
Upgrading of Bio-Syngas via Steam-CO2 Reforming Using Rh/Alumina Monolith Catalysts

Abstract
1. Introduction
2. Experimental
2.1. Catalysts and a Model Bio-Syngas
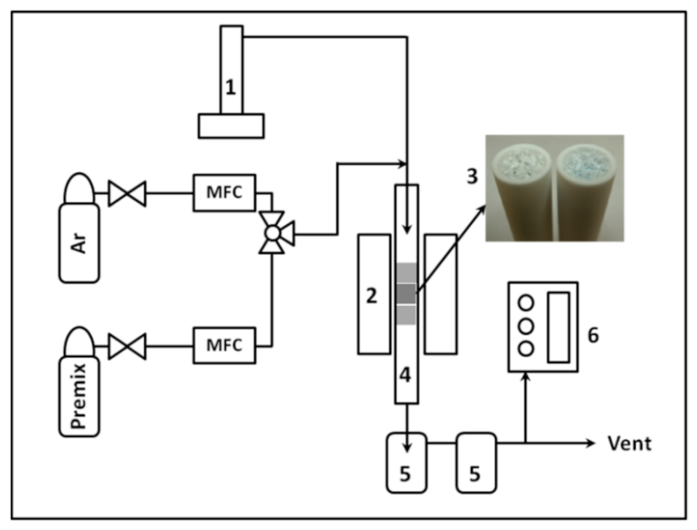
2.2. Catalytic Upgrading of Bio-Syngas
2.3. Thermodynamic Analysis
3. Results and Discussion
3.1. Predicted Gas Composition from Thermodynamic Simulation
3.2. Change in the Main Reaction Pathways with Reaction Temperature
3.3. Activation Energy and Transition Temperature
3.4. Effect of Gas Velocity
3.5. Effect of Concentration of Steam in the Feed
3.6. Stability of Rh/Alumina Foam Monolith
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carlu, E.; Truong, T.; Kundevski, M. Biogas Opportunities for Australia; ENEA Consulting: Melbourne, Australia, 2019. [Google Scholar]
- Korberg, A.D.; Skov, I.R.; Mathiesen, B.V. The role of biogas and biogas-derived fuels in a 100% renewable energy system in Denmark. Energy 2020, 199, 117426. [Google Scholar] [CrossRef]
- International Energy Agency. Global Energy & CO2 Status Report 2018; International Energy Agency: Paris, France, 2018. [Google Scholar]
- Intergovernmental Panel on Climate Change. Special Report on Global Warming of 1.5C; Intergovernmental Panel on Climate Change: Geneva, Switzerland, 2018. [Google Scholar]
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of transportation fuels from biomass: Chemistry, catalysts, and engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [PubMed]
- Zinoviev, S.; Muller-Langer, F.; Das, P.; Bertero, N.; Fornasiero, P.; Kaltschmitt, M.; Centi, G.; Miertus, S. Next-Generation Biofuels: Survey of Emerging Technologies and Sustainability Issues. ChemSusChem 2010, 3, 1106–1133. [Google Scholar] [CrossRef]
- Alonso, D.M.; Bond, J.Q.; Dumesic, J.A. Catalytic conversion of biomass to biofuels. Green Chem. 2010, 12, 1493–1513. [Google Scholar] [CrossRef]
- Li, C.Z. Special issue-Gasification: A route to clean energy. Process Saf. Environ. Prot. 2006, 84, 407–408. [Google Scholar] [CrossRef]
- Zhang, R. Catalytic destruction of tar in biomass derived producer gas. Energy Convers. Manag. 2004, 45, 995–1014. [Google Scholar] [CrossRef]
- Coll, R.; Salvado, J.; Farriol, X.; Montane, D. Steam reforming model compounds of biomass gasification tars: Conversion at different operating conditions and tendency towards coke formation. Fuel Process. Technol. 2001, 74, 19–31. [Google Scholar] [CrossRef]
- Corella, J.; Toledo, J.M.; Padilla, R. Catalytic hot gas cleaning with monoliths in biomass gasification in fluidized beds. 3. Their effectiveness for ammonia elimination. Ind. Eng. Chem. Res. 2005, 44, 2036–2045. [Google Scholar] [CrossRef]
- Corella, J.; Toledo, M.; Padilla, R. Catalytic hot gas cleaning with monoliths in biomass gasification in fluidized beds. 1. Their effectiveness for tar elimination. Ind. Eng. Chem. Res. 2004, 43, 2433–2445. [Google Scholar] [CrossRef]
- Anis, S.; Zainal, Z.A. Tar reduction in biomass producer gas via mechanical, catalytic and thermal methods: A review. Renew. Sustain. Energy Rev. 2011, 15, 2355–2377. [Google Scholar] [CrossRef]
- Abdoulmoumine, N.; Adhikari, S.; Kulkarni, A.; Chattanathan, S. A review on biomass gasification syngas cleanup. Appl. Energy 2015, 155, 294–307. [Google Scholar] [CrossRef]
- Chattanathan, S.A.; Adhikari, S.; Taylor, S. Conversion of carbon dioxide and methane in biomass synthesis gas for liquid fuels production. Int. J. Hydrogen Energy 2012, 37, 18031–18039. [Google Scholar] [CrossRef]
- Wang, C.G.; Wang, T.J.; Ma, L.L.; Gao, Y.; Wu, C.Z. Steam reforming of biomass raw fuel gas over NiO-MgO solid solution cordierite monolith catalyst. Energy Convers. Manag. 2010, 51, 446–451. [Google Scholar] [CrossRef]
- Choudhary, V.R.; Mondal, K.C. CO2 reforming of methane combined with steam reforming or partial oxidation of methane to syngas over NdCoO3 perovskite-type mixed metal-oxide catalyst. Appl. Energy 2006, 83, 1024–1032. [Google Scholar] [CrossRef]
- Koo, K.Y.; Roh, H.S.; Jung, U.H.; Yoon, W.L. CeO2 Promoted Ni/Al2O3 Catalyst in Combined Steam and Carbon Dioxide Reforming of Methane for Gas to Liquid (GTL) Process. Catal. Lett. 2009, 130, 217–221. [Google Scholar] [CrossRef]
- Jang, W.J.; Jeong, D.W.; Shim, J.O.; Kim, H.M.; Roh, H.S.; Son, I.H.; Lee, S.J. Combined steam and carbon dioxide reforming of methane and side reactions: Thermodynamic equilibrium analysis and experimental application. Appl. Energy 2016, 173, 80–91. [Google Scholar] [CrossRef]
- Haryanto, A.; Fernando, S.D.; Pordesimo, L.O.; Adhikari, S. Upgrading of syngas derived from biomass gasification: A thermodynamic analysis. Biomass Bioenergy 2009, 33, 882–889. [Google Scholar] [CrossRef]
- Artz, J.; Muller, T.E.; Thenert, K.; Kleinekorte, J.; Meys, R.; Sternberg, A.; Bardow, A.; Leitner, W. Sustainable Conversion of Carbon Dioxide: An Integrated Review of Catalysis and Life Cycle Assessment. Chem. Rev. 2018, 118, 434–504. [Google Scholar] [CrossRef]
- Giuntoli, J.; Agostini, A.; Edwards, R.; Marelli, L. Solid and Gaseous Bioenergy Pathways: Input Values and GHG Emissions. Calculated According to COM(2016) 767, Ver 2; EUR 27215 EN; European Commission, Joint Research Centre: Ispra, Italy, 2017. [Google Scholar]
- Cybulski, A.; Moulijn, J.A. Monoliths in heterogeneous catalysis. Catal. Rev.-Sci. Eng. 1994, 36, 179–270. [Google Scholar] [CrossRef]
- Twigg, M.V.; Richardson, J.T. Fundamentals and applications of structured ceramic foam catalysts. Ind. Eng. Chem. Res. 2007, 46, 4166–4177. [Google Scholar] [CrossRef]
- Renken, A.; Kiwi-Minsker, L. Microstructured Catalytic Reactors. Adv. Catal. 2010, 53, 47–122. [Google Scholar]
- Mills, P.L.; Quiram, D.J.; Ryley, J.F. Microreactor technology and process miniaturization for catalytic reactions-A perspective on recent developments and emerging technologies. Chem. Eng. Sci. 2007, 62, 6992–7010. [Google Scholar] [CrossRef]
- Richardson, J.T.; Garrait, M.; Hung, J.K. Carbon dioxide reforming with Rh and Pt-Re catalysts dispersed on ceramic foam supports. Appl. Catal. A-Gen. 2003, 255, 69–82. [Google Scholar] [CrossRef]
- Nacken, M.; Ma, L.; Heidenreich, S.; Verpoort, F.; Baron, G.V. Development of a catalytic ceramic foam for efficient tar reforming of a catalytic filter for hot gas cleaning of biomass-derived syngas. Appl. Catal. B-Environ. 2012, 125, 111–119. [Google Scholar] [CrossRef]
- Wang, C.G.; Wang, T.J.; Ma, L.L.; Gao, Y.; Wu, C.Z. Partial oxidation reforming of biomass fuel gas over nickel-based monolithic catalyst with naphthalene as model compound. Korean J. Chem. Eng. 2008, 25, 738–743. [Google Scholar] [CrossRef]
- Rostrupnielsen, J.R.; Hansen, J.H.B. CO2-Reforming of methane over transition-metals. J. Catal. 1993, 144, 38–49. [Google Scholar] [CrossRef]
- Wang, Y.; Chin, Y.H.; Rozmiarek, R.T.; Johnson, B.R.; Gao, Y.; Watson, J.; Tonkovich, A.Y.L.; Vander Wiel, D.P. Highly active and stable Rh/MgO-Al2O3 catalysts for methane steam reforming. Catal. Today 2004, 98, 575–581. [Google Scholar] [CrossRef]
- Meille, V. Review on methods to deposit catalysts on structured surfaces. Appl. Catal. A-Gen. 2006, 315, 1–17. [Google Scholar] [CrossRef]
- Richardson, J.T.; Paripatyadar, S.A. Carbon dioxide reforming of methane with supported rhodium. Appl. Catal. 1990, 61, 293–309. [Google Scholar] [CrossRef]
- Zhang, K.; Jiang, X. An investigation of fuel variability effect on bio-syngas combustion using uncertainty quantification. Fuel 2018, 220, 283–295. [Google Scholar] [CrossRef]
- Zhang, K.; Lupo, G.; Duwig, C. Investigation of wet combustion instability due to bio-syngas fuel variability. Fuel 2021, 285, 119120. [Google Scholar] [CrossRef]
- Lu, Y.J.; Lee, T. Influence of the feed gas composition on the Fischer-Tropsch synthesis in commercial operations. J. Nat. Gas Chem. 2007, 16, 329–341. [Google Scholar] [CrossRef]
- Yin, X.L.; Leung, D.Y.C.; Chang, J.; Wang, J.F.; Fu, Y.; Wu, C.Z. Characteristics of the synthesis of methanol using biomass-derived syngas. Energy Fuels 2005, 19, 305–310. [Google Scholar] [CrossRef]
- Cullis, C.F.; Willatt, B.M. Oxidation of methane over supported precious metal catalysts. J. Catal. 1983, 83, 267–285. [Google Scholar] [CrossRef]
- Bruno, T.; Beretta, A.; Groppi, G.; Roderi, M.; Forzatti, P. A study of methane partial oxidation in annular reactor: Activity of Rh/alpha-Al2O3 and Rh/ZrO2 catalysts. Catal. Today 2005, 99, 89–98. [Google Scholar] [CrossRef]
- Kolaczkowski, S.T.; Thomas, W.J.; Titiloye, J.; Worth, D.J. Catalytic combustion of methane in a monolith reactor: Heat and mass transfer under laminar flow and pseudo-steady-state reaction conditions. Combust. Sci. Technol. 1996, 118, 79–100. [Google Scholar] [CrossRef]
- Hayes, R.E.; Kolaczkowski, S.T.; Thomas, W.J.; Titiloye, J. Transient experiments and modeling of the catalytic combustion of methane in a monolith reactor. Ind. Eng. Chem. Res. 1996, 35, 406–414. [Google Scholar] [CrossRef]
- Wei, J.M.; Iglesia, E. Structural requirements and reaction pathways in methane activation and chemical conversion catalyzed by rhodium. J. Catal. 2004, 225, 116–127. [Google Scholar] [CrossRef]
- Tonkovich, A.L.Y.; Yang, B.; Perry, S.T.; Fitzgerald, S.P.; Wang, Y. From seconds to milliseconds to microseconds through tailored microchannel reactor design of a steam methane reformer. Catal. Today 2007, 120, 21–29. [Google Scholar] [CrossRef]
- Kuznetsov, V.V.; Vitovsky, O.V.; Gasenko, O.A. Methane steam reforming in an annular microchannel with Rh/Al2O3 catalyst. J. Eng. Thermophys. 2009, 18, 187–196. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Kondarides, D.I.; Verykios, X.E. Selective methanation of CO over supported noble metal catalysts: Effects of the nature of the metallic phase on catalytic performance. Appl. Catal. A-Gen. 2008, 344, 45–54. [Google Scholar] [CrossRef]
- Qin, D.Y.; Lapszewicz, J.; Jiang, X.Z. Comparison of partial oxidation and steam-CO2 mixed reforming of CH4 to syngas on MgO-supported metals. J. Catal. 1996, 159, 140–149. [Google Scholar] [CrossRef]
- Oyama, S.T.; Hacarlioglu, P.; Gu, Y.; Lee, D. Dry reforming of methane has no future for hydrogen production: Comparison with steam reforming at high pressure in standard and membrane reactors. Int. J. Hydrogen Energy 2012, 37, 10444–10450. [Google Scholar] [CrossRef]
- Ozkara-Aydinoglu, S. Thermodynamic equilibrium analysis of combined carbon dioxide reforming with steam reforming of methane to synthesis gas. Int. J. Hydrogen Energy 2010, 35, 12821–12828. [Google Scholar] [CrossRef]
- Avraam, D.G.; Halkides, T.I.; Liguras, D.K.; Bereketidou, O.A.; Goula, M.A. An experimental and theoretical approach for the biogas steam reforming reaction. Int. J. Hydrogen Energy 2010, 35, 9818–9827. [Google Scholar] [CrossRef]
- Fiedorow, R.M.J.; Wanke, S.E. Effect of gamma-alumina crystallinity on the state of rhodium during high-temperature treatments in oxygen and hydrogen. Appl. Catal. B-Environ. 1997, 14, 249–259. [Google Scholar] [CrossRef]
- Ziegelbauer, J.M.; Gulla, A.F.; O’Laoire, C.; Urgeghe, C.; Allen, R.J.; Mukerjee, S. Chalcogenide electrocatalysts for oxygen-depolarized aqueous hydrochloric acid electrolysis. Electrochim. Acta 2007, 52, 6282–6294. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alumina foam | 60 mm (L) × 10 mm (ø) |
| Void fraction | 0.93 |
| Reactor | α-SiC; 1000 mm in length |
| Composition of model bio-syngas | CO/CO2/CH4/H2/He = 28/25/12/33/2 vol % |
| Temperature range | 500–1000 °C |
| Flow rate range | up to 1500 mL min−1 |
| Steam addition | 0–40 vol % |
| Steam, % | CH4 Conv.,% | Changes in Mole Fraction | H2/CO 1 (overall 2) | Overall CO/CO2 3 | |||
|---|---|---|---|---|---|---|---|
| ΔCH4 | ΔCO | ΔCO2 | ΔH2(ΔCH4x2) | ||||
| 0 | 51.9 | −6.2 | 17.0 | −11.9 | 6.6 (12.3) | 0.39 (0.87) | 3.10 |
| 20 | 55.5 | −6.6 | 10.7 | −4.8 | 13.8 (13.2) | 1.29 (1.19) | 1.79 |
| 40 | 81.1 | −9.7 | 7.4 | 2.5 | 28.0 (19.4) | 3.78 (1.70) | 1.24 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, W.J.; Li, C.; Patel, J. Upgrading of Bio-Syngas via Steam-CO2 Reforming Using Rh/Alumina Monolith Catalysts. Catalysts 2021, 11, 180. https://doi.org/10.3390/catal11020180
Lee WJ, Li C, Patel J. Upgrading of Bio-Syngas via Steam-CO2 Reforming Using Rh/Alumina Monolith Catalysts. Catalysts. 2021; 11(2):180. https://doi.org/10.3390/catal11020180
Chicago/Turabian StyleLee, Woo Jin, Chaoen Li, and Jim Patel. 2021. "Upgrading of Bio-Syngas via Steam-CO2 Reforming Using Rh/Alumina Monolith Catalysts" Catalysts 11, no. 2: 180. https://doi.org/10.3390/catal11020180
APA StyleLee, W. J., Li, C., & Patel, J. (2021). Upgrading of Bio-Syngas via Steam-CO2 Reforming Using Rh/Alumina Monolith Catalysts. Catalysts, 11(2), 180. https://doi.org/10.3390/catal11020180