
TiO2 Photocatalysis for the Transformation of Aromatic Water Pollutants into Fuels





Abstract
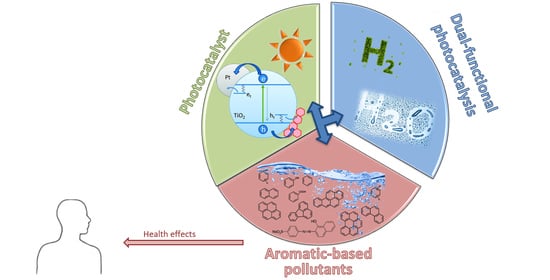
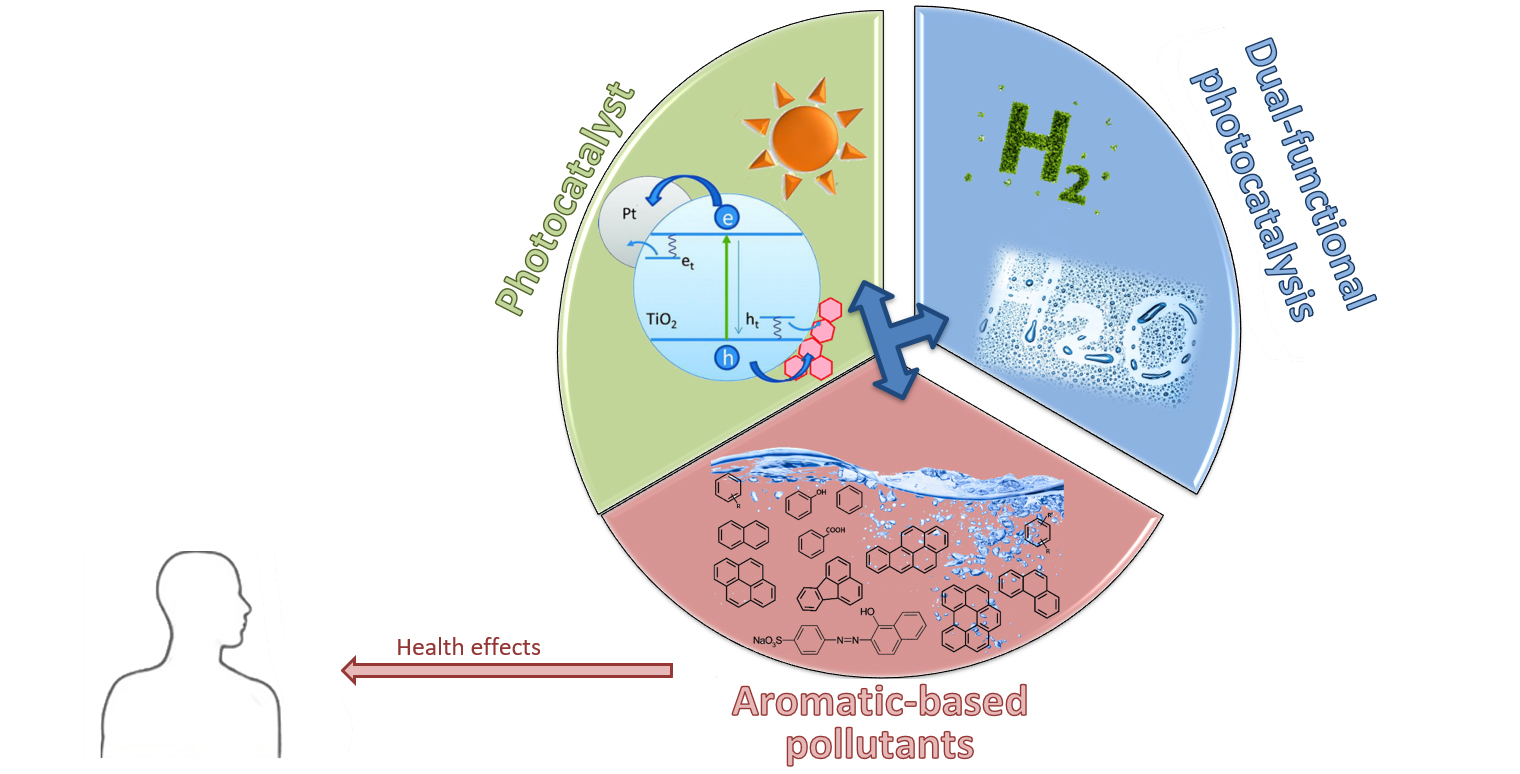
1. Introduction
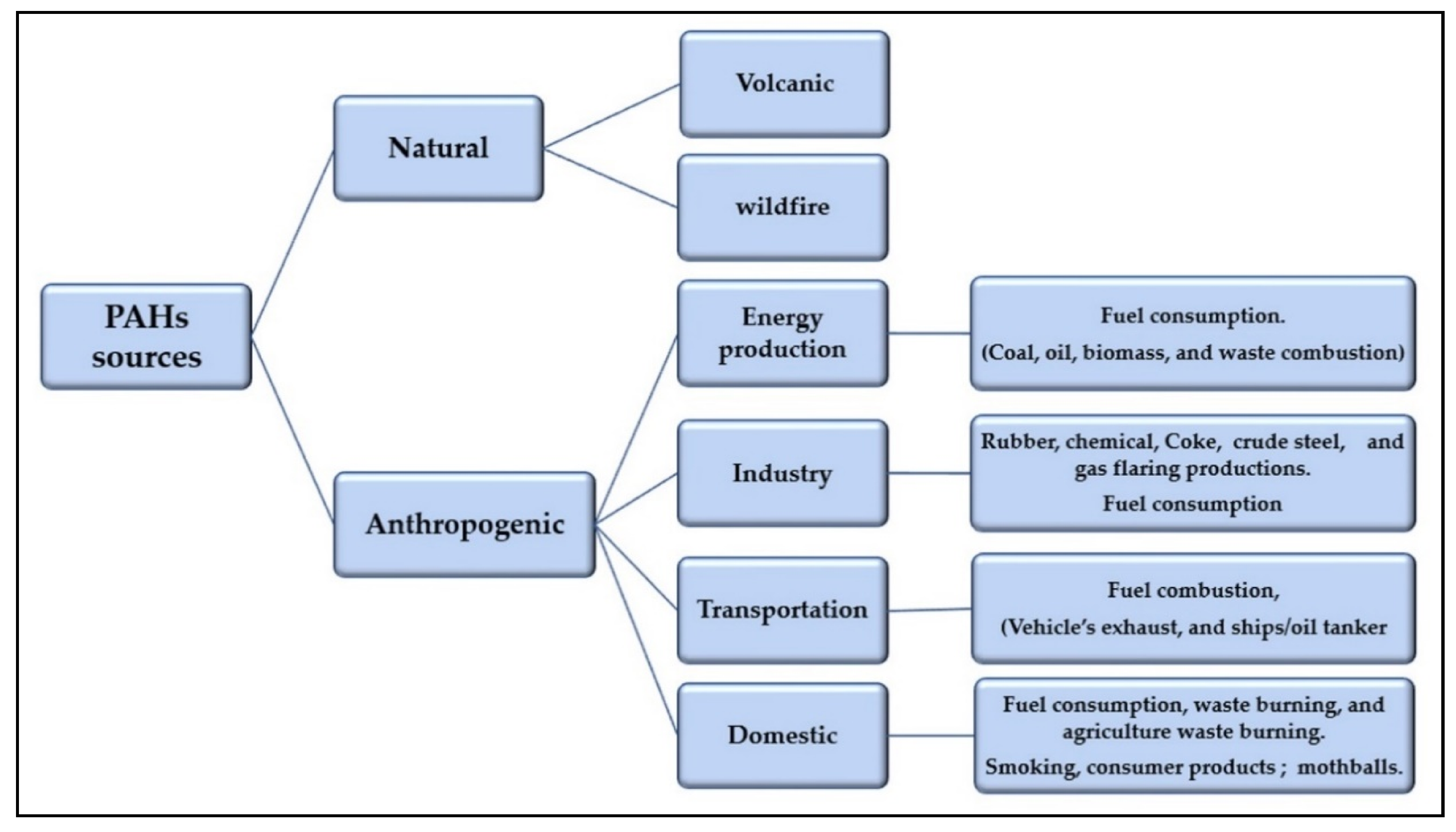
2. Aromatic Hydrocarbons as Water Pollutants
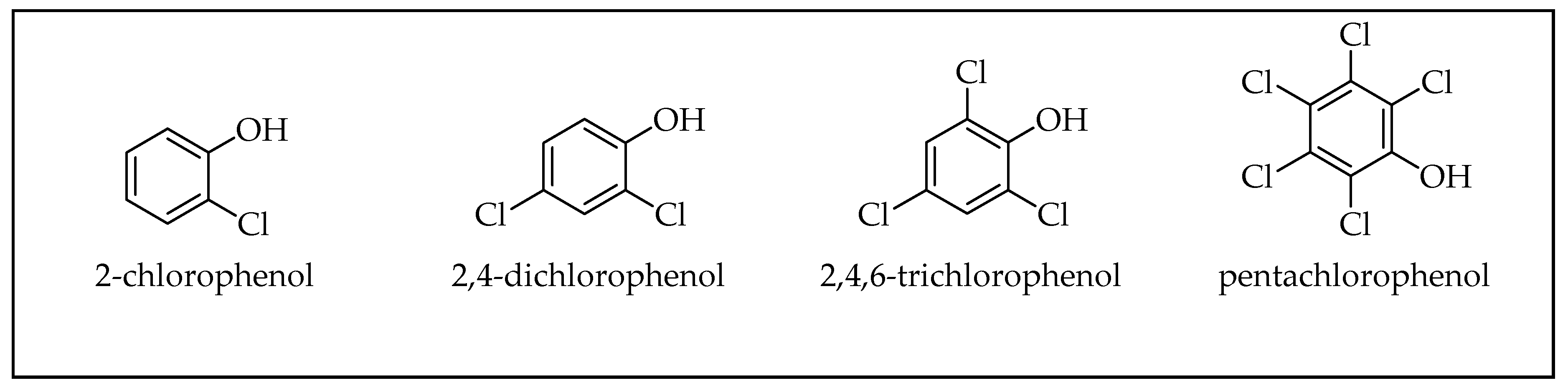
2.1. Phenols
2.2. Polyaromatic Hydrocarbons
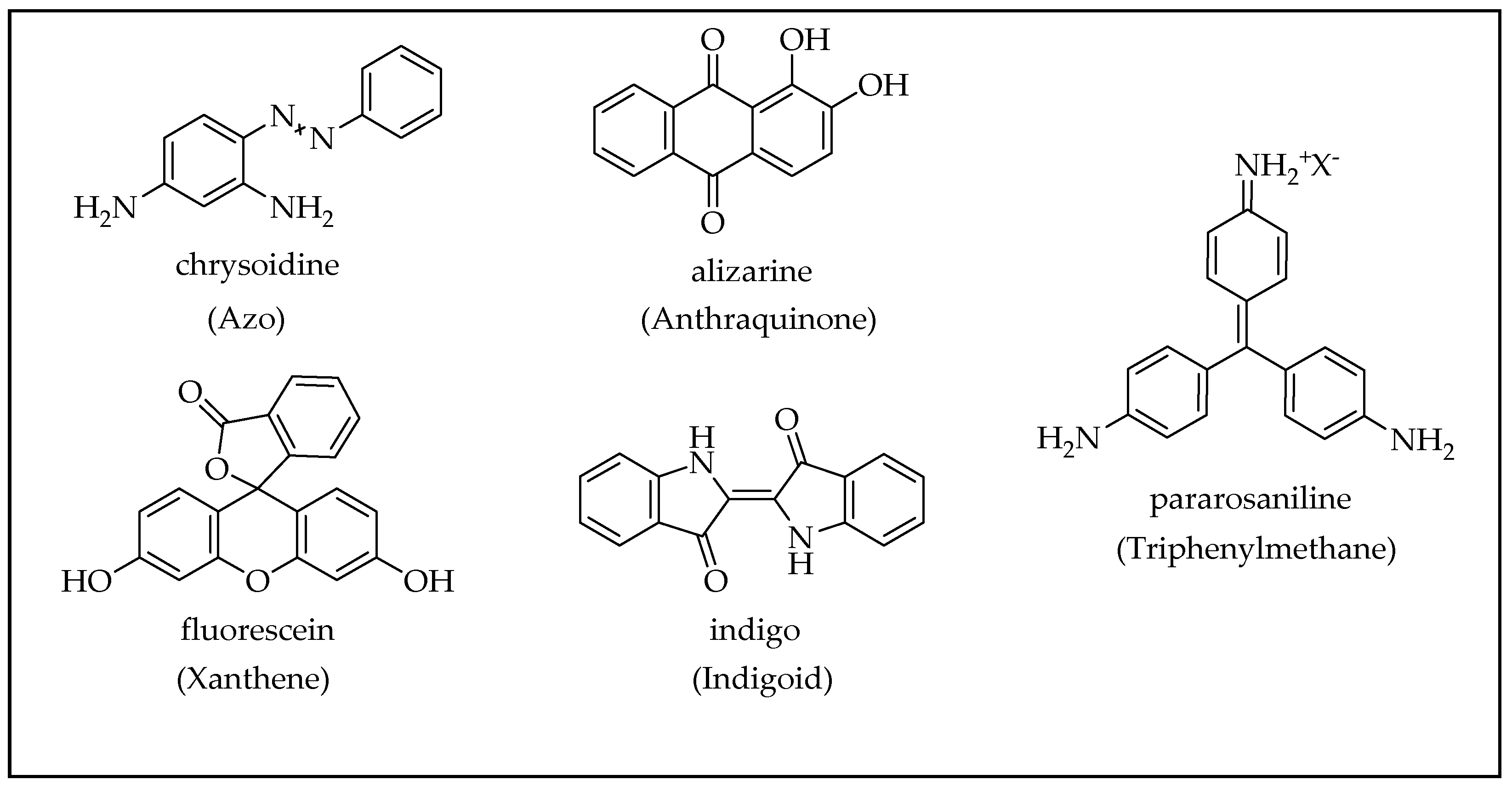
2.3. Organic Dyes
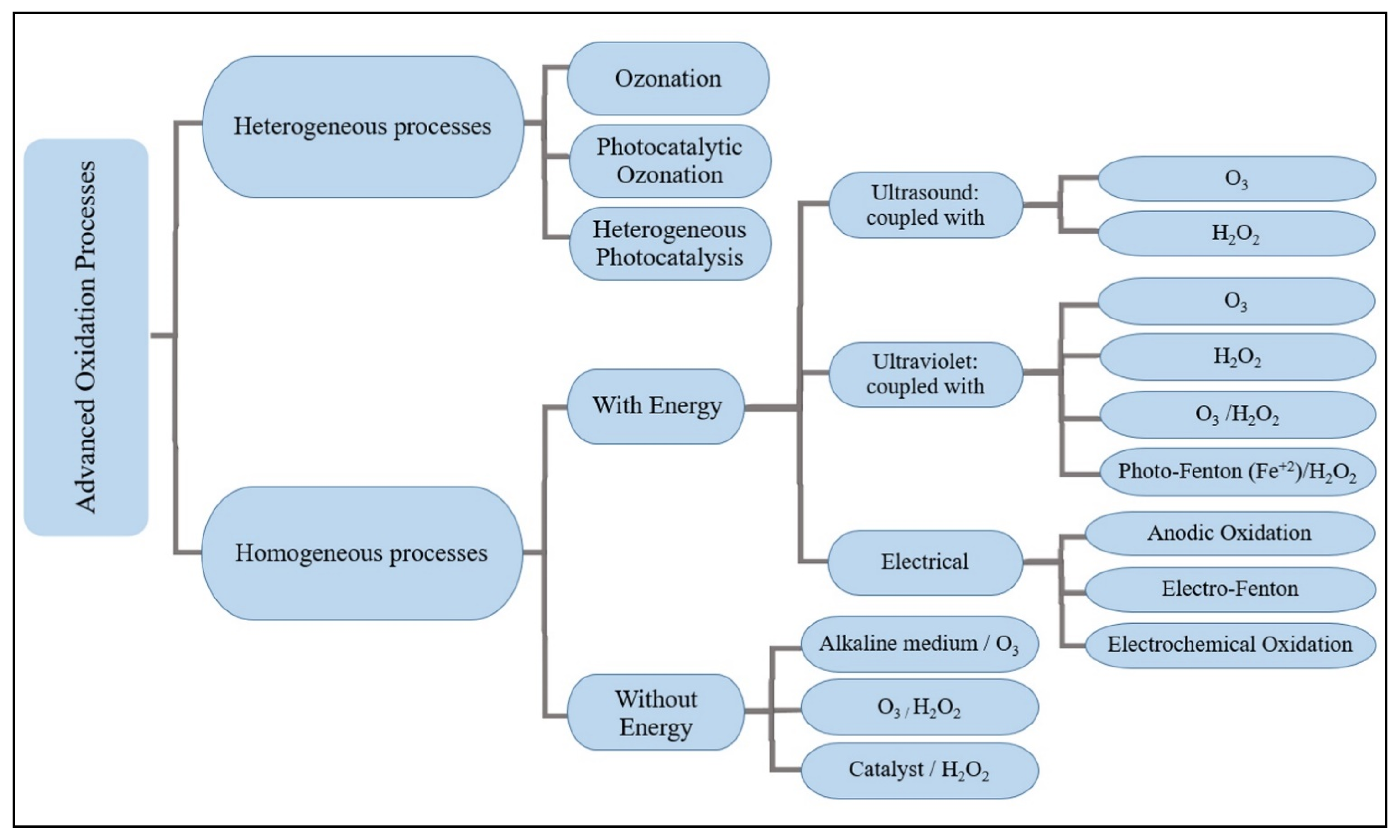
3. Methods of Treatment
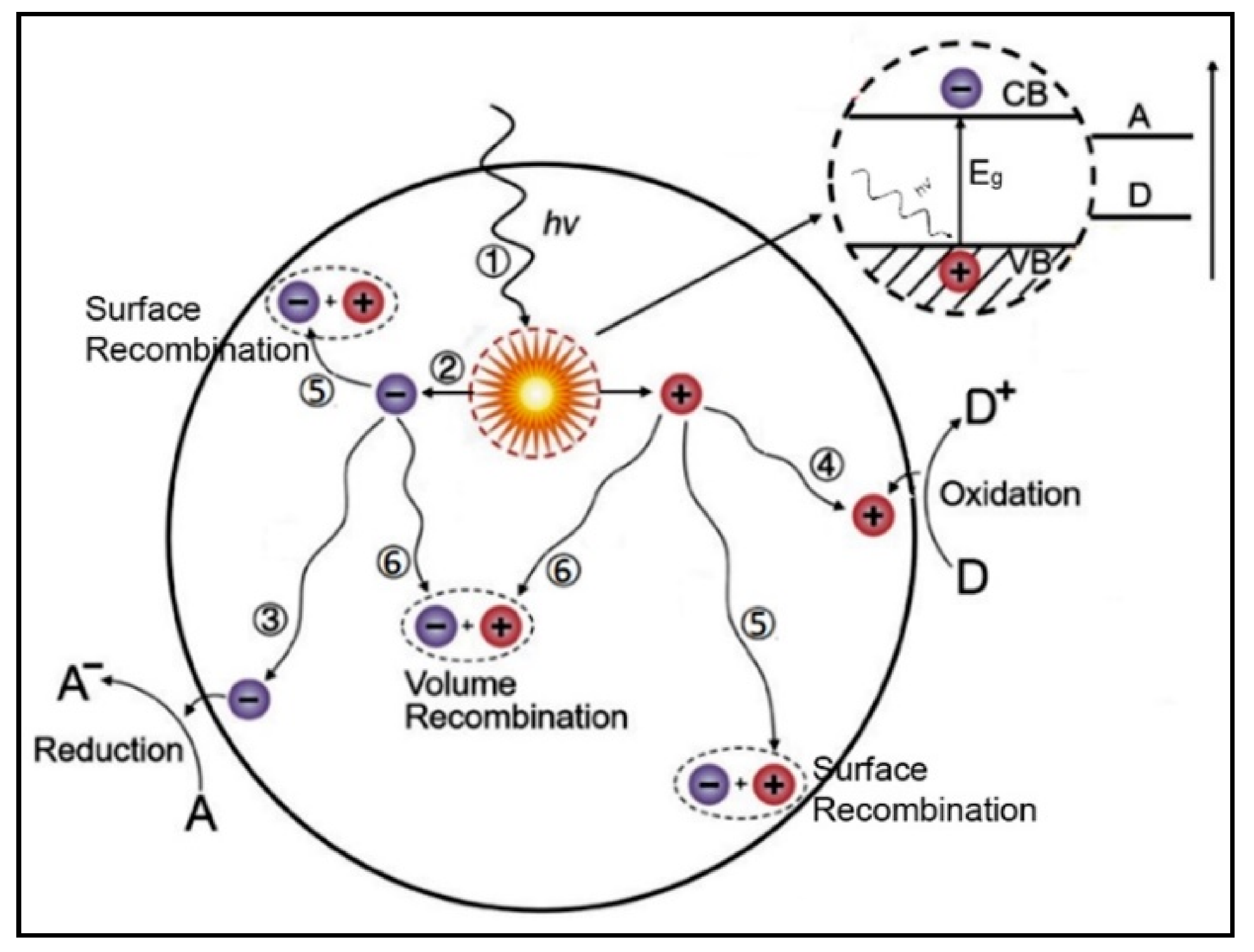
4. Semiconductor-Based Heterogeneous Photocatalysis
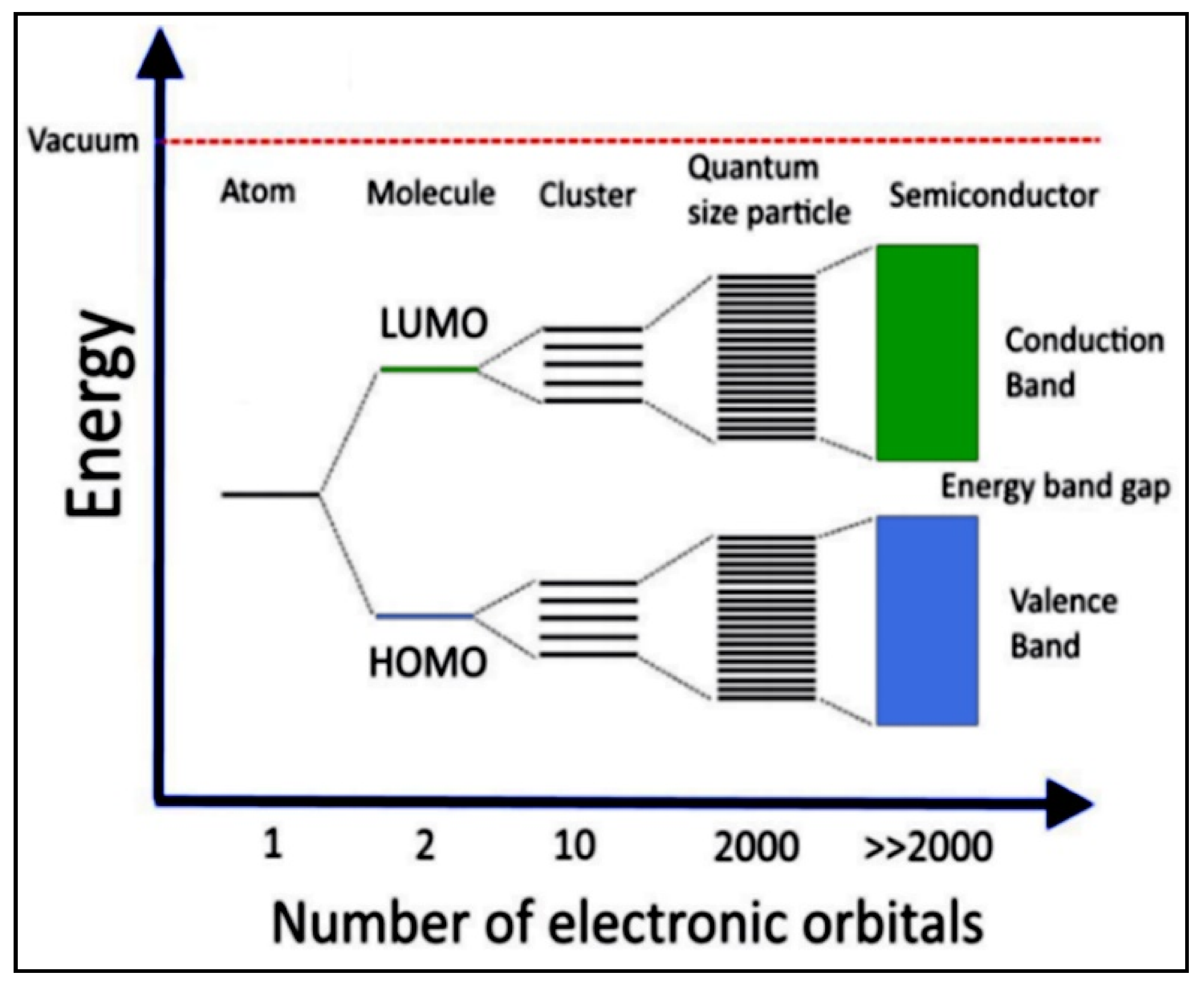
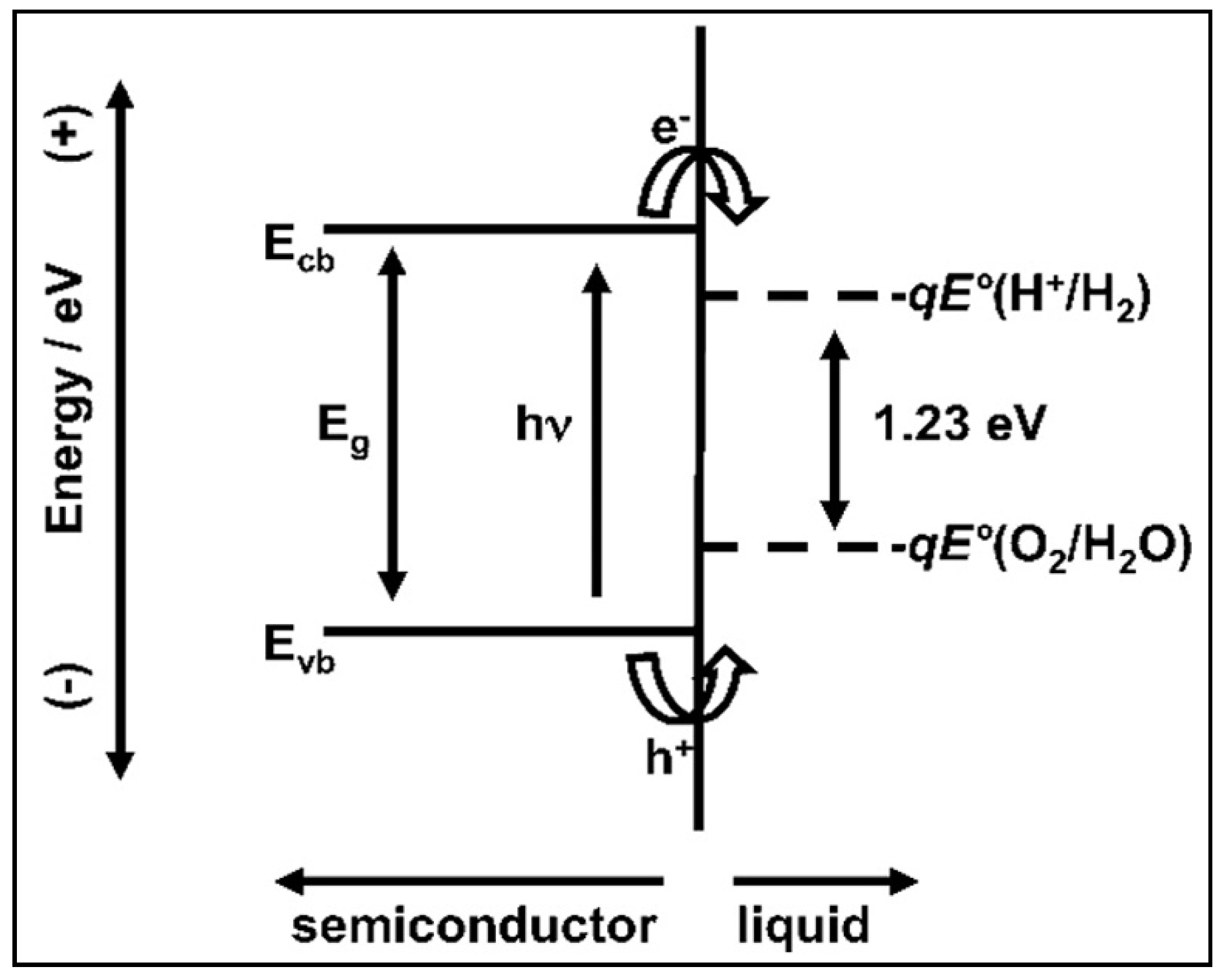
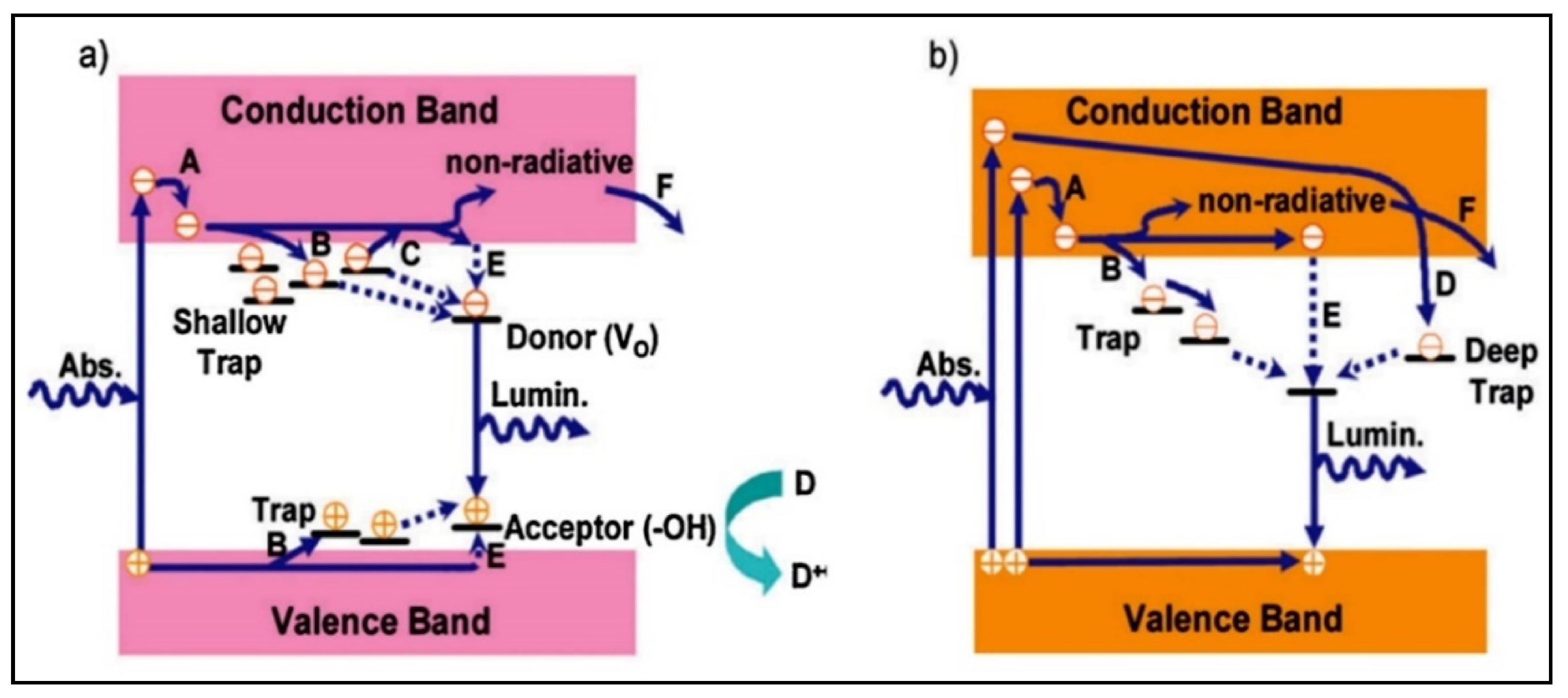
4.1. The Electronic Structure of a Semiconductor–Photocatalyst
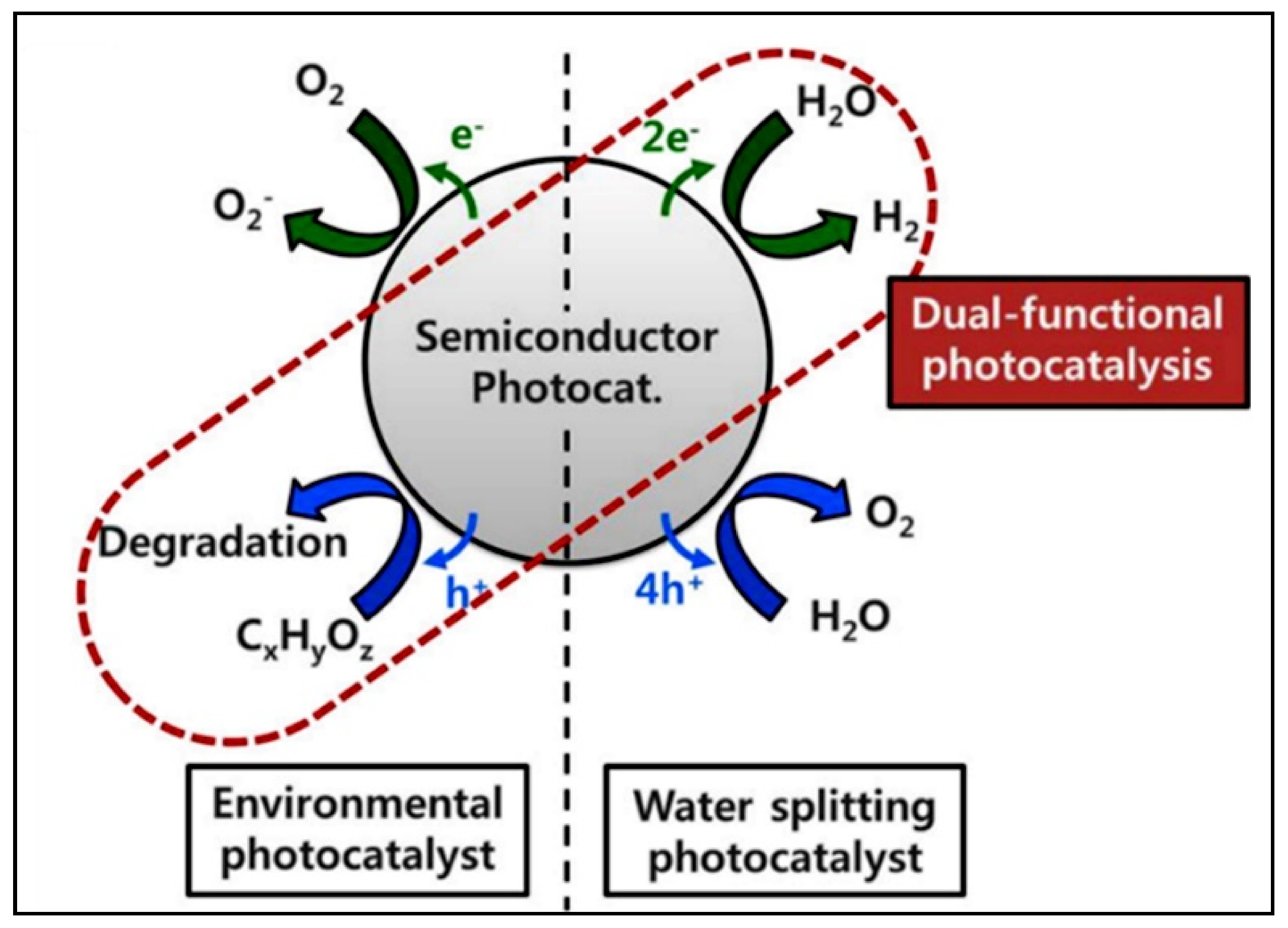
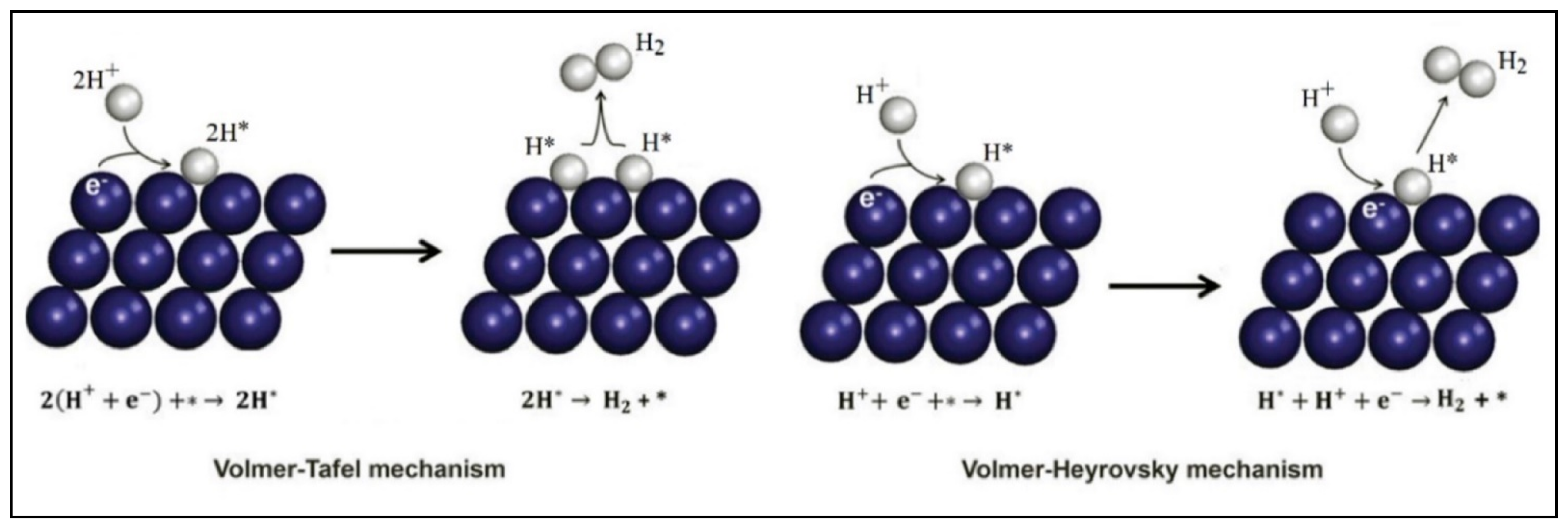
4.2. Photocatalytic Water Splitting vs. Photocatalytic Reforming
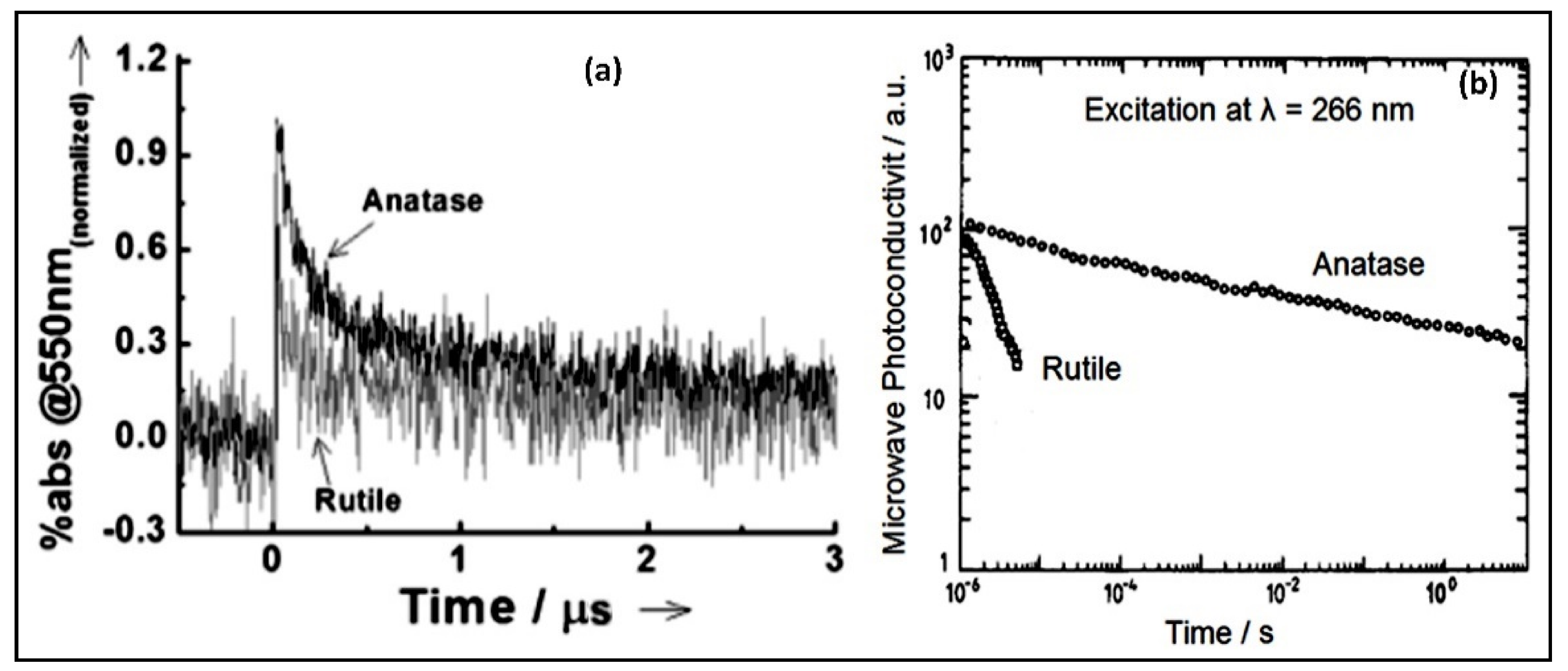
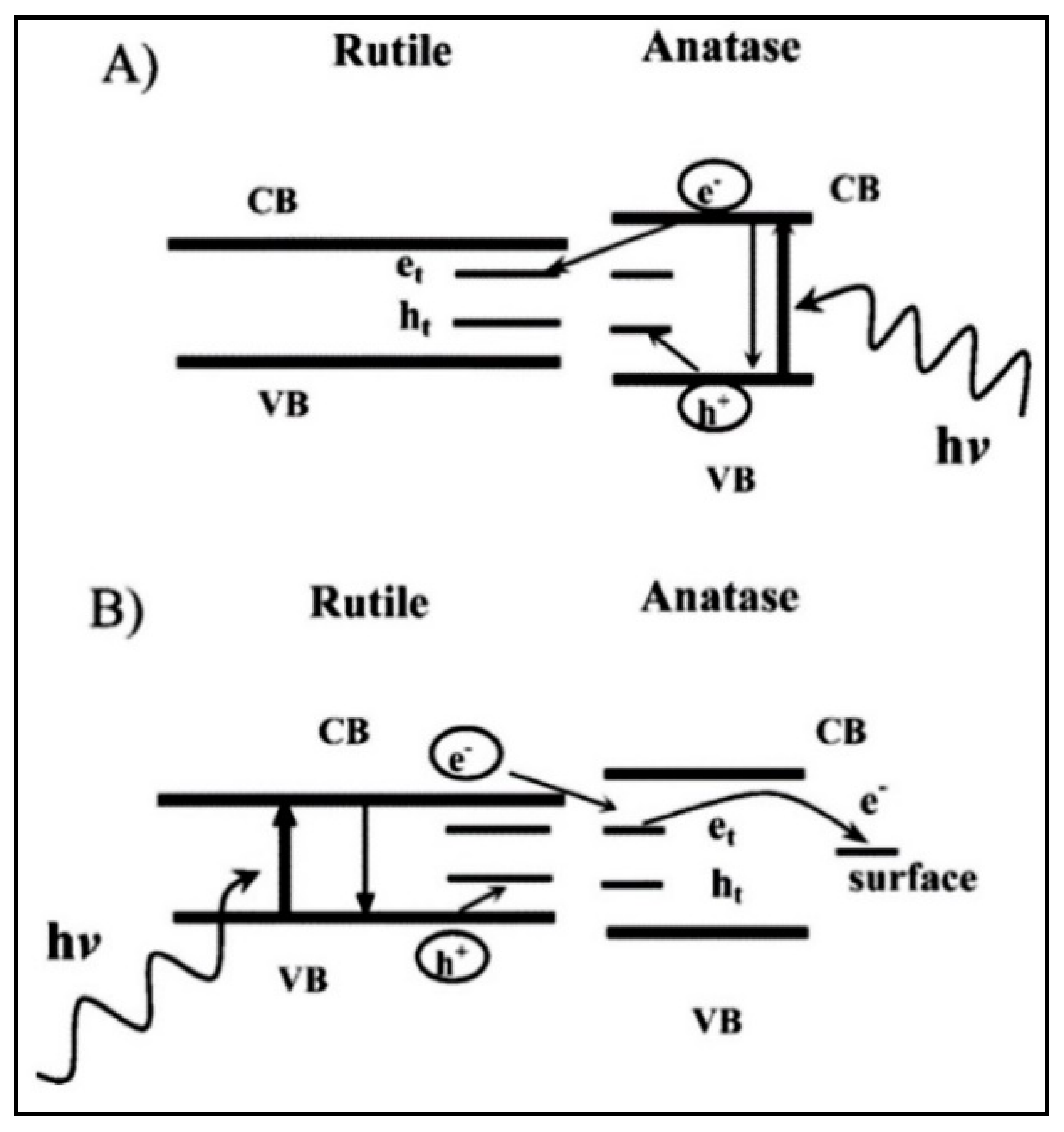
4.3. Titanium Dioxide (TiO2) as a Photocatalyst
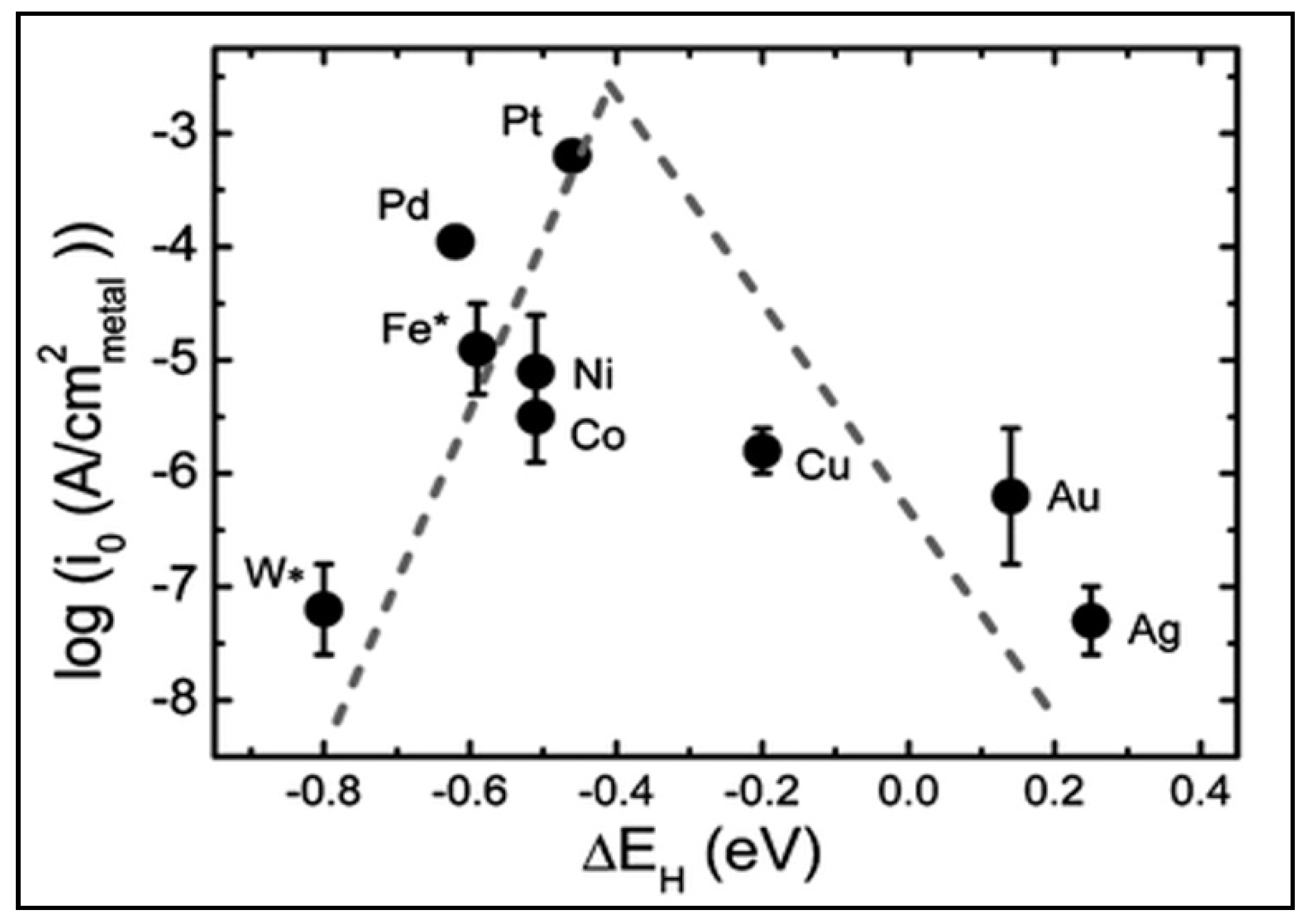
4.4. Enhancing the Performance of Pristine TiO2
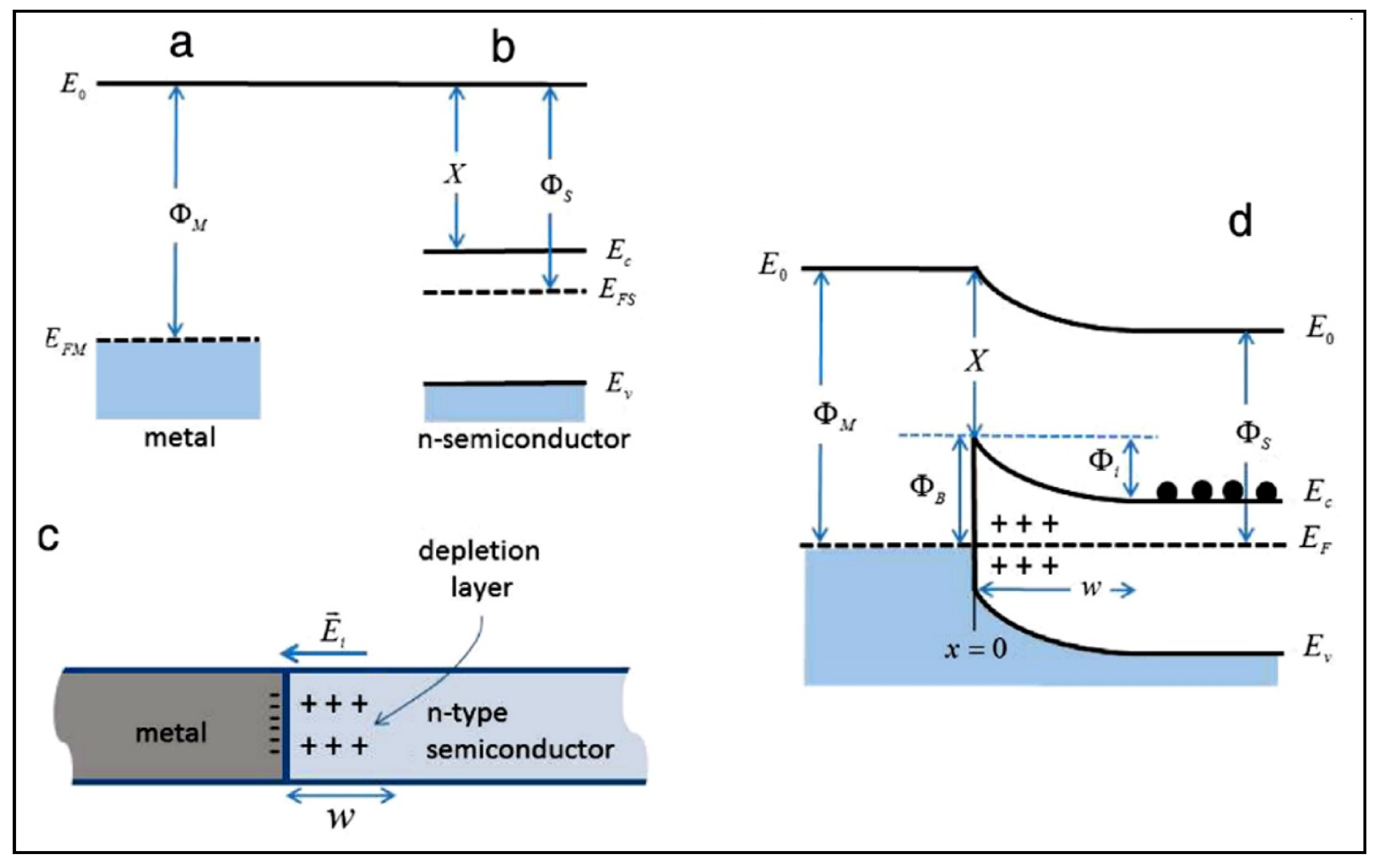
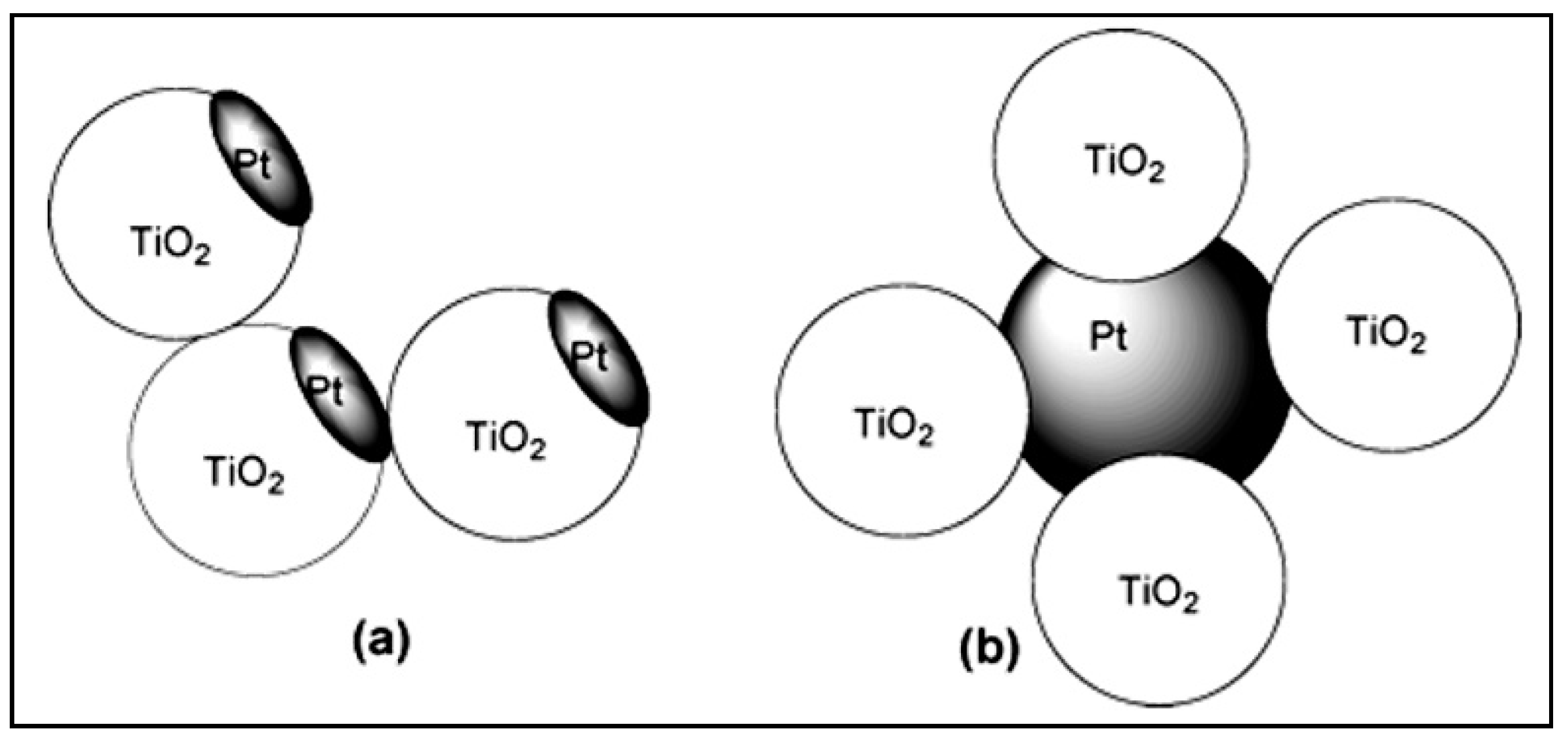
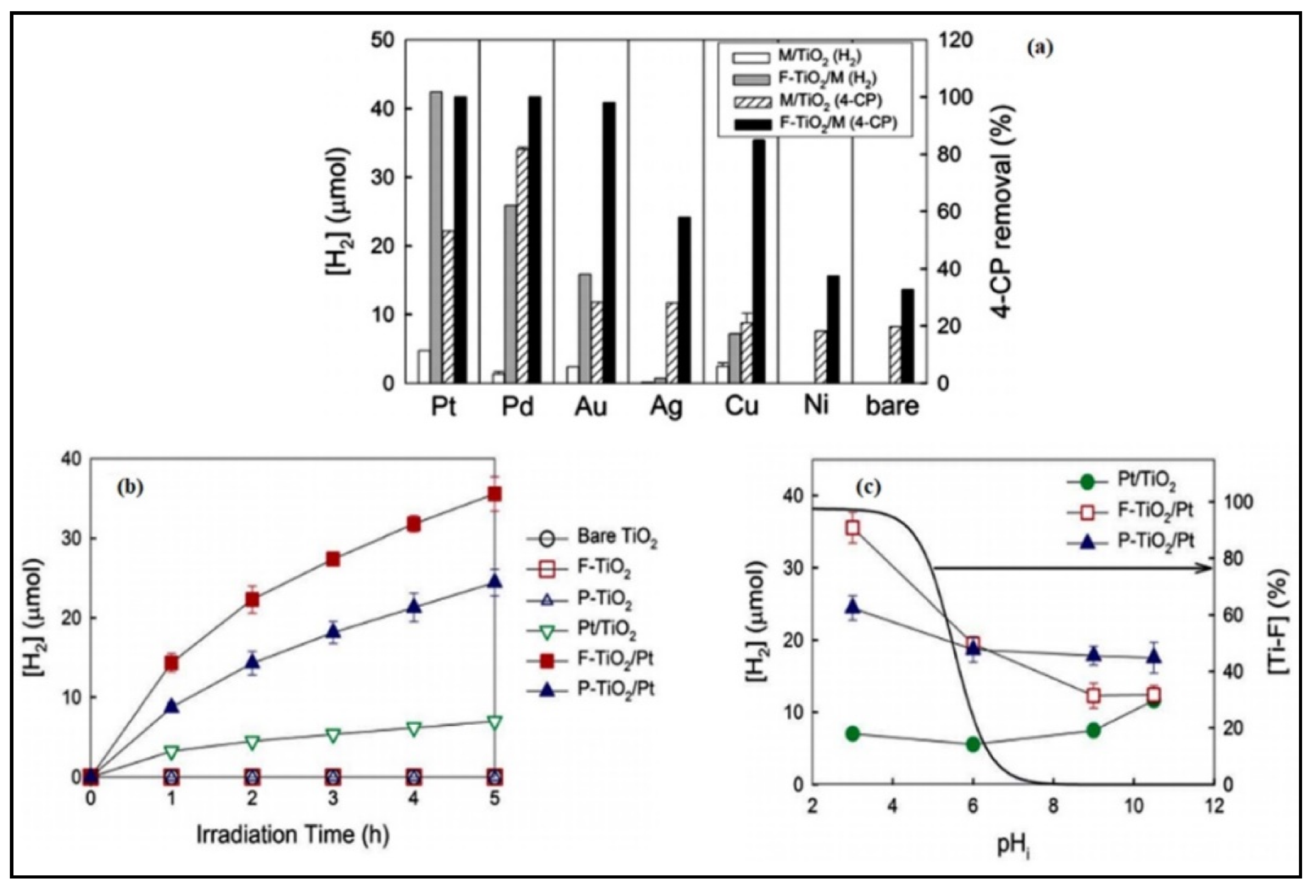
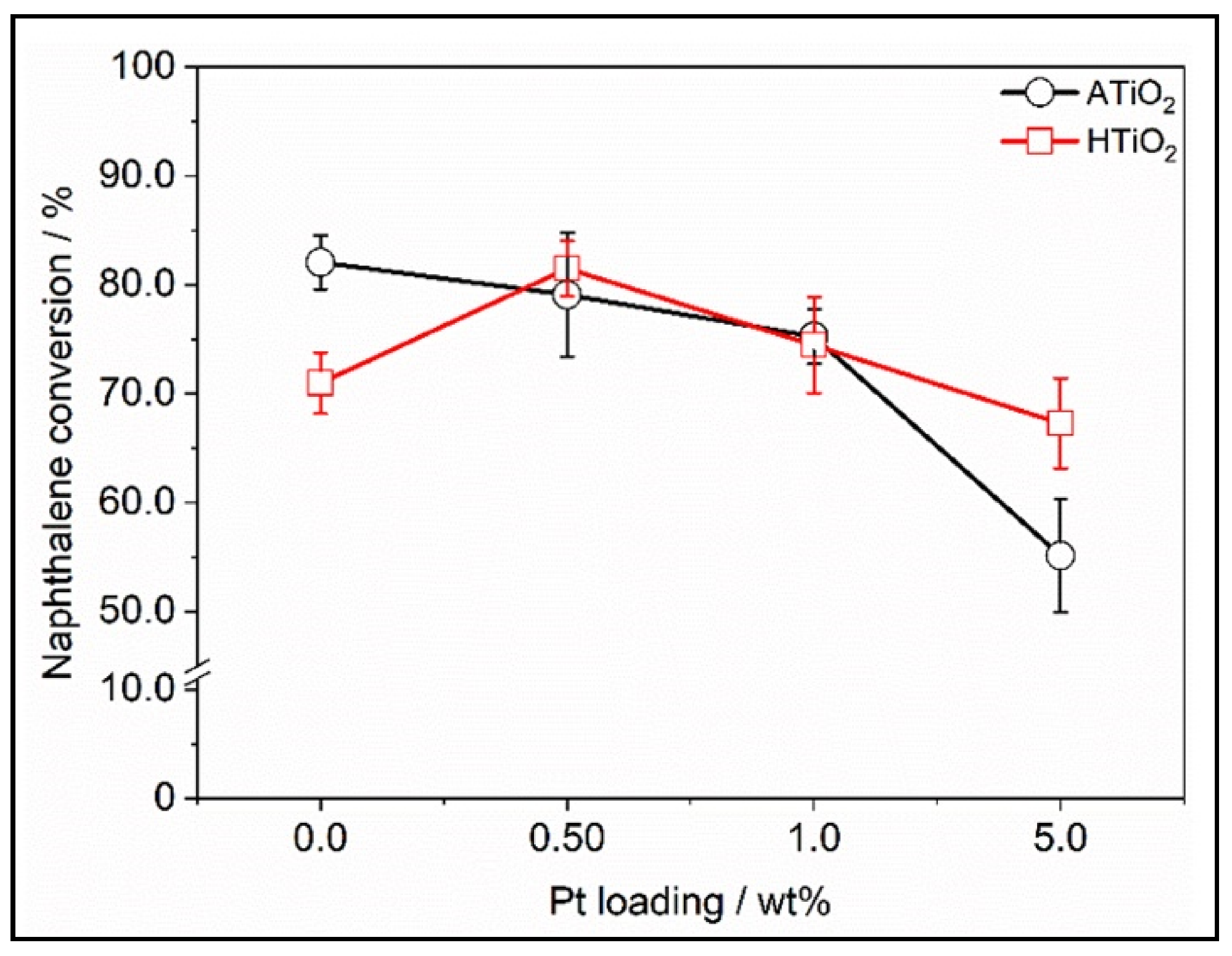
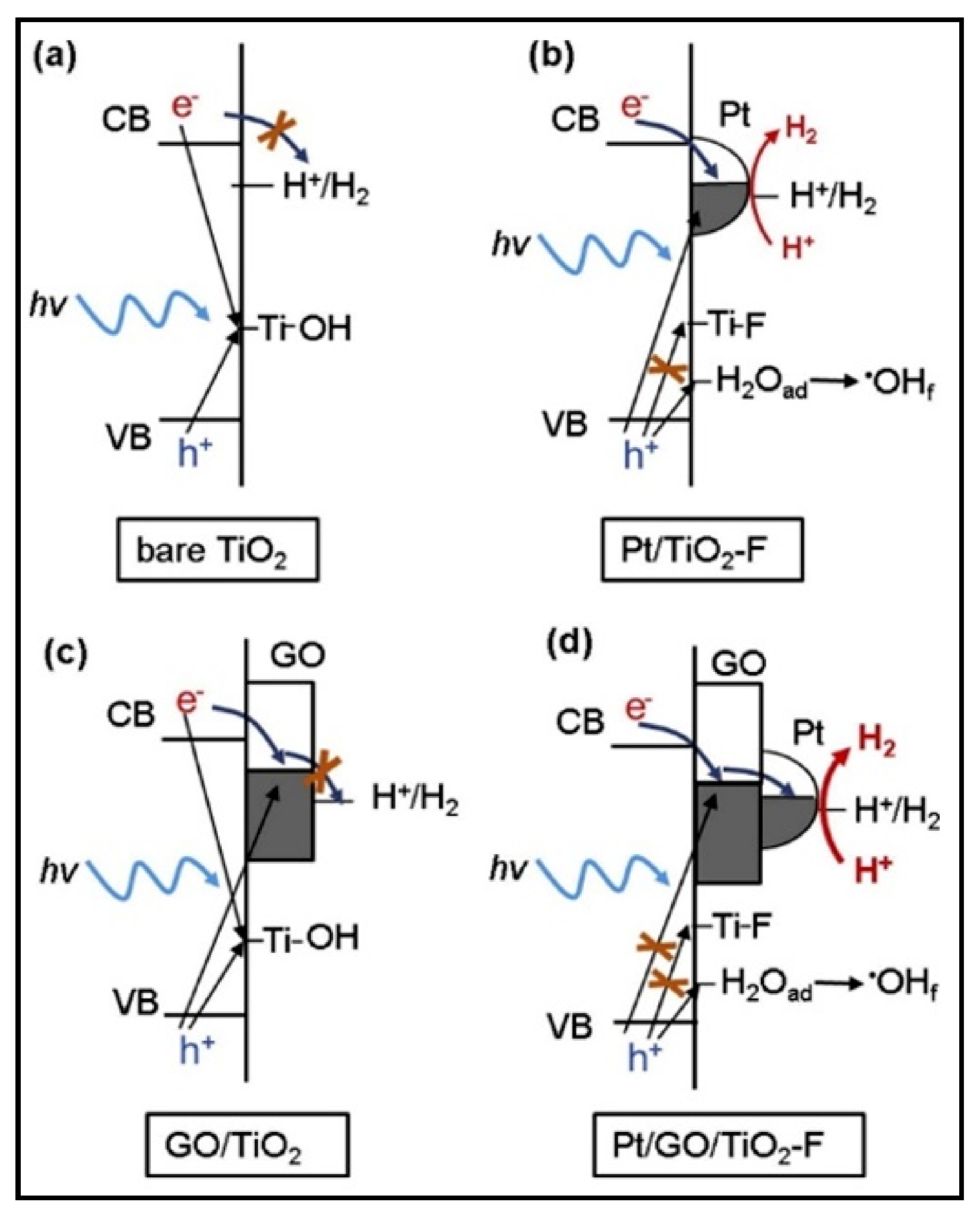
4.5. Effect of the Loading Method on H2 Production
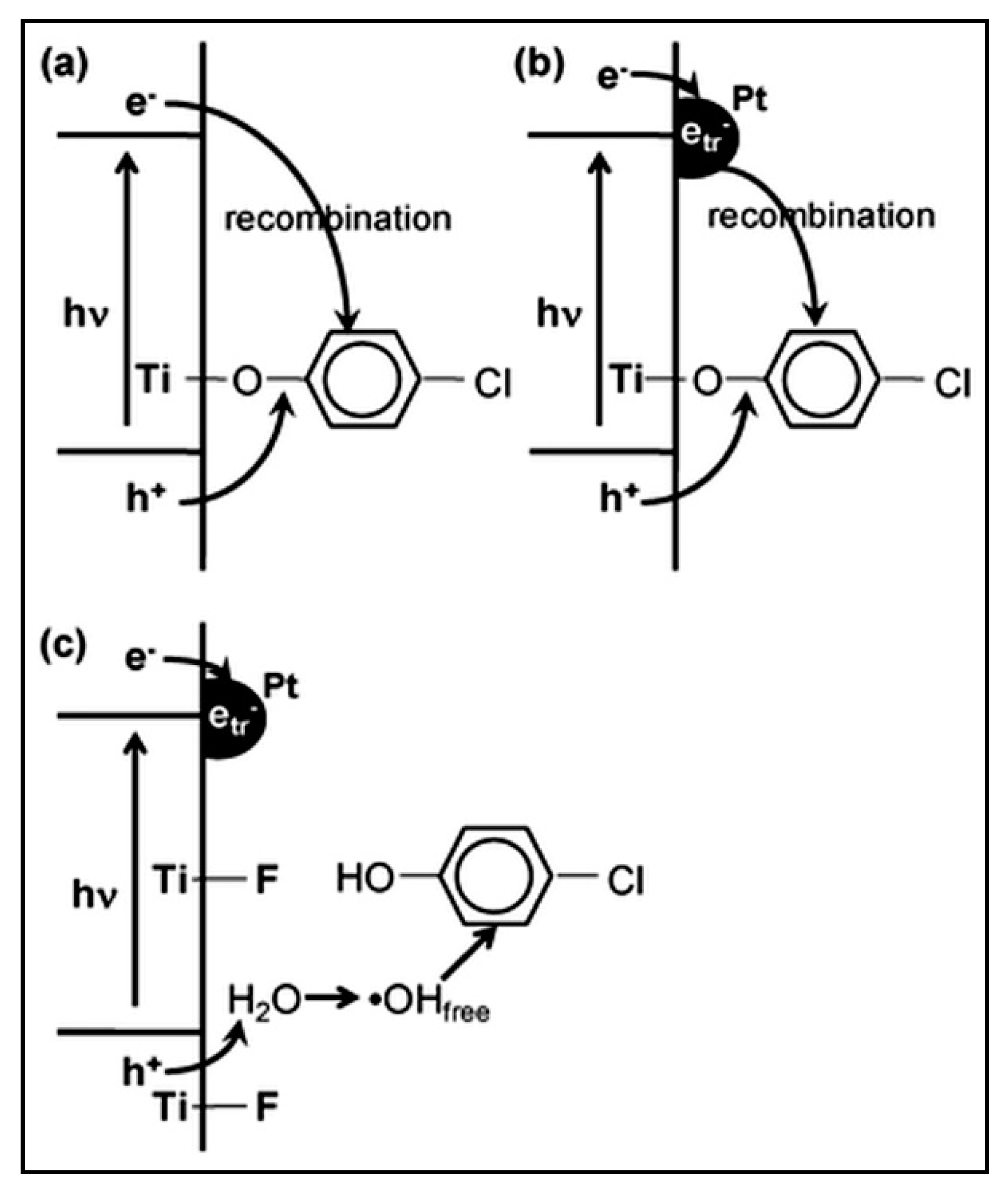
5. Photocatalytic Reforming of Aromatic Compounds
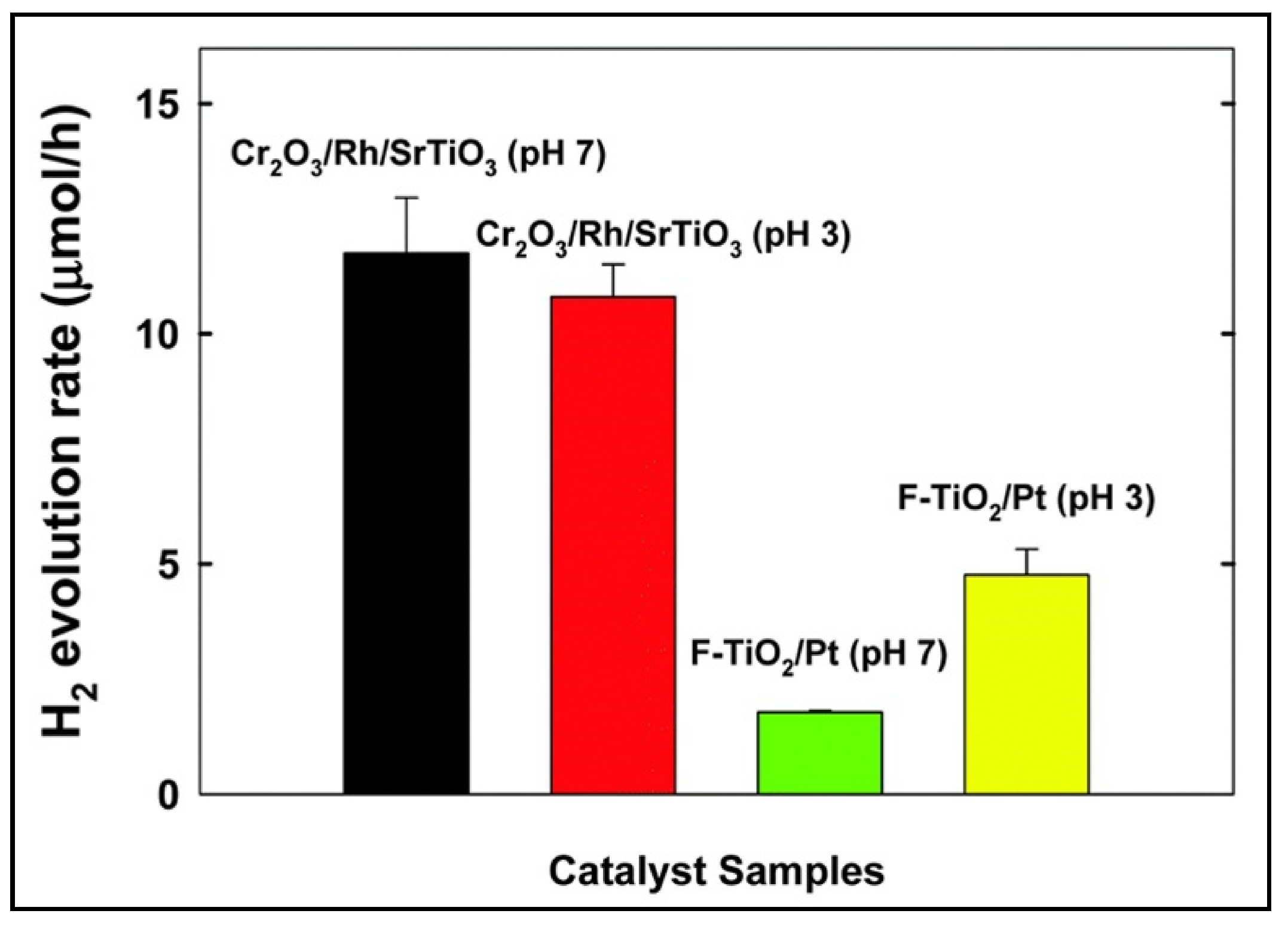
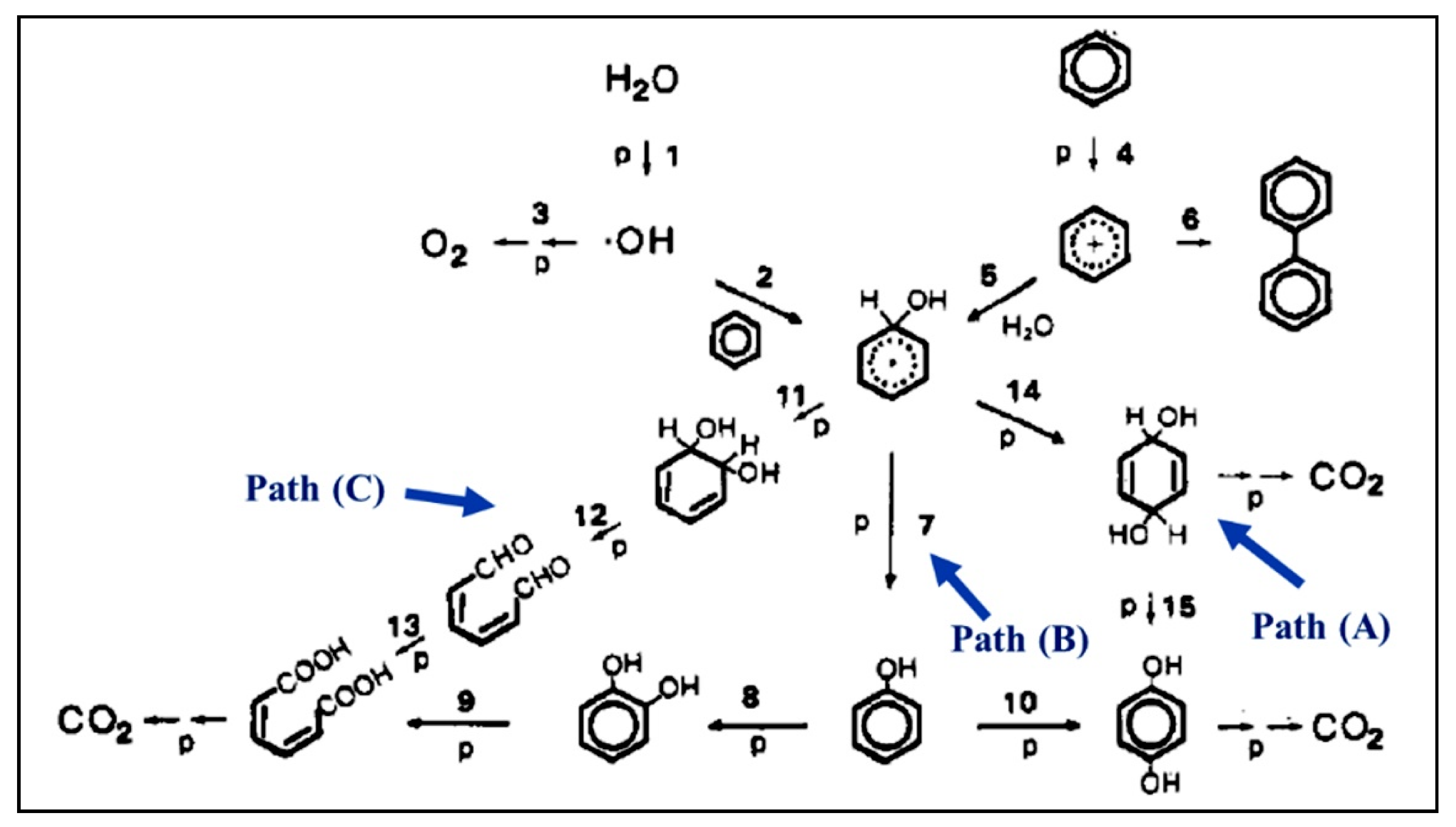
5.1. Monoaromatic and Phenolic-Based Compounds
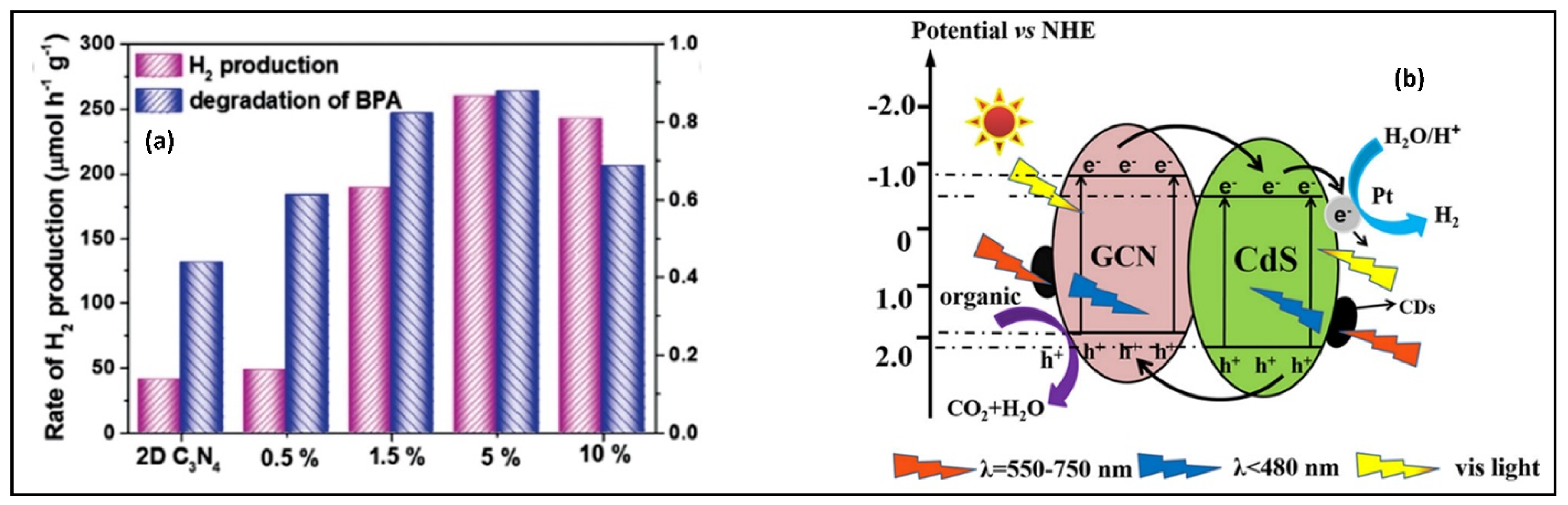
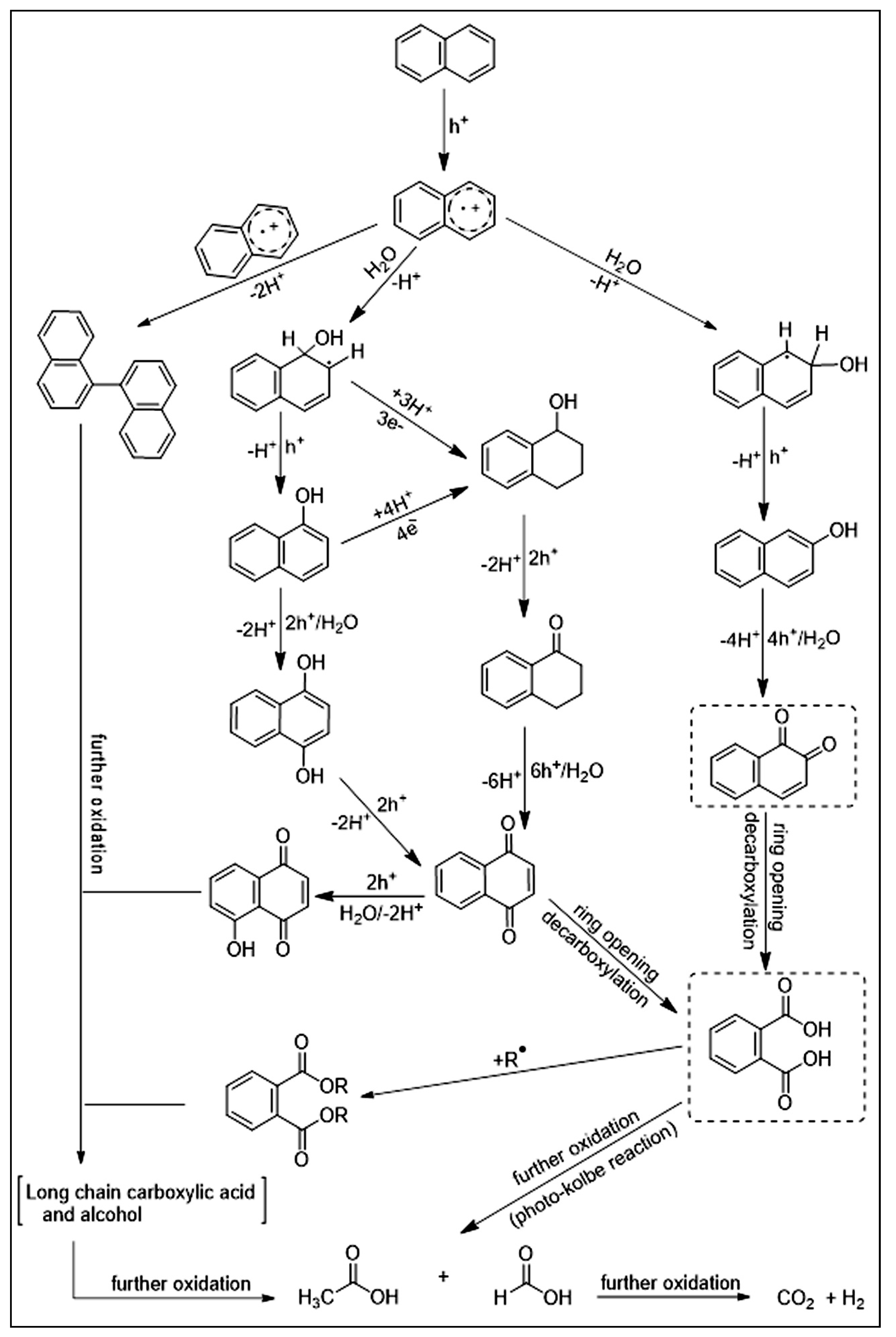
5.2. Dyes and Polyaromatic-Based Pollutants




6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liew, W.T.; Adhitya, A.; Srinivasan, R. Sustainability trends in the process industries: A text mining-based analysis. Comput. Ind. 2014, 65, 393–400. [Google Scholar] [CrossRef]
- Mechhoud, E.; Rouainia, M.; Rodriguez, M. A new tool for risk analysis and assessment in petrochemical plants. Alex. Eng. J. 2016, 55, 2919–2931. [Google Scholar] [CrossRef]
- Rovira, E.; Cuadras, A.; Aguilar, X.; Esteban, L.; Borràs-Santos, A.; Zock, J.-P.; Sunyer, J. Asthma, respiratory symptoms and lung function in children living near a petrochemical site. Environ. Res. 2014, 133, 156–163. [Google Scholar] [CrossRef]
- Villanueva, F.; Tapia, A.; Lara, S.; Amo-Salas, M. Indoor and outdoor air concentrations of volatile organic compounds and NO2 in schools of urban, industrial and rural areas in Central-Southern Spain. Sci. Total Environ. 2018, 622–623, 222–235. [Google Scholar] [CrossRef]
- Bari, M.A.; Kindzierski, W.B. Ambient volatile organic compounds (VOCs) in communities of the Athabasca oil sands region: Sources and screening health risk assessment. Environ. Pollut. 2018, 235, 602–614. [Google Scholar] [CrossRef]
- Matthews, R.W. Purification of water with near—u.v. illuminated suspensions of titanium dioxide. Water Res. 1990, 24, 653–660. [Google Scholar] [CrossRef]
- Matthews, R.W. An adsorption water purifier with in situ photocatalytic regeneration. J. Catal. 1988, 113, 549–555. [Google Scholar] [CrossRef]
- Grigoryan, H.; Edmands, W.M.B.; Lan, Q.; Carlsson, H.; Vermeulen, R.; Zhang, L.; Yin, S.N.; Li, G.L.; Smith, M.T.; Rothman, N.; et al. Adductomic signatures of benzene exposure provide insights into cancer induction. Carcinogenesis 2018, 39, 661–668. [Google Scholar] [CrossRef]
- He, C.; Cheng, J.; Zhang, X.; Douthwaite, M.; Pattisson, S.; Hao, Z. Recent advances in the catalytic oxidation of volatile organic compounds: A review based on pollutant sorts and sources. Chem. Rev. 2019, 119, 4471–4568. [Google Scholar] [CrossRef]
- Alegría, M.; Aliaga, J.; Ballesteros, L.; Sotomayor-Torres, C.; González, G.; Benavente, E. Layered nanocomposite 2D-TiO2 with Cu2O nanoparticles as an efficient photocatalyst for 4-Chlorophenol degradation and hydrogen evolution. Top. Catal. 2020. [Google Scholar] [CrossRef]
- Kim, J.; Monllor-Satoca, D.; Choi, W. Simultaneous production of hydrogen with the degradation of organic pollutants using TiO2 photocatalyst modified with dual surface components. Energy Environ. Sci. 2012, 5, 7647–7656. [Google Scholar] [CrossRef]
- Montoya, J.F.; Bahnemann, D.W.; Peral, J.; Salvador, P. Catalytic role of TiO2 terminal oxygen atoms in liquid-phase photocatalytic reactions: Oxidation of aromatic compounds in anhydrous acetonitrile. ChemPhysChem 2014, 15, 2311–2320. [Google Scholar] [CrossRef] [PubMed]
- Montoya, J.F.; Ivanova, I.; Dillert, R.; Bahnemann, D.W.; Salvador, P.; Peral, J. Catalytic role of surface oxygens in TiO2 photooxidation reactions: Aqueous benzene photooxidation with Ti18O2 under anaerobic conditions. J. Phys. Chem. Lett. 2013, 4, 1415–1422. [Google Scholar] [CrossRef]
- Teras, L.R.; Diver, W.R.; Deubler, E.L.; Krewski, D.; Flowers, C.R.; Switchenko, J.M.; Gapstur, S.M. Residential ambient benzene exposure in the United States and subsequent risk of hematologic malignancies. Int. J. Cancer 2019, 145, 2647–2660. [Google Scholar] [CrossRef]
- Mustieles, V.; Fernández, M.F.; Martin-Olmedo, P.; González-Alzaga, B.; Fontalba-Navas, A.; Hauser, R.; Olea, N.; Arrebola, J.P. Human adipose tissue levels of persistent organic pollutants and metabolic syndrome components: Combining a cross-sectional with a 10-year longitudinal study using a multi-pollutant approach. Environ. Int. 2017, 104, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Gao, B.; Creamer, A.E.; Cao, C.; Li, Y. Adsorption of VOCs onto engineered carbon materials: A review. J. Hazard. Mater. 2017, 338, 102–123. [Google Scholar] [CrossRef]
- Liu, L.; Li, J.; Zhang, H.; Li, L.; Zhou, P.; Meng, X.; Guo, M.; Jia, J.; Sun, T. In situ fabrication of highly active γ-MnO2/SmMnO3 catalyst for deep catalytic oxidation of gaseous benzene, ethylbenzene, toluene, and o-xylene. J. Hazard. Mater. 2019, 362, 178–186. [Google Scholar] [CrossRef]
- Mosmeri, H.; Gholami, F.; Shavandi, M.; Dastgheib, S.M.M.; Alaie, E. Bioremediation of benzene-contaminated groundwater by calcium peroxide (CaO2) nanoparticles: Continuous-flow and biodiversity studies. J. Hazard. Mater. 2019, 371, 183–190. [Google Scholar] [CrossRef]
- Markham, S.C. Photocatalytic properties of oxides. J. Chem. Educ. 1955, 32, 540. [Google Scholar] [CrossRef]
- Fujishima, A.; Honda, K. Electrochemical photolysis of water at a semiconductor electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef]
- Sabarinathan, M.; Harish, S.; Archana, J.; Navaneethan, M.; Ikeda, H.; Hayakawa, Y. Highly efficient visible-light photocatalytic activity of MoS2–TiO2 mixtures hybrid photocatalyst and functional properties. RSC Adv. 2017, 7, 24754–24763. [Google Scholar] [CrossRef]
- Liu, X.; Zhai, H.; Wang, P.; Zhang, Q.; Wang, Z.; Liu, Y.; Dai, Y.; Huang, B.; Qin, X.; Zhang, X. Synthesis of a WO3 photocatalyst with high photocatalytic activity and stability using synergetic internal Fe3+ doping and superficial Pt loading for ethylene degradation under visible-light irradiation. Catal. Sci. Technol. 2019, 9, 652–658. [Google Scholar] [CrossRef]
- Ramadan, W.; Shaikh, P.A.; Ebrahim, S.; Ramadan, A.; Hannoyer, B.; Jouen, S.; Sauvage, X.; Ogale, S. Highly efficient photocatalysis by BiFeO3/α(γ)-Fe2O3 ferromagnetic nano p/n junctions formed by dopant-induced phase separation. J. Nanopart. Res. 2013, 15, 1848. [Google Scholar] [CrossRef]
- Mishra, M.; Chun, D.-M. α-Fe2O3 as a photocatalytic material: A review. Appl. Catal. A 2015, 498, 126–141. [Google Scholar] [CrossRef]
- Ebrahim, S.; Ramadan, W.; Ali, M. Structural, optical and ferromagnetic properties of cobalt doped CdTe quantum dots. J. Mater. Sci. Mater. Electron. 2016, 27, 3826–3833. [Google Scholar] [CrossRef]
- Kaur, R.; Rana, A.; Singh, R.K.; Chhabra, V.A.; Kim, K.-H.; Deep, A. Efficient photocatalytic and photovoltaic applications with nanocomposites between CdTe QDs and an NTU-9 MOF. RSC Adv. 2017, 7, 29015–29024. [Google Scholar] [CrossRef]
- Huang, C.-W.; Nguyen, B.-S.; Wu, J.C.S.; Nguyen, V.-H. A current perspective for photocatalysis towards the hydrogen production from biomass-derived organic substances and water. Int. J. Hydrog. Energy 2020, 45, 18144–18159. [Google Scholar] [CrossRef]
- Serpone, N. Is the band gap of pristine TiO2 narrowed by anion- and cation-doping of titanium dioxide in second-generation photocatalysts? J. Phys. Chem. B 2006, 110, 24287–24293. [Google Scholar] [CrossRef]
- Fouda, A.; Hassan, S.E.-D.; Saied, E.; Azab, M.S. An eco-friendly approach to textile and tannery wastewater treatment using maghemite nanoparticles (γ-Fe2O3-NPs) fabricated by Penicillium expansum strain (K-w). J. Environ. Chem. Eng. 2021, 9, 104693. [Google Scholar] [CrossRef]
- Angaru, G.K.R.; Choi, Y.L.; Lingamdinne, L.P.; Choi, J.S.; Kim, D.S.; Koduru, J.R.; Yang, J.K.; Chang, Y.Y. Facile synthesis of economical feasible fly ash-based zeolite-supported nano zerovalent iron and nickel bimetallic composite for the potential removal of heavy metals from industrial effluents. Chemosphere 2021, 267, 128889. [Google Scholar] [CrossRef]
- Al-Madanat, O.; Jiries, A.; Batarseh, M.; Al-Nasir, F. Indoor and outdoor pollution with heavy metals in Al-Karak city, Jordan. J. Int. Environ. Appl. Sci. 2017, 12, 131–139. [Google Scholar]
- Al-Nasir, F.M.; Jiries, A.G.; Al-Rabadi, G.J.; Alu’datt, M.H.; Tranchant, C.C.; Al-Dalain, S.A.; Alrabadi, N.; Madanat, O.Y.; Al-Dmour, R.S. Determination of pesticide residues in selected citrus fruits and vegetables cultivated in the Jordan Valley. LWT 2020, 123, 109005. [Google Scholar] [CrossRef]
- Lei, M.; Gao, Q.; Zhou, K.; Gogoi, P.; Liu, J.; Wang, J.; Song, H.; Wang, S.; Liu, X. Catalytic degradation and mineralization mechanism of 4-chlorophenol oxidized by phosphomolybdic acid/H2O2. Sep. Purif. Technol. 2021, 257, 117933. [Google Scholar] [CrossRef]
- Kuttiani Ali, J.; Maher Chabib, C.; Abi Jaoude, M.; Alhseinat, E.; Teotia, S.; Patole, S.; Hussain Anjum, D.; Qattan, I. Enhanced removal of aqueous phenol with polyimide ultrafiltration membranes embedded with deep eutectic solvent-coated nanosilica. Chem. Eng. J. 2021, 408, 128017. [Google Scholar] [CrossRef]
- Al Nasir, F.; Batarseh, M.I. Agricultural reuse of r 456545eclaimed water and uptake of organic compounds: Pilot study at Mutah University wastewater treatment plant, Jordan. Chemosphere 2008, 72, 1203–1214. [Google Scholar] [CrossRef]
- Torres-Pinto, A.; Sampaio, M.J.; Silva, C.G.; Faria, J.L.; Silva, A.M.T. Metal-free carbon nitride photocatalysis with in situ hydrogen peroxide generation for the degradation of aromatic compounds. Appl. Catal. B 2019, 252, 128–137. [Google Scholar] [CrossRef]
- Alrabadi, G.; Al-Nasir, F.; Jiries, A.; Al-Dmour, R.; Madanat, O.; Al-Dalain, S. Polychlorinated biphenyls residue in citrus and vegetables in the Jordan Valley, Jordan. JJEES 2019, 10, 247–251. [Google Scholar]
- Lin, Z.; Li, L.; Yu, L.; Li, W.; Yang, G. Dual-functional photocatalysis for hydrogen evolution from industrial wastewaters. Phys. Chem. Chem. Phys. 2017, 19, 8356–8362. [Google Scholar] [CrossRef]
- Puga, A.V.; Barka, N.; Imizcoz, M. Simultaneous H2 production and bleaching via solar photoreforming of model dye-polluted wastewaters on metal/titania. ChemCatChem 2020. [Google Scholar] [CrossRef]
- AlSalka, Y.; Karabet, F.; Hashem, S. Development and optimisation of quantitative analytical method to determine BTEX in environmental water samples using HPLC-DAD. Anal. Methods 2010, 2, 1026–1035. [Google Scholar] [CrossRef]
- Villegas, L.G.C.; Mashhadi, N.; Chen, M.; Mukherjee, D.; Taylor, K.E.; Biswas, N. A short review of techniques for phenol removal from wastewater. Curr. Pollut. Rep. 2016, 2, 157–167. [Google Scholar] [CrossRef]
- Michałowicz, J.; Duda, W. Phenols—Sources and toxicity. Pol. J. Environ. Stud. 2007, 16, 347–362. [Google Scholar]
- Zango, Z.U.; Sambudi, N.S.; Jumbri, K.; Ramli, A.; Abu Bakar, N.H.H.; Saad, B.; Rozaini, M.N.H.; Isiyaka, H.A.; Osman, A.M.; Sulieman, A. An overview and evaluation of highly porous adsorbent materials for polycyclic aromatic hydrocarbons and phenols removal from wastewater. Water 2020, 12, 2921. [Google Scholar] [CrossRef]
- Anku, W.W.; Mamo, M.A.; Govender, P.P. Phenolic Compounds in Water: Sources, Reactivity, Toxicity and Treatment Methods, Phenolic Compounds—Natural Sources, Importance and Applications; IntechOpen-Open Science Open Minds: London, UK, 2017. [Google Scholar] [CrossRef]
- Igbinosa, E.O.; Odjadjare, E.E.; Chigor, V.N.; Igbinosa, I.H.; Emoghene, A.O.; Ekhaise, F.O.; Igiehon, N.O.; Idemudia, O.G. Toxicological profile of chlorophenols and their derivatives in the environment: The public health perspective. Sci. World J. 2013, 2013, 460215. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.Z.; Huang, C.P. Effect of chlorine content of chlorinated phenols on their oxidation kinetics by Fenton’s reagent. Chemosphere 1996, 33, 1621–1635. [Google Scholar] [CrossRef]
- Alsalka, Y.; Karabet, F.; Hashem, S. Evaluation of electrochemical processes for the removal of several target aromatic hydrocarbons from petroleum contaminated water. J. Environ. Monit. 2011, 13, 605–613. [Google Scholar] [CrossRef]
- Drwal, E.; Rak, A.; Gregoraszczuk, E.L. Review: Polycyclic aromatic hydrocarbons (PAHs)-Action on placental function and health risks in future life of newborns. Toxicology 2019, 411, 133–142. [Google Scholar] [CrossRef]
- Patel, A.B.; Shaikh, S.; Jain, K.R.; Desai, C.; Madamwar, D. Polycyclic aromatic hydrocarbons: Sources, toxicity, and remediation approaches. Front. Microbiol. 2020, 11, 562813. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Shafy, H.I.; Mansour, M.S.M. A review on polycyclic aromatic hydrocarbons: Source, environmental impact, effect on human health and remediation. Egypt. J. Pet. 2016, 25, 107–123. [Google Scholar] [CrossRef]
- Mastral, A.M.; Callén, M.S. A review on polycyclic aromatic hydrocarbon (PAH) emissions from energy generation. Environ. Sci. Technol. 2000, 34, 3051–3057. [Google Scholar] [CrossRef]
- Jiries, A.G.; Hussein, H.H.; Lintelmann, J. Polycyclic aromatic hydrocarbon in rain and street runoff in Amman, Jordan. J. Environ. Sci. 2003, 15, 848–853. [Google Scholar]
- Forsgren, A.J. Wastewater Treatment: Occurrence and Fate of Polycyclic Aromatic Hydrocarbons (PAHs), 1st ed.; CRC Press: Boca Raton, FL, USA, 2015. [Google Scholar] [CrossRef]
- Huang, H.; Buekens, A. Chlorinated dioxins and furans as trace products of combustion: Some theoretical aspects. Toxicol. Environ. Chem. 2000, 74, 179–193. [Google Scholar] [CrossRef]
- Novotna, B.; Topinka, J.; Solansky, I.; Chvatalova, I.; Lnenickova, Z.; Sram, R.J. Impact of air pollution and genotype variability on DNA damage in Prague policemen. Toxicol. Lett. 2007, 172, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Batarseh, M.I.; Kreuzig, R.; Bahadir, M. Residue analysis of organic pollutants in sediments from the Amman/Zarqa area in Jordan. Part 1: Development of analytical methods and distribution patterns of PAHs. Fresenius Environ. Bull. 2003, 12, 972–978. [Google Scholar]
- Majumdar, D.; Rajaram, B.; Meshram, S.; Suryawanshi, P.; Chalapati Rao, C.V. Worldwide distribution of polyclyclic aromatic hydrocarbons in urban road dust. Int. J. Environ. Sci. Technol. 2016, 14, 397–420. [Google Scholar] [CrossRef]
- Jiries, A.; Hussain, H.; Lintelmann, J. Determination of polycyclic aromatic hydrocarbons in wastewater, sediments, sludge and plants in Karak Province, Jordan. Water Air Soil Pollut. 2000, 121, 217–228. [Google Scholar] [CrossRef]
- Tian, W.; Bai, J.; Liu, K.; Sun, H.; Zhao, Y. Occurrence and removal of polycyclic aromatic hydrocarbons in the wastewater treatment process. Ecotoxicol. Environ. Saf. 2012, 82, 1–7. [Google Scholar] [CrossRef]
- Wang, X.; Xi, B.; Huo, S.; Sun, W.; Pan, H.; Zhang, J.; Ren, Y.; Liu, H. Characterization, treatment and releases of PBDEs and PAHs in a typical municipal sewage treatment plant situated beside an urban river, East China. J. Environ. Sci. 2013, 25, 1281–1290. [Google Scholar] [CrossRef]
- Sun, H.M.; Tian, W.J.; Wang, Y.M. Occurrence and fate of polycyclic aromatic hydrocarbons in the anaerobic-anoxic-oxic wastewater treatment process. Adv. Mater. Res. 2012, 610–613, 1722–1725. [Google Scholar] [CrossRef]
- Jia, C.; Batterman, S. A critical review of naphthalene sources and exposures relevant to indoor and outdoor air. Int. J. Environ. Res. Public. Health 2010, 7, 2903–2939. [Google Scholar] [CrossRef]
- Robles, H. Naphthalene. In Encyclopedia of Toxicology; Wexler, P., Ed.; Academic Press: Oxford, UK, 2014; pp. 437–439. [Google Scholar] [CrossRef]
- Sudakin, D.L.; Stone, D.L.; Power, L. Naphthalene mothballs: Emerging and recurring issues and their relevance to environmental health. Curr. Top. Toxicol. 2011, 7, 13–19. [Google Scholar] [PubMed]
- Arizavi, A.; Mirbagheri, N.S.; Hosseini, Z.; Chen, P.; Sabbaghi, S. Efficient removal of naphthalene from aqueous solutions using a nanoporous kaolin/Fe3O4 composite. Int. J. Environ. Sci. Technol. 2020, 17, 1991–2002. [Google Scholar] [CrossRef]
- Nesterenko-Malkovskaya, A.; Kirzhner, F.; Zimmels, Y.; Armon, R. Eichhornia crassipes capability to remove naphthalene from wastewater in the absence of bacteria. Chemosphere 2012, 87, 1186–1191. [Google Scholar] [CrossRef]
- Pereira, L.; Alves, M. Dyes—Environmental impact and remediation. In Environmental Protection Strategies for Sustainable Development; Malik, A., Grohmann, E., Eds.; Springer: Dordrecht, The Netherlands, 2012; pp. 111–162. [Google Scholar] [CrossRef]
- Hunger, K.; Gregory, P.; Miederer, P.; Berneth, H.; Heid, C.; Mennicke, W. Important chemical chromophores of dye classes. In Industrial Dyes, Hunger, K., Ed.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2002; pp. 13–112. [Google Scholar] [CrossRef]
- Benkhaya, S.; M’ rabet, S.; El Harfi, A. A review on classifications, recent synthesis and applications of textile dyes. Inorg. Chem. Commun. 2020, 115, 107891. [Google Scholar] [CrossRef]
- Berradi, M.; Hsissou, R.; Khudhair, M.; Assouag, M.; Cherkaoui, O.; El Bachiri, A.; El Harfi, A. Textile finishing dyes and their impact on aquatic environs. Heliyon 2019, 5, e02711. [Google Scholar] [CrossRef] [PubMed]
- Forgacs, E.; Cserháti, T.; Oros, G. Removal of synthetic dyes from wastewaters: A review. Environ. Int. 2004, 30, 953–971. [Google Scholar] [CrossRef]
- Yan, J.; Wang, L.; Fu, P.P.; Yu, H. Photomutagenicity of 16 polycyclic aromatic hydrocarbons from the US EPA priority pollutant list. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2004, 557, 99–108. [Google Scholar] [CrossRef]
- Ravindra, K.; Sokhi, R.; Van Grieken, R. Atmospheric polycyclic aromatic hydrocarbons: Source attribution, emission factors and regulation. Atmos. Environ. 2008, 42, 2895–2921. [Google Scholar] [CrossRef]
- Ghodke, S.A.; Sonawane, S.H.; Bhanvase, B.A.; Potoroko, I. Advanced engineered nanomaterials for the treatment of wastewater. In Handbook of Nanomaterials for Industrial Applications; Mustansar Hussain, C., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 959–970. [Google Scholar] [CrossRef]
- Naushad, M.; Alqadami, A.A.; AlOthman, Z.A.; Alsohaimi, I.H.; Algamdi, M.S.; Aldawsari, A.M. Adsorption kinetics, isotherm and reusability studies for the removal of cationic dye from aqueous medium using arginine modified activated carbon. J. Mol. Liq. 2019, 293, 111442. [Google Scholar] [CrossRef]
- Kadirvelu, K.; Kavipriya, M.; Karthika, C.; Radhika, M.; Vennilamani, N.; Pattabhi, S. Utilization of various agricultural wastes for activated carbon preparation and application for the removal of dyes and metal ions from aqueous solutions. Bioresour. Technol. 2003, 87, 129–132. [Google Scholar] [CrossRef]
- Katheresan, V.; Kansedo, J.; Lau, S.Y. Efficiency of various recent wastewater dye removal methods: A review. J. Environ. Chem. Eng. 2018, 6, 4676–4697. [Google Scholar] [CrossRef]
- Pai, S.; Kini, M.S.; Selvaraj, R. A review on adsorptive removal of dyes from wastewater by hydroxyapatite nanocomposites. Environ. Sci. Pollut. Res. Int. 2019. [Google Scholar] [CrossRef]
- Kiriakidou, F.; Kondarides, D.I.; Verykios, X.E. The effect of operational parameters and TiO2-doping on the photocatalytic degradation of azo-dyes. Catal. Today 1999, 54, 119–130. [Google Scholar] [CrossRef]
- Adeola, A.O.; Forbes, P.B.C. Advances in water treatment technologies for removal of polycyclic aromatic hydrocarbons: Existing concepts, emerging trends, and future prospects. Water Environ. Res. 2020. [Google Scholar] [CrossRef]
- Amin, M.T.; Alazba, A.A.; Manzoor, U. A review of removal of pollutants from water/wastewater using different types of nanomaterials. Adv. Mater. Sci. Eng. 2014, 2014, 1–24. [Google Scholar] [CrossRef]
- Hlongwane, G.N.; Sekoai, P.T.; Meyyappan, M.; Moothi, K. Simultaneous removal of pollutants from water using nanoparticles: A shift from single pollutant control to multiple pollutant control. Sci. Total Environ. 2019, 656, 808–833. [Google Scholar] [CrossRef] [PubMed]
- Rajasekhar, B.; Nambi, I.M.; Govindarajan, S.K. Investigating the degradation of nC12 to nC23 alkanes and PAHs in petroleum- contaminated water by electrochemical advanced oxidation process using an inexpensive Ti/Sb-SnO2/PbO2 anode. Chem. Eng. J. 2021, 404, 125268. [Google Scholar] [CrossRef]
- Singa, P.K.; Isa, M.H.; Lim, J.W.; Ho, Y.C.; Krishnan, S. Photo-Fenton process for removal of polycyclic aromatic hydrocarbons from hazardous waste landfill leachate. Int. J. Environ. Sci. Technol. 2020. [Google Scholar] [CrossRef]
- Wilson, S.C.; Jones, K.C. Bioremediation of soil contaminated with polynuclear aromatic hydrocarbons (PAHs): A review. Environ. Pollut. 1993, 81, 229–249. [Google Scholar] [CrossRef]
- Manariotis, I.D.; Karapanagioti, H.K.; Chrysikopoulos, C.V. Degradation of PAHs by high frequency ultrasound. Water Res. 2011, 45, 2587–2594. [Google Scholar] [CrossRef]
- Khulbe, K.C.; Matsuura, T. Removal of heavy metals and pollutants by membrane adsorption techniques. Appl. Water Sci. 2018, 8, 19. [Google Scholar] [CrossRef]
- Trojanowicz, M. Removal of persistent organic pollutants (POPs) from waters and wastewaters by the use of ionizing radiation. Sci. Total. Environ. 2020, 718, 134425. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wei, C.; An, G. Distribution, partition and removal of polycyclic aromatic hydrocarbons (PAHs) during coking wastewater treatment processes. Environ. Sci. Process Impacts 2015, 17, 975–984. [Google Scholar] [CrossRef]
- Fatone, F.; Di Fabio, S.; Bolzonella, D.; Cecchi, F. Fate of aromatic hydrocarbons in Italian municipal wastewater systems: An overview of wastewater treatment using conventional activated-sludge processes (CASP) and membrane bioreactors (MBRs). Water Res. 2011, 45, 93–104. [Google Scholar] [CrossRef]
- Zhu, X.; Ni, J.; Lai, P. Advanced treatment of biologically pretreated coking wastewater by electrochemical oxidation using boron-doped diamond electrodes. Water Res. 2009, 43, 4347–4355. [Google Scholar] [CrossRef] [PubMed]
- Sakulthaew, C.; Comfort, S.; Chokejaroenrat, C.; Harris, C.; Li, X. A combined chemical and biological approach to transforming and mineralizing PAHs in runoff water. Chemosphere 2014, 117, 1–9. [Google Scholar] [CrossRef]
- Anjum, M.; Miandad, R.; Waqas, M.; Gehany, F.; Barakat, M.A. Remediation of wastewater using various nano-materials. Arab. J. Chem. 2019, 12, 4897–4919. [Google Scholar] [CrossRef]
- Wacławek, S.; Padil, V.V.T.; Černík, M. Major advances and challenges in heterogeneous catalysis for environmental applications: A review. Ecol. Chem. Eng. S 2018, 25, 9–34. [Google Scholar] [CrossRef]
- Rasalingam, S.; Peng, R.; Koodali, R.T. Removal of hazardous pollutants from wastewaters: Applications of TiO2-SiO2 mixed oxide materials. J. Nanomater. 2014, 2014, 1–42. [Google Scholar] [CrossRef]
- Crini, G.; Lichtfouse, E. Advantages and disadvantages of techniques used for wastewater treatment. Environ. Chem. Lett. 2019, 17, 145–155. [Google Scholar] [CrossRef]
- Oturan, M.A.; Aaron, J.-J. Advanced oxidation processes in water/wastewater treatment: Principles and applications. A review. Crit. Rev. Env. Sci. Tec. 2014, 44, 2577–2641. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Park, S.-J. TiO2 photocatalyst for water treatment applications. J. Ind. Eng. Chem. 2013, 19, 1761–1769. [Google Scholar] [CrossRef]
- Mousavi, M.; Habibi-Yangjeh, A.; Pouran, S.R. Review on magnetically separable graphitic carbon nitride-based nanocomposites as promising visible-light-driven photocatalysts. J. Mater. Sci. Mater. Electron. 2018, 29, 1719–1747. [Google Scholar] [CrossRef]
- Mokhbi, Y.; Korichi, M.; Akchiche, Z. Combined photocatalytic and Fenton oxidation for oily wastewater treatment. Appl. Water Sci. 2019, 9, 35. [Google Scholar] [CrossRef]
- Ramadan, W.; Dillert, R.; Koch, J.; Tegenkamp, C.; Bahnemann, D.W. Changes in the solid-state properties of bismuth iron oxide during the photocatalytic reformation of formic acid. Catal. Today 2019, 326, 22–29. [Google Scholar] [CrossRef]
- AlSalka, Y.; Granone, L.I.; Ramadan, W.; Hakki, A.; Dillert, R.; Bahnemann, D.W. Iron-based photocatalytic and photoelectrocatalytic nano-structures: Facts, perspectives, and expectations. Appl. Catal. B 2019, 244, 1065–1095. [Google Scholar] [CrossRef]
- Raza, W.; Haque, M.M.; Muneer, M.; Bahnemann, D. Synthesis of visible light driven TiO2 coated carbon nanospheres for degradation of dyes. Arab. J. Chem. 2019, 12, 3534–3545. [Google Scholar] [CrossRef]
- Al-Madanat, O.; AlSalka, Y.; Curti, M.; Dillert, R.; Bahnemann, D.W. Mechanistic insights into hydrogen evolution by photocatalytic reforming of naphthalene. ACS Catal. 2020, 10, 7398–7412. [Google Scholar] [CrossRef]
- AlSalka, Y.; Al-Madanat, O.; Curti, M.; Hakki, A.; Bahnemann, D.W. Photocatalytic H2 evolution from oxalic acid: Effect of cocatalysts and carbon dioxide radical anion on the surface charge transfer mechanisms. ACS Appl. Energy Mater. 2020, 3, 6678–6691. [Google Scholar] [CrossRef]
- Megatif, L.; Dillert, R.; Bahnemann, D.W. Reaction rate study of the photocatalytic degradation of dichloroacetic acid in a black body reactor. Catalysts 2019, 9, 635. [Google Scholar] [CrossRef]
- Faycal Atitar, M.; Ismail, A.A.; Dillert, R.; Bahnemann, D.W. Photodegradation of herbicide imazapyr and phenol over mesoporous bicrystalline phases TiO2: A Kinetic study. Catalysts 2019, 9, 640. [Google Scholar] [CrossRef]
- Jagadale, T.; Kulkarni, M.; Pravarthana, D.; Ramadan, W.; Thakur, P. Photocatalytic degradation of azo dyes using Au:TiO2, gamma-Fe2O3:TiO2 functional nanosystems. J. Nanosci. Nanotechnol. 2012, 12, 928–936. [Google Scholar] [CrossRef]
- Fox, M.A.; Dulay, M.T. Heterogeneous photocatalysis. Chem. Rev. 1993, 93, 341–357. [Google Scholar] [CrossRef]
- Schneider, J.; Matsuoka, M.; Takeuchi, M.; Zhang, J.; Horiuchi, Y.; Anpo, M.; Bahnemann, D.W. Understanding TiO2 photocatalysis: Mechanisms and materials. Chem. Rev. 2014, 114, 9919–9986. [Google Scholar] [CrossRef] [PubMed]
- Shayegan, Z.; Lee, C.-S.; Haghighat, F. TiO2 photocatalyst for removal of volatile organic compounds in gas phase—A review. Chem. Eng. J. 2018, 334, 2408–2439. [Google Scholar] [CrossRef]
- Pelaez, M.; Nolan, N.T.; Pillai, S.C.; Seery, M.K.; Falaras, P.; Kontos, A.G.; Dunlop, P.S.M.; Hamilton, J.W.J.; Byrne, J.A.; O’Shea, K.; et al. A review on the visible light active titanium dioxide photocatalysts for environmental applications. Appl. Catal. B 2012, 125, 331–349. [Google Scholar] [CrossRef]
- Choquette-Labbé, M.; Shewa, W.; Lalman, J.; Shanmugam, S. Photocatalytic degradation of phenol and phenol derivatives using a nano-TiO2 catalyst: Integrating quantitative and qualitative factors using response surface methodology. Water 2014, 6, 1785–1806. [Google Scholar] [CrossRef]
- Kamat, P.V. Meeting the clean energy demand: Nanostructure architectures for solar energy conversion. J. Phys. Chem. C 2007, 111, 2834–2860. [Google Scholar] [CrossRef]
- Etacheri, V.; Di Valentin, C.; Schneider, J.; Bahnemann, D.; Pillai, S.C. Visible-light activation of TiO2 photocatalysts: Advances in theory and experiments. J. Photochem. Photobiol. C 2015, 25, 1–29. [Google Scholar] [CrossRef]
- Rachna; Rani, M.; Shanker, U. Sunlight mediated improved photocatalytic degradation of carcinogenic benz[a]anthracene and benzo[a]pyrene by zinc oxide encapsulated hexacyanoferrate nanocomposite. J. Photochem. Photobiol. A 2019, 381, 111861. [Google Scholar] [CrossRef]
- Fu, J.; Kyzas, G.Z.; Cai, Z.; Deliyanni, E.A.; Liu, W.; Zhao, D. Photocatalytic degradation of phenanthrene by graphite oxide-TiO2-Sr(OH)2/SrCO3 nanocomposite under solar irradiation: Effects of water quality parameters and predictive modeling. Chem. Eng. J. 2018, 335, 290–300. [Google Scholar] [CrossRef]
- El-Mekkawi, D.M.; Abdelwahab, N.A.; Mohamed, W.A.A.; Taha, N.A.; Abdel-Mottaleb, M.S.A. Solar photocatalytic treatment of industrial wastewater utilizing recycled polymeric disposals as TiO2 supports. J. Clean. Prod. 2020, 249, 119430. [Google Scholar] [CrossRef]
- Horikoshi, S.; Serpone, N. Can the photocatalyst TiO2 be incorporated into a wastewater treatment method? Background and prospects. Catal. Today 2020, 340, 334–346. [Google Scholar] [CrossRef]
- Zhai, S.; Zhu, G.; Wei, X.; Ge, M. Enhanced catalytic degradation of polyvinyl alcohol from aqueous solutions by novel synthesis of MnCoO3@γ-Al2O3 nanocomposites: Performance, degradation intermediates and mechanism. J. Mol. Liq. 2021, 323, 114569. [Google Scholar] [CrossRef]
- Sola, A.C.; Ramírez de la Piscina, P.; Homs, N. Behaviour of Pt/TiO2 catalysts with different morphological and structural characteristics in the photocatalytic conversion of ethanol aqueous solutions. Catal. Today 2020, 341, 13–20. [Google Scholar] [CrossRef]
- Ivanova, I.; Schneider, J.; Gutzmann, H.; Kliemann, J.-O.; Gärtner, F.; Klassen, T.; Bahnemann, D.; Mendive, C.B. Photocatalytic degradation of oxalic and dichloroacetic acid on TiO2 coated metal substrates. Catal. Today 2013, 209, 84–90. [Google Scholar] [CrossRef]
- Hasan, I.; Shekhar, C.; Bin, S., II; Khan, R.A.; Alsalme, A. Ecofriendly green synthesis of the ZnO-doped CuO@Alg bionanocomposite for efficient oxidative degradation of p-nitrophenol. ACS Omega 2020, 5, 32011–32022. [Google Scholar] [CrossRef]
- Wu, S.; Li, X.; Tian, Y.; Lin, Y.; Hu, Y.H. Excellent photocatalytic degradation of tetracycline over black anatase-TiO2 under visible light. Chem. Eng. J. 2021, 406, 126747. [Google Scholar] [CrossRef]
- Miao, Y.; Xu, X.; Liu, K.; Yu, S.; Wang, Y.; Yang, S. Preparation and activity evaluation of the novel Cu/TiO2 nanometer photocatalytic materials. Sci. Adv. Mater. 2020, 12, 1027–1033. [Google Scholar] [CrossRef]
- Rani, M.; Shanker, U.; Yadav, J.; Keshu. Degradation of pesticides residue by engineered nanomaterials. In Sustainable Agriculture Reviews 48; Inamuddin, Ahamed, M.I., Lichtfouse, E., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 259–310. [Google Scholar] [CrossRef]
- Park, H.; Kim, H.-i.; Moon, G.-h.; Choi, W. Photoinduced charge transfer processes in solar photocatalysis based on modified TiO2. Energy Environ. Sci. 2016, 9, 411–433. [Google Scholar] [CrossRef]
- Kumaravel, V.; Mathew, S.; Bartlett, J.; Pillai, S.C. Photocatalytic hydrogen production using metal doped TiO2: A review of recent advances. Appl. Catal. B 2019, 244, 1021–1064. [Google Scholar] [CrossRef]
- Pellegrino, F.; Sordello, F.; Mino, L.; Minero, C.; Hodoroaba, V.-D.; Martra, G.; Maurino, V. Formic acid photoreforming for hydrogen production on shap-controlled anatase TiO2 nanoparticles: Assessment of the role of fluorides, {101}/{001} surfaces ratio, and platinization. ACS Catal. 2019, 9, 6692–6697. [Google Scholar] [CrossRef]
- Kampouri, S.; Stylianou, K.C. Dual-functional photocatalysis for simultaneous hydrogen production and oxidation of organic substances. ACS Catal. 2019, 9, 4247–4270. [Google Scholar] [CrossRef]
- AlSalka, Y.; Hakki, A.; Fleisch, M.; Bahnemann, D.W. Understanding the degradation pathways of oxalic acid in different photocatalytic systems: Towards simultaneous photocatalytic hydrogen evolution. J. Photochem. Photobiol. A 2018, 366, 81–90. [Google Scholar] [CrossRef]
- Hakki, A.; AlSalka, Y.; Mendive, C.B.; Ubogui, J.; dos Santos Claro, P.C.; Bahnemann, D. Hydrogen production by heterogeneous photocatalysis. In Encyclopedia of Interfacial Chemistry; Wandelt, K., Ed.; Elsevier: Oxford, UK, 2018; pp. 413–419. [Google Scholar] [CrossRef]
- Sivula, K.; Van De Krol, R. Semiconducting materials for photoelectrochemical energy conversion. Nat. Rev. Mater. 2016, 1, 15010. [Google Scholar] [CrossRef]
- Takanabe, K.; Domen, K. Toward visible light response: Overall water splitting using heterogeneous photocatalysts. Green 2011, 1, 313–322. [Google Scholar] [CrossRef]
- Maeda, K.; Domen, K. Photocatalytic water splitting: Recent progress and future challenges. J. Phys. Chem. Lett. 2010, 1, 2655–2661. [Google Scholar] [CrossRef]
- Cao, W. Semiconductor Photocatalysis: Materials, Mechanisms and Applications; InTech: Rijeka, Croatia, 2016. [Google Scholar] [CrossRef]
- Jiang, C.; Moniz, S.J.; Wang, A.; Zhang, T.; Tang, J. Photoelectrochemical devices for solar water splitting–materials and challenges. Chem. Soc. Rev. 2017, 46, 4645–4660. [Google Scholar] [CrossRef]
- Kisch, H. Semiconductor Photocatalysis Principle and Applications; Wiley-VCH: Hoboken, NJ, USA, 2015. [Google Scholar]
- Friehs, E.; AlSalka, Y.; Jonczyk, R.; Lavrentieva, A.; Jochums, A.; Walter, J.-G.; Stahl, F.; Scheper, T.; Bahnemann, D. Toxicity, phototoxicity and biocidal activity of nanoparticles employed in photocatalysis. J. Photochem. Photobiol. C 2016, 29, 1–28. [Google Scholar] [CrossRef]
- Hernández-Ramírez, A.; Medina-Ramírez, I. Semiconducting materials. In Photocatalytic Semiconductors; Hernández-Ramírez, A., Medina-Ramírez, I., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 1–40. [Google Scholar] [CrossRef]
- Barr, A. Electrical properties of semiconductors. In Electronic Materials; Miller, L.S., Mullin, J.B., Eds.; Springer: Boston, MA, USA, 1991; pp. 19–24. [Google Scholar] [CrossRef]
- Fonash, S.J. Structures, materials, and scale. In Solar Cell Device Physics; Fonash, S.J., Ed.; Academic Press: Boston, MA, USA, 2010; pp. 67–120. [Google Scholar] [CrossRef]
- Hoffmann, M.R.; Martin, S.T.; Choi, W.; Bahnemann, D.W. Environmental applications of semiconductor photocatalysis. Chem. Rev. 1995, 95, 69–96. [Google Scholar] [CrossRef]
- Mills, A.; Le Hunte, S. An overview of semiconductor photocatalysis. J. Photochem. Photobiol. A 1997, 108, 1–35. [Google Scholar] [CrossRef]
- Linsebigler, A.L.; Lu, G.; Yates, J.T., Jr. Photocatalysis on TiO2 surfaces: Principles, mechanisms, and selected results. Chem. Rev. 1995, 95, 735–758. [Google Scholar] [CrossRef]
- Wang, F.; Li, Q.; Xu, D. Recent progress in semiconductor-based nanocomposite photocatalysts for solar-to-chemical energy conversion. Adv. Energy Mater. 2017, 7, 1700529. [Google Scholar] [CrossRef]
- Takanabe, K. Photocatalytic water splitting: Quantitative approaches toward photocatalyst by design. ACS Catal. 2017, 7, 8006–8022. [Google Scholar] [CrossRef]
- Peter, L.M. Photoelectrochemistry: From basic principles to photocatalysis. In Photocatalysis: Fundamentals and Perspectives; The Royal Society of Chemistry: London, UK, 2016; pp. 1–28. [Google Scholar]
- Habisreutinger, S.N.; Schmidt-Mende, L.; Stolarczyk, J.K. Photocatalytic reduction of CO2 on TiO2 and other semiconductors. Angew. Chem. Int. Ed. 2013, 52, 7372–7408. [Google Scholar] [CrossRef]
- AlSalka, Y.; Hakki, A.; Schneider, J.; Bahnemann, D.W. Co-catalyst-free photocatalytic hydrogen evolution on TiO2: Synthesis of optimized photocatalyst through statistical material science. Appl. Catal. B 2018, 238, 422–433. [Google Scholar] [CrossRef]
- Walter, M.G.; Warren, E.L.; McKone, J.R.; Boettcher, S.W.; Mi, Q.; Santori, E.A.; Lewis, N.S. Solar water splitting cells. Chem. Rev. 2010, 110, 6446–6473. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.G.; Warren, E.L.; McKone, J.R.; Boettcher, S.W.; Mi, Q.; Santori, E.A.; Lewis, N.S. Correction to solar water splitting cells. Chem. Rev. 2011, 111, 5815. [Google Scholar] [CrossRef]
- Pichat, P. A brief overview of photocatalytic mechanisms and pathways in water. Water Sci. Technol. 2007, 55, 167–173. [Google Scholar] [CrossRef]
- Ni, M.; Leung, M.K.; Leung, D.Y.; Sumathy, K. A review and recent developments in photocatalytic water-splitting using TiO2 for hydrogen production. Renew. Sust. Energ. Rev. 2007, 11, 401–425. [Google Scholar] [CrossRef]
- Schneider, J.; Kandiel, T.A.; Bahnemann, D.W. Solar photocatalytic hydrogen production: Current status and future challenges. In Materials and Processes for Solar Fuel Production; Viswanathan, B., Subramanian, V., Lee, J.S., Eds.; Springer: New York, NY, USA, 2014; pp. 41–74. [Google Scholar] [CrossRef]
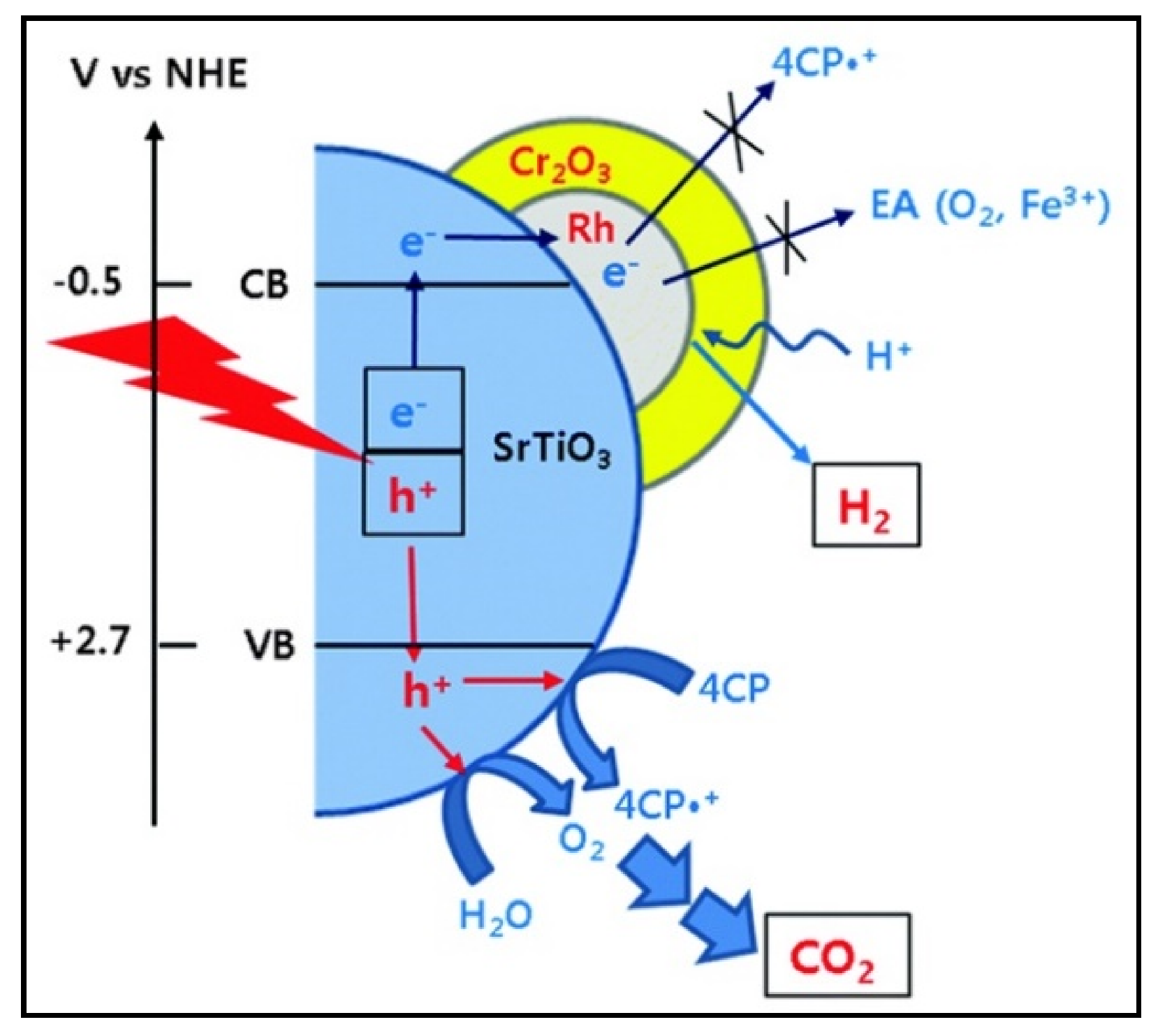
- Cho, Y.J.; Moon, G.H.; Kanazawa, T.; Maeda, K.; Choi, W. Selective dual-purpose photocatalysis for simultaneous H2 evolution and mineralization of organic compounds enabled by a Cr2O3 barrier layer coated on Rh/SrTiO3. Chem. Commun. 2016, 52, 9636–9639. [Google Scholar] [CrossRef]
- Shimura, K.; Yoshida, H. Heterogeneous photocatalytic hydrogen production from water and biomass derivatives. Energy Environ. Sci. 2011, 4, 2467–2481. [Google Scholar] [CrossRef]
- Jeon, T.H.; Koo, M.S.; Kim, H.; Choi, W. Dual-functional photocatalytic and photoelectrocatalytic systems for energy- and resource-recovering water treatment. ACS Catal. 2018, 8, 11542–11563. [Google Scholar] [CrossRef]
- Ohtani, B. Photocatalysis A to Z—What we know and what we do not know in a scientific sense. J. Photochem. Photobiol. C 2010, 11, 157–178. [Google Scholar] [CrossRef]
- Balayeva, N.O.; Zheng, N.; Dillert, R.; Bahnemann, D.W. Visible-light-mediated photocatalytic aerobic dehydrogenation of N-heterocycles by surface-grafted TiO2 and 4-amino-TEMPO. ACS Catal. 2019, 9, 10694–10704. [Google Scholar] [CrossRef]
- Zhang, L.; Jiang, D.; Irfan, R.M.; Tang, S.; Chen, X.; Du, P. Highly efficient and selective photocatalytic dehydrogenation of benzyl alcohol for simultaneous hydrogen and benzaldehyde production over Ni-decorated Zn0.5Cd0.5S solid solution. J. Energy Chem. 2019, 30, 71–77. [Google Scholar] [CrossRef]
- Zheng, Y.W.; Chen, B.; Ye, P.; Feng, K.; Wang, W.; Meng, Q.Y.; Wu, L.Z.; Tung, C.H. Photocatalytic hydrogen-evolution cross-couplings: Benzene C-H amination and hydroxylation. J. Am. Chem. Soc. 2016, 138, 10080–10083. [Google Scholar] [CrossRef]
- Puga, A.V. Photocatalytic production of hydrogen from biomass-derived feedstocks. Coord. Chem. Rev. 2016, 315, 1–66. [Google Scholar] [CrossRef]
- Schneider, J.; Bahnemann, D.W. Undesired role of sacrificial reagents in photocatalysis. J. Phys. Chem. Lett. 2013, 4, 3479–3483. [Google Scholar] [CrossRef]
- Chen, X.; Shen, S.; Guo, L.; Mao, S.S. Semiconductor-based photocatalytic hydrogen generation. Chem. Rev. 2010, 110, 6503–6570. [Google Scholar] [CrossRef]
- Kandiel, T.A.; Ivanova, I.; Bahnemann, D.W. Long-term investigation of the photocatalytic hydrogen production on platinized TiO2: An isotopic study. Energy Environ. Sci. 2014, 7, 1420–1425. [Google Scholar] [CrossRef]
- Al-Madanat, O.; AlSalka, Y.; Dillert, R.; Bahnemann, D. Photocatalytic H2 production from naphthalene by various TiO2 photocatalysts: Impact of Pt loading and formation of intermediates. Catalysts 2021, 11, 107. [Google Scholar] [CrossRef]
- Bahruji, H.; Bowker, M.; Davies, P.R.; Morgan, D.J.; Morton, C.; Egerton, T.; Kennedy, J.; Jones, W. Rutile TiO2–Pd photocatalysts for hydrogen gas production from methanol reforming. Top. Catal. 2015, 58, 70–76. [Google Scholar] [CrossRef]
- Ismail, A.A.; Robben, L.; Bahnemann, D.W. Study of the efficiency of UV and visible-light photocatalytic oxidation of methanol on mesoporous RuO2–TiO2 nanocomposites. ChemPhysChem 2011, 12, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Prairie, M.R.; Evans, L.R.; Stange, B.M.; Martinez, S.L. An investigation of titanium dioxide photocatalysis for the treatment of water contaminated with metals and organic chemicals. Environ. Sci. Technol. 1993, 27, 1776–1782. [Google Scholar] [CrossRef]
- Mo, S.D.; Ching, W.Y. Electronic and optical properties of three phases of titanium dioxide: Rutile, anatase, and brookite. Phys. Rev. B 1995, 51, 13023–13032. [Google Scholar] [CrossRef] [PubMed]
- Enke, C.G. Nonstoichiometry, diffusion, and electrical conductivity in binary metal oxides. (Wiley series on the science and technology of materials). P. Kofstad. 160 Abb. 11 Table XI, 382 S. Ca. 1060 Schrifttumshinweise. Format 15.5 × 23 cm. Wiley Interscience (J. Wiley & Sons, Inc.) New York-London-Sydney-Toronto,1972. Gebunden ca. DM 52. Werkst. Korros. 1974, 25, 801–802. [Google Scholar] [CrossRef]
- Chretien, S.; Metiu, H. Electronic structure of partially reduced rutile TiO2 (110) surface: Where are the unpaired electrons located? J. Phys. Chem. C 2011, 115, 4696–4705. [Google Scholar] [CrossRef]
- Diebold, U. The surface science of titanium dioxide. Surf. Sci. Rep. 2003, 48, 53–229. [Google Scholar] [CrossRef]
- Qian, R.; Zong, H.; Schneider, J.; Zhou, G.; Zhao, T.; Li, Y.; Yang, J.; Bahnemann, D.W.; Pan, J.H. Charge carrier trapping, recombination and transfer during TiO2 photocatalysis: An overview. Catal. Today 2019, 335, 78–90. [Google Scholar] [CrossRef]
- Kohtani, S.; Kawashima, A.; Miyabe, H. Reactivity of trapped and accumulated electrons in titanium dioxide photocatalysis. Catalysts 2017, 7, 303. [Google Scholar] [CrossRef]
- Serpone, N.; Lawless, D.; Khairutdinov, R. Size effects on the photophysical properties of colloidal anatase TiO2 particles: Size quantization versus direct transitions in this indirect semiconductor? J. Phys. Chem. 1995, 99, 16646–16654. [Google Scholar] [CrossRef]
- Stevanovic, A.; Yates Jr, J.T. Probe of NH3 and CO adsorption on the very outermost surface of a porous TiO2 adsorbent using photoluminescence spectroscopy. Langmuir 2012, 28, 5652–5659. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, T.; Katoh, R.; Furube, A.; Tamaki, Y.; Murai, M.; Hara, K.; Murata, S.; Arakawa, H.; Tachiya, M. Identification of reactive species in photoexcited nanocrystalline TiO2 films by wide-wavelength-range (400−2500 nm) transient absorption spectroscopy. J. Phys. Chem. B 2004, 108, 3817–3823. [Google Scholar] [CrossRef]
- Howe, R.F.; Gratzel, M. EPR study of hydrated anatase under UV irradiation. J. Phys. Chem. 1987, 91, 3906–3909. [Google Scholar] [CrossRef]
- Howe, R.F.; Gratzel, M. EPR observation of trapped electrons in colloidal titanium dioxide. J. Phys. Chem. 1985, 89, 4495–4499. [Google Scholar] [CrossRef]
- Wu, C.-Y.; Tu, K.-J.; Deng, J.-P.; Lo, Y.-S.; Wu, C.-H. Markedly enhanced surface hydroxyl groups of TiO2 nanoparticles with superior water-dispersibility for photocatalysis. Materials 2017, 10, 566. [Google Scholar] [CrossRef]
- Augustynski, J. The role of the surface intermediates in the photoelectrochemical behaviour of anatase and rutile TiO2. Electrochim. Acta 1993, 38, 43–46. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, P.; Liu, J.; Yu, J. New understanding of the difference of photocatalytic activity among anatase, rutile and brookite TiO2. PCCP 2014, 16, 20382–20386. [Google Scholar] [CrossRef]
- Sachs, M.; Pastor, E.; Kafizas, A.; Durrant, J.R. Evaluation of surface state mediated charge recombination in anatase and rutile TiO2. J. Phys. Chem. Lett. 2016, 7, 3742–3746. [Google Scholar] [CrossRef] [PubMed]
- Knorr, F.J.; Mercado, C.C.; McHale, J.L. Trap-state distributions and carrier transport in pure and mixed-phase TiO2: Influence of contacting solvent and interphasial electron transfer. J. Phys. Chem. C 2008, 112, 12786–12794. [Google Scholar] [CrossRef]
- Wang, X.; Feng, Z.; Shi, J.; Jia, G.; Shen, S.; Zhou, J.; Li, C. Trap states and carrier dynamics of TiO2 studied by photoluminescence spectroscopy under weak excitation condition. PCCP 2010, 12, 7083–7090. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kafizas, A.; Li, X.; Moniz, S.J.A.; Reardon, P.J.T.; Tang, J.; Parkin, I.P.; Durrant, J.R. Transient absorption spectroscopy of anatase and rutile: The impact of morphology and phase on photocatalytic activity. J. Phys. Chem. C 2015, 119, 10439–10447. [Google Scholar] [CrossRef]
- Kim, W.; Tachikawa, T.; Moon, G.H.; Majima, T.; Choi, W. Molecular-level understanding of the photocatalytic activity difference between anatase and rutile nanoparticles. Angew. Chem. Int. Ed. 2014, 53, 14036–14041. [Google Scholar] [CrossRef]
- Schindler, K.M.; Kunst, M. Charge-carrier dynamics in titania powders. J. Phys. Chem. 1990, 94, 8222–8226. [Google Scholar] [CrossRef]
- Hurum, D.C.; Agrios, A.G.; Gray, K.A.; Rajh, T.; Thurnauer, M.C. Explaining the enhanced photocatalytic activity of degussa P25 mixed-phase TiO2 using EPR. J. Phys. Chem. B 2003, 107, 4545–4549. [Google Scholar] [CrossRef]
- Choi, W.; Termin, A.; Hoffmann, M.R. The role of metal ion dopants in quantum-sized TiO2: Correlation between photoreactivity and charge carrier recombination dynamics. J. Phys. Chem. 1994, 98, 13669–13679. [Google Scholar] [CrossRef]
- Asahi, R.; Morikawa, T.; Ohwaki, T.; Aoki, K.; Taga, Y. Visible-light photocatalysis in nitrogen-doped titanium oxides. Science 2001, 293, 269–271. [Google Scholar] [CrossRef]
- Weber, M.F.; Dignam, M.J. Efficiency of splitting water with semiconducting photoelectrodes. J. Electrochem. Soc. 1984, 131, 1258–1265. [Google Scholar] [CrossRef]
- Bahnemann, D.; Henglein, A.; Lilie, J.; Spanhel, L. Flash photolysis observation of the absorption spectra of trapped positive holes and electrons in colloidal titanium dioxide. J. Phys. Chem. 1984, 88, 709–711. [Google Scholar] [CrossRef]
- Erdey-Grúz, T.; Volmer, M. Zur theorie der wasserstoff überspannung. Z. Phys. Chem. 1930, 150, 203–213. [Google Scholar] [CrossRef]
- Kasarevic-Popovic, Z.; Behar, D.; Rabani, J. Role of excess electrons in TiO2 nanoparticles coated with Pt in reduction reactions studied in radiolysis of aqueous solutions. J. Phys. Chem. B 2004, 108, 20291–20295. [Google Scholar] [CrossRef]
- Hu, C.; Lv, C.; Liu, S.; Shi, Y.; Song, J.F.; Zhang, Z.; Cai, J.G.; Watanabe, A. Nickel phosphide electrocatalysts for hydrogen evolution reaction. Catalysts 2020, 10, 188. [Google Scholar] [CrossRef]
- Michaelson, H.B. The work function of the elements and its periodicity. J. Appl. Phys. 1977, 48, 4729–4733. [Google Scholar] [CrossRef]
- Xiong, G.; Shao, R.; Droubay, T.C.; Joly, A.G.; Beck, K.M.; Chambers, S.A.; Hess, W.P. Photoemission electron microscopy of TiO2 anatase films embedded with rutile nanocrystals. Adv. Funct. Mater. 2007, 17, 2133–2138. [Google Scholar] [CrossRef]
- Di Bartolomeo, A. Graphene Schottky diodes: An experimental review of the rectifying graphene/semiconductor heterojunction. Phys. Rep. 2016, 606, 1–58. [Google Scholar] [CrossRef]
- Iwata, K.; Takaya, T.; Hamaguchi, H.-o.; Yamakata, A.; Ishibashi, T.-a.; Onishi, H.; Kuroda, H. Carrier dynamics in TiO2 and Pt/TiO2 powders observed by femtosecond time-resolved near-infrared spectroscopy at a spectral region of 0.9−1.5 μm with the direct absorption method. J. Phys. Chem. B 2004, 108, 20233–20239. [Google Scholar] [CrossRef]
- Yamakata, A.; Ishibashi, T.-a.; Kato, H.; Kudo, A.; Onishi, H. Photodynamics of NaTaO3 catalysts for efficient water splitting. J. Phys. Chem. B 2003, 107, 14383–14387. [Google Scholar] [CrossRef]
- Yamakata, A.; Ishibashi, T.-A.; Onishi, H. Water-and oxygen-induced decay kinetics of photogenerated electrons in TiO2 and Pt/TiO2: A time-resolved infrared absorption study. J. Phys. Chem. B 2001, 105, 7258–7262. [Google Scholar] [CrossRef]
- Fu, X.; Long, J.; Wang, X.; Leung, D.; Ding, Z.; Wu, L.; Zhang, Z.; Li, Z.; Fu, X. Photocatalytic reforming of biomass: A systematic study of hydrogen evolution from glucose solution. Int. J. Hydrog. Energy 2008, 33, 6484–6491. [Google Scholar] [CrossRef]
- Jang, J.S.; Ji, S.M.; Bae, S.W.; Son, H.C.; Lee, J.S. Optimization of CdS/TiO2 nano-bulk composite photocatalysts for hydrogen production from Na2S/Na2SO3 aqueous electrolyte solution under visible light (λ ≥ 420 nm). J. Photochem. Photobiol. A 2007, 188, 112–119. [Google Scholar] [CrossRef]
- Sabatier, P. Hydrogénations et déshydrogénations par catalyse. Ber. Dtsch. Chem. Ges. 1911, 44, 1984–2001. [Google Scholar] [CrossRef]
- Trasatti, S. Work function, electronegativity, and electrochemical behaviour of metals: III. Electrolytic hydrogen evolution in acid solutions. J. Electroanal. Chem. Interfacial Electrochem. 1972, 39, 163–184. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Logadottir, A.; Kitchin, J.; Chen, J.G.; Pandelov, S.; Stimming, U. Trends in the exchange current for hydrogen evolution. J. Electrochem. Soc. 2005, 152, J23–J26. [Google Scholar] [CrossRef]
- Bamwenda, G.R.; Tsubota, S.; Nakamura, T.; Haruta, M. Photoassisted hydrogen production from a water-ethanol solution: A comparison of activities of Au-TiO2 and Pt-TiO2. J. Photochem. Photobiol. A 1995, 89, 177–189. [Google Scholar] [CrossRef]
- Sheng, W.; Myint, M.; Chen, J.G.; Yan, Y. Correlating the hydrogen evolution reaction activity in alkaline electrolytes with the hydrogen binding energy on monometallic surfaces. Energy Environ. Sci. 2013, 6, 1509–1512. [Google Scholar] [CrossRef]
- Naldoni, A.; D’Arienzo, M.; Altomare, M.; Marelli, M.; Scotti, R.; Morazzoni, F.; Selli, E.; Dal Santo, V. Pt and Au/TiO2 photocatalysts for methanol reforming: Role of metal nanoparticles in tuning charge trapping properties and photoefficiency. Appl. Catal. B 2013, 130–131, 239–248. [Google Scholar] [CrossRef]
- Park, H.; Reddy, D.A.; Kim, Y.; Lee, S.; Ma, R.; Kim, T.K. Synthesis of ultra-small palladium nanoparticles deposited on CdS nanorods by pulsed laser ablation in liquid: Role of metal nanocrystal size in the photocatalytic hydrogen production. Chemistry 2017, 23, 13112–13119. [Google Scholar] [CrossRef] [PubMed]
- Wenderich, K.; Mul, G. Methods, mechanism, and applications of photodeposition in photocatalysis: A review. Chem. Rev. 2016, 116, 14587–14619. [Google Scholar] [CrossRef]
- Pei, Z.; Weng, S.; Liu, P. Enhanced photocatalytic activity by bulk trapping and spatial separation of charge carriers: A case study of defect and facet mediated TiO2. Appl. Catal. B 2016, 180, 463–470. [Google Scholar] [CrossRef]
- Saravanan, R.; Gracia, F.; Stephen, A. Basic principles, mechanism, and challenges of photocatalysis. In Nanocomposites for Visible Light-Induced Photocatalysis; Khan, M.M., Pradhan, D., Sohn, Y., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 19–40. [Google Scholar] [CrossRef]
- Al-Azri, Z.H.N.; Chen, W.-T.; Chan, A.; Jovic, V.; Ina, T.; Idriss, H.; Waterhouse, G.I.N. The roles of metal co-catalysts and reaction media in photocatalytic hydrogen production: Performance evaluation of M/TiO2 photocatalysts (M = Pd, Pt, Au) in different alcohol–water mixtures. J. Catal. 2015, 329, 355–367. [Google Scholar] [CrossRef]
- Al-Azri, Z.H.N.; AlOufi, M.; Chan, A.; Waterhouse, G.I.N.; Idriss, H. Metal particle size effects on the photocatalytic hydrogen ion reduction. ACS Catal. 2019, 9, 3946–3958. [Google Scholar] [CrossRef]
- Bamwenda, G.R.; Tsubota, S.; Nakamura, T.; Haruta, M. The influence of the preparation methods on the catalytic activity of platinum and gold supported on TiO2 for CO oxidation. Catal. Lett. 1997, 44, 83–87. [Google Scholar] [CrossRef]
- Kozlova, E.A.; Lyubina, T.P.; Nasalevich, M.A.; Vorontsov, A.V.; Miller, A.V.; Kaichev, V.V.; Parmon, V.N. Influence of the method of platinum deposition on activity and stability of Pt/TiO2 photocatalysts in the photocatalytic oxidation of dimethyl methylphosphonate. Catal. Commun. 2011, 12, 597–601. [Google Scholar] [CrossRef]
- Marzun, G.; Streich, C.; Jendrzej, S.; Barcikowski, S.; Wagener, P. Adsorption of colloidal platinum nanoparticles to supports: Charge transfer and effects of electrostatic and steric interactions. Langmuir 2014, 30, 11928–11936. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-y.; Pagel, R.; Bahnemann, D.W.; Dohrmann, J.K. Quantum yield of formaldehyde formation in the presence of colloidal TiO2-based photocatalysts: Effect of intermittent illumination, platinization, and deoxygenation. J. Phys. Chem. B 2004, 108, 14082–14092. [Google Scholar] [CrossRef]
- Haselmann, G.M.; Eder, D. Early-stage deactivation of platinum-loaded TiO2 using in situ photodeposition during photocatalytic hydrogen evolution. ACS Catal. 2017, 7, 4668–4675. [Google Scholar] [CrossRef]
- Li, F.B.; Li, X.Z. The enhancement of photodegradation efficiency using Pt–TiO2 catalyst. Chemosphere 2002, 48, 1103–1111. [Google Scholar] [CrossRef]
- Siuzdak, K.; Sawczak, M.; Klein, M.; Nowaczyk, G.; Jurga, S.; Cenian, A. Preparation of platinum modified titanium dioxide nanoparticles with the use of laser ablation in water. Phys. Chem. Chem. Phys. 2014, 16, 15199–15206. [Google Scholar] [CrossRef]
- Murcia, J.J.; Navío, J.A.; Hidalgo, M.C. Insights towards the influence of Pt features on the photocatalytic activity improvement of TiO2 by platinisation. Appl. Catal. B 2012, 126, 76–85. [Google Scholar] [CrossRef]
- Farsinezhad, S.; Sharma, H.; Shankar, K. Interfacial band alignment for photocatalytic charge separation in TiO2 nanotube arrays coated with CuPt nanoparticles. Phys. Chem. Chem. Phys. 2015, 17, 29723–29733. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yin, L.; Zhang, L.; Liu, N.; Lun, N.; Qi, Y. Platinum-nanoparticle-modified TiO2 nanowires with enhanced photocatalytic property. ACS Appl. Mater. Interfaces 2010, 2, 3373–3377. [Google Scholar] [CrossRef]
- Kang, J.-G.; Sohn, Y. Interfacial nature of Ag nanoparticles supported on TiO2 photocatalysts. J. Mater. Sci. 2011, 47, 824–832. [Google Scholar] [CrossRef]
- Zhang, L.; Mohamed, H.H.; Dillert, R.; Bahnemann, D. Kinetics and mechanisms of charge transfer processes in photocatalytic systems: A review. J. Photochem. Photobiol. C 2012, 13, 263–276. [Google Scholar] [CrossRef]
- Mohamed, H.H.; Bahnemann, D.W. The role of electron transfer in photocatalysis: Fact and fictions. Appl. Catal. B 2012, 128, 91–104. [Google Scholar] [CrossRef]
- Wang, Z.; Li, C.; Domen, K. Recent developments in heterogeneous photocatalysts for solar-driven overall water splitting. Chem. Soc. Rev. 2019, 48, 2109–2125. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Janczarek, M.; Endo, M.; Wang, K.; Balcytis, A.; Nitta, A.; Mendez-Medrano, M.G.; Colbeau-Justin, C.; Juodkazis, S.; Ohtani, B.; et al. Noble metal-modified faceted anatase titania photocatalysts: Octahedron versus decahedron. Appl. Catal. B 2018, 237, 574–587. [Google Scholar] [CrossRef] [PubMed]
- Sreethawong, T.; Yoshikawa, S. Impact of Pt loading methods over mesoporous-assembled TiO2–ZrO2 mixed oxide nanocrystal on photocatalytic dye-sensitized H2 production activity. Mater. Res. Bull. 2012, 47, 1385–1395. [Google Scholar] [CrossRef]
- Kraeutler, B.; Bard, A.J. Heterogeneous photocatalytic preparation of supported catalysts. Photodeposition of platinum on titanium dioxide powder and other substrates. J. Am. Chem. Soc. 1978, 100, 4317–4318. [Google Scholar] [CrossRef]
- Kumar, S.G.; Rao, K.S.R.K. Comparison of modification strategies towards enhanced charge carrier separation and photocatalytic degradation activity of metal oxide semiconductors (TiO2, WO3 and ZnO). Appl. Surf. Sci. 2017, 391, 124–148. [Google Scholar] [CrossRef]
- Chen, H.W.; Ku, Y.; Kuo, Y.L. Effect of Pt/TiO2 characteristics on temporal behavior of o-cresol decomposition by visible light-induced photocatalysis. Water Res. 2007, 41, 2069–2078. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xu, J.; Dai, W.-L.; Fan, K. Dependence of Ag deposition methods on the photocatalytic activity and surface state of TiO2 with twistlike helix structure. J. Phys. Chem. C 2009, 113, 8343–8349. [Google Scholar] [CrossRef]
- Elghniji, K.; Hentati, O.; Mlaik, N.; Mahfoudh, A.; Ksibi, M. Photocatalytic degradation of 4-chlorophenol under P-modified TiO2/UV system: Kinetics, intermediates, phytotoxicity and acute toxicity. J. Environ. Sci. 2012, 24, 479–487. [Google Scholar] [CrossRef]
- Yoshida, H. Photocatalytic organic syntheses. In Environmentally Benign Photocatalysts; Anpo, M., Kamat, P.V., Eds.; Springer: New York, NY, USA, 2010; pp. 647–669. [Google Scholar] [CrossRef]
- Szczepanik, B. Photocatalytic degradation of organic contaminants over clay-TiO2 nanocomposites: A review. Appl. Clay Sci. 2017, 141, 227–239. [Google Scholar] [CrossRef]
- Vasseghian, Y.; Khataee, A.; Dragoi, E.-N.; Moradi, M.; Nabavifard, S.; Oliveri Conti, G.; Mousavi Khaneghah, A. Pollutants degradation and power generation by photocatalytic fuel cells: A comprehensive review. Arab. J. Chem. 2020, 13, 8458–8480. [Google Scholar] [CrossRef]
- Benz, D.; Felter, K.M.; Koser, J.; Thoming, J.; Mul, G.; Grozema, F.C.; Hintzen, H.T.; Kreutzer, M.T.; van Ommen, J.R. Assessing the role of Pt clusters on TiO2 (P25) on the photocatalytic degradation of Acid Blue 9 and Rhodamine B. J. Phys. Chem. C 2020, 124, 8269–8278. [Google Scholar] [CrossRef]
- Ajmal, A.; Majeed, I.; Malik, R.N.; Idriss, H.; Nadeem, M.A. Principles and mechanisms of photocatalytic dye degradation on TiO2 based photocatalysts: A comparative overview. RSC Adv. 2014, 4, 37003–37026. [Google Scholar] [CrossRef]
- Qourzal, S.; Barka, N.; Tamimi, M.; Assabbane, A.; Nounah, A.; Ihlal, A.; Ait-Ichou, Y. Sol–gel synthesis of TiO2–SiO2 photocatalyst for β-naphthol photodegradation. Mater. Sci. Eng. C 2009, 29, 1616–1620. [Google Scholar] [CrossRef]
- Antharjanam, S.; Philip, R.; Suresh, D. Photocatalytic degradation of wastewater pollutants: Titanium dioxide mediated degradation of methyl orange and beta-naphthol orange. Ann. Chim. 2003, 93, 719–728. [Google Scholar]
- Hashimoto, K.; Kawai, T.; Sakata, T. Photocatalytic reactions of hydrocarbons and fossil-fuels with water—Hydrogen production and oxidation. J. Phys. Chem. 1984, 88, 4083–4088. [Google Scholar] [CrossRef]
- Kim, J.; Choi, W. Hydrogen producing water treatment through solar photocatalysis. Energy Environ. Sci. 2010, 3, 1042. [Google Scholar] [CrossRef]
- Yuzawa, H.; Kumagai, J.; Yoshida, H. Reaction mechanism of aromatic ring amination of benzene and substituted benzenes by aqueous ammonia over platinum-loaded titanium oxide photocatalyst. J. Phys. Chem. C 2013, 117, 11047–11058. [Google Scholar] [CrossRef]
- Yuzawa, H.; Aoki, M.; Otake, K.; Hattori, T.; Itoh, H.; Yoshida, H. Reaction mechanism of aromatic ring hydroxylation by water over platinum-loaded titanium oxide photocatalyst. J. Phys. Chem. C 2012, 116, 25376–25387. [Google Scholar] [CrossRef]
- Yoshida, H.; Yuzawa, H.; Aoki, M.; Otake, K.; Itoh, H.; Hattori, T. Photocatalytic hydroxylation of aromatic ring by using water as an oxidant. Chem. Commun. 2008, 4634–4636. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, A.; Sacco, O.; Sannino, D.; Venditto, V.; Vaiano, V. One-step catalytic or photocatalytic oxidation of benzene to phenol: Possible alternative routes for phenol synthesis? Catalysts 2020, 10, 1424. [Google Scholar] [CrossRef]
- Kim, J.; Lee, J.; Choi, W. Synergic effect of simultaneous fluorination and platinization of TiO2 surface on anoxic photocatalytic degradation of organic compounds. Chem. Commun. 2008, 756–758. [Google Scholar] [CrossRef]
- Sola, A.C.; Homs, N.; Ramírez de la Piscina, P. Photocatalytic H2 production from ethanol(aq) solutions: The effect of intermediate products. Int. J. Hydrog. Energy 2016, 41, 19629–19636. [Google Scholar] [CrossRef]
- Cho, Y.-J.; Kim, H.-i.; Lee, S.; Choi, W. Dual-functional photocatalysis using a ternary hybrid of TiO2 modified with graphene oxide along with Pt and fluoride for H2-producing water treatment. J. Catal. 2015, 330, 387–395. [Google Scholar] [CrossRef]
- Zhang, X.; Deng, J.; Yan, J.; Song, Y.; Mo, Z.; Qian, J.; Wu, X.; Yuan, S.; Li, H.; Xu, H. Cryo-mediated liquid-phase exfoliated 2D BP coupled with 2D C3N4 to photodegradate organic pollutants and simultaneously generate hydrogen. Appl. Surf. Sci. 2019, 490, 117–123. [Google Scholar] [CrossRef]
- Jiang, X.-H.; Wang, L.-C.; Yu, F.; Nie, Y.-C.; Xing, Q.-J.; Liu, X.; Pei, Y.; Zou, J.-P.; Dai, W.-L. Photodegradation of organic pollutants coupled with simultaneous photocatalytic evolution of hydrogen using quantum-dot-modified g-C3N4 catalysts under visible-light irradiation. ACS Sustain. Chem. Eng. 2018, 6, 12695–12705. [Google Scholar] [CrossRef]
- Mogyorósi, K.; Kmetykó, Á.; Czirbus, N.; Veréb, G.; Sipos, P.; Dombi, A. Comparison of the substrate dependent performance of Pt−-, Au- and Ag-doped TiO2 photocatalysts in H2-production and in decomposition of various organics. React. Kinet. Catal. Lett. 2009, 98, 215–225. [Google Scholar] [CrossRef]
- Vaiano, V.; Iervolino, G. Photocatalytic removal of methyl orange azo dye with simultaneous hydrogen production using Ru-modified ZnO photocatalyst. Catalysts 2019, 9, 964. [Google Scholar] [CrossRef]
- Chu, K.H.; Ye, L.; Wang, W.; Wu, D.; Chan, D.K.L.; Zeng, C.; Yip, H.Y.; Yu, J.C.; Wong, P.K. Enhanced photocatalytic hydrogen production from aqueous sulfide/sulfite solution by ZnO0.6S0.4 with simultaneous dye degradation under visible-light irradiation. Chemosphere 2017, 183, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Patsoura, A.; Kondarides, D.I.; Verykios, X.E. Enhancement of photoinduced hydrogen production from irradiated Pt/TiO2 suspensions with simultaneous degradation of azo-dyes. Appl. Catal. B 2006, 64, 171–179. [Google Scholar] [CrossRef]
- Wu, C.; Yin, M.; Zhang, R.; Li, Z.; Zou, Z.; Li, Z. Further studies of photodegradation and photocatalytic hydrogen production over Nafion-coated Pt/P25 sensitized by rhodamine B. Int. J. Hydrog. Energy 2020, 45, 22700–22710. [Google Scholar] [CrossRef]
- Mills, A.; Lee, S.K. Platinum group metals and their oxides in semiconductor photosensitisation. Platinum Met. Rev. 2003, 47, 2–12. [Google Scholar]
- Montoya, J.F.; Velásquez, J.A.; Salvador, P. The direct–indirect kinetic model in photocatalysis: A reanalysis of phenol and formic acid degradation rate dependence on photon flow and concentration in TiO2 aqueous dispersions. Appl. Catal. B 2009, 88, 50–58. [Google Scholar] [CrossRef]
- Monllor-Satoca, D.; Gómez, R.; González-Hidalgo, M.; Salvador, P. The “Direct–Indirect” model: An alternative kinetic approach in heterogeneous photocatalysis based on the degree of interaction of dissolved pollutant species with the semiconductor surface. Catal. Today 2007, 129, 247–255. [Google Scholar] [CrossRef]
- Kim, J.; Park, Y.; Park, H. Solar hydrogen production coupled with the degradation of a dye pollutant using TiO2 modified with platinum and nafion. Int. J. Photoenergy 2014, 2014, 1–9. [Google Scholar] [CrossRef]
- Howsam, M.; Jones, K.C. Sources of PAHs in the environment. In PAHs and Related Compounds; Neilson, A.H., Ed.; Springer: Berlin/Heidelberg, Germany, 1998; pp. 137–174. [Google Scholar] [CrossRef]
- Soana, F.; Sturini, M.; Cermenati, L.; Albini, A. Titanium dioxide photocatalyzed oxygenation of naphthalene and some of its derivatives. J. Chem. Soc. Perkin Trans. 2000, 699–704. [Google Scholar] [CrossRef]
- Rubio-Clemente, A.; Torres-Palma, R.A.; Penuela, G.A. Removal of polycyclic aromatic hydrocarbons in aqueous environment by chemical treatments: A review. Sci. Total Environ. 2014, 478, 201–225. [Google Scholar] [CrossRef] [PubMed]
- Hykrdová, L.; Jirkovský, J.; Mailhot, G.; Bolte, M. Fe(III) photoinduced and Q-TiO2 photocatalysed degradation of naphthalene: Comparison of kinetics and proposal of mechanism. J. Photochem. Photobiol. A 2002, 151, 181–193. [Google Scholar] [CrossRef]
- Lair, A.; Ferronato, C.; Chovelon, J.-M.; Herrmann, J.-M. Naphthalene degradation in water by heterogeneous photocatalysis: An investigation of the influence of inorganic anions. J. Photochem. Photobiol. A 2008, 193, 193–203. [Google Scholar] [CrossRef]
- Al-Madanat, O.; Curti, M.; Günnemann, C.; Alsalka, Y.; Dillert, R.; Bahnemann, D.W. TiO2 photocatalysis: Impact of the platinum loading method on reductive and oxidative half-reactions. Catal. Today. (Under Review).


























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Chemical Structure | Chemical Formula | Number of Rings | Molecular Weight (g\mol) | Melting Point (°C) | Boiling Point (°C) | Aqueous Solubility (mg/L) | Vapor Pressure (Pa) | Log Kow |
|---|---|---|---|---|---|---|---|---|---|
| Naphthalene |  | C10H8 | 2 | 128.17 | 80.26 | 218 | 31 | 1.0 × 102 | 3.37 |
| Acenaphthene |  | C12H10 | 3 | 154.21 | 93.4 | 279 | 3.8 | 3.0 × 10−1 | 3.92 |
| Acenaphthylene |  | C12H8 | 3 | 152.19 | 92–93 | 265–275 | 16 | 9.0 × 10−1 | 4.00 |
| Fluorene |  | C13H10 | 3 | 166.22 | 116–117 | 295 | 1.9 | 9.0 × 10−2 | 4.18 |
| Anthracene |  | C14H10 | 3 | 178.23 | 218 | 340–342 | 0.045 | 1.0 × 10−3 | 4.54 |
| Phenanthrene |  | C14H10 | 3 | 178.23 | 100 | 340 | 1.1 | 2.0 × 10−2 | 4.57 |
| Fluoranthene |  | C16H10 | 4 | 202.25 | 110.8 | 375 | 0.26 | 1.2 × 10−3 | 5.22 |
| Pyrene |  | C16H10 | 4 | 202.25 | 156 | 393–404 | 0.13 | 6.0 × 10−4 | 5.18 |
| Benzo[a]anthracene |  | C20H12 | 4 | 228.29 | 158 | 438 | 0.011 | 2.8 × 10−5 | 5.91 |
| Chrysene |  | C18H12 | 4 | 228.29 | 254 | 448 | 0.006 | 5.7 × 10−7 | 5.91 |
| Benzo[b]fluoranthene |  | C20H12 | 5 | 252.31 | 168.3 | No data | 0.0015 | - | 5.80 |
| Benzo[k]fluoranthene |  | C20H12 | 5 | 252.31 | 215.7 | 480 | 0.0008 | 5.2 × 10−8 | 6.00 |
| Benzo[a]pyrene |  | C20H12 | 5 | 252.31 | 179–179.3 | 495 | 0.0038 | 7.0 × 10−7 | 5.91 |
| Dibenzo[a,h]anthracene |  | C22H14 | 6 | 278.35 | 262 | No data | 0.0006 | 3.7 × 10−10 | 6.75 |
| Benzo[ghi]perylene |  | C22H12 | 6 | 276.33 | 273 | 550 | 0.00026 | 1.4 × 10−8 | 6.50 |
| Indeno[1,2,3-cd]pyrene |  | C22H12 | 6 | 276.33 | 163.6 | 530 | 0.00019 | - | 6.50 |
| Removal Techniques | Advantage(s) | Disadvantage(s) |
|---|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Madanat, O.; AlSalka, Y.; Ramadan, W.; Bahnemann, D.W. TiO2 Photocatalysis for the Transformation of Aromatic Water Pollutants into Fuels. Catalysts 2021, 11, 317. https://doi.org/10.3390/catal11030317
Al-Madanat O, AlSalka Y, Ramadan W, Bahnemann DW. TiO2 Photocatalysis for the Transformation of Aromatic Water Pollutants into Fuels. Catalysts. 2021; 11(3):317. https://doi.org/10.3390/catal11030317
Chicago/Turabian StyleAl-Madanat, Osama, Yamen AlSalka, Wegdan Ramadan, and Detlef W. Bahnemann. 2021. "TiO2 Photocatalysis for the Transformation of Aromatic Water Pollutants into Fuels" Catalysts 11, no. 3: 317. https://doi.org/10.3390/catal11030317
APA StyleAl-Madanat, O., AlSalka, Y., Ramadan, W., & Bahnemann, D. W. (2021). TiO2 Photocatalysis for the Transformation of Aromatic Water Pollutants into Fuels. Catalysts, 11(3), 317. https://doi.org/10.3390/catal11030317