
Vinylogous and Arylogous Stereoselective Base-Promoted Phase-Transfer Catalysis



Abstract
{kind=link}
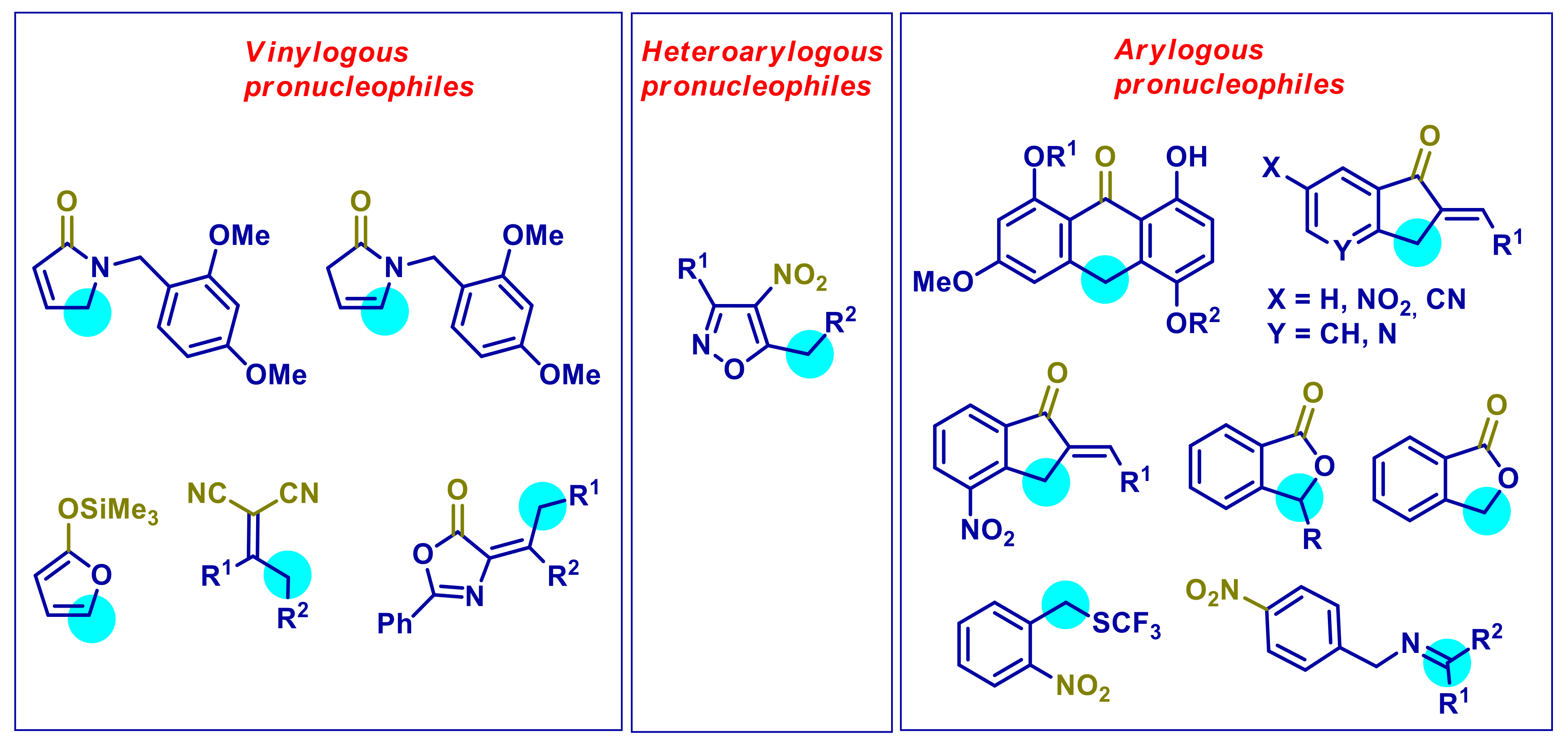{kind=link}
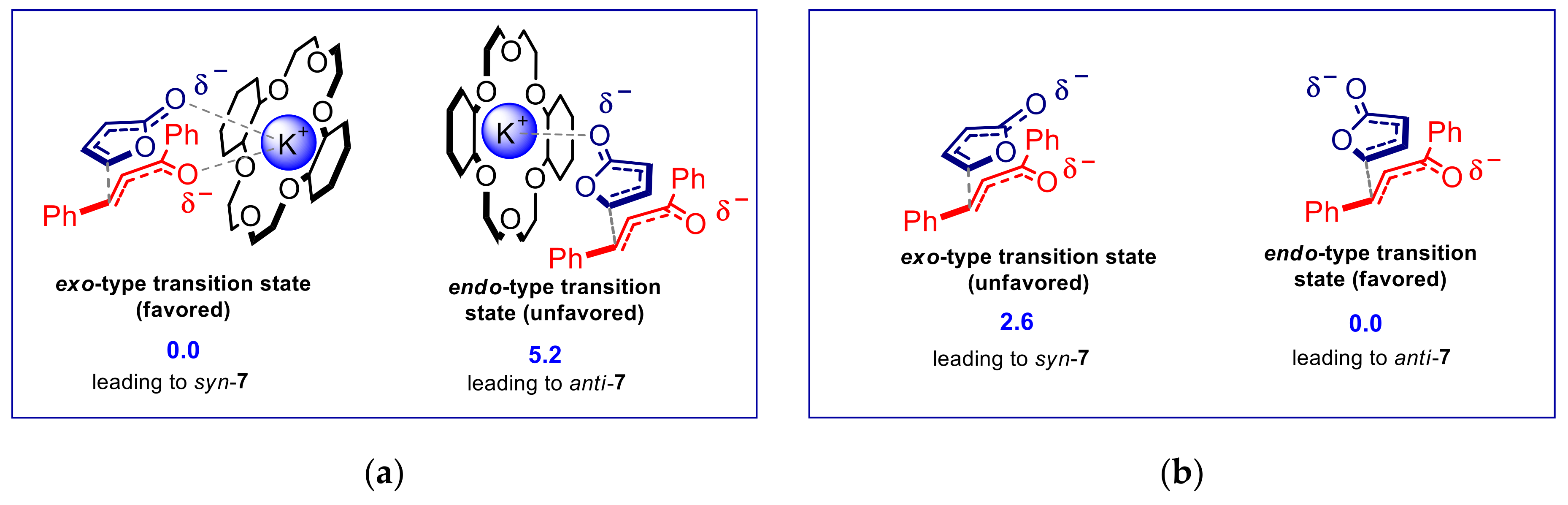{kind=link}
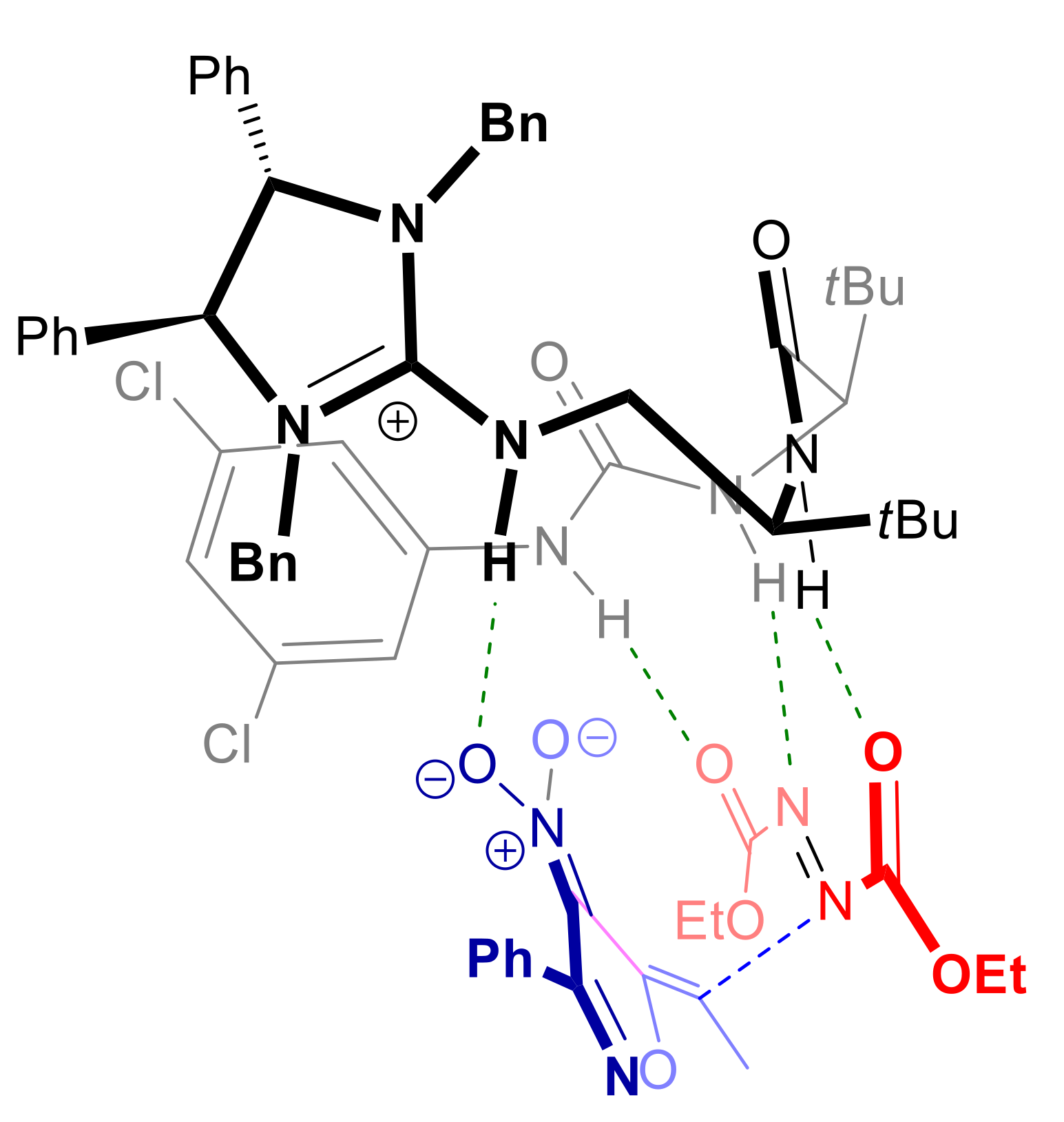{kind=link}
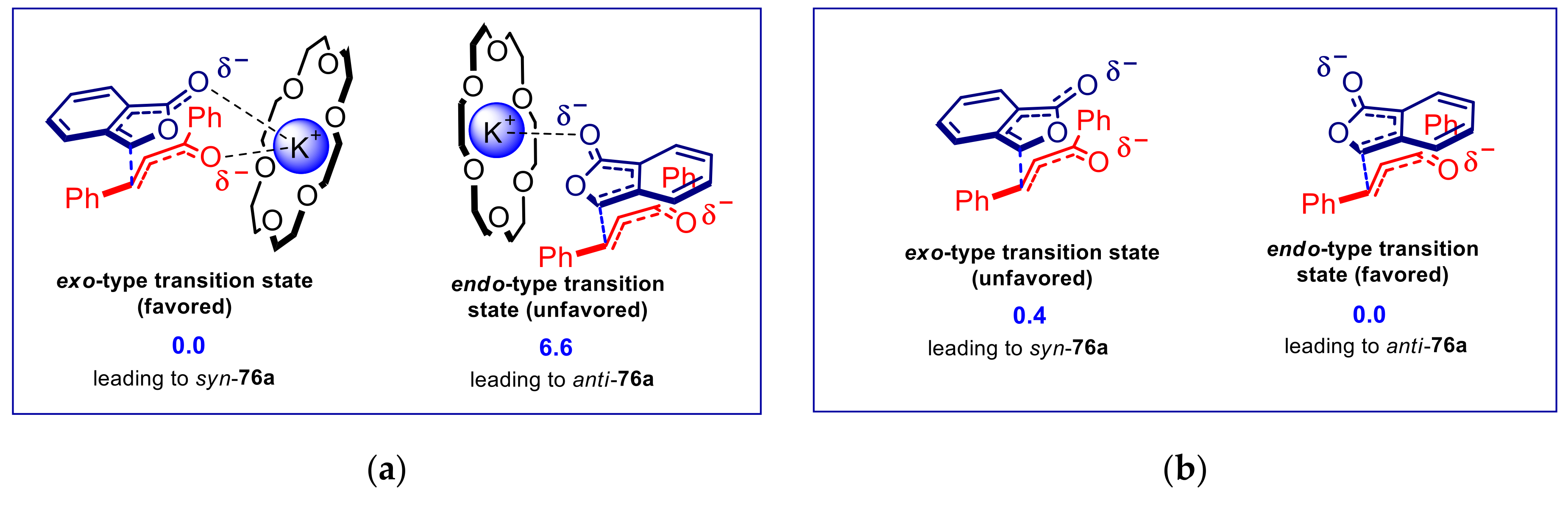{kind=link}
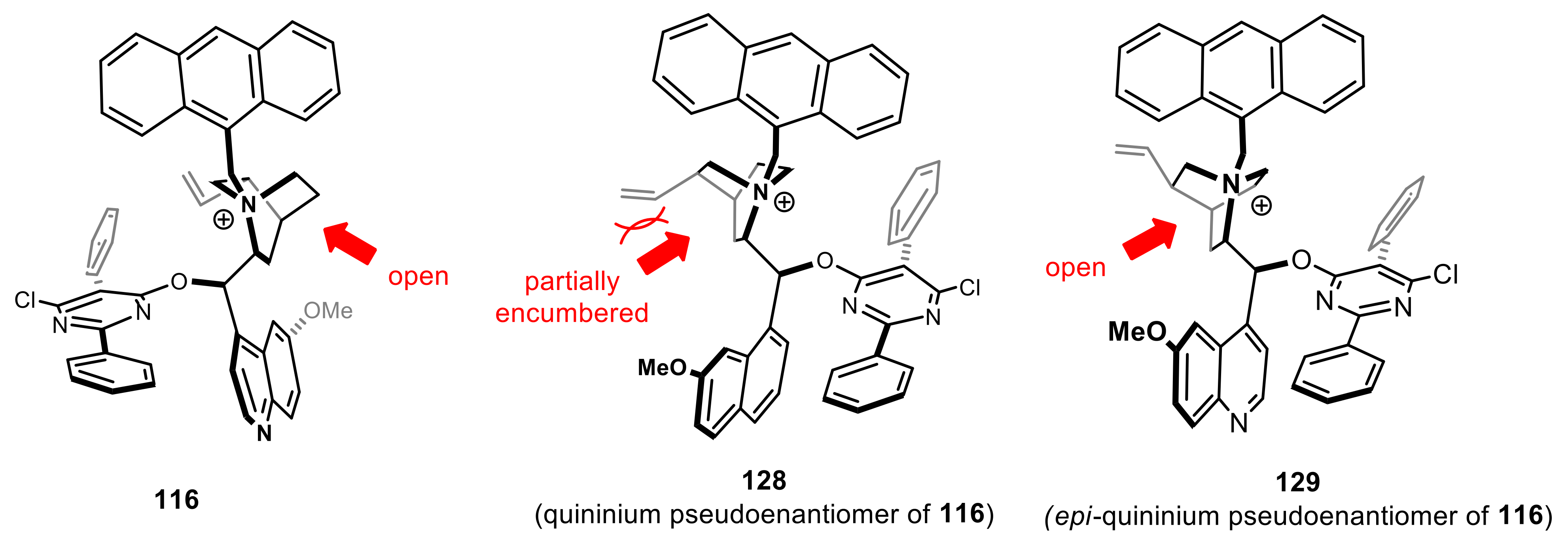{kind=link}
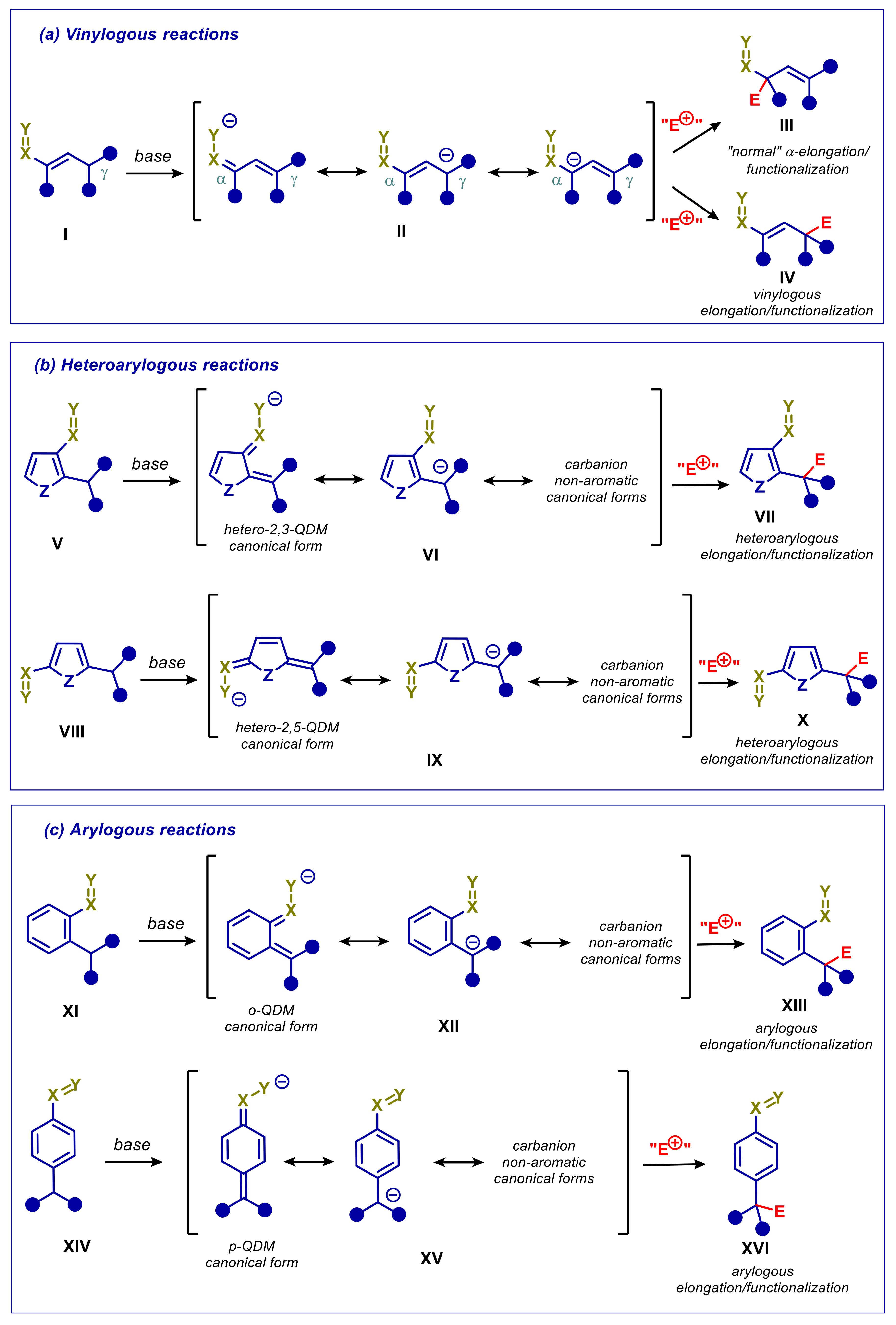{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
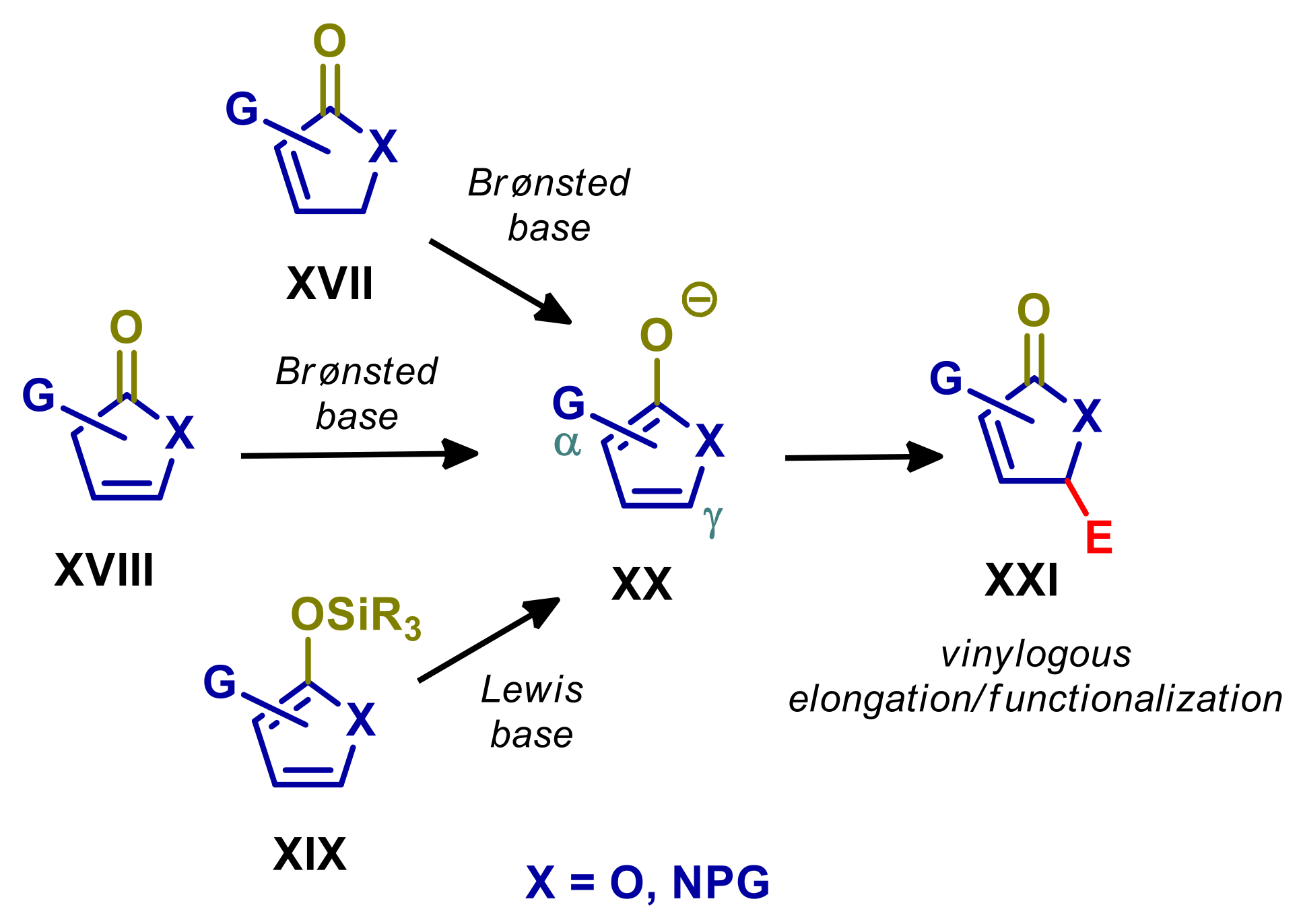
1. Introduction
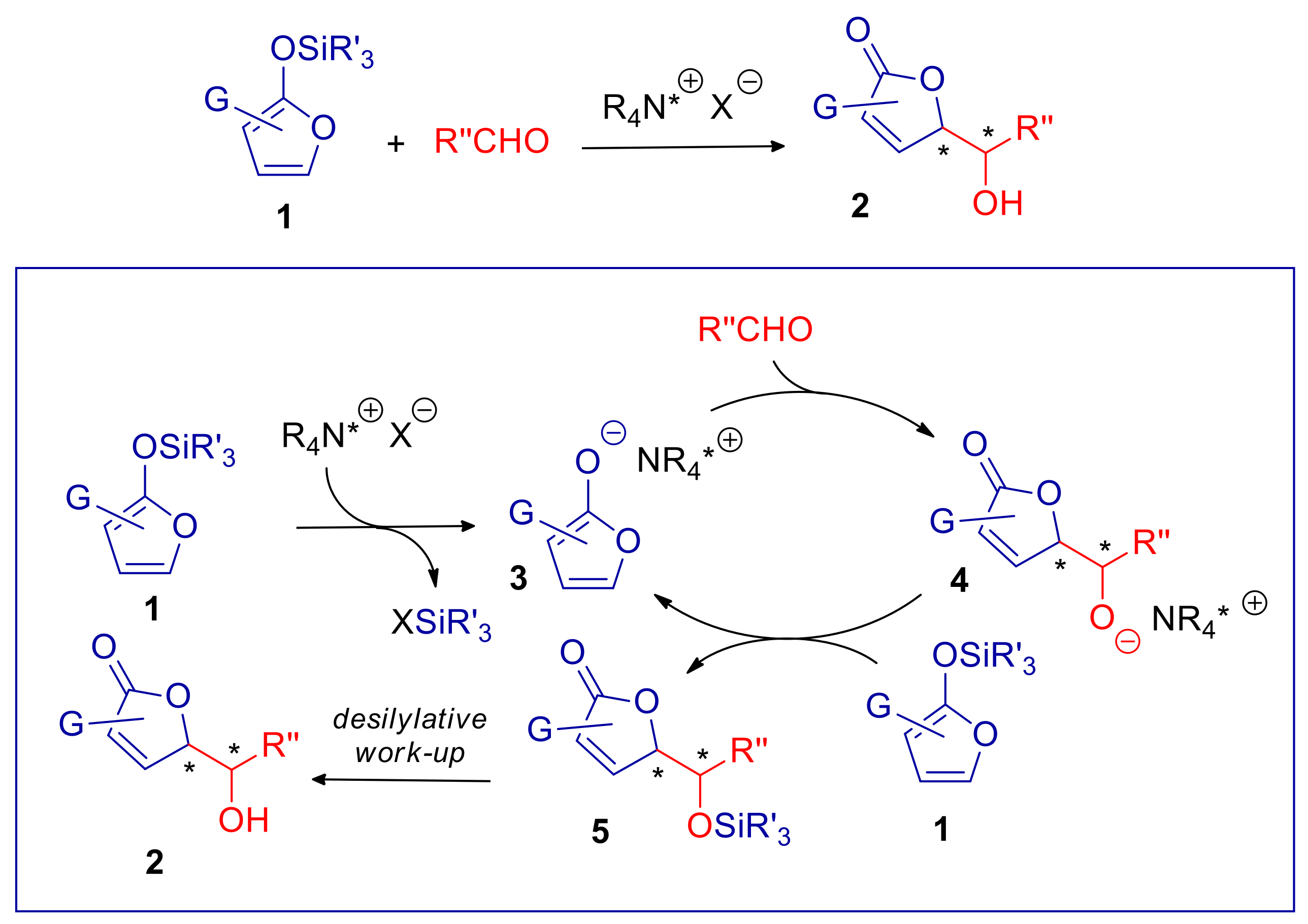
2. Reactions Involving Vinylogous Nucleophiles
2.1. γ-Butenolides and 2-Pyrrolinones
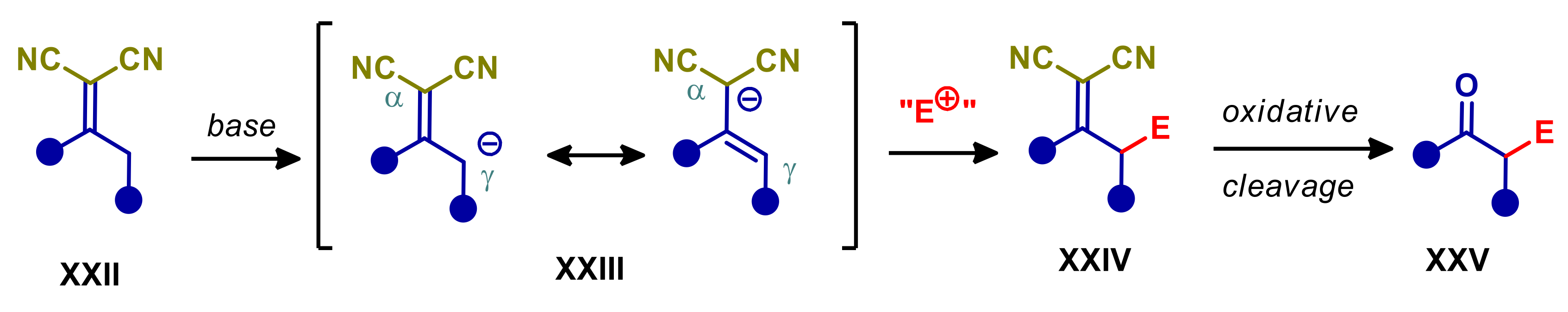
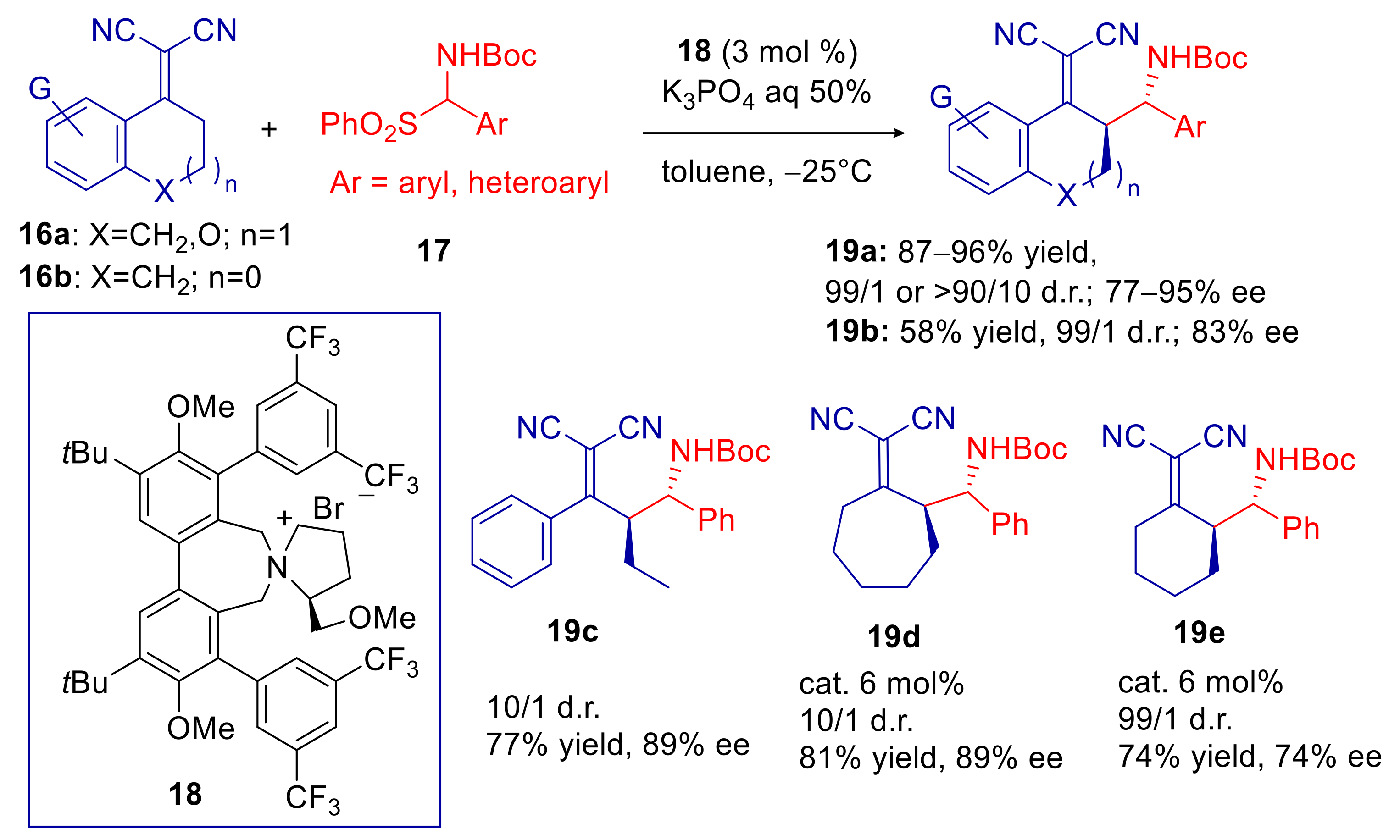
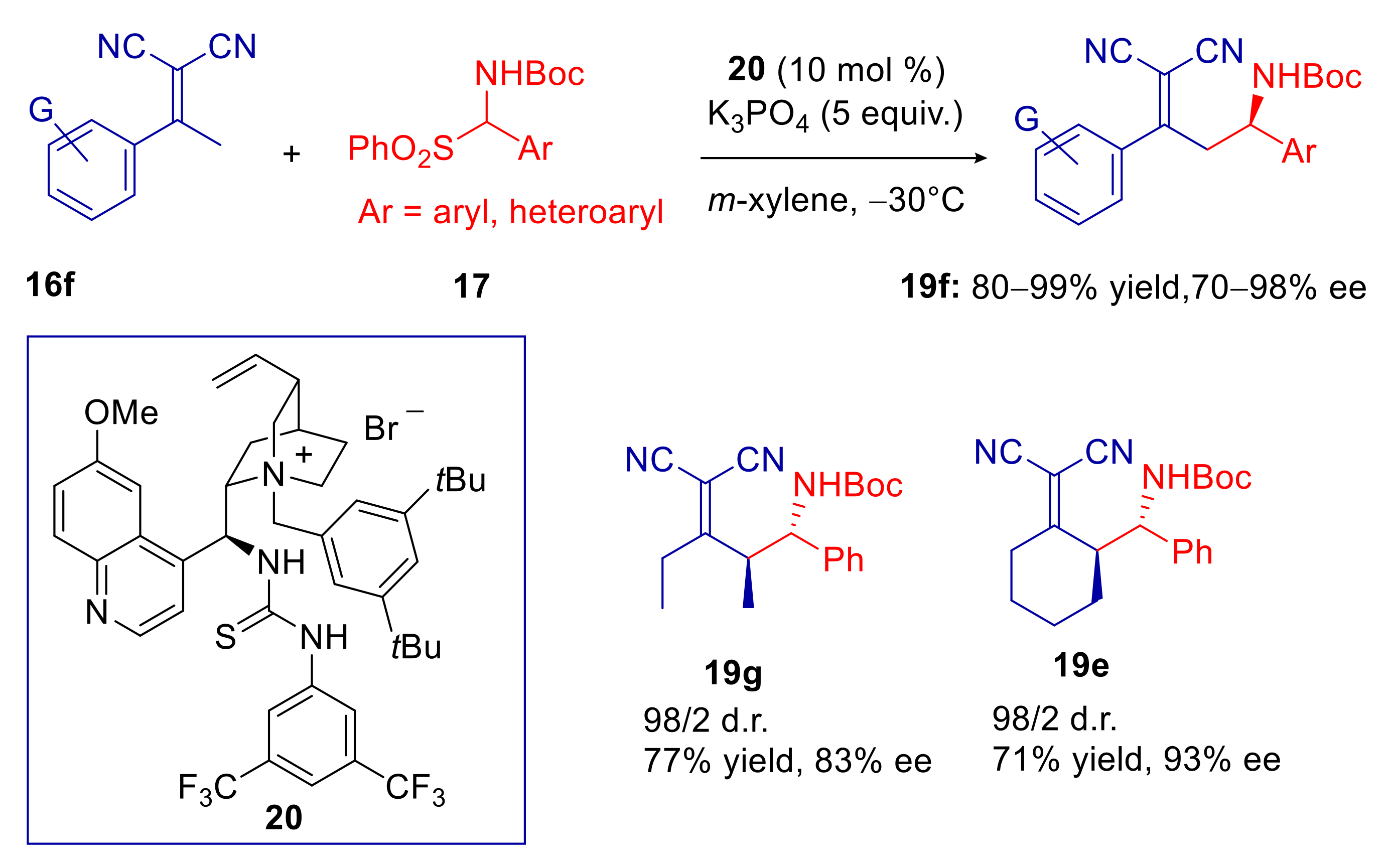
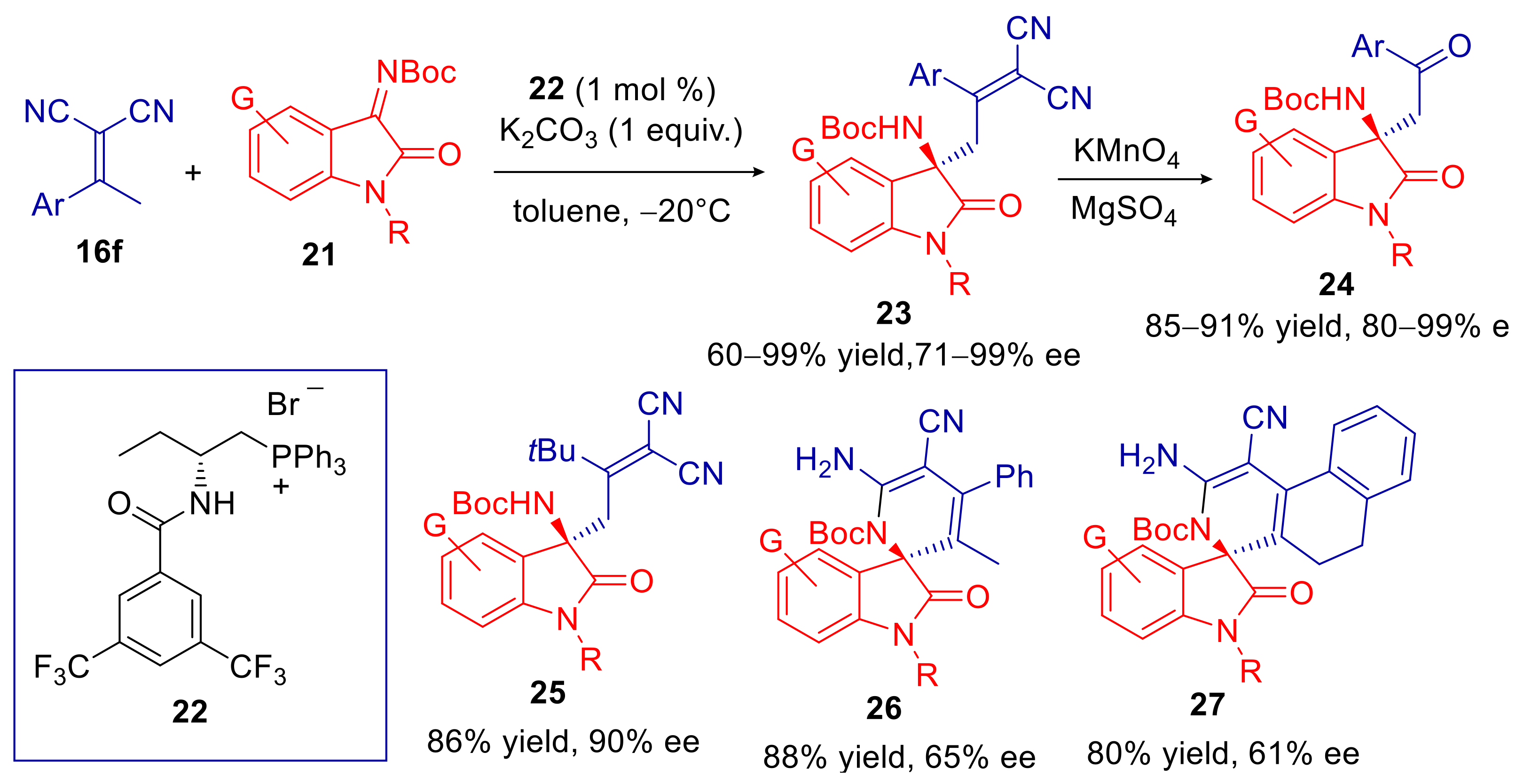
2.2. α,α-Dicyanoalkylidenes
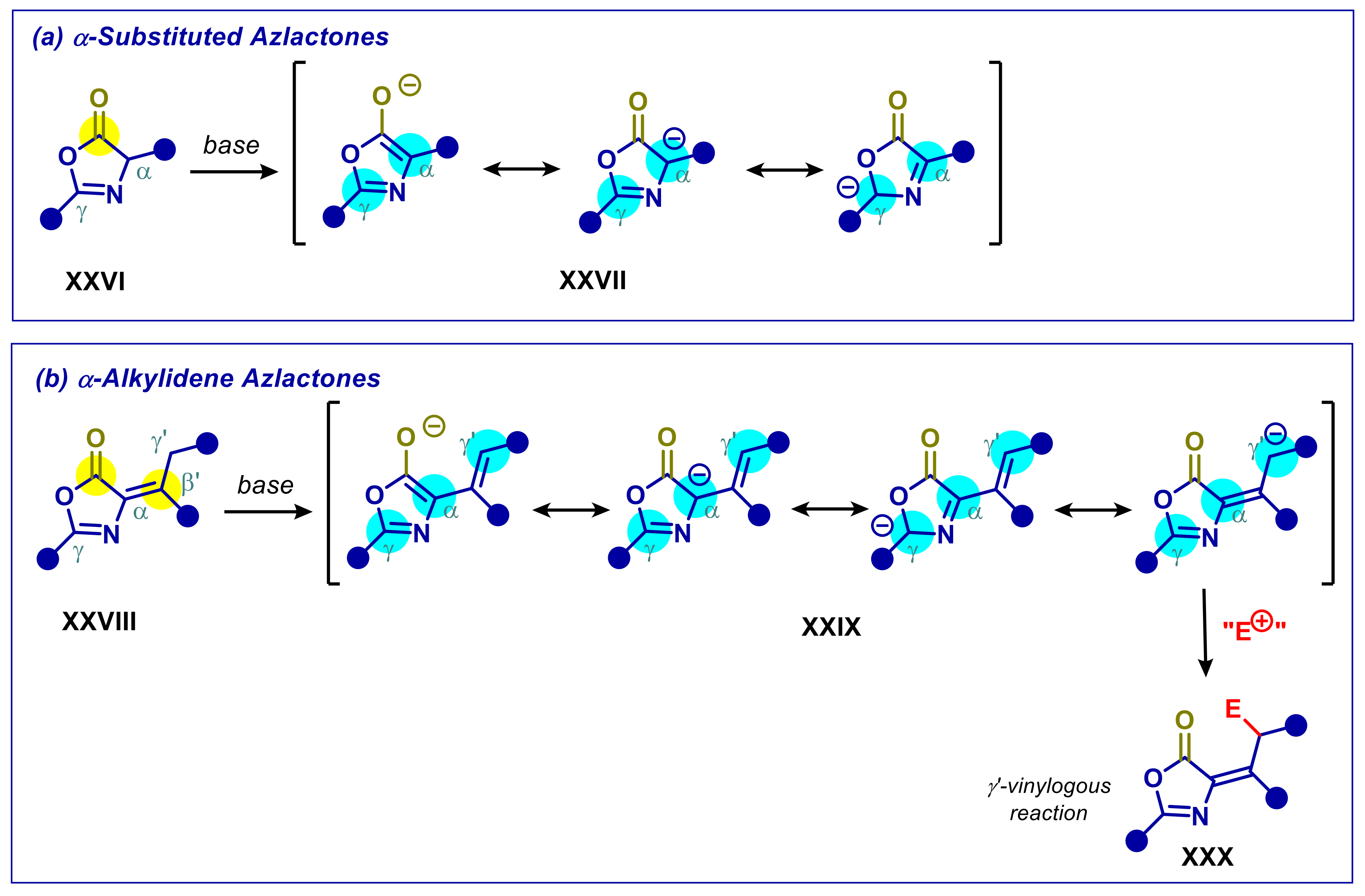
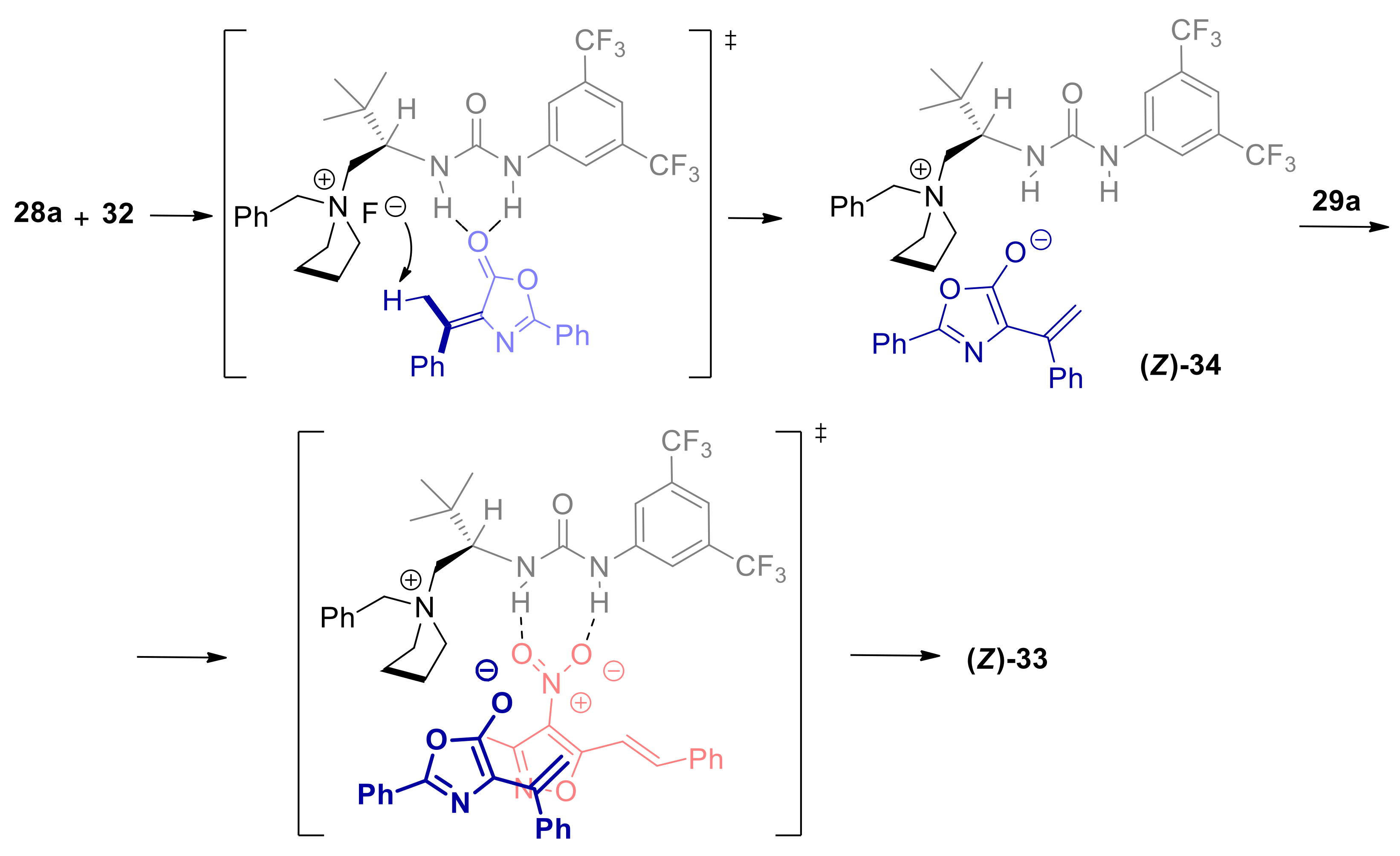
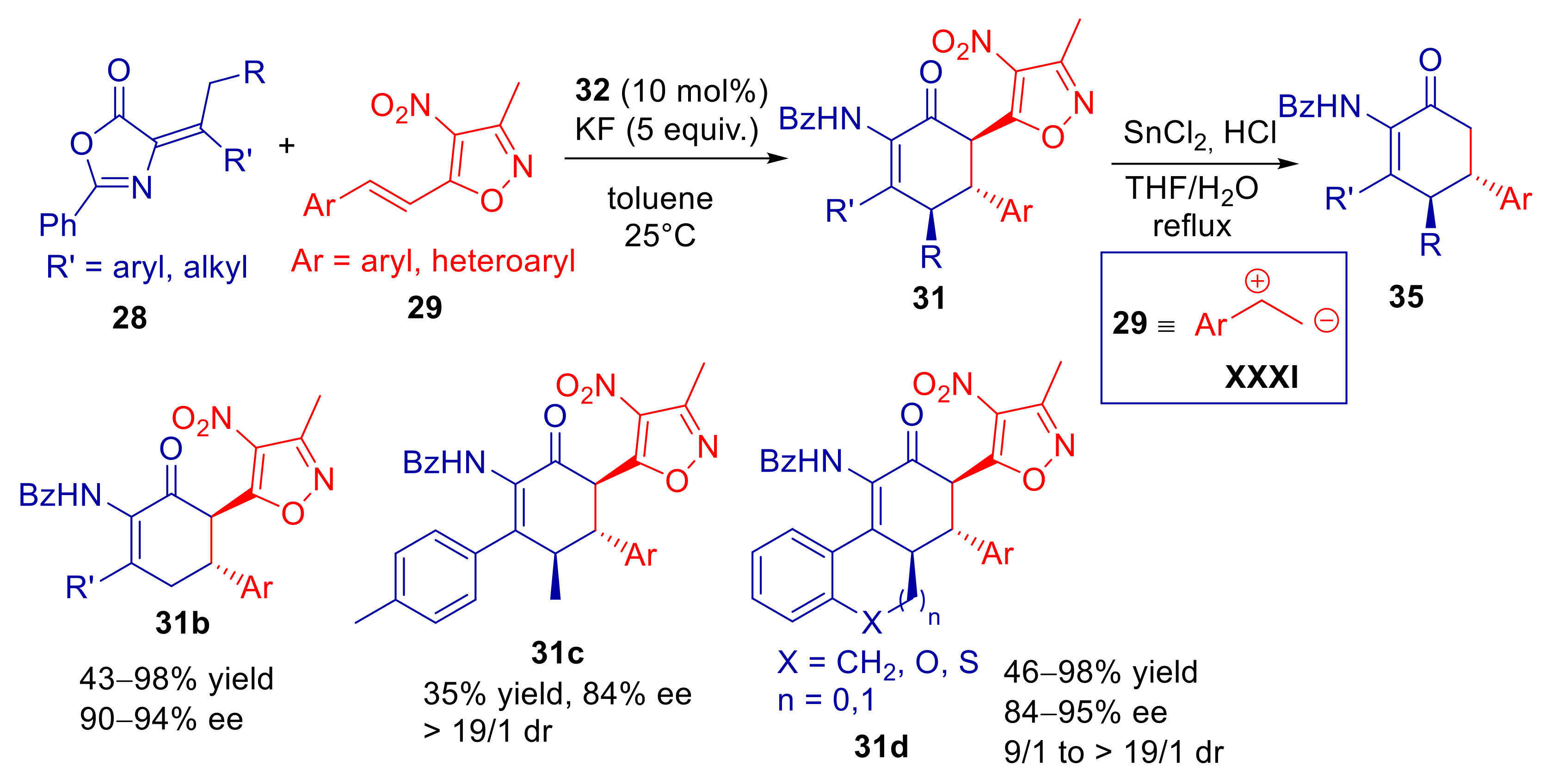
2.3. α-Alkylidene Azlactones
3. Reactions Involving Heteroarylogous Nucleophiles
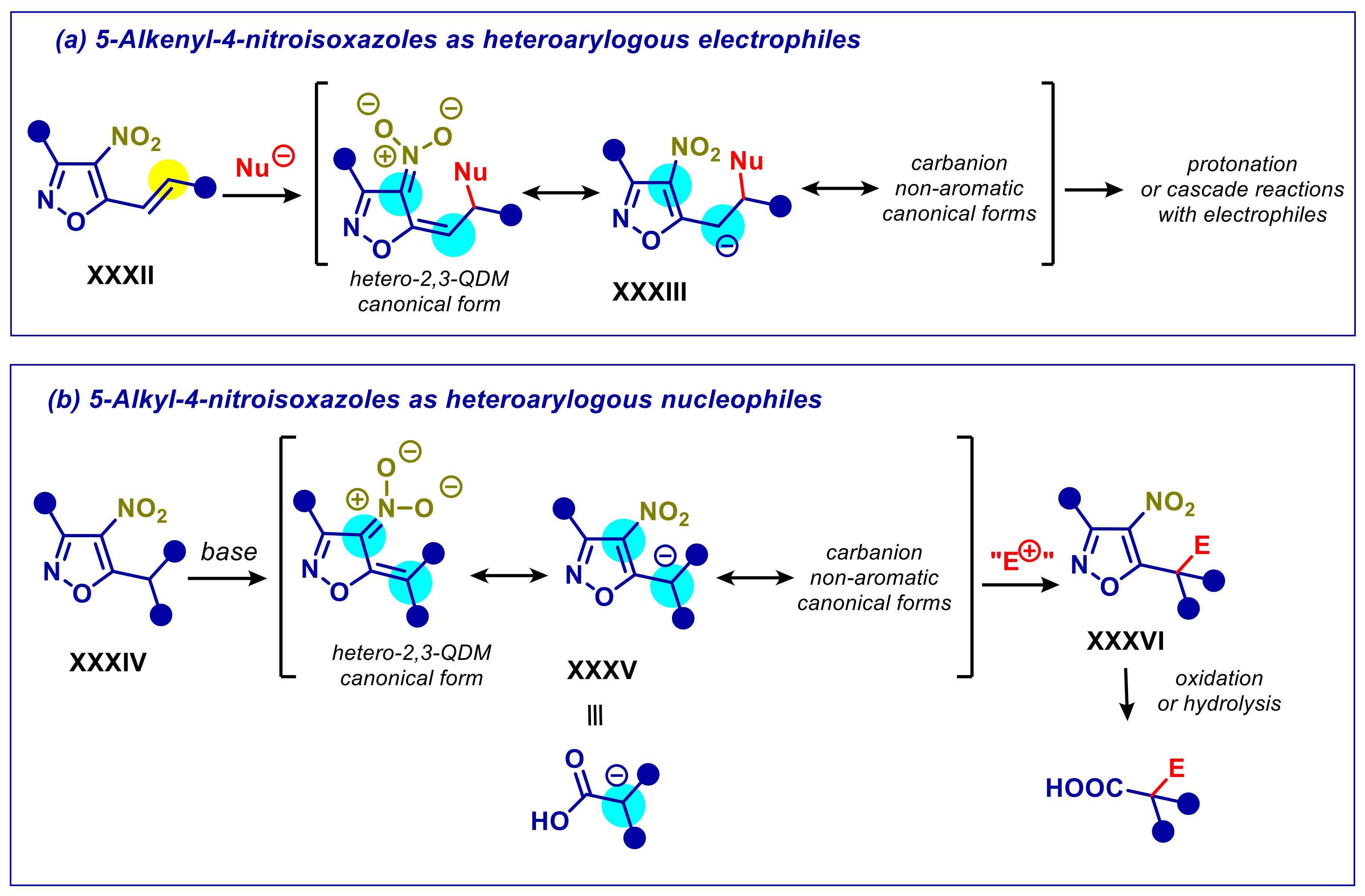
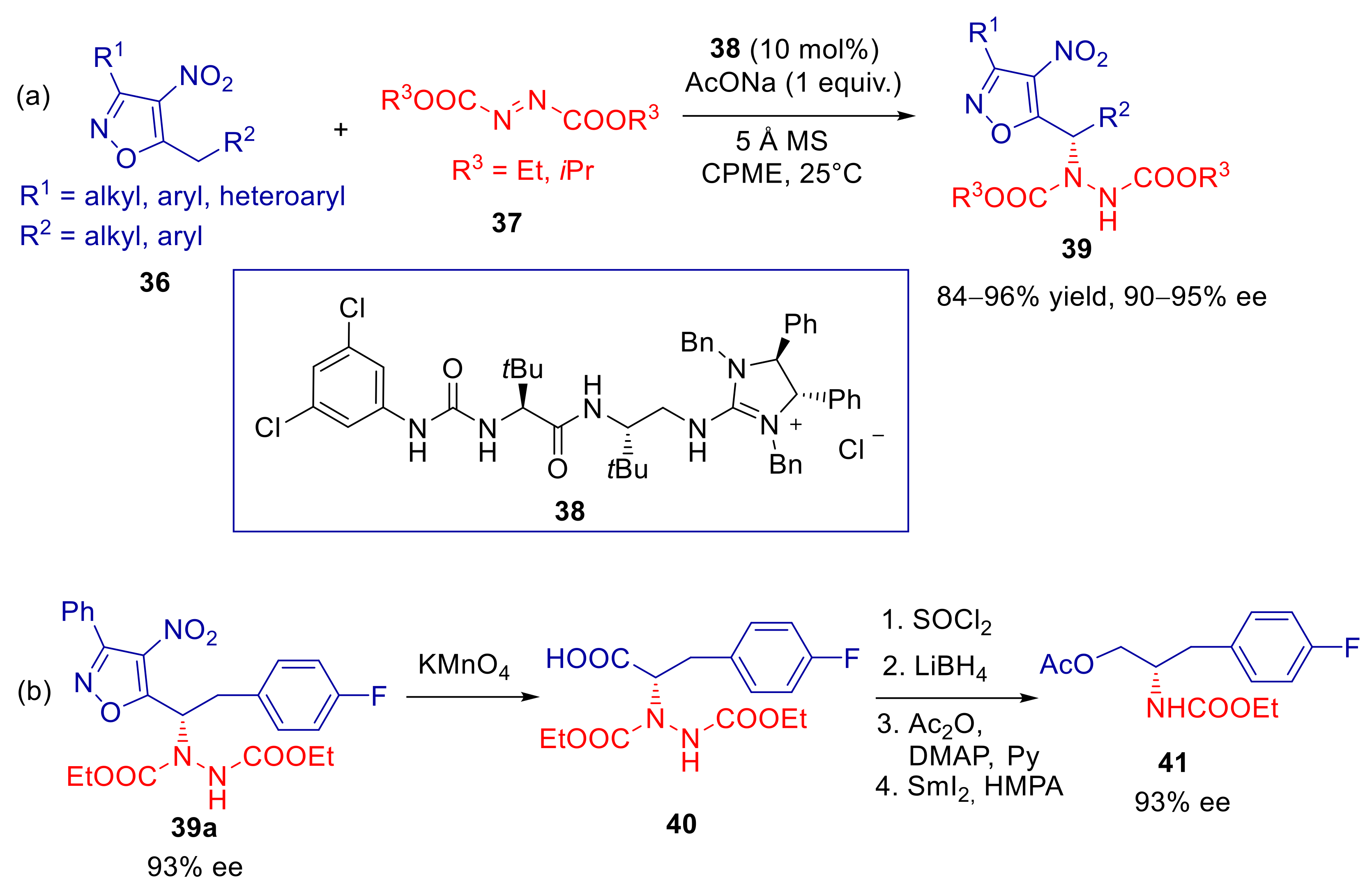
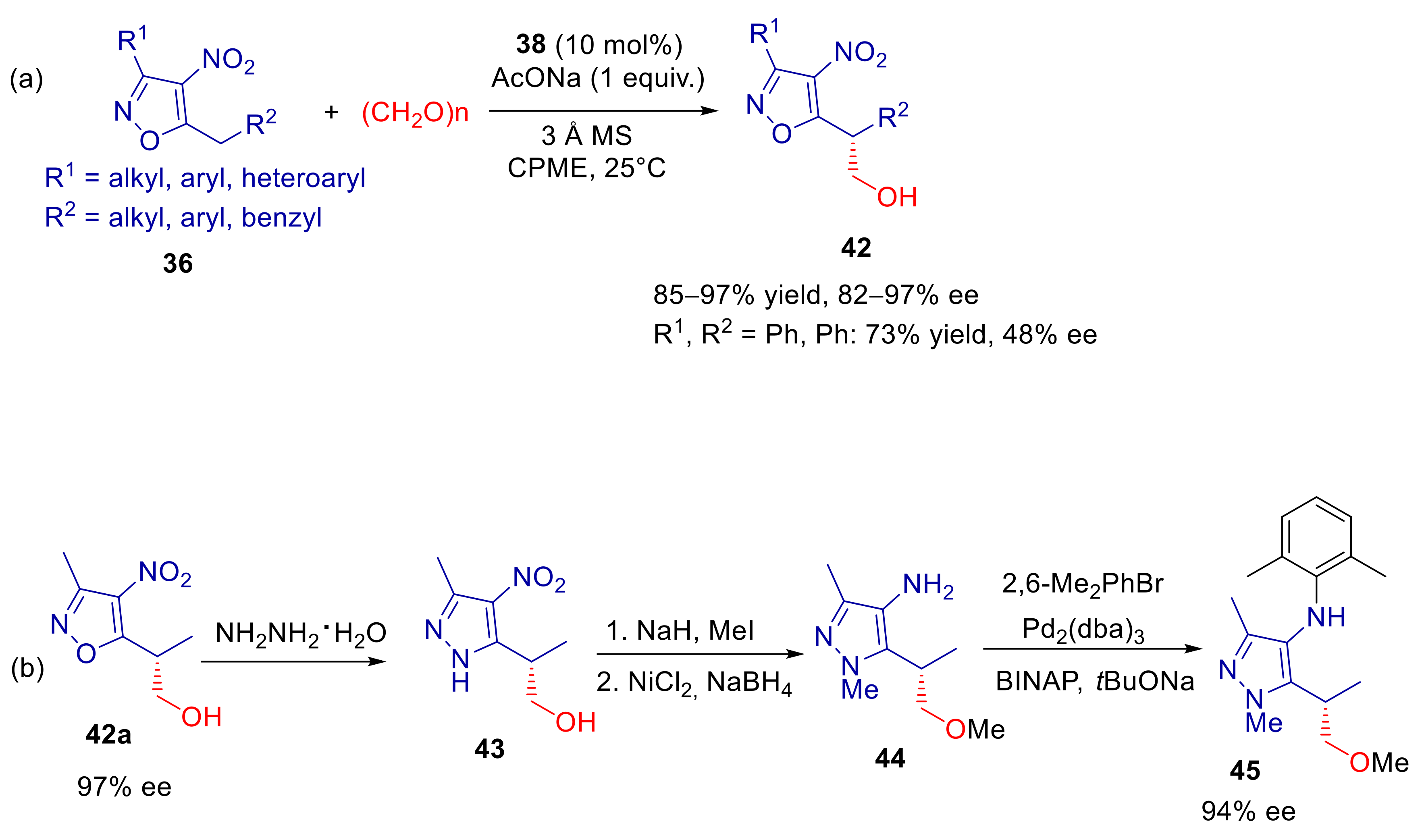
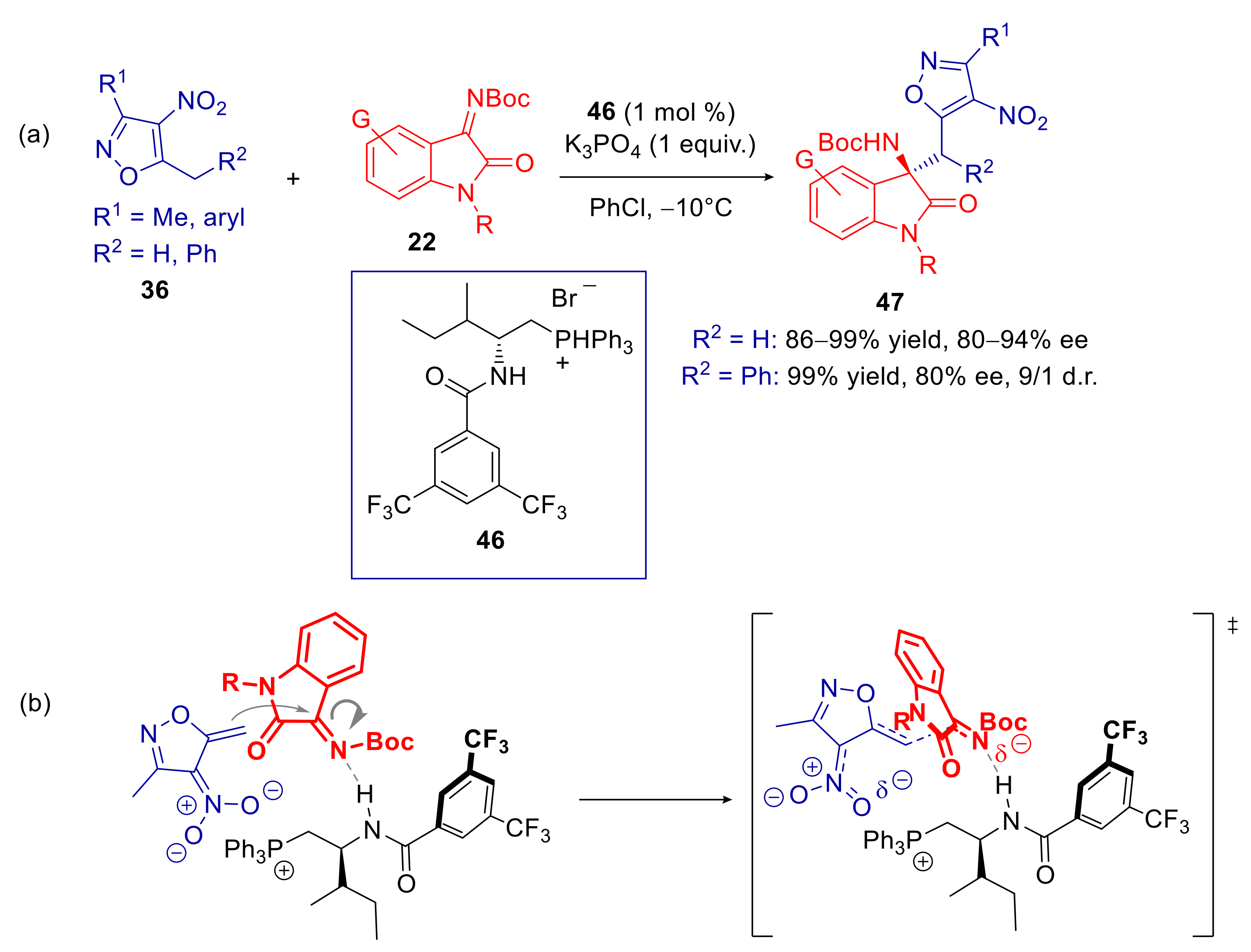
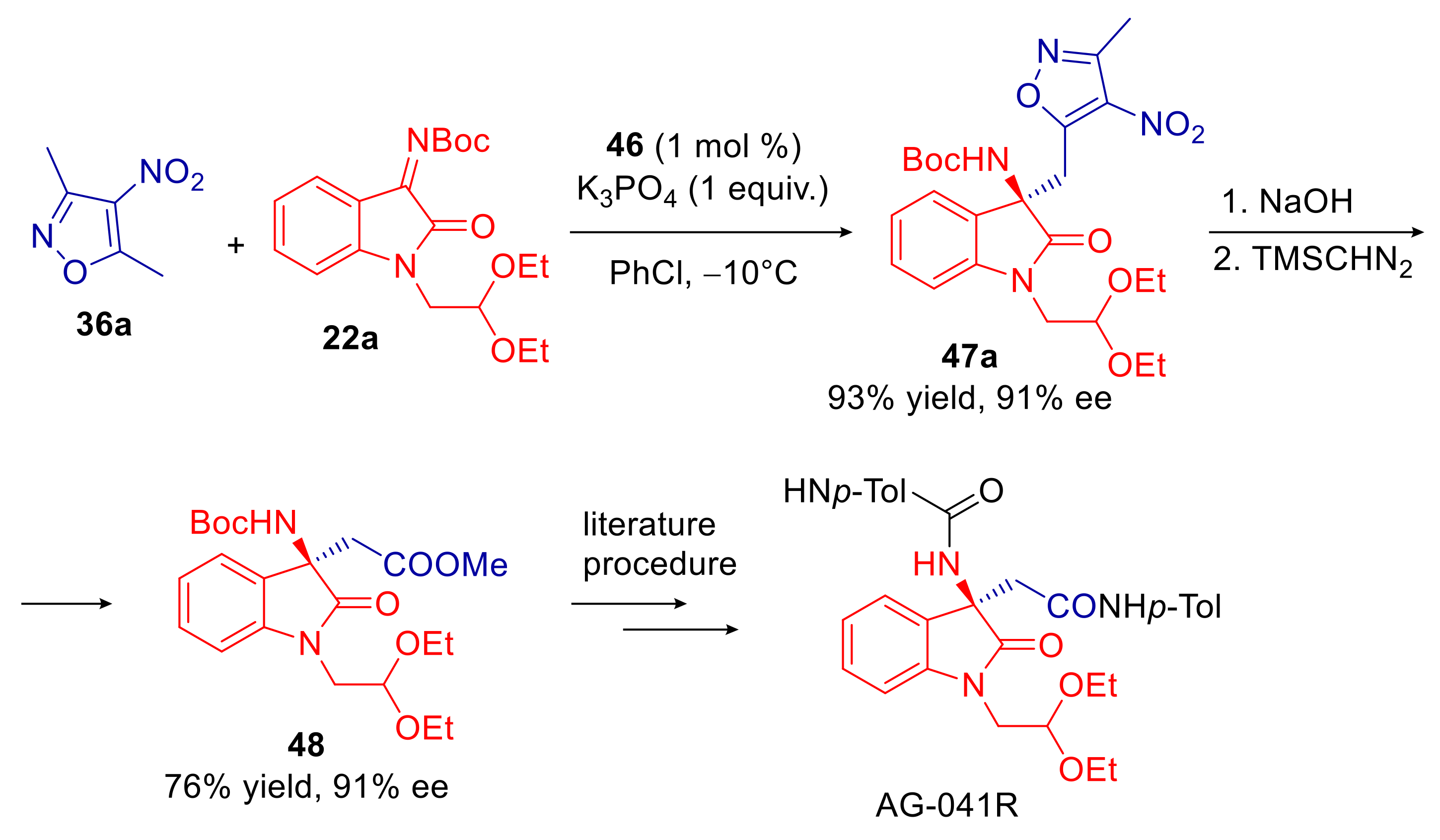
3.1. 5-Alkyl-4-nitroisoxazoles
4. Reactions Involving Arylogous Nucleophiles
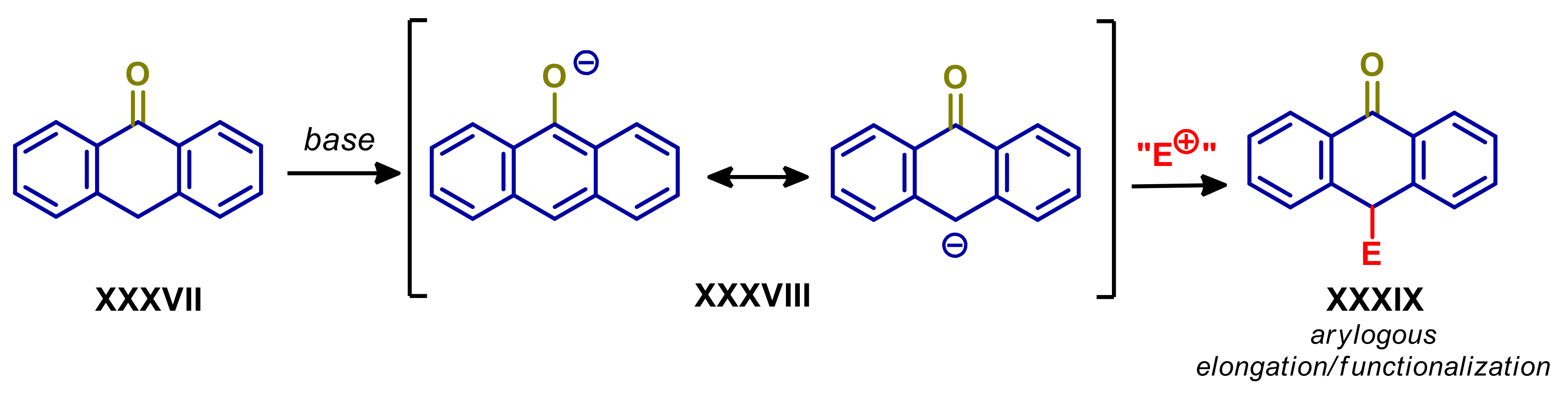
4.1. Anthrones
4.2. 2-Alkylidene-1-indanones
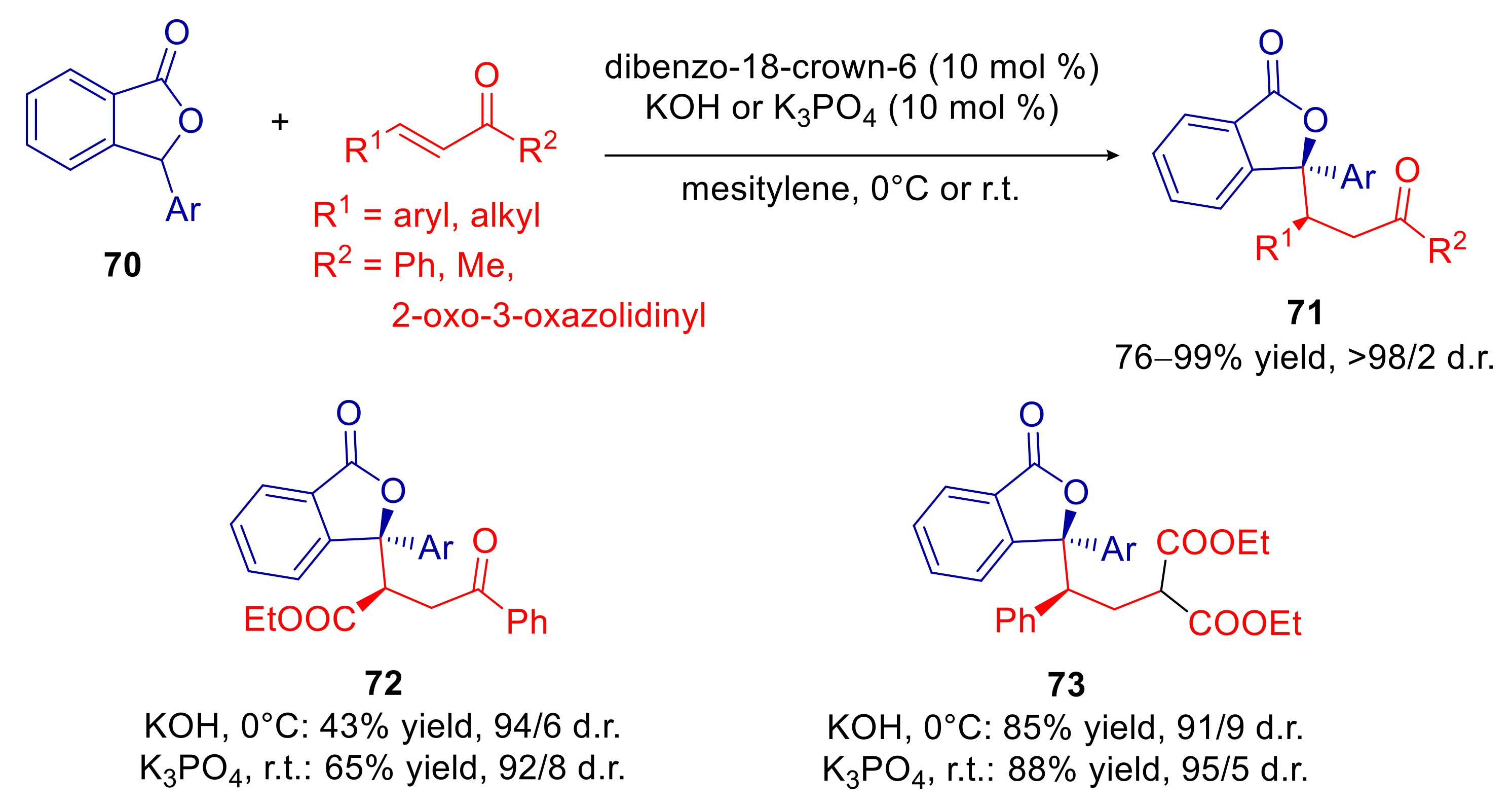
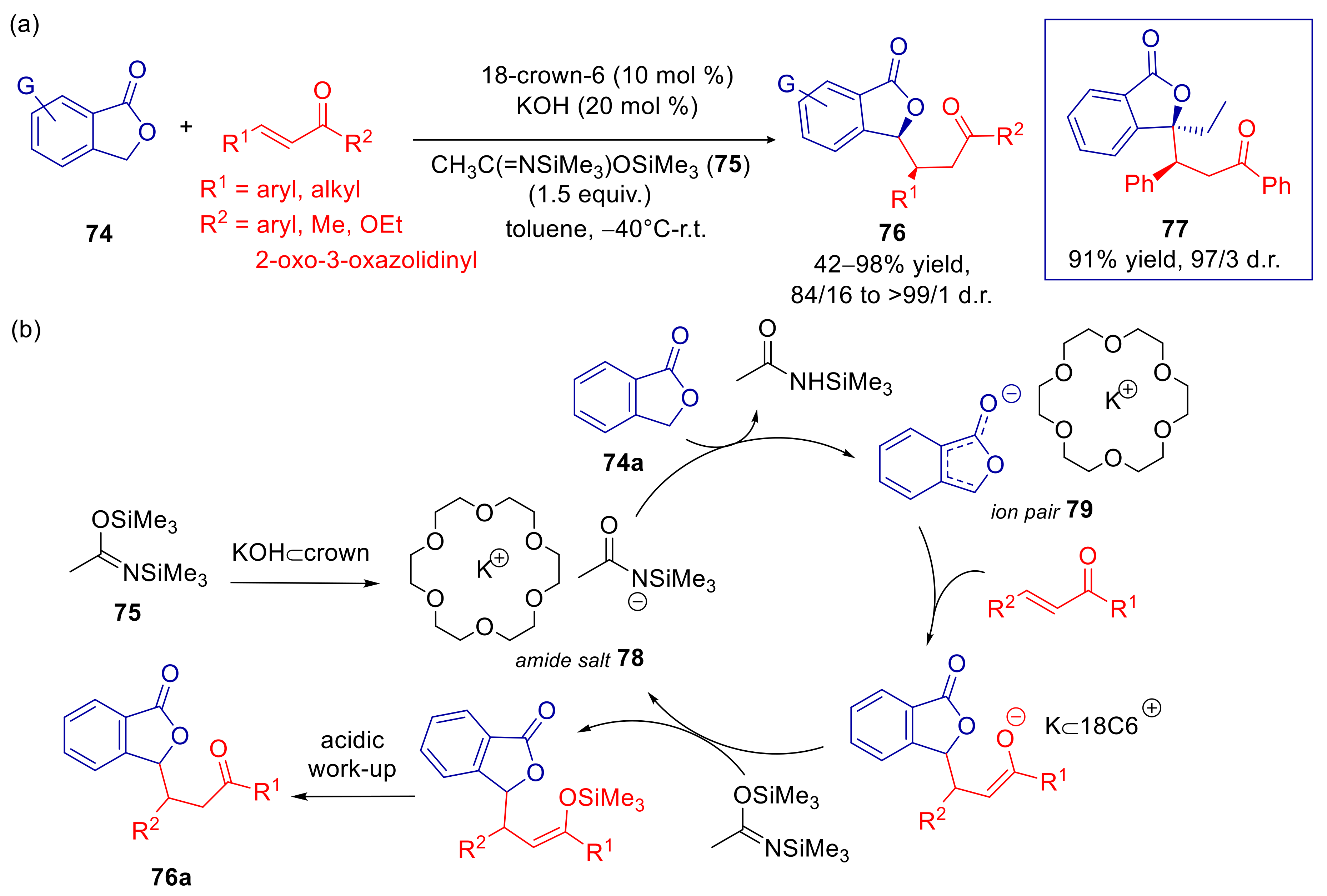
4.3. Phthalides
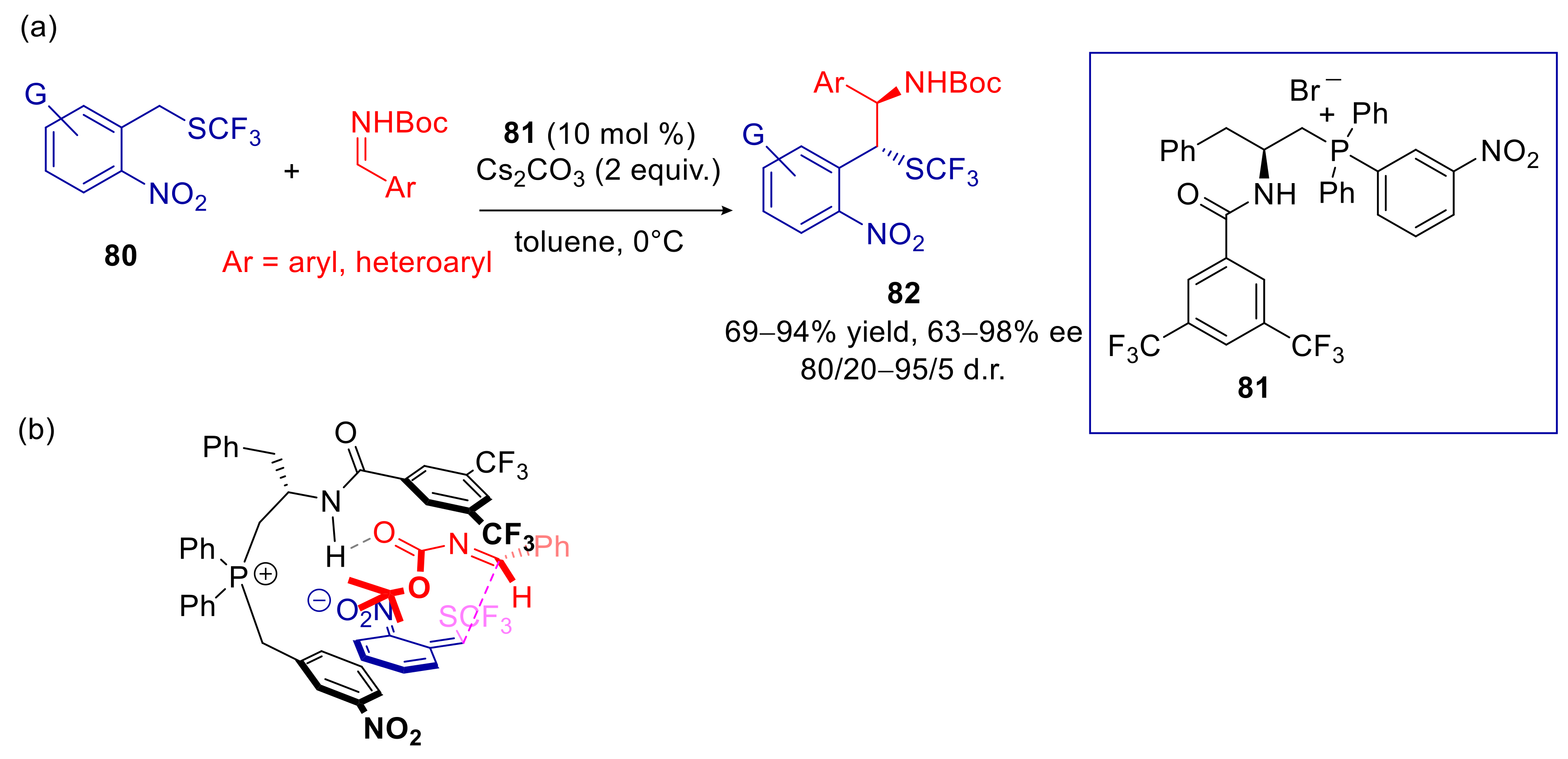
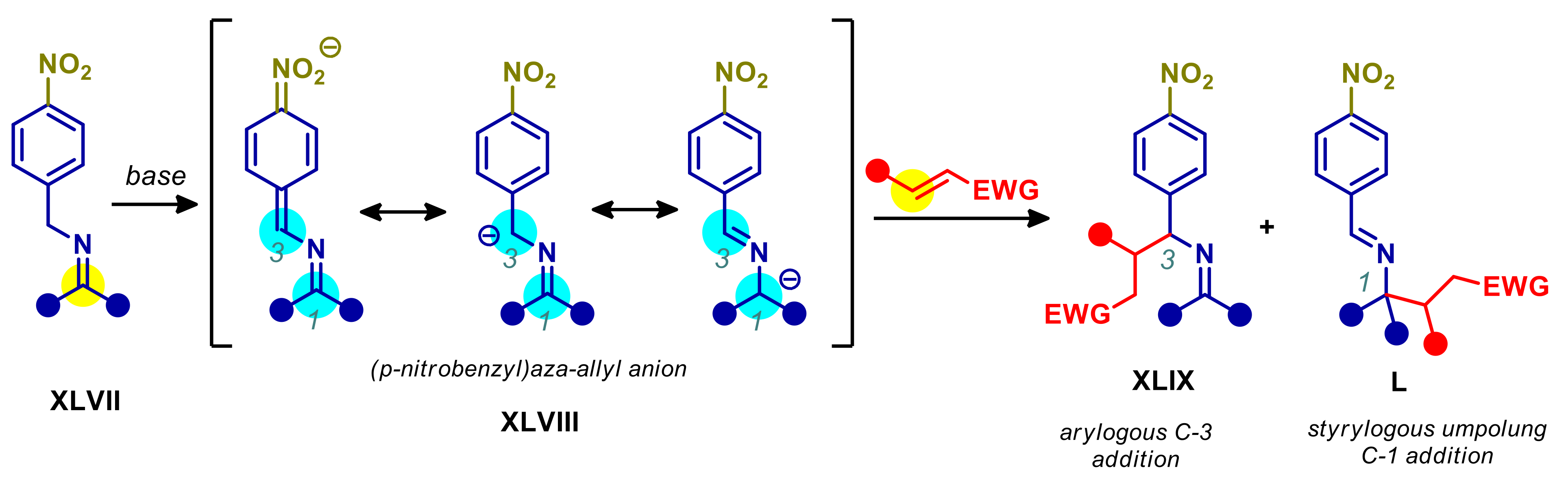
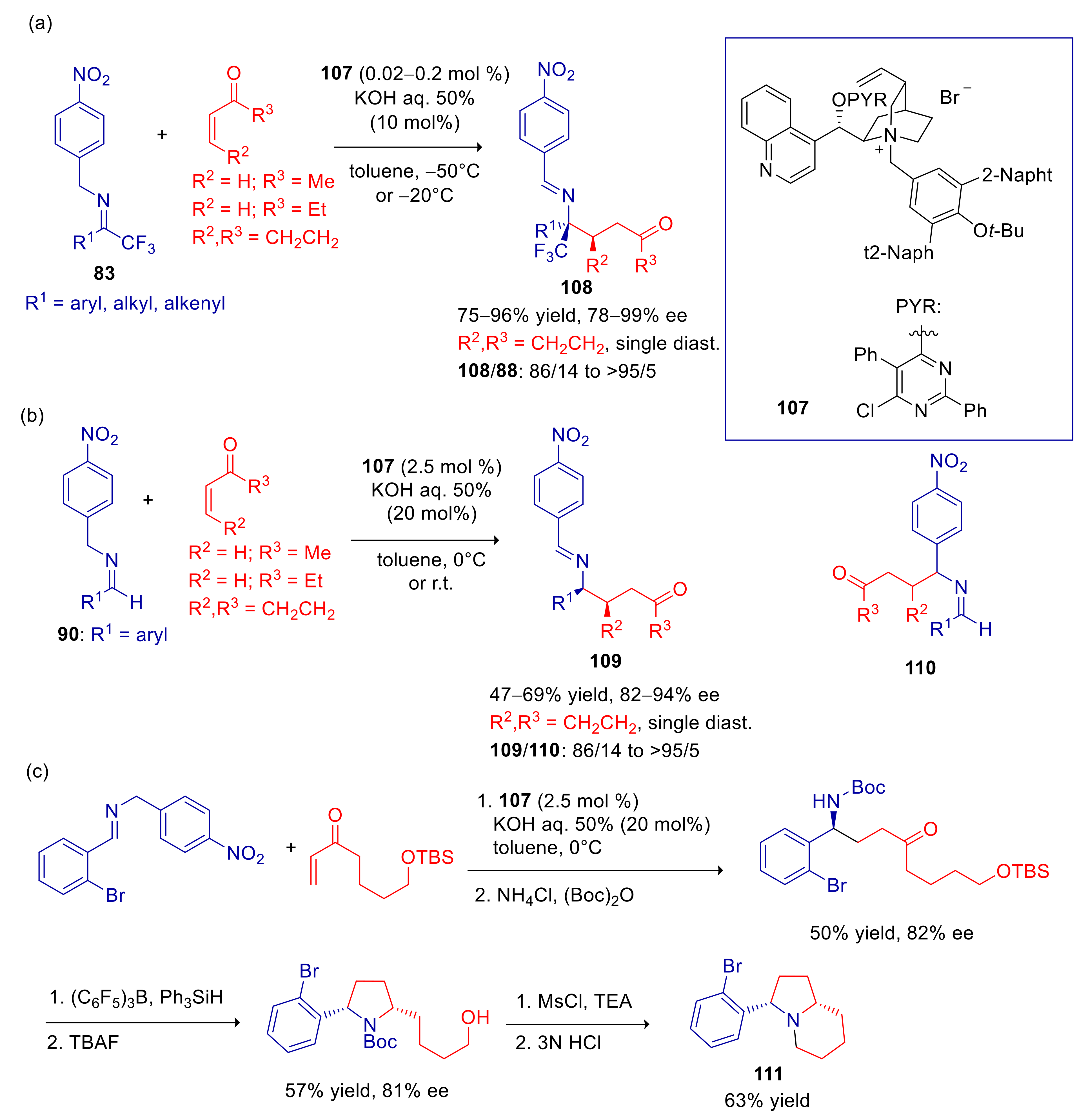
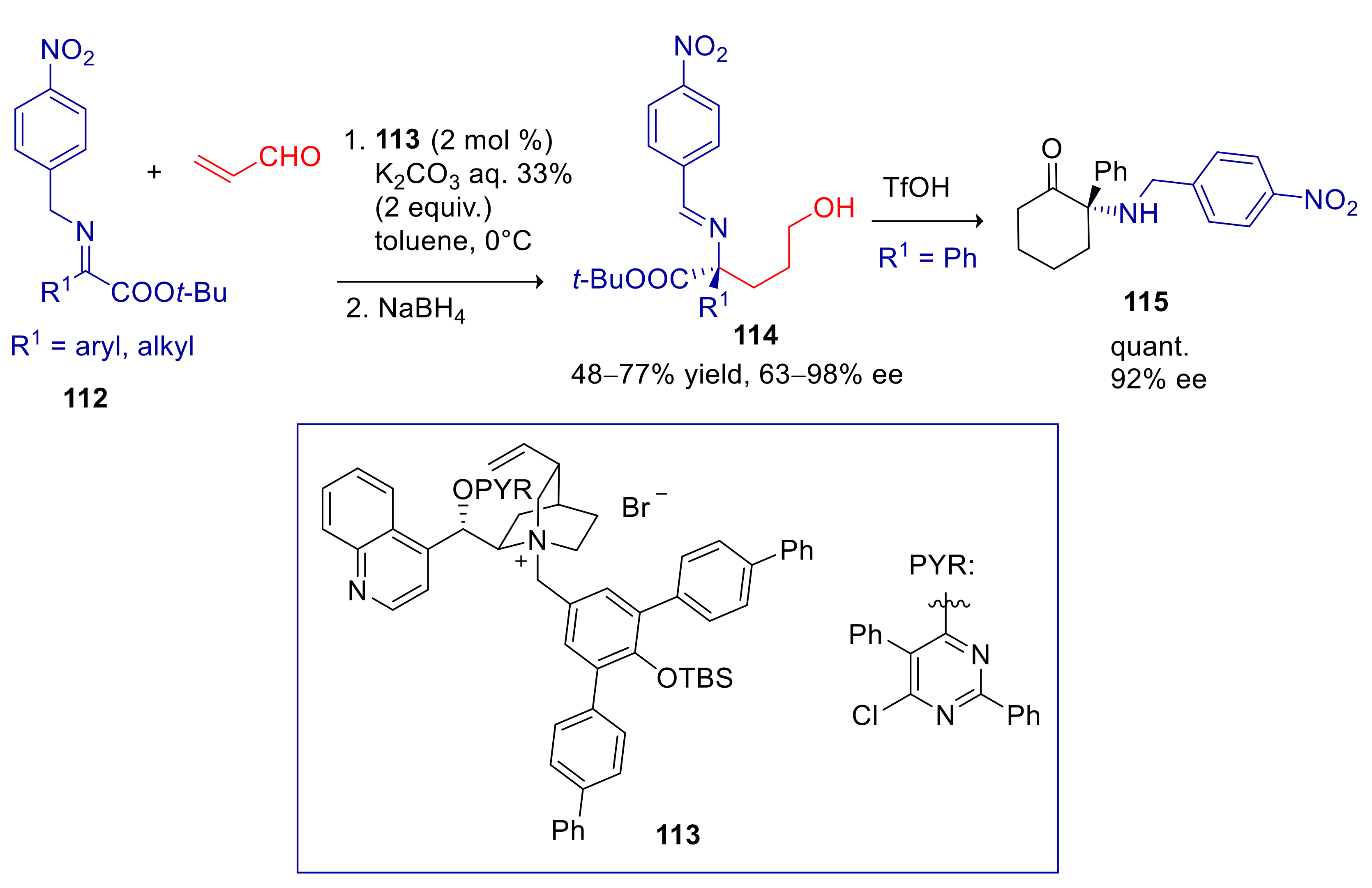
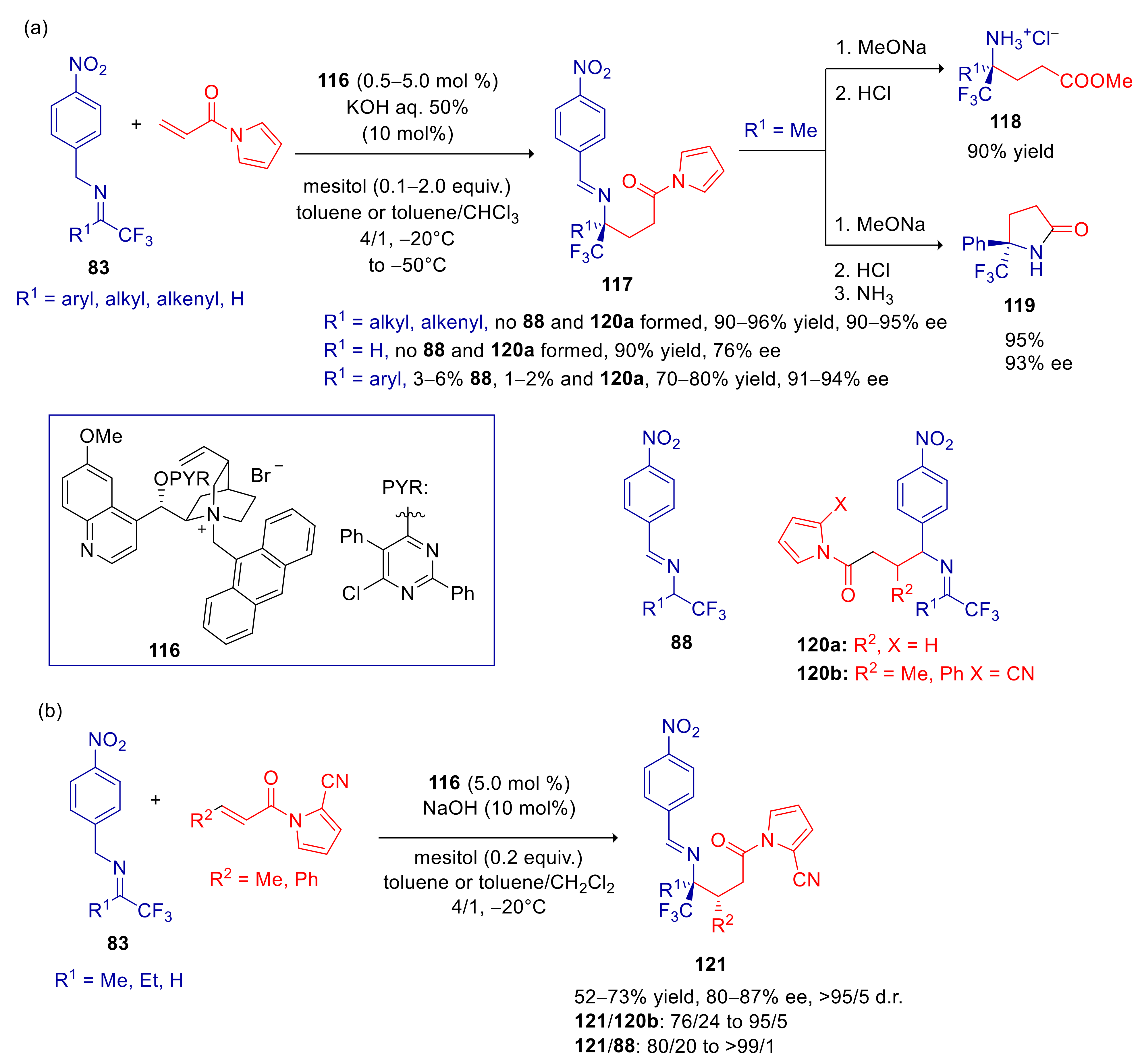
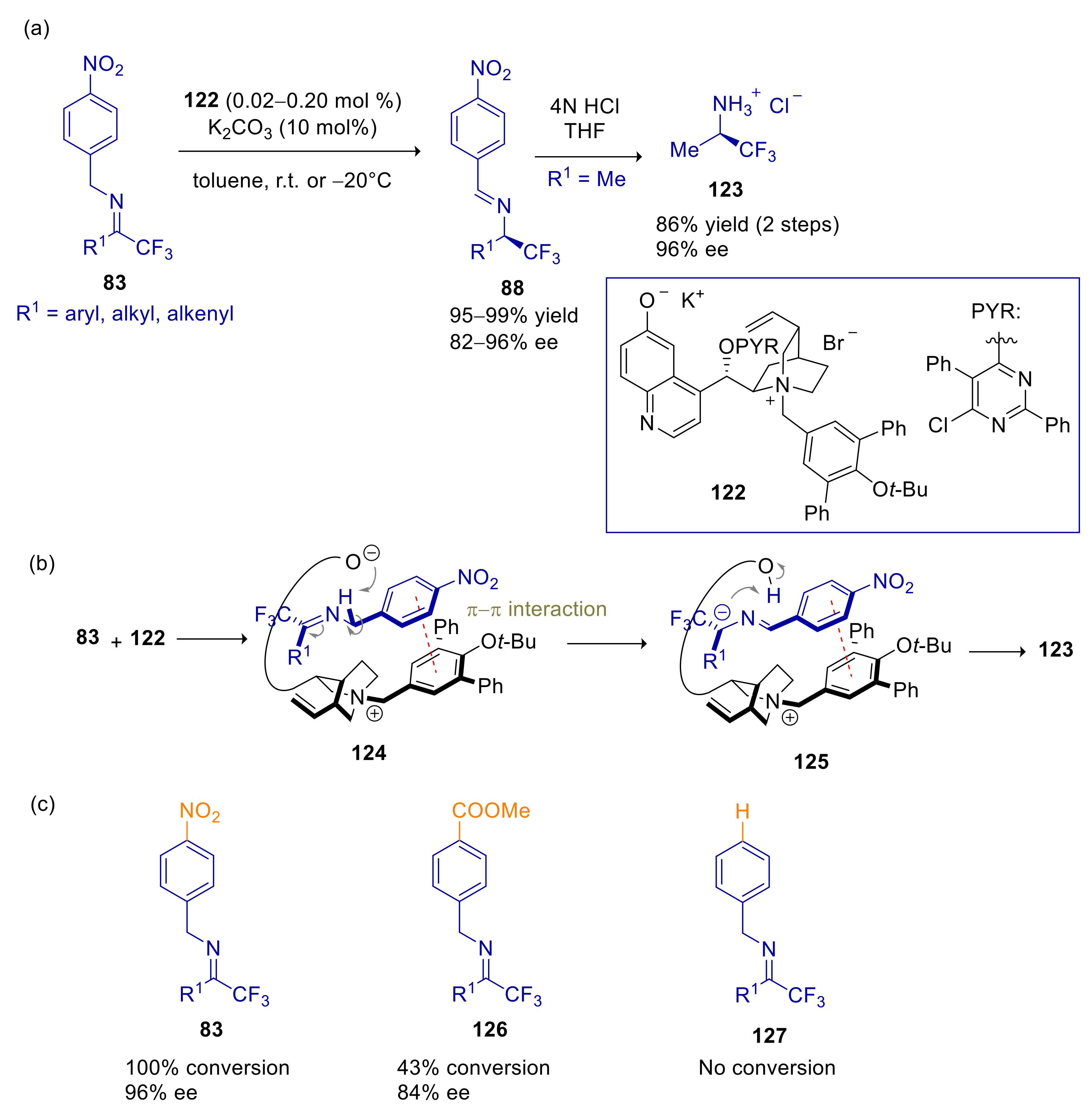
4.4. 2- and 4-Alkylnitroarenes
5. Conclusions and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Fuson, R.C. The principle of vinylogy. Chem. Rev. 1935, 16, 1–27. [Google Scholar] [CrossRef]
- Krishnamurthy, J. The principle of vinylogy. J. Chem. Ed. 1982, 59, 543–547. [Google Scholar] [CrossRef]
- Curti, C.; Battistini, L.; Sartori, A.; Zanardi, F. New Developments of the Principle of Vinylogy as Applied to π-Extended Enolate-Type Donor Systems. Chem. Rev. 2020, 120, 2448–2612. [Google Scholar] [CrossRef]
- Battistini, L.; Curti, C.; Rassu, G.; Sartori, A.; Zanardi, F. Enolizable Alkylidene Heterocyclic and Carbocyclic Carbonyl Systems: Valuable Vinylogous Donor Substrates in Synthesis. Synthesis 2017, 49, 2297–2336. [Google Scholar] [CrossRef]
- Casiraghi, G.; Battistini, L.; Curti, C.; Rassu, G.; Zanardi, F. The Vinylogous Aldol and Related Addition Reactions: Ten Years of Progress. Chem. Rev. 2011, 111, 3076–3154. [Google Scholar] [CrossRef]
- Przydacz, A.; Skrzyńska, A.; Albrecht, Ł. Breaking Aromaticity with Aminocatalysis: A Convenient Strategy for Asymmetric Synthesis. Angew. Chem. Int. Ed. 2019, 58, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Cheng, Y.; Cheng, J.; Li, R.; Li, P. Organocatalytic Asymmetric Benzylation and Aldol-Hemiacetalization of α,β-Unsaturated Trifluoromethyl Ketones: Efficient Enantioselective Construction of 3,4-Dihydroisocoumarins. Chem. Eur. J. 2017, 23, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Leng, H.J.; Yang, X.Y.; Han, B.; Rao, C.L.; Liu, L.; Peng, C.; Huang, W. Efficient Synthesis of Tetrahydronaphthalene- or Isochroman-Fused Spirooxindoles Using Tandem Reactions. RSC Adv. 2015, 5, 88272–88276. [Google Scholar] [CrossRef]
- Li, X.; Wang, S.; Li, T.; Li, J.; Li, H.; Wang, W. Formation of Dihydronaphthalenes via Organocatalytic Enatioselective Michael-Aldol Cascade Reactions with Arylalkanes. Org. Lett. 2013, 15, 5634–5637. [Google Scholar] [CrossRef]
- Raja, A.; Hong, B.-C.; Lee, G.-H. Organocatalytic Enantioselective Michael−Michael−Michael−Aldol Condensation Reactions: Control of Five Stereocenters in a Quadruple-Cascade Asymmetric Synthesis of Highly Functionalized Hexahydrophenanthrenes. Org. Lett. 2014, 16, 5756–5759. [Google Scholar] [CrossRef]
- Dell’Amico, L.; Companyõ, X.; Naicker, T.; Brauer, T.M.; Jørgensen, K.A. Asymmetric Organocatalytic Benzylation of α,β-Unsaturated Aldehydes with Toluenes. Eur. J. Org. Chem. 2013, 2013, 5262–5265. [Google Scholar] [CrossRef]
- Li, T.; Zhu, J.; Wu, D.; Li, X.; Wang, S.; Li, H.; Li, J.; Wang, W. A Strategy Enabling Enantioselective Direct Conjugate Addition of Inert Aryl Methane Nucleophiles to Enals with a Chiral Amine Catalyst under Mild Conditions. Chem. Eur. J. 2013, 19, 9147–9150. [Google Scholar] [CrossRef]
- Hiltebrandt, K.; Elies, K.; D’hooge, D.R.; Blinco, J.P.; Barner-Kowollik, C. A Light-Activated Reaction Manifold. J. Am. Chem. Soc. 2016, 138, 7048–7054. [Google Scholar] [CrossRef]
- Cuadros, S.; Dell’Amico, L.; Melchiorre, P. Forging Fluorine-Containing Quaternary Stereocenters by a Light-Driven Organocatalytic Aldol Desymmetrization Process. Angew. Chem. Int. Ed. 2017, 56, 11875–11879. [Google Scholar] [CrossRef]
- Dell’Amico, L.; Vega-Peñaloza, A.; Cuadros, S.; Melchiorre, P. Enantioselective Organocatalytic Diels-Alder Trapping of Photochemically Generated Hydroxy-o-Quinodimethanes. Angew. Chem. Int. Ed. 2016, 55, 3313–3317. [Google Scholar] [CrossRef]
- Dell’Amico, L.; Fernández-Alvarez, V.M.; Maseras, F.; Melchiorre, P. Light-Driven Enantioselective Organocatalytic β-Benzylation of Enals. Angew. Chem. Int. Ed. 2017, 56, 3304–3308. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Dong, S.; Liu, Z.; Wu, G.; Zou, C.; Ye, J. Enantioselective Michael Addition of Photogenerated o-Quinodimethanes to Enones Catalyzed by Chiral Amino Acid Esters. Org. Lett. 2017, 19, 2322–2325. [Google Scholar] [CrossRef] [PubMed]
- Masuda, Y.; Ishida, N.; Murakami, M. Light-Driven Carboxylation of o-Alkylphenyl Ketones with CO2. J. Am. Chem. Soc. 2015, 137, 14063–14066. [Google Scholar] [CrossRef]
- Starks, C.M.; Liotta, C.L.; Halpern, M.E. Phase-Transfer Catalysis, Fundamentals, Applications, and Industrial Perspectives; Chapman & Hall: New York, NY, USA, 1994. [Google Scholar] [CrossRef]
- Halpern, M.E. (Ed.) Phase-Transfer Catalysis, Fundamentals, Mechanisms and Syntheses; American Chemical Society: Washington, DC, USA, 1997. [Google Scholar] [CrossRef]
- Sasson, Y.; Neumann, R. (Eds.) Handbook of Phase-Transfer Catalysis; Chapman & Hall: London, UK, 1997. [Google Scholar] [CrossRef]
- Albanese, D. Liquid–Liquid Phase Transfer Catalysis: Basic Principles and Synthetic Applications. Catal. Rev. Sci. Eng. 2003, 45, 369–395. [Google Scholar] [CrossRef]
- Dehmlow, E.V.; Dehmlow, S.S. Phase Transfer Catalysis, 3rd ed.; Wiley-VCH: New York, NY, USA, 1993; ISBN 3527284087. [Google Scholar]
- Ikunaka, M. PTC in OPRD: An Illustrative Overview. Org. Process Res. Dev. 2008, 12, 698–709. [Google Scholar] [CrossRef]
- Freedman, H.H. Industrial applications of phase transfer catalysis (PTC): Past, present and future. Pure Appl. Chem. 1986, 58, 857–868. [Google Scholar] [CrossRef]
- Sharma, M. Application of phase transfer catalysis in the chemical industry. In Handbook of Phase-Transfer Catalysis; Sasson, Y., Neumann, R., Eds.; Chapman & Hall: London, UK, 1997; pp. 168–199. [Google Scholar] [CrossRef]
- Mąkosza, M. Phase-transfer catalysis. A general green methodology in organic synthesis. Pure Appl. Chem. 2000, 72, 1399–1403. [Google Scholar] [CrossRef]
- Tan, J.; Yasuda, N. Contemporary Asymmetric Phase Transfer Catalysis: Large-Scale Industrial Applications. Org. Process Res. Dev. 2015, 19, 1731–1746. [Google Scholar] [CrossRef]
- Maruoka, K. (Ed.) Asymmetric Phase Transfer Catalysis; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar] [CrossRef]
- Li, S.; Ma, J.-A. Asymmetric Phase-Transfer Catalysis in Organic Synthesis. In Bridging Heterogeneous and Homogeneous Catalysis: Concepts, Strategies, and Applications; Li, C., Liu, Y., Eds.; Wiley-VCH: Weinheim, Germany, 2014; pp. 425–468. [Google Scholar] [CrossRef]
- Shirakawa, S.; Maruoka, K. Chiral Onium Salts (Phase-Transfer Reactions). In Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions and Applications; Dalko, P., Ed.; Wiley-VCH: Weinheim, Germany, 2013; Volume 2, pp. 365–379. [Google Scholar] [CrossRef]
- Shirakawa, S.; Maruoka, K. Recent Developments in Asymmetric Phase-Transfer Reactions. Angew. Chem. Int. Ed. 2013, 52, 4312–4348. [Google Scholar] [CrossRef]
- Kaneko, S.; Kumatabara, Y.; Shirakawa, S. A new generation of chiral phase-transfer catalysts. Org. Biomol. Chem. 2016, 14, 5367–5376. [Google Scholar] [CrossRef]
- Schettini, R.; Sicignano, M.; De Riccardis, F.; Izzo, I.; Della Sala, G. Macrocyclic Hosts in Asymmetric Phase-Transfer Catalyzed Reactions. Synthesis 2018, 50, 4777–4795. [Google Scholar] [CrossRef]
- Gururaja, G.N.; Waser, M. Asymmetric Phase-Transfer Catalysis as a Powerful Tool in the Synthesis of Biologically Active Chiral Complex Natural Products. In Studies of Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elsevier: Oxford, UK, 2014; Volume 43, pp. 409–435. [Google Scholar] [CrossRef]
- Starks, C.M.; Liotta, C.L.; Halpern, M.E. Phase-Transfer Catalysis Reaction with Strong Bases. In Phase-Transfer Catalysis, Fundamentals, Applications, and Industrial Perspectives; Chapman & Hall: New York, NY, USA, 1994; pp. 383–451. [Google Scholar] [CrossRef]
- Brak, K.; Jacobsen, E.N. Asymmetric Ion-Pairing Catalysis. Angew. Chem. Int. Ed. 2013, 52, 534–561. [Google Scholar] [CrossRef] [PubMed]
- Qian, D.; Sun, J. Recent Progress in Asymmetric Ion-Pairing Catalysis with Ammonium Salts. Chem. Eur. J. 2019, 25, 3740–3751. [Google Scholar] [CrossRef] [PubMed]
- Zong, L.; Tan, C.-H. Phase-Transfer and Ion-Pairing Catalysis of Pentanidiums and Bisguanidiniums. Acc. Chem. Res. 2017, 50, 842–856. [Google Scholar] [CrossRef]
- Waser, M.; Novacek, J.; Gratzer, K. Cooperative Catalysis Involving Chiral Ion Pair Catalysts. In Cooperative Catalysis: Designing Efficient Catalysts for Synthesis; Peters, R., Ed.; Wiley-VCH: Weinheim, Germany, 2015; pp. 197–226. [Google Scholar] [CrossRef]
- Uraguchi, D.; Ooi, T. Site-Selective Conjugate Addition through Catalytic Generation of Ion-Pairing Intermediates. In Site-Selective Catalysis; Kawabata, T., Ed.; Topics in Current Chemistry; Springer: Cham, Switzerland, 2016; Volume 372. [Google Scholar] [CrossRef]
- Mao, B.; Fañanás-Mastral, M.; Feringa, B.L. Catalytic Asymmetric Synthesis of Butenolides and Butyrolactones. Chem. Rev. 2017, 117, 10502–10566. [Google Scholar] [CrossRef] [PubMed]
- Kitson, R.R.A.; Millemaggi, A.; Taylor, R.J.K. The Renaissance of α-Methylene-γ-butyrolactones: New Synthetic Approaches. Angew. Chem. Int. Ed. 2009, 48, 9426–9451. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.-D.; Ye, L.-W. Chiral γ-lactam synthesis via asymmetric C–H amidation. Nat. Catal. 2019, 2, 182–183. [Google Scholar] [CrossRef]
- Lebedev, A. Preparation of Chiral 4-Substituted γ-Lactams and the Corresponding γ-Aminobutyric Acids. Chem. Heterocycl. Compd. 2007, 43, 673–684. [Google Scholar] [CrossRef]
- Mardjan, M.I.D.; Parrain, J.L.; Commeiras, L. Strategies to Access γ-Hydroxy-γ-butyrolactams. Synthesis 2018, 50, 1175–1198. [Google Scholar] [CrossRef]
- Choudhury, A.R.; Mukjerjee, S. Deconjugated butenolide: A versatile build block for asymmetric catalysis. Chem. Soc. Rev. 2020, 49, 6755–6788. [Google Scholar] [CrossRef]
- Yan, L.; Wu, X.; Liu, H.; Xie, L.; Jiang, Z. Catalytic Asymmetric Synthesis of γ-Butenolides by Direct Vinylogous Reactions. Mini-Rev. Med. Chem. 2013, 13, 845–853. [Google Scholar] [CrossRef]
- Schneider, C.; Abels, F. Catalytic, enantioselective vinylogous Michael reactions. Org. Biomol. Chem. 2014, 12, 3531–3543. [Google Scholar] [CrossRef] [PubMed]
- Jusseau, X.; Chabaud, L.; Guillou, C. Synthesis of γ-butenolides and α,β-unsaturated γ-butyrolactams by addition of vinylogous nucleophiles to Michael acceptors. Tetrahedron 2014, 70, 2595–2615. [Google Scholar] [CrossRef]
- Casiraghi, G.; Zanardi, F.; Battistini, L.; Rassu, G. Advances in Exploring Heterocyclic Dienoxysilane Nucleophiles in Asymmetric Synthesis. Synlett 2009, 10, 1525–1542. [Google Scholar] [CrossRef]
- Rassu, G.; Zanardi, F.; Battistini, L.; Casiraghi, G. The synthetic utility of furan-, pyrrole- and thiophene-based 2-silyloxy dienes. Chem. Soc. Rev. 2000, 29, 109–118. [Google Scholar] [CrossRef]
- Rassu, G.; Zanardi, F.; Battistini, L.; Casiraghi, G. The Vinylogous Aldol Addition of Heterocyclic Silyloxy Dienes: Application in Synthesis. Synlett 1999, 1999, 1333–1350. [Google Scholar] [CrossRef]
- Yin, Y.; Jiang, Z. Organocatalytic Asymmetric Vinylogous Michael Reactions. ChemCatChem 2017, 9, 4306–4318. [Google Scholar] [CrossRef]
- Nagao, H.; Yamane, Y.; Mukaiyama, T. Effective Synthesis of 5-Substituted Butenolide Derivatives by Using Cinchonidine-derived Quaternary Ammonium Phenoxide Catalyst. Chem. Lett. 2007, 36, 8–9. [Google Scholar] [CrossRef]
- Singh, R.P.; Foxman, B.M.; Deng, L. Asymmetric Vinylogous Aldol Reaction of Silyloxy Furans with a Chiral Organic Salt. J. Am. Chem. Soc. 2010, 132, 9558–9560. [Google Scholar] [CrossRef]
- Fukuyama, T.; Goto, S. Synthetic Approaches Toward FR-900482. I. Stereoselective Synthesis of a Pentacyclic Model Compound. Tetrahedron Lett. 1989, 30, 6491–6494. [Google Scholar] [CrossRef]
- Fukuyama, T.; Yang, L. Synthetic Approaches Toward Mitomycins. I. Stereoselective Synthesis of a Tetracyclic Intermediate. Tetrahedron Lett. 1986, 27, 6299–6300. [Google Scholar] [CrossRef]
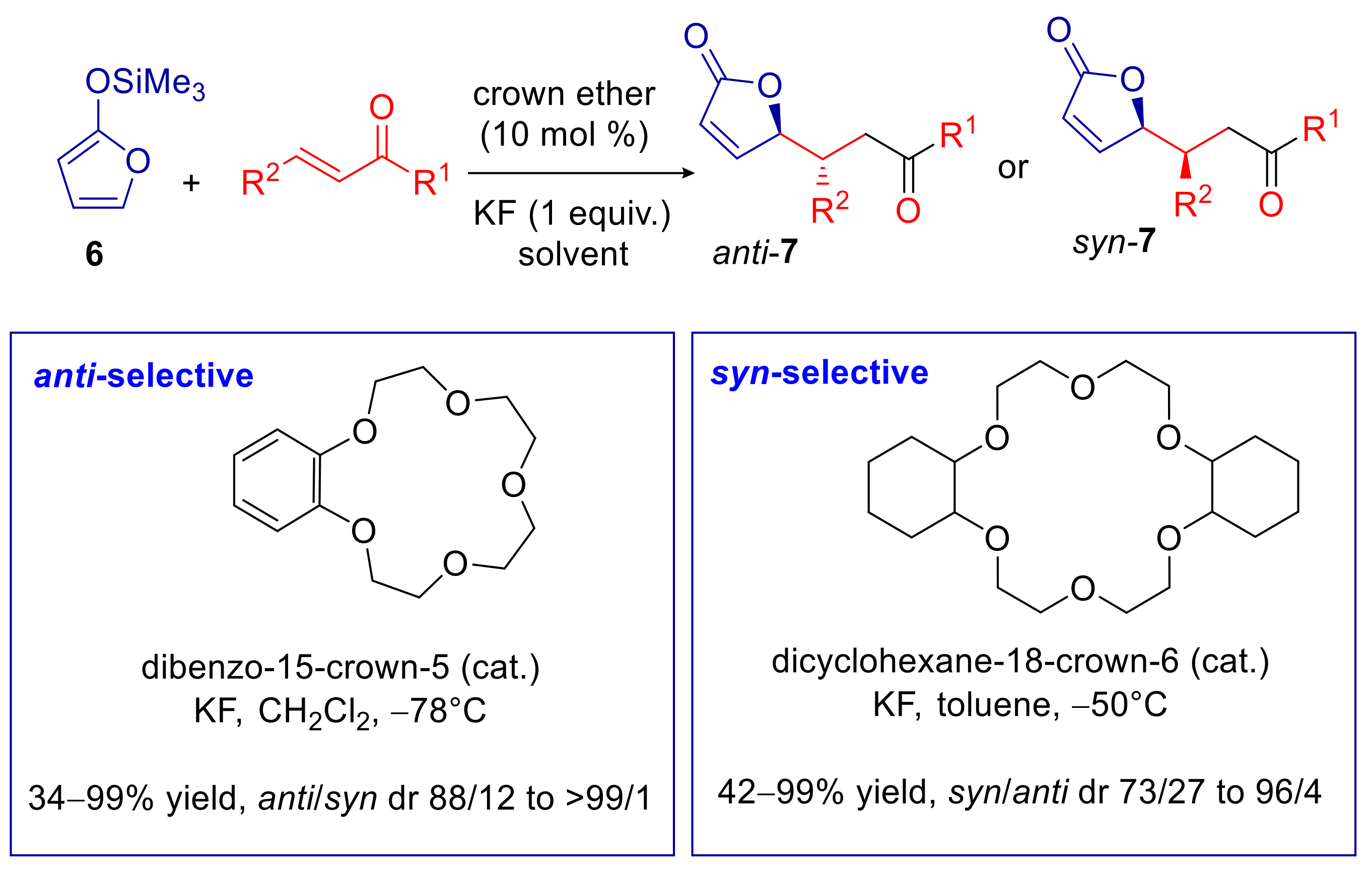
- Della Sala, G.; Sicignano, M.; Schettini, R.; De Riccardis, F.; Cavallo, L.; Minenkov, Y.; Batisse, C.; Hanquet, C.; Leroux, F.; Izzo, I. Switchable Diastereoselectivity in the Fluoride-Promoted Vinylogous Mukaiyama−Michael Reaction of 2-[(Trimethylsilyl)oxy]furan Catalyzed by Crown Ethers. J. Org. Chem. 2017, 82, 6629–6637. [Google Scholar] [CrossRef]
- Sicignano, M.; Schettini, R.; Sica, L.; Pierri, G.; De Riccardis, F.; Izzo, I.; Maity, B.; Minenkov, Y.; Cavallo, L.; Della Sala, G. Unprecedented Diastereoselective Arylogous Michael Addition of Unactivated Phthalides. Chem. Eur. J. 2019, 25, 7131–7141. [Google Scholar] [CrossRef]
- Claraz, A.; Oudeyer, S.; Levacher, V. Chiral Quaternary Ammonium Aryloxide/N,O-Bis(trimethylsilyl)acetamide Combination as Efficient Organocatalytic System for the Direct Vinylogous Aldol Reaction of (5H)-Furan-2-one Derivatives. Adv. Synth. Catal. 2013, 55, 841–846. [Google Scholar] [CrossRef]
- Arlt, A.; Toyama, H.; Takada, K.; Hashimoto, T.; Maruoka, K. Phase-transfer catalyzed asymmetric synthesis of α,β-unsaturated γ, γ-disubstituted γ-lactams. Chem. Commun. 2017, 53, 4779–4782. [Google Scholar] [CrossRef]
- Cui, H.-L.; Chen, Y.-C. α,α-Dicyanoalkenes: Versatile vinylogous nucleophiles for organic synthesis. Chem. Commun. 2009, 4479–4486. [Google Scholar] [CrossRef]
- Curti, C.; Sartori, A.; Battistini, L.; Zanardi, F. Exploring the Remote Reactivity of π-Extended Carbonyl Compounds: The Vinylogous Alkylidene Malononitrile Activation Strategy. Synlett 2018, 29, 266–281. [Google Scholar] [CrossRef]
- Niess, B.; Jørgensen, K.A. The asymmetric vinylogous Mannich reaction of dicyanoalkylidenes with α-amido sulfones under phase-transfer conditions. Chem. Commun. 2007, 1620–1622. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.-Y.; Cui, H.-L.; Long, J.; Li, B.-J.; Wu, Y.; Ding, L.-S.; Chen, Y.-C. Organocatalytic and Highly Stereoselective Direct Vinylogous Mannich Reaction. J. Am. Chem. Soc. 2007, 129, 1878–1879. [Google Scholar] [CrossRef]
- Fang, Y.; Wei, Z.; Wang, Y.; Liu, S.; Cao, J.; Liang, D.; Lin, Y.; Duan, H. The asymmetric vinylogous Mannich reaction of noncyclic dicyanoolefins catalyzed by a bifunctional thiourea–ammonium salt phase transfer catalyst. New J. Chem. 2019, 43, 10012–10016. [Google Scholar] [CrossRef]
- Lu, J.; Chen, W.-Y. Organocatalytic and Enantioselective Mannich Reaction of Dicyanoolefins with α-Amido Sulfones. Bull. Korean Chem. Soc. 2012, 33, 3175–3176. [Google Scholar] [CrossRef][Green Version]
- Cheng, C.; Lu, X.; Ge, L.; Chen, J.; Cao, W.; Zhao, G. Effective asymmetric vinylogous Mannich reaction of isatin imines with α,α-dicyanoolefins in the presence of a simple chiral amide phosphonium bifunctional phase transfer catalyst. Org. Chem. Front. 2017, 4, 101–114. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, Y.; Meng, Q.; Li, X. An organocatalytic enantioselective vinylogous Mannich reaction of α,α-dicyanoolefins with isatin N-Boc ketimines. Org. Chem. Front. 2016, 3, 709–713. [Google Scholar] [CrossRef]
- De Castro, P.P.; Carpanez, A.G.; Amarante, G.W. Azlactone Reaction Developments. Chem. Eur. J. 2016, 22, 10294–10318. [Google Scholar] [CrossRef] [PubMed]
- Marra, I.F.S.; de Castro, P.P.; Amarante, G.W. Recent Advances in Azlactone Transformations. Eur. J. Org. Chem. 2019, 5830–5855. [Google Scholar] [CrossRef]
- Alba, A.-N.R.; Rios, R. Oxazolones in Organocatalysis, New Tricks for an Old Reagent. Chem. Asian J. 2011, 6, 720–734. [Google Scholar] [CrossRef]
- Fisk, J.S.; Mosey, R.A.; Tepe, J.J. The diverse chemistry of oxazol-5-(4H)-ones. Chem. Soc. Rev. 2007, 36, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Dell’Amico, L.; Albrecht, Ł.; Naicker, T.; Poulsen, P.H.; Jørgensen, K.A. Beyond Classical Reactivity Patterns: Shifting from 1,4- to 1,6-Additions in Regio- and Enantioselective Organocatalyzed Vinylogous Reactions of Olefinic Lactones with Enals and 2,4-Dienals. J. Am. Chem. Soc. 2013, 135, 8063–8070. [Google Scholar] [CrossRef]
- Zhao, S.; Zhao, Y.-Y.; Lin, J.-B.; Xie, T.; Liang, Y.-M.; Xu, P.-F. Organocatalyzed Asymmetric Vinylogous Allylic−Allylic Alkylation of Morita−Baylis−Hillman Carbonates with Olefinic Azlactones: Facile Access to Chiral Multifunctional α-Amino Acid Derivatives. Org. Lett. 2015, 17, 3206–3209. [Google Scholar] [CrossRef]
- Gao, T.-P.; Lin, J.-B.; Hu, X.-Q.; Xu, P.-F. A catalytic asymmetric hetero-Diels–Alder reaction of olefinic azlactones and isatins: Facile access to chiral spirooxindole dihydropyranones. Chem. Commun. 2014, 50, 8934–8936. [Google Scholar] [CrossRef]
- Gao, T.-P.; Liu, D.; Lin, J.-B.; Hu, X.-Q.; Wang, Z.-Y.; Xu, P.-F. Direct construction of chiral quaternary dihydropyranones through highly enantioselective organocatalytic hetero-Diels–Alder reactions of olefinic azlactones. Org. Chem. Front. 2016, 3, 598–602. [Google Scholar] [CrossRef]
- Zhu, B.; Lu, B.; Zhang, H.; Xu, X.; Jiang, Z.; Chang, J. Phase-Transfer-Catalyzed, Enantioselective Vinylogous Conjugate Addition−Cyclization of Olefinic Azlactones To Access Multifunctionalized Chiral Cyclohexenones. Org. Lett. 2019, 21, 3271–3275. [Google Scholar] [CrossRef]
- Baschieri, A.; Bernardi, L.; Ricci, A.; Suresh, S.; Adamo, M.F.A. Catalytic Asymmetric Conjugate Addition of Nitroalkanes to 4-Nitro-5-styrylisoxazoles. Angew. Chem. Int. Ed. 2009, 48, 9342–9345. [Google Scholar] [CrossRef]
- Del Fiandra, C.; Piras, L.; Fini, F.; Disetti, P.; Moccia, M.; Adamo, M.F.A. Phase transfer catalyzed enantioselective cyclopropanation of 4-nitro-5-styrylisoxazoles. Chem. Commun. 2012, 48, 3863–3865. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Xiong, Y.; Wang, Z.-F.; Tao, H.-Y.; Wang, C.-J. Ligand-controlled stereodivergent 1,3-dipolar cycloaddition of azomethine ylides with 3-methyl-4-nitro-5-styrylisoxazoles. Chem. Commun. 2016, 52, 9458–9461. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, X.; Ma, X.; Wang, R. Organocatalyzed asymmetric vinylogous Michael addition of α,β-unsaturated γ-butyrolactam. Chem. Commun. 2013, 49, 9329–9331. [Google Scholar] [CrossRef]
- Liu, X.-L.; Han, W.-Y.; Zhang, X.-M.; Wang, W.-C. Highly Efficient and Stereocontrolled Construction of 3,3′-Pyrrolidonyl Spirooxindoles via Organocatalytic Domino Michael/Cyclization Reaction. Org. Lett. 2013, 15, 1246–1249. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; López-Delgado, F.J.; Jørgensen, D.K.B.; Nielsen, R.P.; Jiang, H.; Jørgensen, K.A. Trienamine-mediated asymmetric [4+2]-cycloaddition of α,β-unsaturated ester surrogates applying 4-nitro-5-styrylisoxazoles. Chem. Commun. 2014, 50, 15689–15691. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Pei, W.; Wang, J.; Liu, J.; Wang, J.; Zhang, M.-l.; Chen, Z.; Liu, L. Organocatalytic asymmetric synthesis of compounds bearing both isoxazole and pyrazole moieties via 1,6-addition of pyrazol-5-ones to 3-methyl-4-nitro-5-alkenylisoxazoles. Org. Chem. Front. 2018, 5, 1342–1347. [Google Scholar] [CrossRef]
- Kowalczyk-Dworak, D.; Kwit, M.; Albrecht, Ł. Allylic−Allylic Alkylation with 3,5-Dimethyl-4-nitroisoxazole: A Route to Dicarboxylic Acid Derivatives. J. Org. Chem. 2020, 85, 2938–2944. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wei, B.; Lin, H.; Cui, W.; Zeng, X.; Fan, X. Organocatalyzed Asymmetric Vinylogous Michael Reactions of 3,5-Dialkyl-Substituted 4-Nitroisoxazoles: A Direct Method for the Synthesis of Chiral Isoxazole Derivatives. Adv. Synth. Catal. 2015, 357, 1299–1304. [Google Scholar] [CrossRef]
- Zhu, B.; Lee, R.; Yin, Y.; Li, F.; Coote, M.L.; Jiang, Z. Enantioselective Vinylogous Amination of 5-Alkyl-4-nitroisoxazoles with a Dipeptide-Based Guanidinium Phase-Transfer Catalyst. Org. Lett. 2018, 20, 429–432. [Google Scholar] [CrossRef]
- Zhu, B.; Li, F.; Lu, B.; Chang, J.; Jiang, Z. Organocatalytic Enantioselective Vinylogous Aldol Reaction of 5-Alkyl-4-Nitroisoxazoles to Paraformaldehyde. J. Org. Chem. 2018, 83, 11350–11358. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Zhu, Q.; Wang, J.; Chen, J.; Cao, W.; Zhu, B.; Wu, X. Direct Asymmetric Vinylogous Mannich Addition of 3,5-Disubstituted-4-nitroisoxazoles to Isatin-Derived Imines Catalyzed by a Bifunctional Phase-Transfer-Catalyst. J. Org. Chem. 2018, 83, 14617–14625. [Google Scholar] [CrossRef]
- Shaqura, I.I.; Bule, M.; Khan, F.; Niaz, K. Phloroglucinols, xanthones and anthrones. In Recent Advances in Natural Products Analysis, 1st ed.; Nabavi, S.M., Saeedi, M., Nabavi, S.F., Silva, A.S., Eds.; Elsevier: Amsterdam, The Netherland, 2020; pp. 175–197. [Google Scholar]
- Kadarkaraisamy, M.; Sykes, A.G. The chemistry of constrained crown ring systems and fluorescence sensor applications. J. Incl. Phenom. Macrocycl. Chem. 2013, 75, 23–30. [Google Scholar] [CrossRef]
- Shen, J.; Nguyen, T.T.; Goh, Y.-P.; Ye, W.; Fu, X.; Xu, J.; Tan, C.-H. Chiral Bicyclic Guanidine-Catalyzed Enantioselective Reactions of Anthrones. J. Am. Chem. Soc. 2006, 128, 13692–13693. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lei, Z.-Y.; Zhao, M.-X.; Shi, J.-W. A highly efficient asymmetric Michael addition of anthrone to nitroalkenes with cinchona organocatalysts. Tetrahedron Lett. 2007, 48, 5743–5746. [Google Scholar] [CrossRef]
- Alba, A.-N.; Bravo, N.; Moyano, A.; Rios, R. Enantioselective addition of anthrones to α,β-unsaturated aldehydes. Tetrahedron Lett. 2009, 50, 3067–3069. [Google Scholar] [CrossRef]
- Wu, C.; Li, W.; Yang, J.; Liang, X.; Ye, J. Asymmetric organocatalytic Michael addition of anthrone to enone. Org. Biomol. Chem. 2010, 8, 3244–3250. [Google Scholar] [CrossRef]
- Zhao, H.; Xiao, M.; Xu, L.; Wang, L.; Xiao, J. Bifunctional thiourea catalyzed asymmetric Michael addition of anthrone to methyleneindolinones. RSC Adv. 2016, 6, 38558–38562. [Google Scholar] [CrossRef]
- Ceban, V.; Tauchman, J.; Meazza, M.; Gallagher, G.; Light, M.E.; Gergelitsová, I.; Veselý, J.; Rios, R. Expanding the scope of MetalFree enantioselective allylic substitutions: Anthrones. Sci. Rep. 2015, 5, 16886. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, K.C.; Chattopadhyay, S.K.; Khan, A.T. Phase-transfer-catalyzed Alkylation of Anthrone and 10-Propargylanthrone. Synthesis 1988, 552–553. [Google Scholar] [CrossRef]
- Nickel, H.C.; Schmidt, P.; Böhm, K.J.; Baasner, S.; Müller, K.; Gerlach, M.; Unger, E.; Günther, E.G.; Prinz, H. Synthesis, antiproliferative activity and inhibition of tubulin polymerization by 1,5- and 1,8-disubstituted 10H-anthracen-9-ones bearing a 10-benzylidene or 10-(2-oxo-2-phenylethylidene) moiety. Eur. J. Med. Chem. 2010, 45, 3420–3438. [Google Scholar] [CrossRef]
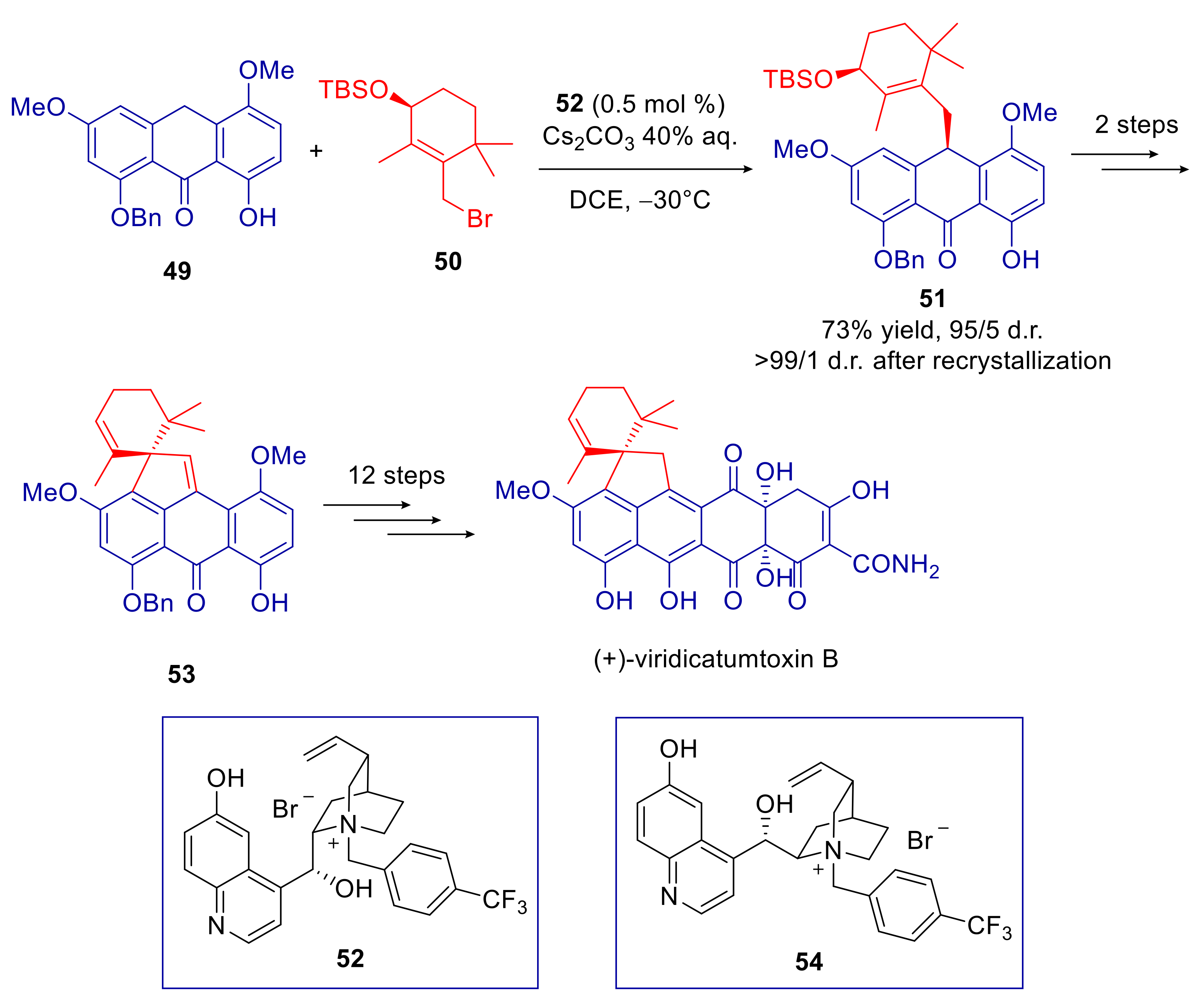
- Nicolaou, K.C.; Liu, G.; Beabout, K.; McCurry, M.D.; Shamoo, Y. Asymmetric Alkylation of Anthrones, Enantioselective Total Synthesis of (−)- and (+)-Viridicatumtoxins B and Analogues Thereof: Absolute Configuration and Potent Antibacterial Agents. J. Am. Chem. Soc. 2017, 139, 3736–3746. [Google Scholar] [CrossRef]
- Donslund, B.S.; Monleón, A.; Palazzo, T.A.; Christensen, M.L.; Dahlgaard, A.; Erickson, J.D.; Jørgensen, K.A. Organocatalytic Enantioselective Higher-Order Cycloadditions of In Situ Generated Amino Isobenzofulvenes. Angew. Chem. Int. Ed. 2018, 57, 1246–1250. [Google Scholar] [CrossRef]
- Donslund, B.S.; Jessen, N.I.; Bertuzzi, G.; Giardinetti, M.; Palazzo, T.A.; Christensen, M.L.; Jørgensen, K.A. Catalytic Enantioselective [10+4] Cycloadditions. Angew. Chem. Int. Ed. 2018, 57, 13182–13186. [Google Scholar] [CrossRef] [PubMed]
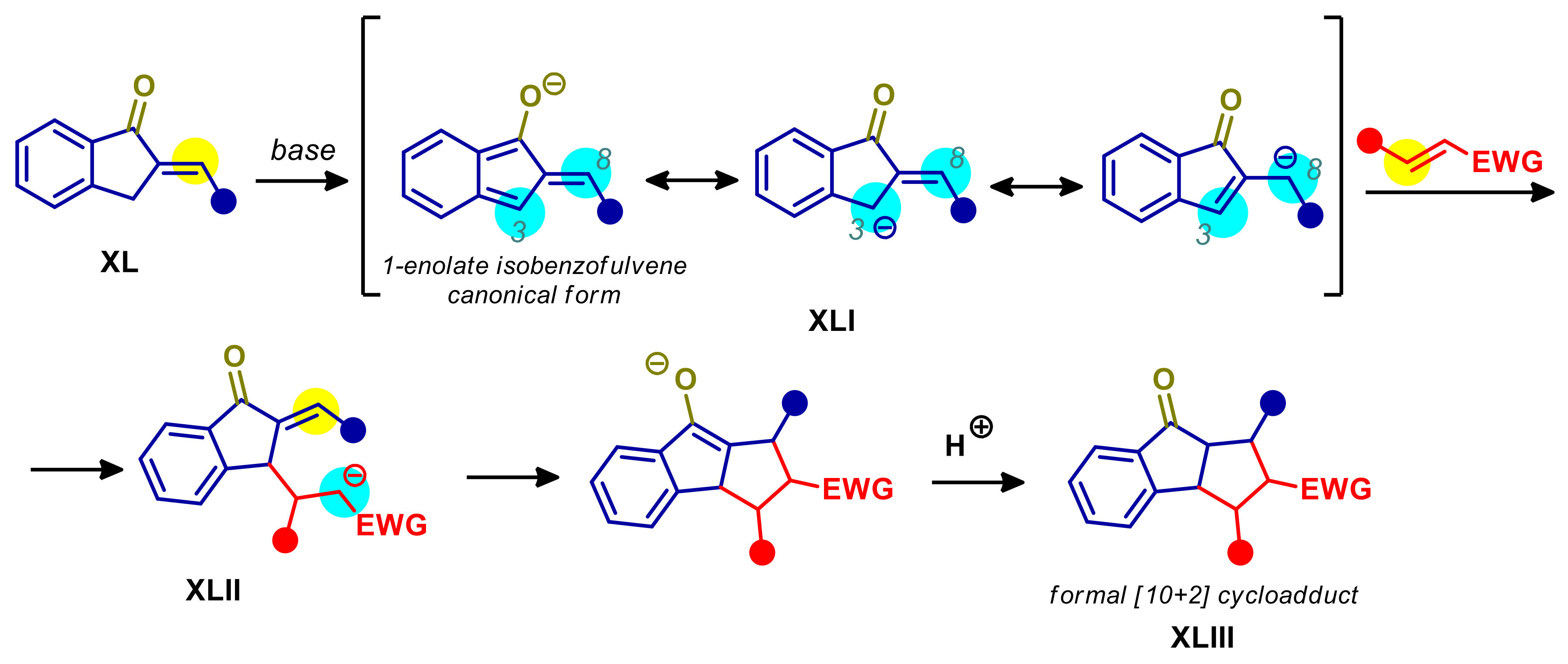
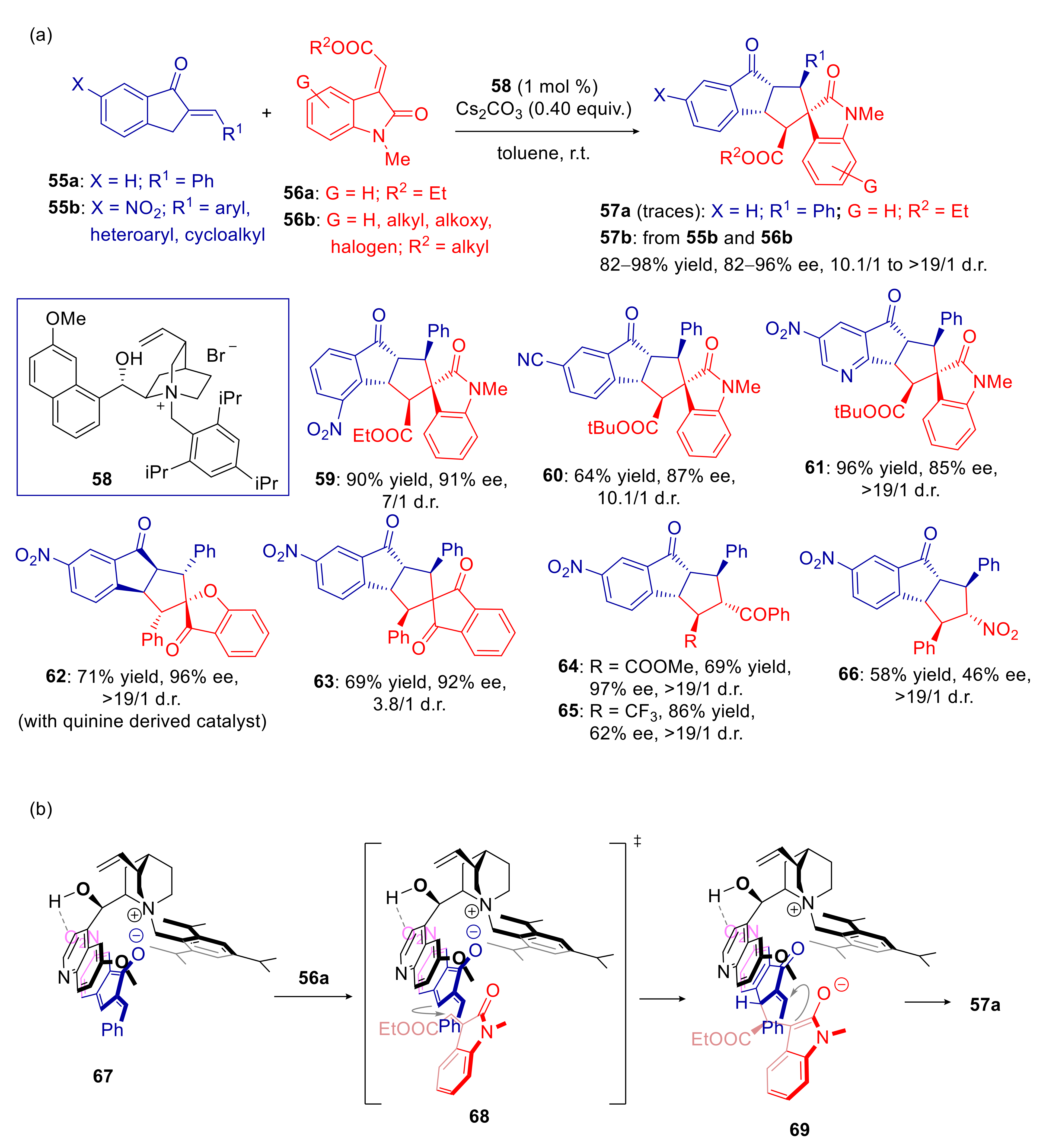
- Yang, Y.; Jiang, Y.; Du, W.; Chen, Y.-C. Asymmetric Cross [10+2] Cycloadditions of 2-Alkylidene-1-indanones and Activated Alkenes under Phase-Transfer Catalysis. Chem. Eur. J. 2020, 26, 1754–1758. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Wang, Z.-X.; Zhou, Y.-C.; Xiao, W.; Ouwyang, Q.; Du, W.; Chen, Y.-C. Switchable regioselectivity in amine-catalysed asymmetric cycloadditions. Nat. Chem. 2017, 9, 590–594. [Google Scholar] [CrossRef]
- Berthelette, C.; McCooye, C.; Leblanc, Y.; Trimble, L.A.; Tsou, N.N. Studies on the Dimerization of 2-Benzylidene-1-indanone. J. Org. Chem. 1997, 62, 4339–4342. [Google Scholar] [CrossRef]
- Camps, P.; Domingo, L.R.; Formosa, X.; Galdeano, C.; González, D.; Muñoz-Torrero, D.; Segalés, S.; Font-Bardia, M.; Solans, X. Highly Diastereoselective One-Pot Synthesis of Spiro{cyclopenta[a]indene-2,2’-indene}diones from 1-Indanones and Aromatic Aldehydes. J. Org. Chem. 2006, 71, 3464–3471. [Google Scholar] [CrossRef]
- Karmakar, R.; Pahari, P.; Mal, D. Phthalides and Phthalans: Synthetic Methodologies and Their Applications in the Total Synthesis. Chem. Rev. 2014, 114, 6213–6284. [Google Scholar] [CrossRef]
- Awasthi, A.; Singh, M.; Rathee, G.; Chandra, R. Recent advancements in synthetic methodologies of 3-substituted phthalides and their application in the total synthesis of biologically active natural products. RSC Adv. 2020, 10, 12626–12652. [Google Scholar] [CrossRef]
- Janowski, W.K.; Prager, R.H. The Chemistry of Phthalide-3-carboxylic Acid. III Decarboxylation of Salts in the Presence of α,β-Unsaturated Ketones. Aust. J. Chem. 1985, 38, 921–929. [Google Scholar] [CrossRef]
- Ciufolini, M.A.; Browne, M.E. Efficient palladium-mediated synthesis of a spirocyclic model for fredericamycin A. Tetrahedron Lett. 1987, 28, 171–174. [Google Scholar] [CrossRef]
- Ogawa, Y.; Hosaka, K.; Chin, M.; Mitsuhashi, H. Synthesis of (Z)-3-Butylidene-4-hydroxyphthalide. Heterocycles 1991, 32, 1737–1744. [Google Scholar] [CrossRef]
- Mali, R.S.; Babu, K.N. Reactions of 3-(1-Hydroxyalkyl)phthalides with Acids: Synthesis of (Z)-3-Alkylidenephthalides and 3-Alkyl-8-hydroxyisocoumarins. J. Org. Chem. 1998, 63, 2488–2492. [Google Scholar] [CrossRef]
- Luo, J.; Jiang, C.; Wang, H.; Xu, L.-W.; Lu, Y. Direct asymmetric Michael addition of phthalide derivatives to chalcones. Tetrahedron Lett. 2013, 54, 5261–5265. [Google Scholar] [CrossRef]
- Luo, J.; Wang, H.; Zhong, F.; Kwiatkowski, J.; Xu, L.-W.; Lu, Y. Highly diastereoselective and enantioselective direct Michael addition of phthalide derivatives to nitroolefins. Chem. Commun. 2013, 49, 5775–5777. [Google Scholar] [CrossRef]
- Luo, J.; Wang, H.; Zhong, F.; Kwiatkowski, J.; Xu, L.-W.; Lu, Y. Direct asymmetric Mannich reaction of phthalides: Facile access to chiral substituted isoquinolines and isoquinolinones. Chem. Commun. 2012, 48, 4707–4709. [Google Scholar] [CrossRef]
- Zhong, F.; Luo, J.; Chen, G.-Y.; Dou, X.; Lu, Y. Highly Enantioselective Regiodivergent Allylic Alkylations of MBH Carbonates with Phthalides. J. Am. Chem. Soc. 2012, 134, 10222–10227. [Google Scholar] [CrossRef]
- Wang, J.; Li, X.; Cheng, J.-P. Quinine-derived thiourea promoted enantioselective Michael addition reactions of 3-substituted phthalides to maleimides. Sci. China Chem. 2019, 62, 649–652. [Google Scholar] [CrossRef]
- Liu, W.; Hu, Z.-P.; Yan, Y.; Liao, W.-W. Highly diastereo- and enantioselective construction of phthalideoxindole hybrids bearing vicinal quaternary chiral centers via an organocatalytic allylic alkylation. Tetrahedron Lett. 2018, 59, 3132–3135. [Google Scholar] [CrossRef]
- Zhuang, Z.; Hu, Z.-P.; Liao, W.-W. Asymmetric Synthesis of Functionalized Dihydronaphthoquinones Containing Quaternary Carbon Centers via a Metal-Free Catalytic Intramolecular Acylcyanation of Activated Alkenes. Org. Lett. 2014, 16, 3380–3383. [Google Scholar] [CrossRef]
- Hu, Z.-P.; Zhuang, Z.; Liao, W.-W. Asymmetric Synthesis of Dihydronaphthoquinones Containing Adjacent Stereocenters via a Sulfa-Michael Addition Triggered Ring-Expansion Approach. J. Org. Chem. 2015, 80, 4627–4637. [Google Scholar] [CrossRef]
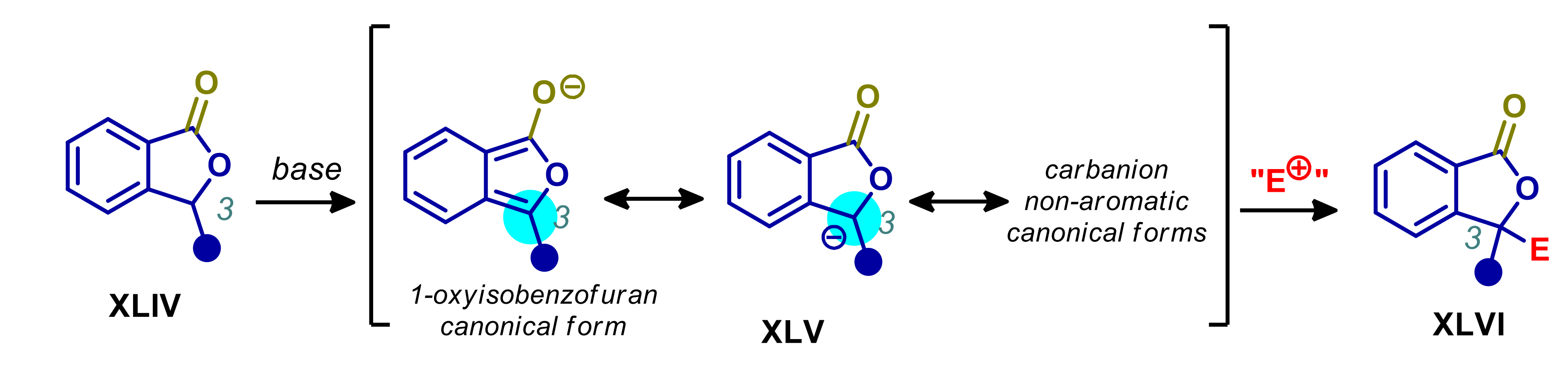
- Sicignano, M.; Schettini, R.; Pierri, G.; Marino, M.L.; Izzo, I.; De Riccardis, F.; Della Sala, G. An Entry to Enantioenriched 3,3-Disubstituted Phthalides through Asymmetric Phase-Transfer-Catalyzed γ-Alkylation. J. Org. Chem. 2020, 85, 7476–7484. [Google Scholar] [CrossRef] [PubMed]
- Sicignano, M.; Dentoni Litta, A.; Schettini, R.; De Riccardis, F.; Pierri, G.; Tedesco, C.; Izzo, I.; Della Sala, G. Highly Diastereoselective Crown Ether Catalyzed Arylogous Michael Reaction of 3-Aryl Phthalides. Org. Lett. 2017, 19, 4383–4386. [Google Scholar] [CrossRef]
- Autuori, G. Addizione di Michael di Ftalidi Promossa da Superbasi Organiche. Bachelor’s Thesis, Università degli Studi di Salerno, Fisciano, Italy, 17 May 2021. [Google Scholar]
- Ripin, D.H.B. pKa. In Practical Synthetic Organic Chemistry—Reactions, Principles, and Techniques; Caron, S., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2011; pp. 771–803. [Google Scholar]
- Specht, A.; Thomann, J.-S.; Alarcon, K.; Wittayanan, W.; Ogden, D.; Furuta, T.; Kurakawa, Y.; Goeldner, M. New Photoremovable Protecting Groups for Carboxylic Acids with High Photolytic Efficiencies at Near-UV Irradiation. Application to the Photocontrolled Release of L-Glutamate. ChemBioChem 2006, 7, 1690–1695. [Google Scholar] [CrossRef]
- Bléger, D.; Ciesielski, A.; Samorì, P.; Hecht, S. Photoswitching Vertically Oriented Azobenzene Self-Assembled Monolayers at the Solid–Liquid Interface. Chem. Eur. J. 2010, 16, 14256–14260. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.; Jwa, D.G.; You, A.; Park, S.; Kim, J.G.; Kang, S.M.; Kim, M. Photochemical Control of Polydopamine Coating in an Aprotic Organic Solvent. Asian J. Org. Chem. 2019, 8, 1610–1612. [Google Scholar] [CrossRef]
- Artamkina, G.A.; Grinfel’d, A.A.; Beletskaya, I.P. Oxidation of triaryl- and diarylmethanes by oxygen in the system KOH-dimethoxyethane-18-crown-6-ether and cleavage of triarylcarbinol and diaryl ketone intermediates. Russ. Chem. Bull. 1983, 32, 345–352. [Google Scholar] [CrossRef]
- Chupp, J.P.; Grabiak, R.C.; Leschinsky, K.L.; Neumann, T.L. Substituted Benzotrichloride Synthesis by Phase Transfer-Catalyzed Chlorination. Synthesis 1986, 224–226. [Google Scholar] [CrossRef]
- Taha, N.; Sasson, Y.; Chidambaram, M. Phase transfer methodology for the synthesis of substituted stilbenes under Knoevenagel condensation condition. Appl. Catal. A Gen. 2008, 350, 217–224. [Google Scholar] [CrossRef]
- Ye, F.; Li, Y.; Fu, Y.; Gao, S.; Zhao, L.-X. Microwave-assisted synthesis and crystal structure of novel 2-dichloromethyl-1,3-dioxolanes. Heterocycles 2013, 87, 407–415. [Google Scholar] [CrossRef]
- Ye, F.; Sun, C.-Y.; Fu, Y. Synthesis and Crystal Structure of 2-(Dichloromethyl)-2-(4-nitrophenyl)-1,3-dioxane. J. Chem. 2013, 2013, 974174. [Google Scholar] [CrossRef]
- Hardy, M.A.; Chachignon, H.; Cahard, D. Advances in Asymmetric Di- and Trifluoromethylthiolation, and Di- and Trifluoromethoxylation Reactions. Asian J. Org. Chem. 2019, 8, 591–609. [Google Scholar] [CrossRef]
- Yang, X.; Wu, T.; Phipps, R.J.; Toste, F.D. Advances in Catalytic Enantioselective Fluorination, Mono-, Di-, and Trifluoromethylation, and Trifluoromethylthiolation Reactions. Chem. Rev. 2015, 115, 826–870. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.; Puglisi, A.; Raimondi, L.; Benaglia, M. Synthesis of Alpha-trifluoromethylthio Carbonyl Compounds: A Survey of the Methods for the Direct Introduction of the SCF3 Group on to Organic Molecules. ChemCatChem 2018, 10, 2717–2733. [Google Scholar] [CrossRef]
- Xu, L.; Yu, L.; Liu, J.; Wang, H.; Zheng, C.; Zhao, G. Enantioselective Vinylogous Mannich-Type Reactions to Construct CF3S-Containing Stereocenters Catalysed by Chiral Quaternary Phosphonium Salts. Adv. Synth. Catal. 2020, 362, 1851–1857. [Google Scholar] [CrossRef]
- Wu, Y.; Deng, L. Asymmetric Synthesis of Trifluoromethylated Amines via Catalytic Enantioselective Isomerization of Imines. J. Am. Chem. Soc. 2012, 134, 14334–14337. [Google Scholar] [CrossRef]
- Tang, S.; Zhang, X.; Sun, J.; Niu, D.; Chruma, J.J. 2-Azaallyl Anions, 2-Azaallyl Cations, 2-Azaallyl Radicals, and Azomethine Ylides. Chem. Rev. 2018, 118, 10393–10457. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Hu, L.; Li, Z.; Deng, L. Catalytic asymmetric umpolung reactions of imines. Nature 2015, 523, 445–450. [Google Scholar] [CrossRef]
- Li, Z.; Hu, B.; Wu, Y.; Fei, C.; Deng, L. Control of chemoselectivity in asymmetric tandem reactions: Direct synthesis of chiral amines bearing nonadjacent stereocenters. Proc. Natl. Acad. Sci. USA 2018, 115, 1730–1735. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Wu, Y.; Li, Z.; Deng, L. Catalytic Asymmetric Synthesis of Chiral γ-Amino Ketones via Umpolung Reactions of Imines. J. Am. Chem. Soc. 2016, 138, 15817–15820. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Mino, T.; Sakamoto, M. Organocatalytic Highly Regio- and Enantioselective Umpolung Michael Addition Reaction of α-Imino Esters. Chem. Eur. J. 2017, 23, 12749–12753. [Google Scholar] [CrossRef]
- Hu, B.; Deng, L. Catalytic Asymmetric Synthesis of Trifluoromethylated γ-Amino Acids through the Umpolung Addition of Trifluoromethyl Imines to Carboxylic Acid Derivatives. Angew. Chem. Int. Ed. 2018, 57, 2233–2237. [Google Scholar] [CrossRef]
- Zhou, X.; Wu, Y.; Deng, L. Cinchonium Betaines as Efficient Catalysts for Asymmetric Proton Transfer Catalysis: The Development of a Practical Enantioselective Isomerization of Trifluoromethyl Imines. J. Am. Chem. Soc. 2016, 138, 12297–12302. [Google Scholar] [CrossRef]
- Hu, B.; Bezpalko, M.W.; Fei, C.; Dickie, D.A.; Foxman, B.M.; Deng, L. Origin of and a Solution for Uneven Efficiency by Cinchona Alkaloid-Derived, Pseudoenantiomeric Catalysts for Asymmetric Reactions. J. Am. Chem. Soc. 2018, 138, 13913–13920. [Google Scholar] [CrossRef]
- Hu, B.; Deng, L. Direct Catalytic Asymmetric Synthesis of Trifluoromethylated γ-Amino Esters/Lactones via Umpolung Strategy. J. Org. Chem. 2019, 84, 994–1005. [Google Scholar] [CrossRef] [PubMed]








































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Amato, A.; Della Sala, G. Vinylogous and Arylogous Stereoselective Base-Promoted Phase-Transfer Catalysis. Catalysts 2021, 11, 1545. https://doi.org/10.3390/catal11121545
D’Amato A, Della Sala G. Vinylogous and Arylogous Stereoselective Base-Promoted Phase-Transfer Catalysis. Catalysts. 2021; 11(12):1545. https://doi.org/10.3390/catal11121545
Chicago/Turabian StyleD’Amato, Assunta, and Giorgio Della Sala. 2021. "Vinylogous and Arylogous Stereoselective Base-Promoted Phase-Transfer Catalysis" Catalysts 11, no. 12: 1545. https://doi.org/10.3390/catal11121545
APA StyleD’Amato, A., & Della Sala, G. (2021). Vinylogous and Arylogous Stereoselective Base-Promoted Phase-Transfer Catalysis. Catalysts, 11(12), 1545. https://doi.org/10.3390/catal11121545