
One-Pot Tandem Catalytic Epoxidation—CO2 Insertion of Monounsaturated Methyl Oleate to the Corresponding Cyclic Organic Carbonate




Abstract
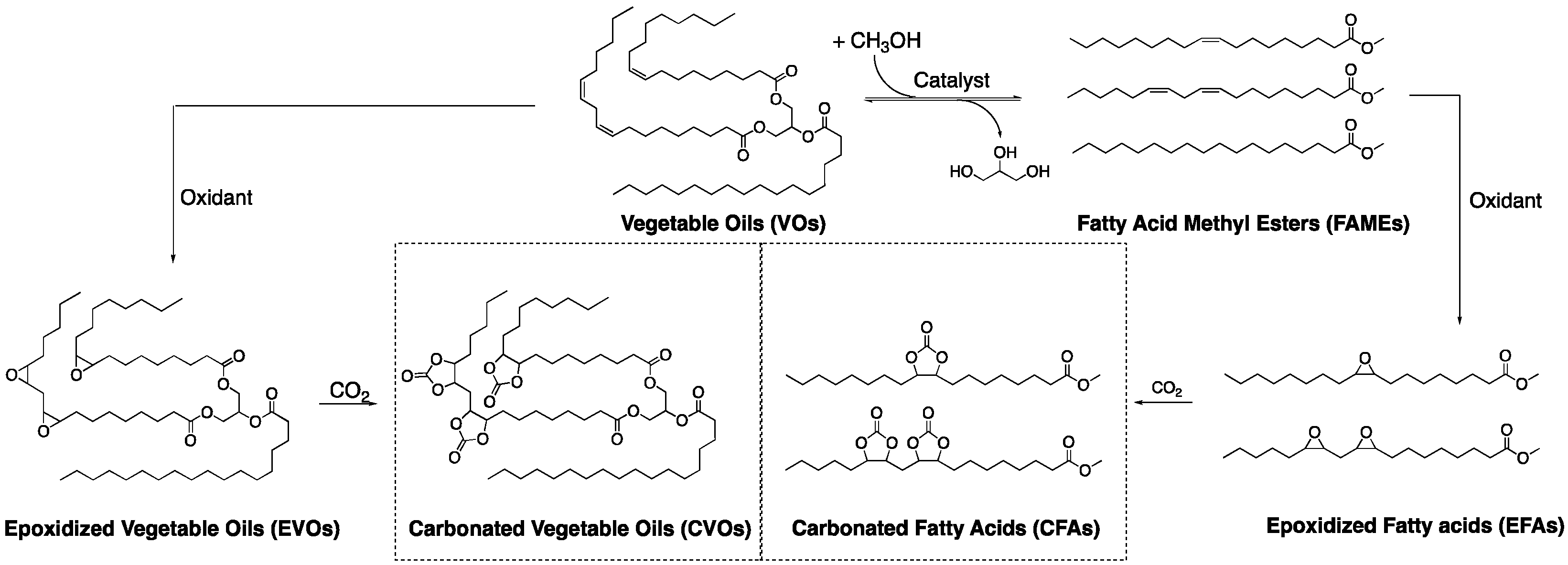
:1. Introduction
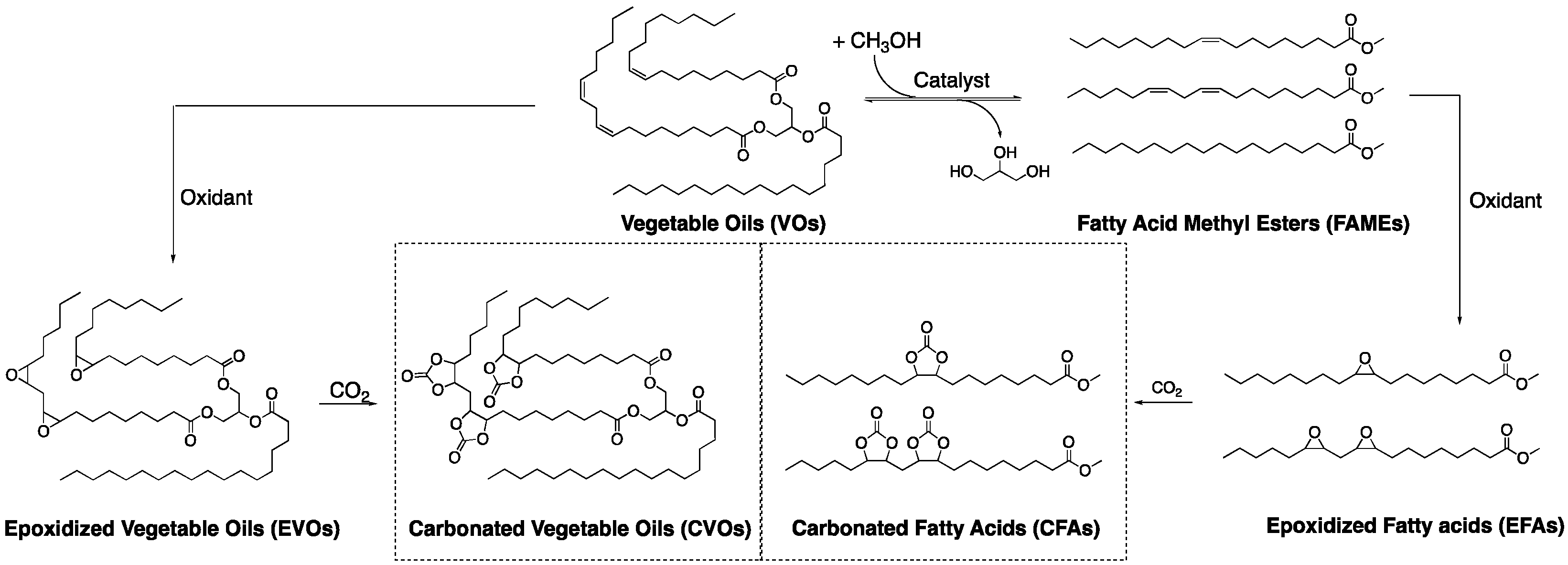
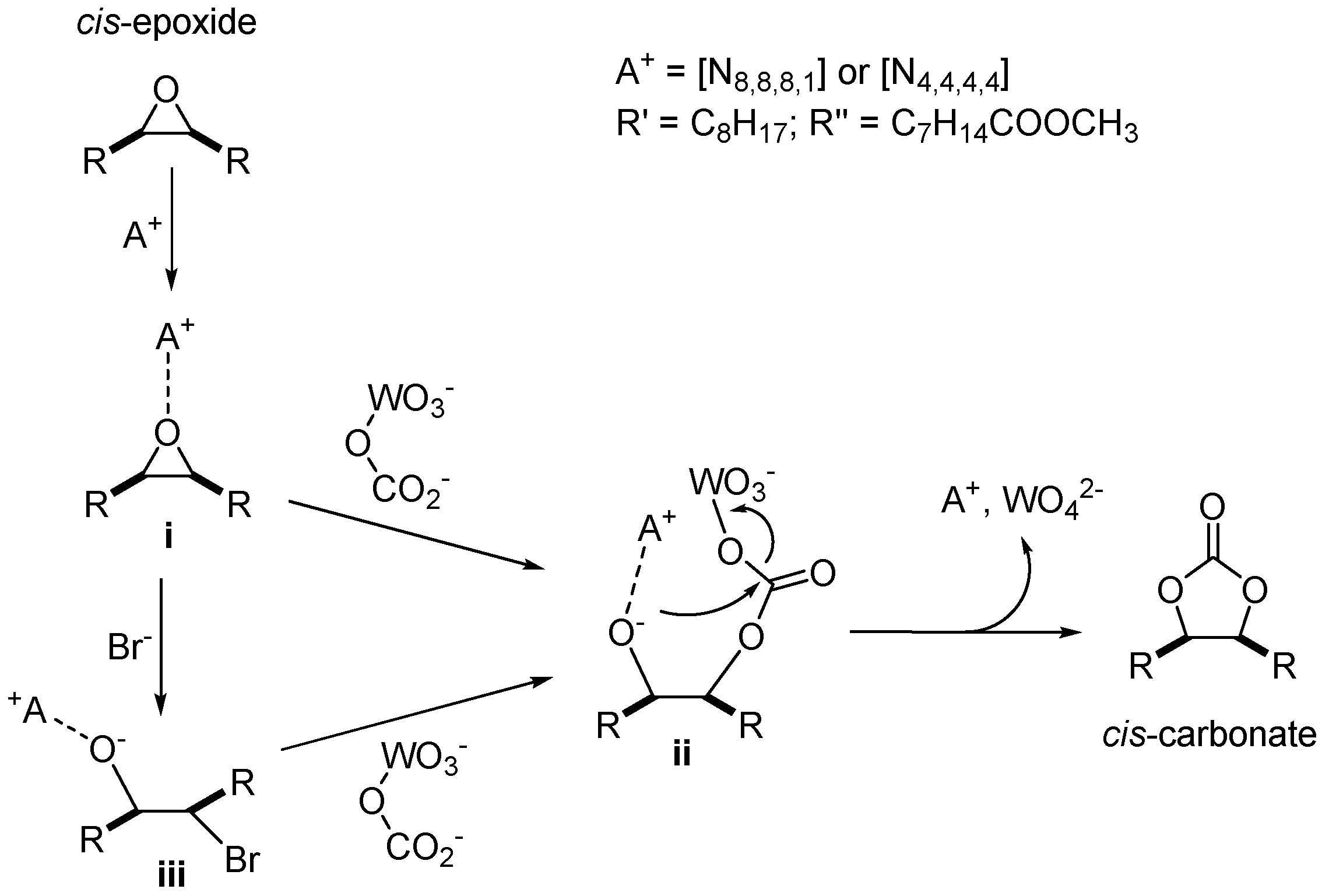
2. Results and Discussions
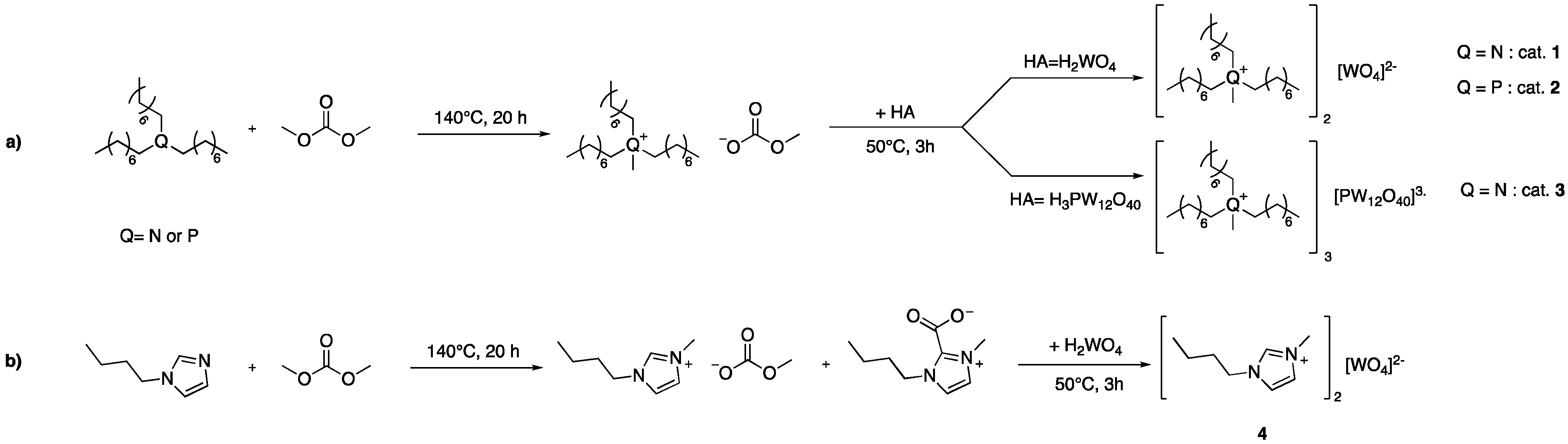
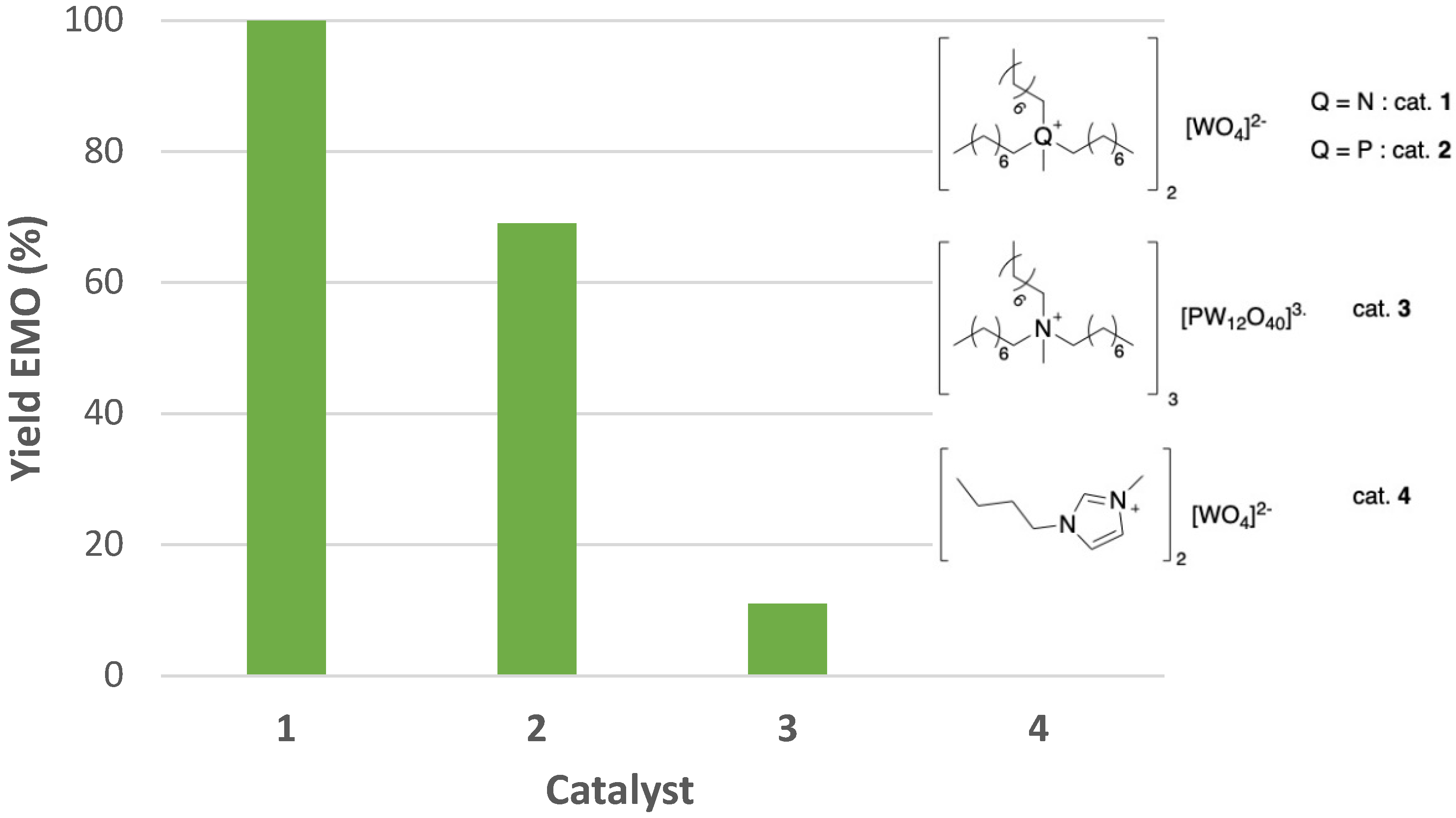
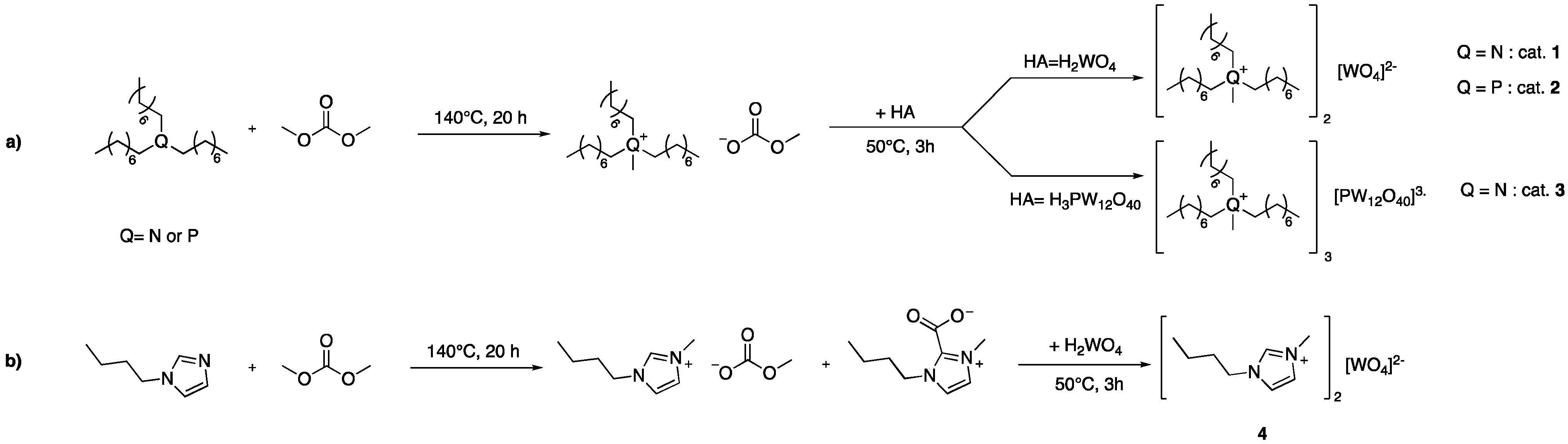
2.1. Synthesis of Tungstate-Based Ionic Liquids
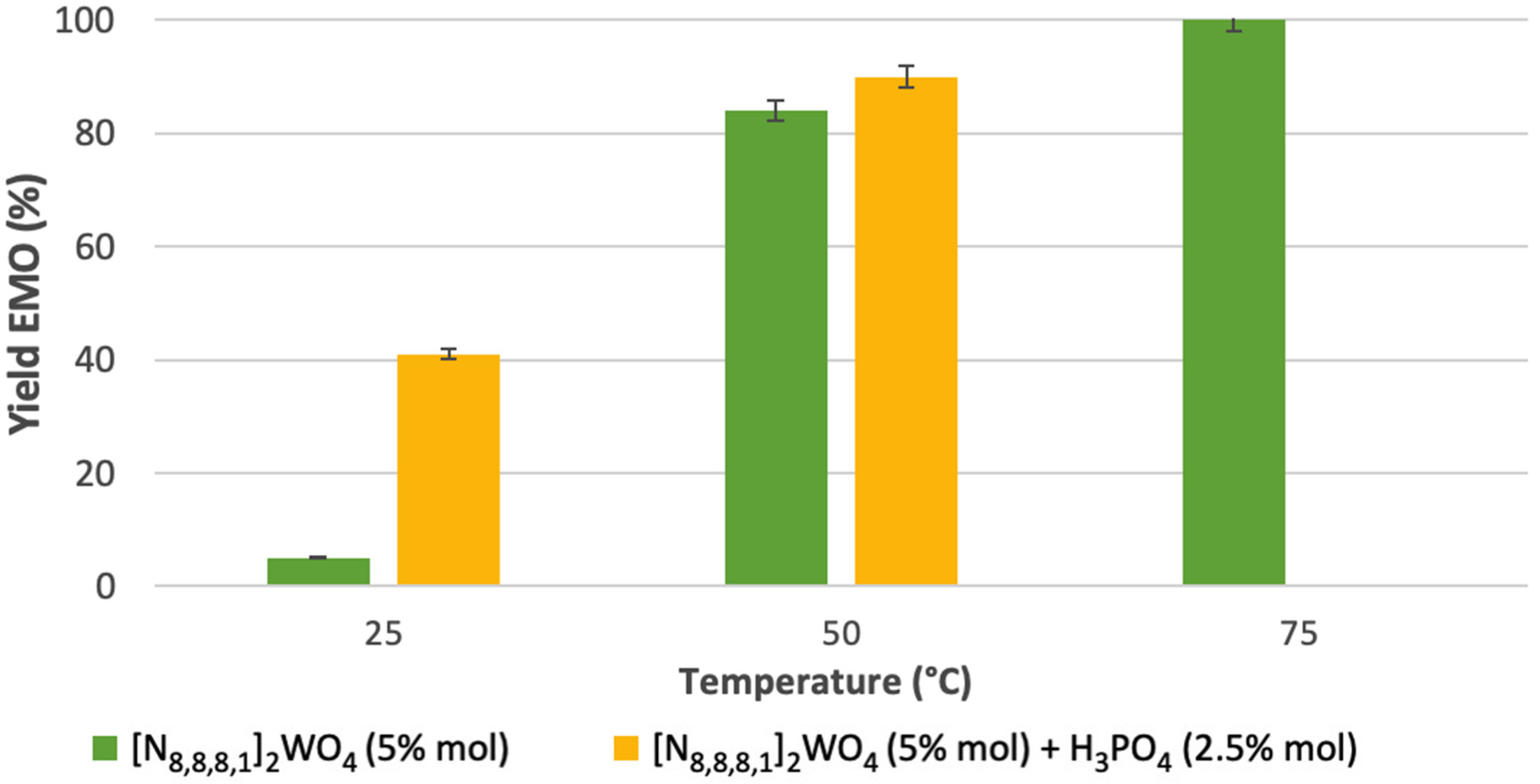
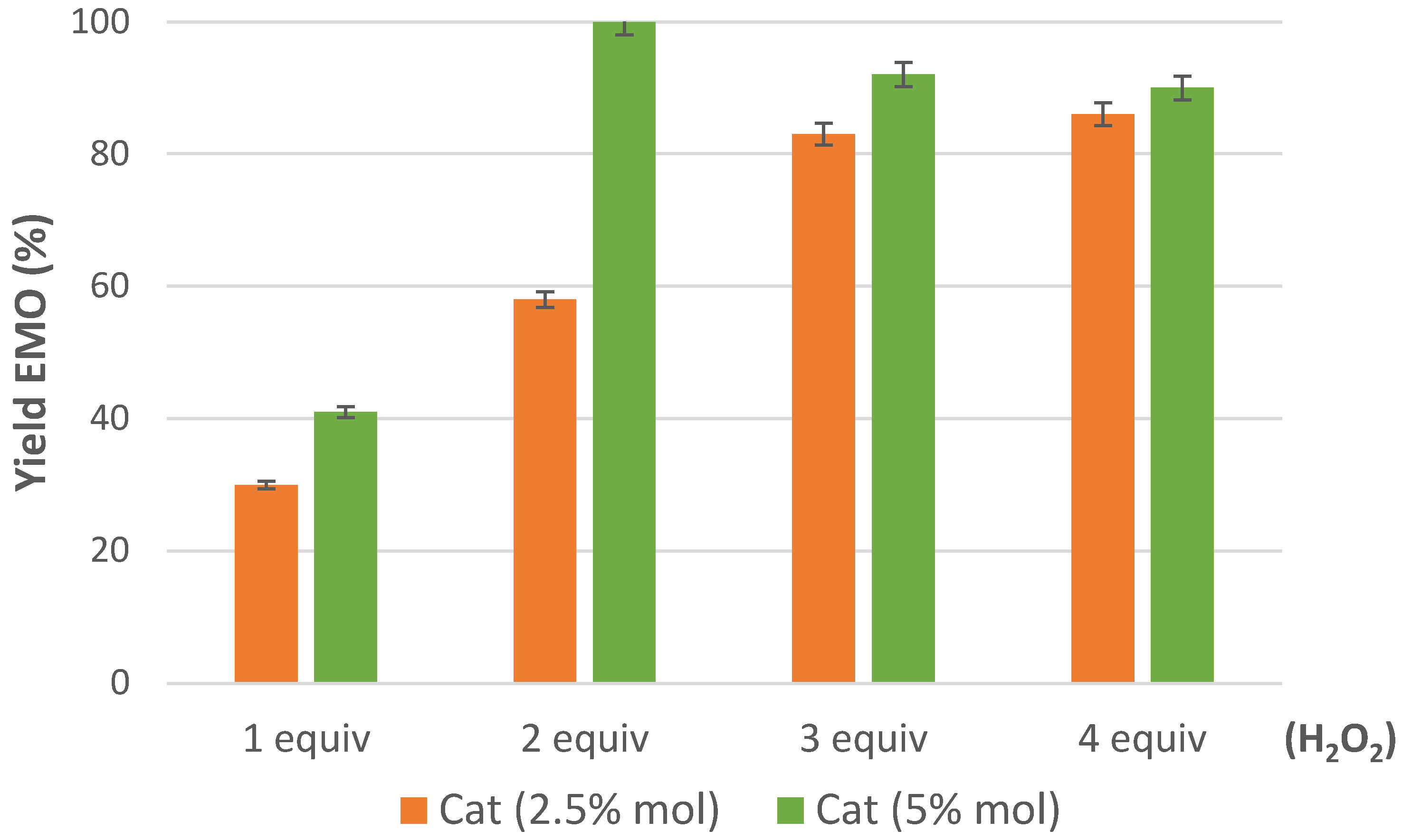
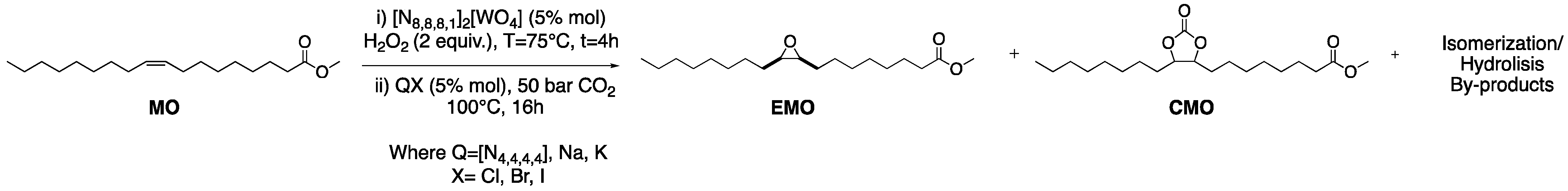
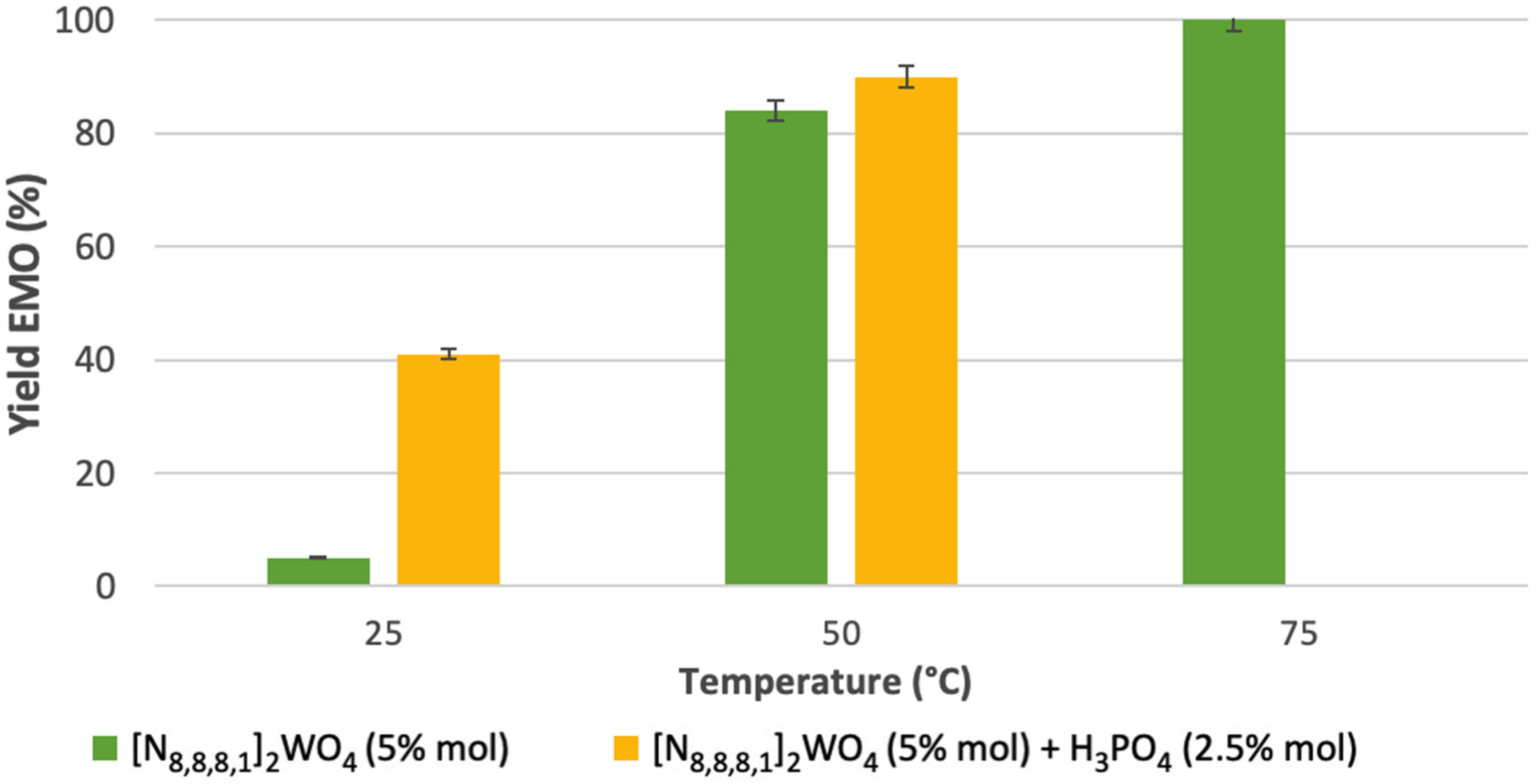
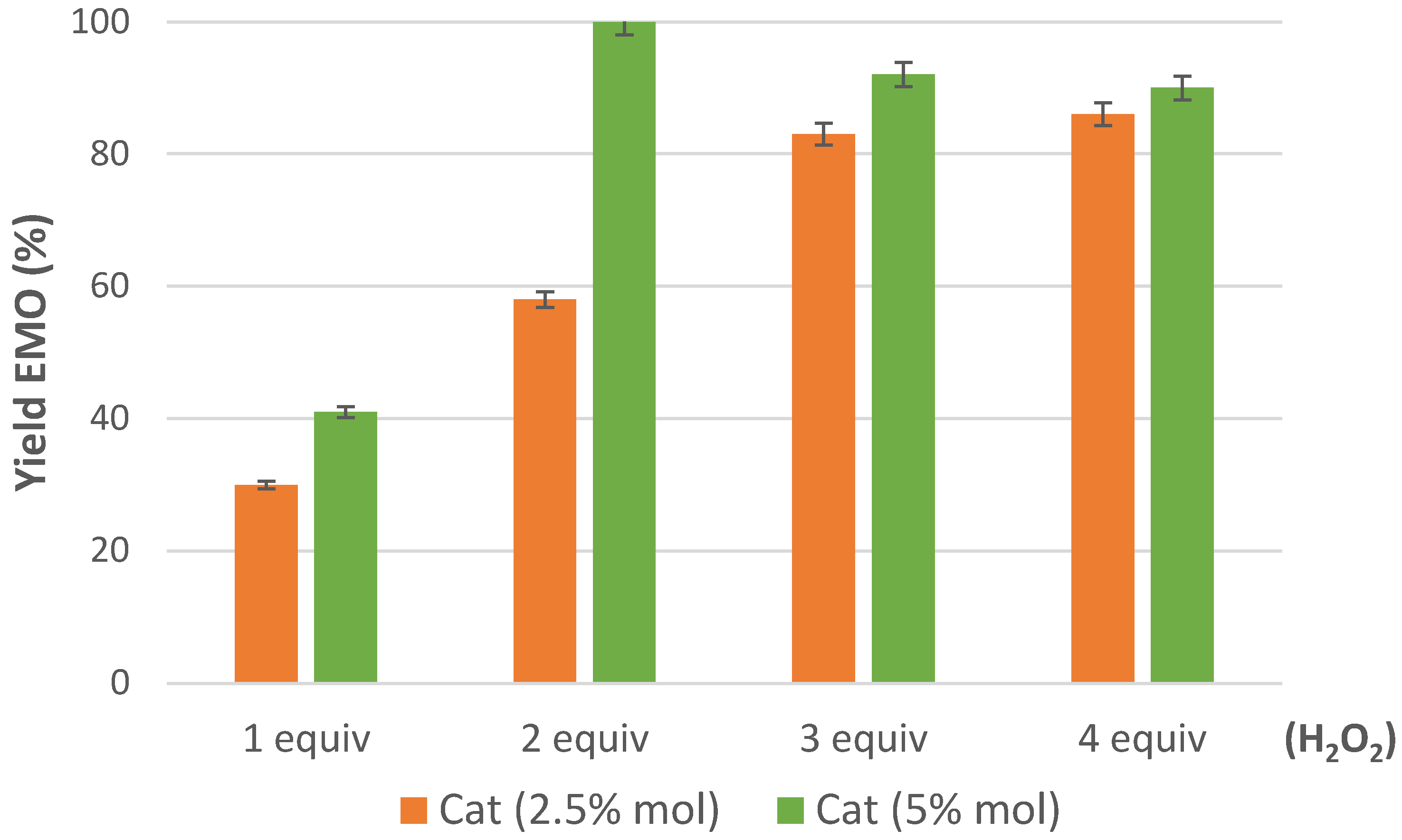
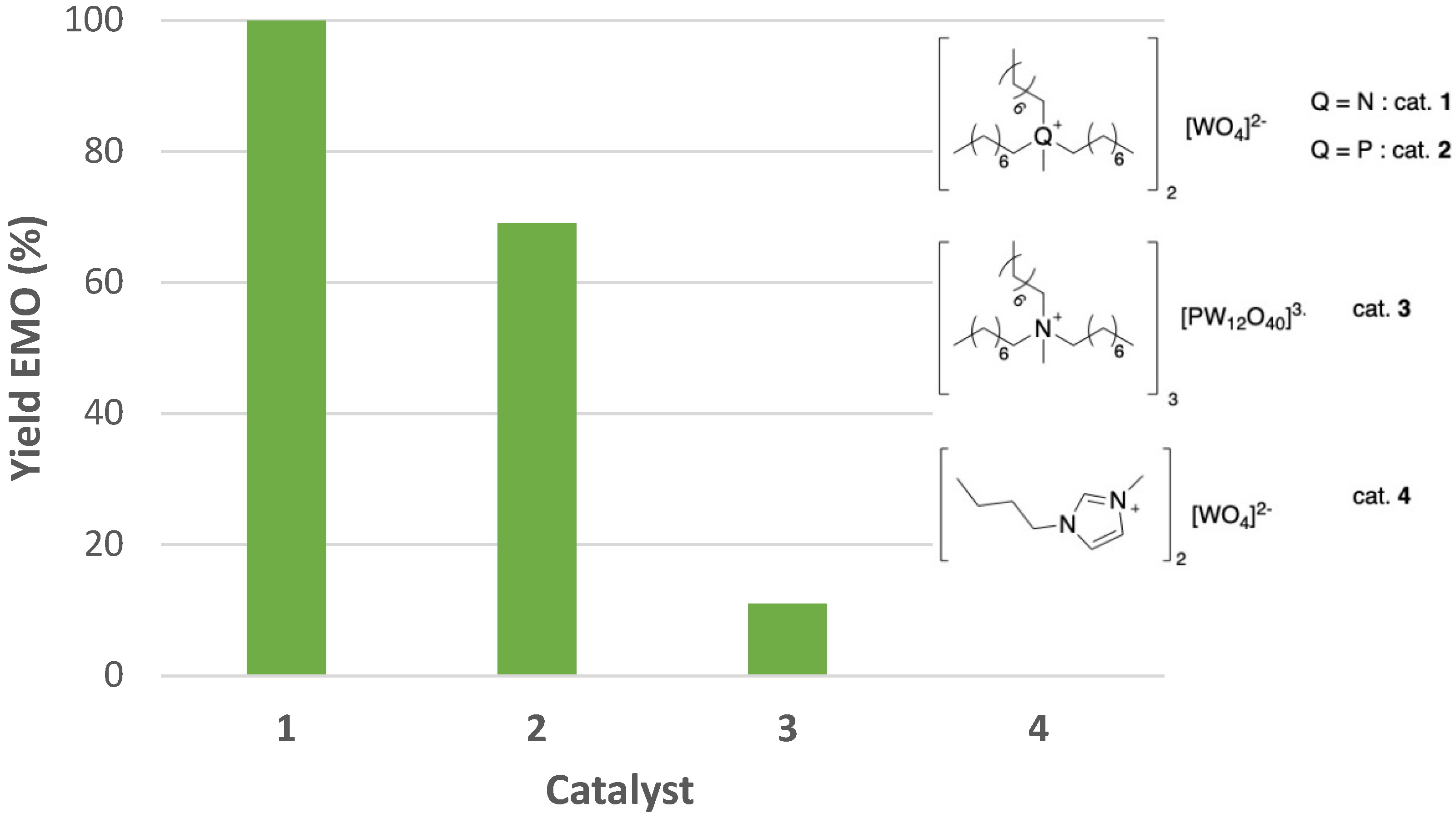
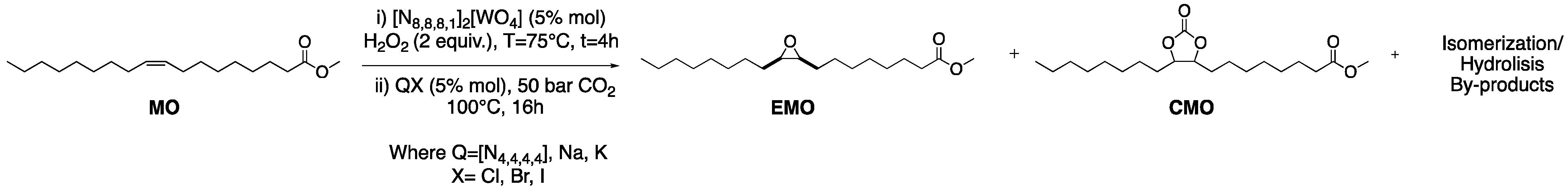
2.2. Epoxidation of Methyl Oleate
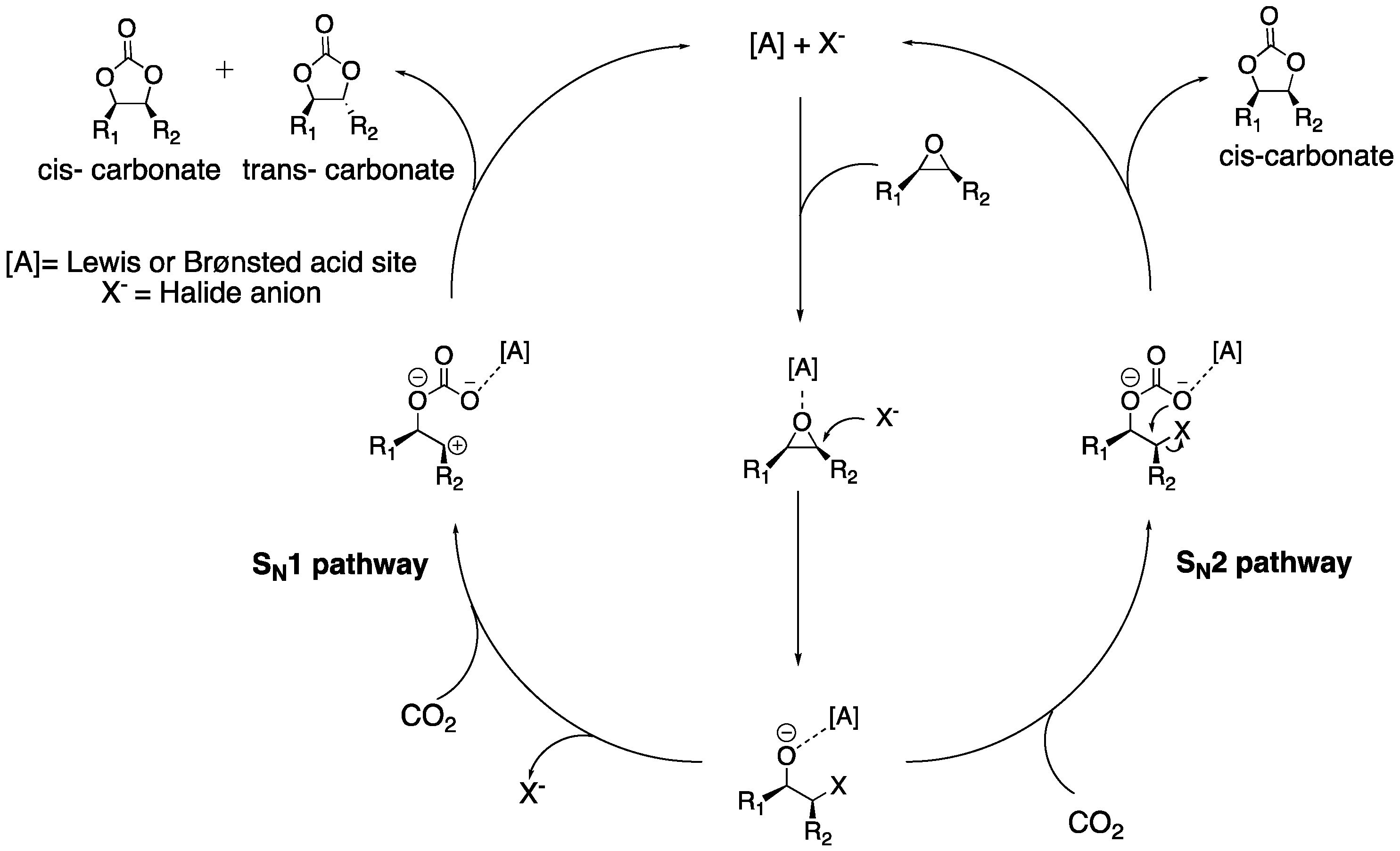
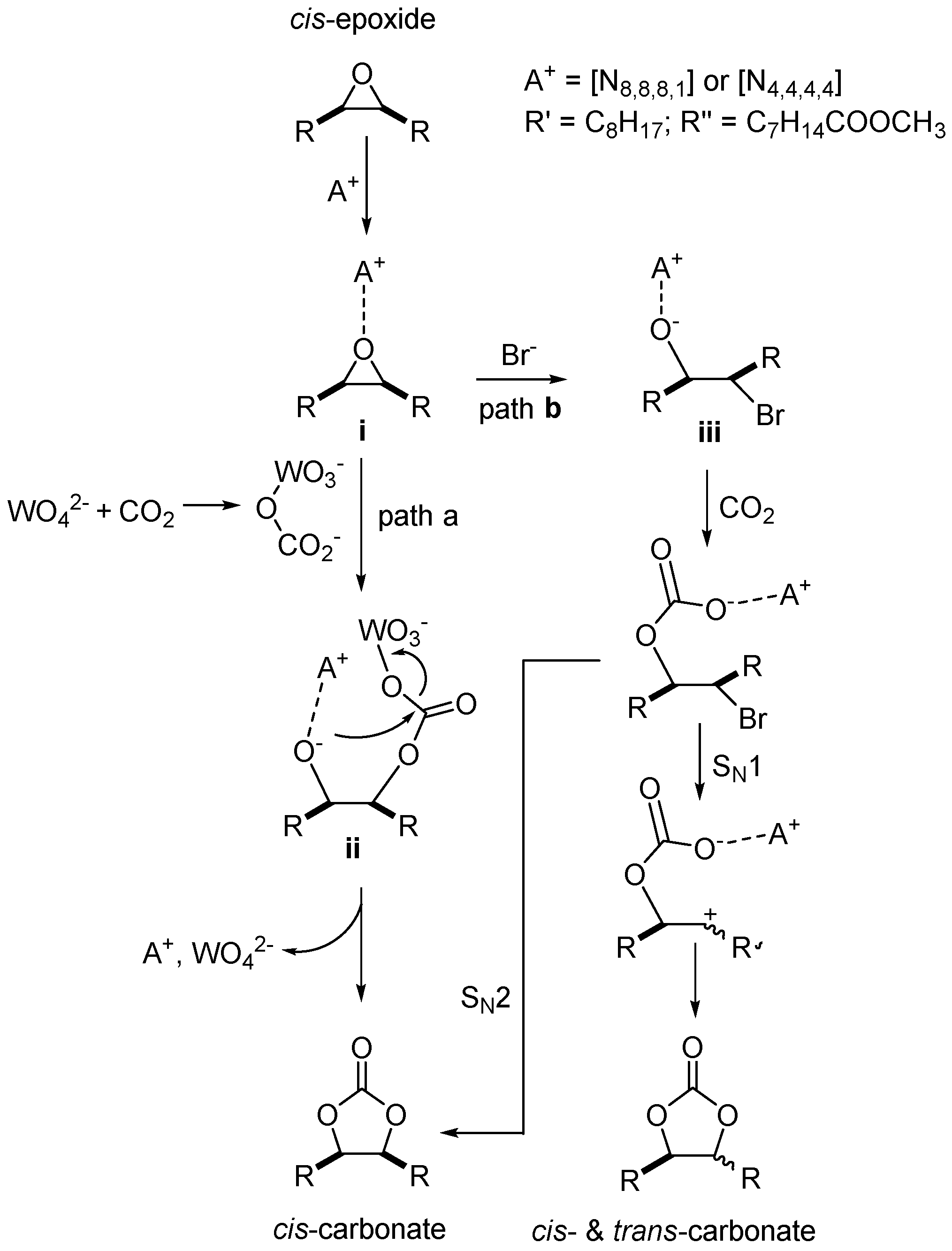
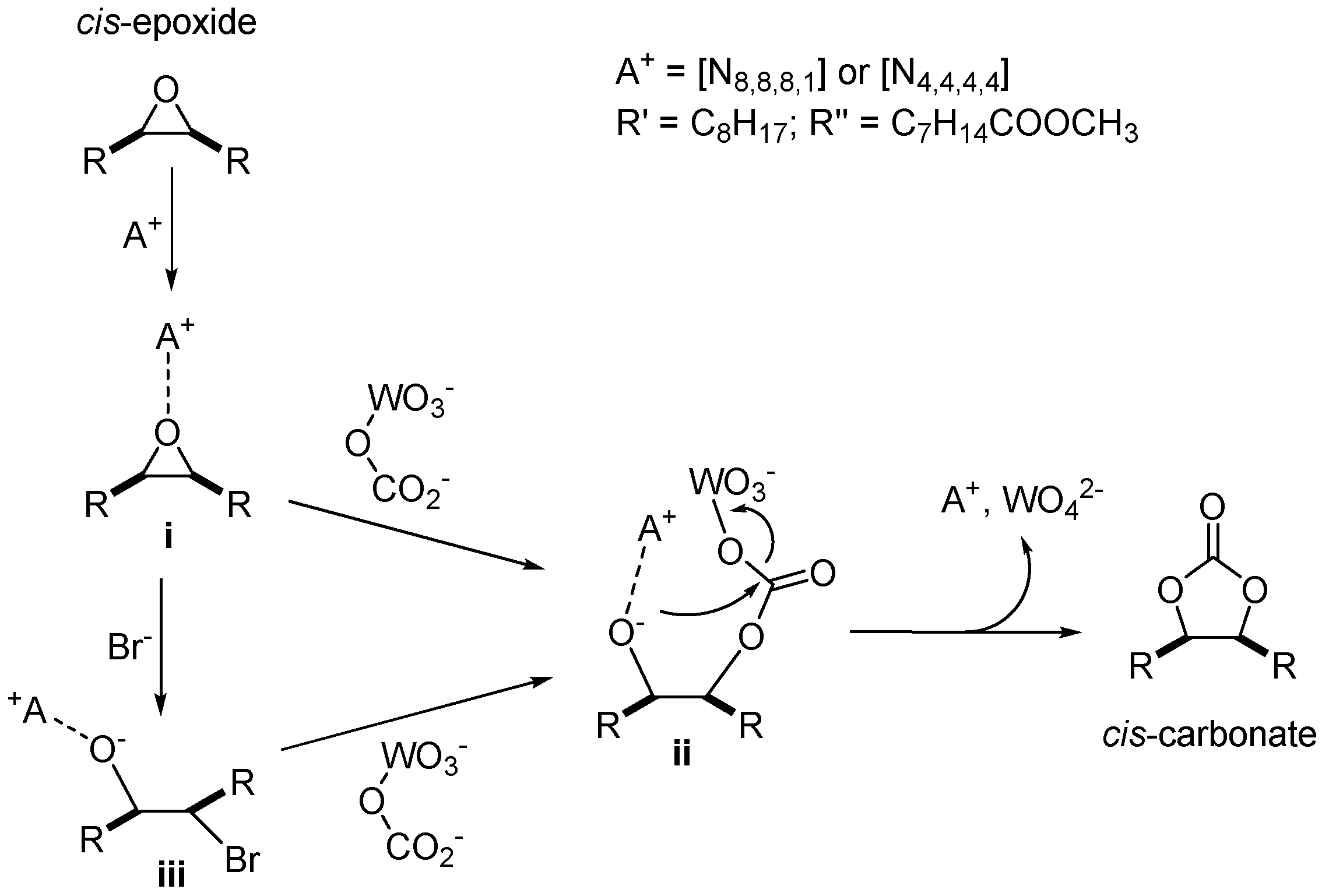
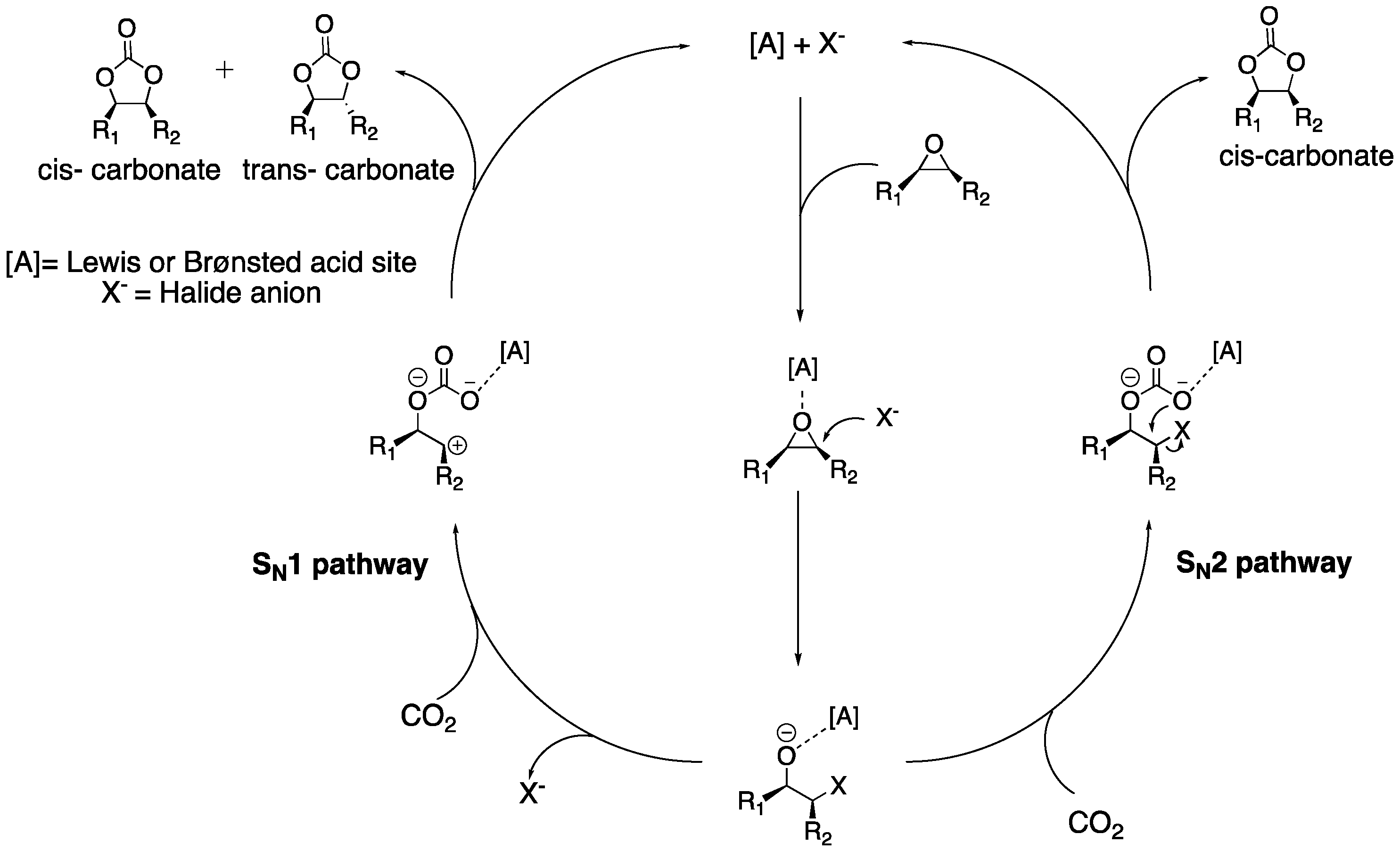
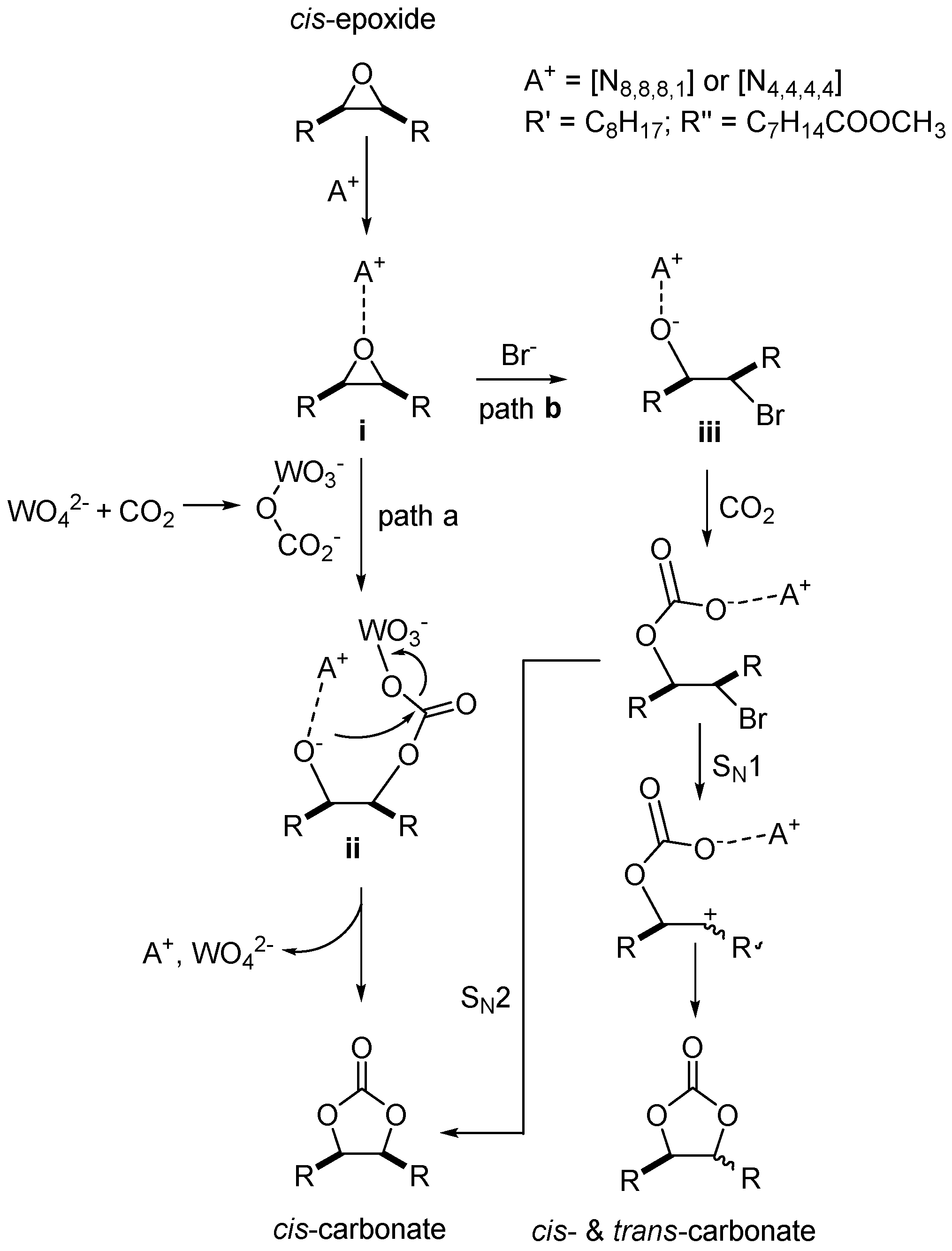
2.3. CO2 Insertion Step
2.4. Tandem Reaction
3. Conclusions
4. Materials and Methods
4.1. General Reagents
4.2. Analyses
4.3. Synthesis of Catalysts
4.4. Syntheses of Ionic Liquids with Dimethyl Carbonate (DMC)
4.5. Syntheses of Tungstate-Based Ionic Liquids (TILCs)
4.6. Reaction Procedures
4.6.1. Synthesis of Methyl Oleate
4.6.2. Typical Procedure for the Epoxidation of Methyl Oleate
4.7. Test for the Scalability of 1-Decene Epoxidation
4.8. Typical Procedure for CO2 Fixation Reaction into Epoxidized Methyl Oleate
4.9. Typical Procedure for the Assisted One-Pot Tandem Reaction
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mac Dowell, N.; Fennell, P.S.; Shah, N.; Maitland, G.C. The role of CO2 capture and utilization in mitigating climate change. Nat. Clim. Chang. 2017, 7, 243–249. [Google Scholar] [CrossRef] [Green Version]
- Poliakoff, M.; Leitner, W.; Streng, E.S. The twelve principles of CO2 chemistry. Faraday Discuss. 2015, 183, 9–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selva, M.; Perosa, A.; Fiorani, G.; Cattelan, L. CO2 and Organic Carbonates for the Sustainable Valorization of Renewable Compounds. In Green Synthetic Processes and Procedures; Royal Society of Chemistry: London, UK, 2019; pp. 319–342. [Google Scholar]
- Aomchad, V.; Cristòfol, À.; Della Monica, F.; Limburg, B.; D’Elia, V.; Kleij, A.W.J.G.C. Recent progress in the catalytic transformation of carbon dioxide into biosourced organic carbonates. Green Chem. 2021, 23, 1077–1113. [Google Scholar] [CrossRef]
- OECD; FAO. Oilseeds and Oilseed Products. In OECD-FAO Agricultural Outlook 2018–2027; OECD Publishing: Paris, France; Food and Agriculture Organization of the United Nations: Rome, Italy, 2018. [Google Scholar]
- Ahmad, M.U. Fatty Acids: Chemistry, Synthesis, and Applications; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Karmakar, G.; Ghosh, P.; Sharma, B.K.J.L. Chemically modifying vegetable oils to prepare green lubricants. Lubricants 2017, 5, 44. [Google Scholar] [CrossRef] [Green Version]
- Schäffner, B.; Blug, M.; Kruse, D.; Polyakov, M.; Köckritz, A.; Martin, A.; Rajagopalan, P.; Bentrup, U.; Brückner, A.; Jung, S. Synthesis and Application of Carbonated Fatty Acid Esters from Carbon Dioxide Including a Life Cycle Analysis. ChemSusChem 2014, 7, 1133–1139. [Google Scholar] [CrossRef] [PubMed]
- Carré, C.; Ecochard, Y.; Caillol, S.; Avérous, L. From the synthesis of biobased cyclic carbonate to polyhydroxyurethanes: A promising route towards renewable NonIsocyanate Polyurethanes. ChemSusChem 2019, 12, 3410–3430. [Google Scholar] [CrossRef] [PubMed]
- Danov, S.; Kazantsev, O.; Esipovich, A.; Belousov, A.; Rogozhin, A.; Kanakov, E. Recent advances in the field of selective epoxidation of vegetable oils and their derivatives: A review and perspective. Catal. Sci. Technol. 2017, 7, 3659–3675. [Google Scholar] [CrossRef]
- Santacesaria, E.; Renken, A.; Russo, V.; Turco, R.; Tesser, R.; Di Serio, M. Biphasic model describing soybean oil epoxidation with H2O2 in continuous reactors. Ind. Eng. Chem. Res. 2012, 51, 8760–8767. [Google Scholar] [CrossRef]
- Köckritz, A.; Martin, A. Oxidation of unsaturated fatty acid derivatives and vegetable oils. Eur. J. Lipid. Sci. Technol. 2008, 110, 812–824. [Google Scholar] [CrossRef]
- Buettner, H.; Longwitz, L.; Steinbauer, J.; Wulf, C.; Werner, T. Recent developments in the synthesis of cyclic carbonates from epoxides and CO2. In Chemical Transformations of Carbon Dioxide; Springer: Cham, Switzerland, 2017; pp. 89–144. [Google Scholar]
- Tenhumberg, N.; Büttner, H.; Schäffner, B.; Kruse, D.; Blumenstein, M.; Werner, T. Cooperative catalyst system for the synthesis of oleochemical cyclic carbonates from CO2 and renewables. Green Chem. 2016, 18, 3775–3788. [Google Scholar] [CrossRef]
- Carrodeguas, L.P.; Cristòfol, À.; Fraile, J.M.; Mayoral, J.A.; Dorado, V.; Herrerías, C.I.; Kleij, A. Fatty acid based biocarbonates: Al-mediated stereoselective preparation of mono-, di-and tricarbonates under mild and solvent-less conditions. Green Chem. 2017, 19, 3535–3541. [Google Scholar] [CrossRef]
- Langanke, J.; Greiner, L.; Leitner, W. Substrate dependent synergetic and antagonistic interaction of ammonium halide and polyoxometalate catalysts in the synthesis of cyclic carbonates from oleochemical epoxides and CO2. Green Chem. 2013, 15, 1173–1182. [Google Scholar] [CrossRef] [Green Version]
- Bobbink, F.D.; Vasilyev, D.; Hulla, M.; Chamam, S.; Menoud, F.; Laurenczy, G.b.; Katsyuba, S.; Dyson, P.J. Intricacies of cation–anion combinations in imidazolium salt-catalyzed cycloaddition of CO2 into epoxides. ACS Catal. 2018, 8, 2589–2594. [Google Scholar] [CrossRef] [Green Version]
- Steinbauer, J.; Kubis, C.; Ludwig, R.; Werner, T. Mechanistic Study on the Addition of CO2 to Epoxides Catalyzed by Ammonium and Phosphonium Salts: A Combined Spectroscopic and Kinetic Approach. ACS Sust. Chem. Eng. 2018, 6, 10778–10788. [Google Scholar] [CrossRef]
- Fankhauser-Noti, A.; Fiselier, K.; Biedermann-Brem, S.; Grob, K. Assessment of epoxidized soy bean oil (ESBO) migrating into foods: Comparison with ESBO-like epoxy fatty acids in our normal diet. Food Chem. Toxicol. 2006, 44, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Greene, J.F.; Newman, J.W.; Williamson, K.C.; Hammock, B.D. Toxicity of epoxy fatty acids and related compounds to cells expressing human soluble epoxide hydrolase. Chem. Res. Toxicol. 2000, 13, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Calmanti, R.; Selva, M.; Perosa, A. Direct oxidative carboxylation of terminal olefins to cyclic carbonates by tungstate assisted-tandem catalysis. Green Chem. 2021, 23, 7609–7619. [Google Scholar] [CrossRef]
- Calmanti, R.; Selva, M.; Perosa, A.J.G.C. Tandem catalysis: One-pot synthesis of cyclic organic carbonates from olefins and carbon dioxide. Green Chem. 2021, 23, 1921–1941. [Google Scholar] [CrossRef]
- Fabris, M.; Lucchini, V.; Noè, M.; Perosa, A.; Selva, M. Ionic liquids made with dimethyl carbonate: Solvents as well as boosted basic catalysts for the Michael reaction. Chem.-Eur. J. 2009, 15, 12273–12282. [Google Scholar] [CrossRef] [Green Version]
- Holbrey, J.D.; Rogers, R.D.; Shukla, S.S.; Wilfred, C.D. Optimised microwave-assisted synthesis of methylcarbonate salts: A convenient methodology to prepare intermediates for ionic liquid libraries. Green Chem. 2010, 12, 407–413. [Google Scholar] [CrossRef]
- Busca, G. Differentiation of mono-oxo and polyoxo and of monomeric and polymeric vanadate, molybdate and tungstate species in metal oxide catalysts by IR and Raman spectroscopy. J. Raman Spectrosc. 2002, 33, 348–358. [Google Scholar] [CrossRef]
- Calmanti, R.; Selva, M.; Perosa, A. Tungstate ionic liquids as catalysts for CO2 fixation into epoxides. Mol. Catal. 2020, 486, 110854. [Google Scholar] [CrossRef]
- Chen, Y.-G.; Gong, J.; Qu, L.-Y. Tungsten-183 nuclear magnetic resonance spectroscopy in the study of polyoxometalates. Coord. Chem. Rev. 2004, 248, 245–260. [Google Scholar] [CrossRef]
- Ueda, T.; Kodani, K.; Ota, H.; Shiro, M.; Guo, S.-X.; Boas, J.F.; Bond, A.M. Voltammetric and Spectroscopic Studies of α-and β-[PW12O40] 3–Polyoxometalates in Neutral and Acidic Media: Structural Characterization as Their [(n-Bu4N)3][PW12O40] Salts. Inorg. Chem. 2017, 56, 3990–4001. [Google Scholar] [CrossRef] [PubMed]
- Poli, E.; Clacens, J.-M.; Barrault, J.; Pouilloux, Y. Solvent-free selective epoxidation of fatty esters over a tungsten-based catalyst. Catal. Today 2009, 140, 19–22. [Google Scholar] [CrossRef]
- Clayden, J.; Greeves, N.; Warren, S.; Wothers, P. Organic Chemistry; Oxford University Press: Oxford, UK, 2014. [Google Scholar]
- Poli, E.; Bion, N.; Barrault, J.; Casciato, S.; Dubois, V.; Pouilloux, Y.; Clacens, J.-M. Selective epoxidation of unsaturated fatty esters over peroxophosphotungstic catalysts (POW) under solvent free conditions: Study of the POW catalyst’s mechanism. Catal. Today 2010, 157, 371–377. [Google Scholar] [CrossRef]
- Resul, M.F.M.G.; Fernández, A.M.L.; Rehman, A.; Harvey, A.P. Development of a selective, solvent-free epoxidation of limonene using hydrogen peroxide and a tungsten-based catalyst. React. Chem. Eng. 2018, 3, 747–756. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.-L.; Huang, T.-H. Kinetic study of the epoxidation of 1, 7-octadiene under phase-transfer-catalysis conditions. Ind. Eng. Chem. Res. 2004, 43, 675–681. [Google Scholar] [CrossRef]
- Mizuno, N.; Yamaguchi, K.; Kamata, K. Epoxidation of olefins with hydrogen peroxide catalyzed by polyoxometalates. Coord. Chem. Rev. 2005, 249, 1944–1956. [Google Scholar] [CrossRef]
- Pai, Z.P.; Khlebnikova, T.B.; Mattsat, Y.V.; Parmon, V.N. Catalytic oxidation of fatty acids. I. Epoxidation of unsaturated fatty acids. React. Kinet. Catal. Lett. 2009, 98, 1–8. [Google Scholar] [CrossRef]
- Musik, M.; Janus, E.; Pełech, R.; Sałaciński, Ł. Effective Epoxidation of Fatty Acid Methyl Esters with Hydrogen Peroxide by the Catalytic System H3PW12O40/Quaternary Phosphonium Salts. Catalysts 2021, 11, 1058. [Google Scholar] [CrossRef]
- Deferm, C.; Van den Bossche, A.; Luyten, J.; Oosterhof, H.; Fransaer, J.; Binnemans, K. Thermal stability of trihexyl (tetradecyl) phosphonium chloride. Phys. Chem. Chem. Phys. 2018, 20, 2444–2456. [Google Scholar] [CrossRef]
- Galvan, M.; Selva, M.; Perosa, A.; Noè, M. Toward the Design of Halide-and Metal-Free Ionic-Liquid Catalysts for the Cycloaddition of CO2 to Epoxides. Asian J. Org. Chem. 2014, 3, 504–513. [Google Scholar] [CrossRef]
- Blundell, R.K.; Licence, P. Quaternary ammonium and phosphonium based ionic liquids: A comparison of common anions. Phys. Chem. Chem. Phys. 2014, 16, 15278–15288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, P.J.; Ventura, S.P.; Batista, M.L.; Schröder, B.; Gonçalves, F.; Esperança, J.; Mutelet, F.; Coutinho, J.A. Understanding the impact of the central atom on the ionic liquid behavior: Phosphonium vs ammonium cations. J. Chem. Phys. 2014, 140, 064505. [Google Scholar] [CrossRef] [PubMed]
- Doll, K.M.; Erhan, S.Z. The improved synthesis of carbonated soybean oil using supercritical carbon dioxide at a reduced reaction time. Green Chem. 2005, 7, 849–854. [Google Scholar] [CrossRef]
- Natongchai, W.; Pornpraprom, S.; D’Elia, V. Synthesis of Bio-Based Cyclic Carbonates Using a Bio-Based Hydrogen Bond Donor: Application of Ascorbic Acid to the Cycloaddition of CO2 to Oleochemicals. Asian J. Org. Chem. 2020, 9, 801–810. [Google Scholar] [CrossRef]
- Cai, T.; Liu, J.; Cao, H.; Cui, C. Synthesis of bio-based cyclic carbonate from vegetable oil methyl ester by CO2 fixation with acid-base pair MOFs. Ind. Crops Prod. 2020, 145, 112155. [Google Scholar] [CrossRef]
- Kimura, T.; Kamata, K.; Mizuno, N. A bifunctional tungstate catalyst for chemical fixation of CO2 at atmospheric pressure. Angew. Chem. Int. Ed. 2012, 51, 6700–6703. [Google Scholar] [CrossRef]
- Sels, B.; De Vos, D.; Buntinx, M.; Pierard, F.; Kirsch-De Mesmaeker, A.; Jacobs, P. Layered double hydroxides exchanged with tungstate as biomimetic catalysts for mild oxidative bromination. Nature 1999, 400, 855–857. [Google Scholar] [CrossRef]
- Podgoršek, A.; Zupan, M.; Iskra, J. Oxidative halogenation with “green” oxidants: Oxygen and hydrogen peroxide. Angew. Chem. Int. Ed. 2009, 48, 8424–8450. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Lu, G.; Xiao, B.; Xie, C. Potassium iodide–polyethylene glycol catalyzed cycloaddition reaction of epoxidized soybean oil fatty acid methyl esters with CO2. RSC Adv. 2018, 8, 30860–30867. [Google Scholar] [CrossRef] [Green Version]
- Longwitz, L.; Steinbauer, J.; Spannenberg, A.; Werner, T. Calcium-based catalytic system for the synthesis of bio-derived cyclic carbonates under mild conditions. ACS Catal. 2018, 8, 665–672. [Google Scholar] [CrossRef]
- Alassmy, Y.A.; Pescarmona, P.P. The Role of Water Revisited and Enhanced: A Sustainable Catalytic System for the Conversion of CO2 into Cyclic Carbonates under Mild Conditions. ChemSusChem 2019, 12, 3856–3863. [Google Scholar] [CrossRef] [PubMed]
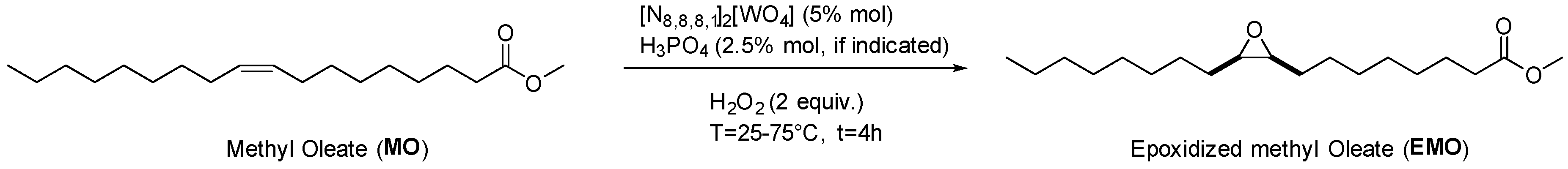
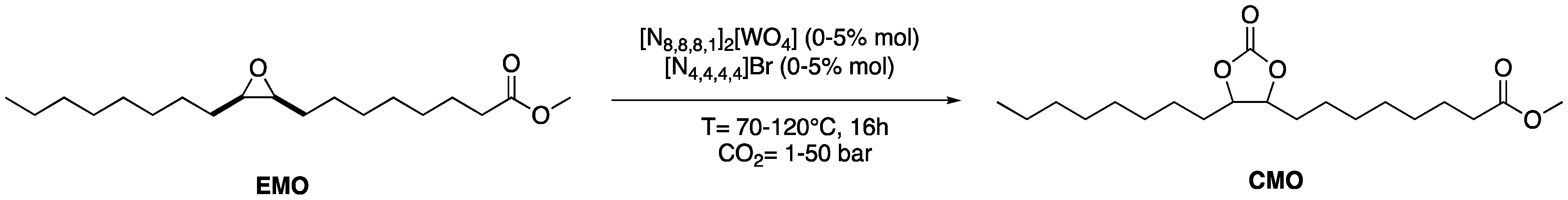

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
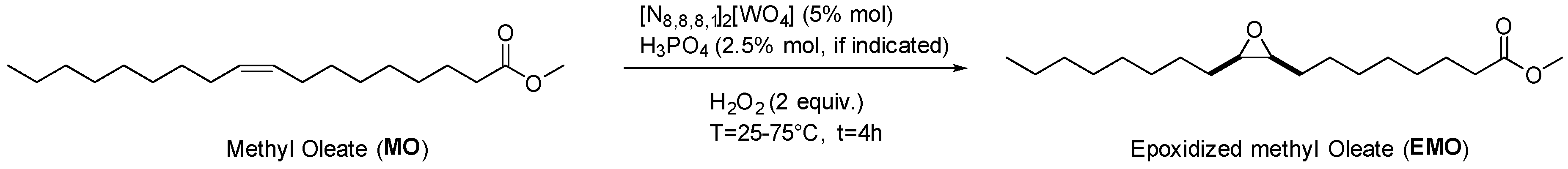{kind=link}
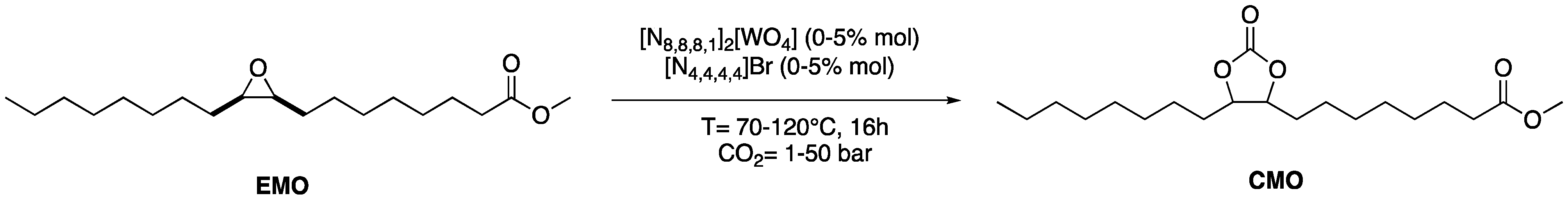{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | p(CO2) Bar | T (°C) | (N8,8,8,1)2-WO4 (% mol) | [N4,4,4,4]Br (% mol) | Conversion EMO | Selectivity CMO | Cis:Trans CMO Ratio |
|---|---|---|---|---|---|---|---|
| 1 | 50 | 100 | 5 | - | 28 | 64 | 99:1 |
| 2 | 50 | 100 | 2.5 | 80 | 91 | 56:44 | |
| 3 | 50 | 100 | 5 | 2.5 | 87 | 94 | 99:1 |
| 4 | 1 | 100 | 5 | 2.5 | 7 | 20 | 99:1 |
| 5 | 10 | 100 | 5 | 2.5 | 59 | 77 | 90:10 |
| 6 | 50 | 70 | 5 | 2.5 | 10 | 100 | 99:1 |
| 7 | 50 | 120 | 5 | 2.5 | 50 | 50 | 70:30 |
| 8 | 50 | 100 | 5 | 1.25 | 79 | 87 | 99:1 |
| 9 | 50 | 100 | 5 | 5 | 94 | 95 | 99:1 |
| 10 a | 50 | 100 | 5 | 5 | 47 | 100 | 90:10 |
| 11 a | 50 | 100 | - | 5 | 0 | 0 | - |
| Entry | Co-Catalyst (5% mol) | Product Distribution a,b | CMO Cis:Trans Ratio | ||
|---|---|---|---|---|---|
| EMO (%) | CMO (%) | By-Products c (%) | |||
| 1 d | [N4,4,4,4]Br | 67 | 33 | 0 | 99:1 |
| 2 | [N4,4,4,4]Cl | 80 | 20 | 0 | 99:1 |
| 3 | [N4,4,4,4]Br | 55 | 45 | 0 | 99:1 |
| 4 | [N4,4,4,4]I | 35 | 65 | 0 | 90:10 |
| 5 | NaCl | 78 | 22 | 0 | 99:1 |
| 6 | NaBr | 54 | 46 | 0 | 99:1 |
| 7 | NaI | 23 | 67 | 10 | 56:44 |
| 8 | KCl | 77 | 23 | 0 | 99:1 |
| 9 | KBr | 30 | 70 | 0 | 95:5 |
| 10 | KI | 35 | 65 | 0 | 96:4 |
| 11 e | NaBr | 46 | 38 | 16 | 93:7 |
| 12 e | NaI | 20 | 62 | 18 | 42:58 |
| 13 e | KBr | 1 | 99 | 0 | 92:8 |
| 14 e | KI | 7 | 87 | 6 | 67:34 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calmanti, R.; Sargentoni, N.; Selva, M.; Perosa, A. One-Pot Tandem Catalytic Epoxidation—CO2 Insertion of Monounsaturated Methyl Oleate to the Corresponding Cyclic Organic Carbonate. Catalysts 2021, 11, 1477. https://doi.org/10.3390/catal11121477
Calmanti R, Sargentoni N, Selva M, Perosa A. One-Pot Tandem Catalytic Epoxidation—CO2 Insertion of Monounsaturated Methyl Oleate to the Corresponding Cyclic Organic Carbonate. Catalysts. 2021; 11(12):1477. https://doi.org/10.3390/catal11121477
Chicago/Turabian StyleCalmanti, Roberto, Nicola Sargentoni, Maurizio Selva, and Alvise Perosa. 2021. "One-Pot Tandem Catalytic Epoxidation—CO2 Insertion of Monounsaturated Methyl Oleate to the Corresponding Cyclic Organic Carbonate" Catalysts 11, no. 12: 1477. https://doi.org/10.3390/catal11121477
APA StyleCalmanti, R., Sargentoni, N., Selva, M., & Perosa, A. (2021). One-Pot Tandem Catalytic Epoxidation—CO2 Insertion of Monounsaturated Methyl Oleate to the Corresponding Cyclic Organic Carbonate. Catalysts, 11(12), 1477. https://doi.org/10.3390/catal11121477