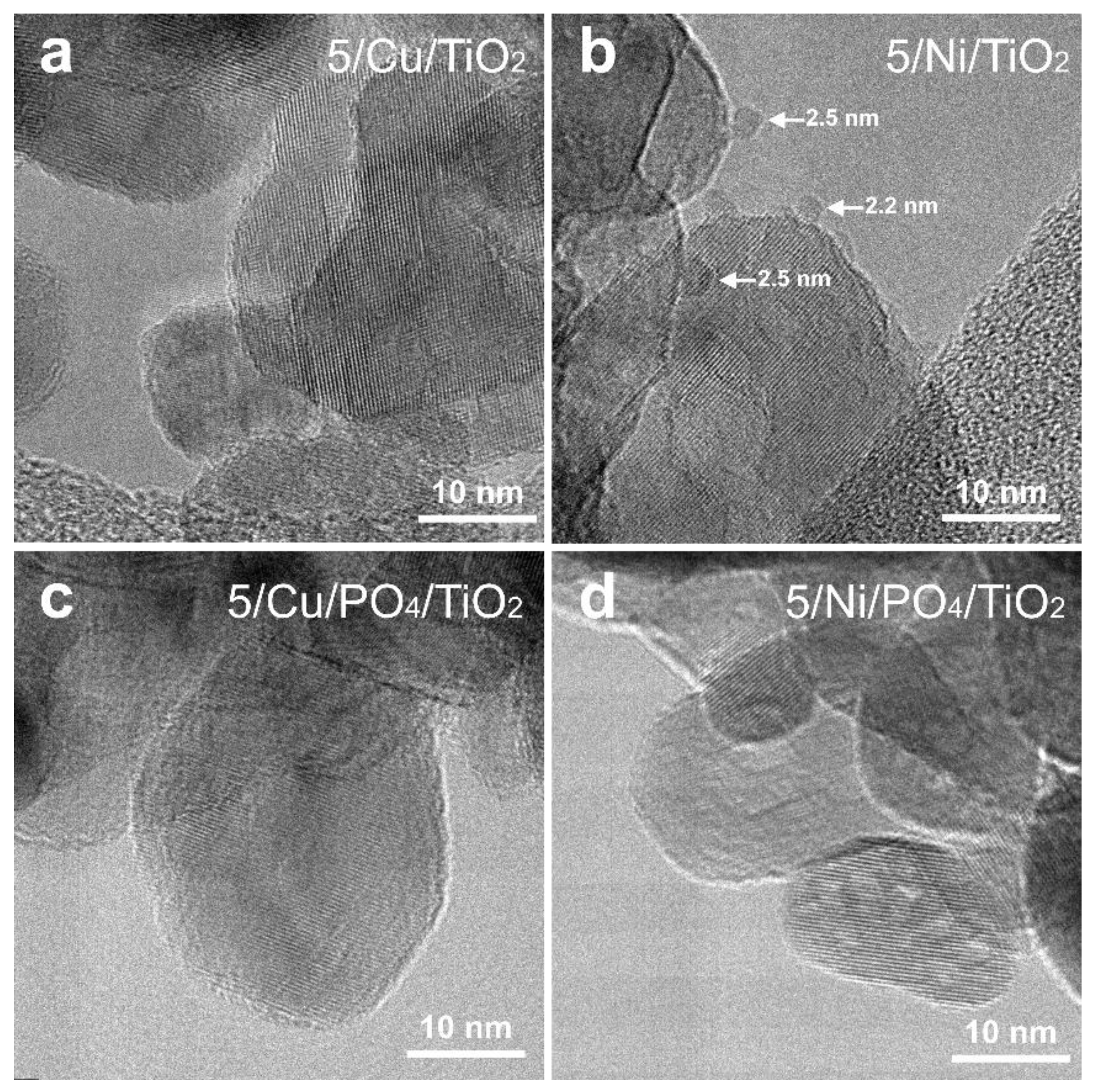
2.2.3. Morphology and Structure
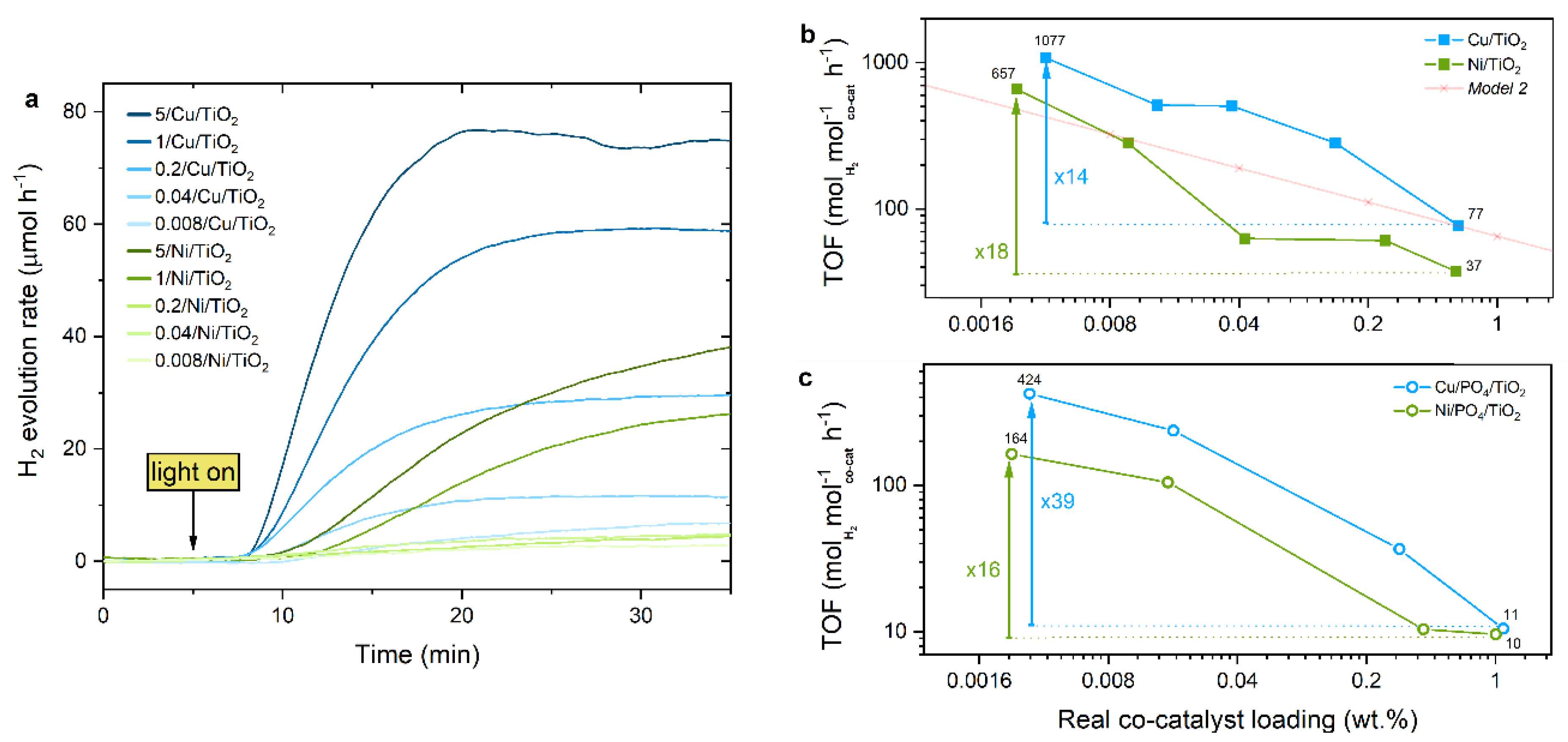
2.3. Photocatalytic Performance
2.3.1. TOF Evaluation
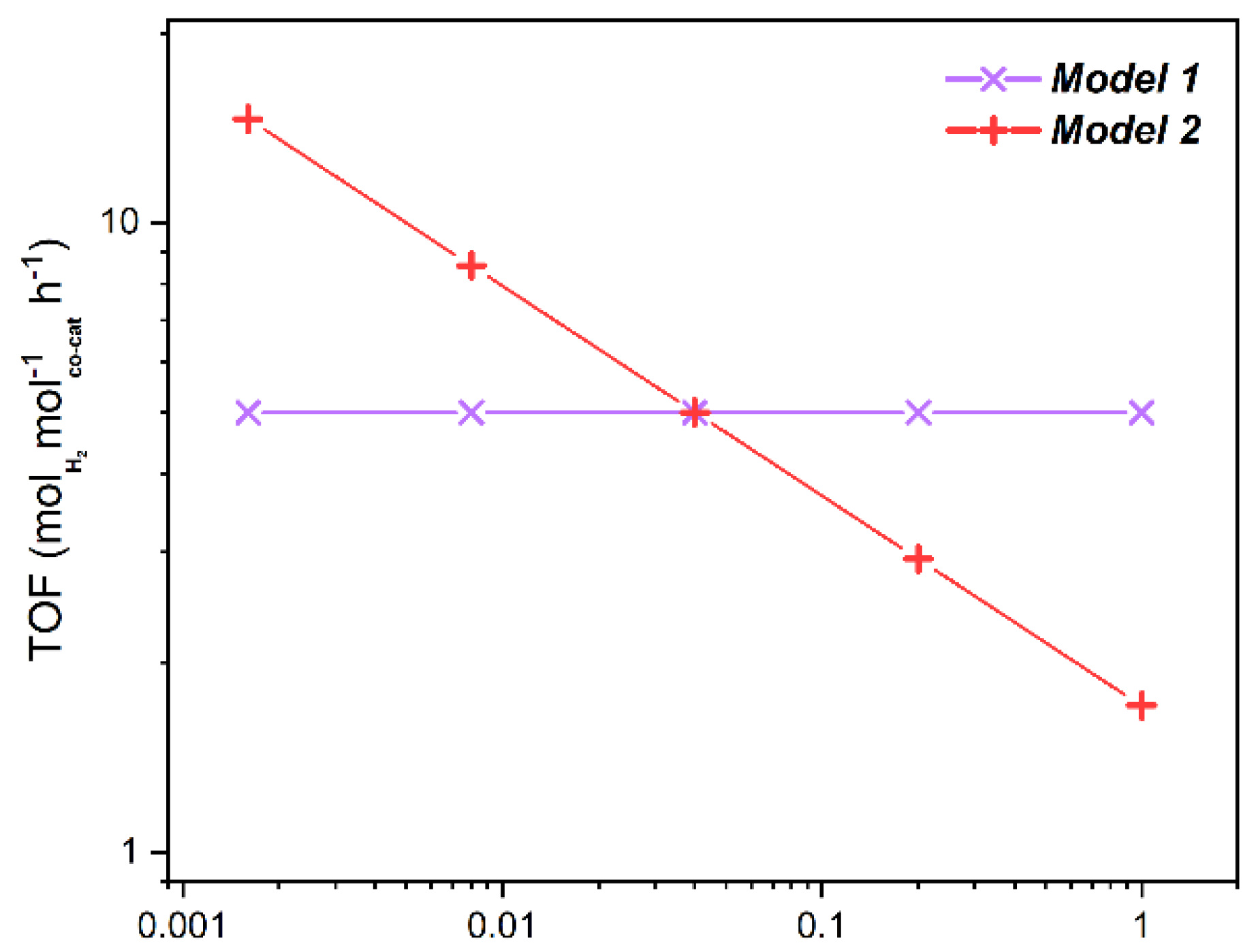
2.3.2. Geometrical Models
2.3.3. Effect of the Support
3. Materials and Methods
3.1. List of Chemicals
3.2. Synthesis of Phosphate-Modified TiO2
3.3. Co-Catalyst Modification of TiO2 (Impregnation Process)
3.4. Characterization Methods
3.4.1. Attenuated-Total Reflection Fourier-Transformed Infrared Spectroscopy (ATR-FTIR)
3.4.2. Diffuse Reflectance Spectroscopy (DRS)
3.4.3. Total Reflection X-Ray Fluorescence Spectroscopy (TXRF)
3.4.4. Powder X-Ray Diffraction (XRD)
3.4.5. Scanning Electron Microscopy (SEM)
3.4.6. Transmission Electron Microscopy (TEM)
3.5. Photocatalytic Tests
Efficiency of a Photocatalyst
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Model Calculations


References
- Detz, R.J.; Reek, J.N.H.; van der Zwaan, B.C.C. The Future of Solar Fuels: When Could They Become Competitive? Energy Environ. Sci. 2018, 11, 1653–1669. [Google Scholar] [CrossRef]
- Wang, Z.; Li, C.; Domen, K. Recent Developments in Heterogeneous Photocatalysts for Solar-Driven Overall Water Splitting. Chem. Soc. Rev. 2019, 48, 2109–2125. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Teramura, K.; Lu, D.; Takata, T.; Saito, N.; Inoue, Y.; Domen, K. Characterization of Rh−Cr Mixed-Oxide Nanoparticles Dispersed on (Ga1-XZnx)(N1-XOx) as a Cocatalyst for Visible-Light-Driven Overall Water Splitting. J. Phys. Chem. B 2006, 110, 13753–13758. [Google Scholar] [CrossRef] [PubMed]
- Kanan, M.W.; Nocera, D.G. In Situ Formation of an Oxygen-Evolving Catalyst in Neutral Water Containing Phosphate and Co2+. Science 2008, 321, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Takata, T.; Jiang, J.; Sakata, Y.; Nakabayashi, M.; Shibata, N.; Nandal, V.; Seki, K.; Hisatomi, T.; Domen, K. Photocatalytic Water Splitting with a Quantum Efficiency of Almost Unity. Nature 2020, 581, 411–414. [Google Scholar] [CrossRef]
- Cui, X.; Li, W.; Ryabchuk, P.; Junge, K.; Beller, M. Bridging Homogeneous and Heterogeneous Catalysis by Heterogeneous Single-Metal-Site Catalysts. Nat. Catal. 2018, 1, 385–397. [Google Scholar] [CrossRef]
- Wang, A.; Li, J.; Zhang, T. Heterogeneous Single-Atom Catalysis. Nat. Rev. Chem. 2018, 2, 65–81. [Google Scholar] [CrossRef]
- Zecchina, A.; Bordiga, S.; Groppo, E. The Structure and Reactivity of Single and Multiple Sites on Heterogeneous and Homogeneous Catalysts: Analogies, Differences, and Challenges for Characterization Methods. In Selective Nanocatalysts and Nanoscience; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2011; pp. 1–27. ISBN 978-3-527-63568-9. [Google Scholar]
- Wang, B.; Cai, H.; Shen, S. Single Metal Atom Photocatalysis. Small Methods 2019, 3, 1800447. [Google Scholar] [CrossRef]
- Li, Y.H.; Xing, J.; Yang, X.H.; Yang, H.G. Cluster Size Effects of Platinum Oxide as Active Sites in Hydrogen Evolution Reactions. Chem. Eur. J. 2014, 20, 12377–12380. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Chen, J.F.; Li, Y.H.; Yuan, W.T.; Zhou, Y.; Zheng, L.R.; Wang, H.F.; Hu, P.; Wang, Y.; Zhao, H.J.; et al. Stable Isolated Metal Atoms as Active Sites for Photocatalytic Hydrogen Evolution. Chem. Eur. J. 2014, 20, 2138–2144. [Google Scholar] [CrossRef]
- Li, X.; Bi, W.; Zhang, L.; Tao, S.; Chu, W.; Zhang, Q.; Luo, Y.; Wu, C.; Xie, Y. Single-Atom Pt as Co-Catalyst for Enhanced Photocatalytic H2 Evolution. Adv. Mater. 2016, 28, 2427–2431. [Google Scholar] [CrossRef]
- Gao, G.; Jiao, Y.; Waclawik, E.R.; Du, A. Single Atom (Pd/Pt) Supported on Graphitic Carbon Nitride as an Efficient Photocatalyst for Visible-Light Reduction of Carbon Dioxide. J. Am. Chem. Soc. 2016, 138, 6292–6297. [Google Scholar] [CrossRef]
- Fang, X.; Shang, Q.; Wang, Y.; Jiao, L.; Yao, T.; Li, Y.; Zhang, Q.; Luo, Y.; Jiang, H.-L. Single Pt Atoms Confined into a Metal–Organic Framework for Efficient Photocatalysis. Adv. Mater. 2018, 30, 1705112. [Google Scholar] [CrossRef]
- Liu, S.; Wang, Y.; Wang, S.; You, M.; Hong, S.; Wu, T.-S.; Soo, Y.-L.; Zhao, Z.; Jiang, G.; Qiu, J.; et al. Photocatalytic Fixation of Nitrogen to Ammonia by Single Ru Atom Decorated TiO2 Nanosheets. ACS Sustain. Chem. Eng. 2019, 7, 6813–6820. [Google Scholar] [CrossRef]
- Yang, Y.; Li, F.; Chen, J.; Fan, J.; Xiang, Q. Single Au Atoms Anchored on Amino-Group-Enriched Graphitic Carbon Nitride for Photocatalytic CO2 Reduction. ChemSusChem 2020, 13, 1979–1985. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Chen, S.; Luo, Q.; Yan, H.; Lin, Y.; Liu, W.; Cao, L.; Lu, J.; Yang, J.; Yao, T.; et al. Atomic-Level Insight into Optimizing the Hydrogen Evolution Pathway over a Co1-N4 Single-Site Photocatalyst. Angew. Chem. Int. Ed. 2017, 56, 12191–12196. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Sun, J.; Li, S.; Huang, C.; Yao, W.; Chen, W.; Zeng, T.; Wu, Q.; Xu, Q. Single Nickel Atoms Anchored on Nitrogen-Doped Graphene as a Highly Active Cocatalyst for Photocatalytic H2 Evolution. ACS Catal. 2018, 8, 11863–11874. [Google Scholar] [CrossRef]
- Huang, P.; Huang, J.; Pantovich, S.A.; Carl, A.D.; Fenton, T.G.; Caputo, C.A.; Grimm, R.L.; Frenkel, A.I.; Li, G. Selective CO2 Reduction Catalyzed by Single Cobalt Sites on Carbon Nitride under Visible-Light Irradiation. J. Am. Chem. Soc. 2018, 140, 16042–16047. [Google Scholar] [CrossRef]
- Lee, B.-H.; Park, S.; Kim, M.; Sinha, A.K.; Lee, S.C.; Jung, E.; Chang, W.J.; Lee, K.-S.; Kim, J.H.; Cho, S.-P.; et al. Reversible and Cooperative Photoactivation of Single-Atom Cu/TiO2 Photocatalysts. Nat. Mater. 2019, 18, 620–626. [Google Scholar] [CrossRef]
- Li, Y.; Li, B.; Zhang, D.; Cheng, L.; Xiang, Q. Crystalline Carbon Nitride Supported Copper Single Atoms for Photocatalytic CO2 Reduction with Nearly 100% CO Selectivity. ACS Nano 2020, 14, 10552–10561. [Google Scholar] [CrossRef]
- Xiao, X.; Gao, Y.; Zhang, L.; Zhang, J.; Zhang, Q.; Li, Q.; Bao, H.; Zhou, J.; Miao, S.; Chen, N.; et al. A Promoted Charge Separation/Transfer System from Cu Single Atoms and C3N4 Layers for Efficient Photocatalysis. Adv. Mater. 2020, 32, 2003082. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Heil, T.; Zhu, B.; Tung, C.-W.; Yu, J.; Chen, H.M.; Antonietti, M.; Cao, S. A Single Cu-Center Containing Enzyme-Mimic Enabling Full Photosynthesis under CO2 Reduction. ACS Nano 2020, 14, 8584–8593. [Google Scholar] [CrossRef] [PubMed]
- Schubert, J.S.; Popovic, J.; Haselmann, G.M.; Nandan, S.P.; Wang, J.; Giesriegl, A.; Cherevan, A.S.; Eder, D. Immobilization of Co, Mn, Ni and Fe Oxide Co-Catalysts on TiO2 for Photocatalytic Water Splitting Reactions. J. Mater. Chem. A 2019, 7, 18568–18579. [Google Scholar] [CrossRef]
- Ji, S.; Chen, Y.; Wang, X.; Zhang, Z.; Wang, D.; Li, Y. Chemical Synthesis of Single Atomic Site Catalysts. Chem. Rev. 2020. [Google Scholar] [CrossRef]
- Han, X.; Ling, X.; Wang, Y.; Ma, T.; Zhong, C.; Hu, W.; Deng, Y. Generation of Nanoparticle, Atomic-Cluster, and Single-Atom Cobalt Catalysts from Zeolitic Imidazole Frameworks by Spatial Isolation and Their Use in Zinc–Air Batteries. Angew. Chem. Int. Ed. 2019, 58, 5359–5364. [Google Scholar] [CrossRef]
- Hadjiivanov, K.I.; Klissurski, D.G.; Davydov, A.A. Study of Phosphate-Modified TiO2 (Anatase). J. Catal. 1989, 116, 498–505. [Google Scholar] [CrossRef]
- Zhao, D.; Chen, C.; Wang, Y.; Ji, H.; Ma, W.; Zang, L.; Zhao, J. Surface Modification of TiO2 by Phosphate: Effect on Photocatalytic Activity and Mechanism Implication. J. Phys. Chem. C 2008, 112, 5993–6001. [Google Scholar] [CrossRef]
- Liu, X.; Li, Y.; Peng, S.; Lu, G.; Li, S. Modification of TiO2 with Sulfate and Phosphate for Enhanced Eosin Y-Sensitized Hydrogen Evolution under Visible Light Illumination. Photochem. Photobiol. Sci. 2013, 12, 1903–1910. [Google Scholar] [CrossRef]
- Möllers, F.; Tolle, H.J.; Memming, R. On the Origin of the Photocatalytic Deposition of Noble Metals on TiO2. J. Electrochem. Soc. 1974, 121, 1160. [Google Scholar] [CrossRef]
- Diebold, U. The Surface Science of Titanium Dioxide. Surface Sci. Rep. 2003, 48, 53–229. [Google Scholar] [CrossRef]
- Qiu, S.R.; Wood, B.C.; Ehrmann, P.R.; Demos, S.G.; Miller, P.E.; Schaffers, K.I.; Suratwala, T.I.; Brow, R.K. Origins of Optical Absorption Characteristics of Cu2+ Complexes in Aqueous Solutions. Phys. Chem. Chem. Phys. 2015, 17, 18913–18923. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yang, L.; Zhao, W.; Cao, L.; Sun, Z.; Zhang, F. A Facile Synthesis of Copper Nanoparticles Supported on an Ordered Mesoporous Polymer as an Efficient and Stable Catalyst for Solvent-Free Sonogashira Coupling Reactions. Green Chem. 2017, 19, 1949–1957. [Google Scholar] [CrossRef]
- Chuang, C.-C.; Lin, C.-K.; Wang, T.T.; Srinivasadesikan, V.; Raghunath, P.; Lin, M.C. Computational and Experimental Studies on the Effect of Hydrogenation of Ni-Doped TiO2 Anatase Nanoparticles for the Application of Water Splitting. RSC Adv. 2015, 5, 81371–81377. [Google Scholar] [CrossRef]
- Meftah, A.M.A.; Saion, E.; Abd Moksin, M.M.; Zainuddin, H. Absorbance of Nickel Nanoparticles/Polyaniline Composite Films Prepared by Radiation Technique. Solid State Sci. Technol. 2009, 17, 167–174. [Google Scholar]
- Lewis, F.D.; Salvi, G.D.; Kanis, D.R.; Ratner, M.A. Electronic Structure and Spectroscopy of Nickel(II), Palladium(II), and Platinum(II) Acetylacetonate Complexes. Inorg. Chem. 1993, 32, 1251–1258. [Google Scholar] [CrossRef]
- Mikuriya, M.; Schumacher, M.; Kawano, C.; Akihara, T.; Ono, K.; Yoshioka, D.; Sakiyama, H.; Handa, M. Dinuclear Nickel(II) Pivalate with Μ-Aqua and Di-Μ-Pivalato Bridges Showing a Ferromagnetic Interaction. Chem. J. Mold. 2014, 9, 62–66. [Google Scholar] [CrossRef]
- González, E.; Rodrigue-Witchel, A.; Reber, C. Absorption Spectroscopy of Octahedral Nickel(II) Complexes: A Case Study of Interactions between Multiple Electronic Excited States. Coord. Chem. Rev. 2007, 251, 351–363. [Google Scholar] [CrossRef]
- Zhu, S.; Wang, L.M.; Zu, X.T.; Xiang, X. Optical and Magnetic Properties of Ni Nanoparticles in Rutile Formed by Ni Ion Implantation. Appl. Phys. Lett. 2006, 88, 043107. [Google Scholar] [CrossRef]
- Cromer, D.T.; Herrington, K. The Structures of Anatase and Rutile. J. Am. Chem. Soc. 1955, 77, 4708–4709. [Google Scholar] [CrossRef]
- Bertolotti, F.; Vivani, A.; Moscheni, D.; Ferri, F.; Cervellino, A.; Masciocchi, N.; Guagliardi, A. Structure, Morphology, and Faceting of TiO2 Photocatalysts by the Debye Scattering Equation Method. The P25 and P90 Cases of Study. Nanomaterials 2020, 10, 743. [Google Scholar] [CrossRef]
- Korzhak, A.V.; Ermokhina, N.I.; Stroyuk, A.L.; Bukhtiyarov, V.K.; Raevskaya, A.E.; Litvin, V.I.; Kuchmiy, S.Y.; Ilyin, V.G.; Manorik, P.A. Photocatalytic Hydrogen Evolution over Mesoporous TiO2/Metal Nanocomposites. J. Photochem. Photobiol. A Chem. 2008, 198, 126–134. [Google Scholar] [CrossRef]
- Huerta-Flores, A.M.; Torres-Martínez, L.M.; Moctezuma, E.; Ceballos-Sanchez, O. Enhanced Photocatalytic Activity for Hydrogen Evolution of SrZrO3 Modified with Earth Abundant Metal Oxides (MO, M = Cu, Ni, Fe, Co). Fuel 2016, 181, 670–679. [Google Scholar] [CrossRef]
- Soto-Arreola, A.; Huerta-Flores, A.M.; Mora-Hernández, J.M.; Torres-Martínez, L.M. Comparative Study of the Photocatalytic Activity for Hydrogen Evolution of MFe2O4 (M = Cu, Ni) Prepared by Three Different Methods. J. Photochem. Photobiol. A Chem. 2018, 357, 20–29. [Google Scholar] [CrossRef]
- Trasatti, S. Work Function, Electronegativity, and Electrochemical Behaviour of Metals: III. Electrolytic Hydrogen Evolution in Acid Solutions. J. Electroanal. Chem. Interfacial Electrochem. 1972, 39, 163–184. [Google Scholar] [CrossRef]
- Jenkins, H.D.B.; Thakur, K.P. Reappraisal of Thermochemical Radii for Complex Ions. J. Chem. Educ. 1979, 56, 576. [Google Scholar] [CrossRef]
- Manku, G.S. Theoretical Principles of Inorganic Chemistry; Tata McGraw-Hill Education: New York, NY, USA, 1980; ISBN 978-0-07-096500-3. [Google Scholar]
- Ramer, G.; Lendl, B. Attenuated Total Reflection Fourier Transform Infrared Spectroscopy. In Encyclopedia of Analytical Chemistry; American Cancer Society: Atlanta, GA, USA, 2013; ISBN 978-0-470-02731-8. [Google Scholar]
- Wobrauschek, P. Total Reflection X-Ray Fluorescence Analysis—A Review. X-Ray Spectrom. 2007, 36, 289–300. [Google Scholar] [CrossRef]
- Prost, J.; Wobrauschek, P.; Streli, C. Dual Energy-Band Excitation from a Low Power Rh Anode X-Ray Tube for the Simultaneous Determination of Low Z and High Z Elements (Na-U) Using Total-Reflection X-Ray Fluorescence Analysis (TXRF). Rev. Sci. Instrum. 2018, 89, 093108. [Google Scholar] [CrossRef] [PubMed]
- Kozuch, S.; Martin, J.M.L. “Turning Over” Definitions in Catalytic Cycles. ACS Catal. 2012, 2, 2787–2794. [Google Scholar] [CrossRef]
- Ye, S.; Ding, C.; Li, C. Chapter One—Artificial photosynthesis systems for catalytic water oxidation. In Advances in Inorganic Chemistry; van Eldik, R., Hubbard, C.D., Eds.; Water Oxidation Catalysts; Academic Press: Cambridge, MA, USA, 2019; Volume 74, pp. 3–59. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cu/TiO2 | Real Loading (wt.%) * | Cu/PO4/TiO2 | Real Loading (wt.%) |
| 5/Cu/TiO2 | 0.6126 ± 0.0072 | 5/Cu/PO4/TiO2 | 1.1004 ± 0.0090 |
| 1/Cu/TiO2 | 0.1326 ± 0.0036 | 1/Cu/PO4/TiO2 | 0.3012 ± 0.0048 |
| 0.2/Cu/TiO2 | 0.0366 ± 0.0024 | 0.2/Cu/PO4/TiO2 | 0.0180 ± 0.0018 |
| 0.04/Cu/TiO2 | 0.0144 ± 0.0018 | 0.04/Cu/PO4/TiO2 | 0.003 ± 0.0012 |
| 0.008/Cu/TiO2 | 0.0036 ± 0.0018 | 0.008/Cu/PO4/TiO2 | <0.0018 ** |
| Ni/TiO2 | Real Loading (wt.%) * | Ni/PO4/TiO2 | Real Loading (wt.%) |
| 5/Ni/TiO2 | 0.5976 ± 0.0084 | 5/Ni/PO4/TiO2 | 1.0038 ± 0.0102 |
| 1/Ni/TiO2 | 0.2472 ± 0.0054 | 1/Ni/PO4/TiO2 | 0.4068 ± 0.0060 |
| 0.2/Ni/TiO2 | 0.0429 ± 0.0043 | 0.2/Ni/PO4/TiO2 | 0.0168 ± 0.0018 |
| 0.04/Ni/TiO2 | 0.0096 ± 0.0018 | 0.04/Ni/PO4/TiO2 | 0.0024 ± 0.0002 |
| 0.008/Ni/TiO2 | 0.0024 ± 0.0018 | 0.008/Ni/PO4/TiO2 | <0.0018 ** |
| Sample | Si * (a, Å) | Anatase (a, Å) | Anatase (c, Å) |
|---|---|---|---|
| 5/Cu/TiO2 | 5.431 | 3.789 | 9.495 |
| 5/Ni/TiO2 | 5.431 | 3.789 | 9.511 |
| 5/Cu/PO4/TiO2 | 5.431 | 3.788 | 9.511 |
| 5/Ni/PO4/TiO2 | 5.431 | 3.789 | 9.511 |
| Chemical Formula | Name | CAS Number | Purity | Brand |
|---|---|---|---|---|
| CH3OH | Methanol | 67-56-1 | absolute | VWR chemicals |
| TiO2 | Anatase | 1317-70-0 | 99.7% | Sigma-Aldrich |
| H3PO4 | Phosphoric acid | 7664-38-2 | extra pure | Acros organics |
| Ni(Ac)2·4H2O | Nickel(II) acetate tetrahydrate | 6018-89-9 | 99% | Sigma-Aldrich |
| Ni(AcAc)2 | Nickel(II) acetylacetonate | 3264-82-2 | 96% | Sigma-Aldrich |
| Cu(Ac)2·H2O | Copper(II) acetate monohydrate | 6046-93-1 | 99% | Fluka |
| HAuCl4·3H2O | Tetrachloroauric(III) acid trihydrate | 16961-25-4 | 99.99% | Alfa Aesar |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ayala, P.; Giesriegl, A.; Nandan, S.P.; Myakala, S.N.; Wobrauschek, P.; Cherevan, A. Isolation Strategy towards Earth-Abundant Single-Site Co-Catalysts for Photocatalytic Hydrogen Evolution Reaction. Catalysts 2021, 11, 417. https://doi.org/10.3390/catal11040417
Ayala P, Giesriegl A, Nandan SP, Myakala SN, Wobrauschek P, Cherevan A. Isolation Strategy towards Earth-Abundant Single-Site Co-Catalysts for Photocatalytic Hydrogen Evolution Reaction. Catalysts. 2021; 11(4):417. https://doi.org/10.3390/catal11040417
Chicago/Turabian StyleAyala, Pablo, Ariane Giesriegl, Sreejith P. Nandan, Stephen Nagaraju Myakala, Peter Wobrauschek, and Alexey Cherevan. 2021. "Isolation Strategy towards Earth-Abundant Single-Site Co-Catalysts for Photocatalytic Hydrogen Evolution Reaction" Catalysts 11, no. 4: 417. https://doi.org/10.3390/catal11040417
APA StyleAyala, P., Giesriegl, A., Nandan, S. P., Myakala, S. N., Wobrauschek, P., & Cherevan, A. (2021). Isolation Strategy towards Earth-Abundant Single-Site Co-Catalysts for Photocatalytic Hydrogen Evolution Reaction. Catalysts, 11(4), 417. https://doi.org/10.3390/catal11040417