2.1. The Basics
The first stage of building up a thermodynamic model is to consider the simplest system possible. This is a system with a single redox cofactor that engages in reduction or oxidation by a single electron.
This simple system could be used to describe an iron-sulfur cluster in a ferredoxin or the H-cluster in [FeFe] hydrogenase maturated with a [2Fe] precursor containing PDT. The latter has been argued to have a pH dependent redox potential [
36]. However, provided that we do not change the pH of the redox titration, this simple one-electron redox model is sufficient. This simple redox couple is described by the well-known Nernst equation [
39]. Assuming that the activity coefficients of the species
Ox and
Red approach 1 (a valid assumption when dealing with low concentrations as is the case for experiments with enzymes), the activity can be approximated with concentrations.
where
E is the applied potential in V vs. the Standard Hydrogen Electrode (SHE),
EO is the standard redox potential of the
Ox/
Red couple in V vs. SHE, R is the molar gas constant (8.314 J K
−1 mol
−1), T is the temperature in K, n is the number of electrons transferred (in this case n = 1), F is the Faraday constant (96,485 C mol
−1), and [
Red] and [
Ox] are the concentrations of the reduced and oxidised species in M units, respectively.
To find how the species
Ox and
Red vary with the potential they experience (this can either be from an electrode, or from a chemical redox couple, e.g., 2H
+/H
2 in the case of hydrogenase), we need to rearrange the Nernst equation and define the total concentration of both
Ox and
Red. Throughout this review, we will define the sum of the concentration of the involved species as 1 (when dealing with real concentrations of the enzyme, the fraction in a given state will need to be scaled to the total concentration). Therefore:
Rearranging the Nernst Equation (1), we get:
Substituting [
Red] with (2), we get:
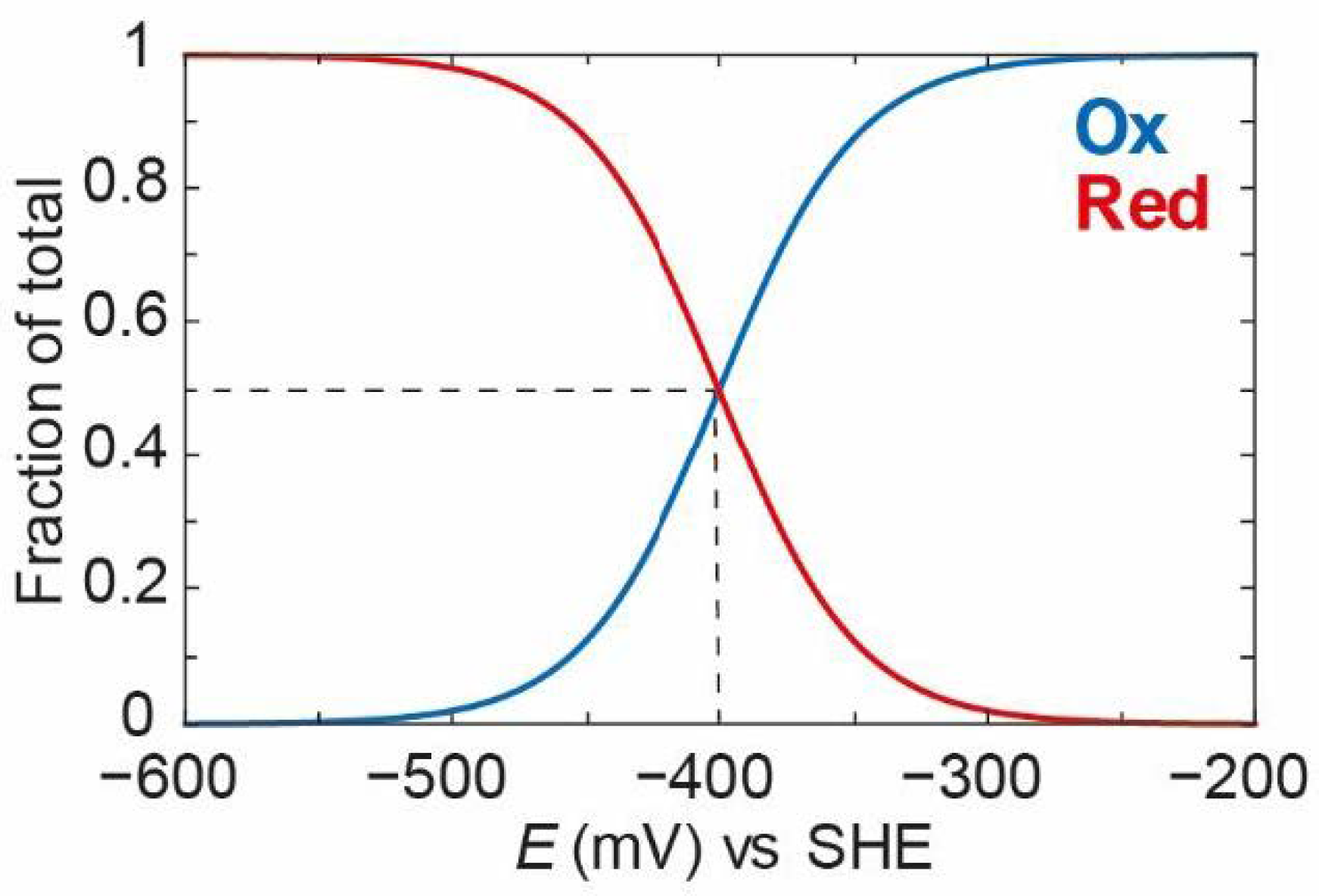
Plotting [
Ox] (Equation (3)) and [
Red] (Equation (2)) against the applied potential using an
EO value of −400 mV vs. SHE (chosen as an arbitrary potential close to the thermodynamic potential of the 2H
+/H
2 couple at physiological pH) and n = 1 (indicating a single electron transferred), it then gives the curves shown in
Figure 2. It can clearly be seen that, at a high applied potential (−200 mV)
Ox dominates, and, as the applied potential decreases,
Ox is converted gradually to
Red with a middle point (the midpoint potential) of −400 mV.
Next, instead of assuming a redox event, we will assume a deprotonation event of a single cofactor with a single proton.
This situation is explained nicely by a simple acid dissociation constant (
Ka):
where
Ka is the acid dissociation constant, [
Ox] is the concentration of the deprotonated species, [
H+] is the concentration of protons, and [
OxH] is the concentration of the protonated species in M units.
Rearranging Equation (4), we get:
Then [
OxH] is substituted with Equation (5):
Finally, this can be rearranged to express [
Ox] in terms of the difference between the
pKa and the pH:
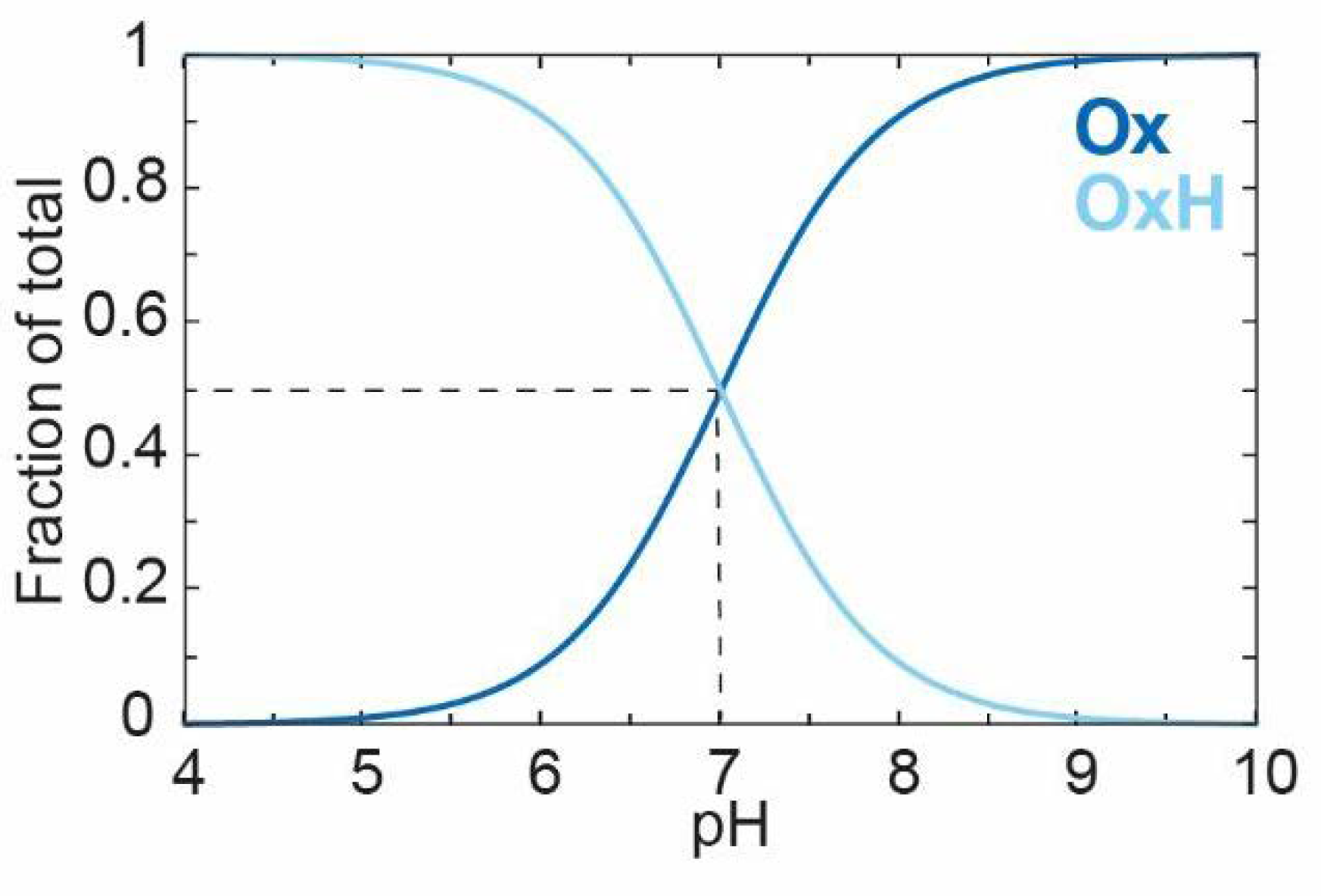
Plotting [
Ox] (Equation (6)) and [
OxH] (Equation (5)) against the pH using a
Ka value of 10
−7 (or a
pKa of 7) gives the curves shown in
Figure 3. At a high pH, the deprotonated species
Ox dominates, and, as the pH decreases,
Ox converts gradually to
OxH with a
pKa of 7.
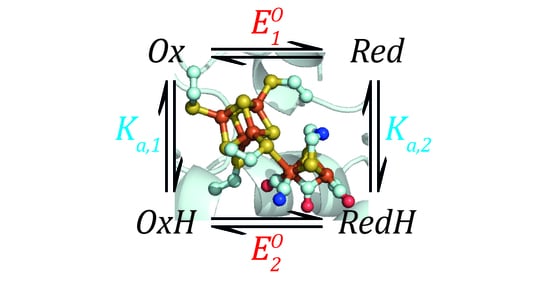
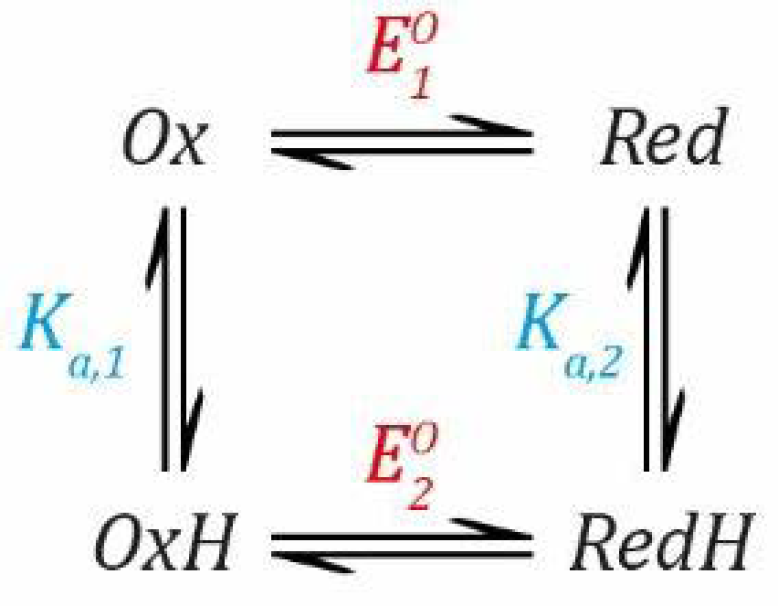
2.2. Proton Coupled Electron Transfer
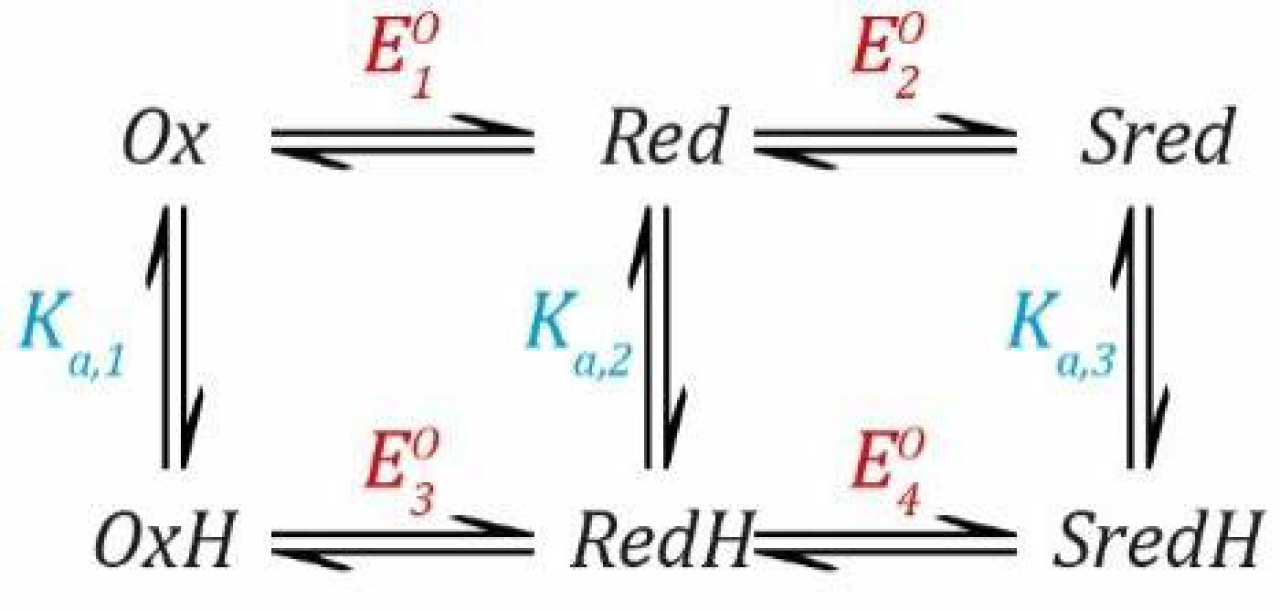
Now, we can combine both events into a thermodynamic model describing a species that can be protonated and reduced by one electron simultaneously, as shown in
Scheme 1.
Now, we need to define two
EO values based on the Nernst Equation (1), including one for the deprotonated states (
Ox and
Red) and one for the protonated states (
OxH and
RedH), and two
Ka values based on the simple acid dissociation constant Equation (4), one for the oxidised states (
Ox and
OxH) and one for the reduced states (
Red and
RedH). Here, as before,
E refers to the applied potential to the system and, therefore, all redox couples are in equilibrium with this.
These equations can be rearranged to get everything in terms of [
Ox] as follows.
where:
where:
Again, we define the sum of the concentration of all states as 1.
Next, we substitute [
OxH] and [
RedH] from Equations (11) and (12) into Equation (13).
followed by substitution of [
Red] from Equation (7) into Equation (14).
As for Equation (6) from
Section 2.1, this can be rearranged to express [
H+],
Ka,1, and
Ka,2 as pH,
pKa,1, and
pKa,2.
Importantly, in order to arrive at this result, we only need to consider the two protonation events defined by
Ka,1 and
Ka,2 and the reduction event defined by
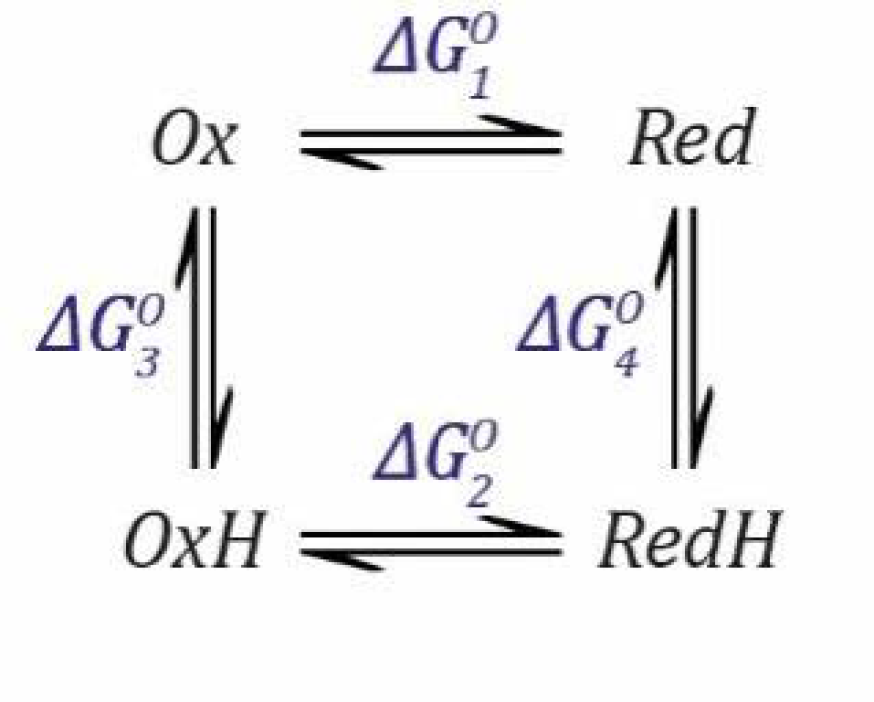
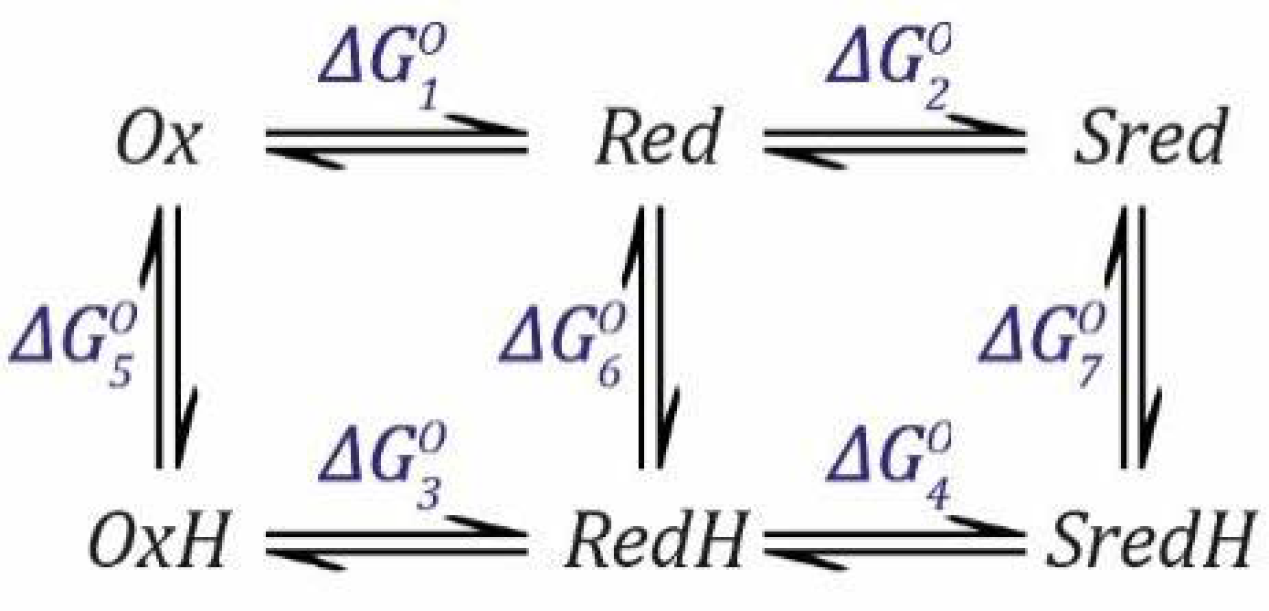
. We can calculate
by considering that the free energy change (ΔG) when going from
Ox to
RedH is the same, whether we go via
Red or via
OxH. We can define the situation (
Scheme 2) as follows (remember that
Ka,1 and
Ka,2 are dissociation constants, so the free energy change Δ
G is defined for this direction and must be subtracted when we go in the opposite direction).
The total free energy change when going from
Ox to
RedH is the same whether we go via
Red or
OxH (
and
are subtracted because they are defined for the deprotonation process).
Since:
we can define the free energy changes for the reduction and deprotonation steps as follows.
which can then be substituted into Equation (16) and rearranged for
.
Using this simple model, we can plot the dependence of each of the species
Ox,
Red,
OxH, and
RedH as a function of either the applied potential at a given pH (
Figure 4A) or as a function of the pH at a given applied potential (
Figure 4B).
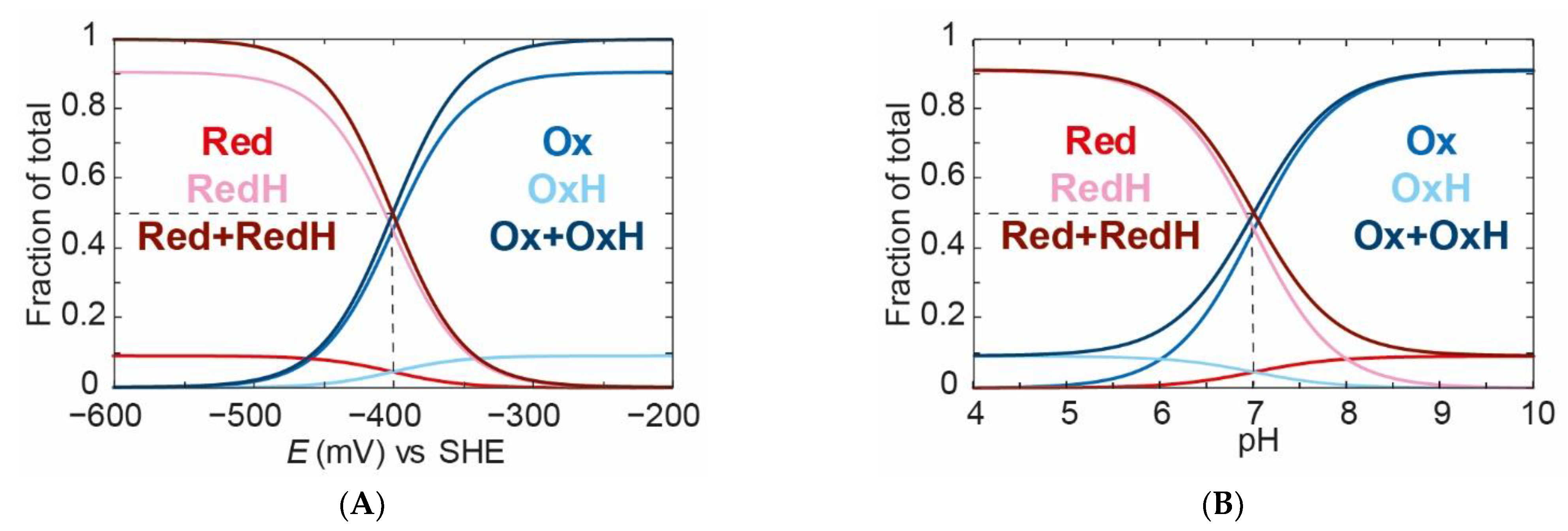
We can see that, at a fixed pH value of 7 (
Figure 4A), when we titrate the applied potential from high (less negative) to low (more negative), we go from about 90%
Ox and 10%
OxH to about 10%
Red and 90%
RedH. A crucial observation needs to be made at this stage: all species titrate with an identical “apparent” midpoint potential of −400 mV. This is because the redox and protonation events are coupled. Importantly, the
Ox:
OxH ratio and the
Red:
RedH ratio do not change with the applied potential. This may seem clear, but a lack of understanding of this concept could lead to erroneous conclusions.
Similarly, if we titrate the pH from high to low at a constant applied potential of −400 mV (
Figure 4B), we can see that we go from a mixture of 90%
Ox and 10%
Red to a mixture of 10%
OxH and 90%
RedH with an “apparent”
pKa of 7. This is an important observation in which we have shown that the redox state is dependent on the pH. This indicates the presence of a proton coupled electron transfer (PCET). The reason for this is that we have chosen different
pKa values for the oxidised and reduced species. If we had identical
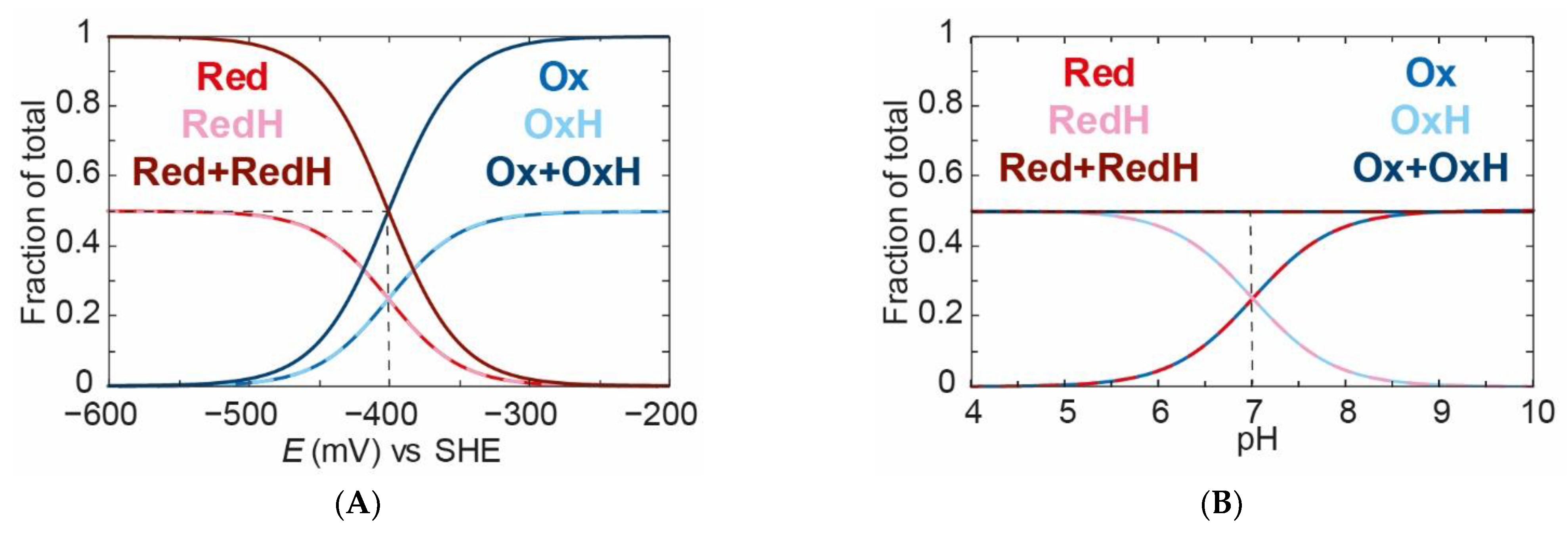
pKa values for the oxidised and reduced species, then no PCET would occur. This situation is shown in
Figure 5A,B, where the
pKa values are both 7. Since the applied potential is titrated from high to low, a 50% mixture of
Ox and
OxH converts to a 50% mixture of
Red and
RedH with an “apparent” midpoint potential of −400 mV vs. SHE. In
Figure 5B, the total oxidised species (
Ox +
OxH) to the total reduced species (
Red +
RedH) ratio does not change with pH, indicating no PCET. However, it can be seen that
Ox titrates to
OxH and
Red titrated to
RedH as the pH is changed from high to low.
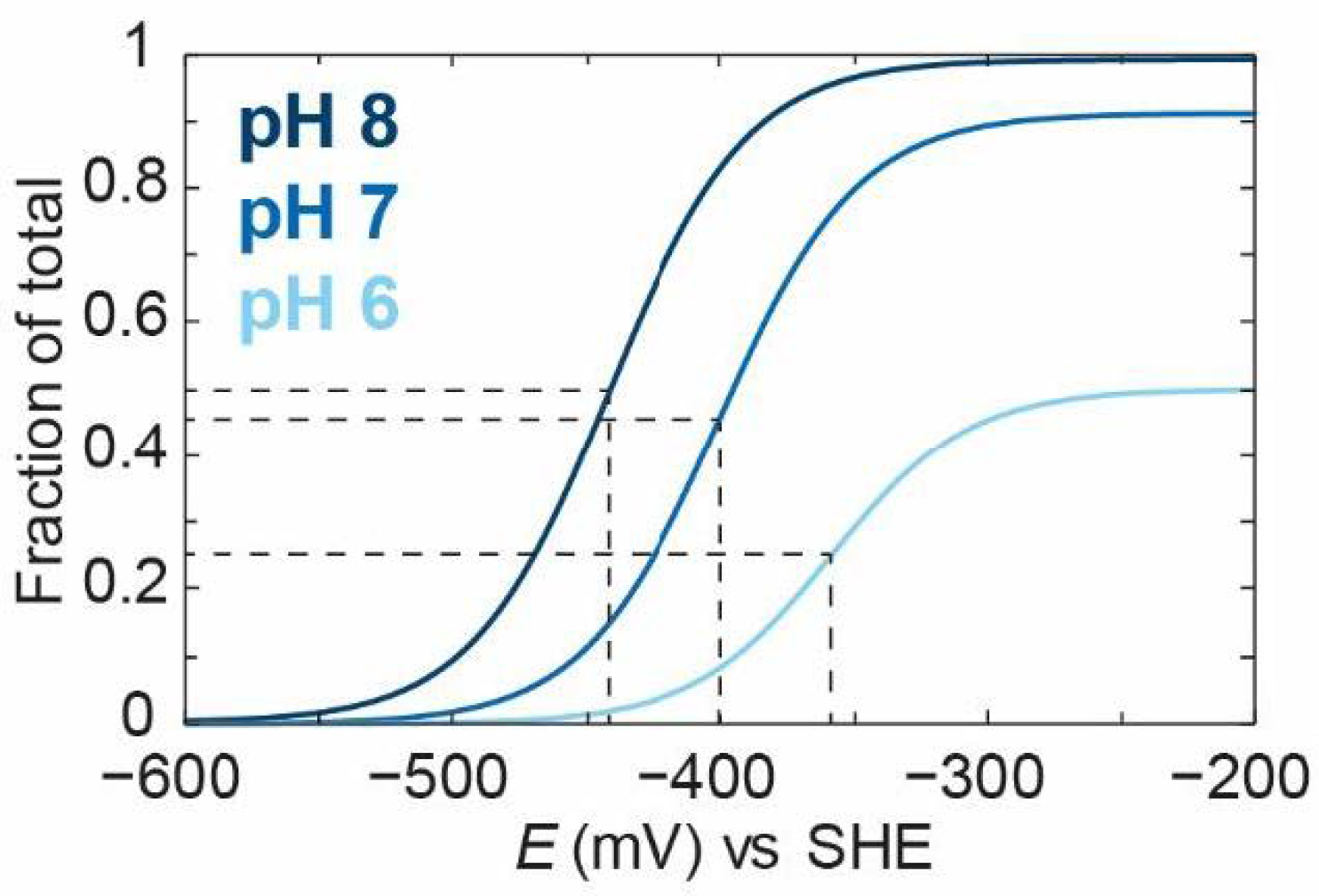
To emphasise the crucial observation that “apparent” redox potentials can shift with pH for the case where we have different
pKa values for
Ox and Red, one can compare the titrations of the
Ox species at three pH values (
Figure 6).
As can be seen from
Figure 6, the “apparent” midpoint potential for the titration of
Ox shifts with pH, even if the change of
Ox to
Red itself does not directly involve a proton. However, the fundamental point is that the ratio between the
Ox and
OxH species will be the same at every value of the applied potential (as long as the pH is fixed). This is because the ratio between
Ox and
OxH is determined only by the acid dissociation constant
Ka,1. Thus, if the process of proton-coupled reduction of
Ox to
RedH can occur, then the titration of the total amount of the oxidised species (
Ox +
OxH) and the titration of the total amount of the reduced species (
Red +
RedH), will change with pH. It must, therefore, be the case that the titration of the component states changes with pH. To illustrate this,
Figure 7 shows the titration of Ox,
OxH, and
Ox +
OxH at pH 6 and pH 8. It is clear that, at pH 6, the
Ox state titrates with a more positive “apparent” redox potential than at pH 8. The components
Ox and
OxH species titrate with the same potential at pH 6. At pH 8, only the
Ox state can be observed since its
pKa is too low to observe
OxH at this pH.
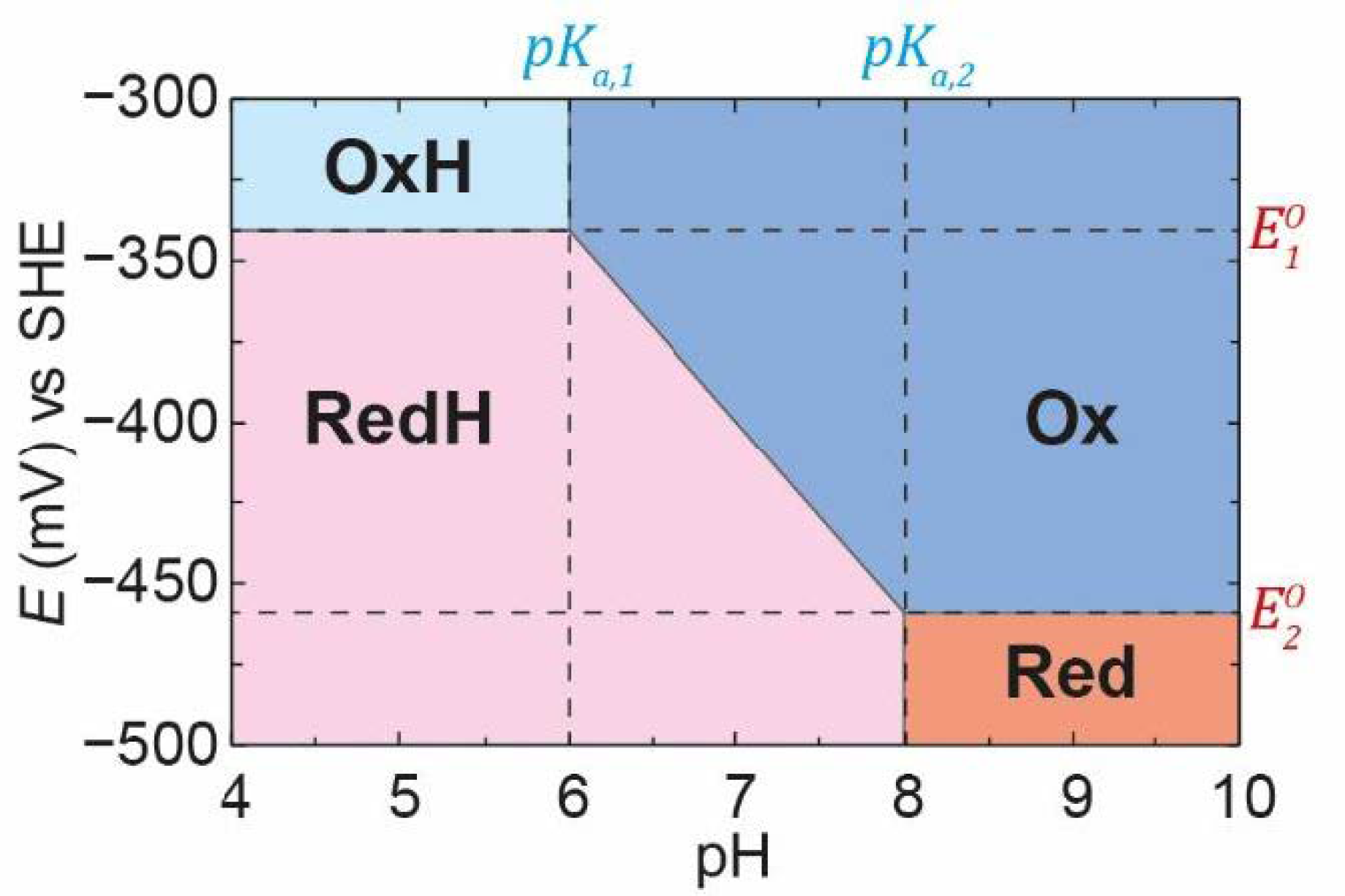
Using the
,
,
pKa,1, and
pKa,2 values defined in
Figure 4, for a PCET process, we can plot the famous Pourbaix diagram [
40] in which the regions of stability of each of the species
Ox,
OxH,
Red, and
RedH are defined (
Figure 8). It should be recognised, however, that the boundaries between each of these regions do not represent step functions but represent the smooth titrations between each state. For example, it can be seen in
Figure 4 that, at pH 7 and
E = −400 mV (a point right in the middle of the Pourbaix diagram in
Figure 8), we have about 45%
Ox, 45%
RedH, 5%
OxH, and 5%
Red. Thus, we still observe
OxH and
Red even though we are outside of the regions where these states are defined as stable in the Pourbaix diagram.
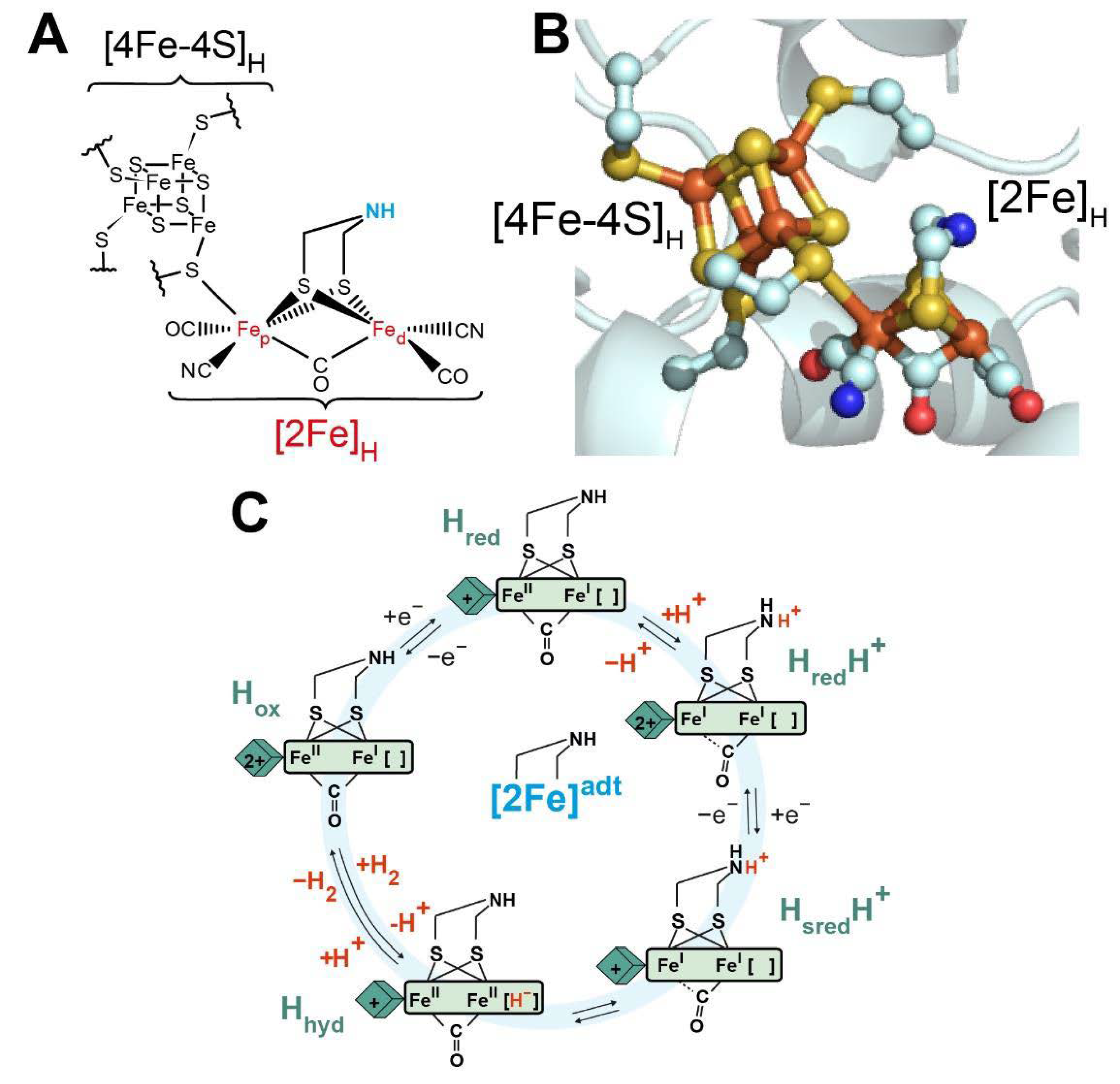
2.3. A Simple Model for the [FeFe] Hydrogenase
Experimental data show that the simplest [FeFe] hydrogenase,
CrHydA1, containing only the H-cluster and no additional F-clusters, can be reduced by two electrons. The most oxidised state
Hox gets reduced by one electron to form
Hred, and
Hred can subsequently be reduced by a second electron to yield
Hsred [
41,
42].
It was shown that photoexcitation of frozen samples of the
Hred state gave a different form of
Hred, which was named
Hred’ [
43]. Around the same time, it was noted that
Hred’ dominated at high pH, while
Hred dominated at low pH [
35]. Furthermore, it was observed that only the
Hred’ state formed in the PDT variant of
CrHydA1 (which lacks a protonatable dithiolate ligand in [2Fe]
H) [
38]. The simplest interpretation of the data was that the electron could reside mostly on the [4Fe-4S]
H subcluster, or mostly on the [2Fe]
H subcluster, and that protonation of the ADT bridging nitrogen from NH to NH
2+ was the event that triggered the electronic rearrangement. Thus, the
Hred’ and
Hred states were renamed
Hred and
HredH+, respectively [
35]. Moreover, based on the fact that
Hsred cannot be formed in the PDT variant of
CrHydA1 [
38], it was assumed that
Hsred should be in a protonated state, and was named
HsredH+. To describe this situation, a more complex model is required including two redox steps and one protonation step.
In
Section 2.2, we described a simple model for proton coupled electron transfer in which four states exist:
Ox,
OxH,
Red, and
RedH. Now, we will extend this model to include a second redox event (
Scheme 3).
This creates a total of six states connected by seven steps. In this model,
Ox is the most oxidised state,
Red is the one electron reduced state, and
Sred is the two-electron reduced state or “super-reduced” state. Each of these unprotonated states has a protonated counterpart:
OxH,
RedH, and
SredH. Importantly, nothing about this model indicates the location of redox or (de)protonation events because we have a system that can accept two electrons and one proton. One could also consider a model involving one redox and two (de)protonation events, or even two redox and two (de)protonation events. However, the former does not fit at all with experimentally observed spectro-electrochemical titrations, as two redox steps are clearly discerned, while the latter leads to a reactive state that can release H
2 [
35,
42]. To make a 2e
−/2H
+ model reversible, an H
2 binding/dissociation step is required, which would complicate the model. Nevertheless, the simple 2e
−/1H
+ model has been shown to accurately describe pH-dependent spectro-electrochemical titrations [
35]. The equations describing this model can be derived using the Nernst Equation (1) and the equation for a simple acid dissociation constant (4) by analogy to those in
Section 2.2.
Although we did not need to use all the parameters to arrive at the results (
Ka,2 and
Ka,3 were not used), we can use the conjoined thermodynamic cycles in this model to derive further equations for these parameters. We can define the situation (
Scheme 4) as follows.
The rules still apply so that the free energy change is the same regardless which pathway is taken. Thus, going from
Ox to
RedH via
Red or via
OxH have the same overall free energy change, as does going from
Red to
SredH via
Sred or via
RedH. Therefore, we can define the following equations.
where:
These equations can then be combined to find expressions for
Ka,2 and
Ka,3 (see
SI Section S1 for complete derivations). The final results are shown here:
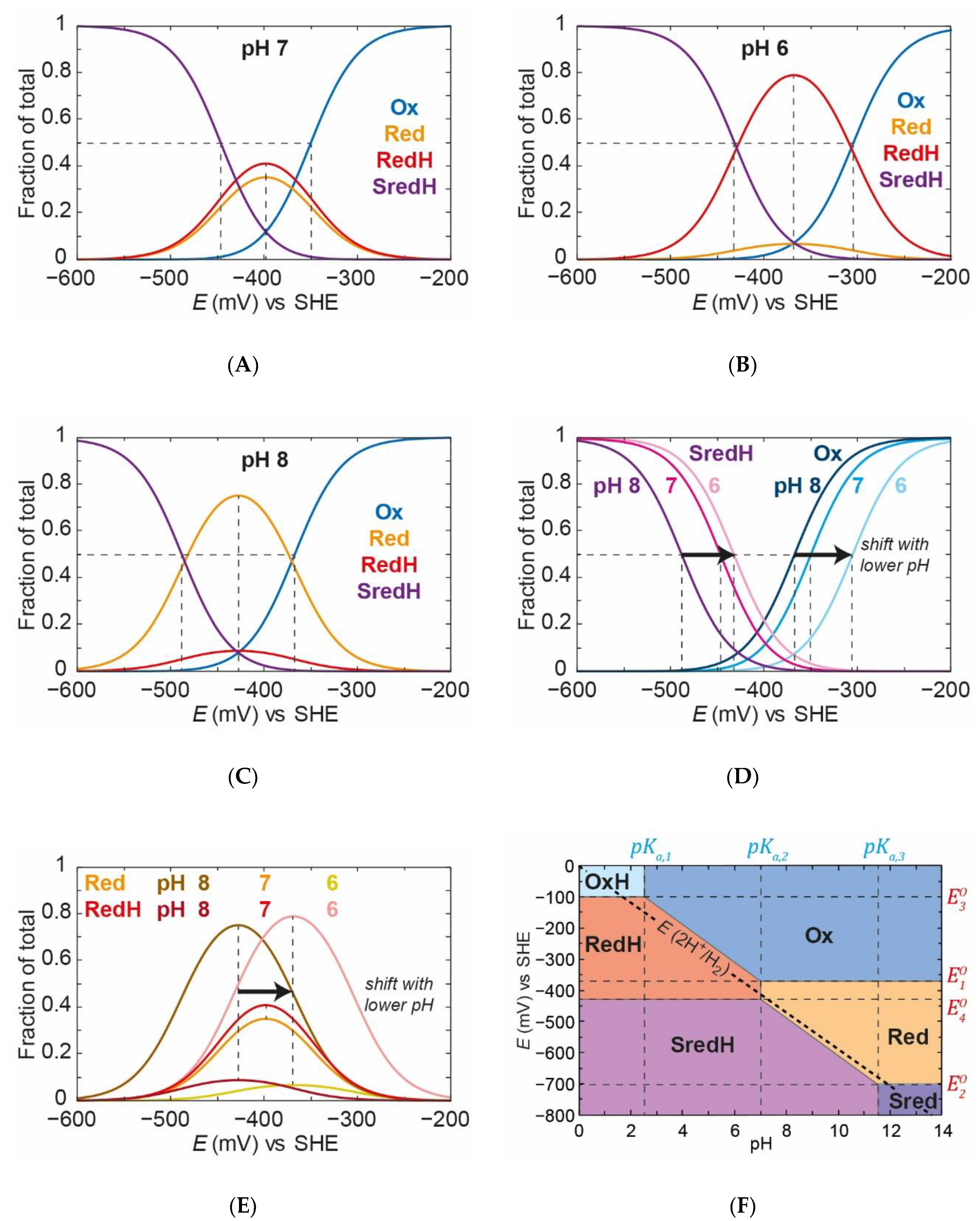
Using this model, the populations of all the states can be varied as a function of applied potential at various pH values (
Figure 9). As can be seen from
Figure 9A, for pH 7, at a high applied potential, only the
Ox state exists. The
Ox/
OxH transition has such a low
pKa value (2.5) that, at pH 7, the
OxH state is essentially completely deprotonated and only
Ox exists. As the applied potential is decreased, the
Ox state decreases in population (with an apparent midpoint potential of −350 mV) and the
Red and
RedH states come up with roughly equal proportions, reaching a maximum concentration at around −400 mV vs. SHE. In this case, the
pKa for the
Red/
RedH transition is ≈7.1, which is why the amount of
RedH is slightly higher than that of
Red. As the applied potential decreases even further,
Red and
RedH are both converted to
SredH (with an apparent midpoint potential of −450 mV). The
pKa for the
Sred/
SredH transition is ≈11.6, which means that the
Sred state is essentially completely protonated to
SredH at pH 7.
At pH 6 (
Figure 9B) and 8 (
Figure 9C), similar behaviour can be observed. At pH 6, the
Ox state is converted to
Red and
RedH with a higher apparent midpoint potential (−300 mV) than at pH 7 (−350 mV). The
Red state is mostly protonated to
RedH, and the
Red and
RedH states are reduced to
SredH with a higher apparent midpoint potential (−430 mV) than at pH 7 (−450 mV). At pH 8, the
Ox state is converted to
Red and
RedH with a lower apparent midpoint potential (−370 mV) than at pH 7 (−350 mV). The
RedH state is mostly deprotonated to Red, and the
Red and
RedH states are reduced to
SredH with a lower apparent midpoint potential (−480 mV) than at pH 7 (−450 mV).
In
Figure 9D, the behaviours of
Ox and
SredH are compared at the three pH values. There is a clear observation that the titrations of both species are affected by pH. This is intuitive because the reduction of
Ox can occur with or without protonation to give
RedH and
Red, respectively, with the former requiring a proton. Likewise, reduction of
Red to
SredH requires protonation, while reduction of
RedH to
SredH does not. In
Figure 9E, the behaviours of
Red and
RedH are compared at the three pH values. Again, both species appear to be affected by pH. The appearance of
RedH and the disappearance of
Red should be pH dependent. What is possibly less intuitive is that the appearance of
Red and the disappearance of
RedH also appear to be pH dependent. This is caused by the fact that
Red and
RedH are in an equilibrium set by the
pKa,2 value and the pH. Thus, whatever happens to
Red must also apply to
RedH and vice versa. This means that a simple analysis of how the maximum populations of
Red and
RedH shift with pH (see Supplementary Discussion in Reference [
36]) could erroneously lead to the conclusion that there are two separate proton coupled electron transfer events, including one to make
Red and one to make
RedH. This can be seen even more clearly in
Figure 9F, where we have plotted a Pourbaix diagram for this model. Between the limits of the
pKa,1 (2.5) and
pKa,2 (7.1), the midpoint potential of
Ox and
OxH converting to
Red and
RedH is pH dependent. Likewise, between the limits of
pKa,2 and
pKa,3, the midpoint potential of
Red and
RedH converting to
Sred and
SredH is also dependent on pH.
In
Figure 9, the
pKa values and intrinsic redox potentials have been set to ensure a situation where the
OxH state is always deprotonated to
Ox, while the
Sred state is always protonated to
SredH and the intermediate
Red(H) state is a mixture of the protonated (
RedH) and deprotonated (
Red) forms (around physiological pH). This situation appears to most accurately reflect the behaviour of the [FeFe] hydrogenase from
CrHydA1 [
35]. In
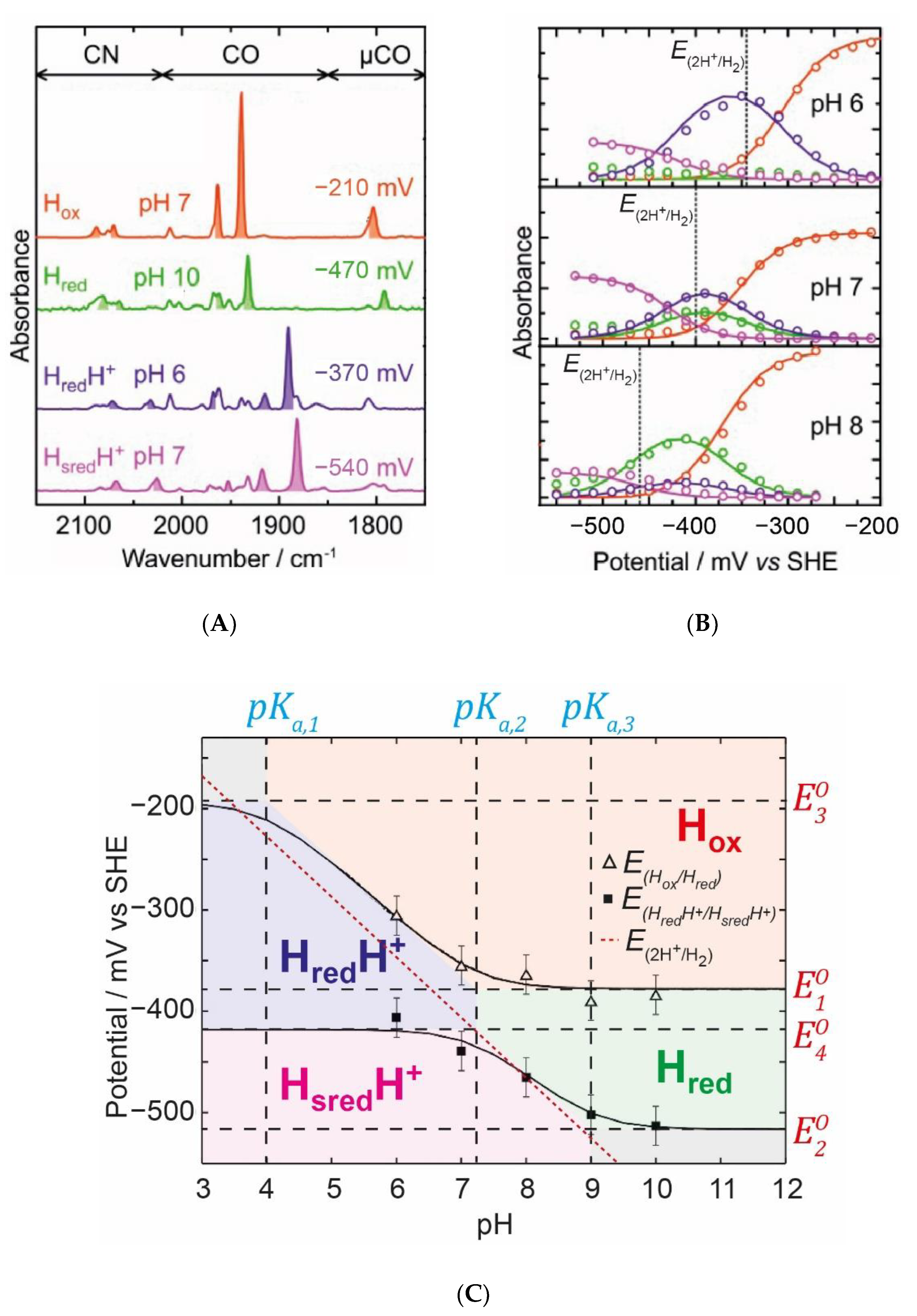
Figure 10, we exemplify how this thermodynamic model was employed by Sommer et al. to fit the IR spectro-electrochemical data obtained for the
CrHydA1 hydrogenase at various pH values [
35].
Figure 10A shows typical IR spectra under specific conditions that highlight the main catalytic states:
Hox,
Hred,
HredH+, and
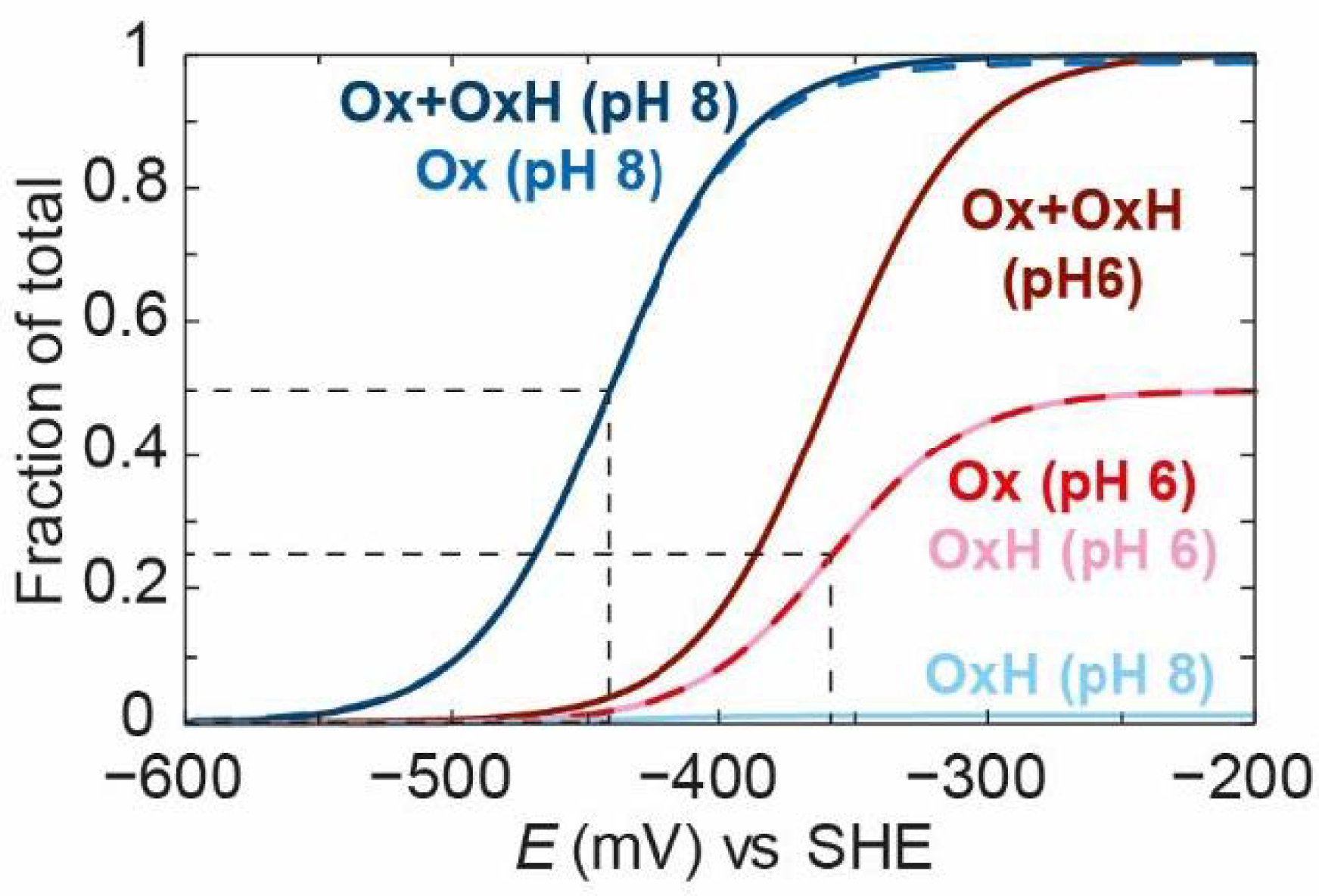
HsredH+.
Figure 10B shows electrochemical titrations of the most dominant IR band in each of these states at pH 6, 7, and 8. It can clearly be seen that, as the pH increases, the titration curves shift to a more negative potential. The
HredH+ state becomes less abundant and the
Hred state becomes more abundant. This behaviour seems very logical and fits the requirements of a reversible enzyme. Around neutral pH and the thermodynamic 2H
+/H
2 potential, the enzyme is in a roughly equal mixture of both
Red (
Hred) and
RedH (
HredH+) states, maximising the concentrations of both and, thereby, maximising the rates of oxidation of
Red to
Ox and reduction of
RedH to
SredH. If, instead, we imagine a situation where all redox states are deprotonated and only
Ox,
Red, and
Sred exist, then one expects H
2 oxidation to be efficient, but H
+ reduction will be inefficient. Likewise, if all states are protonated, H
+ reduction is efficient but H
2 oxidation is not.
Effectively, the enzyme is optimised so that the potentials of the
Ox(H)/
Red(H) and
Red(H)/
Sred(H) are as close as possible to the thermodynamic 2H
+/H
2 potential. This can be seen again in the Pourbaix diagram (
Figure 9F for the model and
Figure 10C for the model applied to
CrHydA1). The thermodynamic 2H
+/H
2 potential is shown as a dotted line. It is clear that, at a neutral pH, both transitions (
Ox(H)/
Red(H) and
Red(H)/
Sred(H)) are close to the thermodynamic 2H
+/H
2 potential. In
Figure 10C, the midpoint potential of the
Hox/
Hred and
Hred/
Hsred transitions are plotted against the pH and are fitted with curves based on the model described above, with areas coloured to indicate where the various states dominate (in analogy to
Figure 9F). Ideally, both curves would fit perfectly to the 2H
+/H
2 potential at all pH values. However, using this model, it is not possible to generate such curves. One expects, therefore, at extreme pH values, to start to observe deviations of the catalytic efficiency in one direction. At a low pH, we expect a slight overpotential from H
+ reduction, while, at a high pH, we expect a slight overpotential for H
2 oxidation. This seems to fit well with the known behaviour of the [FeFe] hydrogenase from
CrHydA1 in IR spectro-electro-chemical titrations [
33,
35,
44,
45], but, interestingly, not with those from more complex enzymes including accessory iron-sulfur clusters [
32,
34]. In order to understand the behaviour in these more complex enzymes, we first need to deal with the concept of redox anti-cooperativity.
2.4. Redox Anticooperativity Model
In
Section 2.2, we dealt with what happens when we can oxidise/reduce and protonate/deprotonate simultaneously, but we could also have two simultaneous oxidation/reduction events. Such a situation could be used to describe two iron-sulfur clusters located close to one another. These two redox events may be independent (i.e., their redox potentials do not influence one another) or they may be dependent (i.e., their redox potentials influence one another) [
46]. This influence can be positive or negative. If reduction of one cluster makes it easier to reduce the second cluster, this would give us redox cooperativity. If reduction of one cluster makes it more difficult to reduce the second cluster, this would give us redox anti-cooperativity.
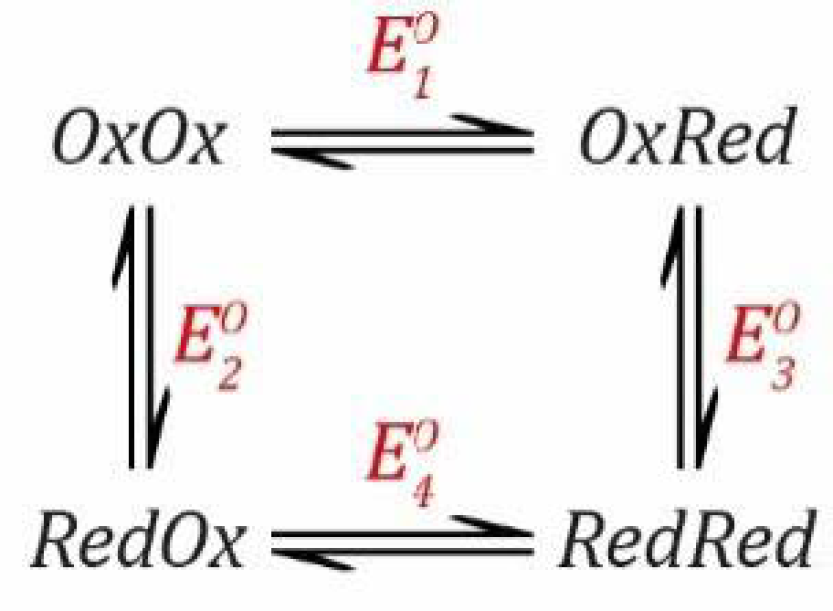
Scheme 5 shows a simple model.
This situation is somewhat similar to the PCET scenario described in
Section 2.2 except that it involves two electrons rather than one electron and one proton. All steps are redox steps and defined by the Nernst equation.
These equations can be rearranged to get everything in terms of [
OxOx] (see
SI Section S2 for a complete derivation).
where:
As before, in order to arrive at this result, we only need to consider the three reduction events defined by
,
and
.
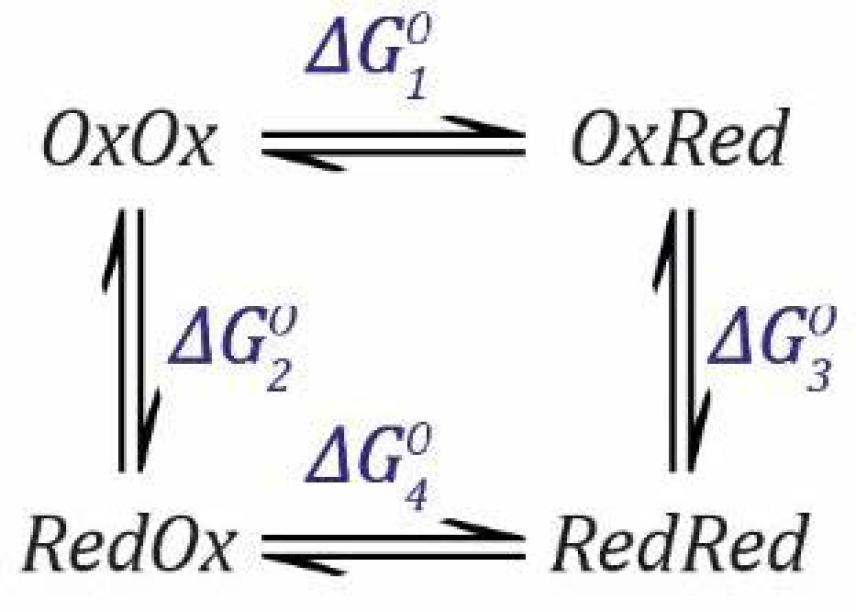
can be calculated considering the thermodynamic square (
Scheme 6) as follows.
The rules still apply so that the free energy change is the same regardless which pathway is taken. Thus, going from
OxOx to
RedRed via
OxRed or via
RedOx have the same overall free energy change. Therefore, we can define the following equation.
where:
These equations can then be substituted into Equation (33) and rearranged to give
.
Now, we can set up two different situations, one in which we have two independent redox events (i.e.,
=
and
=
) and one in which the redox events are dependent (i.e.,
≠
and
≠
). In
Figure 11, the independent situation is shown where
=
= −390 mV and
=
= −410 mV (the average
EO is −400 mV).
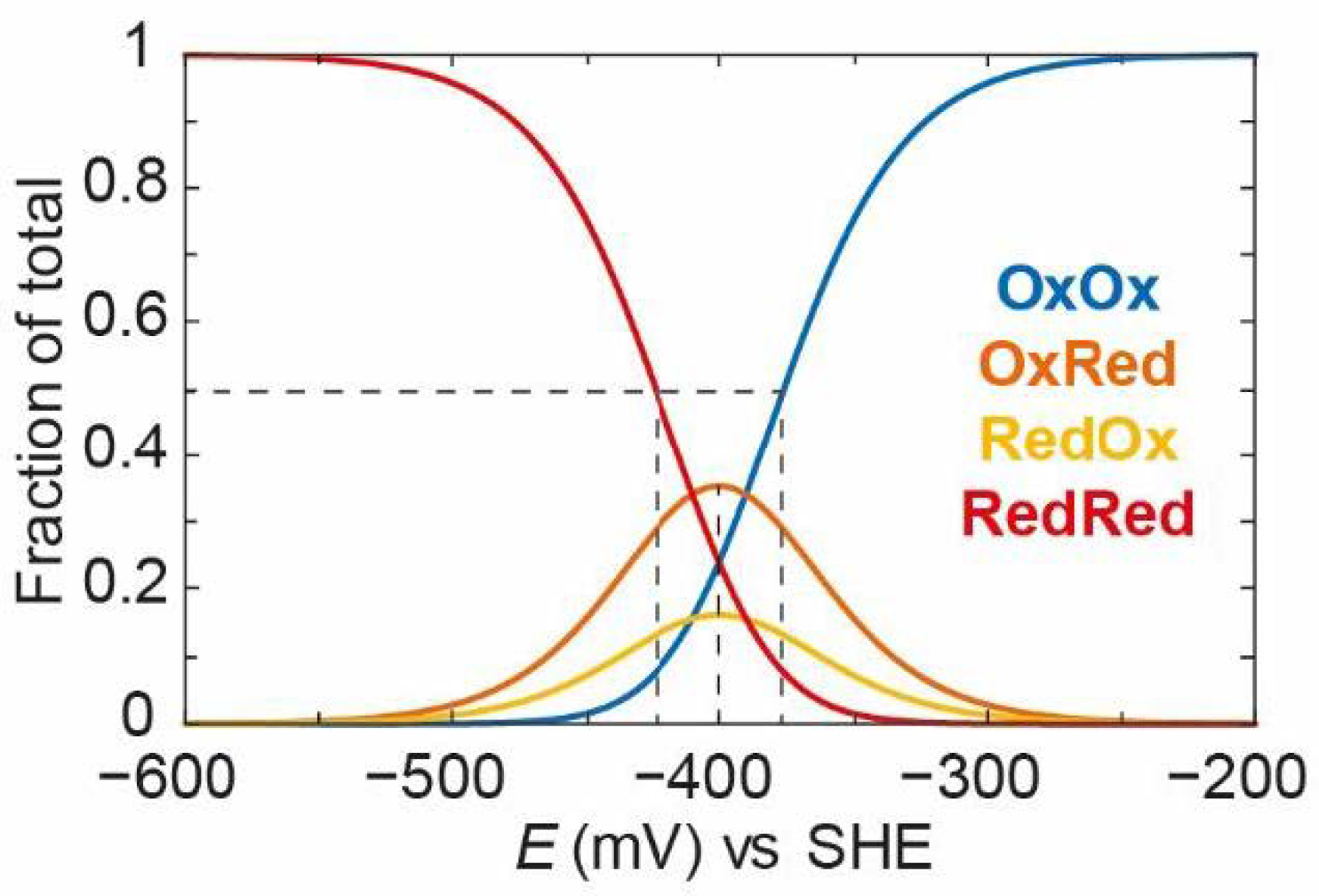
It can be seen that, as the potential goes from positive to negative, OxOx decreases and both OxRed and RedOx increase. One can think of this as a single electron entering the enzyme and equilibrating between both clusters depending on their intrinsic redox potentials. In this case, OxRed is more dominant because it has a more positive redox potential (−390 mV vs. −410 mV). It should also be noted that the decrease in the OxOx state follows a titration with an “apparent” midpoint potential more positive than −390 mV. In fact, the “apparent” midpoint potential is −380 mV. This is because both the OxOx/OxRed and OxOx/RedOx couples are titrating at the same time. This means that the OxOx species is lost at slightly more positive potentials than the values of and . The take-home message here is that these systems often behave in ways that, at first glance, seem non-intuitive, and it is only by studying the models that it is possible to fully understand their behaviour. As the second electron enters the enzyme, it will equilibrate among the remaining oxidised clusters and both OxRed and RedOx start to decrease as RedRed increases. RedRed increases with an “apparent” midpoint potential of −420 mV, which is slightly more negative than the values of and .
Next, we can apply the same model but introduce some redox anti-cooperativity. In
Figure 12, we have defined
=
− 100 mV, i.e., there is 100 mV of redox anti-cooperativity, or the redox potential of one cluster is 100 mV more negative when the other cluster is reduced. Because of the thermodynamic square, this also means that
=
− 100 mV, and the redox potential of the second cluster is also 100 mV more negative when the other cluster is reduced. We have redefined the intrinsic redox potentials
= −340 mV and
= −360 mV so that the average
EO is still −400 mV, as in
Figure 11.
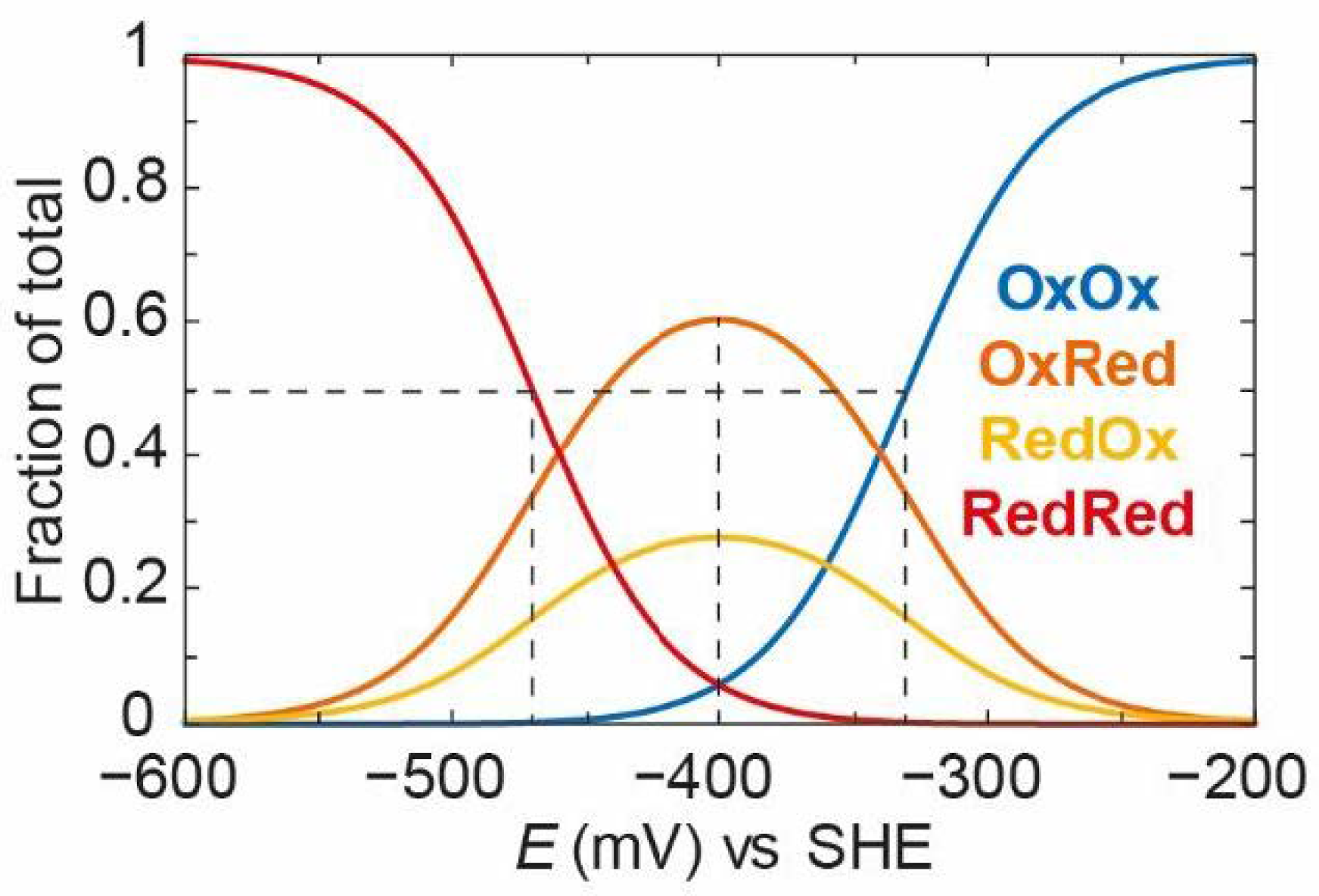
As can be observed,
OxOx decreases with a very positive “apparent” midpoint potential (−330 mV).
OxRed and
RedOx increase to a much greater extent and persist for longer until the
RedRed state starts to increase with an “apparent” midpoint potential of −470 mV. These “apparent” midpoint potentials are 50 mV more positive and more negative, respectively, than those observed in
Figure 11, reflecting the 100 mV redox anti-cooperativity.
This model was used to fit IR spectro-electrochemistry data from the [FeFe] hydrogenase from
Desulfovibrio desulfuricans (
DdHydAB) [
34]. For the sake of brevity, we have not reproduced the fitting of the experimental data in this review. However, the interested reader is highly recommended to read Reference [
34], where the simple anti-cooperativity model was applied to
DdHydAB with PDT in the [2Fe] cluster. This enzyme possesses the active site H-cluster as well as two accessory iron-sulfur clusters (F-clusters). Unusual behaviour was observed in IR spectro-electrochemical redox titrations of this enzyme in which the active site had been artificially matured with a diiron cluster containing PDT. Since PDT cannot be protonated at the bridgehead atom, reduction only occurs at the [4Fe-4S]
H subcluster of the H-cluster giving small (5–10 cm
−1) shifts in the CO and CN
− bands to lower energy. These titrations showed non-Nernstian behaviour that indicated reduction of the proximal F-cluster caused the redox potential of the [4Fe-4S]
H to become more negative. In fact, the reduction of the proximal F-cluster could be observed directly as minute (<2 cm
−1) shifts in the IR bands of both the
Hox and
Hred states. These data were fitted using the redox anti-cooperativity model described above and yielded a value of 118 mV for the interaction between the two clusters. This demonstrates that the modelling approach described in this case is not only illustrative but also practically useful for understanding spectro-electrochemical data and studying enzyme mechanisms. Since the PDT cofactor cannot be protonated, this simplified the number of redox states observed, and allowed the redox anti-cooperativity model to be investigated. However, to fully understand the behaviour of the natural H-cluster containing ADT in an F-cluster containing [FeFe] hydrogenase, it is necessary to incorporate redox anti-cooperativity into the full H-cluster model described in
Section 2.3. In the next section, this is exactly what we will do.
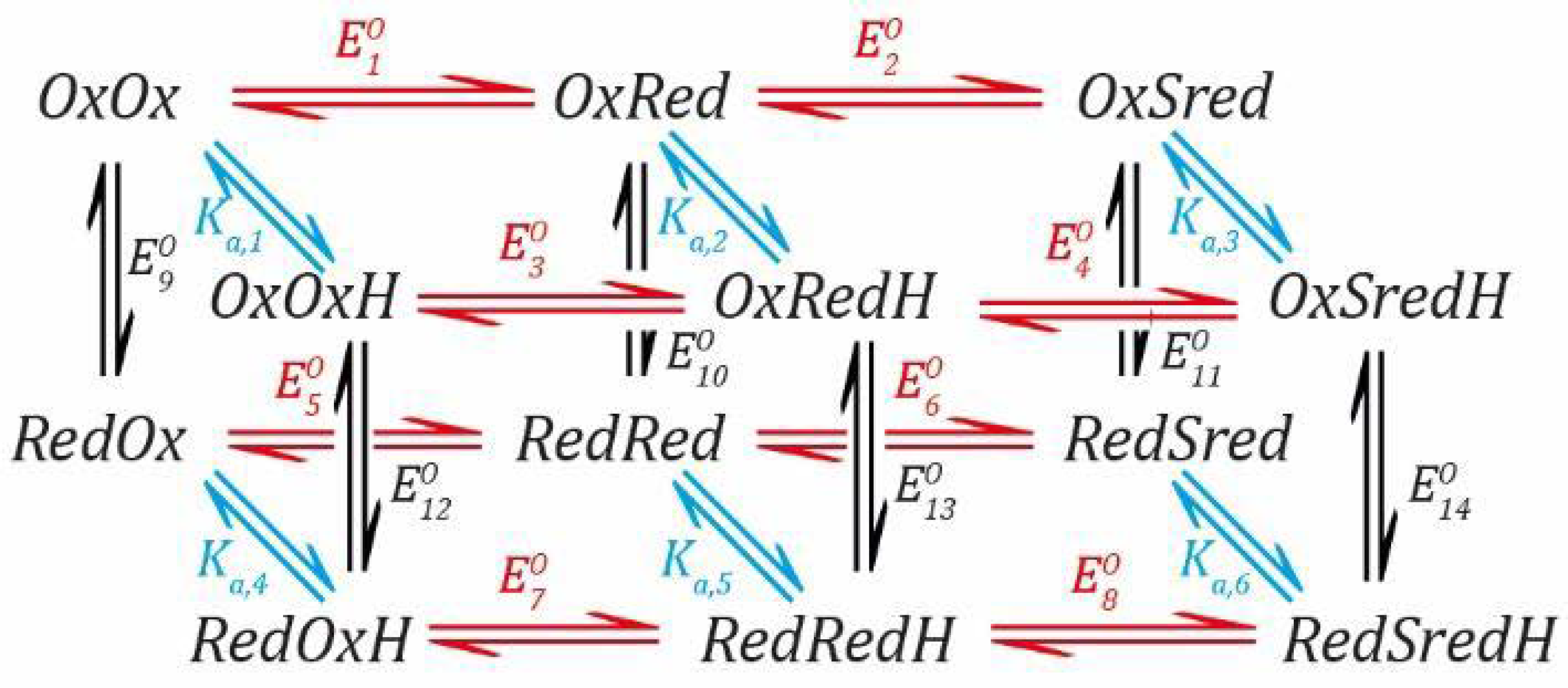
2.5. Redox Anticooperativity Model for the Active [FeFe] Hydrogenase
In
Section 2.1, we laid the foundations, showing the behaviour of a simple one electron redox and (de)protonation events. In
Section 2.2, we combined redox and (de)protonation into a proton-coupled electron transfer model. Then, in
Section 2.3, we added a second redox step to build a simple model that can be used to describe thermodynamic titrations of the H-cluster in the F-cluster free [FeFe] hydrogenase
CrHydA1. In
Section 2.4, we took a detour to look at what happens when two redox events occur adjacent to one another and can influence each other by redox anti-cooperativity. Now, we will combine what we have learned to build up a highly complex model containing both PCET and redox anti-cooperativity. Each species can now exist in three redox states of the H-cluster (
Ox,
Red, and
Sred), two protonation states, and also two different redox states of the proximal F-cluster, making the model “three-dimensional” (
Scheme 7).
As before, each
EO value defines the Nernst relationship between a reduced and an oxidised species and each
Ka value defines the relationship between a protonated and a deprotonated species. Once all the equations are described for each pathway, they can all be rearranged to get everything in terms of [
OxOx] (for the full derivation, please refer to the
Supplementary Materials Section S3). Here, we will simply present the final equation for [
OxOx] in terms of
EO values and
Ka values, in all its complexity.
where
As with the previous models, it was only necessary to use a limited number of the parameters. The rest can be calculated by considering the thermodynamic cycles. We used
to
as well as
Ka,1 and
Ka,4. Therefore, we need to calculate
to
as well as
Ka,2,
Ka,3,
Ka,5, and
Ka,6 (see
SI Section S3 for derivations of the equations).
Now these equations can be used to plot how the concentrations of intermediates vary with the applied potential as we did before for the simpler situation. In
Figure 13, the same parameters have been used as for
Figure 9A to describe the situation when the proximal F-cluster is oxidised. Then redox anti-cooperativity is added, such that
RedOx/
RedRed and
RedRedH/
RedSredH transitions are 100 mV more negative than the
OxOx/
OxRed and
OxRedH/
OxSredH transitions, respectively (affecting the steps that involve reduction of the proximal F-cluster and [4Fe-4S]
H). The intrinsic redox potential of the proximal F-cluster was set to −370 mV (i.e., the
OxOx/
RedOx transition has a redox potential of −370 mV). For
Figure 13, it has been assumed that states in which the proximal F-cluster is reduced cannot be spectroscopically distinguished from those in which the F-cluster is oxidised so that their populations are added together, i.e., [
Ox] = [
OxOx] + [
RedOx], etc. Lastly, states with essentially zero population across the potential range are not shown.
From
Figure 13, it can be seen that, as the applied potential becomes more negative, the
Ox state disappears and is replaced by a mixture of
Red and
RedH. However, as the potential becomes even more negative,
Red decreases while
RedH increases further. At the most negative potentials,
RedH also decreases and
SredH appears. This behaviour almost exactly reproduces the observations made during IR spectro-electrochemical titrations of
DdHydAB maturated with the ADT-containing cofactor [
34]. At first, this behaviour seemed entirely inexplicable. However, consideration of redox anti-cooperativity, which is clearly observed in the case of the PDT variant, allowed this behaviour to be understood. As the potential decreases, an electron enters the enzyme and equilibrates between the clusters, giving a mixture of
RedOx (F-cluster reduced),
OxRed ([4Fe-4S]
H subcluster reduced), and
OxRedH ([2Fe]
H subcluster reduced and protonated). As the applied potential becomes even more negative, a second electron enters the enzyme and fills the empty spaces, i.e.,
RedOx and
OxRed are converted to
RedRed, and
OxRedH is converted to
RedRedH and a small amount of
OxSredH. However,
RedRed is very unstable due to redox anti-cooperativity between the proximal F-cluster and the [4Fe-4S]
H subcluster, and prefers to make
RedRedH by protonation of the [2Fe]
H subcluster followed by an electronic rearrangement. Thus, at more negative potentials,
RedRedH accumulates. Now the question is: why is
RedRedH more stable than
OxSredH? This is likely because formation of the
SredH state requires a large amount of electron density on the H-cluster, while
RedRedH allows the two electrons to be stored on separate cofactors, one on the H-cluster and a second on the proximal F-cluster. Effectively though, the
RedRedH state in
DdHydAB has the same ability to produce H
2 as the
SredH state in
CrHydA1 (both have two electrons and one proton). However,
RedRedH can be formed at more positive potentials than
SredH, allowing H
+ reduction to start even closer to the thermodynamic 2H
+/H
2 potential. Likewise, the
Ox state is formed at slightly more negative potentials than in
CrHydA1, allowing H
2 oxidation to start even closer to the thermodynamic 2H
+/H
2 potential. Overall, this behaviour of the F-cluster containing [FeFe] hydrogenases may help to explain their excellent efficiency and reversibility for 2H
+/H
2 interconversion when compared with the F-cluster free
CrHydA1 [
44,
47].
Although we hypothesise that redox anti-cooperativity is purely an electrostatic effect from repulsion of electrons on the two reduced clusters, the factors governing this behaviour remain unclear. Furthermore, in addition to the effects described above, redox anti-cooperativity may also be involved in determining the catalytic bias of [FeFe] hydrogenases (i.e., whether the enzyme favours H
2 oxidation or H
+ reduction). It should be noted, however, that the factors determining catalytic bias are complex and include interactions between the active site and the protein matrix, which are essential for stabilising the various states of the H-cluster [
48]. Deletion of the F-cluster domain in a number of [FeFe] hydrogenases has a direct influence on the catalytic bias, possibly supporting a role for redox anti-cooperativity [
49,
50].
It should be noted that, in this model, as well as the previous model from
Section 2.4, we have only considered the interaction between the proximal F-cluster and the [4Fe-4S]
H subcluster. In
DdHydAB, a distal F-cluster is also present. However, the spectro-electrochemical titrations of this enzyme could be well reproduced without considering this cluster. This could indicate that the potential of this cluster is very positive in relation to the proximal F-cluster, and so it is essentially in the reduced state throughout the titrations, or that the redox anti-cooperativity between the distal and proximal F-clusters, and between the distal F-cluster and the H-cluster, is negligible. In a recent study of the F-cluster containing [FeFe] hydrogenase from
Clostridium pasteurianum (
CpHydA1), it was necessary to include a second redox anti-cooperative interaction into the model [
51]. In this case, the second cluster appears to have a more negative redox potential than the proximal cluster.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



















