Abstract
The pyrolysis of lignocellulosic biomass is one of the most promising methods of alternative fuels production. However, due to the low selectivity of this process, the quality of the obtained bio-oil is usually not satisfactory and does not allow for its direct use as an engine fuel. Therefore, there is a need to apply catalysts able to upgrade the composition of the mixture of pyrolysis products. Unfortunately, despite the increase in the efficiency of the thermal decomposition of biomass, the catalysts undergo relatively fast deactivation and their stability can be considered a bottleneck of efficient pyrolysis of lignocellulosic feedstock. Therefore, solving the problem of catalyst stability is extremely important. Taking that into account, we presented, in this review, the most important reasons for catalyst deactivation, including coke formation, sintering, hydrothermal instability, and catalyst poisoning. Moreover, we discussed the progress in the development of methods leading to an increase in the stability of the catalysts of lignocellulosic biomass pyrolysis and strengthening their resistance to deactivation.
1. Introduction
The pyrolysis of lignocellulosic biomass has been identified as a prospective direction towards obtaining fuels for current infrastructure [1]. This process results in the formation of a bio-oil, char, and gaseous fraction. Pyrolysis products can be obtained during rapid heating as a result of the initial thermal decomposition of lignocellulosic biomass. High-temperature conversion of this type of feedstock leads to the formation of primary intermediates which are subsequently subjected to dehydration, decarbonylation, decarboxylation, oligomerization, and reforming, among others (Scheme 1) [2,3,4,5,6]. It results in the formation of a wide group of hydrocarbon derivatives (oxygenates—phenols, carboxylic acids, aldehydes, ketones, esters, ethers, etc.). That mixture can be enriched with hydrocarbons in the presence of catalysts. However, secondary reactions may also lead to the formation of undesirable coke due to polymerization, condensation, or aromatization of primary pyrolysis products.
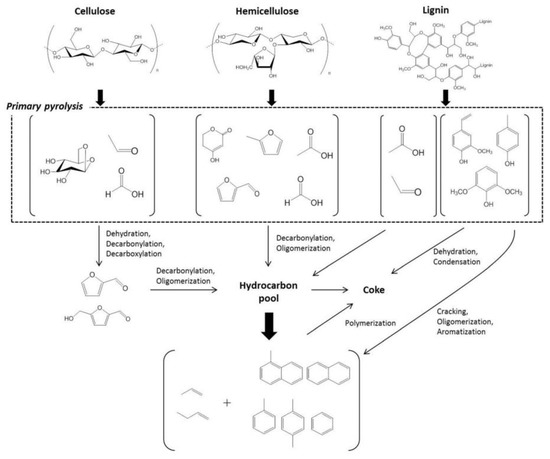
Scheme 1.
Subsequent steps of the pyrolysis process. Republished with the permission of RSC from Catalytic pyrolysis of individual components of lignocellulosic biomass, Wang, K., Kim, K.H., Brown, R.C., Green Chem. 2014, 16, 727–735; permission conveyed through Copyright Clearance Center, Inc [7].
Bio-oil cannot be directly used as engine fuel, as it has low caloric value, high oxygen content, contains water, acids, low storage time, poor chemical stability, and it is immiscible with the fossil fuel due to high polarity [8]. Therefore, as mentioned earlier, the composition of bio-oil should be upgraded by the use of a catalyst. Many catalysts have been developed for the upgrading process including acidic zeolites, recognized as efficient cracking catalysts, which can increase the yield of aromatics that are highly demanded; however, they undergo a fast deactivation process [9,10]. On the other hand, there are mesoporous materials, which are less prone to deactivation as they possess weaker acid sites but in consequence, their activity is lower [11]. On the other side, there is a group of metal-based catalysts including noble metal-based systems such as Pd or Ru and known hydrotreating catalysts mainly based on transition metals such as Ni, Co, or Fe [12,13]. They can also increase the contribution of aromatics, limit water formation, and promote hydrogen transfer and reforming reactions. However, due to the high activity, they might decrease the liquid fraction due to the formation of gases [14]. Moreover, oxides of different chemical character are also often used as catalysts for this process. Generally, acidic oxides promote the formation of carboxylic acids, aromatic components, and furans, but they also enhance the formation of tars. On the other hand, tar formation is lower in the case of basic oxides, but they promote the formation of a gaseous fraction and can inhibit the formation of phenols [15].
Commercial use of those catalysts is, however, strongly limited due to problems with their stability in reaction conditions. Therefore, in recent years, researchers have been increasing their efforts to solve this issue, as evidenced by the increasing number of publications devoted to this topic (Figure 1). It appears that coke formation, sintering, hydrothermal instability, and catalysts poisoning are considered as the main reasons for catalyst deactivation in the pyrolysis process (Figure 2).
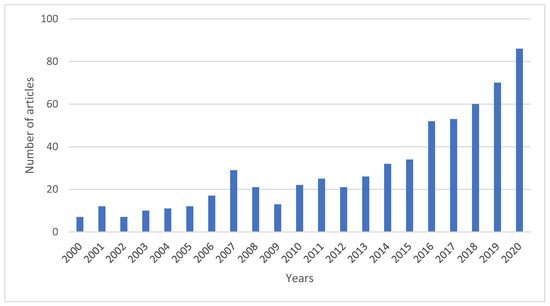
Figure 1.
Increasing number of scientific papers devoted to the problem of catalyst deactivation in pyrolysis process (Scopus 28.12.2020, pyrolysis + catalyst + deactivation).
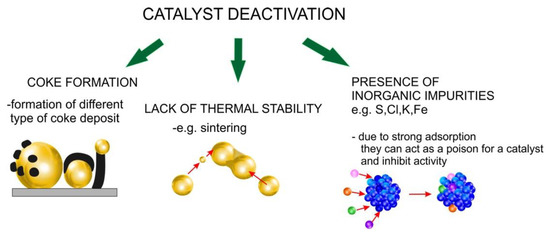
Figure 2.
Types of catalyst deactivation in pyrolysis process.
The formation of coke deposits is the most common reason for severe deactivation of catalysts in the fast pyrolysis of lignocellulosic biomass and bio-oil upgrading [7,16]. Deposition of carbon may result in direct encapsulation of active sites present on the catalyst’s surface, formation of filamentous carbon, plugging of pores (which contributes to blocking the access of reagents to active sites located in the inner pores), and deterioration of catalyst structure. The mechanism of coke formation was previously described in extensive review and research articles [17,18]. Generally, the formation of two main forms of carbon deposit can be observed—encapsulating coke and filamentous coke. The first one is produced from reaction intermediates by subsequent condensation and polymerization, while the second arises due to dissociation of small coke precursors (i.e., carbon oxide, methane, or other light hydrocarbons), which results in the formation of atomic carbon. The carbon atoms can diffuse through the surface of metal nanoparticles to the bulk. Then, their nucleation and precipitation on the basis or surface of metal grains can be observed. This results in the formation of carbon filaments, which lift the metal crystallites on their top. The formation of carbon fibers does not affect the activity of catalysts directly but can be a reason for a decrease in catalytic performance due to loss of an active phase during friction between grains in the catalyst bed or in the regeneration process.
The next reason for the deactivation of catalysts used in the pyrolysis of lignocellulosic biomass is related to the sintering of metals deposited on the catalyst surface [17]. In this case, an increase in the size of metal crystallites due to the migration of atoms located on small metal particles towards larger ones or the migration of smaller crystallites and formation of large metal grains can be observed. Sintering takes place at high reaction temperature and may be facilitated by higher pressure, the presence of steam, or weak metal–support interaction, among others. This phenomenon leads to a decrease in the number of exposed metal active sites on the catalyst surface and a reduction in catalytic activity.
Thermal treatment of the catalyst may also result in the deterioration of its structure. This can be related to sintering of the support grains, phase transformation, or the formation of new compounds (i.e., hydroxides, spinels) on the catalyst surface [19,20]. This leads to a decrease in surface area, collapse of porous structure, or change in chemical composition of the active phase. Degradation of the catalyst structure can be limited by the selection of the materials stable in the reaction conditions or the addition of promoters increasing catalyst stability.
Last but not least, the presence of different kinds of impurities of inorganic nature is considered as a serious obstacle. Those impurities can be present both in the biomass itself, but can be also introduced during catalyst preparation. During the catalytic reaction process, they can be transferred to the catalyst surface and, depending on many factors, such as the nature of the impurity, the contact time, and the reaction conditions, can cause a redhibitory decrease in catalyst lifetime or even the full inhibition of activity.
The above-mentioned factors are crucial when considering catalytic performance and should, in consequence, be taken into consideration for catalyst design. Here, in the current review, we aim at highlighting some of the most crucial problems related to catalyst stability with a focus on the pyrolysis process. Strong emphasis will be put on carbon deposit formation, nanoparticle sintering, and the poisonous effect of impurities, as well as their respective role on the catalyst’s deactivation.
2. Formation of Coke Deposit
Coke deposition rate strongly depends on the physicochemical properties of the catalysts (the presence of functional groups—acid-base character, porosity, the presence of metal and degree of its dispersion, type of support in the case of supported catalysts, incorporation of promoters). Therefore, studies devoted to the increase in the efficiency of biomass pyrolysis and bio-oil upgrading are focused on the design of the catalytic systems possessing the structure and composition which allows maintaining of the satisfying activity with simultaneous reduction in susceptibility to carbon deposit formation (Table 1). Generally, the mechanism of coke formation involves several paths depending on the temperature: arising of metal carbides (150–400 °C), formation of amorphous films with a polymeric character (250–500 °C), production of graphitic carbon (about 500 °C), or growth of carbon filaments (above 300 °C). In the case of heterogeneous catalysts, coke can be located on the surface of metal particles, the metal–support interface, or support (Figure 3). Deactivation of the catalyst may follow through the encapsulation of metal particles due to polymerization or condensation of the primary products of the thermal decomposition of biomass (i.e., oxygenates, aromatics) which usually occur below 500 °C or the formation of carbon fibers at higher temperature range. Encapsulating carbon can be formed due to adsorption of oxygenated and/or nonoxygenated products produced in the initial step of the process on the metal sites. In these conditions, carbon atoms are not only dissolved in the metal crystallites and adsorbed on the bottom of metal grains (at the interface between active phase and the support), but primary decomposition products can be subjected to subsequent condensation or polymerization over the metal particle surface. It results in the formation of a polymeric or amorphous coke film, which covers or encapsulates the metal particles. This limits their capacity for the adsorption of reactants and causes a rapid loss of catalyst activity due to the decrease in active metallic surface [21].

Table 1.
Formation of carbon deposits on the surface of catalysts of high temperature conversion of lignocellulosic biomass.
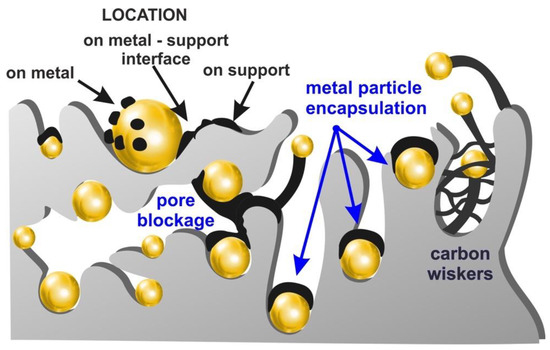
Figure 3.
Formation of coke deposit on the catalyst surface.
In the second case, filamentous carbon can be produced by the transformation of carbon oxide via the Boudouard reaction or its reduction to carbon and the decomposition of light hydrocarbons (mainly methane). This leads to the adsorption and diffusion of atomic carbon through the metal particle and further precipitation on the interface between the metal and the support, resulting in the formation of carbon filaments. Due to the fact that carbon atoms cannot dissolve in noble metal particles, this phenomenon can only be observed for catalysts containing transition metals. Figure 4 presents an exemplary carbon filament formed on the surface of the Ni/Pr2O3-ZrO2 catalyst during pyrolysis of cellulose.
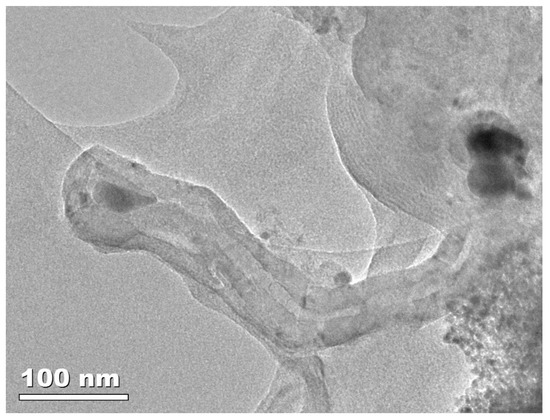
Figure 4.
TEM image of carbon filament formed on the surface of Ni/Pr2O3-ZrO2 catalyst during pyrolysis of cellulose.
The highest temperature favors the formation of pyrolytic coke, which can be produced via the cracking of reaction intermediates (both oxygenates and hydrocarbons). The pyrolytic coke is deposited non-selectively on the entire surface, leading to the total encapsulation of the catalyst. Taking into account the effect of the size of the active phase crystallites on the type of formed carbon deposit, it is suggested that smaller metal particles enhance the formation of encapsulating carbon, leading to faster deactivation of the catalyst by covering of the active phase with an impermeable carbonaceous layer. On the other hand, larger metal crystallites (above 6–7 nm) facilitate the production of filamentous carbon with Ni particles located on the top of formed filaments [22].
The literature shows that HZSM-5 is one of the most widely used catalysts of fast pyrolysis of lignocellulosic biomass [11]. However, this material, in spite of its high activity, suffers from rapid deactivation due to coking. This is related to the relatively small pore size and high surface acidity [23]. The application of mesoporous material, such as SBA-15, possessing weaker acid sites than HZSM-5, may contribute to the limitation of carbon deposition and blockage of active centers. Unfortunately, it also leads to a reduction in catalyst activity. Therefore, Xu et al. [24] decided to create material with developed mesoporosity, but retaining high acidity. They synthesized multilayered ZSM-5 nanosheet zeolite and compared its catalytic behavior in the upgrading of cellulose pyrolysis vapors with commercial HZSM-5. The obtained results showed that the yields of olefins and aromatic hydrocarbons were similar in both cases. Despite the formation of a higher amount of coke, ZSM-5 nanosheets (possessing higher mesoporosity) exhibited a longer lifetime related to the increased accessibility of reagents to the active sites located in the inner pores of zeolite.
Yu et al. [25] limited fast deactivation of HZSM-5 by the formation of MCM-41/ZSM-5 composite with ZSM-5 crystallites encapsulated in the MCM-41 mesoporous matrix. It is known that in the case of ZSM-5, large diameter pyrolysis intermediates may polymerize on the catalyst surface and block access to the micropores, decreasing the yield of valuable products. The application of a composite catalyst allows cracking of the large molecules in the mesoporous matrix to smaller ones, which can be further transferred to ZSM-5 microcrystals and converted into desired compounds. The studies of the catalytic performance of MCM-41/ZSM-5 composite in fast pyrolysis of miscanthus demonstrated that the efficiency of this catalyst in hydrocarbon production is comparable to parent zeolite. Although, the MCM-41 matrix can act as a layer protecting against the rapid deactivation of ZSM-5 due to the fact that a considerable part of coke was deposited on the surface of the mesoporous material.
One of the methods for the selective increase of HZSM-5 activity in fast pyrolysis of lignocellulosic biomass consists of the modification of its structure by the addition of various dopants. Persson et al. [14] demonstrated that introduction of selected transition metals (i.e., Fe or Ni) to the zeolite structure resulted in a noticeable increase in the contribution of monoaromatic hydrocarbons or naphthalenes in the bio-oil produced from softwood sawdust. Unfortunately, the presence of a metallic phase led to the more intense formation of coke whose amount increased from 3.5% to 7.2% in the case of parent and Fe-modified zeolite, respectively. An increase in carbon content was related to a higher concentration of acid sites on the surface of the doped catalyst. On the other hand, the presence of metal was responsible for a decrease in the temperature of coke removal during the regeneration step. Bimetallic Fe-Ni/ZSM-5 exhibited the most interesting features, allowing for the formation of a high amount of aromatic compounds with simultaneous moderate coking of the catalyst.
An increase in the production of aromatics in comparison to parent zeolite was also observed by Zheng et al. [26] who applied mixed CaO/HZSM-5 catalyst in the pyrolysis of Yunnan pine. The introduction of CaO also resulted in a decrease in bio-oil yield due to the formation of a larger amount of gaseous products. Additionally, the presence of calcium oxide led to a noticeable reduction in coke content arising on the catalyst surface (from 8.9% for HZSM-5 to 4.0% in the case of CaO/HZSM-5, respectively). It can be suggested that CaO facilitates the adsorption of CO2 and the oxidation of deposited carbon. A similar phenomenon was noticed in the case of the pyrolysis of cellulose, lignin, and sunflower stalk and in situ upgrading of bio-oil performed in the presence of MgO/Al-MCM-41 [27]. The addition of magnesium promoted deoxygenation and aromatization reactions, leading to the formation of aromatic hydrocarbons. Moreover, the coexistence of acid and basic sites formed on the surface of modified Al-MCM-41 was responsible for higher coking resistance. It was demonstrated that the presence of magnesium limited the formation of carbon deposit via condensation or polymerization and enhanced hydrogen migration due to the activation of C-H groups. Generally, the selectivity to monocyclic aromatic carbons increased and coke content decreased with the increase in the number of base sites on the surface of Mg-doped catalysts. The coke formation rate also depends on the type of used feedstock. Mullen et al. [28] showed that the co-pyrolysis of biomass (switchgrass) and plastic waste (polyethylene) resulted in a reduction in the amount of deposited carbon. Polyethylene changes the reaction mechanism, providing additional hydrogen to the reaction medium. It enhances the formation of aromatic hydrocarbons and limits the polymerization of carbonaceous residues.
The metal/support catalysts are the next group of materials, allowing for the increase in the efficiency of the fast pyrolysis of biomass and bio-oil upgrading. In this case, studies are focused on both optimization of the conditions of catalytic reactions and the development of new materials possessing higher resistance against coking. Arregi et al. [29] confirmed that the main reason for the deactivation of commercial Ni catalyst (G90-LDP) in pyrolysis and in-line steam reforming of wood sawdust was severe coke deposition. It was suggested that coke is formed by the re-polymerization of phenolic oxygenates (being the intermediate products of the first pyrolysis step) and its amount can be limited by the optimization of reaction conditions favoring reforming and water–gas shift reactions. The lowest content of oxygenates (being the main coke precursors) was observed when bio-oil upgrading was performed at 600 °C using a steam to biomass ratio = 3. Further measurements of continuous pyrolysis and in-line catalytic reforming of pine wood and high-density polyethylene (HDPE), conducted with the use of the same catalyst, demonstrated that its deactivation rate depends not only on the amount of deposited carbon but also the type of coke [30]. Two types of carbon deposit were observed—encapsulating coke consisted of amorphous carbon and filamentous coke composed of carbon nanorods or nanotubes. The first one, which originated mainly from the reforming of oxygenates derived from biomass, resulted in faster catalyst deactivation. The contribution of filamentous coke was higher in the presence of HDPE (its formation was stimulated by the reforming of hydrocarbons derived from polyethylene). It does not block nickel active size rapidly, but gradually hinders the contact of reagents with metal particles.
The stability of nickel catalysts, which are one of the most commonly used in the biomass pyrolysis process, strictly depends on the type of the used support. Santamaria et al. [31] evaluated the catalytic performance of Ni introduced on the surface of Al2O3, SiO2, MgO, TiO2, and ZrO2 by wet impregnation method in in-line steam reforming of vapors from the fast pyrolysis of pine. The obtained results demonstrated promising activity of Ni/Al2O3, Ni/MgO, and Ni/ZrO2. However, Ni/Al2O3 possessing high initial activity underwent noticeable coke deposition and progressive deactivation due to the presence of a higher number of acid sites. On the other hand, Ni/MgO and Ni/ZrO2, which were less active in the first stage of the process, revealed higher resistance against carbon deposit formation. This was related to the presence of basic sites on the surface of magnesium oxide and zirconium oxide responsible for gasification of adsorbed coke. It is worth noticing that ZrO2 possessed higher surface area and pore volume in comparison to MgO, resulting in better access to the catalyst active sites by larger reaction intermediates and easier regeneration. On the other hand, lower activity of Ni/TiO2 can be ascribed to the phase transformation of anatase to rutile, leading to a decrease in the dispersion of an active phase. In turn, the low activity of Ni/SiO2 can be related to the microporous structure of silica, hindering the contact between larger oxygenates formed in the initial pyrolysis step and active sites located inside the SiO2 pores. Similar catalytic behavior of supported Ni catalysts in the pyrolysis of cellulose was previously presented by our group in [32,33]. Further measurements confirmed the increased stability of Ni/ZrO2 catalysts [34]. Its redox properties and high oxygen storage capacity hindered the formation of coke deposits. Additionally, zirconium oxide is known for its ability to adsorb H2O molecules present in the reaction mixture and facilitation of their decomposition. This results in the formation of hydroxyl groups on the catalyst surface participating in gasification of the deposited carbon [35].
The studies aimed at the development of catalysts with increased resistance against carbon deposition were not only devoted to the choice of an appropriate support, but also focused on the effect of the introduction of promoters into the support structure [36]. Lanthanides are one of the most popular dopants of Ni catalysts used in fast pyrolysis of biomass and bio-oil upgrading. Bimbela et al. [37] showed that the introduction of Ce to Ni/Mg-Al catalyst of steam reforming of the aqueous fraction of bio-oil resulted in noticeable enhancement of resistance to coke formation. Cerium oxide exhibits high oxygen mobility and possesses remarkable oxygen storage capacity. These features are especially helpful in the removal of deposited carbon from the catalyst surface. CeO2 provides oxygen atoms which are subsequently used in the gasification of coke. On the other hand, it is reported that the use of cerium may lead to a significant decrease in surface area of the modified catalyst. This may lead to a decrease in the dispersion of the active phase and due to that, a drop in the efficiency of the conversion process [38].
The beneficial role of cerium was also emphasized in Refs. [39,40]. Modification of Ni/Al2O3 by Ce resulted in improved stability of the catalyst in pyrolysis and in-line steam reforming of lignocellulosic biomass due to a reduction in coke formation rate. A comparison of the catalytic performance of Ni/Al2O3-CeO2 and Ni/Al2O3-MgO indicated that the catalyst doped by cerium was more efficient than its counterpart modified by magnesium. It was suggested that the lower activity of the latter may be related to the lower reducibility of Ni/Al2O3-MgO due to the formation of a MgAl2O4 spinel phase [37].
Zhang et al. [41] evaluated the effect of the addition of Na, Mg, and La on the activity and stability of the Ni/SiO2 catalyst in a model reaction of the steam reforming of guaiacol. The obtained results showed that introduction of sodium and magnesium led to a decrease in the surface area of modified catalysts, while incorporation of lanthanum allowed keeping the surface area on the same level as for non-modified material. It resulted in the formation of more dispersed nickel crystallites in the case of La-Ni/SiO2. Additionally, the presence of lanthanum facilitated the reduction of nickel oxide leading to the increase in the activity of the modified catalyst. Introduction of La increased the alkalinity of Ni/SiO2 catalyst to a mild extent, while the presence of Na led to the formation of a higher number of strong basic sites. It was suggested that strong basic sites favored the adsorption of oxygen-containing species, which could undergo polymerization to a carbonaceous deposit. The formation of thermally stable amorphous coke may contribute to faster deactivation of the catalyst. On the other hand, mild basicity in combination with the ability of lanthanum oxide to adsorb water molecules and their further dissociation may facilitate the gasification of carbon species adsorbed on the catalyst surface and limit its deactivation rate [42].
Valle et al. [43] confirmed the formation of two types of deposited coke on the surface of La-modified Ni/Al2O3 catalyst used in fast pyrolysis and steam reforming of pine sawdust. Their contribution was strongly influenced by the reaction conditions and composition of the reaction mixture. The encapsulating coke leading to the fast deactivation of the catalyst originated from oxygenates, while the formation of filamentous coke was launched by decomposition of CO or CH4 and dissolution of carbon atoms on the surface of nickel crystallites.
It is reported that pyrolysis and catalytic reforming of biomass (pine sawdust) can be successfully performed in the presence of Ni supported on waste slag-based carriers (magnesium slag, steel slag, blast furnace slag, pyrite cinder, and calcium silicate slag) [44]. The obtained results demonstrated that the activity of Ni deposited on the magnesium slag was the highest among the studied slag supports and even higher than the activity of the commercial Ni/Al2O3 catalyst. The promising catalytic performance of the Ni/magnesium slag can be related to the composition of the support which is composed of the compounds containing Mg, Fe, and Ca. Yu et al. [45] suggested that the presence of calcium can promote the adsorption of water molecules on the surface of the catalysts, which facilitates the removal of deposited carbon species and reduces the coke formation rate. An interaction between nickel and iron may be responsible for the formation of Ni-Fe alloy or NiFe2O4 spinel phase contributing to the more efficient cracking of tar. Additionally, the Ni-Mg-O solid solution may influence nickel dispersion on the catalyst surface and inhibit the growth of the carbon deposit. It is worth noting that the activity of Ni/slag catalysts was related to the character of the coke deposit. The most active material was characterized by the highest contribution of disordered carbon in comparison to less active catalysts possessing a higher content of graphite carbon species.
The literature shows that the application of catalysts in high-temperature conversion of biomass prepared from waste materials is becoming more and more popular. Jahromi and Agblevor [46] used red mud (caustic waste generated in the Bayer process of alumina production consisting of a wide group of metal oxides, such as: Fe2O3, Al2O3, SiO2, MgO, CaO, Na2O, K2O, etc.) for the synthesis of a Ni-based catalyst for hydrodeoxygenation of pinyon-juniper wood pyrolysis oil. Analysis of the obtained results demonstrated that the use of Ni supported on waste material facilitated the production of liquid products (about 68% of liquid fraction) to a greater extent than commercial Ni/SiO2-Al2O3 (not more than 42% of liquids). Moreover, the introduction of Ni on the surface of red mud led to a reduction in coking rate in comparison to the commercial catalyst (4.2 and 7.3% of carbon, respectively). It was demonstrated that the regeneration of Ni/red mud by burning off the coke and reducing with hydrogen allowed for complete restoration of its activity, while in the case of the commercial catalyst, it was impossible. Carbon deposit formation was not the only cause of deactivation of Ni supported on red mud-based material. A decrease in the catalyst activity can also be related to the oxidation of nickel active sites during the reaction or formation of Fe2NiO4 on the catalyst surface. A beneficial effect of the application of waste materials as catalyst or support for a Ni-based catalyst for pyrolysis and catalytic steam reforming of waste wood pellets was also described by Al-Rahbi and Williams [47]. It was suggested that the application of ash originating from coal combustion, waste tires, or refuse-derived fuel allows for the increase in hydrogen-rich gas production due to the presence of metal impurities (such as Al, Ca, Mg, Cu, Fe, K, Na, or Zn), promoting the reforming of primary products formed in the first step of the studied process.
Similar studies, focused on the synthesis of zeolites using fly ash produced by the combustion of coal, were performed in our group [48]. It was reported that an application of those materials as supports for Ni catalysts in high-temperature conversion of cellulose and pine led to the increase in hydrogen-rich gas production in comparison to nickel supported on commercial ZSM-5. Moreover, the modification of the most active Ni/Na-A catalyst by selected rare-earth or transition metals (La, Pr, Ce, Y, Gd, Zr) resulted in a further rise in H2 formation. This phenomenon was related to moderate acidity, more homogeneous dispersion of active phase, higher reducibility of nickel oxide, and limitation of carbon deposit formation in the case of fly ash-based catalysts. It was suggested that an incorporation of dopants into the structure of the support enhanced oxygen atoms mobility and the improved ionic conductivity of the modified catalysts, leading to more efficient decomposition of lignocellulosic feedstock and coke gasification.
3. Sintering and Deterioration of Catalyst Structure
Despite the high importance of carbon deposit formation, the mechanism of deactivation of catalysts in the fast pyrolysis of biomass and bio-oil upgrading may also include metal sintering and deterioration of the catalyst surface [17]. Metal sintering can be observed in the case of both supported and unsupported catalysts. Regardless of the type of used material, it leads to a decrease in the surface of the active phase grains and increase in the size of metal crystallites (Figure 5). The harsh reaction conditions or high pre-treatment temperature are considered the main reasons for catalyst sintering. However, an influence of the reaction atmosphere and the strength of interactions between catalyst components should also be taken into consideration. On the other hand, the high temperature of the pyrolysis process may not only induce the sintering of metal crystallites, but can also be responsible for degradation of the structure of the support including the loss of surface area and deterioration of porous structure leading to the limited access of reaction substrates to catalytically active sites or the change of surface properties of the used catalysts. Examples of research devoted to this area are presented in Table 2.
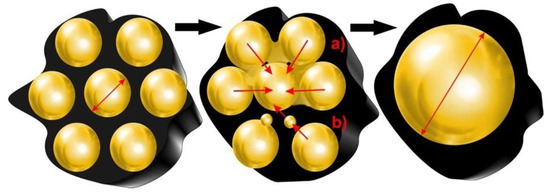
Figure 5.
Growth of metal particles due to the migration of small crystallites (a) or atoms (b) on the catalyst surface.
Table 2.
Sintering and deterioration of catalyst structure during high-temperature conversion of lignocellulosic biomass and products of its processing.
The studies performed by Stefanidis et al. [20] proved that ZSM-5, used as the catalyst in the pyrolysis of biomass, may suffer not only from coke formation but can also be deactivated by the dealumination of the zeolite framework. Dealumination, being one of the reasons for hydrothermal deactivation, results in the removal of aluminum atoms from the zeolite structure, leading to the reduction in both the strength and number of the acid sites. The main reason of dealumination is the presence of hot water vapor in the reaction mixture. The water molecules may originate from moisture in the lignocellulosic feedstock, dehydration of biomass components during initial steps of pyrolysis, and the upgrading of pyrolysis vapors. Additionally, dealumination can also be observed during the regeneration step of the spent catalyst. The high temperature and presence of water formed during the oxidation of coke may result in the subsequent dehydroxylation of the Brønsted acid sites located on the zeolite surface [52]. One of the methods of stability improvement of ZSM-5 zeolite in the catalytic pyrolysis of biomass is the creation of a hierarchical core/shell structure with a mesoporous core (containing ZSM-5) and microporous shell (consisted of Silicalite-1) [53]. The introduction of a Silicalite-1 shell allowed for maintaining of the structure, surface area, and porosity of the zeolite after hydrothermal treatment, which was not the case with parent mesoporous zeolite. On the other hand, the mesoporous core facilitated the diffusion of reagents through the catalyst and increased the accessibility of catalytically active sites. Moreover, the activity tests in the catalytic pyrolysis of pine sawdust showed enhanced shape-selectivity of the modified material, resulting in a noticeable increase in the production of p-xylene, among others.
Similar to in the case of zeolites, the presence of water is considered as a very critical factor for the stability of Ni/Al2O3 catalysts. The structural properties of the alumina-supported catalyst can be strongly modified in the presence of hydrothermal conditions. In the presence of water, alumina is transformed into boehmite AlO(OH) [54,55]. In consequence, the interaction between Ni and the support is modified as well. Metal support interaction in those catalysts depends on the number and strength of Lewis acid sites: the stronger the acidity of the alumina, the stronger the Ni–Al2O3 interaction that is observed. In contrast, the hydration of alumina preferentially affects the medium and strong acid sites which firstly can result in the decrease in the strength of the metal–support interaction [56,57] and additionally, in the leaching of the metal to the reaction media.
Further studies indicated that besides deterioration of the catalyst structure, and especially coking, the metal sintering may be the next reason for the deactivation of the catalysts used in the thermocatalytic conversion of lignocellulosic biomass. Ochoa et al. [58] showed that high reaction temperature and reducing atmosphere in pyrolysis and in-line catalytic steam reforming of pine wood resulted in the continuous increase in the average size of Ni crystallites on the surface of G90LDP (commercial Ni reforming catalyst provided by Süd Chemie and consisted of NiO supported on Al2O3 doped with calcium) from 25 to 39 nm. Initially, the sintering process occurs intensively and subsequently takes place with the rate of about 5–8 nm per hour up to 100 min of the process. The high reaction temperature −600 °C—which is slightly above the Tamman temperature—results in the increased mobility of metal species if the interaction between an active phase and the support, in the case of the supported catalysts, is not strong enough. Unfortunately, the formation of strong metal–support interactions may affect the surface composition of the catalyst (i.e., formation of heavily reducible spinels) and hence, limit catalytic activity.
Sintering of Ni crystallites may also occur during the regeneration of G90LDP used in the mentioned reaction. It was demonstrated that coke combustion carried out at 700 °C resulted in irreversible deactivation of that catalyst due to the increase in the Ni crystallites’ size from 25 to 55 nm after a fifth reaction–regeneration cycle [59]. However, it is worth noting that sintering was less intense in successive regeneration cycles and the catalyst structure reached stability after four of them. Additionally, it was observed that the increased size of Ni crystallites facilitated the formation of amorphous coke at the expense of filamentous carbon, contributing to a further drop in catalytic activity.
The studies performed by Santamaria et al. [37] showed that sintering depends on the type of the used support. Analysis of the results obtained for Ni supported on Al2O3, MgO-Al2O3, and CeO2-Al2O3 demonstrated only a slight increase in the metal crystallite size during steam reforming of biomass pyrolysis vapors in the first two cases (from 10 to 13 nm and from 15 to 19 nm, respectively), while nickel particles supported on CeO2-Al2O3 became much larger (increase from 18 to 31 nm). The increase in the size of Ni nanoparticles was related to the poor dispersion of ceria crystallites, which in consequence could not prevent metal sintering in the reaction conditions. Despite the sintering of nickel on the surface of CeO2-Al2O3, this catalyst revealed the highest stability in the studied reaction. The higher stability of Ni/CeO2-Al2O3 resulted from the high oxygen storage capacity and redox properties promoting decomposition, and the removal of accumulated coke being the main cause of catalyst deactivation. On the other hand, the lower activity of Ni/MgO-Al2O3 in comparison to Ni/Al2O3 was caused by the formation of MgAl2O4 spinel which hindered the reduction in nickel oxide species [60].
It was reported that sintering of Ni contributed also to the deactivation of the catalyst derived from bulk NiAl2O4 spinel used in the oxidative steam reforming of raw bio-oil produced in flash pyrolysis of pine sawdust [61]. However, its deactivation was reversible and regeneration of the spent catalyst by the combustion of coke, deposited on its surface during the reaction at 850 °C in the air, resulted in total recovery of the initial structure of NiAl2O4 characterized by homogeneous distribution of nickel species.
One of the methods for limiting the sintering of metal involves modification of the composition of the catalyst’s active phase. Wang et al. [62] showed that the synthesis of bimetallic Ni-Co/LaFeO3 catalyst allowed for better dispersion of metals than that observed in the case of monometallic Ni/LaFeO3 and Co/LaFeO3 counterparts (10 vs. 20 and 27 nm, respectively). Despite the application of these catalysts to the model reaction of steam reforming of ethanol resulting in the increase in the size of metal crystallites, the smallest nanoparticles were still observed for the bimetallic sample (22 vs. 44 and 43 nm for monometallic catalysts). The anti-sintering ability of Ni-Co/LaFeO3 was related to the possibility of restructuring the interface between the active phase and the support at high temperature. In the case of the bimetallic sample, this led to lower interface energy and a stronger interaction between metal species and perovskite support.
An analysis of the deactivation of Rh/CeO2-ZrO2 catalyst in steam reforming of bio-oil coming from flash pyrolysis of pine sawdust demonstrated that sintering of rhodium was not a significant cause of activity decrease [63]. Although, the performed studies indicated that the catalyst underwent structural changes during the reaction involving support aging and partial occlusion of Rh species. The most noticeable decrease in surface area and pore volume of Rh/CeO2-ZrO2 was observed during the initial stage of the process, which may be due to high temperature and the presence of water in the reaction medium. Aging of the support led to the increase in pore diameter, which suggested a collapse of the narrow pores during the reaction. This resulted in the partial occlusion of metal nanoparticles in the catalyst structure. The high water content in the reaction mixture also led to progressive oxidation of rhodium species being the next reason for catalyst deactivation.
The oxidation of metal species was also observed in the case of Fe/hydrochar catalysts used in ex situ microwave-assisted pyrolysis of rice husk and corn cob [64]. α-Fe and Fe3C phases present on the catalyst surface were completely oxidized to Fe3O4 during the reaction, which led to the decrease in the activity of the catalyst. Simultaneously, sintering of the active phase was observed. On the other hand, no metal leaching was noted. This was related to the encapsulation of iron crystallites in the biochar matrix. A decrease in surface area and micropore volume was mainly caused by coke deposition.
4. Contamination of Bio-Oil during Pyrolysis
Soils are very prone to any type of contamination, coming from both hydrological and atmospheric sources. In the case of highly polluted soils such as from post mine or post-industrial areas, they might contain many impurities of different nature such as heavy metals (e.g., As, Cd, Hg, Mo, Pb). Soil pollution is among the serious environmental concerns of today’s world. It is a growing problem causing vast areas of land to become unexploited and hazardous for both wildlife and human beings [65].
The accumulation of inorganics by plants from polluted soil allows, in a large time frame, a decrease in the number of pollutants in the soil, but due to the high concentration of the impurities by plants, their use is limited; a number of strategies are currently being developed to use them as feedstock for multiple syntheses. The process in which living plants are used to reduce the toxicity or volume of pollution in soil is called phytoremediation. Plants may clean up many kinds of pollutions such as heavy metals, pesticides, or oil. On the one side, this increases the sustainability aspect but, on the other hand, brings new challenges to be solved related to inorganic types of impurities [56].
The problem of impurities present in the feedstock is of high importance, as impurities can easily migrate to the bio-oil. This is particularly important when the most demanding feedstocks containing large amounts of impurities are used such as in post phytoremediation, municipal waste, or sludge. Sludge and municipal waste might also contain significant amounts of impurities, among them heavy metals, such as lead, zinc, cadmium, copper, chromium, and nickel. Impurities that are present in large quantities such as Al, Ca, Cu, and Zn in biomass can easily migrate to the bio-oil. This process depends on many factors—the conditions of the pyrolysis process, the kind and amount of impurities present in the feedstock, and the biomass species [66].
Despite that it has been shown that inorganic species can remain in the char [67,68], a significant part of them may vaporize at the pyrolysis process temperature of 500–600 °C. In several works, it was also observed that inorganic impurities can also be transferred to bio-oil at much lower temperatures [69,70,71,72]. The vaporized impurities may block the active sites and poison the catalyst, leading to its deactivation [20]. There are a series of metal catalysts that are proved to be efficient in the deoxygenation of bio-oil derived from sludge or other polluted feedstocks; however, catalysts are usually based on metals (Ni, Co, Mo, Zn, Fe, Cu, Cs, Mg, and noble metal-based catalysts) [73,74] which are highly sensitive to the presence of the impurities. Therefore, the stability problem is an important issue and efficient solutions are highly demanded [75]. One of the strategies allowing for mitigation of the negative effect of those impurities present in highly polluted feedstock such as sludge is to blend it with other non-polluted biomass before high-temperature treatment [76].
5. Effect of Impurities Presence on the Catalytic Performance
5.1. Metal-Based Catalysts
The main types of inorganic impurities that can be accumulated in metal-based catalysts are presented in Figure 6.
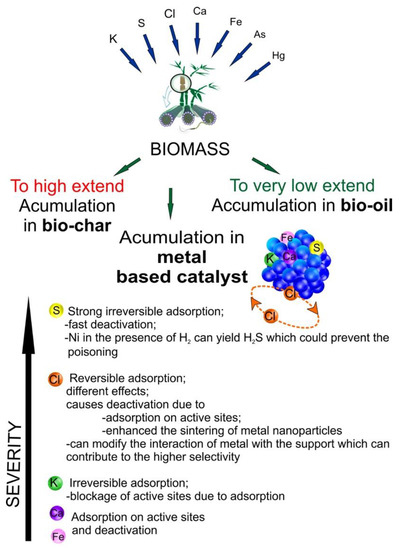
Figure 6.
Main types of inorganic impurities that can be accumulated in metal-based catalysts.
Metal impurities such as potassium and magnesium were considered as a poison for zirconia-supported iron-oxide catalysts used in catalytic upgrading of pyroligneous acid derived from the pyrolysis of woody biomass such as Japanese cedar. Pyroligneous acid is an aqueous fraction where volatile matters produced during slow pyrolysis are condensed. In contrast to pyrolysis oil, this fraction is not upgraded to biofuels but to higher value chemicals. Those inorganic impurities were identified at concentrations of 580 and 430 ppm in the pyroligneous acid. This concentration was sufficient to block the active sites of the catalysts. In consequence, this concentration inhibited the ketonization reaction that was used for the upgrading of hydroxyacetone and carboxylic acids (acetic and propionic acids) into aliphatic ketones [77].
The stability and resistance of a typical metal catalyst used for bio-oil upgrading, namely Ni/ZrO2, was investigated by Mortensen et al. [78]. The reaction was performed using a model component such as guaiacol in 1-octanol as a solvent, in the flow system. The effect of several typical inorganic impurities such as S, Cl, and K on the activity performance of nickel catalyst was investigated. A typical amount of impurities which can be present in bio-oil was added separately to the reaction mixture in a controlled way, which allow understanding of their influence on the tested catalysts. Sulfur was considered as the most severe poison, as Ni/ZrO2 was fully deactivated in a relatively short reaction time. Both reactions—hydrogenation and deoxygenation—were inhibited. This behavior concerning Ni catalysts was already observed in the literature in the different processes and is usually related to the strong irreversible adsorption of S on the Ni active sites which causes the complete loss of activity [79,80]. It was shown as well that the deactivation depends strongly on the reaction conditions; in the presence of H2, metallic nickel can also convert C–S bonds and generate metastable nickel sulfide, which, in the presence of hydrogen, can yield in H2S, which is easily removed from the reaction mixture and can prevent Ni poisoning [81]. Sulphur can also be intentionally introduced to Ni catalysts in order to improve isomerization selectivity in the fat oil hydrogenation or in steam reforming to minimize coking due to the partial poisoning and activity decrease [71].
In contrast, a different poisonous effect was observed on the Ni catalyst when chlorine was added to the reaction feed. Primarily, the activity of deoxygenation steadily decreased after exposure to this impurity in the flow reactor. This deactivation was, however, reversible when chlorine was removed from the feed. This was explained by the preferential adsorption of chlorine on the low coordinated sites of Ni catalysts and the formation of an equilibrium surface layer. It is difficult, therefore, to unequivocally classify the role of chlorine in the case of Ni-based catalysts. On one side, it is considered as a poison blocking active sites and, therefore, inhibiting the activity of the catalyst. On the other hand, its positive effect is recognized. Chlorine presence during the catalyst’s synthesis can enhance the reducibility of Ni and improve its dispersion in the final catalyst [82]. On the other hand, the presence of chlorine in the reaction feed during the upgrade of pyrolysis products enhanced the sintering of nickel nanoparticles on the catalyst, which, in consequence, contributed to the activity decrease [62].
Reversible chlorine adsorption was also identified in the case of Ni-Mo catalysts. The stability of Ni-MoS2/ZrO2 during the hydrodeoxygenation of phenol in 1-octanol as a bio-oil model in a continuous flow reactor was investigated by Mortensen et al. [83]. The resistance to impurities of the catalysts was evaluated in the presence of several elements such as potassium or organically bound chlorine. In order to understand the effect of the chlorine’s presence, 1-chlorooctane was added to the bio-oil, which resulted in an activity decrease, which probably was related to the competitive adsorption of the chlorine on the metal sites. This adsorption was, however, reversible, as after the removal of 1-chlorooctane from the reaction environment, the catalyst shows the same activity as without the presence of the poison. Furthermore, the presence of K also has a negative effect on the Ni catalysts and mainly affects deoxygenation activity, whereas hydrogenation activity was not significantly inhibited [69].
The presence of K was also harmful for other bimetallic, Ni-based catalysts. In order to check its effect, Ni-MoS2/ZrO2 was impregnated with the KNO3 in a molar ratio of K/(Ni + Mo) of 1. This resulted in a strong decrease in activity in the hydrodeoxygenation process. This deactivation was permanent, and it was probably related to the blockage of vacancy sites of MoS2 by potassium, which resulted in the inhibition of active sites for HDO (hydrodeoxygenation) [74].
The influence of different supports (ZrO2, SiO2) in Ni and Ni-Cu catalysts was evaluated in the work of Schmitt et al. [84] for the hydrodeoxygenation of bio-oil obtained using beech wood. The authors confirmed the presence of sulfur in all catalysts after the reaction. Other impurities such as calcium and iron were deposited in a more selective way, only on zirconia-supported catalysts. The presence of calcium influences the mobility of metal centers and, therefore, can act twofold—limiting the sintering of metal nanoparticles or acting as a poison and reducing the redispersion of metal during regeneration [80].
The influence of the impurities present in the bio-oil obtained from wheat straw on the Ni-Cu/Al2O3 and Ru/C catalysts’ activity was also investigated. A relatively high content of sulfur present in the feedstock had a negative effect on both tested catalysts and even led to the formation of bulk Ni3S2 in the Ni-based catalysts. This probably caused the sintering of nanoparticles. Additionally, the environment of bio-oil related to the presence of water changed the phase of alumina to boehmite. Besides sulfur species, nitrogen-based impurities were also identified on the surface of the catalysts, which could also contribute to surface poisoning [85].
5.2. Zeolites Used as Catalysts
The main inorganic impurities that are accumulated in zeolites are presented in Figure 7. The poisoning of zeolites that typically have acid sites can mainly occur due to the presence alkali and alkaline earth metals in the feedstock [86].
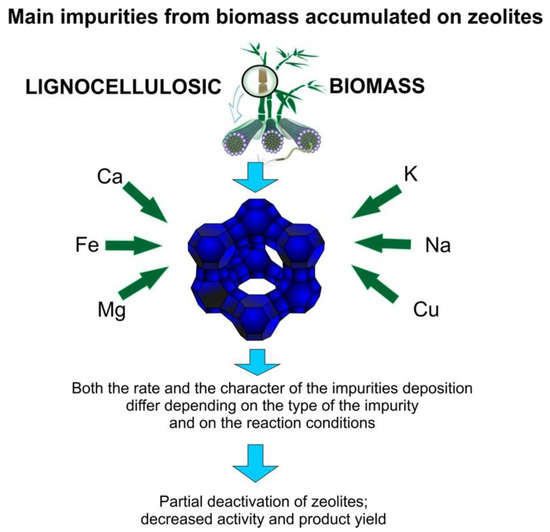
Figure 7.
Main inorganic impurities that are accumulated in zeolites.
The influence of inorganic impurities that have origin from biomass on the activity of ZSM-5 in fast pyrolysis of pine wood sawdust was investigated by the group of Huber [87]. It was found that inorganic impurities such as calcium, potassium, magnesium, and manganese from the biomass were identified on zeolite catalyst after the reaction. The concentration of acid sites on the zeolite was, however, not affected. The situation was different in a long-term study of fast pyrolysis of pine sawdust with HZSM-5 catalyst. The linear deposition of alkali metals present in the feedstock was observed. Despite the catalyst retaining its activity in the studied period of 4 days, poisoning of the acid sites by impurities was noted, which, in consequence, significantly reduces the catalyst’s lifetime [88].
On the other hand, Mullen and Boateng [89] in their work concerning the catalytic pyrolysis of switchgrass in a fluidized bed with HZSM-5 studied the mechanism of the impurities’ deposition. It was identified that Ca, Mg, and K were the most abundant in the switchgrass and were found in relatively high concentration within the range 884–1654 mg kg−1. Besides that, the other inorganic impurities such as Fe, P, Cu Na, and Mg were identified in a slightly lower concentration as well. Despite most of the impurities accumulating in the biochar, several were selectively trapped by the catalyst. Interestingly, only a small number of impurities was found in the bio-oil. Potassium was deposited very rapidly on the zeolite—more than Ca that was the most abundant—however, the rate of the deposition decreased with the biomass loading. In contrast, for calcium, the rate of deposition did not decrease with the biomass loading but Ca deposition was slower. Mg, Cu, and Fe deposited almost linearly on the zeolite catalyst. The Fe concentration on the zeolite increased rapidly in comparison to the availability of this element in the feedstock. Fe trapped by the catalysts accounted for 42% of the amount of Fe in the switchgrass. Respectively, its lowest contribution was found in the biochar. This shows that Fe was trapped by the zeolite in the most selective way in comparison to other elements. Consequently, together with the deposition of inorganic impurities on the HZSM-5, the decrease in the deoxygenation of the products of pyrolysis oil as well as the decrease in selectivity for aromatic hydrocarbons were observed.
The stability of commercial ZSM-5 zeolite and its deactivation by inorganic impurities was also tested by the group of Lappas [20] in pyrolysis conducted in a bubbling fluidized bed. Similar to previous work, it was observed that the accumulation rate of impurities on the zeolite depends on the metal type. Ca, Na, and K showed a linear accumulation rate on the catalyst. Moreover, K and Na deposited very selectively on the surface of zeolite but not as much as K. In agreement with the previous work, it was identified that potassium was deposited in a significant amount and that deposition was fast, while the deposition of Ca and Mg was noted in the lower rate and not so selectively as K. Exposure of a hydrothermally deactivated catalyst to inorganic impurities allowed an organic fraction with similar oxygen content to be obtained, but at reduced yields.
In contrast to zeolite materials, alkaline earth oxides could be a possible alternative. Magnesium oxide was found to constitute an efficient alternative to zeolitic catalysts which, as described above, can be easily deactivated in the presence of alkali metals. Contrary to zeolite materials, no deposition of alkali metals could be identified on the surface of basic oxides [47]. It is, however, known that even the presence of a small amount of water can easily deactivate alkaline earth metal oxides, and depending on the reaction conditions, transform them into colloids (basic environment) or, due to leaching, transform to ions [90].
6. Summary
The deactivation of catalysts during lignocellulosic biomass pyrolysis is a serious problem that should be overcome in order to increase the competitiveness of this process in comparison to current methods of the production of fuels. Coke formation, sintering, hydrothermal instability, and poisoning are considered the main reasons for a decrease in the activity of the catalysts of thermal conversion of biomass. Deposited carbon species can be removed during catalyst regeneration. Sintering and poisoning of the catalysts are usually irreversible, although there are reports in the literature showing exceptions (such as reversible chlorine contamination). Numerous strategies allowing the limiting of catalyst coking, sintering, or the influence of impurities are reported in the literature. Among them, there are the incorporation of dopants, support modification, or the change of metal–support interaction. Moreover, structure change, as in the case of mesoporous materials or zeolites, can also be proposed. Those modifications result in the change of metal electronic structure, acid-base properties of the catalyst, increased oxygen mobility, or the development of porosity, among others. The most typical approaches are summarized in Figure 8.
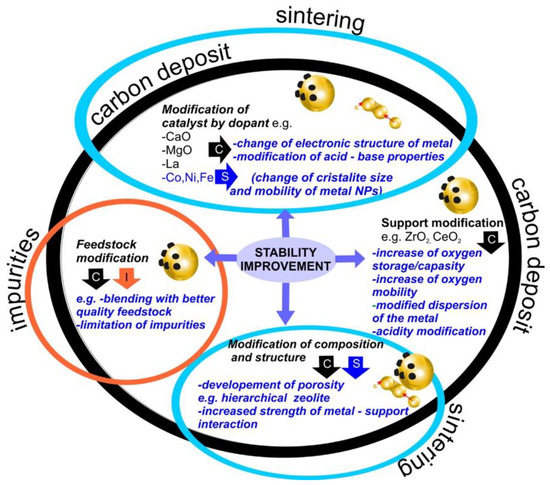
Figure 8.
The most typical strategies allowing the limiting of catalyst coking, sintering, or the influence of impurities.
Generally, it is more favorable to protect the catalysts against deactivation than regenerate them. Therefore, a thorough understanding of the factors affecting catalyst stability in biomass pyrolysis is essential. There are two possible ways to increase the efficiency of the thermal conversion of lignocellulosic feedstock. The first of them is related to the prevention of catalyst deactivation by the design of a new synthesis protocol of the catalysts (including new methods of catalyst preparation, optimization of its composition (choice of metal, support, introduction of dopants), and pretreatment conditions, or regeneration procedure). On the other hand, efforts should be directed towards optimization of the reaction conditions (composition of the reaction mixture, reaction temperature, mass and energy transfer, design of reactors). A reduction in the adsorption rate of impurities on the catalyst surface seems to be difficult; however, it can be achieved by pretreatment or initial purification of the used feedstock.
Author Contributions
Conceptualization, J.G. and A.M.R.; writing—original draft preparation, J.G. and A.M.R.; writing—review and editing, J.G. and A.M.R.; visualization, A.M.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bridgwater, A.V. Review of fast pyrolysis of biomass and product upgrading. Biomass Bioenergy 2012, 38, 68–94. [Google Scholar] [CrossRef]
- Rahman, M.; Liu, R.; Cai, J. Catalytic fast pyrolysis of biomass over zeolites for high quality bio-oil—A review. Fuel Process. Technol. 2018, 180, 32–46. [Google Scholar] [CrossRef]
- Mullen, C.A.; Boateng, A.A. Production of aromatic hydrocarbons via catalytic pyrolysis of biomass over fe-modified HZSM-5 zeolites. ACS Sustain. Chem. Eng. 2015, 3, 1623–1631. [Google Scholar] [CrossRef]
- Yildiz, G.; Ronsse, F.; Van Duren, R.; Prins, W. Challenges in the design and operation of processes for catalytic fast pyrolysis of woody biomass. Renew. Sustain. Energy Rev. 2016, 57, 1596–1610. [Google Scholar] [CrossRef]
- Navarro, R.M.; Peña, A.M.A.; Fierro, J.L.G. Hydrogen production reactions from carbon feedstocks: Fossil fuels and biomass. Chem. Rev. 2007, 107, 3952–3991. [Google Scholar] [CrossRef] [PubMed]
- Collard, F.-X.; Blin, J. A review on pyrolysis of biomass constituents: Mechanisms and composition of the products obtained from the conversion of cellulose, hemicelluloses and lignin. Renew. Sustain. Energy Rev. 2014, 38, 594–608. [Google Scholar] [CrossRef]
- Wang, K.; Kim, K.H.; Brown, R.C. Catalytic pyrolysis of individual components of lignocellulosic biomass. Green Chem. 2014, 16, 727–735. [Google Scholar] [CrossRef]
- Kabir, G.; Hameed, B. Recent progress on catalytic pyrolysis of lignocellulosic biomass to high-grade bio-oil and bio-chemicals. Renew. Sustain. Energy Rev. 2017, 70, 945–967. [Google Scholar] [CrossRef]
- Tanksale, A.; Beltramini, J.; Lu, G.Q.M. A review of catalytic hydrogen production processes from biomass. Renew. Sustain. Energy Rev. 2010, 14, 166–182. [Google Scholar] [CrossRef]
- Fan, Y.; Cai, Y.; Li, X.; Yin, H.; Xia, J. Coking characteristics and deactivation mechanism of the HZSM-5 zeolite employed in the upgrading of biomass-derived vapors. J. Ind. Eng. Chem. 2017, 46, 139–149. [Google Scholar] [CrossRef]
- Grams, J.; Ruppert, A.M. Development of heterogeneous catalysts for thermo-chemical conversion of lignocellulosic biomass. Energies 2017, 10, 545. [Google Scholar] [CrossRef]
- Bulushev, D.A.; Ross, J.R. Catalysis for conversion of biomass to fuels via pyrolysis and gasification: A review. Catal. Today 2011, 171, 1–13. [Google Scholar] [CrossRef]
- Liu, C.; Wang, H.; Karim, A.M.; Sun, J.; Wang, Y. Catalytic fast pyrolysis of lignocellulosic biomass. Chem. Soc. Rev. 2014, 43, 7594–7623. [Google Scholar] [CrossRef] [PubMed]
- Persson, H.; Duman, I.; Wang, S.; Pettersson, L.J.; Yang, W. Catalytic pyrolysis over transition met-al-modified zeolites: A comparative study between catalyst activity and deactivation. J. Anal. Appl. Pyrol. 2019, 138, 54–61. [Google Scholar] [CrossRef]
- Zhang, C.; Hu, X.; Guo, H.; Wei, T.; Dong, D.; Hu, G.; Hu, S.; Xiang, J.; Liu, Q.; Wang, Y. Pyrolysis of poplar, cellulose and lignin: Effects of acidity and alkalinity of the metal oxide catalysts. J. Anal. Appl. Pyrolysis 2018, 134, 590–605. [Google Scholar] [CrossRef]
- Ryczkowski, R.; Chałupka, K.; Kwapiński, W.; Przybysz, K.; Fridrichová, D.; Grams, J. Modification of Ni/ZrO2 catalyst by selected rare earth metals as a promising way for increase in the efficiency of thermocatalytic conversion of lignocellulosic biomass to hydrogen-rich gas. Fuel 2020, 276, 118110. [Google Scholar] [CrossRef]
- Ochoa, A.; Bilbao, J.; Gayubo, A.G.; Castaño, P. Coke formation and deactivation during catalytic reforming of biomass and waste pyrolysis products: A review. Renew. Sustain. Energy Rev. 2020, 119, 109600. [Google Scholar] [CrossRef]
- Liu, N.; Xie, H.; Cao, H.; Shi, L.; Meng, X. Multi-technique characterization of recycled acetylene carbonylation catalyst CuY: Deactivation and coke analysis. Fuel 2019, 242, 617–623. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, L.; Yang, Z.; He, Z. An experiment study of biomass steam gasification over NiO/Dolomite for hydrogen-rich gas production. Int. J. Hydrog. Energy 2017, 42, 76–85. [Google Scholar] [CrossRef]
- Stefanidis, S.D.; Kalogiannis, K.G.; Pilavachi, P.A.; Fougret, C.M.; Jordan, E.; Lappas, A.A. Catalyst hydrother-mal deactivation and metal contamination during the in situ catalytic pyrolysis of biomass. Catal. Sci. Technol. 2016, 6, 2807–2819. [Google Scholar] [CrossRef]
- Bartholomew, C.H. Mechanisms of catalyst deactivation. Appl. Catal. A Gen. 2001, 212, 17–60. [Google Scholar] [CrossRef]
- Liang, W.; Yan, H.; Chen, C.; Lin, D.; Tan, K.; Feng, X.; Liu, Y.; Chen, X.; Yang, C.; Shan, H. Revealing the ef-fect of nickel particle size on carbon formation type in the methane decomposition reaction. Catalysts 2020, 10, 890. [Google Scholar] [CrossRef]
- Veses, A.; Puértolas, B.; Callén, M.; García, T. Catalytic upgrading of biomass derived pyrolysis vapors over metal-loaded ZSM-5 zeolites: Effect of different metal cations on the bio-oil final properties. Microporous Mesoporous Mater. 2015, 209, 189–196. [Google Scholar] [CrossRef]
- Xu, M.; Mukarakate, C.; Iisa, K.; Budhi, S.; Menart, M.; Davidson, M.; Robichaud, D.J.; Nimlos, M.R.; Trewyn, B.G.; Richards, R.M. Deactivation of multilayered MFI nanosheet zeolite during upgrading of biomass pyrolysis vapors. ACS Sustain. Chem. Eng. 2017, 5, 5477–5484. [Google Scholar] [CrossRef]
- Yu, L.; Farinmade, A.; Ajumobi, O.; Su, Y.; John, V.T.; Valla, J.A. MCM-41/ZSM-5 composite particles for the catalytic fast pyrolysis of biomass. Appl. Catal. A Gen. 2020, 602, 117727. [Google Scholar] [CrossRef]
- Zheng, Y.; Tao, L.; Huang, Y.; Liu, C.; Wang, Z.; Zheng, Z. Improving aromatic hydrocarbon content from catalytic pyrolysis upgrading of biomass on a CaO/HZSM-5 dual-catalyst. J. Anal. Appl. Pyrolysis 2019, 140, 355–366. [Google Scholar] [CrossRef]
- Karnjanakom, S.; Suriya-Umporn, T.; Bayu, A.; Kongparakul, S.; Samart, C.; Fushimi, C.; Abudula, A.; Guan, G. High selectivity and stability of Mg-doped Al-MCM-41 for in-situ catalytic upgrading fast pyrolysis bio-oil. Energy Convers. Manag. 2017, 142, 272–285. [Google Scholar] [CrossRef]
- Mullen, C.A.; Dorado, C.; Boateng, A.A. Catalytic co-pyrolysis of switchgrass and polyethylene over HZSM-5: Catalyst deactivation and coke formation. J. Anal. Appl. Pyrolysis 2018, 129, 195–203. [Google Scholar] [CrossRef]
- Arregi, A.; Lopez, G.; Amutio, M.; Artetxe, M.; Barbarias, I.; Bilbao, J.; Olazar, M. Role of operating conditions in the catalyst deactivation in the in-line steam reforming of volatiles from biomass fast pyrolysis. Fuel 2018, 216, 233–244. [Google Scholar] [CrossRef]
- Arregi, A.; Amutio, M.; Lopez, G.; Artetxe, M.; Alvarez, J.; Bilbao, J.; Olazar, M. Hydrogen-rich gas production by continuous pyrolysis and in-line catalytic reforming of pine wood waste and HDPE mixtures. Energy Convers. Manag. 2017, 136, 192–201. [Google Scholar] [CrossRef]
- Santamaria, L.; Lopez, G.; Arregi, A.; Amutio, M.; Artetxe, M.; Bilbao, J.; Olazar, M. Stability of different Ni supported catalysts in the in-line steam reforming of biomass fast pyrolysis volatiles. Appl. Catal. B Environ. 2019, 242, 109–120. [Google Scholar] [CrossRef]
- Grams, J.; Niewiadomski, M.; Ruppert, A.M.; Kwapiński, W. Influence of Ni catalyst support on the product distribution of cellulose fast pyrolysis vapors upgrading. J. Anal. Appl. Pyrolysis 2015, 113, 557–563. [Google Scholar] [CrossRef]
- Matras, J.; Niewiadomski, M.; Ruppert, A.M.; Grams, J. Activity of Ni catalysts for hydrogen production via biomass pyrolysis. Kinet. Catal. 2012, 53, 565–569. [Google Scholar] [CrossRef]
- Santamaria, L.; Arregi, A.; Alvarez, J.; Artetxe, M.; Amutio, M.; Lopez, G.; Bilbao, J.; Olazar, M. Performance of a Ni/ZrO2 catalyst in the steam reforming of the volatiles derived from biomass pyrolysis. J. Anal. Appl. Pyrolysis 2018, 136, 222–231. [Google Scholar] [CrossRef]
- Grams, J.; Ryczkowski, R.; Chalupka, K.A.; Sobczak, I.; Rzeznicka, I.I.; Przybysz, K. Impact of support (MCF, ZrO2, ZSM-5) on the efficiency of ni catalyst in high-temperature conversion of lignocellulosic biomass to hydrogen-rich gas. Materials 2019, 12, 3792. [Google Scholar] [CrossRef]
- Dong, L.; Wu, C.; Ling, H.; Shi, J.; Williams, P.T.; Huang, J. Promoting hydrogen production and minimizing catalyst deactivation from the pyrolysis-catalytic steam reforming of biomass on nanosized NiZnAlOx catalysts. Fuel 2017, 188, 610–620. [Google Scholar] [CrossRef]
- Bimbela, F.; Ábrego, J.; Puerta, R.; García, L.; Arauzo, J. Catalytic steam reforming of the aqueous fraction of bio-oil using Ni-Ce/Mg-Al catalysts. Appl. Catal. B Environ. 2017, 209, 346–357. [Google Scholar] [CrossRef]
- Grams, J.; Niewiadomski, M.; Ryczkowski, R.; Ruppert, A.M.; Kwapinski, W. Activity and characterization of Ni catalyst supported on CeO2–ZrO2 for thermo-chemical conversion of cellulose. Int. J. Hydrog. Energy 2016, 41, 8679–8687. [Google Scholar] [CrossRef]
- Santamaria, L.; Artetxe, M.; Lopez, G.; Cortazar, M.; Amutio, M.; Bilbao, J.; Olazar, M. Effect of CeO2 and MgO promoters on the performance of a Ni/Al2O3 catalyst in the steam reforming of biomass pyrolysis volatiles. Fuel Process. Technol. 2020, 198, 106223. [Google Scholar] [CrossRef]
- Bizkarra, K.; Bermudez, J.; Arcelus-Arrillaga, P.; Barrio, V.; Cambra, J.; Millan, M. Nickel based monometallic and bimetallic catalysts for synthetic and real bio-oil steam reforming. Int. J. Hydrog. Energy 2018, 43, 11706–11718. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, X.; Zhang, L.; Wang, Y.; Li, X.; Zhang, S.; Liu, Q.; Wei, T.; Gao, G.; Hu, X. Steam reforming of guaiacol over Ni/SiO2 catalyst modified with basic oxides: Impacts of alkalinity on properties of coke. Energy Convers. Manag. 2020, 205, 112301. [Google Scholar] [CrossRef]
- Santamaria, L.; Arregi, A.; Lopez, G.; Artetxe, M.; Amutio, M.; Bilbao, J.; Olazar, M. Effect of La2O3 promotion on a Ni/Al2O3 catalyst for H2 production in the in-line biomass pyrolysis-reforming. Fuel 2020, 262, 116593. [Google Scholar] [CrossRef]
- Valle, B.; Aramburu, B.; Olazar, M.; Bilbao, J.; Gayubo, A.G. Steam reforming of raw bio-oil over Ni/La2O3-αAl2O3: Influence of temperature on product yields and catalyst deactivation. Fuel 2018, 216, 463–474. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, H.; Liu, J.; Chen, D. Catalytic characteristics of innovative Ni/slag catalysts for syngas production and tar removal from biomass pyrolysis. Int. J. Hydrog. Energy 2019, 44, 11848–11860. [Google Scholar] [CrossRef]
- Yu, H.; Liu, Y.; Liu, J.; Chen, D. High catalytic performance of an innovative Ni/magnesium slag catalyst for the syngas production and tar removal from biomass pyrolysis. Fuel 2019, 254, 115622. [Google Scholar] [CrossRef]
- Jahromi, H.; Agblevor, F.A. Hydrodeoxygenation of pinyon-juniper catalytic pyrolysis oil using red mud-supported nickel catalysts. Appl. Catal. B Environ. 2018, 236, 1–12. [Google Scholar] [CrossRef]
- Al-Rahbi, A.S.; Williams, P.T. Waste ashes as catalysts for the pyrolysis–catalytic steam reforming of biomass for hydrogen-rich gas production. J. Mater. Cycles Waste Manag. 2019, 21, 1224–1231. [Google Scholar] [CrossRef]
- Ryczkowski, R.; Goscianska, J.; Panek, R.; Franus, W.; Przybysz, K.; Grams, J. Sustainable nickel catalyst for the conversion of lignocellulosic biomass to H2-rich gas. Int. J. Hydrog. Energy 2021, in press. [Google Scholar] [CrossRef]
- Heracleous, E.; Pachatouridou, E.; Hernández-Giménez, A.; Hernando, H.; Fakin, T.; Paioni, A.; Baldus, M.; Serrano, D.; Bruijnincx, P.; Weckhuysen, B.; et al. Characterization of deactivated and regenerated zeolite ZSM-5-based catalyst extrudates used in catalytic pyrolysis of biomass. J. Catal. 2019, 380, 108–122. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, L.; Li, Q.; Wang, Y.; Liu, Q.; Wei, T.; Dong, D.; Salavati, S.; Gholizadeh, M.; Hu, X. Catalytic pyrolysis of poplar wood over transition metal oxides: Correlation of catalytic behaviors with physiochemical properties of the oxides. Biomass- Bioenergy 2019, 124, 125–141. [Google Scholar] [CrossRef]
- Yi, L.; Liu, H.; Li, M.; Man, G.; Yao, H. Prevention of CaO deactivation using organic calcium precursor during multicyclic catalytic upgrading of bio-oil. Fuel 2020, 271, 117692. [Google Scholar] [CrossRef]
- Kalogiannis, K.G.; Stefanidis, S.D.; Karakoulia, S.A.; Triantafyllidis, K.S.; Yiannoulakis, H.; Michailof, C.; Lappas, A.A. First pilot scale study of basic vs. acidic catalysts in biomass pyrolysis: Deoxygenation mecha-nisms and catalyst deactivation. Appl. Catal. B Environ. 2018, 238, 346–357. [Google Scholar] [CrossRef]
- Li, M.; Hu, Y.; Fang, Y.; Tan, T. Coating mesoporous ZSM-5 by thin microporous Silicalite-1 shell: Formation of core/shell structure, improved hydrothermal stability and outstanding catalytic performance. Catal. Today 2020, 339, 312–320. [Google Scholar] [CrossRef]
- Paluch, P.; Potrzebowska, N.; Ruppert, A.M.; Potrzebowski, M.J. Application of 1H and 27Al magic angle spinning solid state NMR at 60 kHz for studies of Au and Au-Ni catalysts supported on boehmite/alumina. Solid State Nucl. Magn. Reson. 2017, 84, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xu, Y.; Gao, C.; Zhao, Y. Structural and textural evolution of Ni/γ-Al2O3 catalyst under hydrothermal conditions. Catal. Today 2010, 158, 475–480. [Google Scholar] [CrossRef]
- Sehested, J.; Gelten, J.A.P.; Helveg, S. Sintering of nickel catalysts: Effects of time, atmosphere, temperature, nickel–carrier interactions, and dopants. Appl. Catal. A Gen. 2006, 309, 237–246. [Google Scholar] [CrossRef]
- Liu, X. DRIFTS Study of Surface of γ-Alumina and Its Dehydroxylation. J. Phys. Chem. C 2008, 112, 5066–5073. [Google Scholar] [CrossRef]
- Ochoa, A.; Arregi, A.; Amutio, M.; Gayubo, A.G.; Olazar, M.; Bilbao, J.; Castaño, P. Coking and sintering progress of a Ni supported catalyst in the steam reforming of biomass pyrolysis volatiles. Appl. Catal. B Environ. 2018, 233, 289–300. [Google Scholar] [CrossRef]
- Arregi, A.; Lopez, G.; Amutio, M.; Barbarias, I.; Santamaria, L.; Bilbao, J.; Olazar, M. Regenerability of a Ni catalyst in the catalytic steam reforming of biomass pyrolysis volatiles. J. Ind. Eng. Chem. 2018, 68, 69–78. [Google Scholar] [CrossRef]
- Santamaria, L.; Lopez, G.; Arregi, A.; Amutio, M.; Artetxe, M.; Bilbao, J.; Olazar, M. Effect of calcination conditions on the performance of Ni/MgO–Al2O3 catalysts in the steam reforming of biomass fast pyrolysis volatiles. Catal. Sci. Technol. 2019, 9, 3947–3963. [Google Scholar] [CrossRef]
- Arandia, A.; Remiro, A.; Valle, B.; Bilbao, J.; Gayubo, A.G. Deactivation of Ni spinel derived catalyst during the oxidative steam reforming of raw bio-oil. Fuel 2020, 276, 117995. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, C.; Chen, S.; Liu, Y. Co–Ni bimetal catalyst supported on perovskite-type oxide for steam reforming of ethanol to produce hydrogen. Int. J. Hydrog. Energy 2014, 39, 5644–5652. [Google Scholar] [CrossRef]
- Remiro, A.; Ochoa, A.; Arandia, A.; Castaño, P.; Bilbao, J.; Gayubo, A.G. On the dynamics and reversibility of the deactivation of a Rh/CeO2ZrO2 catalyst in raw bio-oil steam reforming. Int. J. Hydrog. Energy 2019, 44, 2620–2632. [Google Scholar] [CrossRef]
- Dai, L.; Zeng, Z.; Yang, Q.; Yang, S.; Wang, Y.; Liu, Y.; Ruan, R.; He, C.; Yu, Z.; Jiang, L. Synthesis of iron nanoparticles-based hydrochar catalyst for ex-situ catalytic microwave-assisted pyrolysis of lignocellulosic biomass to renewable phenols. Fuel 2020, 279, 118532. [Google Scholar] [CrossRef]
- Ma, L.; Xiao, T.; Ning, Z.; Liu, Y.; Chen, H.; Peng, J. Pollution and health risk assessment of toxic metal(loid)s in soils under different land use in sulphide mineralized areas. Sci. Total. Environ. 2020, 724, 138176. [Google Scholar] [CrossRef]
- Velghe, I.; Carleer, R.; Yperman, J.; Schreurs, S. Study of the pyrolysis of municipal solid waste for the production of valuable products. J. Anal. Appl. Pyrol. 2011, 92, 366–375. [Google Scholar] [CrossRef]
- Wise, J.; Vietor, D.; Provin, T.; Capareda, S.C.; Munster, C.; Boateng, A. Mineral nutrient recovery from pyrolysis systems. Environ. Prog. Sustain. Energy 2012, 31, 251–255. [Google Scholar] [CrossRef]
- Mullen, C.A.; Boateng, A.A.; Goldberg, N.M.; Lima, I.M.; Laird, D.A.; Hicks, K.B. Bio-oil and bio-char pro-duction from corn cobs and stover by fast pyrolysis. Biomass Bioenerg. 2010, 34, 67–74. [Google Scholar] [CrossRef]
- Olsson, J.G.; Jäglid, U.; Pettersson, J.B.C.; Hald, P. Alkali metal emission during pyrolysis of biomass. Energy Fuels 1997, 11, 779–784. [Google Scholar] [CrossRef]
- Jensen, P.A.; Frandsen, F.J.; Dam-Johansen, A.K.; Sander, B. Experimental investigation of the transformation and release to gas phase of potassium and chlorine during straw pyrolysis. Energy Fuels 2000, 14, 1280–1285. [Google Scholar] [CrossRef]
- Davidsson, K.O.; Stojkova, B.J.; Pettersson, J.B.C. Alkali emission from birchwood particles during rapid py-rolysis. Energy Fuels 2002, 16, 1033–1039. [Google Scholar] [CrossRef]
- Kowalski, T.; Ludwig, C.; Wokaun, A. Qualitative Evaluation of Alkali Release during the Pyrolysis of Bio-mass. Energy Fuels 2007, 21, 3017–3022. [Google Scholar] [CrossRef]
- Tu, Y.; Tian, S.; Kong, L.; Xiong, Y. Co-catalytic effect of sewage sludge-derived char as the support of Fen-ton-like catalyst. Chem. Eng. J. 2012, 185–186, 44–51. [Google Scholar] [CrossRef]
- Lup, A.N.K.; Abnisa, F.; Daud, W.M.A.W.; Aroua, M.K. A review on reactivity and stability of heterogeneous metal catalysts for deoxygenation of bio-oil model compounds. J. Ind. Eng. Chem. 2017, 56, 1–34. [Google Scholar] [CrossRef]
- Gao, N.; Kamran, K.; Quan, C.; Wiliams, P.T. Thermochemical conversion of sewage sludge: A critical re-view. Prog. Energy Combust. Sci. 2020, 79, 100843. [Google Scholar] [CrossRef]
- Yang, T.; Huang, H.; Lai, F.-Y. Pollution hazards of heavy metals in sewage sludge from four wastewater treatment plants in Nanchang, China. Bioresour. Technol. 2016, 200, 991–998. [Google Scholar] [CrossRef]
- Mansur, D.; Yoshikawa, T.; Norinaga, K.; Hayashi, J.-I.; Tago, T.; Masuda, T. Production of ketones from py-roligneous acid of woody biomass pyrolysis over an iron-oxide catalyst. Fuel 2013, 103, 130–134. [Google Scholar] [CrossRef]
- Mortensen, P.M.; Gardini, D.; De Carvalho, H.W.P.; Damsgaard, C.D.; Grunwaldt, J.-D.; Jensen, P.A.; Wagner, J.B.; Jensen, A.D. Stability and resistance of nickel catalysts for hydrodeoxygenation: Carbon deposition and effects of sulfur, potassium, and chlorine in the feed. Catal. Sci. Technol. 2014, 4, 3672–3686. [Google Scholar] [CrossRef]
- Rostrup-Nielsen, J.R. Steam reforming. In Handbook of Heterogeneous Catalysis; John Wiley & Sons, Inc.: New York, NY, USA, 2008; pp. 2882–2905. [Google Scholar]
- Argyle, M.D.; Bartholomew, C.H. Heterogeneous catalyst deactivation and regeneration: A review. Catalysts 2015, 5, 145–269. [Google Scholar] [CrossRef]
- Song, Q.; Wang, F.; Xu, J. Hydrogenolysis of lignosulfonate into phenols over heterogeneous nickel catalysts. Chem. Commun. 2012, 48, 7019–7021. [Google Scholar] [CrossRef]
- Soszka, E.; Reijneveld, H.M.; Jędrzejczyk, M.; Rzeznicka, I.I.; Grams, J.; Ruppert, A.M. Chlorine influence on palladium doped nickel catalysts in levulinic acid hydrogenation with formic acid as hydrogen source. ACS Sustain. Chem. Eng. 2018, 6, 14607–14613. [Google Scholar] [CrossRef]
- Mortensen, P.M.; Gardini, D.; Damsgaard, C.D.; Grunwaldt, J.-D.; Jensen, P.A.; Wagner, J.B.; Jensen, A.D. Deactivation of Ni-MoS2 by bio-oil impurities during hydrodeoxygenation of phenol and octanol. Appl. Catal. A Gen. 2016, 523, 159–170. [Google Scholar] [CrossRef]
- Schmitt, C.C.; Reolon, M.B.G.; Zimmermann, M.; Raffelt, K.; Grunwaldt, J.-D.; Dahmen, N. Synthesis and regeneration of nickel-based catalysts for hydrodeoxygenation of beech wood fast pyrolysis bio-oil. Catalysts 2018, 8, 449. [Google Scholar] [CrossRef]
- Boscagli, C.; Morgano, M.T.; Raffelt, K.; Leibold, H.; Grunwaldt, J.-D. Influence of feedstock, catalyst, pyrol-ysis and hydrotreatment temperature on the composition of upgraded oils from intermediate pyrolysis. Biomass Bioenergy 2018, 116, 236–248. [Google Scholar] [CrossRef]
- Gastelu, N.; Lopez-Urionabarrenechea, A.; Acha, E.; Caballero, B.M.; de Marco, I. Evaluation of HZSM-5 zeolite as cracking catalyst for upgrading the vapours generated in the pyrolysis of an epoxy-carbon fibre waste composite. Top. Catal. 2019, 62, 479–490. [Google Scholar] [CrossRef]
- Carlson, T.R.; Cheng, Y.-T.; Jae, J.; Huber, G.W. Production of green aromatics and olefins by catalytic fast pyrolysis of wood sawdust. Energy Environ. Sci. 2010, 4, 145–161. [Google Scholar] [CrossRef]
- Paasikallio, V.; Lindfors, C.; Kuoppala, E.; Solantausta, Y.; Oasmaa, A.; Lehto, J.; Lehtonen, J. Product quality and catalyst deactivation in a four day catalytic fast pyrolysis production run. Green Chem. 2014, 16, 3549–3559. [Google Scholar] [CrossRef]
- Mullen, C.A.; Boateng, A.A. Accumulation of inorganic impurities on HZSM-5 zeolites during catalytic fast pyrolysis of switchgrass. Ind. Eng. Chem. Res. 2013, 52, 17156–17161. [Google Scholar] [CrossRef]
- Ruppert, A.M.; Meeldijk, J.D.; Kuipers, B.W.M.; Erné, B.H.; Weckhuysen, B.M. Glycerol etherification over highly active CaO-based materials: New mechanistic aspects and related colloidal particle formation. Chem. Eur. J. 2008, 14, 2016–2024. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).