
Thermochemical and Catalytic Conversion Technologies for the Development of Brazilian Biomass Utilization

 ,
, 
Abstract
:1. Introduction
2. Biomass and Lignocellulosic Biomass
2.1. Renewable Energy and Biomass Availability in Brazil
2.2. Sugarcane

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biorefineries | Location | Net Profit (R$ Million) | Cogeneration |
|---|---|---|---|
| Grupo São Martinho [70] | Pradópolis/SP | 288.3 | Fuel: biomass Fertilizer: vinasse, filter cake |
| Usina Colombo [70] | Ariranha/SP | 118.3 | - |
| Grupo Santa Terezinha Participações [71] | Maringá/PR | 112.8 | Fuel: biomass Fertilizer: vinasse, filter cake, ashes |
| Usina Ipiranga [72] | Mococa/SP | 68.9 | Fuel: biomass Fertilizer: vinasse, filter cake, ashes |
| Usina São Manoel [73] | São Manuel/SP | 61.7 | Fuel: biomass Fertilizer: vinasse, filter cake, ashes Fermentation: second-generation ethanol |
| Usina da Pedra [70] | Serrana/SP | 48.6 | Fuel: biomass Fertilizer: vinasse, filter cake, ashes |
| Cia Melhoramentos do Norte do Paraná [70] | Jussara/PR | 38.4 | Fuel: biomass Fertilizer: vinasse, filter cake, ashes |
| Usina Batatais [74] | Batatais/SP | 30.7 | Fuel: biomass Fertilizer: vinasse, filter cake, ashes |
| Grupo Balbo [75] | Sertãozinho/SP | 25.3 | Fuel: biomass Fertilizer: vinasse, filter cake |
| Adecoagro [76] | São Paulo/SP | 17.1 | Fuel: biomass Fertilizer: vinasse, filter cake |
| Usina São João [77] | Araras/SP | 12.1 | Fuel: biomass |
3. Thermochemical Conversion Routes of Lignocellulosic Biomass
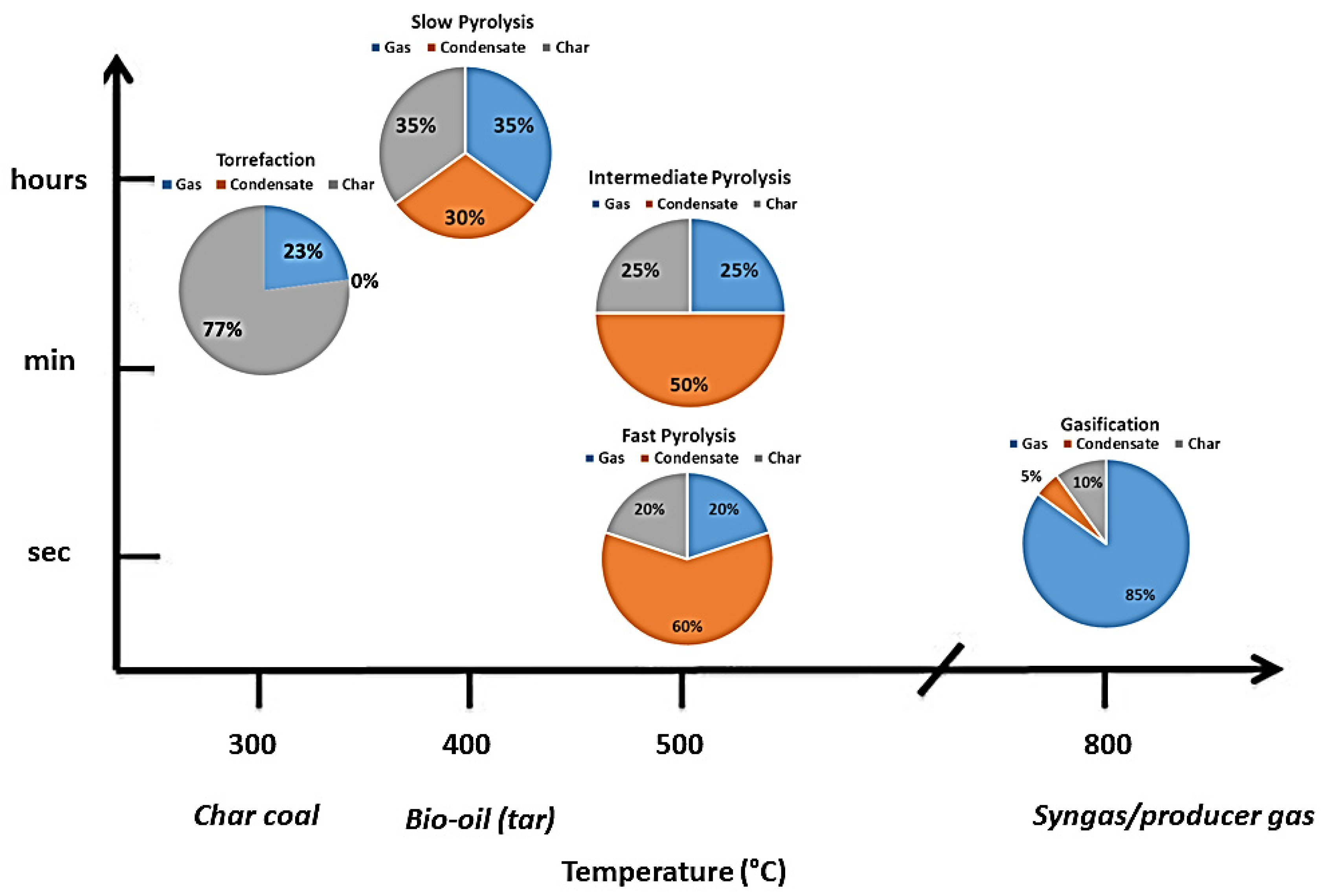
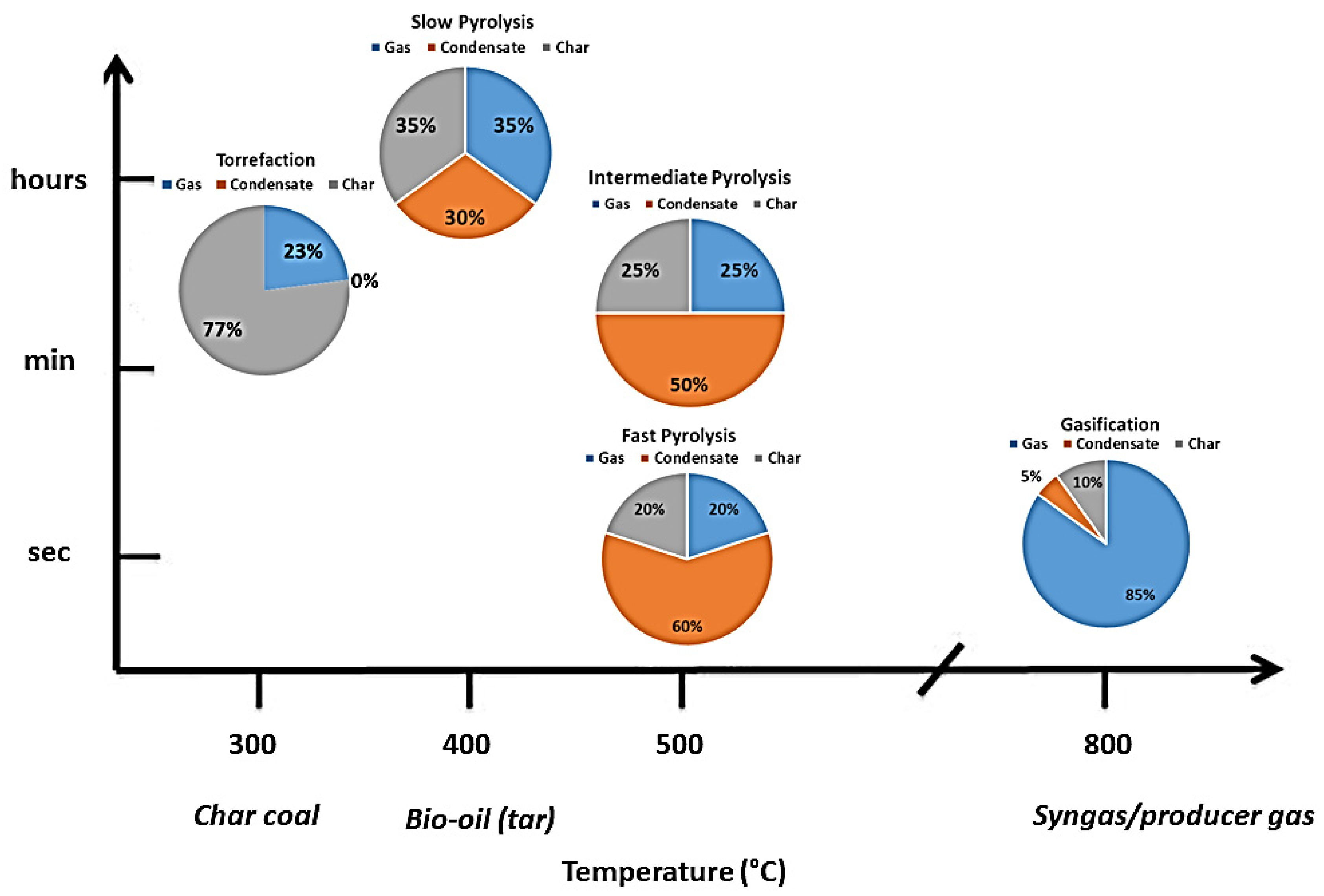
3.1. Pyrolysis
| Heating Rate | Temperature | Solid Residence Time | Vapor Residence Time | |
|---|---|---|---|---|
| Slow pyrolysis | Low (<1 °C/s) | Low (>400 °C) | Long or very long (minutes to days) | Long (>seconds) |
| Intermediate pyrolysis | Moderate | Moderate | Long (minutes) | Moderate (some seconds) |
| Fast pyrolysis | Very high (>100 °C/s) | Moderate or High (425–600 °C) | Short (a few seconds or less) | Very short (<seconds) |
3.2. Factors Affecting the Behavior of Pyrolysis
3.2.1. Degradation Reactions and Their Kinetics
3.2.2. Influence of the Feedstock Properties
3.2.3. Feedstock Moisture Content and Vapor Residence Time
3.2.4. Effect of Condensation and Aging
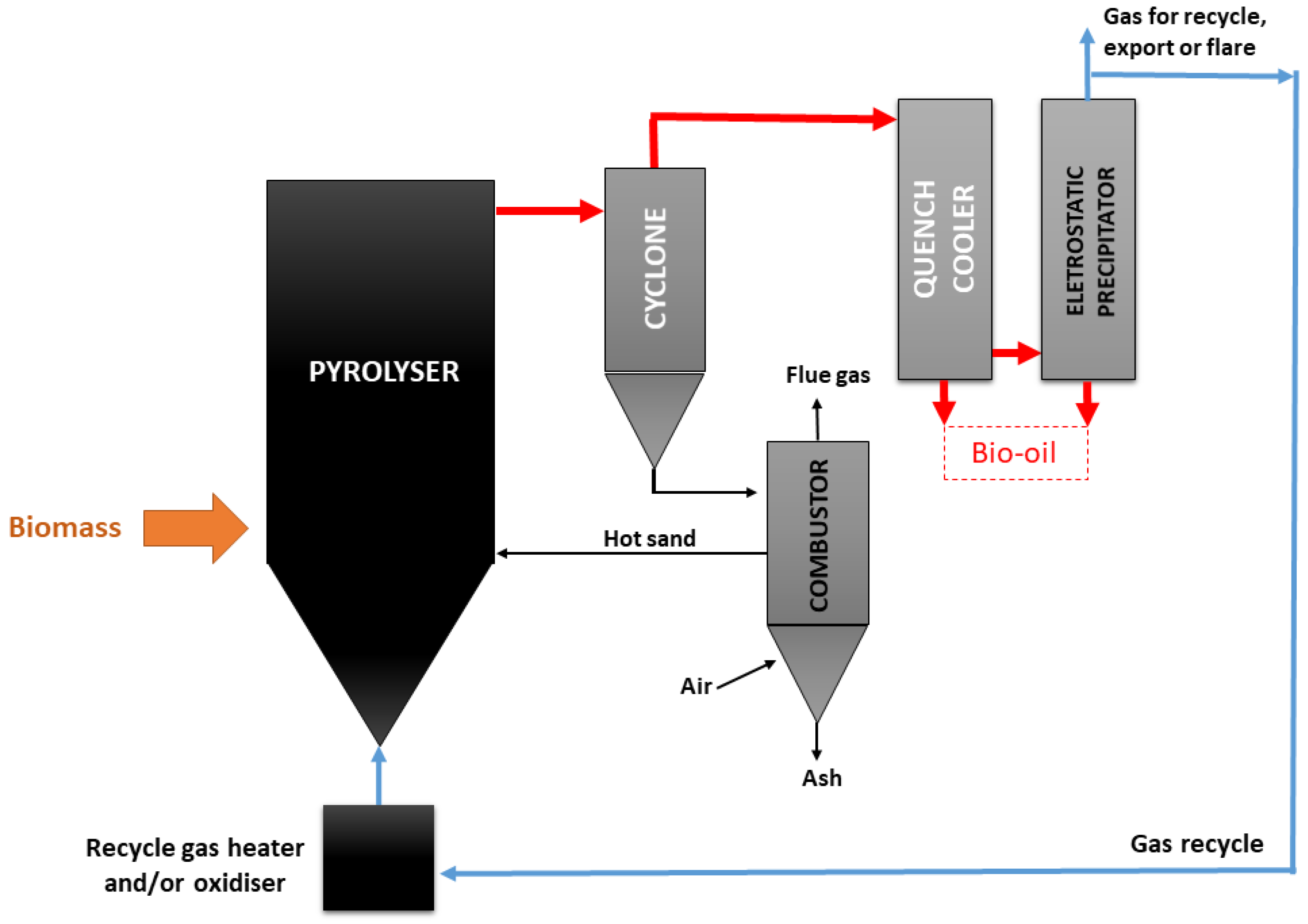
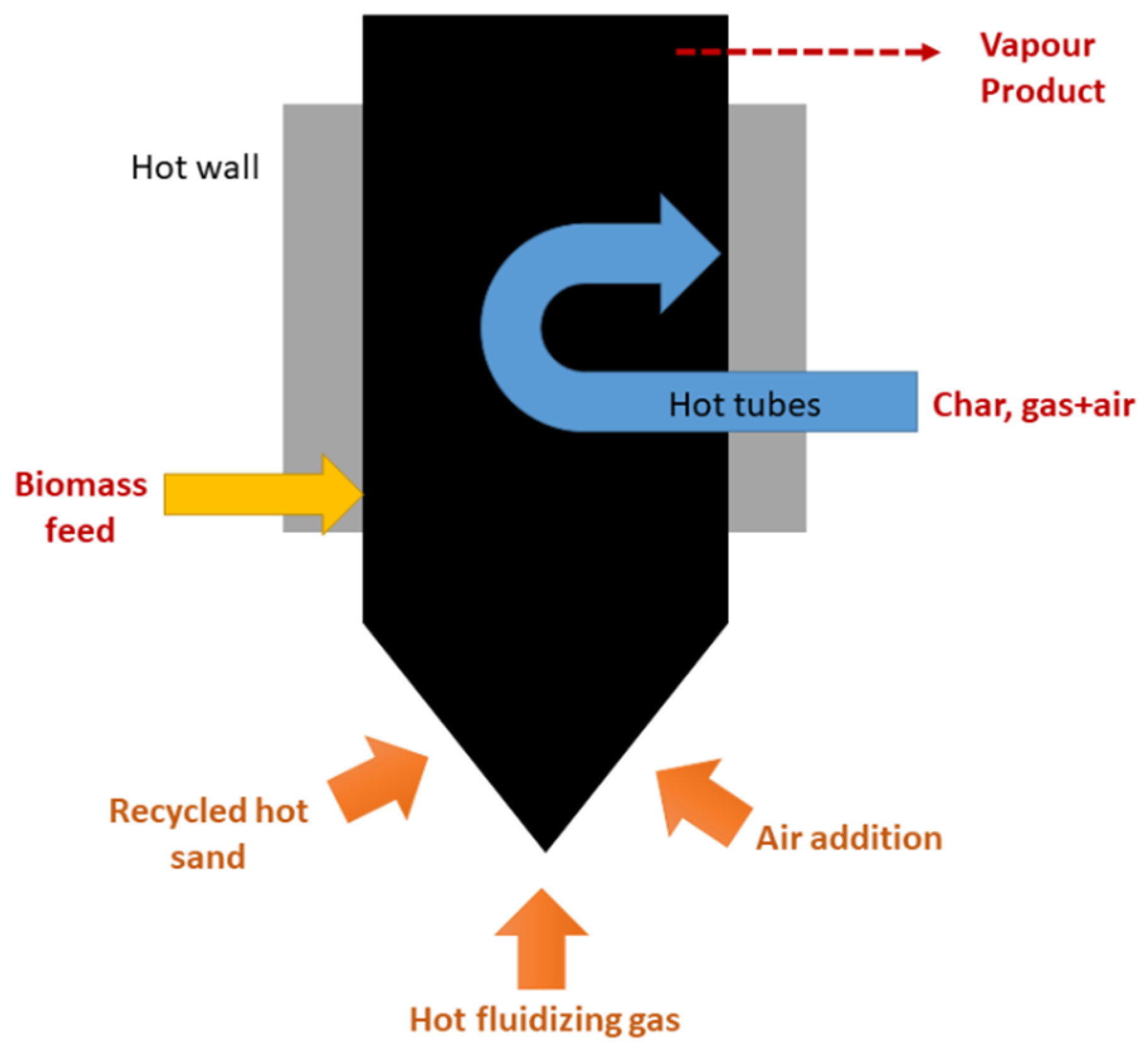
3.2.5. Reactor Design
- Entrained flow reactors
- Stationary fluidized bed reactors
- Circulating fluidized bed reactors
- Screw reactors
- Rotating cone reactors
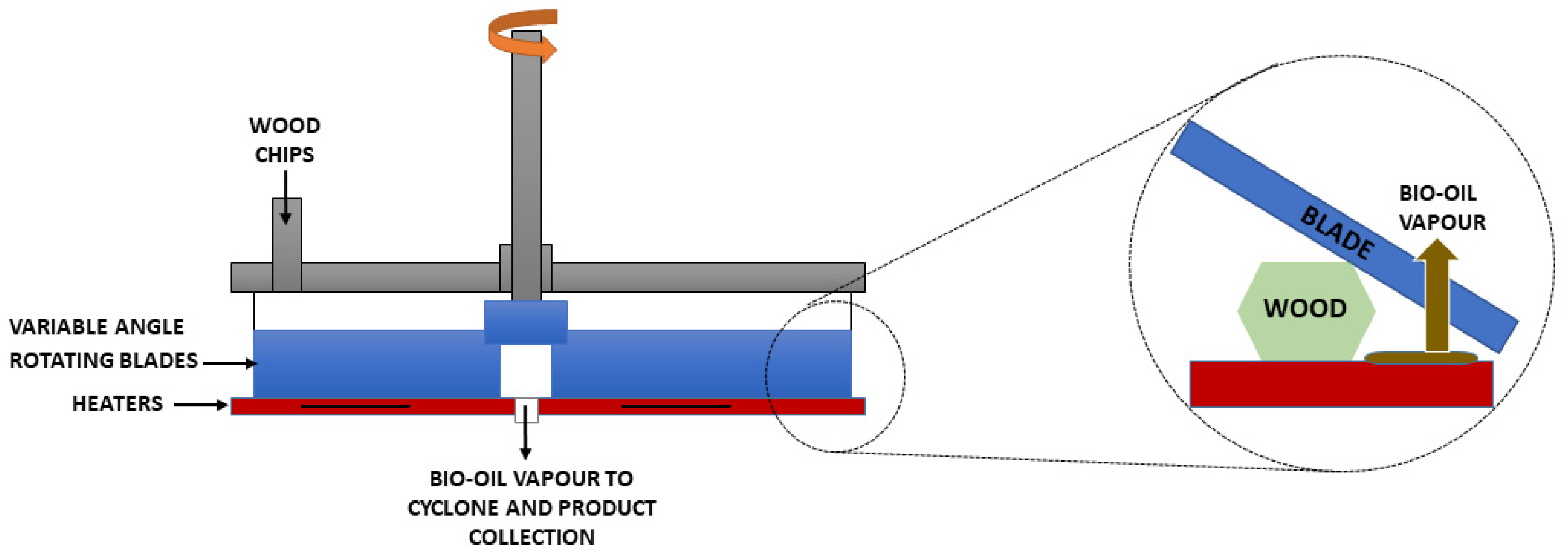
- Ablative pyrolysis
- Vacuum reactors
- Vortex reactors
- Rotating kiln reactors
3.3. Research Facilities and Large-Scale Materializations
4. The Pyrolysis Liquid, i.e., the Bio-Oil
| Wheat Straw [13] | Wheat Straw FPBO [193] | Generic Wood FPBO [194] | Crude Oil [194] | |
|---|---|---|---|---|
| Carbon (wt.%) | 42.8 | 45.9 | 40–50 | 85 |
| Hydrogen (wt.%) | 5.4 | 7.69 | 6.0–7.6 | 11–13 |
| Oxygen (wt.%) | 38.9 | 41.3 | 36–52 | 0.1–1.0 |
| Sulfur (wt.%) | n/a | n/a | 0.00–0.02 | 1.0–1.8 |
| Nitrogen (wt.%) | n/a | 2.19 | 0.00–0.15 | 0.1 |
| Water (wt.%) | 5.7 | 28.4 | 17–30 | 0.02–0.1 |
| Solid (wt.%) | n/a | n/a | 0.03–0.7 | 1 |
| pH | n/a | n/a | 2.4–2.8 | n/a |
| Viscosity at 323 K (cP) | n/a | n/a | 13–30 | 180 |
| HHV (MJ/Kg) | 16.6 | 21.0 | 16–20 | 40 |
| Density (kg/m3) | n/a | n/a | 1.2–1.3 | 0.9–1.0 |
4.1. Bio-Oil Upgrading
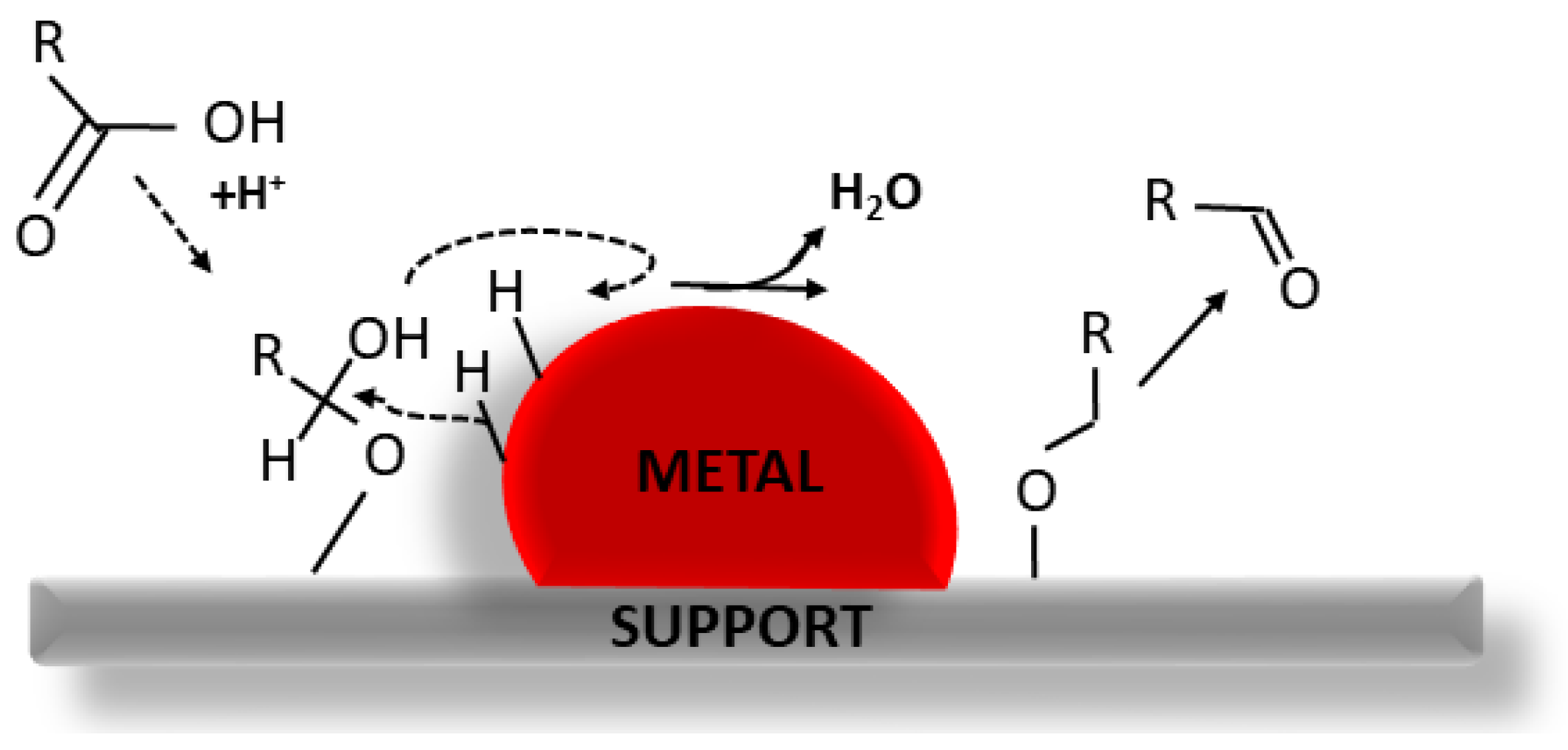
4.2. Bio-Oil Catalytic Hydrotreatment
4.2.1. Parameters Impacting the Bio-Oil Hydrotreatment Reactions
- Temperature
- Hydrogen pressure and consumption
- Heating rate
- Reaction duration time in batch reactions
- Catalysts
4.2.2. Catalysts: Sulfided Catalysts
4.2.3. Catalysts: Noble Metal Catalysts
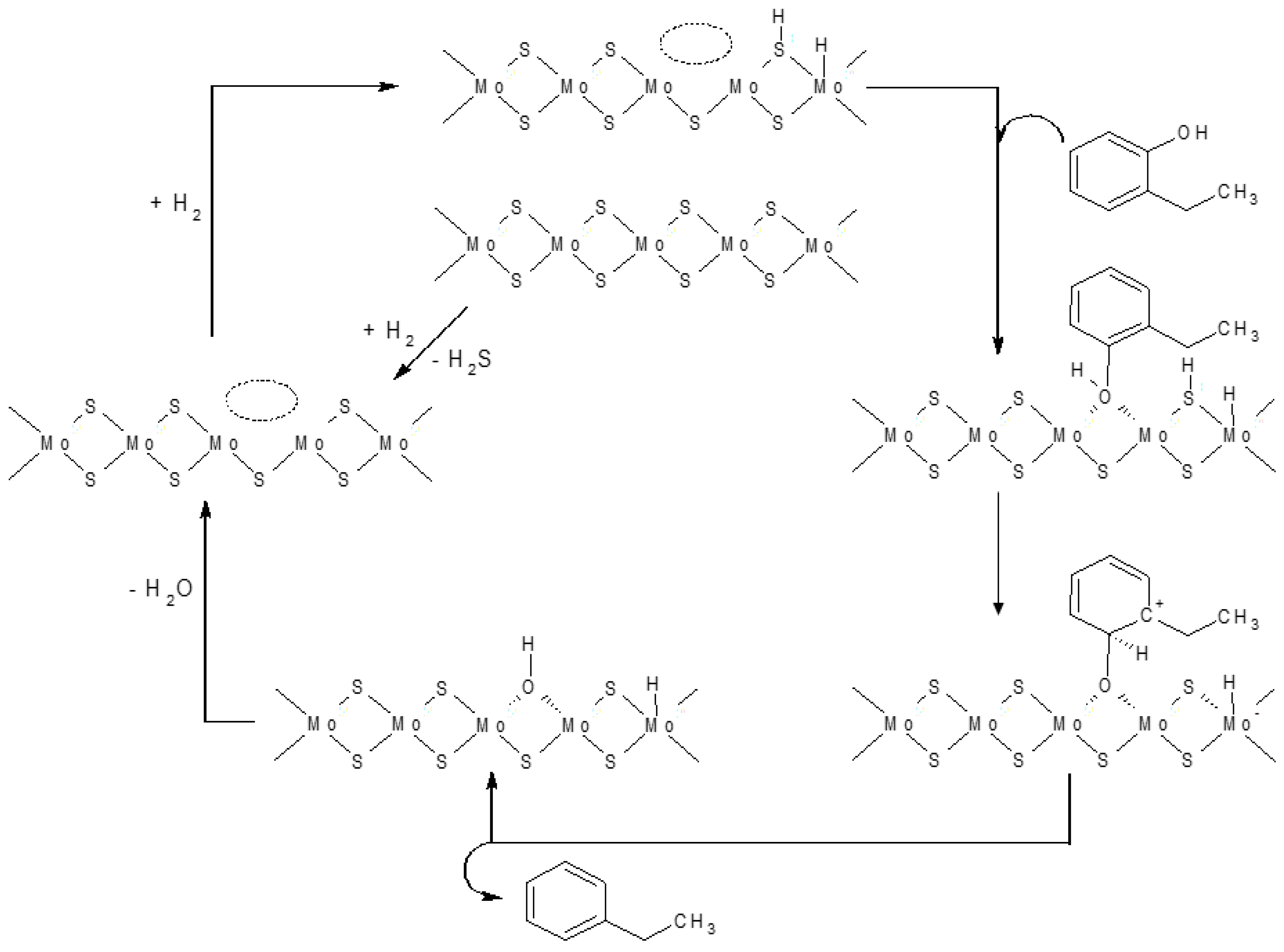
4.2.4. Niobium-Based Catalysts
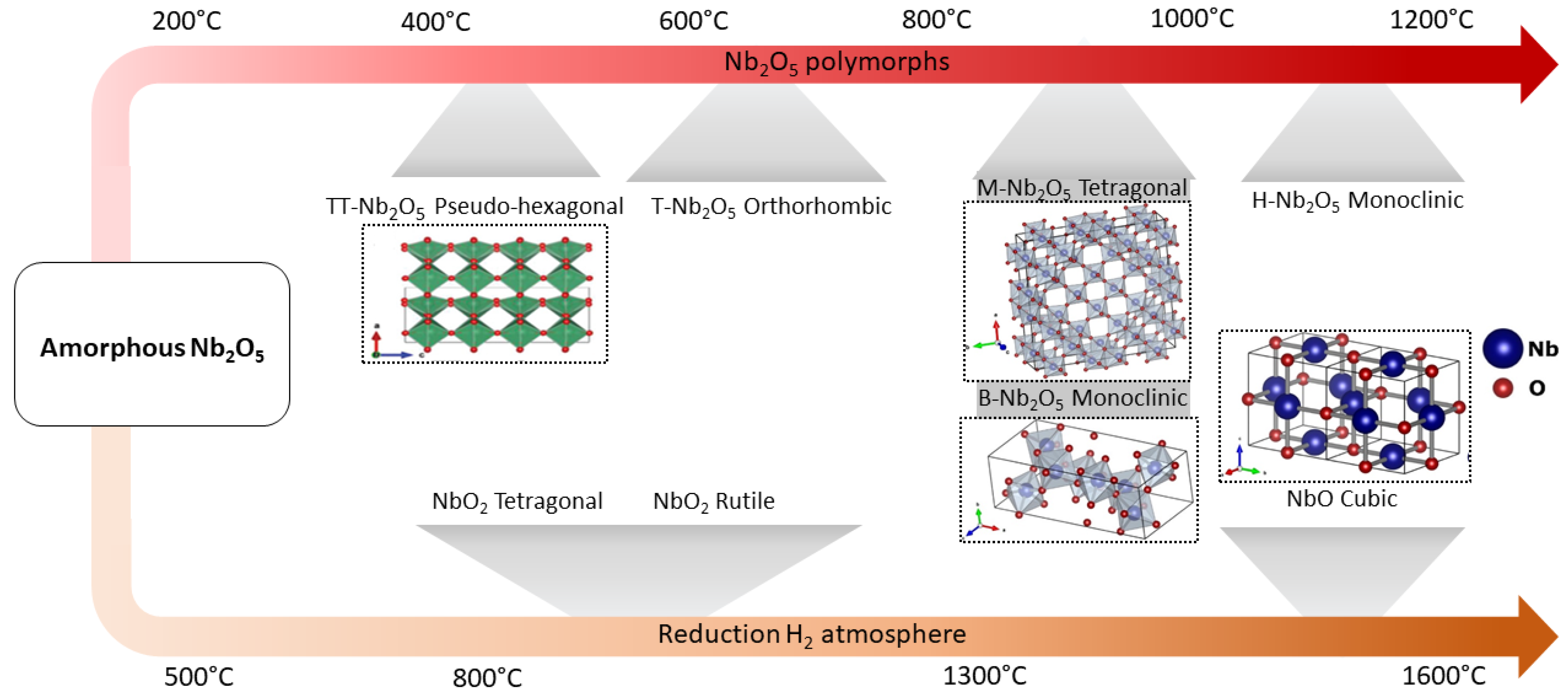
4.2.5. Niobium Catalysts for Hydrotreatment Reactions
4.2.6. HDT of Model Compounds with Niobium-Containing Catalysts
| Niobium Function | Author | Catalysts | HDT Feed |
|---|---|---|---|
| Support | Jing et al. (2019) [245] | Pd supported on CaO, MgO, MgAl-HT, ZrO2, Al2O3, and Nb2O5. | Furfural and 4-heptanone |
| Support | Barrios et al. (2018) [240] | Pd/SiO2 and Pd/Nb2O5 | Phenol |
| Support | Rezende et al. (2018) [246] | Ni/Nb2O5, Ni/CeO2, Ni/Ce0.30Nb0.70O2, Ni/Ce0.80Nb0.20O2, | Phenol |
| Promoter | Xue et al. (2018) [248] | Pd-Nb/SBA-15 | 2,5-dimethyltetrahydrofuran |
| Promoter | Jin et al. (2017) [247] | Nb-Ni oxide catalyst | Anisole |
| Promoter | Guan et al. (2019) [251] | Pt/CN with HNbWO6, HNbMoO6, HTaWO6 nanosheets | Diphenyl ether |
| Promoter | Infantes-Molina et al. (2016) [250] | Pd-Nb/SiO2 and Pd-Nb/PPH | Dibenzofurane |
| Promoter | Shao et al. (2015) [249] | Pd/Nb2O5/SiO2 and Ni/Nb2O5/SiO2 | 4-(2-furyl)-3-buten-2-one; palmitic acid; tristearin and diphenyl eter |
| Promoter | Zhang et al. (2019) [252] | Ru/Nb2O5/micromesoporous carbon | Phenol |
| Promoter | Jeon et al. (2018) [253] | Ru-Nb/Al2O3 | Glycerol |
4.2.7. HDT of Biomass-Derived Feedstock with Nb-Containing Catalysts
4.2.8. Catalysts: Nickel-Based Catalysts
4.2.9. Catalysts: Supports
4.3. Catalysts: Deactivation Mechanisms of Hydrotreatment Catalysts
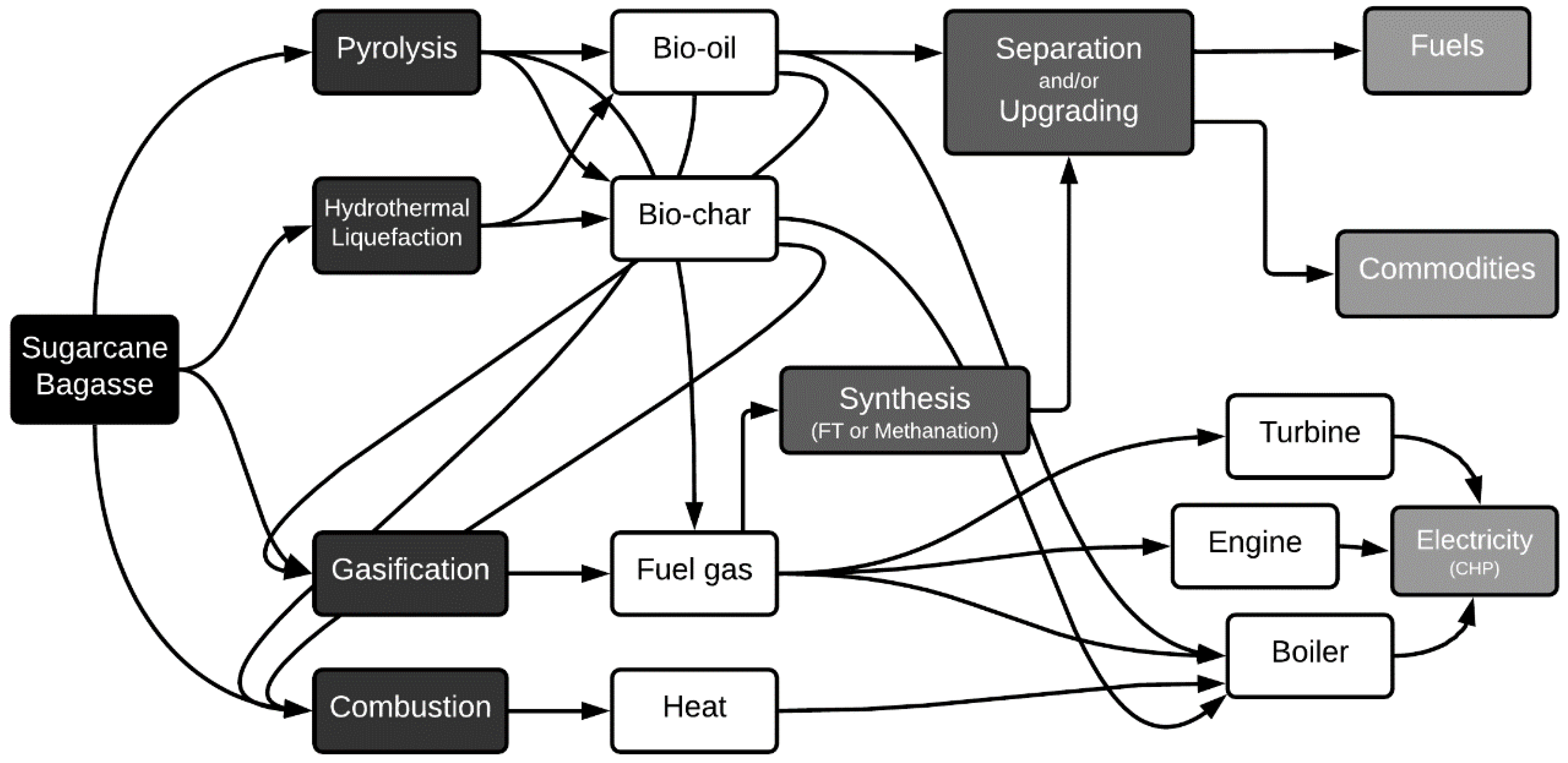
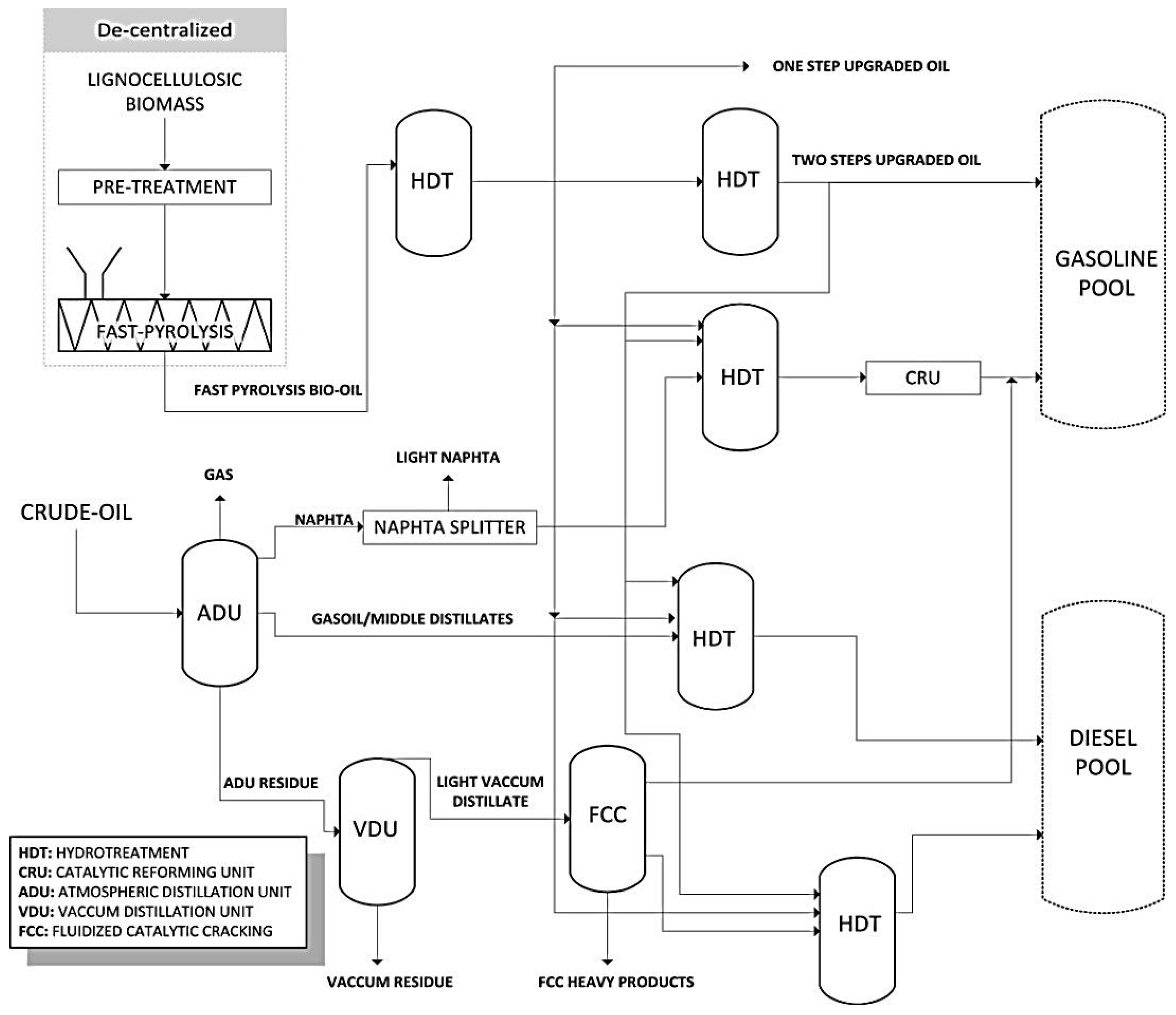
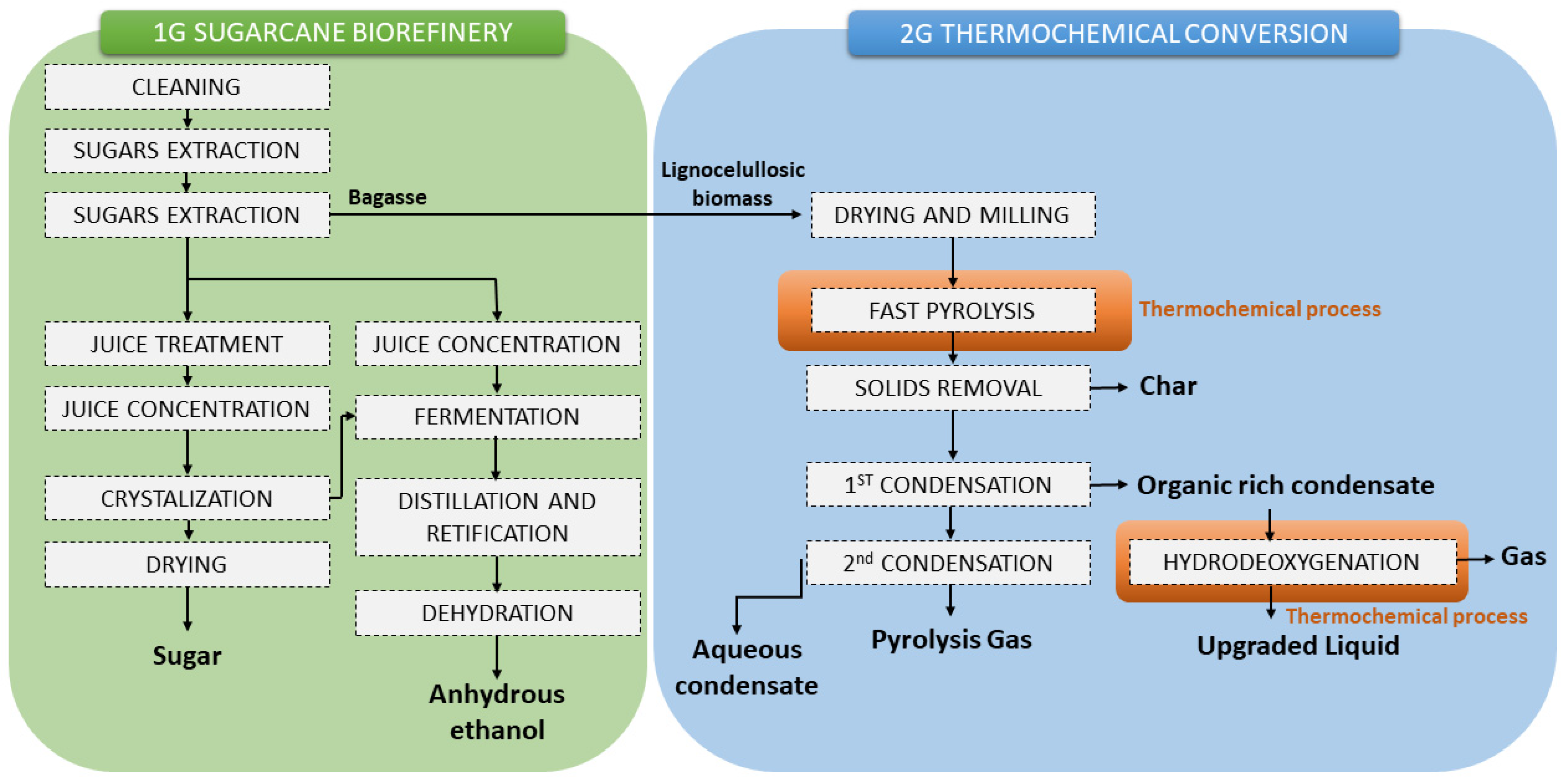
5. Biorefineries
Pyrolysis-Centered Biorefineries
6. Summary and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- BBC COVID Map: Coronavirus Cases, Deaths, Vaccinations by Country. Available online: https://www.bbc.com/news/world-51235105 (accessed on 3 December 2021).
- Desemprego vai Crescer no Mundo, Mas Subirá Ainda Mais no Brasil. Available online: https://www.cnnbrasil.com.br/business/desemprego-vai-crescer-no-mundo-mas-subira-ainda-mais-no-brasil/ (accessed on 9 November 2021).
- Dickon, P.; Rogers, M.; Samandari, H. Quarterly Reports: Addressing Climate Change Post-Coronavirus. Available online: https://www.mckinsey.com/business-functions/sustainability/our-insights/addressing-climate-change-in-a-post-pandemic-world# (accessed on 9 November 2021).
- Chiaramonti, D.; Maniatis, K. Security of supply, strategic storage and COVID19: Which lessons learnt for renewable and recycled carbon fuels, and their future role in decarbonizing transport? Appl. Energy 2020, 271, 115216. [Google Scholar] [CrossRef]
- Magalhães, A.I.; Carvalho, J.C.; Melo Pereira, G.V.; Karp, S.G.; Câmara, M.C.; Medina, J.D.C.; Soccol, C.R. Lignocellulosic biomass from agro-industrial residues in South America: Current developments and perspectives. Biofuels Bioprod. Biorefining 2019, 13, 1505–1519. [Google Scholar] [CrossRef]
- United Nations Green Economy Could Create 24 Million New Jobs. Available online: https://www.un.org/sustainabledevelopment/blog/2019/04/green-economy-could-create-24-million-new-jobs/ (accessed on 9 November 2021).
- IRENA; IEA Bioenergy. FAO Bioenergy for Sustainable Development. 2017. Available online: https://www.ieabioenergy.com/blog/publications/bioenergy-for-sustainable-development/ (accessed on 4 December 2021).
- Anwar, Z.; Gulfraz, M.; Irshad, M. Agro-industrial lignocellulosic biomass a key to unlock the future bio-energy: A brief review. J. Radiat. Res. Appl. Sci. 2014, 7, 163–173. [Google Scholar] [CrossRef]
- Mohan, D.; Pittman, C.U.; Steele, P.H. Pyrolysis of wood/biomass for bio-oil: A critical review. Energy Fuels 2006, 20, 848–889. [Google Scholar] [CrossRef]
- Xu, F.; Yu, J.; Tesso, T.; Dowell, F.; Wang, D. Qualitative and quantitative analysis of lignocellulosic biomass using infrared techniques: A mini-review. Appl. Energy 2013, 104, 801–809. [Google Scholar] [CrossRef] [Green Version]
- Oasmaa, A.; Fonts, I.; Pelaez-Samaniego, M.R.; Garcia-Perez, M.E.M.; Garcia-Perez, M.E.M. Pyrolysis Oil Multiphase Behavior and Phase Stability: A Review. Energy Fuels 2016, 30, 6179–6200. [Google Scholar] [CrossRef]
- Collard, F.X.; Blin, J. A review on pyrolysis of biomass constituents: Mechanisms and composition of the products obtained from the conversion of cellulose, hemicelluloses and lignin. Renew. Sustain. Energy Rev. 2014, 38, 594–608. [Google Scholar] [CrossRef]
- Funke, A.; Tomasi Morgano, M.; Leibold, H.; Dahmen, N.; Leibold, H. Experimental comparison of two bench scale units for fast and intermediate pyrolysis. J. Anal. Appl. Pyrolysis 2017, 124, 504–514. [Google Scholar] [CrossRef]
- James, A.K.; Thring, R.W.; Helle, S.; Ghuman, H.S. Ash management review-applications of biomass bottom ash. Energies 2012, 5, 3856–3873. [Google Scholar] [CrossRef]
- Hayes, D.J.M. Second-generation biofuels: Why they are taking so long. Wiley Interdiscip. Rev. Energy Environ. 2013, 2, 304–334. [Google Scholar] [CrossRef]
- Tripathi, M.; Sahu, J.N.; Ganesan, P. Effect of process parameters on production of biochar from biomass waste through pyrolysis: A review. Renew. Sustain. Energy Rev. 2016, 55, 467–481. [Google Scholar] [CrossRef]
- Alam, F.; Mobin, S.; Chowdhury, H. Third Generation Biofuel from Algae. Procedia Eng. 2015, 105, 763–768. [Google Scholar] [CrossRef]
- Goyal, H.B.; Seal, D.; Saxena, R.C. Bio-fuels from thermochemical conversion of renewable resources: A review. Renew. Sustain. Energy Rev. 2008, 12, 504–517. [Google Scholar] [CrossRef]
- Welfle, A. Balancing growing global bioenergy resource demands—Brazil’s biomass potential and the availability of resource for trade. Biomass Bioenergy 2017, 105, 83–95. [Google Scholar] [CrossRef]
- Bonassa, G.; Schneider, L.T.; Canever, V.B.; Cremonez, P.A.; Frigo, E.P.; Dieter, J.; Teleken, J.G. Scenarios and prospects of solid biofuel use in Brazil. Renew. Sustain. Energy Rev. 2018, 82, 2365–2378. [Google Scholar] [CrossRef]
- IBGE. Levantamento Sistemático da Produção Agrícola. 2020. Available online: https://www.ibge.gov.br/estatisticas/economicas/agricultura-e-pecuaria/9201-levantamento-sistematico-da-producao-agricola.html (accessed on 4 December 2021).
- Brainer, M.S.C.P. Produção de Coco: O Nordeste é Destaque Nacional. 2018. Available online: https://www.bnb.gov.br/documents/80223/4296541/61_coco.pdf/c172dd8f-3044-f1db-5d0c-a94c5eb735e0 (accessed on 4 December 2021).
- Kumari, S.; Das, D. Biohythane production from sugarcane bagasse and water hyacinth: A way towards promising green energy production. J. Clean. Prod. 2019, 207, 689–701. [Google Scholar] [CrossRef]
- Philippini, R.R.; Martiniano, S.E.; Chandel, A.K.; de Carvalho, W.; da Silva, S.S. Pretreatment of Sugarcane Bagasse from Cane Hybrids: Effects on Chemical Composition and 2G Sugars Recovery. Waste Biomass Valorization 2019, 10, 1561–1570. [Google Scholar] [CrossRef]
- Dai, L.; Wang, Y.; Liu, Y.; Ruan, R.; Duan, D.; Zhao, Y.; Yu, Z.; Jiang, L. Catalytic fast pyrolysis of torrefied corn cob to aromatic hydrocarbons over Ni-modified hierarchical ZSM-5 catalyst. Bioresour. Technol. 2019, 272, 407–414. [Google Scholar] [CrossRef]
- Stachowiak-Wencek, A.; Zborowska, M.; Waliszewska, H.; Waliszewska, B. Chemical changes in Lignocellulosic Biomass (Corncob) influenced by pretreatment and Anaerobic Digestion (AD). BioResources 2019, 14, 8082–8099. [Google Scholar]
- Shao, X.; Wang, J.; Liu, Z.; Hu, N.; Liu, M.; Xu, Y. Preparation and Characterization of Porous Microcrystalline Cellulose from Corncob. Ind. Crops Prod. 2020, 151, 1–6. [Google Scholar] [CrossRef]
- Cutrim, F.M.; Ramos, E.C.S.S.; Abreu, M.C.C.; Godinho, A.S.; Maciel, A.P.; Mendonça, C.J.S.; Cavalcante, K.S.B. A study of chemical composition and enzymatic hydrolysis of solid organic waste from agrosilvopastoral systems. J. Braz. Chem. Soc. 2019, 30, 1955–1963. [Google Scholar] [CrossRef]
- de Paranhos, A.G.O.; Adarme, O.F.H.; Barreto, G.F.; de Silva, S.Q.; de Aquino, S.F. Methane production by co-digestion of poultry manure and lignocellulosic biomass: Kinetic and energy assessment. Bioresour. Technol. 2020, 300, 122588. [Google Scholar] [CrossRef] [PubMed]
- Pocan, P.; Bahcegul, E.; Oztop, M.H.; Hamamci, H. Enzymatic Hydrolysis of Fruit Peels and Other Lignocellulosic Biomass as a Source of Sugar. Waste Biomass Valorization 2018, 9, 929–937. [Google Scholar] [CrossRef]
- Borges, T.E.; Almeida, J.H.S.; Amico, S.C.; Amado, F.D.R. Hollow glass microspheres/piassava fiber-reinforced homo- and co-polypropylene composites: Preparation and properties. Polym. Bull. 2017, 74, 1979–1993. [Google Scholar] [CrossRef]
- d’Almeida, J.R.M.; Aquino, R.C.M.P.; Monteiro, S.N. Tensile mechanical properties, morphological aspects and chemical characterization of piassava (Attalea funifera) fibers. Compos. Part A Appl. Sci. Manuf. 2006, 37, 1473–1479. [Google Scholar] [CrossRef]
- Daud, Z.; Sari, A.; Kassim, M.; Aripin, A.M.; Awang, H.; Hatta, Z.M.; Education, V.; Tun, U.; Onn, H.; Pahat, B. Chemical Composition and Morphological of Cocoa Pod Husks and Cassava Peels for Pulp and Paper Production. Aust. J. Basic Appl. Sci. 2013, 7, 406–411. [Google Scholar]
- Aripin, A.M. Cassava Peels for Alternative Fibre in Pulp and Paper Industry: Chemical Properties and Morphology Characterization. Int. J. Integr. Eng. 2013, 5, 30–33. [Google Scholar]
- Abiaziem, C.V.; Williams, A.B.; Inegbenebor, A.I.; Onwordi, C.T.; Petrik, L.F.; Land, C.; State, O.; Federal, T.; Ilaro, P.; State, O.; et al. Preparation, Characterisation and Physicochemical Properties of Cellulose Nanocrystals from Cassava Peel. In Proceedings of the 14th International Conference on Materials Chemistry (MC14), Birmingham, UK, 8–11 July 2019. [Google Scholar]
- Cheng, J.; Zhang, J.; Lin, R.; Liu, J.; Zhang, L.; Cen, K. Ionic-liquid pretreatment of cassava residues for the cogeneration of fermentative hydrogen and methane. Bioresour. Technol. 2017, 228, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Huang, T.; Geng, W.; Yang, L. Comparison of sodium carbonate pretreatment for enzymatic hydrolysis of wheat straw stem and leaf to produce fermentable sugars. Bioresour. Technol. 2013, 137, 294–301. [Google Scholar] [CrossRef]
- Mancini, G.; Papirio, S.; Lens, P.N.L.; Esposito, G. Increased biogas production from wheat straw by chemical pretreatments. Renew. Energy 2018, 119, 608–614. [Google Scholar] [CrossRef]
- Lu, X.; Li, C.; Zhang, S.; Wang, X.; Zhang, W.; Wang, S.; Xia, T. Enzymatic sugar production from elephant grass and reed straw through pretreatments and hydrolysis with addition of thioredoxin-His-S. Biotechnol. Biofuels 2019, 12, 1–11. [Google Scholar] [CrossRef]
- Espinosa, E.; Sánchez, R.; Otero, R.; Domínguez-Robles, J.; Rodríguez, A. A comparative study of the suitability of different cereal straws for lignocellulose nanofibers isolation. Int. J. Biol. Macromol. 2017, 103, 990–999. [Google Scholar] [CrossRef]
- Gao, Y.; Guo, X.; Liu, Y.; Fang, Z.; Zhang, M.; Zhang, R.; You, L.; Li, T.; Liu, R.H. A full utilization of rice husk to evaluate phytochemical bioactivities and prepare cellulose nanocrystals. Sci. Rep. 2018, 8, 1–8. [Google Scholar] [CrossRef]
- Sajith, S.; Arumugam, V.; Dhakal, H.N. Comparison on mechanical properties of lignocellulosic flour epoxy composites prepared by using coconut shell, rice husk and teakwood as fillers. Polym. Test. 2017, 58, 60–69. [Google Scholar] [CrossRef] [Green Version]
- Rambo, M.K.D.; Schmidt, F.L.; Ferreira, M.M.C. Analysis of the lignocellulosic components of biomass residues for biorefinery opportunities. Talanta 2015, 144, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Gomes, H.A.R.; da Silva, A.J.; Gómez-Mendoza, D.P.; dos Santos Júnior, A.C.M.; de di Cologna, N.M.; Almeida, R.M.; Miller, R.N.G.; Fontes, W.; de Sousa, M.V.; Ricart, C.A.O.; et al. Identification of multienzymatic complexes in the Clonostachys byssicola secretomes produced in response to different lignocellulosic carbon sources. J. Biotechnol. 2017, 254, 51–58. [Google Scholar] [CrossRef]
- Orozco, R.S.; Hernández, P.B.; Morales, G.R.; Núñez, F.U.; Villafuerte, J.O.; Lugo, V.L.; Ramírez, N.F.; Díaz, C.E.B.; Vázquez, P.C. Characterization of lignocellulosic fruit waste as an alternative feedstock for bioethanol production. BioResources 2014, 9, 1873–1885. [Google Scholar]
- Arisht, S.N.; Abdul, P.M.; Liu, C.M.; Lin, S.K.; Maaroff, R.M.; Wu, S.Y.; Jahim, J.M. Biotoxicity assessment and lignocellulosic structural changes of phosphoric acid pre-treated young coconut husk hydrolysate for biohydrogen production. Int. J. Hydrogen Energy 2019, 44, 5830–5843. [Google Scholar] [CrossRef]
- Teixeira, J.N.; Silva, D.W.; Vilela, A.P.; Savastano Junior, H.; de Siqueira Brandão Vaz, L.E.V.; Mendes, R.F. Lignocellulosic Materials for Fiber Cement Production. Waste Biomass Valorization 2020, 11, 2193–2200. [Google Scholar] [CrossRef]
- Toscan, A.; Fontana, R.C.; Andreaus, J.; Camassola, M.; Lukasik, R.M.; Dillon, A.J.P. New two-stage pretreatment for the fractionation of lignocellulosic components using hydrothermal pretreatment followed by imidazole delignification: Focus on the polysaccharide valorization. Bioresour. Technol. 2019, 285, 121346. [Google Scholar] [CrossRef] [PubMed]
- da Silva, S.B.; Arantes, M.D.C.; de Andrade, J.K.B.; Andrade, C.R.; de Carneiro, A.C.O.; de Protásio, T.P. Influence of physical and chemical compositions on the properties and energy use of lignocellulosic biomass pellets in Brazil. Renew. Energy 2020, 147, 1870–1879. [Google Scholar] [CrossRef]
- Vintila, T.; Negrea, A.; Barbu, H.; Sumalan, R.; Kovacs, K. Metal distribution in the process of lignocellulosic ethanol production from heavy metal contaminated sorghum biomass. J. Chem. Technol. Biotechnol. 2016, 91, 1607–1614. [Google Scholar] [CrossRef]
- Pengilly, C.; García-Aparicio, M.; Swart, J.P.J.; Görgens, J.F. Micro-assay method for enzymatic saccharification of industrially relevant lignocellulose substrates. Biomass Convers. Biorefinery 2020. Available online: https://link.springer.com/article/10.1007/s13399-020-00700-6 (accessed on 4 December 2020). [CrossRef]
- Chen, B.; Luo, Z.; Chen, H.; Chen, C.; Cai, D.; Qin, P.; Cao, H.; Tan, T. Wood Plastic Composites from the Waste Lignocellulosic Biomass Fibers of Bio-Fuels Processes: A Comparative Study on Mechanical Properties and Weathering Effects. Waste Biomass Valorization 2020, 11, 1701–1710. [Google Scholar] [CrossRef]
- Guerfali, M.; Ayadi, I.; Belhassen, A.; Gargouri, A.; Belghith, H. Single cell oil production by Trichosporon cutaneum and lignocellulosic residues bioconversion for biodiesel synthesis. Process Saf. Environ. Prot. 2018, 113, 292–304. [Google Scholar] [CrossRef]
- Cai, J.; He, Y.; Yu, X.; Banks, S.W.; Yang, Y.; Zhang, X.; Yu, Y.; Liu, R.; Bridgwater, A.V. Review of physicochemical properties and analytical characterization of lignocellulosic biomass. Renew. Sustain. Energy Rev. 2017, 76, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Companhia Nacional de Abastecimento—CONAB. Acompanhamento de Safra Brasileira de Cana-de-Açúcar; CONAB: Brasília, Brazil, 2020; Volume 7. Available online: https://www.conab.gov.br/info-agro/safras/cana (accessed on 3 November 2020).
- Santucci, B.S. Estudo dos Efeitos dos Tratamentos Físico- Mecânicos Na Hidrólise Da Celulose do Bagaço de Cana-de-Açúcar; Universidade de São Paulo: São Paulo, Brasil, 2018. [Google Scholar]
- Santos, F.; Diola, V. Physiology. In Sugarcane: Agricultural Production, Bioenergy and Ethanol; Fernando, S., Aluízio Borém, C.C., Eds.; Academic Press: Cambridge, MA, USA, 2015; pp. 13–33. ISBN 9780128022399. [Google Scholar]
- Cortez, L.A.B.; Baldassin, R.; De Almeida, E. Energy from sugarcane. In Sugarcane Biorefinery, Technology and Perspectives; Fernando, S., Sarita, C.R., De Matos Mario, P.E., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 117–139. ISBN 9780128142363. [Google Scholar]
- Santos, F.; Eichler, P.; Machado, G.; De Mattia, J.; De Souza, G. By-products of the sugarcane industry. In Sugarcane Biorefinery, Technology and Perspectives; Santos, F., Rabelo, S.C., De Matos, M., Eichler, P., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 21–48. ISBN 9780128142363. [Google Scholar]
- Assad, L. Aproveitamento de resíduos do setor sucroalcooleiro desafia empresas e pesquisadores. Cienc. Cult. 2017, 69, 13–16. [Google Scholar] [CrossRef] [Green Version]
- Hojo, L.Y.C.P.; Ajala, E.; Neto, A.G.; Martins, C.H.; de Angelis Neto, C.G.; Regina, G.T. Diagóstico do gerenciamento dos resíduos em uma indústria sucroalcooleira. In Proceedings of the Anais do III Simpósio de Pós-Graduação em Engenharia Urbana (SIMPGEU), Maringá, Brazil, 7–8 November 2012; pp. 1–9. [Google Scholar]
- Antonio Bizzo, W.; Lenço, P.C.; Carvalho, D.J.; Veiga, J.P.S. The generation of residual biomass during the production of bio-ethanol from sugarcane, its characterization and its use in energy production. Renew. Sustain. Energy Rev. 2014, 29, 589–603. [Google Scholar] [CrossRef]
- Díaz Pérez, Á.A.; Escobar Palacio, J.C.; Venturini, O.J.; Martínez Reyes, A.M.; Rúa Orozco, D.J.; Silva Lora, E.E.; del Olmo Almazán, O.A. Thermodynamic and economic evaluation of reheat and regeneration alternatives in cogeneration systems of the Brazilian sugarcane and alcohol sector. Energy 2018, 152, 247–262. [Google Scholar] [CrossRef]
- Alves, M. Estudo de Sistemas de Cogeração em Usinas de Açúcar e Álcool, Com Utilização do Bagaço e Palha da Cana; Universidade Estadual de Campinas: Campinas, Brazil, 2011. [Google Scholar]
- Smithers, J. Review of sugarcane trash recovery systems for energy cogeneration in South Africa. Renew. Sustain. Energy Rev. 2014, 32, 915–925. [Google Scholar] [CrossRef]
- Mutran, V.M.; Ribeiro, C.O.; Nascimento, C.A.O.; Chachuat, B. Risk-conscious optimization model to support bioenergy investments in the Brazilian sugarcane industry. Appl. Energy 2020, 258, 113978. [Google Scholar] [CrossRef]
- Kanwal, S.; Chaudhry, N.; Munir, S.; Sana, H. Effect of torrefaction conditions on the physicochemical characterization of agricultural waste (sugarcane bagasse). Waste Manag. 2019, 88, 280–290. [Google Scholar] [CrossRef]
- Gong, S.H.; Im, H.S.; Um, M.; Lee, H.W.; Lee, J.W. Enhancement of waste biomass fuel properties by sequential leaching and wet torrefaction. Fuel 2019, 239, 693–700. [Google Scholar] [CrossRef]
- Cahyanti, M.N.; Doddapaneni, T.R.K.C.; Kikas, T. Biomass torrefaction: An overview on process parameters, economic and environmental aspects and recent advancements. Bioresour. Technol. 2020, 301, 122737. [Google Scholar] [CrossRef] [PubMed]
- Redação Jornal da Cana Das 20 Maiores Usinas de Cana, só 9 Têm Lucro Positive. Available online: https://jornalcana.com.br/das-20-maiores-usinas-de-cana-so-9-tem-lucro-positivo/ (accessed on 19 May 2021).
- Relatório de Sustentabilidade-Comunicação de Progresso Safra 2019/2020, Maringá, PA, Brazil. 2020. Available online: https://www.usacucar.com.br/conteudos.php?local=index&id=32 (accessed on 4 December 2021).
- Copersucar Relatório de Sustentabilidade 2016–2018. Available online: http://relatorios.copersucar.com.br/2018/inicio1.html (accessed on 19 May 2021).
- Relatório de Sustentabilidade 2018, São Manoel, SP, Brazil. 2019. Available online: https://www.saomanoel.com.br (accessed on 9 June 2021).
- Usina Batatais Processo Produtivo–Reutilização de Recursos. Available online: http://www.usinabatatais.com.br/programas-socioambientais-e-culturais/processo-produtivo-reutilizacao-de-recursos.html (accessed on 17 May 2021).
- Plano de Gestão de Impacto Ambiental Grupo Balbo. Available online: https://www.canaverde.com.br/wp-content/uploads/2020/09/Usina-Uberaba-EIMP-Resumo-2020-1.pdf (accessed on 9 June 2021).
- Relatório de Sustentabilidade 2019 Adecoagro. Available online: https://sustainability.adecoagro.com/storage/documents/2019-br.pdf (accessed on 9 June 2021).
- Usina São João Technical Report. Available online: https://site.usj.com.br/site (accessed on 9 June 2021).
- Terán Hilares, R.; Swerts, M.P.; Ahmed, M.A.; Ramos, L.; da Silva, S.S.; Santos, J.C. Organosolv Pretreatment of Sugar Cane Bagasse for Bioethanol Production. Ind. Eng. Chem. Res. 2017, 56, 3833–3838. [Google Scholar] [CrossRef]
- Mesa, L.; González, E.; Cara, C.; González, M.; Castro, E.; Mussatto, S.I. The effect of organosolv pretreatment variables on enzymatic hydrolysis of sugarcane bagasse. Chem. Eng. J. 2011, 168, 1157–1162. [Google Scholar] [CrossRef] [Green Version]
- da Gomes, A.C.; Rodrigues, M.I.; de França Passos, D.; Machado de Castro, A.; Maria Mello Santa Anna, L.; Pereira, N. Acetone–butanol–ethanol fermentation from sugarcane bagasse hydrolysates: Utilization of C5 and C6 sugars. Electron. J. Biotechnol. 2019, 42, 16–22. [Google Scholar] [CrossRef]
- de Guilherme, A.A.; Dantas, P.V.F.; de Padilha, C.E.A.; dos Santos, E.S.; de Macedo, G.R. Ethanol production from sugarcane bagasse: Use of different fermentation strategies to enhance an environmental-friendly process. J. Environ. Manage. 2019, 234, 44–51. [Google Scholar] [CrossRef]
- Salomão, G.S.B.; Agnezi, J.C.; Paulino, L.B.; Hencker, L.B.; de Lira, T.S.; Tardioli, P.W.; Pinotti, L.M. Production of cellulases by solid state fermentation using natural and pretreated sugarcane bagasse with different fungi. Biocatal. Agric. Biotechnol. 2019, 17, 1–6. [Google Scholar] [CrossRef]
- Berenguer, R.A.; Capraro, A.P.B.; de Medeiros, M.H.F.; Carneiro, A.M.P.; De Oliveira, R.A. Sugar cane bagasse ash as a partial substitute of Portland cement: Effect on mechanical properties and emission of carbon dioxide. J. Environ. Chem. Eng. 2020, 8, 103655. [Google Scholar] [CrossRef]
- de Mello, L.C.A.; dos Anjos, M.A.S.; de Sá, M.V.V.A.; de Souza, S.L.N.; de Farias, E.C. Effect of high temperatures on self-compacting concrete with high levels of sugarcane bagasse ash and metakaolin. Constr. Build. Mater. 2020, 248, 118715. [Google Scholar] [CrossRef]
- Oliveira, J.A.; Cunha, F.A.; Ruotolo, L.A.M. Synthesis of zeolite from sugarcane bagasse fly ash and its application as a low-cost adsorbent to remove heavy metals. J. Clean. Prod. 2019, 229, 956–963. [Google Scholar] [CrossRef]
- Jacob, M.M.; Ponnuchamy, M.; Kapoor, A.; Sivaraman, P. Bagasse based biochar for the adsorptive removal of chlorpyrifos from contaminated water. J. Environ. Chem. Eng. 2020, 8, 103904. [Google Scholar] [CrossRef]
- Akinfalabi, S.-I.; Rashid, U.; Ngamcharussrivichai, C.; Nehdi, I.A. Synthesis of reusable biobased nano-catalyst from waste sugarcane bagasse for biodiesel production. Environ. Technol. Innov. 2020, 18, 100788. [Google Scholar] [CrossRef]
- Bridgwater, A. Renewable fuels and chemicals by thermal processing of biomass. Chem. Eng. J. 2003, 91, 87–102. [Google Scholar] [CrossRef]
- Dahmen, N.; Lewandowski, I.; Zibek, S.; Weidtmann, A. Integrated lignocellulosic value chains in a growing bioeconomy: Status quo and perspectives. GCB Bioenergy 2019, 11, 107–117. [Google Scholar] [CrossRef] [Green Version]
- Xiu, S.; Shahbazi, A. Bio-oil production and upgrading research: A review. Renew. Sustain. Energy Rev. 2012, 16, 4406–4414. [Google Scholar] [CrossRef]
- Kan, T.; Strezov, V.; Evans, T.J. Lignocellulosic biomass pyrolysis: A review of product properties and effects of pyrolysis parameters. Renew. Sustain. Energy Rev. 2016, 57, 126–1140. [Google Scholar] [CrossRef]
- Bridgwater, A.V.V. Review of fast pyrolysis of biomass and product upgrading. Biomass Bioenergy 2012, 38, 68–94. [Google Scholar] [CrossRef]
- Pfitzer, C.; Dahmen, N.; Tröger, N.; Weirich, F.; Sauer, J.; Günther, A.; Müller-Hagedorn, M. Fast Pyrolysis of Wheat Straw in the Bioliq Pilot Plant. Energy Fuels 2016, 30, 8047–8054. [Google Scholar] [CrossRef]
- Polin, P.J.; Carr, D.H.; Whitmer, L.E.; Smith, R.G.; Brown, R.C. Conventional and autothermal pyrolysis of corn stover: Overcoming the processing challenges of high-ash agricultural residues. J. Anal. Appl. Pyrolysis 2019, 143, 104679. [Google Scholar] [CrossRef]
- Basu, P. Biomass Gasification and Pyrolysis: Practical Design and Theory; Elsevier: Amsterdam, The Netherlands, 2010; ISBN 978-0-12-374988-8. [Google Scholar]
- Van de Velden, M.; Baeyens, J.; Brems, A.; Janssens, B.; Dewil, R. Fundamentals, kinetics and endothermicity of the biomass pyrolysis reaction. Renew. Energy 2010, 35, 232–242. [Google Scholar] [CrossRef]
- Guedes, R.E.; Luna, A.S.; Torres, A.R. Operating parameters for bio-oil production in biomass pyrolysis: A review. J. Anal. Appl. Pyrolysis 2018, 129, 134–149. [Google Scholar] [CrossRef]
- Fonseca, F.G.; Funke, A.; Niebel, A.; Soares Dias, A.P.; Dahmen, N. Moisture content as a design and operational parameter for fast pyrolysis. J. Anal. Appl. Pyrolysis 2019, 139, 73–86. [Google Scholar] [CrossRef]
- Demirbas, A.; Arin, G. An Overview of Biomass Pyrolysis. Energy Sources 2002, 24, 471–482. [Google Scholar] [CrossRef]
- IEA Bioenergy Pyrolysis Reactors. Available online: http://task34.ieabioenergy.com/pyrolysis-reactors/ (accessed on 4 December 2019).
- Park, J.; Meng, J.; Lim, K.H.; Rojas, O.J.; Park, S. Transformation of lignocellulosic biomass during torrefaction. J. Anal. Appl. Pyrolysis 2013, 100, 199–206. [Google Scholar] [CrossRef]
- Dahmen, N. Lecture: Energy from Biomass; Karlsruhe Institute of Technology: Karlsruhe, Germany, 2019. [Google Scholar]
- Oasmaa, A.; Solantausta, Y.; Arpiainen, V.; Kuoppala, E.; Sipilä, K. Fast pyrolysis bio-oils from wood and agricultural residues. Energy Fuels 2010, 24, 1380–1388. [Google Scholar] [CrossRef]
- Fahmi, R.; Bridgwater, A.V.; Donnison, I.; Yates, N.; Jones, J.M. The effect of lignin and inorganic species in biomass on pyrolysis oil yields, quality and stability. Fuel 2008, 87, 1230–1240. [Google Scholar] [CrossRef]
- Di Blasi, C. Modeling chemical and physical processes of wood and biomass pyrolysis. Prog. Energy Combust. Sci. 2008, 34, 47–90. [Google Scholar] [CrossRef]
- Yang, H.; Yan, R.; Chen, H.; Lee, D.H.; Zheng, C. Characteristics of hemicellulose, cellulose and lignin pyrolysis. Fuel 2007, 86, 1781–1788. [Google Scholar] [CrossRef]
- Carrier, M.; Windt, M.; Ziegler, B.; Appelt, J.; Saake, B.; Meier, D.; Bridgwater, A. Quantitative Insights into the Fast Pyrolysis of Extracted Cellulose, Hemicelluloses, and Lignin. ChemSusChem 2017, 10, 3212–3224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lédé, J. Cellulose pyrolysis kinetics: An historical review on the existence and role of intermediate active cellulose. J. Anal. Appl. Pyrolysis 2012, 94, 17–32. [Google Scholar] [CrossRef]
- Gollakota, A.R.K.; Reddy, M.; Subramanyam, M.D.; Kishore, N. A review on the upgradation techniques of pyrolysis oil. Renew. Sustain. Energy Rev. 2016, 58, 1543–1568. [Google Scholar] [CrossRef]
- Westerhof, R.J.M.M.; Nygård, H.S.; Van Swaaij, W.P.M.M.; Kersten, S.R.A.A.; Brilman, D.W.F.F. Effect of particle geometry and microstructure on fast pyrolysis of beech wood. Energy Fuels 2012, 26, 2274–2280. [Google Scholar] [CrossRef]
- Scott, D.S.; Piskorz, J. The flash pyrolysis of aspen-poplar wood. Can. J. Chem. Eng. 1982, 60, 666–674. [Google Scholar] [CrossRef]
- Wang, X.; Kersten, S.R.A.; Prins, W.; Van Swaaij, W.P.M. Biomass pyrolysis in a fluidized bed reactor. Part 2: Experimental validation of model results. Ind. Eng. Chem. Res. 2005, 44, 8786–8795. [Google Scholar] [CrossRef]
- Shen, J.; Wang, X.-S.; Garcia-Perez, M.; Mourant, D.; Rhodes, M.J.; Li, C.-Z. Effects of particle size on the fast pyrolysis of oil mallee woody biomass. Fuel 2009, 88, 1810–1817. [Google Scholar] [CrossRef]
- Salehi, E.; Abedi, J.; Harding, T. Bio-oil from Sawdust: Effect of Operating Parameters on the Yield and Quality of Pyrolysis Products. Energy Fuels 2011, 25, 4145–4154. [Google Scholar] [CrossRef]
- Demirbas, A. Effects of temperature and particle size on bio-char yield from pyrolysis of agricultural residues. J. Anal. Appl. Pyrolysis 2004, 72, 243–248. [Google Scholar] [CrossRef]
- Luangkiattikhun, P.; Tangsathitkulchai, C.; Tangsathitkulchai, M. Non-isothermal thermogravimetric analysis of oil-palm solid wastes. Bioresour. Technol. 2008, 99, 986–997. [Google Scholar] [CrossRef]
- Pütün, A.E.; Koçkar, Ö.M.; Yorgun, S.; Gerçel, H.F.; Andresen, J.; Snape, C.E.; Pütün, E. Fixed-bed pyrolysis and hydropyrolysis of sunflower bagasse: Product yields and compositions. Fuel Process. Technol. 1996, 46, 49–62. [Google Scholar] [CrossRef]
- Kersten, S.R.A.; Wang, X.; Prins, W.; van Swaaij, W.P.M. Biomass Pyrolysis in a Fluidized Bed Reactor. Part 1: Literature Review and Model Simulations. Ind. Eng. Chem. Res. 2005, 44, 8773–8785. [Google Scholar] [CrossRef]
- Di Blasi, C.; Branca, C. Temperatures of wood particles in a hot sand bed fluidized by nitrogen. Energy Fuels 2003, 17, 247–254. [Google Scholar] [CrossRef]
- Chaiwat, W.; Hasegawa, I.; Tani, T.; Sunagawa, K.; Mae, K. Analysis of cross-linking behavior during pyrolysis of cellulose for elucidating reaction pathway. Energy Fuels 2009, 23, 5765–5772. [Google Scholar] [CrossRef]
- Manyà, J.J.; Ruiz, J.; Arauzo, J. Some peculiarities of conventional pyrolysis of several agricultural residues in a packed bed reactor. Ind. Eng. Chem. Res. 2007, 46, 9061–9070. [Google Scholar] [CrossRef]
- Font, R.; Marcilla, A.; Devesa, J.; Verdú, E. Gaseous hydrocarbons from flash pyrolysis of almond shells. Ind. Eng. Chem. Res. 1988, 27, 1143–1149. [Google Scholar] [CrossRef]
- Haykiri-Acma, H. The role of particle size in the non-isothermal pyrolysis of hazelnut shell. J. Anal. Appl. Pyrolysis 2006, 75, 211–216. [Google Scholar] [CrossRef]
- Jayaweera, S.A.A.; Moss, J.H.; Thwaites, M.W. The effect of particle size on the combustion of weardale coal. Thermochim. Acta 1989, 152, 215–225. [Google Scholar] [CrossRef]
- Funazukuri, T.; Hudgins, R.R.; Silveston, P.L. Product distribution in pyrolysis of cellulose in a microfluidized bed. J. Anal. Appl. Pyrolysis 1986, 9, 139–158. [Google Scholar] [CrossRef]
- Westerhof, R.J.M.; Brilman, D.W.F.; van Swaaij, W.P.M.; Kersten, S.R.A. Effect of Temperature in Fluidized Bed Fast Pyrolysis of Biomass: Oil Quality Assessment in Test Units. Ind. Eng. Chem. Res. 2010, 49, 1160–1168. [Google Scholar] [CrossRef]
- Haas, T.J.; Nimlos, M.R.; Donohoe, B.S. Real-time and post-reaction microscopic structural analysis of biomass undergoing pyrolysis. Energy Fuels 2009, 23, 3810–3817. [Google Scholar] [CrossRef]
- Teixeira, A.R.; Mooney, K.G.; Kruger, J.S.; Williams, C.L.; Suszynski, W.J.; Schmidt, L.D.; Schmidt, D.P.; Dauenhauer, P.J. Aerosol generation by reactive boiling ejection of molten cellulose. Energy Environ. Sci. 2011, 4, 4306. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Garcia-Perez, M.; Pecha, B.; Kersten, S.R.A.; McDonald, A.G.; Westerhof, R.J.M. Effect of the fast pyrolysis temperature on the primary and secondary products of lignin. Energy Fuels 2013, 27, 5867–5877. [Google Scholar] [CrossRef]
- Bridgeman, T.G.; Darvell, L.I.; Jones, J.M.; Williams, P.T.; Fahmi, R.; Bridgwater, A.V.; Barraclough, T.; Shield, I.; Yates, N.; Thain, S.C.; et al. Influence of particle size on the analytical and chemical properties of two energy crops. Fuel 2007, 86, 60–72. [Google Scholar] [CrossRef]
- Krutof, A.; Hawboldt, K.A. Upgrading of biomass sourced pyrolysis oil review: Focus on co-pyrolysis and vapour upgrading during pyrolysis. Biomass Convers. Biorefinery 2018, 8, 775–787. [Google Scholar] [CrossRef]
- Hoekstra, E.; Westerhof, R.J.M.; Brilman, W.; Van Swaaij, W.P.M.; Kersten, S.R.A.; Hogendoorn, K.J.A.; Windt, M. Heterogeneous and homogeneous reactions of pyrolysis vapors from pine wood. AIChE J. 2012, 58, 2830–2842. [Google Scholar] [CrossRef]
- Kim, P.; Weaver, S.; Labbé, N. Effect of sweeping gas flow rates on temperature-controlled multistage condensation of pyrolysis vapors in an auger intermediate pyrolysis system. J. Anal. Appl. Pyrolysis 2016, 118, 325–334. [Google Scholar] [CrossRef]
- Papari, S.; Hawboldt, K. A review on condensing system for biomass pyrolysis process. Fuel Process. Technol. 2018, 180, 1–13. [Google Scholar] [CrossRef]
- Williams, P.T.; Brindle, A.J. Temperature selective condensation of tyre pyrolysis oils to maximise the recovery of single ring aromatic compounds. Fuel 2003, 82, 1023–1031. [Google Scholar] [CrossRef]
- Johansson, A.C.; Iisa, K.; Sandström, L.; Ben, H.; Pilath, H.; Deutch, S.; Wiinikka, H.; Öhrman, O.G.W. Fractional condensation of pyrolysis vapors produced from Nordic feedstocks in cyclone pyrolysis. J. Anal. Appl. Pyrolysis 2017, 123, 244–254. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, C.C.; Moreira, R.; Neves, R.C.; Richter, D.; Funke, A.; Raffelt, K.; Grunwaldt, J.-D.; Dahmen, N. From agriculture residue to upgraded product: The thermochemical conversion of sugarcane bagasse for fuel and chemical products. Fuel Process. Technol. 2020, 197, 106199. [Google Scholar] [CrossRef]
- Kornmayer, C. Verfahrenstechnik Untersuchungen zur Schnellpyrolyse von Lignocellulose im Doppenschnecken-Mischreaktor; Universität Fridericiana Karlsruhe: Karlsruhe, Germany, 2009. [Google Scholar]
- Bridgwater, A. Fast pyrolysis processes for biomass. Renew. Sustain. Energy Rev. 2000, 4, 1–73. [Google Scholar] [CrossRef]
- Lappas, A.A.; Samolada, M.C.; Iatridis, D.K.; Voutetakis, S.S.; Vasalos, I.A. Biomass pyrolysis in a circulating fluid bed reactor for the production of fuels and chemicals. Fuel 2002, 81, 2087–2095. [Google Scholar] [CrossRef]
- Lakshmanan, C.M.; Gal-Or, B.; Hoelscher, H.E. Production of Levoglucosan by Pyrolysis of Carbohydrates Pyrolysis. Ind. Eng. Chem. Prod. Res. Dev. 1969, 8, 261–264. [Google Scholar]
- Campuzano, F.; Brown, R.C.; Martínez, J.D. Auger reactors for pyrolysis of biomass and wastes. Renew. Sustain. Energy Rev. 2019, 102, 372–409. [Google Scholar] [CrossRef]
- Henrich, E.; Dahmen, N.; Weirich, F.; Reimert, R.; Kornmayer, C. Fast pyrolysis of lignocellulosics in a twin screw mixer reactor. Fuel Process. Technol. 2016, 143, 151–161. [Google Scholar] [CrossRef]
- Badger, P.C.; Fransham, P. Use of mobile fast pyrolysis plants to densify biomass and reduce biomass handling costs—A preliminary assessment. Biomass Bioenergy 2006, 30, 321–325. [Google Scholar] [CrossRef]
- Schulzke, T.; Conrad, S.; Westermeyer, J. Fractionation of flash pyrolysis condensates by staged condensation. Biomass Bioenergy 2016, 95, 287–295. [Google Scholar] [CrossRef]
- Bech, N. In Situ Flash Pyrolysis of Straw; Technical University of Denmark: Copenhagen, Denmark, 2008. [Google Scholar]
- Glaser, B. Prehistorically modified soils of central Amazonia: A model for sustainable agriculture in the twenty-first century. Philos. Trans. R. Soc. B Biol. Sci. 2007, 362, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Anderson, P.S. Introduction to the Rotatable Covered Cavity (RoCC) Kiln for Medium-Size Production of Pyrolytic Biochar and Thermal Energy. Available online: https://woodgas.com/resources/ (accessed on 15 July 2020).
- Worldwide, W.H. WarmHeart Worldwide. Available online: https://warmheartworldwide.org/ (accessed on 4 December 2021).
- Juntos NFP. Juntos NFP. Available online: https://juntosnfp.org/ (accessed on 4 December 2021).
- Meier, D.; Van De Beld, B.; Bridgwater, A.V.; Elliott, D.C.; Oasmaa, A.; Preto, F. State-of-the-art of fast pyrolysis in IEA bioenergy member countries. Renew. Sustain. Energy Rev. 2013, 20, 619–641. [Google Scholar] [CrossRef]
- Bridgwater, A.V. The production of biofuels and renewable chemicals by fast pyrolysis of biomass. Int. J. Glob. Energy Issues 2007, 27, 160. [Google Scholar] [CrossRef]
- Venderbosch, R.; Prins, W. Fast pyrolysis technology development. Biofuels Bioprod. Biorefining 2010, 4, 178–208. [Google Scholar] [CrossRef]
- Garcia-Nunez, J.A.; Pelaez-Samaniego, M.R.; Garcia-Perez, M.E.; Fonts, I.; Abrego, J.; Westerhof, R.J.M.; Garcia-Perez, M. Historical Developments of Pyrolysis Reactors: A Review. Energy Fuels 2017, 31, 5751–5775. [Google Scholar] [CrossRef]
- Oasmaa, A.; Peacocke, C. Properties and Fuel Use of Biomass-Derived Fast Pyrolysis Liquids. A Guide; VTT Publications: Espoo, Finland, 2010; Volume 731, ISBN 9789513873844. [Google Scholar]
- Pirowiki Welcome to PyroWiki. Available online: http://pyrowiki.pyroknown.eu/index.php/Welcome_to_PyroWiki (accessed on 21 November 2021).
- Peacocke, G.V.C. Ablative Pyrolysis of Biomass; Aston University: Birmingham, UK, 1994; Volume 7. [Google Scholar]
- Austrian Federal Ministry of Education Science and Research Fixed bed lab-scale reactor. Available online: https://forschungsinfrastruktur.bmbwf.gv.at/en/fi/fixed-bed-lab-scale-reactor_3026 (accessed on 14 June 2021).
- Soares Dias, A.P.; Rego, F.; Fonseca, F.; Casquilho, M.; Rosa, F.; Rodrigues, A. Catalyzed pyrolysis of SRC poplar biomass. Alkaline carbonates and zeolites catalysts. Energy 2019, 183, 1114–1122. [Google Scholar] [CrossRef]
- Kaminsky, W.; Predel, M.; Sadiki, A. Feedstock recycling of polymers by pyrolysis in a fluidised bed. Polym. Degrad. Stab. 2004, 85, 1045–1050. [Google Scholar] [CrossRef]
- Knight, J.A.; Gorton, C.W.; Kovac, R.J. Oil production by entrained flow pyrolysis of biomass. Biomass 1984, 6, 69–76. [Google Scholar] [CrossRef]
- Funke, A.; Richter, D.; Niebel, A.; Dahmen, N.; Sauer, J. Fast Pyrolysis of Biomass Residues in a Twin-screw Mixing Reactor. J. Vis. Exp. 2016, 115, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Santana, K.V.R.; Apolônio, F.C.S.O.; Wisniewski, A., Jr. Valorization of cattle manure by thermoconversion process in a rotary kiln reactor to produce environmentally friendly products. BioEnergy Res. 2020, 13, 605–617. [Google Scholar] [CrossRef]
- Berruti, F.F.; Briens, C.; Ferrante, L. A Mobile Pyrolyzer for Converting Agricultural and Forestry Residues into Liquid Bio-Oil and Bio-Char. In Proceedings of the CO2 Summit: Technology and Opportunity, Vail, CO, USA, 6–10 June 2010; Zhu, F., Ed.; UOP: Des Plaines, IL, USA, 2010. [Google Scholar]
- Li, J.; Li, N.; Qiao, Y.; Zong, P.; Wang, C.; Tian, Y.; Qin, S. Biomass Pyrolysis Liquefaction Technique: State of Research and Development Trends. IOP Conf. Ser. Earth Environ. Sci. 2020, 558, 022016. [Google Scholar] [CrossRef]
- Kaminsky, W. The Hamburg Fluidized-bed Pyrolysis Process to Recycle Polymer Wastes and Tires. In Feedstock Recycling and Pyrolysis of Waste Plastics; John Wiley & Sons Ltd.: Chichester, UK, 2006; pp. 475–491. [Google Scholar]
- Gust, S.; McLellan, R.J.; Meier, D.; Oasmaa, A.; Ormrod, D.; Peacocke, G.V.C. Determination of Norms and Standards for Biomass Fast Pyrolysis Liquids as an Alternative Renewable Fuel for Electricity and Heat Production; CPL Press: Cambridge, UK, 2005. [Google Scholar]
- Conner, G.T.; Tyrrell-Baxter, F.J. Apparatus and Method for Processing Biomass (AU2012341144B2). IP Australia. 2017. Available online: https://patents.google.com/patent/AU2012341144B2/en (accessed on 4 December 2021).
- Vancouver Sun DynaMotive’s Words Speak Louder than Its Actions. Available online: http://www.canada.com/story_print.html?id=83be7aa4-c0cb-4ee0-8a2f-9662d6b8e9bb (accessed on 4 December 2021).
- Eric, S.; Gary, W.; Robert, C.B.; Ryan, P.; Desmond, R.; Warren, J. Integrated Pyrolysis Combined Cycle Biomass Power System Concept Definition; Alliant Energy: Madison, WI, USA, 2003. [Google Scholar]
- Maniatis, K.; Baeyens, J.; Peeters, H.; Roggeman, G. The EGEMIN Flash Pyrolysis Process: Commissioning and Initial Results. In Advances in Thermochemical Biomass Conversion; Springer: Dordrecht, The Netherlands, 1993; pp. 1257–1264. [Google Scholar]
- Meier, D.; Eusterbrock, C.; Gannon, B. Ablative fast pyrolysis of biomass: A new demonstration project in California, USA. In ECI Symposium Series, Proceedings of the Pyroliq 2019: Pyrolysis and Liquefaction of Biomass and Wastes, Cork, Ireland, 16–20 June 2019; Berruti, F., Dufour, A., Prins, W., Garcia-Pérez, M., Eds.; ECI Digital Archives: New York, NY, USA, 2019; Available online: https://dc.engconfintl.org/pyroliq_2019/32/ (accessed on 4 December 2021).
- Honeywell UOP RTP Biomass Conversion-Renewable Fuels. Available online: https://uop.honeywell.com/en/industry-solutions/renewable-fuels/rtp-biomass-conversion (accessed on 19 May 2021).
- Ensyn Corporation Ensyn, Arbec and Rémabec Begin Construction of the Cote Nord Biocrude Production Facility in Quebec; Ensyn News Release: Port-Cartier, QC, Canada, 2016; Available online: http://www.ensyn.com/uploads/6/9/7/8/69787119/cn_release_2016.7.13_as_issued.pdf (accessed on 4 December 2021).
- Ensyn Corporation Aracruz, Espirito Santo, Brazil-Initial Biocrude Production Facility under JV with Suzano. Available online: http://www.ensyn.com/brazil.html (accessed on 4 December 2019).
- Autio, J.; Lehto, J.; Oasmaa, A.; Solantausta, Y.; Jokela, P.; Alin, J.; Power, M. A Pyrolysis Pilot Unit Integrated to a Circulating Fluidized Bed Boiler—Experiences From a Pilot Project. In ECI Symposium Series, Proceedings of the 10th International Conference on Circulating Fluidized Beds and Fluidization Technology-CFB-10, Sun River, OR, USA, 1–5 May 2011; Knowlton, T., Ed.; ECI Digital Archives: New York, NY, USA, 2013; Available online: https://dc.engconfintl.org/cfb10/19/ (accessed on 4 December 2021).
- Independent Commodity Intelligence Services Lurgi to Start up Pyrolysis Plant. Available online: https://www.icis.com/explore/resources/news/1999/05/17/80767/lurgi-to-start-up-pyrolysis-plant/ (accessed on 4 December 2021).
- Diebold, J.P.; Scahill, J.W. Improved Vortex Reactor System 1995. Available online: https://www.osti.gov/biblio/46290-improved-vortex-reactor-system (accessed on 4 December 2021).
- Diebold, J.; Power, A. Engineering Aspects of the Vortex Pyrolysis Reactor to Produce Primary Pyrolysis Oil Vapors for Use in Resins and Adhesives. In Research in Thermochemical Biomass Conversion; Springer: Dordrecht, The Netherlands, 1988; pp. 609–628. [Google Scholar]
- Yang, J.; Blanchette, D.; de Caumia, B.; Roy, C. Modelling, Scale-Up and Demonstration of a Vacuum Pyrolysis Reactor. In Progress in Thermochemical Biomass Conversion; Blackwell Science Ltd.: Oxford, UK, 2007; pp. 1296–1311. [Google Scholar]
- Strezov, V.; Evans, T.J. Biomass Processing Technologies; CRC Press: Boca Raton, FL, USA, 2014; ISBN 9781482282603. [Google Scholar]
- Badger, P. Demonstrating Bio-Oil Technology for Poultry Litter Management; ROI Alabama Operations LLC: Florence, AL, USA, 2006; Available online: http://fppcinc.org/Assets/FPPC/PDF/reports_renewableoilinternational.pdf (accessed on 4 December 2021).
- Neumann, J.; Jäger, N.; Apfelbacher, A.; Daschner, R.; Binder, S.; Hornung, A. Upgraded biofuel from residue biomass by Thermo-Catalytic Reforming and hydrodeoxygenation. Biomass Bioenergy 2016, 89, 91–97. [Google Scholar] [CrossRef]
- Williams, P.T. Fuels, Chemicals and Materials from Waste. In Energy, Waste & Resources–Three Sides of the Same Coin; The Royal Society of Chemistry: Environmental Chemistry Group: London, UK, 2012. [Google Scholar]
- BTG Bioliquids. BTG-BTL Empyro Project. Available online: https://www.btg-btl.com/en/company/projects/empyro (accessed on 4 December 2019).
- BTG-BTL Malaysia Plant. Available online: https://www.btg-btl.com/en/company/projects/malaysia (accessed on 4 December 2019).
- Greiner, L. Thermochemical Process Converts Poultry Litter into Bio-Oil to Provide Safer and More Environmental Solution to Waste Disposal. Available online: https://www.eurekalert.org/pub_releases/2007-08/vt-tpc081307.php (accessed on 4 December 2021).
- Elliott, D.C. Biofuel from fast pyrolysis and catalytic hydrodeoxygenation. Curr. Opin. Chem. Eng. 2015, 9, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Dabros, T.M.H.; Stummann, M.Z.; Høj, M.; Jensen, P.A.; Grunwaldt, J.-D.; Gabrielsen, J.; Mortensen, P.M.; Jensen, A.D. Transportation fuels from biomass fast pyrolysis, catalytic hydrodeoxygenation, and catalytic fast hydropyrolysis. Prog. Energy Combust. Sci. 2018, 68, 268–309. [Google Scholar] [CrossRef]
- Albrecht, K.O.; Olarte, M.V.; Wang, H. Upgrading Fast Pyrolysis Liquids. In Thermochemical Processing of Biomass; Brown, R.C., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2019; pp. 207–255. [Google Scholar]
- Si, Z.; Zhang, X.; Wang, C.; Ma, L.; Dong, R. An Overview on Catalytic Hydrodeoxygenation of Pyrolysis Oil and Its Model Compounds. Catalysts 2017, 7, 169. [Google Scholar] [CrossRef] [Green Version]
- Venderbosch, R.H.; Ardiyanti, A.R.; Wildschut, J.; Oasmaa, A.; Heeres, H.J. Stabilization of biomass-derived pyrolysis oils. J. Chem. Technol. Biotechnol. 2010, 85, 674–686. [Google Scholar] [CrossRef]
- Negahdar, L.; Gonzalez-Quiroga, A.; Otyuskaya, D.; Toraman, H.E.; Liu, L.; Jastrzebski, J.T.B.H.; Van Geem, K.M.; Marin, G.B.; Thybaut, J.W.; Weckhuysen, B.M. Characterization and Comparison of Fast Pyrolysis Bio-oils from Pinewood, Rapeseed Cake, and Wheat Straw Using 13C NMR and Comprehensive GC × GC. ACS Sustain. Chem. Eng. 2016, 4, 4974–4985. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Male, J.; Wang, Y. Recent advances in hydrotreating of pyrolysis bio-oil and its oxygen-containing model compounds. ACS Catal. 2013, 3, 1047–1070. [Google Scholar] [CrossRef]
- Saidi, M.; Samimi, F.; Karimipourfard, D.; Nimmanwudipong, T.; Gates, B.C.; Rahimpour, M.R. Upgrading of lignin-derived bio-oils by catalytic hydrodeoxygenation. Energy Environ. Sci. 2014, 7, 103–129. [Google Scholar] [CrossRef]
- Elliott, D.C.; Wang, H.; French, R.; Deutch, S.; Iisa, K. Hydrocarbon liquid production from biomass via hot-vapor-filtered fast pyrolysis and catalytic hydroprocessing of the bio-oil. Energy Fuels 2014, 28, 5909–5917. [Google Scholar] [CrossRef]
- Baldwin, R.M.; Feik, C.J. Bio-oil stabilization and upgrading by hot gas filtration. Energy Fuels 2013, 27, 3224–3238. [Google Scholar] [CrossRef]
- Zhang, Q.; Xu, Y.; Li, Y.; Wang, T.; Zhang, Q.; Ma, L.; He, M.; Li, K. Investigation on the esterification by using supercritical ethanol for bio-oil upgrading. Appl. Energy 2015, 160, 633–640. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, C.; Zhai, Y.; Liu, Y.; Zhang, R.; Tang, X. Upgrading of bio-oil using supercritical 1-butanol over a ru/c heterogeneous catalyst: Role of the solvent. Energy Fuels 2014, 28, 4611–4621. [Google Scholar] [CrossRef]
- Tang, Z.; Lu, Q.; Zhang, Y.; Zhu, X.; Guo, Q. One step bio-oil upgrading through hydrotreatment, esterification, and cracking. Ind. Eng. Chem. Res. 2009, 48, 6923–6929. [Google Scholar] [CrossRef]
- Oasmaa, A.; Czernik, S. Fuel oil quality of biomass pyrolysis oils—State of the art for the end users. Energy Fuels 1999, 13, 914–921. [Google Scholar] [CrossRef]
- Diebold, J.P.P. A Review of the Chemical and Physical Mechanisms of the Storage Stability of Fast Pyrolysis Bio-Oils; National Renewable Energy Laboratory, Lakewood, CO, USA. 2000. Available online: https://www.nrel.gov/docs/fy00osti/27613.pdf (accessed on 4 December 2021).
- Oasmaa, A.; Kuoppala, E.; Selin, J.F.; Gust, S.; Solantausta, Y. Fast pyrolysis of forestry residue and pine. 4. Improvement of the product quality by solvent addition. Energy Fuels 2004, 18, 1578–1583. [Google Scholar] [CrossRef]
- Mercader, F.D.M.; Koehorst, P.J.J.; Heeres, H.J.; Kersten, S.R.A.; Hogendoorn, J.A. Competition Between Hydrotreating and Polymerization Reactions During Pyrolysis Oil Hydrodeoxygenation. AIChE J. 2011, 55, 3160–3170. [Google Scholar] [CrossRef]
- Mercader, F.D.M. Pyrolysis Oil Upgrading for Co-Processing in Standard Refinery Units; University of Twente: Enschede, The Netherlands, 2010; ISBN 9789036530859. [Google Scholar]
- Yin, W.; Venderbosch, R.H.; Heeres, H.J. Recent Developments in the Catalytic Hydrotreatment of Pyrolysis Liquids; Elsevier Ltd.: Amsterdam, The Netherlands, 2018; ISBN 9780081010259. [Google Scholar]
- Zacher, A.H.; Olarte, M.V.; Santosa, D.M.; Elliott, D.C.; Jones, S.B. A review and perspective of recent bio-oil hydrotreating research. Green Chem. 2014, 16, 491–515. [Google Scholar] [CrossRef]
- Mortensen, P.M.; Grunwaldt, J.D.; Jensen, P.A.; Knudsen, K.G.; Jensen, A.D. A review of catalytic upgrading of bio-oil to engine fuels. Appl. Catal. A Gen. 2011, 407, 1–19. [Google Scholar] [CrossRef]
- Boscagli, C.; Yang, C.; Welle, A.; Wang, W.; Behrens, S.; Raffelt, K.; Grunwaldt, J.D. Effect of pyrolysis oil components on the activity and selectivity of nickel-based catalysts during hydrotreatment. Appl. Catal. A Gen. 2017, 544, 161–172. [Google Scholar] [CrossRef]
- Elliott, D.C.; Hart, T.R.; Neuenschwander, G.G.; Rotness, L.J.; Olarte, M.V.; Zacher, A.H.; Solantausta, Y. Catalytic hydroprocessing of fast pyrolysis bio-oil from pine sawdust. Energy Fuels 2012, 26, 3891–3896. [Google Scholar] [CrossRef]
- Wildschut, J.; Iqbal, M.; Mahfud, F.H.; Cabrera, I.M.; Venderbosch, R.H.; Heeres, H.J. Insights in the hydrotreatment of fast pyrolysis oil using a ruthenium on carbon catalyst. Energy Environ. Sci. 2010, 3, 962. [Google Scholar] [CrossRef]
- Talmadge, M.S.; Baldwin, R.M.; Biddy, M.J.; McCormick, R.L.; Beckham, G.T.; Ferguson, G.A.; Czernik, S.; Magrini-Bair, K.A.; Foust, T.D.; Metelski, P.D.; et al. A perspective on oxygenated species in the refinery integration of pyrolysis oil. Green Chem. 2014, 16, 407–453. [Google Scholar] [CrossRef]
- Oasmaa, A.; Kuoppala, E.; Ardiyanti, A.; Venderbosch, R.H.; Heeres, H.J. Characterization of hydrotreated fast pyrolysis liquids. Energy Fuels 2010, 24, 5264–5272. [Google Scholar] [CrossRef]
- Elliott, D.C.; Neuenschwander, G.G. Liquid fuels by low-severity hydrotreating of biocrude. Dev. Thermochem. Biomass Convers. 1996, 1, 611–621. [Google Scholar]
- French, R.J.; Stunkel, J.; Black, S.; Myers, M.; Yung, M.M.; Iisa, K. Evaluate impact of catalyst type on oil yield and hydrogen consumption from mild hydrotreating. Energy Fuels 2014, 28, 3086–3095. [Google Scholar] [CrossRef]
- Joshi, N.; Lawal, A. Hydrodeoxygenation of pyrolysis oil in a microreactor. Chem. Eng. Sci. 2012, 74, 1–8. [Google Scholar] [CrossRef]
- Schmitt, C.C.; Zimina, A.; Fam, Y.; Raffelt, K.; Grunwaldt, J.-D.; Dahmen, N. Evaluation of High-Loaded Ni-Based Catalysts for Upgrading Fast Pyrolysis Bio-Oil. Catalysts 2019, 9, 784. [Google Scholar] [CrossRef] [Green Version]
- Bozell, J.J.; Holladay, J.E.; Johnson, D.; White, J.F. Top Value-Added Chemicals from Biomass Volume II-Results of Screening for Potential Candidates from Biorefinery Lignin PNNL-16983; Pacific Northwest National Laboratory: Richland, WA, USA, 2007; Volume II. Available online: https://www.pnnl.gov/main/publications/external/technical_reports/PNNL-16983.pdf (accessed on 4 December 2021).
- Pires, A.P.P.; Arauzo, J.; Fonts, I.; Domine, M.E.; Arroyo, A.F.; Garcia-Perez, M.E.M.M.E.; Montoya, J.; Chejne, F.; Pfromm, P.; Garcia-Perez, M.E.M.M.E. Challenges and opportunities for bio-oil refining: A review. Energy Fuels 2019, 33, 4683–4720. [Google Scholar] [CrossRef]
- Elliott, D.C. Historical Developments in Hydroprocessing Bio-oils. Energy Fuels 2007, 21, 1792–1815. [Google Scholar] [CrossRef]
- Shafaghat, H.; Rezaei, P.S.; Ashri Wan Daud, W.M. Effective parameters on selective catalytic hydrodeoxygenation of phenolic compounds of pyrolysis bio-oil to high-value hydrocarbons. RSC Adv. 2015, 5, 103999–104042. [Google Scholar] [CrossRef]
- Şenol, O.I.; Viljava, T.R.; Krause, A.O.I. Hydrodeoxygenation of methyl esters on sulphided NiMo/γ-Al2O3 and CoMo/γ-Al2O3 catalysts. Catal. Today 2005, 100, 331–335. [Google Scholar] [CrossRef]
- Auersvald, M.; Shumeiko, B.; Vrtiška, D.; Straka, P.; Staš, M.; Šimáček, P.; Blažek, J.; Kubička, D. Hydrotreatment of straw bio-oil from ablative fast pyrolysis to produce suitable refinery intermediates. Fuel 2019, 238, 98–110. [Google Scholar] [CrossRef]
- Gholizadeh, M.; Gunawan, R.; Hu, X.; Hasan, M.M.; Kersten, S.; Westerhof, R.; Chaitwat, W.; Li, C.Z. Different reaction behaviours of the light and heavy components of bio-oil during the hydrotreatment in a continuous pack-bed reactor. Fuel Process. Technol. 2016, 146, 76–84. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Wang, X. Hydrodeoxygenation of model compounds and catalytic systems for pyrolysis bio-oils upgrading. Catal. Sustain. Energy 2012, 1, 28–52. [Google Scholar] [CrossRef]
- Romero, Y.; Richard, F.; Brunet, S. Hydrodeoxygenation of 2-ethylphenol as a model compound of bio-crude over sulfided Mo-based catalysts: Promoting effect and reaction mechanism. Appl. Catal. B Environ. 2010, 98, 213–223. [Google Scholar] [CrossRef]
- Wan, H.; Chaudhari, R.V.; Subramaniam, B. Aqueous phase hydrogenation of acetic acid and its promotional effect on p-cresol hydrodeoxygenation. Energy Fuels 2013, 27, 487–493. [Google Scholar] [CrossRef]
- Gholizadeh, M.; Gunawan, R.; Hu, X.; De Miguel Mercader, F.; Westerhof, R.; Chaitwat, W.; Hasan, M.M.; Mourant, D.; Li, C.Z. Effects of temperature on the hydrotreatment behaviour of pyrolysis bio-oil and coke formation in a continuous hydrotreatment reactor. Fuel Process. Technol. 2016, 148, 175–183. [Google Scholar] [CrossRef]
- Silva, A.C.; de Barros, M.R.; Macedo, K.S.; Silva, E.M.S. Ferroniobium Alloy Fines Agglomeration through Briquetting. Tecnol. Metal. Mater. Mineração 2019, 16, 414–420. [Google Scholar] [CrossRef]
- Nowak, I.; Ziolek, M. Niobium Compounds: Preparation, Characterization, and Application in Heterogeneous Catalysis. Chem. Rev. 1999, 99, 3603–3624. [Google Scholar] [CrossRef] [PubMed]
- Nico, C.; Monteiro, T.; Graça, M.P.F. Niobium oxides and niobates physical properties: Review and prospects. Prog. Mater. Sci. 2016, 80, 1–37. [Google Scholar] [CrossRef]
- Schäfer, H.; Gruehn, T.; Schulte, F. The Modifications of Niobium Pentoxide. Angew. Chemie-Int. Ed. 1966, 5, 40–52. [Google Scholar] [CrossRef]
- Ko, E.I.; Weissman, J.G. Structures of Niobium Pentoxide and their Implications on Chemical Behavior. Catal. Today 1990, 8, 27–36. [Google Scholar] [CrossRef]
- Rani, R.A.; Zoolfakar, A.S.; O’Mullane, A.P.; Austin, M.W.; Kalantar-Zadeh, K. Thin films and nanostructures of niobium pentoxide: Fundamental properties, synthesis methods and applications. J. Mater. Chem. A 2014, 2, 15683–15703. [Google Scholar] [CrossRef] [Green Version]
- Meng, J.; He, Q.; Xu, L.; Zhang, X.; Liu, F.; Wang, X.; Li, Q.; Xu, X.; Zhang, G.; Niu, C.; et al. Identification of Phase Control of Carbon-Confined Nb 2 O 5 Nanoparticles toward High-Performance Lithium Storage. Adv. Energy Mater. 2019, 9, 18. [Google Scholar] [CrossRef]
- Chan, X.; Pu, T.; Chen, X.; James, A.; Lee, J.; Parise, J.B.; Kim, D.H.; Kim, T. Effect of niobium oxide phase on the furfuryl alcohol dehydration. Catal. Commun. 2017, 97, 65–69. [Google Scholar] [CrossRef] [Green Version]
- Tanabe, K. Catalytic applications of niobium compounds. Niobium. Sci. Technol. 2001, 78, 269–290. [Google Scholar]
- Noronha, F.B.; Frydman, A.; Aranda, D.A.G.; Perez, C.; Soares, R.R.; Morawek, B.; Castner, D.; Campbell, C.T.; Frety, R.; Schmal, M. The promoting effect of noble metal addition on niobia-supported cobalt catalysts. Catal. Today 1996, 28, 147–157. [Google Scholar] [CrossRef]
- Passos, F.B.; Aranda, D.A.G.; Soares, R.R.; Schmal, M. Effect of preparation method on the properties of Nb2O5 promoted platinum catalysts. Catal. Today 1998, 43, 3–9. [Google Scholar] [CrossRef]
- Barrios, A.M.; Teles, C.A.; de Souza, P.M.; Rabelo-Neto, R.C.; Jacobs, G.; Davis, B.H.; Borges, L.E.P.; Noronha, F.B. Hydrodeoxygenation of phenol over niobia supported Pd catalyst. Catal. Today 2018, 302, 115–124. [Google Scholar] [CrossRef]
- Shao, Y.; Xia, Q.; Dong, L.; Liu, X.; Han, X.; Parker, S.F.; Cheng, Y.; Daemen, L.L.; Ramirez-Cuesta, A.J.; Yang, S.; et al. Selective production of arenes via direct lignin upgrading over a niobium-based catalyst. Nat. Commun. 2017, 8, 1–9. [Google Scholar] [CrossRef]
- Dong, L.; Shao, Y.; Han, X.; Liu, X.; Xia, Q.; Parker, S.F.; Cheng, Y.; Daemen, L.L.; Ramirez-Cuesta, A.J.; Wang, Y.; et al. Comparison of two multifunctional catalysts [M/Nb2O5 (M = Pd, Pt)] for one-pot hydrodeoxygenation of lignin. Catal. Sci. Technol. 2018, 8, 6129–6136. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Dong, L.; Liu, X.; Guo, Y.; Wang, Y. Selective production of ethylbenzene from lignin oil over FeOx modified Ru/Nb2O5 catalyst. Appl. Catal. B Environ. 2020, 260, 118143. [Google Scholar] [CrossRef]
- Xin, Y.; Dong, L.; Guo, Y.; Liu, X.; Hu, Y.; Wang, Y. Correlation of the catalytic performance with Nb2O5 surface properties in the hydrodeoxygenation of lignin model compound. J. Catal. 2019, 375, 202–212. [Google Scholar] [CrossRef]
- Jing, Y.; Xin, Y.; Guo, Y.; Liu, X.; Wang, Y. Highly efficient Nb2O5 catalyst for aldol condensation of biomass-derived carbonyl molecules to fuel precursors. Chin. J. Catal. 2019, 40, 1168–1177. [Google Scholar] [CrossRef]
- Resende, K.A.; Braga, A.H.; Noronha, F.B.; Hori, C.E. Hydrodeoxygenation of phenol over Ni/Ce1-xNbxO2 catalysts. Appl. Catal. B Environ. 2019, 245, 100–113. [Google Scholar] [CrossRef]
- Jin, S.; Guan, W.; Tsang, C.W.; Yan, D.Y.S.; Chan, C.Y.; Liang, C. Enhanced Hydroconversion of Lignin-Derived Oxygen-Containing Compounds Over Bulk Nickel Catalysts Though Nb2O5 Modification. Catal. Lett. 2017, 147, 2215–2224. [Google Scholar] [CrossRef]
- Xue, F.; Ma, D.; Tong, T.; Liu, X.; Hu, Y.; Guo, Y.; Wang, Y. Contribution of Different NbOx Species in the Hydrodeoxygenation of 2,5-Dimethyltetrahydrofuran to Hexane. ACS Sustain. Chem. Eng. 2018, 6, 13107–13113. [Google Scholar] [CrossRef]
- Shao, Y.; Xia, Q.; Liu, X.; Lu, G.; Wang, Y. Pd/Nb2O5/SiO2 Catalyst for the Direct Hydrodeoxygenation of Biomass-Related Compounds to Liquid Alkanes under Mild Conditions. ChemSusChem 2015, 8, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- Infantes-Molina, A.; Moretti, E.; Segovia, E.; Lenarda, A.; Rodríguez-Castellón, E. Pd-Nb binfunctional catalysts supported on silica and zirconium phosphate heterostructures for O-removal of dibenzofurane. Catal. Today 2016, 277, 143–151. [Google Scholar] [CrossRef]
- Guan, W.; Chen, X.; Li, C.; Zhang, J.; Tsang, C.W.; Hu, H.; Li, S.; Liang, C. Nb(Ta)-based solid acid modified Pt/CNTs catalysts for hydrodeoxygenation of lignin-derived compounds. Mol. Catal. 2019, 467, 61–69. [Google Scholar] [CrossRef]
- Zhang, C.; Jia, C.; Cao, Y.; Yao, Y.; Xie, S.; Zhang, S.; Lin, H. Water-assisted selective hydrodeoxygenation of phenol to benzene over the Ru composite catalyst in the biphasic process. Green Chem. 2019, 21, 1668–1679. [Google Scholar] [CrossRef]
- Jeon, S.; Park, Y.M.; Park, J.; Saravanan, K.; Jeong, H.K.; Bae, J.W. Synergistic effects of Nb2O5 promoter on Ru/Al2O3 for an aqueous-phase hydrodeoxygenation of glycerol to hydrocarbons. Appl. Catal. A Gen. 2018, 551, 49–62. [Google Scholar] [CrossRef]
- Leal, G.F.; Lima, S.; Graça, I.; Carrer, H.; Barrett, D.H.; Teixeira-Neto, E.; Curvelo, A.A.S.; Rodella, C.B.; Rinaldi, R. Design of Nickel Supported on Water-Tolerant Nb2O5 Catalysts for the Hydrotreating of Lignin Streams Obtained from Lignin-First Biorefining. iScience 2019, 15, 467–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, D.; Lu, S.; Liu, X.; Guo, Y.; Wang, Y. Depolymerization and hydrodeoxygenation of lignin to aromatic hydrocarbons with a Ru catalyst on a variety of Nb-based supports. Chin. J. Catal. 2019, 40, 609–617. [Google Scholar] [CrossRef]
- Teles, C.A.; de Souza, P.M.; Rabelo-Neto, R.C.; Griffin, M.B.; Mukarakate, C.; Orton, K.A.; Resasco, D.E.; Noronha, F.B. Catalytic upgrading of biomass pyrolysis vapors and model compounds using niobia supported Pd catalyst. Appl. Catal. B Environ. 2018, 238, 38–50. [Google Scholar] [CrossRef]
- Mortensen, P.M.; Grunwaldt, J.D.; Jensen, P.A.; Jensen, A.D. Screening of catalysts for hydrodeoxygenation of phenol as a model compound for bio-oil. ACS Catal. 2013, 3, 1774–1785. [Google Scholar] [CrossRef]
- Ardiyanti, A.R.; Bykova, M.V.; Khromova, S.A.; Yin, W.; Venderbosch, R.H.; Yakovlev, V.A.; Heeres, H.J. Ni-Based Catalysts for the Hydrotreatment of Fast Pyrolysis Oil. Energy Fuels 2016, 30, 1544–1554. [Google Scholar] [CrossRef]
- Ardiyanti, A.R.; Khromova, S.A.; Venderbosch, R.H.; Yakovlev, V.A.; Melián-Cabrera, I.V.; Heeres, H.J. Catalytic hydrotreatment of fast pyrolysis oil using bimetallic Ni-Cu catalysts on various supports. Appl. Catal. A Gen. 2012, 449, 121–130. [Google Scholar] [CrossRef]
- Ardiyani, A.R.; Khromova, S.A.; Venderbosch, R.H.; Yakovlev, V.A.; Heeres, H.J. Catalytic hydrotreatment of fast-pyrolysis oil using non-sulfided bimetallic Ni-Cu catalysts on a δ-Al2O3 support. Appl. Catal. B Environ. 2012, 117–118, 105–117. [Google Scholar] [CrossRef]
- Mortensen, P.M.; Gardini, D.; de Carvalho, H.W.P.; Damsgaard, C.D.; Grunwaldt, J.; Jensen, P.A.; Wagner, J.B.; Jensen, A.D. Stability and resistance of nickel catalysts for hydrodeoxygenation: Carbon deposition and effects of sulfur, potassium, and chlorine in the feed. Catal. Sci. Technol. 2014, 4, 3672–3686. [Google Scholar] [CrossRef] [Green Version]
- Mortensen, P.M.; Grunwaldt, J.D.; Jensen, P.A.; Jensen, A.D. Influence on nickel particle size on the hydrodeoxygenation of phenol over Ni/SiO2. Catal. Today 2016, 259, 277–284. [Google Scholar] [CrossRef]
- Boscagli, C.; Raffelt, K.; Zevaco, T.A.; Olbrich, W.; Otto, T.N.; Sauer, J.; Grunwaldt, J.D. Mild hydrotreatment of the light fraction of fast-pyrolysis oil produced from straw over nickel-based catalysts. Biomass Bioenergy 2015, 83, 525–538. [Google Scholar] [CrossRef] [Green Version]
- Argyle, M.D.; Bartholomew, C.H. Heterogeneous Catalyst Deactivation and Regeneration: A Review. Catalysts 2015, 5, 145–269. [Google Scholar] [CrossRef] [Green Version]
- Sultana, A.; Kumar, A. Optimal configuration and combination of multiple lignocellulosic biomass feedstocks delivery to a biorefinery. Bioresour. Technol. 2011, 102, 9947–9956. [Google Scholar] [CrossRef] [PubMed]
- Demirbaş, A. Biomass resource facilities and biomass conversion processing for fuels and chemicals. Energy Convers. Manag. 2001, 42, 1357–1378. [Google Scholar] [CrossRef]
- Jonutis, V. The Biomass Exchange-virtual transmission gird in biomass markets. In Proceedings of the IEA Bioenergy workshop: Developing business models for efficient use of biomass, Tallinn, Estonia, 22 October 2019. [Google Scholar]
- Dimitriou, I. SWOT of bio-hubs in deploying biobased supply chains. In Proceedings of the IEA Bioenergy workshop: Developing business models for efficient use of biomass, Tallinn, Estonia, 22 October 2019. [Google Scholar]
- Annevelink, B. Examples of developing biobased business models: Session conclusions. In Proceedings of the IEA Bioenergy workshop: Developing business models for efficient use of biomass, Tallinn, Estonia, 22 October 2019. [Google Scholar]
- Maung, T.A.; Gustafson, C.R.; Saxowsky, D.M.; Nowatzki, J.; Miljkovic, T.; Ripplinger, D. The logistics of supplying single vs. multi-crop cellulosic feedstocks to a biorefinery in southeast North Dakota. Appl. Energy 2013, 109, 229–238. [Google Scholar] [CrossRef]
- Brown, D.; Rowe, A.; Wild, P. A techno-economic analysis of using mobile distributed pyrolysis facilities to deliver a forest residue resource. Bioresour. Technol. 2013, 150, 367–376. [Google Scholar] [CrossRef]
- Brown, M. Enabling a Regional Bioeconomy: Australian case study for developing biomass supply chains. In Proceedings of the IEA Bioenergy Workshop: Developing Business Models for Efficient Use of Biomass, Tallinn, Estonia, 22 October 2019. [Google Scholar]
- Kärki, J. Outlook on bioenergy combined with carbon capture, utilization and storage (BECCUS). In Proceedings of the IEA Bioenergy Workshop: Developing Business Models for Efficient Use of Biomass, Tallinn, Estonia, 22 October 2019. [Google Scholar]
- Consoli, C. Bioenergy and Carbon Capture and Storage; Global CCS Institute: Melbourne, Australia, 2019. [Google Scholar]
- Lindroos, T.J.; Rydén, M.; Langørgen, Ø.; Pursiheimo, E.; Pikkarainen, T. Robust decision making analysis of BECCS (bio-CLC) in a district heating and cooling grid. Sustain. Energy Technol. Assess. 2019, 34, 157–172. [Google Scholar] [CrossRef]
- van Dyk, S.; Su, J.; Mcmillan, J.D.; Saddler, J. Potential synergies of drop-in biofuel production with further co-processing at oil refineries. Biofuels Bioprod. Biorefining 2019, 13, 760–775. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.C. PY Refinery: Thermal fractionation of lignocellulosic biomass into diverse biobased products. In Proceedings of the Frontiers in Biorefining, St. Simons Island, GA, USA, 21–24 October 2014. [Google Scholar]
- Ceccarelli, C. Bioliq-Startseite. Available online: https://www.bioliq.de/ (accessed on 19 May 2021).
- Venderbosch, R.; Heeres, H. Stabilisation of Biomass derived Pyrolysis Oils by Catalytic Hydrotreatment. InTech 2011. Available online: https://www.intechopen.com/chapters/17490 (accessed on 4 December 2021).
- Valle, B.; Aramburu, B.; Santiviago, C.; Bilbao, J.; Gayubo, A.G. Upgrading of Bio-Oil in a Continuous Process with Dolomite Catalyst. Energy Fuels 2014, 28, 6419–6428. [Google Scholar] [CrossRef]
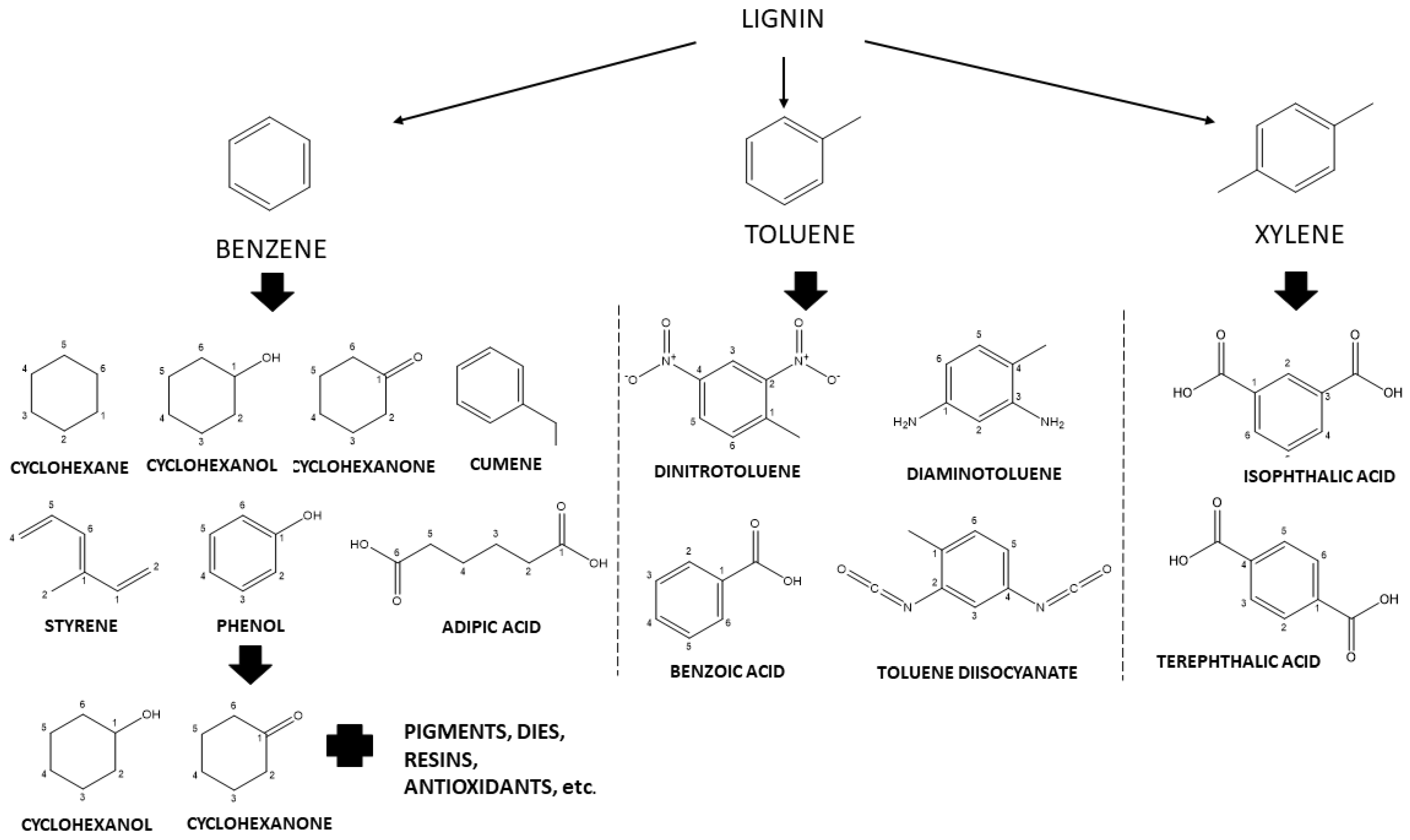
- Graglia, M.; Kanna, N.; Esposito, D. Lignin Refinery: Towards the Preparation of Renewable Aromatic Building Blocks. ChemBioEng. Rev. 2015, 2, 377–392. [Google Scholar] [CrossRef]
- Zakzeski, J.; Bruijnincx, P.C.A.; Jongerius, A.L.; Weckhuysen, B.M. The Catalytic Valorization of Ligning for the Production of Renewable Chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef]
- Sun, Z.; Fridrich, B.; De Santi, A.; Elangovan, S.; Barta, K. Bright Side of Lignin Depolymerization: Toward New Platform Chemicals. Chem. Rev. 2018, 118, 614–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.; Xiao, Z.; Li, C.; Chen, X.; Wang, L.; Xing, J.; Li, W.; Liang, C. Catalytic hydrodeoxygenation of anisole as lignin model compound over supported nickel catalysts. Catal. Today 2014, 234, 125–132. [Google Scholar] [CrossRef]
- Izquierdo, U.; Barrio, V.L.; Cambra, J.F.; Requies, J.; Güemez, M.B.; Arias, P.L.; Kolb, G.; Zapf, R.; Gutiérrez, A.M.; Arraibi, J.R. Hydrogen production from methane and natural gas steam reforming in conventional and microreactor reaction systems. Int. J. Hydrogen Energy 2012, 37, 7026–7033. [Google Scholar] [CrossRef]
- Lap, T.; Benders, R.; Köberle, A.; van der Hilst, F.; Nogueira, L.; Szklo, A.; Schaeffer, R.; Faaij, A. Pathways for a Brazilian biobased economy: Towards optimal utilization of biomass. Biofuels Bioprod. Biorefining 2019, 13, 673–689. [Google Scholar] [CrossRef]
- Lap, T.; Benders, R.; van der Hilst, F.; Faaij, A. How does the interplay between resource availability, intersectoral competition and reliability affect a low-carbon power generation mix in Brazil for 2050? Energy 2020, 195, 116948. [Google Scholar] [CrossRef]
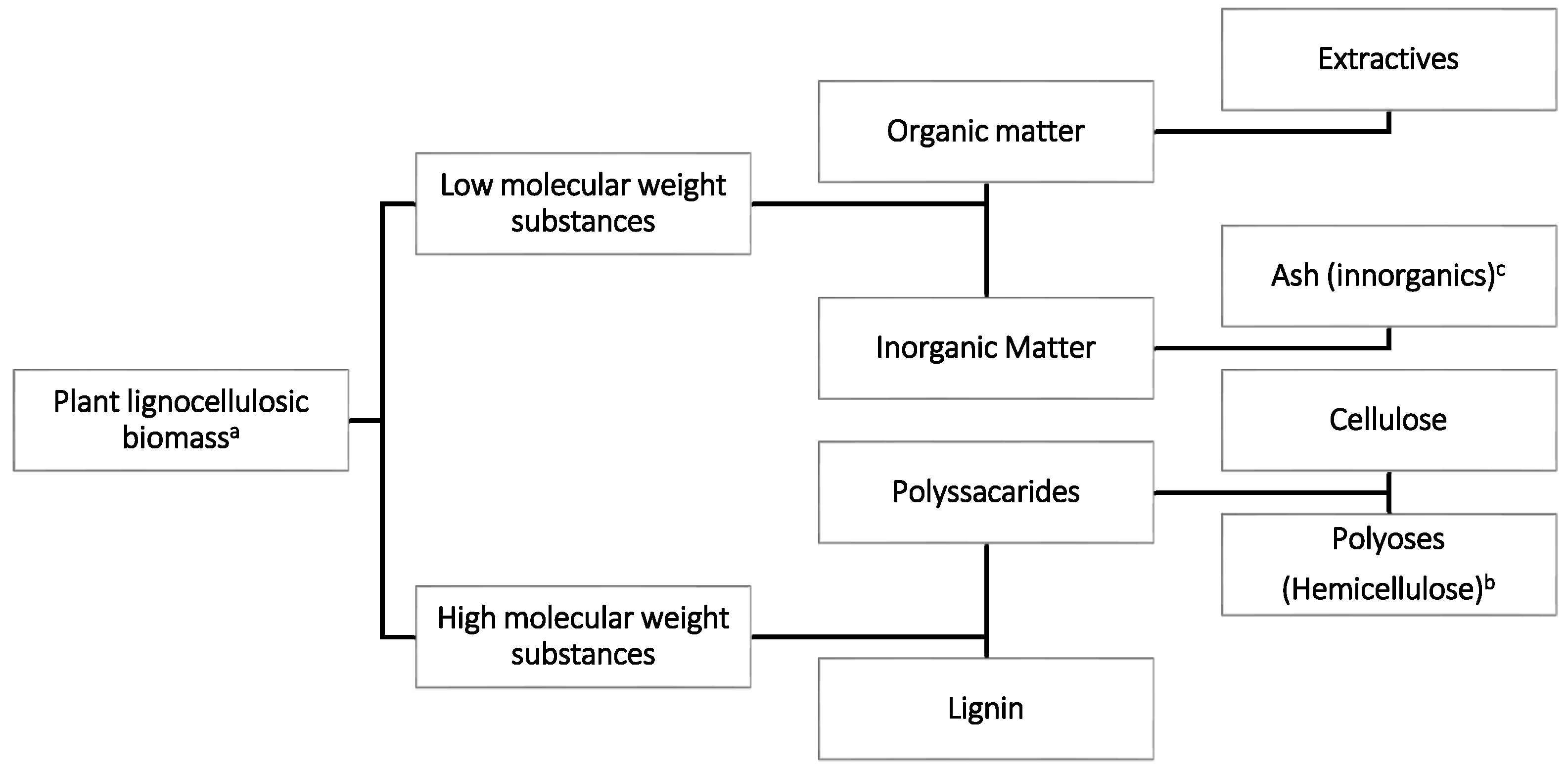
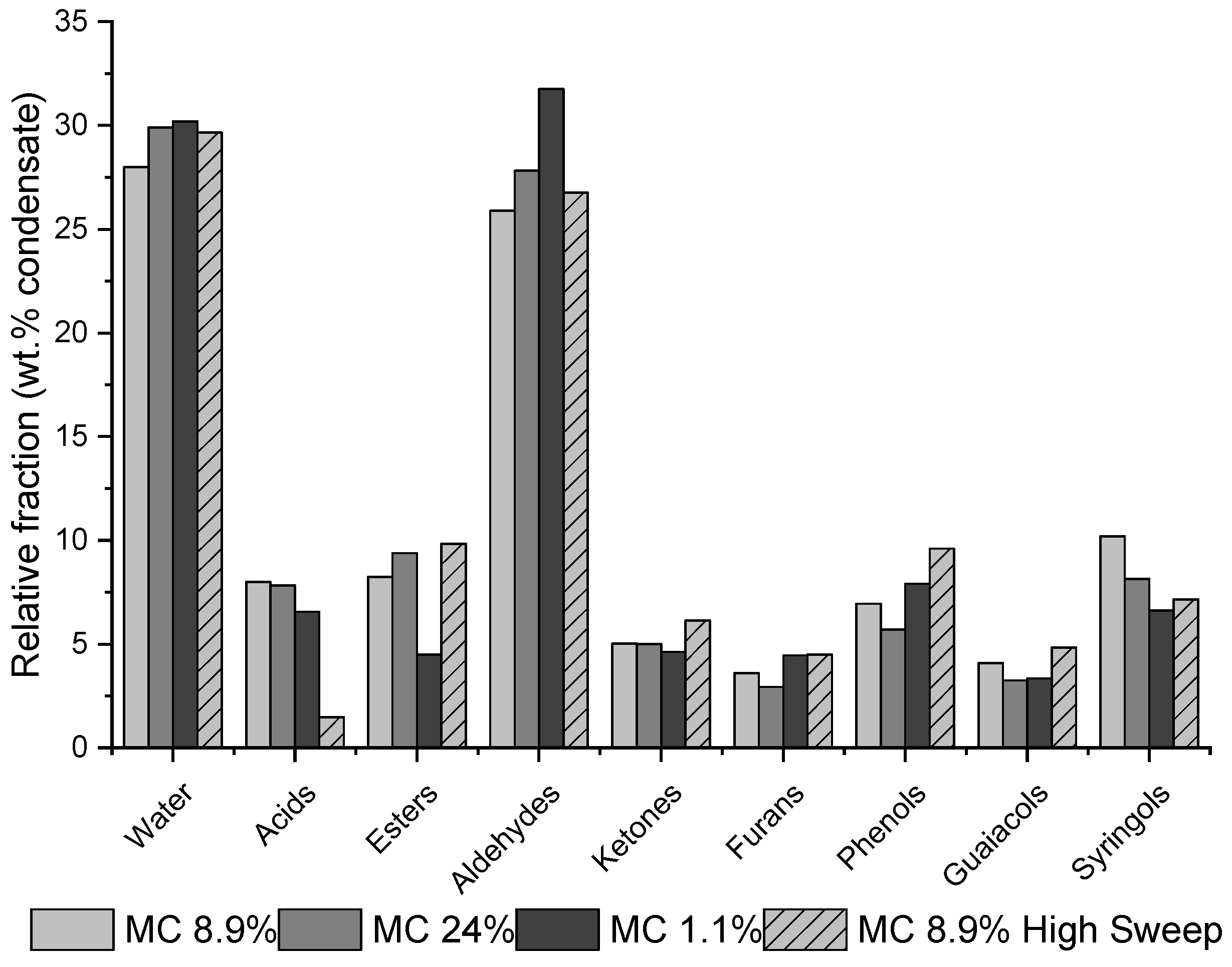
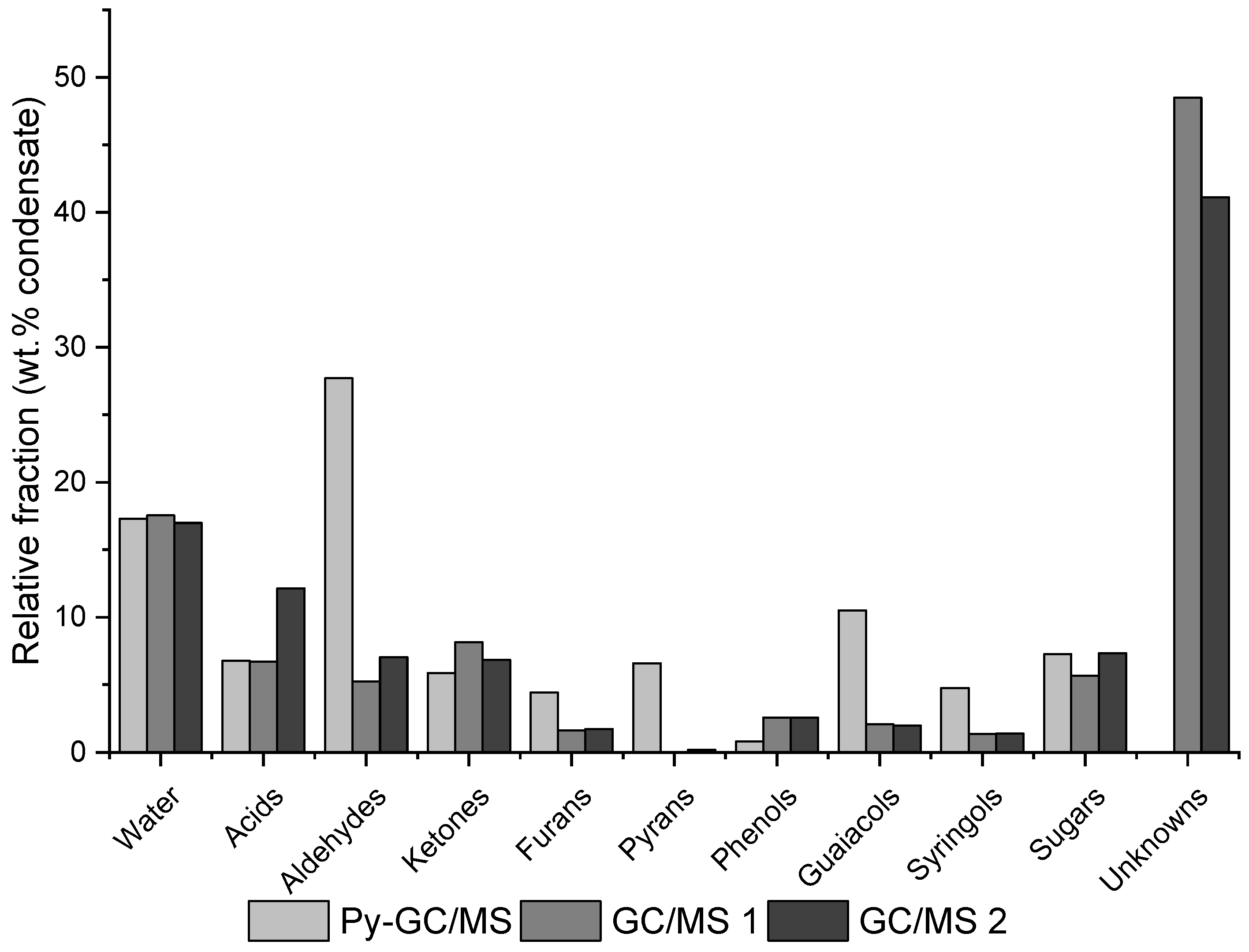
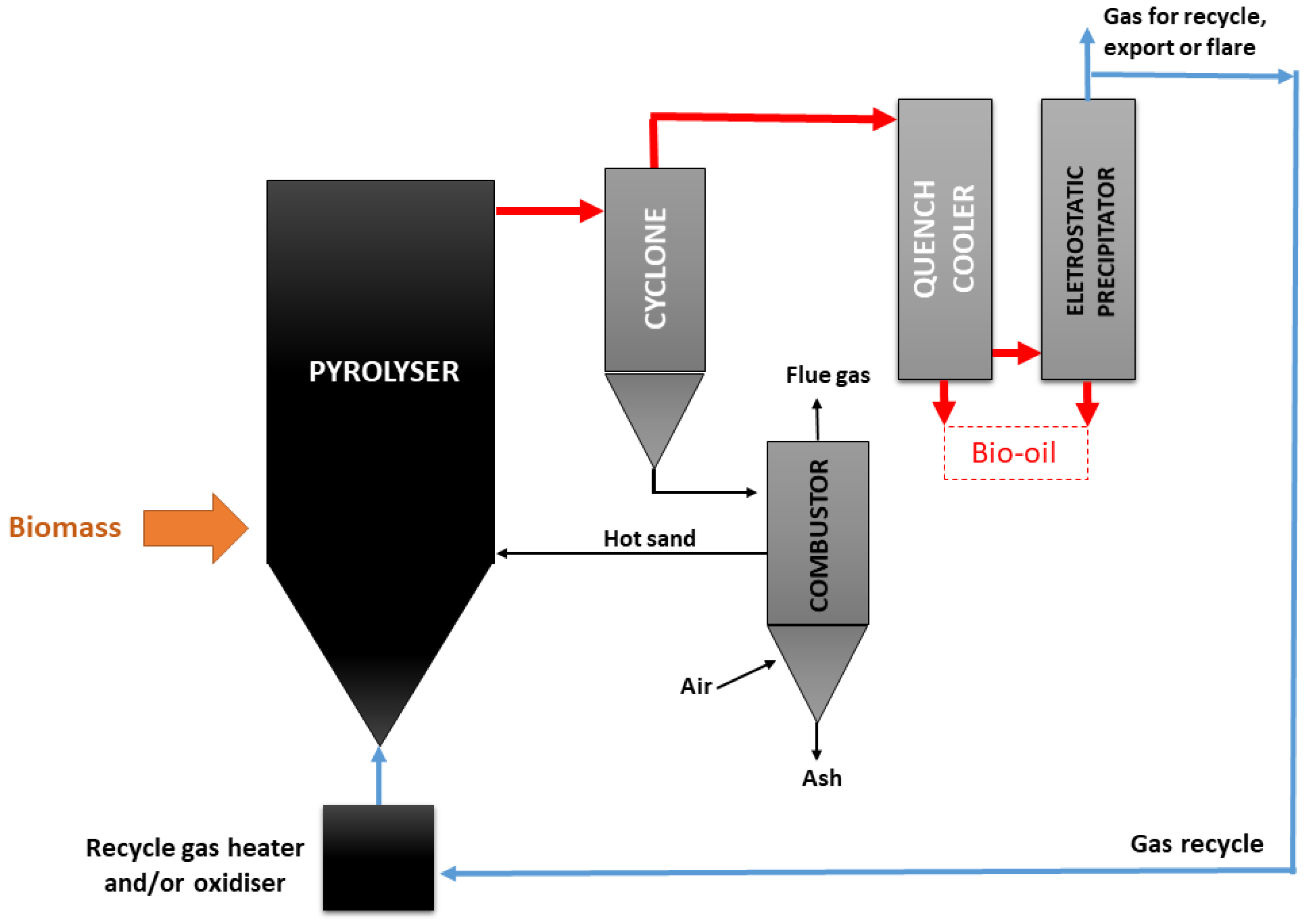

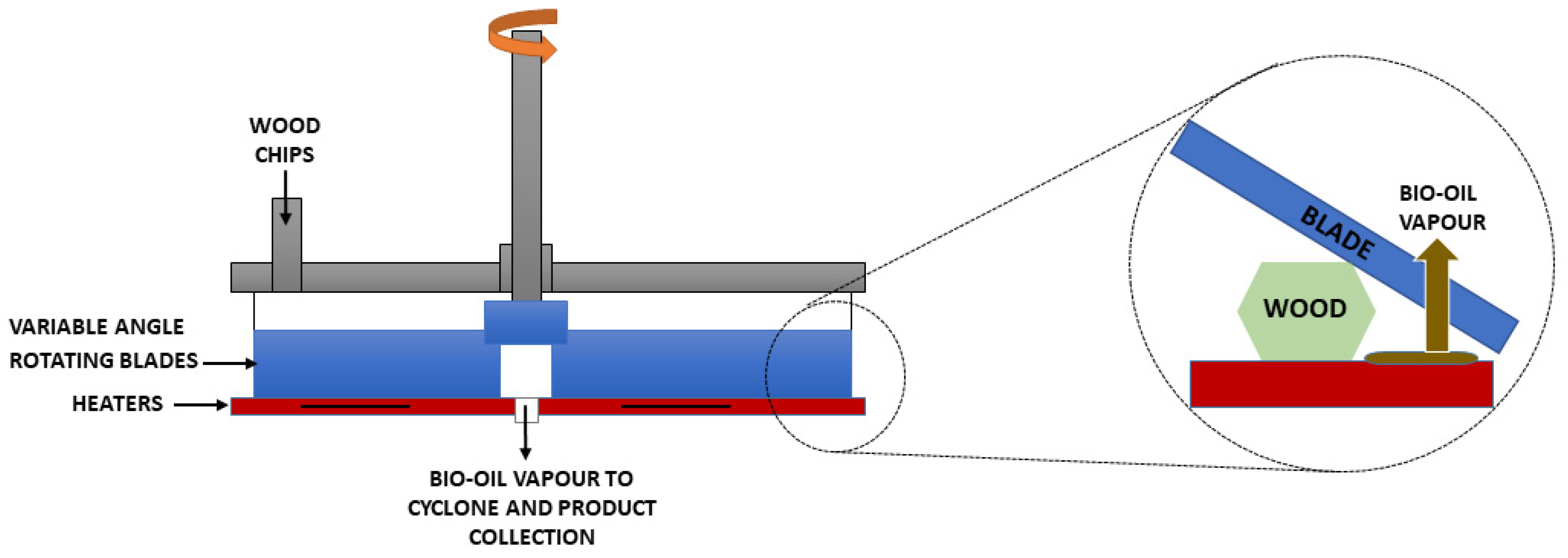
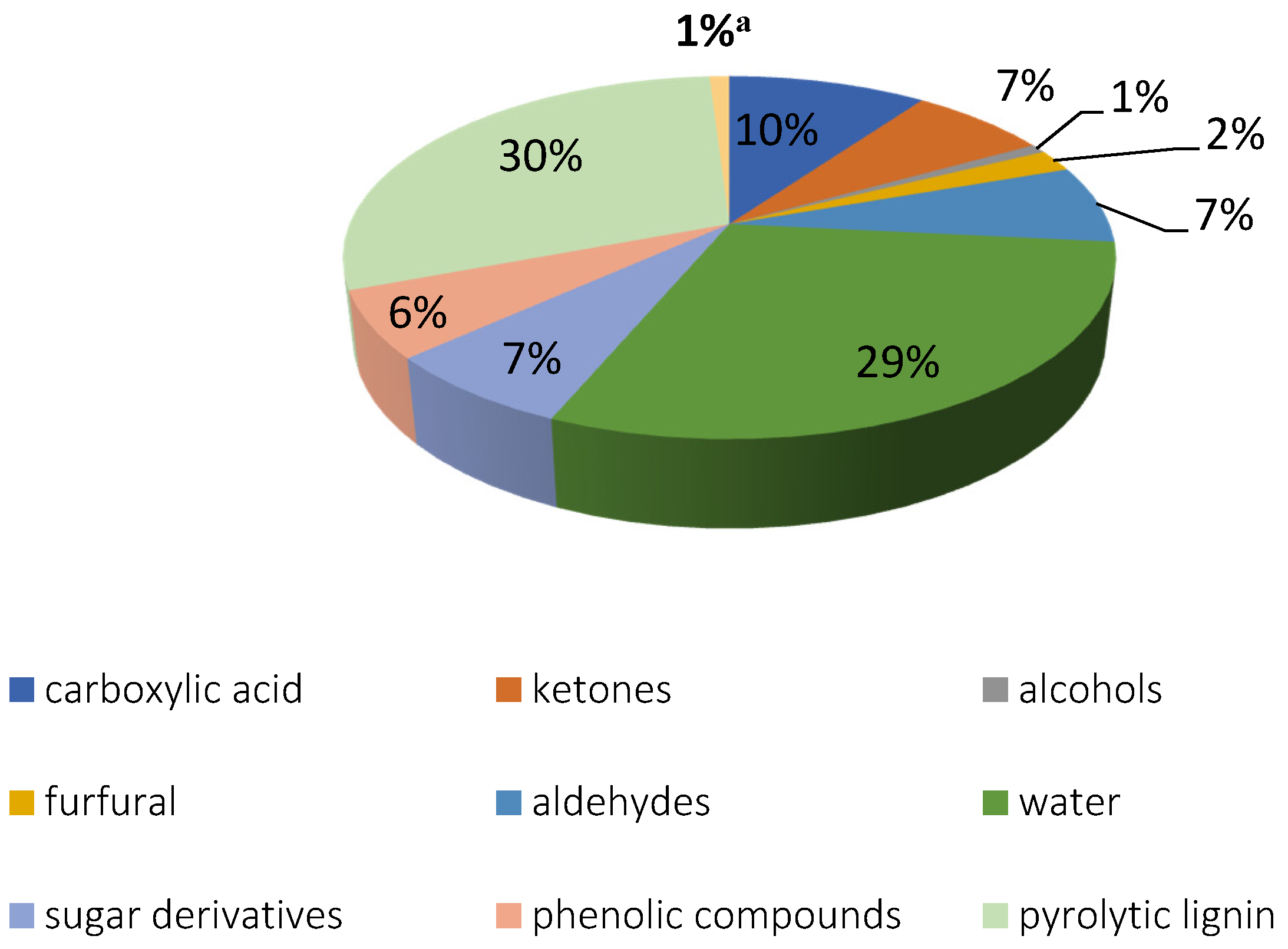
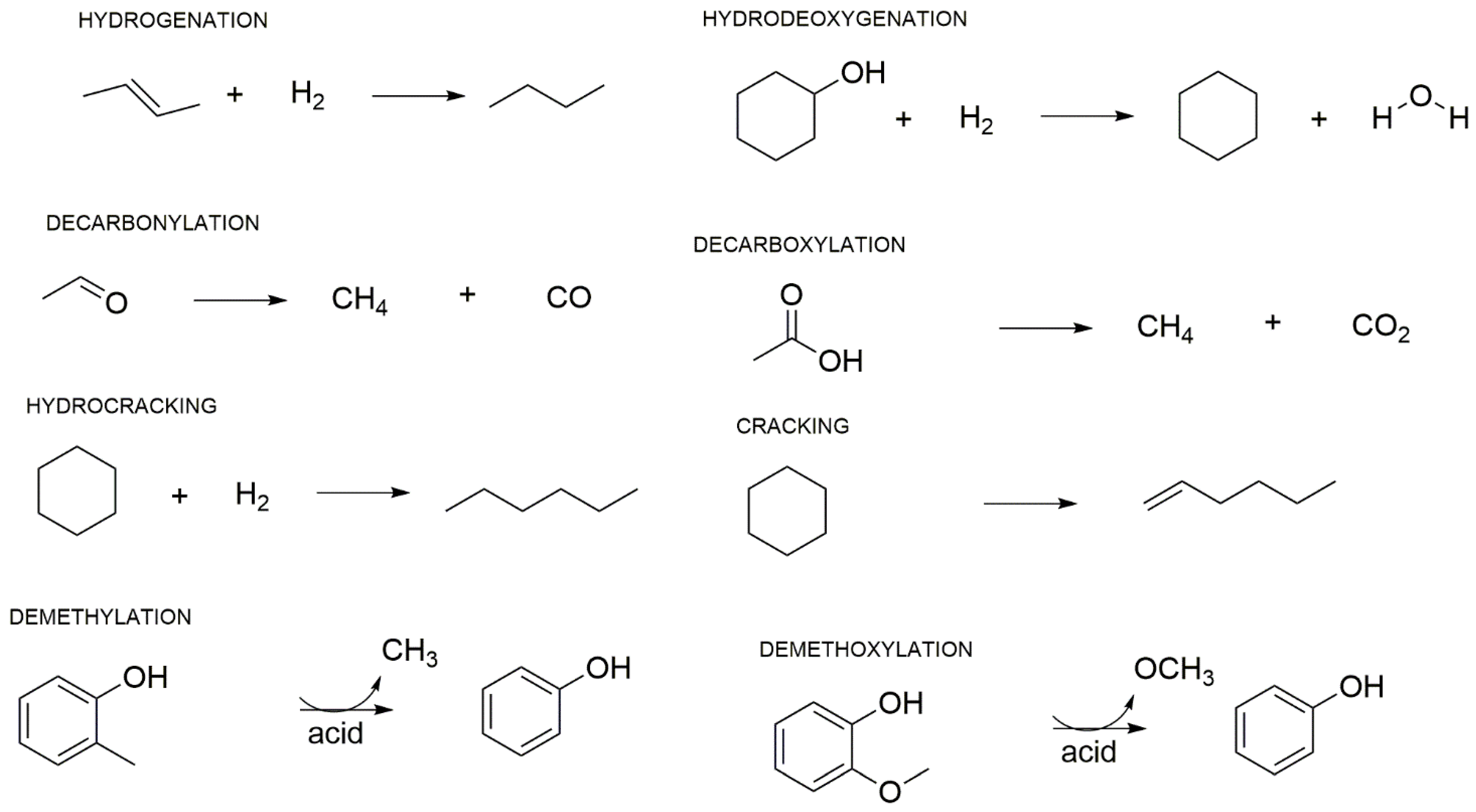


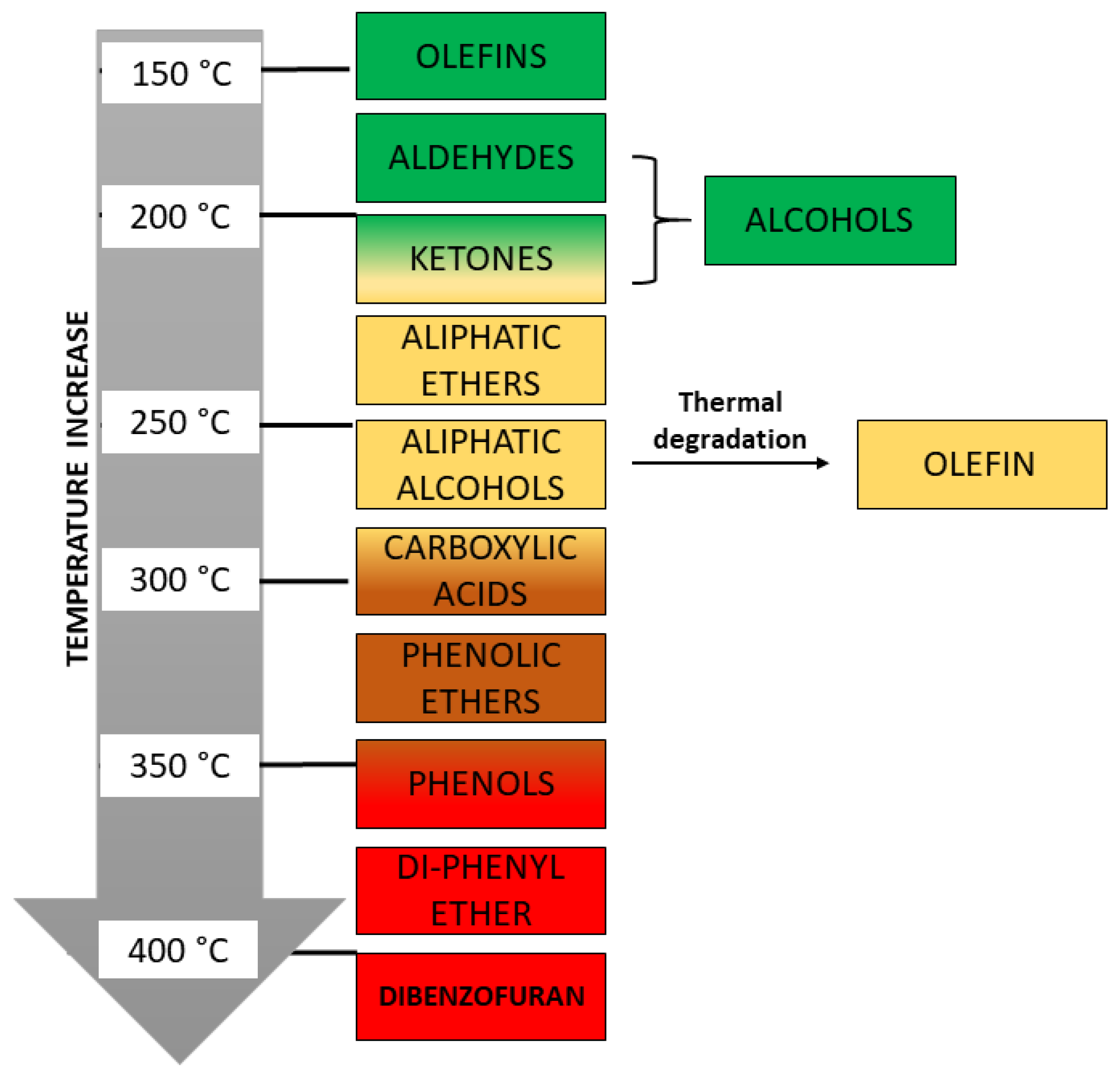
| Residue | Annual Production 2019 Brazil (×106 kg) [21,22] | Cellulose (wt.%) | Hemicellulose (wt.%) | Lignin (wt.%) | Extractives (EtOH) (wt.%) | Others (wt.%) | Ash (wt.%) | Pectin (wt.%) | References | |
|---|---|---|---|---|---|---|---|---|---|---|
| Sugarcane | Sugarcane bagasse | 657,532 | 36.9–42.8 | 21.0–27.2 | 17.4–33.71 | 2.78–12.7 | n/a | 0.68–6.71 | n/a | [23,24] |
| Corn | Corncob | 100,566 | 28.0–44.1 | 26.8–44.4 | 11.9–23.9 | 5.8–10.5 | n/a | 1.1–2.1 | n/a | [25,26,27,28,29,30] |
| Piassava | Piassava fibers | 8.5 (2018) | 29.6–31.6 | 0–11.0 | 45.0–48.4 | n/a | n/a | n/a | n/a | [31,32] |
| Cassava | Cassava peel | 18,990 | 7–37.9 | 4.9–37 | 7.6–65.0 | n/a | 7.6–20.11 | 4.5–8.33 | n/a | [28,33,34,35,36] |
| Wheat | Wheat straw | 5231 | 31–40.4 | 15.5–30.6 | 17.7–21.5 | 4.6–6.5 | - | 7.6–7.7 | n/a | [37,38,39,40] |
| Rice | Rice peel | 10,260 | 30.6–52 | 12–21.0 | 23.9–21 | 2.3–2.6 | - | 10–15.9 | n/a | [28,29,41,42,43] |
| Soy | Soy peel | 113,448 | 22.4–35.8 | 13–23.1 | 7.6–17.5 | 6.8 | - | 1.1–6.7 | n/a | [28,43,44] |
| Citrus (orange) | Orange peel | 17,614 | 11.9–26.1 | 11.9–14.4 | 0.2–4.8 | n/a | - | n/a | 17–21.3 | [30,44,45] |
| Coconut | Coconut hull | 2340 (2017) | 29.2–32.4 | 10.3–17.6 | 36–49.1 | 1.4–14.1 | - | 2.96–8.39 | n/a | [43,46,47] |
| Gramineous Crops | Elephant grass | n/a | 27.5–40.3 | 9.19–28.13 | 15.6–21.7 | 11.5–14.4 | - | 7.3–13 | n/a | [39,43,48,49] |
| Coffee | Coffee husk | 2995 | 35.3–36.0 | 29.4–30.2 | 24.5–31.1 | 4.2–16 | - | 2 | n/a | [29,43] |
| Sorghum | Sorghum bagasse | 2596 | 30–38.0 | 17.3–22.2 | 17.4–21.4 | 7.7 | - | 1.7–9.0 | n/a | [50,51,52] |
| Barley | Barley straw | 400 | 30.6–43 | 24–46.8 | 6.3–16.3 | 8.1–8.9 | - | 3.5–9.5 | n/a | [40,53,54] |
| Açai | Açai seed | n/a | 8.66 | 59.05 | 17.3 | 9.5 | - | 0.69 | n/a | [43] |
| Component | Di Blasi | Yang |
|---|---|---|
| Hemicellulose | 225–325 | 220–315 |
| Cellulose | 325–375 | 315–400 |
| Lignin | 250–500 | Up to 900 |
| Location | Type | Capacity | Reference |
|---|---|---|---|
| Aston University, UK | Ablative | 20 kg/h | [92] |
| Aston University, UK | Ablative | 2 kg/h dry | [157] |
| BBC, Canada | Ablative | 10–25 kg/h | [154] |
| DTU, Denmark | Ablative | 1.5 kg/h | [92] |
| Institute of Engineering Thermophysics, Ukraine | Ablative | 15 kg/h | [92] |
| Latvian State Institute, Latvia | Ablative | 150 g/h | [92] |
| NREL, USA | Ablative | 35 kg/h | [92] |
| PYTEC, Germany | Ablative | 15 kg/h | [154] |
| University of Hamburg, Germany | Ablative | 20 kg/h | [154] |
| Shandong University of Technology, China | Ceramic ball drop flow | 110 kg/h | [92] |
| BEST—Bioenergy and Sustainable Technologies GmbH, Austria | Fixed bed | 100–400 g | [158] |
| Lisbon Tech, Portugal | Fixed bed | 30 g | [159] |
| University of Science & Technology of China | Fixed bed | 500 g | [92] |
| Aston University, Birmingham, UK | Fluidized bed | 5 kg/h | [92] |
| Biomass Engineering Ltd., UK | Fluidized bed | 200 kg/h | [92] |
| Cirad, France | Fluidized bed | 2 kg/h | [92] |
| Curtin University, Australia | Fluidized bed | 2 kg/h | [92] |
| Energy Research Centre of the Netherlands | Fluidized bed | 1 kg/h | [92] |
| Guangzou Institute, China | Fluidized bed | 10 kg/h | [92] |
| Iowa State University, USA | Fluidized bed | 6 kg/h | [92] |
| Karlsruhe Institute of Technology, Germany | Fluidized bed | 100 g/h | a |
| Monash University, Australia | Fluidized bed | 1 kg/h | [92] |
| Netherlands Organisation for Applied Scientific Research (TNO) | Fluidized bed | 10 kg/h | [92] |
| NREL, USA | Fluidized bed | 10 kg/h | [92] |
| PNNL, USA | Fluidized bed | 1 kg/h | [92] |
| RTI, Canada | Fluidized bed | 20 kg/h | [92] |
| Shanghai JiaoTong University, China | Fluidized bed | 1 kg/h | [92] |
| Shenyang University, China | Fluidized bed | 1 kg/h | [92] |
| South East University, China | Fluidized bed | 1 kg/h | [92] |
| Texas A&M University, USA | Fluidized bed | 42 kg/h | [92] |
| Universidade de Campinas, Brazil | Fluidized bed | 100 kg/h | [92] |
| University of Adelaide, Australia | Fluidized bed | 1 kg/h | [92] |
| University of Ghent, Belgium | Fluidized bed | 0.3 kg/h | [92] |
| University of Hamburg, Germany | Fluidized bed | 60–3000 g/h | [160] |
| 30 kg/h | |||
| University of Maine, USA | Fluidized bed | 100 g/h | [92] |
| University of Melbourne, Australia | Fluidized bed | 100 g/h | [92] |
| University of Naples, Italy | Fluidized bed | 1 kg/h | [92] |
| University of Twente, Netherlands | Fluidized bed | 1 kg/h | [92] |
| USDA, ARS, ERRC, USA | Fluidized bed | 1 kg/h | [92] |
| Virginia Tech, USA | Fluidized bed | 100 g/h | [92] |
| vTI, Germany | Fluidized bed | 6 kg/h | [92] |
| VTT, Finland | Fluidized bed | 1 kg/h | [92] |
| Zhejiang University, China | Fluidized bed | 3 kg/h | [92] |
| Zhengzhou University, China | Fluidized bed | 2 kg/h | [92] |
| CPERI, Greece | Circulating fluid bed | 1 kg/h | [92] |
| Anhui U. of Science & Technology, China | Sprouted fluid bed | 5 kg/h | [92] |
| Ikerlan, Spain | Sprouted fluid bed | 10 kg/h | [92] |
| European Biomass Research Institute, Aston University, UK | Stationary fluidized bed | 7 kg/h | [151] |
| Georgia Tech, USA | Entrained flow | 56.7 kg/h | [161] |
| VTT, Finland | Entrained flow | 20 kg/h | [151,155] |
| Auburn U. USA | Screw | 1 kg/h | [92] |
| Michigan State University, USA | Screw | 500 g/h | [92] |
| Mississippi State University, USA | Screw | 2 kg/h | [92] |
| Texas A&M University, USA | Screw | 30 kg/h | [92] |
| European Biomass Research Institute, Aston University, UK | Twin-screw | 20 kg/h; 100 kg/h | [151] |
| Karlsruhe Institute of Technology, Germany | Twin-screw | 10 kg/h | [162] |
| TNO, Netherlands | Vortex | 30 kg/h | [92] |
| Federal University of Sergipe | Rotary kiln | 0.3 kg/h | [163] |
| Enterprise | Location | Type | Feedstock | Product | Maximum Capacity | Status | Reference |
|---|---|---|---|---|---|---|---|
| ABRI-Tech | Canada | Mobile screw | Sewage sludge | Bio-oil | 1 Ton/day | Demonstration, know-how provider | [151] |
| Agri-THERM/Western University | London, Canada | Mobile fluidized bed with novel heating system | Agricultural residues | Bio-oil, char | 10 Ton/day | Demonstration, know-how provider | [155,164] |
| Anhui Yineng Bio-energy Ltd., China | Hefei City, China | Moving bed | Biomass | Bio-oil | 2083 kg/h | Operating | [165] |
| Biogas Energy | Richmond, USA | Ablative | Demolition wood, forestry/agricultural residues | Bio-oil | 500 kg/h | Expected to have started in summer 2020 | [165] |
| bioliq ® | Karlsruhe Institute of Technology, Germany | Twin-screw reactor | Forestry/ agricultural residues | Biosyncrude for posterior gasification | 500 kg/h | Industrial pilot, operating semiannually | [93] |
| BP a | Grangemouth, Scotland | Fluidized bed | Mixed waste plastics | Waxes | 5000 Ton/year | Ceased operation (as of 1991) | [166] |
| Castle Capital b | Halifax, Canada | Ablative | Biomass | Bio-oil | 1–2 Ton/h | Ceased operation (as of 1996) | [167] |
| CarbonScape Ltd. | Several, based in Blenheim, New Zealand | Microwave | Pine sawdust, fossil coal | Coke for graphitization | Variable | Operating, consulting services | [168] |
| DRP/ABB a | Ebenhausen, Germany | Fluidized bed | Waste plastics | Pyrolysis liquid, carbon black | 800 kg/h | Ceased operation (as of 1991) | [166] |
| DRP/ABB a | Grimma, Germany | Fluidized bed | Waste tires | Carbon black | 5000 Ton/year | Ceased operation (as of 1991) | [166] |
| Dynamotive’s Integrated Pyrolysis Combined Cycle Biomass Power System | West Lorne, Canada | Stationary fluidized bed | Lignocellulosic biomass | Bio-oil, blended liquid fuels | 4200 kg/h [156] | Ceased operation (as of 2008 [169]) | [170] |
| Dynamotive’s Integrated Pyrolysis Combined Cycle Biomass Power System | Guelph, Canada | Stationary fluidized bed | Lignocellulosic biomass | Bio-oil, blended liquid fuels | 8400 kg/h [156] | Ceased operation (as of 2008 [169]) | [170] |
| EGEMIN | Belgium | Flash entrained flow | Biomass | Bio-oil | 200 kg/h | Unknown (as of 2021) | [171] |
| Energolesprom c | Kazan, Russia | Ablative | Forestry/agricultural residues | Bio-oil | 500 kg/h | Industrial pilot | [172] |
| Envergent Technologies’ RTP™ (Rapid Thermal Processing) d | Rhinelander, Wisconsin, USA | Circulating fluidized bed | Forestry residues | Liquid smoke, preservatives | 45 Ton/day | Operating | [173] |
| Envergent Technologies’ RTP™ (Rapid Thermal Processing) d | Côte Nord, Canada | Circulating fluidized bed | Forestry residues | Heating fuel | 65,000 dry Ton/year | Operating | [174] |
| Envergent Technologies’ RTP™ (Rapid Thermal Processing) d | Aracruz, ES, Brazil | Circulating fluidized bed | Eucalyptus residues from pulp production | Bio-oil | 2.3 MTon/year | Under project | [175] |
| Fortum Orso® e | Joensuu, Finland | Circulating fluidized bed | Forestry residues | Bio-oil for district heating | 7 Ton/day | Operating | [151,176] |
| Interchem c | Kansas, USA | Ablative | Biomass | Bio-oil | 1360 kg/h | Ceased operation (early 1900s) | [167] |
| Lurgi LR | Ingolstadt, Germany | Twin-screw | Biomass, plastic | Light and heavy bio-oil for catalytic refining | 500 kg/h [92] | Unknown (as of 2021) | [92,177] |
| MRIGlobal b | Lutherville-Timonium, USA | Vortex | Biomass | Bio-oil, coke | 3 cm/s | Patent | [178,179] |
| Pyrovac/Corigin | Jonquière, Canada | Vacuum pyrolysis using horizontal conveyers (molten salt system) | Biomass | Bio-oil | 3000 kg/h dry | Demonstration, know-how provider | [180] |
| PyTEC | Cuxhafen, Germany | Ablative | Biomass | Bio-oil | 250 kg/h [92,156] | Unknown (as of 2021) | [181] |
| Renewable Oil International® | Florence, USA | Augur | Chicken litter | Bio-oil, char | 5 Ton/day | Unknown (as of 2006) | [182] |
| Thermo-Catalytic Reformer (TCR)® | Fraunhofer UMSICHT, Sulzbach-Rosenberg, Germany | Catalytic screw | Sewage sludge, organic waste | Drop-in fuel, fuel oil additive, gasoline | 300 kg/h | Industrial pilot | [183] |
| Toshiba | Sapporo, Japan | Rotating kiln | Mixed plastic waste | Bio-oil, gas, HCl from PVC | 14 kTon/year | Operating | [184] |
| Twence/Empyro BV f | Hengelo, Netherlands | Rotating cone | Forestry residues | Bio-oil | 20 M liters bio-oil/year | Operating | [185] |
| Twence/Empyro BV f | Genting, Malaysia | Rotating cone | Palm tree residues | Bio-oil | 6 wet Ton/h | Dormant | [186] |
| University of Hamburg | Hamburg, Germany | Ablative | Biomass | Bio-oil | 250 kg/h | Research pilot | [154] |
| University of Science & Technology of China | Hefei City, China | Fluidized bed | Biomass | Bio-oil | 650 kg/h | Research pilot | [92] |
| Virginia Tech, USA | Blacksburg, USA | Fluidized bed | Poultry litter | Bio-oil, coke | 250 kg/h [156] | Mobile demonstration pilot | [187] |
| Woodgas Pyrolytics® Rotatable Covered Cavity Kiln | Normal, USA | Rotating kiln | Forestry/agricultural residues | Coke, heat | 250 kg/day | Demonstration, know-how provider | [148] |
| Temperature (°C) | H2 Pressure (Bar) | |
|---|---|---|
| Naphta | 260–350 | 15–35 |
| Light oil | 290–400 | 17–35 |
| Heavy oil | 350–425 | 70–140 |
| Residuum hydrocracking | 400–425 | 140–200 |
| Fast pyrolysis bio-oil | 175–450 | 80–230 |
| Niobium Function | Publication | Catalysts | HDT Feed |
|---|---|---|---|
| Support | Teles et al. (2018) [256] | Pd/SiO2, Pd/Nb2O5, Pd/NbOPO4 | Pine pyrolysis product, phenol, anisole, m-cresol, guaiacol (vapor phase HDT). |
| Support | Li et al. (2020) [243] | RuFe/Nb2O5 | Birch lignin oil, 3-(4-hydroxyphenyl)propanol |
| Support | Dong et al. (2018) [242] | Pd/Nb2O5 and Pt/Nb2O5 | Extracted raw birch lignin, 4-methylphenol |
| Support | Shao et al. (2017) [241] | Ru/Nb2O5 | Extracted birch lignin, 4-methylphenol |
| Support | Xin et al. (2019) [244] | Ru/Nb2O5 | 4-methylphenol, enzymatic lignin. |
| Support | Ma et al. (2019) [255] | Ru/layered Nb2O5, Ru/NbPO, Ru/Nb2O5.H2O (HY-340) | Enzymatic lignin, p-cresol |
| Support | Leal et al. (2019) [254] | Ni/TT- Nb2O5 nanorods | Lignin oil from catalytic upstream biorefining, diphenyl ether |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmitt, C.C.; Fonseca, F.G.; Fraga, M.M.C.; Wisniewski, A., Jr.; Karp, S.; Mello José, Á.H.; Rodrigues, R.C.L.B.; Moreira, R.; Hirayama, D.E.; Raffelt, K.; et al. Thermochemical and Catalytic Conversion Technologies for the Development of Brazilian Biomass Utilization. Catalysts 2021, 11, 1549. https://doi.org/10.3390/catal11121549
Schmitt CC, Fonseca FG, Fraga MMC, Wisniewski A Jr., Karp S, Mello José ÁH, Rodrigues RCLB, Moreira R, Hirayama DE, Raffelt K, et al. Thermochemical and Catalytic Conversion Technologies for the Development of Brazilian Biomass Utilization. Catalysts. 2021; 11(12):1549. https://doi.org/10.3390/catal11121549
Chicago/Turabian StyleSchmitt, Caroline Carriel, Frederico Gomes Fonseca, Mariana M. Campos Fraga, Alberto Wisniewski, Jr., Susan Karp, Álvaro Henrique Mello José, Rita C. L. B. Rodrigues, Renata Moreira, Danilo Eiji Hirayama, Klaus Raffelt, and et al. 2021. "Thermochemical and Catalytic Conversion Technologies for the Development of Brazilian Biomass Utilization" Catalysts 11, no. 12: 1549. https://doi.org/10.3390/catal11121549
APA StyleSchmitt, C. C., Fonseca, F. G., Fraga, M. M. C., Wisniewski, A., Jr., Karp, S., Mello José, Á. H., Rodrigues, R. C. L. B., Moreira, R., Hirayama, D. E., Raffelt, K., & Dahmen, N. (2021). Thermochemical and Catalytic Conversion Technologies for the Development of Brazilian Biomass Utilization. Catalysts, 11(12), 1549. https://doi.org/10.3390/catal11121549