2.2. Activity, Selectivity, and Stability
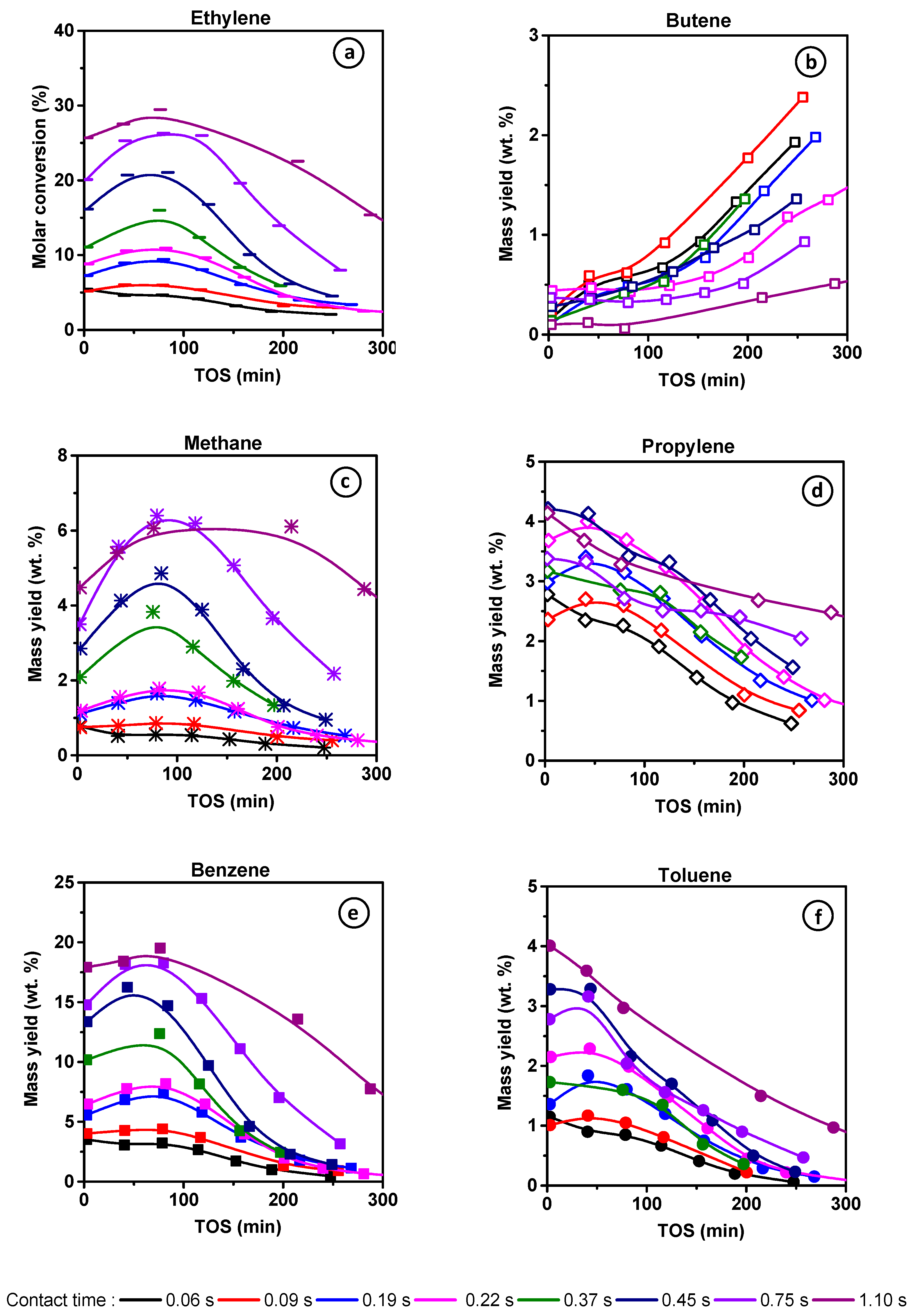
Figure 1a displays the ethylene conversion at a function of time-on-stream (TOS) obtained on M zeolite at 700 °C with different contact times (τ) ranging from 0.06 to 1.10 s. Under such operating conditions, a blank test run without catalyst, thus solely driven by thermal conversion revealed less than 3% conversion of C
2H
4 into butenes (isobutene and but-1-ene), of which a small fraction further cracks into propylene and methane.
The partial ethylene conversion is due to the combination of high temperature, low partial pressure of the reactant, and low acid sites density (Si/Al = 40), which hinders the C
2H
4 oligomerisation on protonic sites [
4,
5,
6,
7,
8,
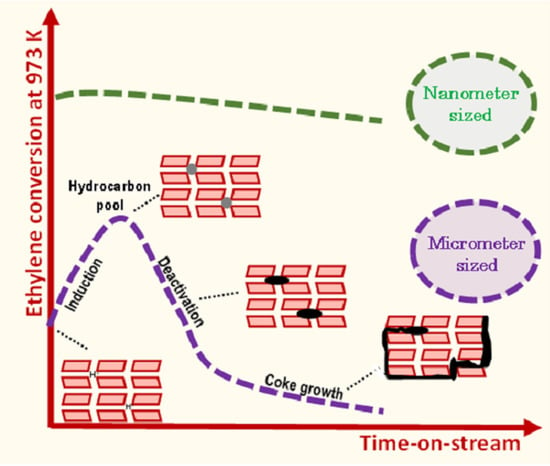
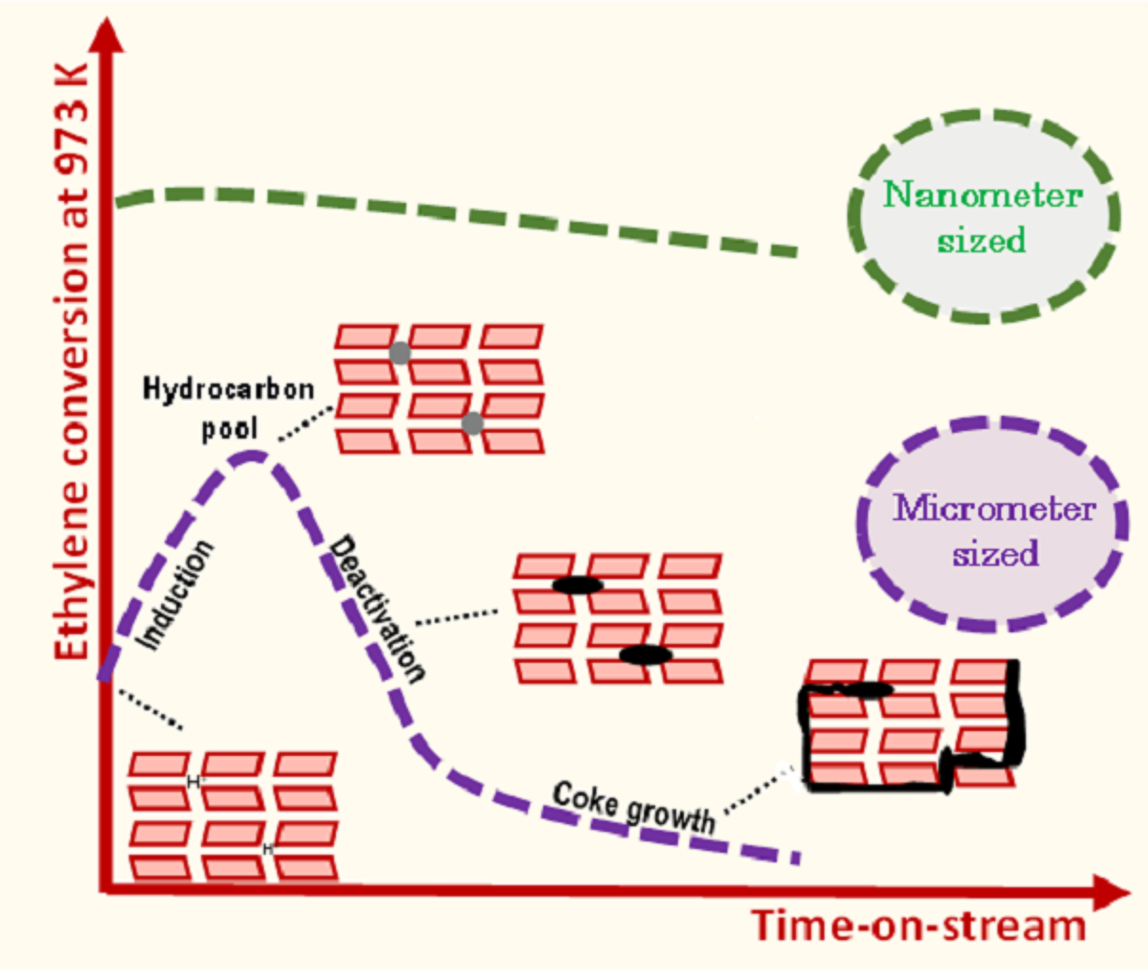
9]. The ethylene conversion increases with contact time (τ). Independently of τ, ethylene conversion achieves a maximal value at approximately 80 min TOS, then decreases to reach the thermal conversion value (3%), indicating a complete catalyst deactivation. Such a behaviour, involving an induction period followed by catalyst deactivation, is similar to what is observed in the MDA reaction on bifunctional Mo/H-ZSM-5 catalyst [
3,
20]. The induction step is defined by the in situ generations of new active species with high catalytic activity. Volmer et al. have also observed an induction period during ethylene conversion at 700 °C on a H-ZSM-5 zeolite with a molar ratio of 40. They explained the catalyst activation by the formation of a hydrocarbon pool [
3].
Among the reaction products presented in
Figure 1b–f, benzene presents the highest molar yield. Similar proportions of propylene, methane, and toluene are detected, followed by butenes to a lesser extent, and traces of naphthalene. The effect of the induction period can be observed for the product mass yields of aromatics, methane, and, to a lesser extent, with a shorter contact time for propylene. Differently, C
4H
8 formation increases significantly at TOS values, which correspond to those of the catalyst deactivation. Butene evolution is hence a deactivation marker.
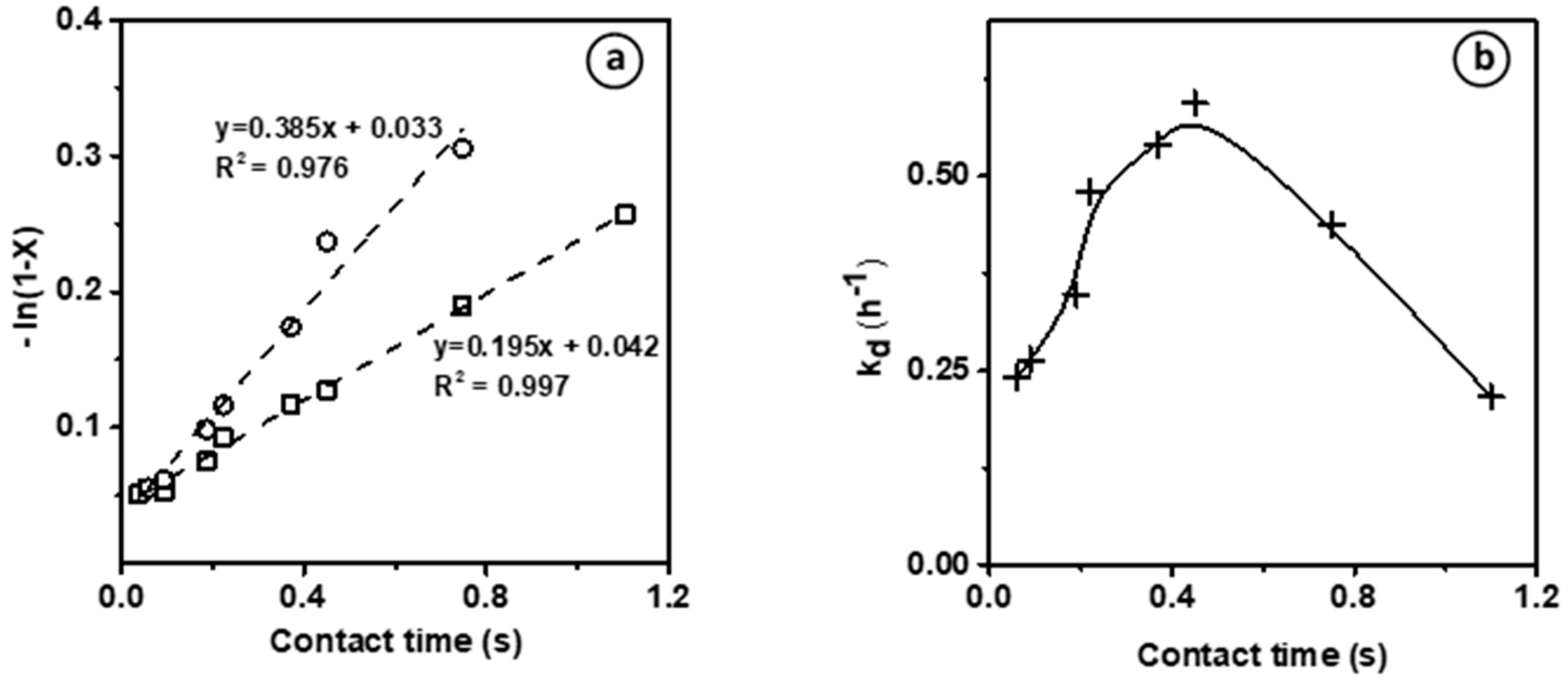
Figure 2a shows the evaluation of the kinetic rate constants for ethylene transformation (assuming a first-order reaction) (i) on the bare Brønsted acid sites at t = 2 min, and (ii) on the new active phase generated in situ during the reaction, at maximal conversion, after ca. 80 min on stream. Independently of the nature of the active sites involved, the plots
vs. contact time, where X is the ethylene conversion, gives a straight line corresponding to the equation:
. For both species, a
value between of 0.03–0.04 is achieved, corresponding to the y-intercept, which is due to the thermal conversion level previously inferred from blank experiments showing 3% of ethylene conversion at 700 °C. These yet feature distinct slopes as a function of the maximal conversion, which is approximately twice as high at short TOS on bare BAS compared to HCP. Hence, two distinct kinetics constants can be evidenced, which strongly supports the existence of two distinct active phases. The first corresponds to the bare BAS and features a moderate kinetic constant of 0.195 s
−1. The second active phase shows a two times higher activity and is generated in situ during the reaction. In accordance with the findings by Uslamin et al. [
5], we assume that these active centers are related to the formation of HCP (hydrocarbon pool) made of small aromatic molecules. Hence, the induction, as mentioned earlier, corresponds to the transformation of ethylene into HCP. The ethylene transformation catalyzed by HCP is hence ca. two times faster than by BAS.
The assumed change of the nature of the active sites during the reaction is expected not only to control the catalyst activity, but also its stability. This latter can be quantified by calculating the catalyst deactivation constant (k
d, h
−1) at end of the induction period after 40–80 min TOS, assuming a pseudo deactivation order of 1 [
21]. The deactivation rate as a function of the contact time shows a bell-shaped curve, in which k
d increases with the contact time, reaches a maximum and then decreases (
Figure 2b). This mitigation of the deactivation could result from the formation at high conversion of reaction products that scavenge the catalyst surface (i.e., H
2) or that are weakly reactive (alkanes, benzene) to acidic sites. Moreover, such behavior supports the hypothesis of the emergence of a new active phase during the reaction.
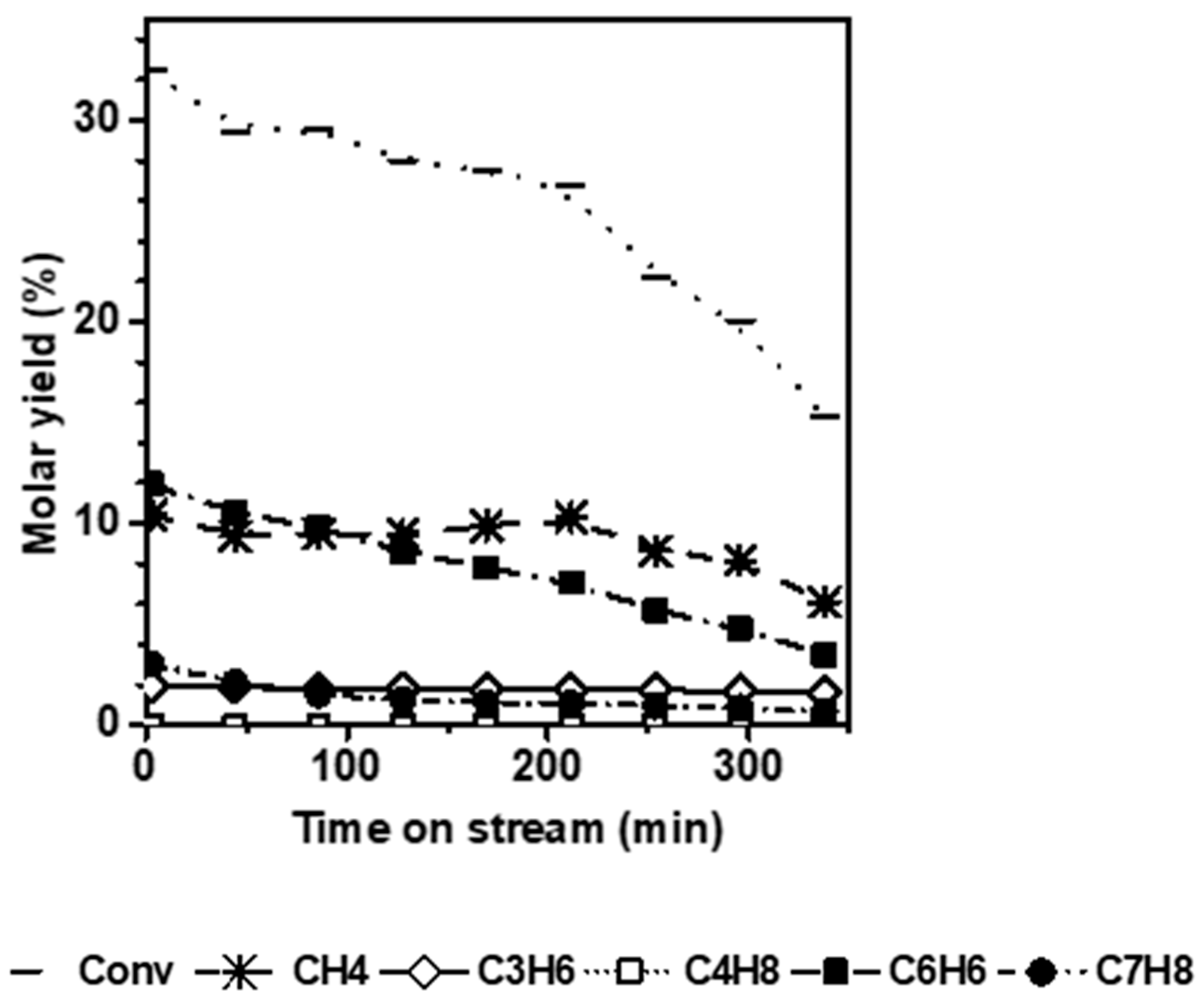
Figure 3 shows the conversion and molar yields as a function of the time-on-stream obtained on N ZSM-5. Unlike the micro-sized counterpart, the short diffusion path length (D
L) characteristic of the small crystals avoids the induction period and significantly mitigates deactivation. Indeed, k
d is about 5 times lower for the N catalyst (0.05 h
−1) compared to the micro-sized sample.
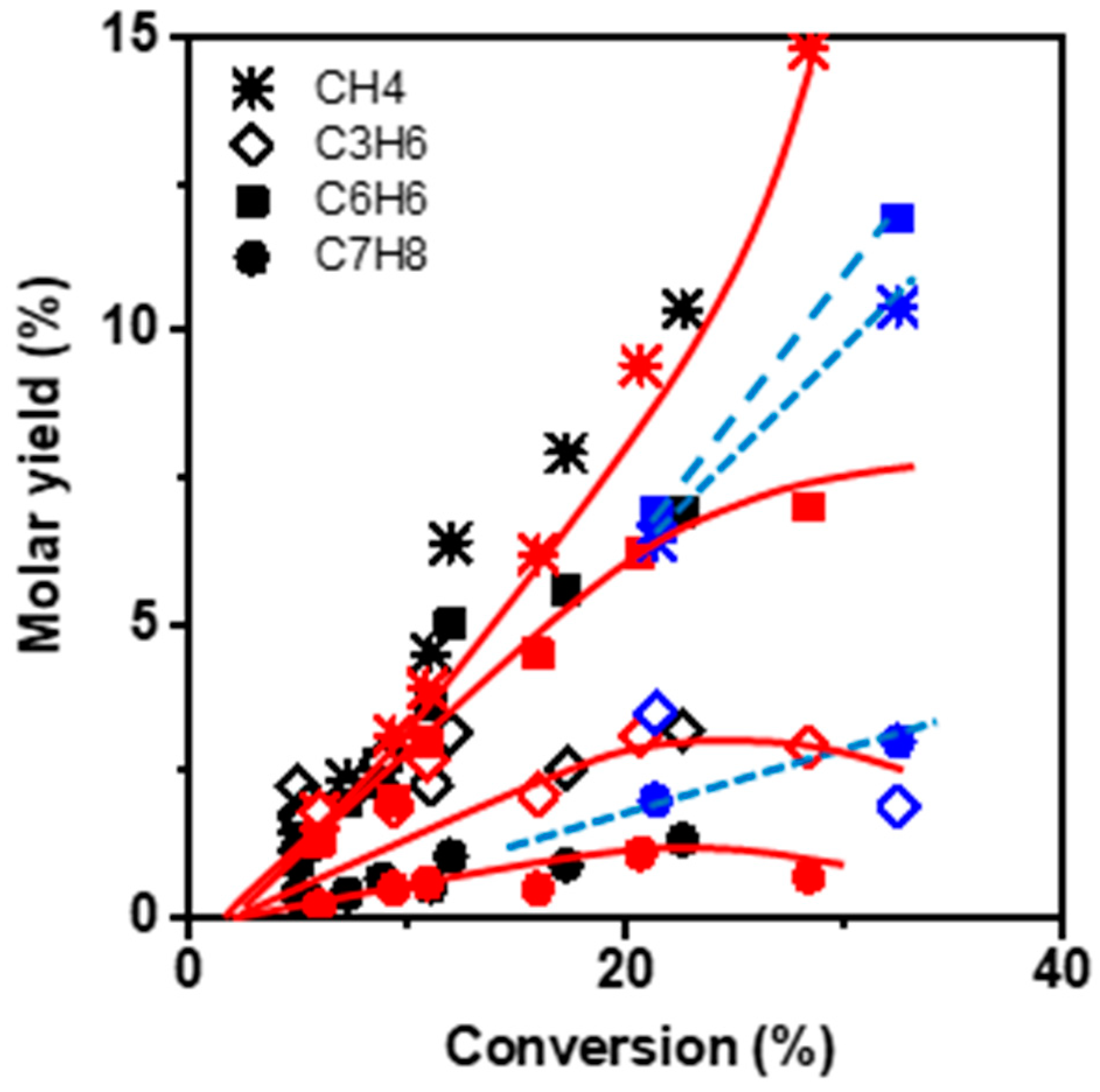
Figure 4 compares the molar yields of the main products: benzene, methane, propylene, and toluene as a function of the conversion (i) at low TOS (operated by zeolite BAS) and (ii) at TOS corresponding to the maximal conversion value, which we assume to be operated by HCP. The product selectivity profile remains identical in both cases, namely short TOS and maximum conversion, revealing that the reaction scheme is the same on BAS and HCP. However, the mechanism of the transformation is necessarily different in so far as light aromatics (HCP) participate in ethylene dimerization via the paring reaction, whereas dimerization on BAS implies the formation of an unstable primary carbenium ion. The alkylation of butene by ethylene or propylene followed by a cyclisation reaction occurs on the acid site and involves secondary carbenium ions.
Methane, benzene, propylene, and toluene appear as secondary products after 2% conversion (extrapolated value). This can be ascribed to the non-catalytic transformation of ethylene and the formation of the primary product butene, resulting from ethylene dimerization. At low conversion, the molar ratio of CH4–C3H6 is slightly lower than one, indicating that only a minor fraction of propylene reacts with butene to form toluene. With increasing conversion, the CH4–C3H6 molar ratio gradually deviates from unity. Indeed, at 20% conversion a bifurcation point is observed for which the methane yield increases with growing conversion, while the slope of C3H6 yield decreases. The methane yield keeps increasing with conversion, even though the BAS density at the zeolite surface significantly lowered during the reaction.
Besides, both propylene and toluene yields achieve a maximum of 20% conversion before stabilizing and finally declining (
Figure 4). Therefore, CH
4 does not result from a cracking mechanism but more likely from the dealkylation of toluene [
2]. The aromatisation requires either an intra-molecular dehydrogenation (DH:
) or an inter-molecular hydrogen transfer (HT:
). The HT mechanism involves alkanes as by-products; thereby, it is possible to identify which mechanism is at work by tracking the production of alkanes. In the present case, the alkane yield at low conversion is low, while at high conversion values, the alkanes (mainly ethane) yield becomes significant and further grows with increasing TOS (
Figure S2, in Supplementary Materials). This change indicates that deactivation results from the formation of less reactive molecules catalysed on BAS.
The reduction in zeolite crystal size impacts the selectivity at high conversion. The methane and benzene yields are lower and higher, respectively, than those obtained on the M zeolite. This difference suggests the limitation of toluene dealkylation (
Figure 4).
Scheme 1 proposes a reaction path by considering the evolution of primary or secondary products and the occurrence of side-reactions (toluene dealkylation and intermolecular hydrogen transfer).
2.3. Characterisation of Carbon Deposits
To investigate the role of carbon deposits in either promoting the catalytic activity (HCP) or causing catalyst deactivation (coke), spent catalysts were collected at key steps of the reaction, namely (i) right after the induction period (20 min), (ii) after achieving the highest conversion value (80 min), (iii) before total deactivation (180 min), and (iv) after complete deactivation (360 min). The reaction was run at 0.45 s contact time (
Figure S3 in Supplementary Materials) and stopped at the desired time. The reactor was then cooled to room temperature under N
2 flow, and the spent catalyst was collected for characterisation. Moreover, the effect of the diffusion path length and mass transfer on the carbon deposit speciation was evaluated through a systematic comparison M and N zeolites.
Figure 5 reports the coke content, the residual micropore volume, the remaining BAS content, and residual microporous volume for each of the reaction steps mentioned above.
In both M and N materials, the coke formation follows the first stage with a high rate and a subsequent slower growth regime as TOS > 80 min. (
Figure 5a,b) At TOS = 80 min, significant coke amounts are formed in both M (5.5 wt.%) and N (10 wt.%) catalysts. At TOS > 80 min linear increase in coke content continues, despite complete deactivation of M. As the highest ethylene conversion value is reached on M zeolite (TOS = 80 min; 5.5 wt.% coke), more than 85% of BAS are poisoned, but the reduction in micropore volume is below 50% (
Figure 5c). This result validates our assumption that the observed induction period is due to the generation of a pool of small aromatic molecules (HCP) adsorbed on the zeolite walls by reaction with BAS sites while having a moderate impact on the overall microporous volume. A complete loss of BAS and V
micro is observed when coke deposit reaches 7wt.% (extrapolated value) and 15 wt.%, respectively (
Figure 5c). The molar weight of the coke (
) constituting the HCP was calculated assuming that one coke molecule neutralized one BAS. As such,
increases from 130 to 220 g mol
−1, which corresponds approximately to the molar weight of naphthalene and pyrene, respectively.
The absence of diffusion limitation on the nano-sized (N) zeolite may seem inconsistent with the higher coking rate and higher coke content measured on N (
Figure 5b) compared to M. (
Figure 5a). This contradiction can be explained by the fact that coke precursors are easily trapped on the numerous silanol groups located on the outer surface of N [
16]. Lakiss et al. have shown that such external coke has little impact on the selectivity and none on the stability of the catalyst.
The faster development of HCP on N zeolite than M zeolite is consistent with the faster reduction in micropore volume (
Figure 5c,d). The induction period on the M catalyst reveals that substantial diffusion limitations occur during the HCP formation. Moreover, these intracrystalline diffusion resistances impact the kinetic desorption of products, which favors the growth of coke, leading to catalyst deactivation. Indeed, Cui et al. have shown the MDA reaction on Mo/ZSM-5 catalysts the existence of three rate-controlling regions. These reaction steps correspond to external mass transfer, intracrystalline diffusion, and kinetic desorption regimes [
22].
Figure 5e,f show the CO
2 profiles resulting from the temperature-controlled coke burn-off on spent M and N zeolites. Note that no CO was detected during the operation. Low intensity of the water signal indicates poor hydrogen content of the coke. The CO
2 profiles of the catalysts collected at TOS = 80 min (at the end of the induction period of M) display a low (M) to moderate intensity (N) peak at ca. 600 °C. Vollmer et al. observed also for EDA on a H-ZSM-5, a coke oxidation peak at ca. 650 °C [
3]. This high temperature is associated with diffusion-limited combustion of coke deposited in the zeolite micropores [
23,
24,
25]. The CO
2 profiles collected after 180 and 360 min TOS show an additional, higher temperature component. This shoulder is particularly noticeable in the CO
2 production profile of M recovered after 180 min TOS (red line,
Figure 5e), showing the co-existence of two components at ~600 and ~700 °C. In general, at TOS ranging from 80 to 120 min, a single peak is detected around 600 °C, which intensity increases with carbon content. At TOS ≥ 180 min, a second peak appears and increases with TOS, while the first peak remains stable. On the N catalyst, the co-existence of distinct coke species is less pronounced. However, a moderate shift towards higher temperatures occurs upon varying the TOS from 20 to 180 min. The peak assignment is possible from the chemical composition of the coke. The coke combustion is more driven by its aromaticity degree of polycyclic hydrocarbons rather than by their accessibility to oxygen. Indeed, the molecules trapped within the micropores burn at a lower temperature compared to heavy coke molecule on the external surface. Therefore, the lower combustion temperature on N zeolite compared to M zeolite (ca. 50 °C) suggests a lower aromaticity degree of the external coke (
Figure 5c–f).
A further investigation of the speciation of the coke trapped inside the M zeolite pores at each step was made using liquid–liquid extraction of the mineralized zeolite matrix and the subsequent analysis of both the organic phase and the solid residual. The organic phase embeds the soluble (C
soluble), while the solid residual contains the insoluble (C
insoluble) coke fraction. GC–MS (Gas chromatography mass spectroscopy) analysis of the soluble coke fraction shows that the formation polyaromatic compounds: naphthalene, anthracene, phenanthrene, pyrene, and benzo(e)pyrene, occurs during the induction period (
Figure S4 in Supplementary Materials). Before and after complete deactivation (3 and 6 h), the fraction of polycyclic hydrocarbons composed of 2 to 5 aromatic cycles disappear. This disappearance occurs despite the increase in the overall coke content (
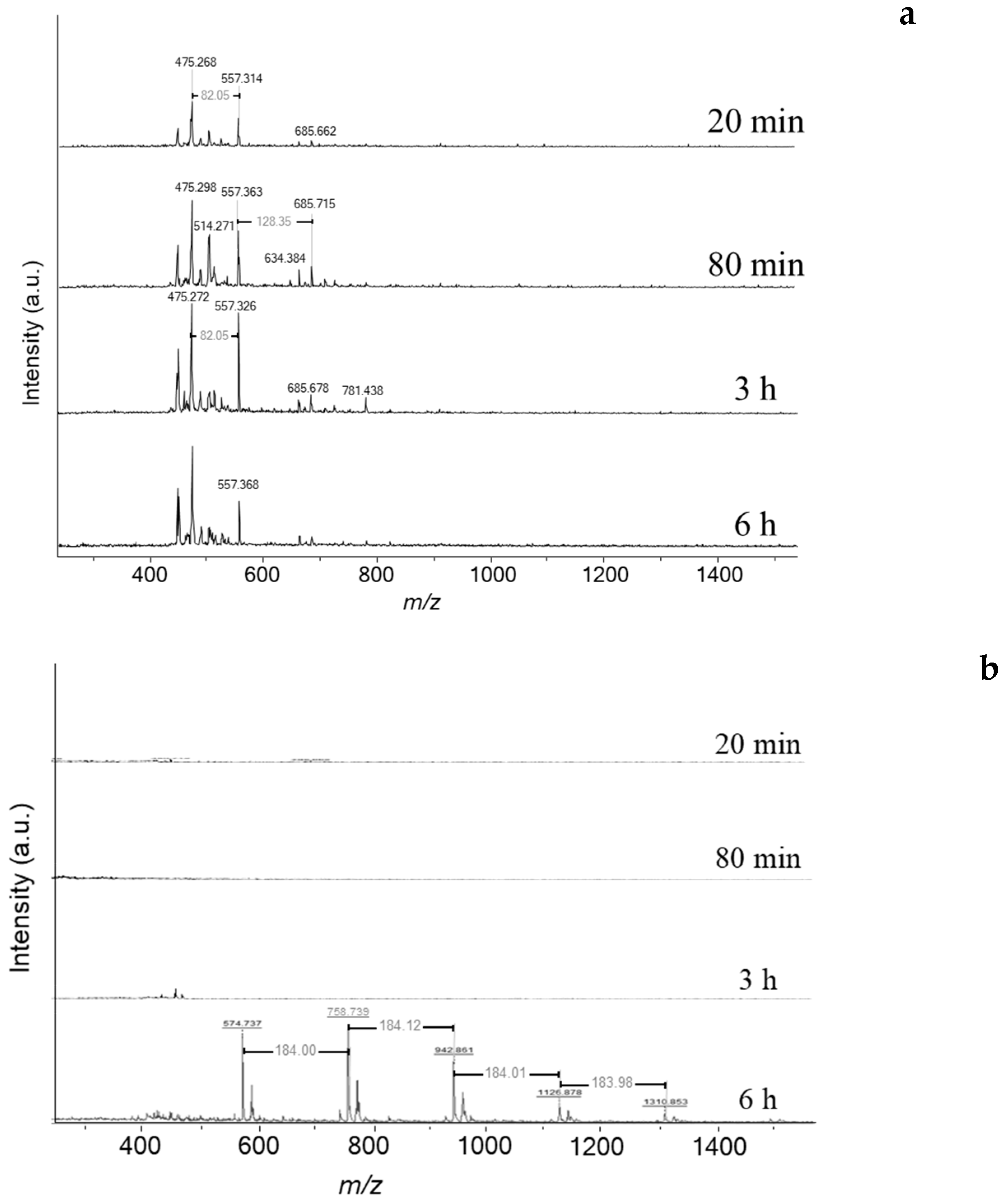
Figure 5a). Therefore, deactivation is likely linked to a drastic change of carbon speciation. The characterisation of soluble coke by MALDI-TOF/MS (Matrix Assisted Laser Desorption Ionization—Time of Flight Mass Spectroscopy) reveals a limited number of heavy molecules (
Figure 6a). It is worth indicating that the odd masses are due to the protonation of aromatic molecules by dithranol (MW: 226 g mol
−1). Traces of heavy coke molecules appear from 80 min of reaction with molar weights of 474, 556, and 684
m/
z, which corresponds to the chemical formulas C
37H
30, C
43H
40, and C
53H
48, respectively. The weight distribution is very different with the one obtained on ethanol transformation at 350 °C [
25]. At low temperature the coke is very alkykated while at high temperature as dealkylation of aromatic rings occurs, the number of alkyl groups is limited.
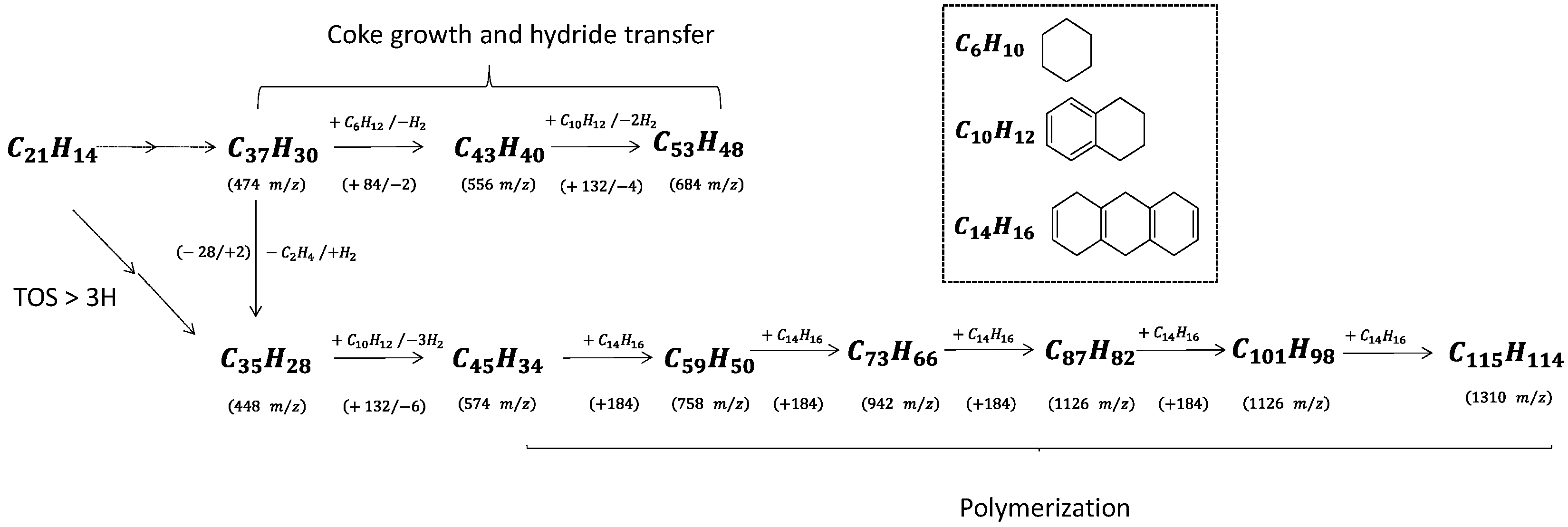
Coke molecules are chemically reactive, and their interaction with reaction products results in continuous growth until they reach the size of the zeolite pores. The trapped molecules participating in HCP, such as pyrene and benzopyrene, react with cyclohexane and tetralin, yielding hydrogen as a by-product (
Scheme 2). Note that cyclohexane and tetralin are present as reaction intermediates to form benzene and naphthalene, respectively. The spent catalyst placed directly onto the mass spectrometer’s target (LDI-TOF) allows characterizing the external coke. In the absence of the matrix (hydroxyanthrone), the molar masses of volatilized molecules are even (
Figure 6b). The external coke appears after 3 h of reaction. The molecules located on the outer surface are heavier than those trapped within the zeolite micropores due to the absence of steric constraints limiting their growth. Their molar weight ranges from 448 up to 1310
m/
z, which might correspond to polycyclic aromatic hydrocarbons with 35 to 115 carbons. Despite the wide range, the number of possible molecules is somewhat limited (5–10). Moreover, the spectrum after 6 h displays a mass loss sequence of 184
m/
z, which could correspond to hexahydroanthracene. This sequence suggests that the coke growth mechanism involves the polymerisation of hexahydroanthracene units, probably via a free-radical mechanism [
26].
As the heavy coke overgrows the external surface of the crystals, it blocks the access of reactant molecules to the zeolite channels, which translates into a loss of accessible microporous volume, as shown in
Figure 5b.
Scheme 2 summarizes the growth sequence of coke molecules within the micropores and on the external surface. To the best of our knowledge, this work provides the first evidence of coke growth on the outer zeolite surface, mostly driven by a condensation mechanism.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}