
A Combination of EPR, Microscopy, Electrophoresis and Theory to Elucidate the Chemistry of W- and N-Doped TiO2 Nanoparticle/Water Interfaces

 , , and
, , and 
Abstract
:1. Introduction
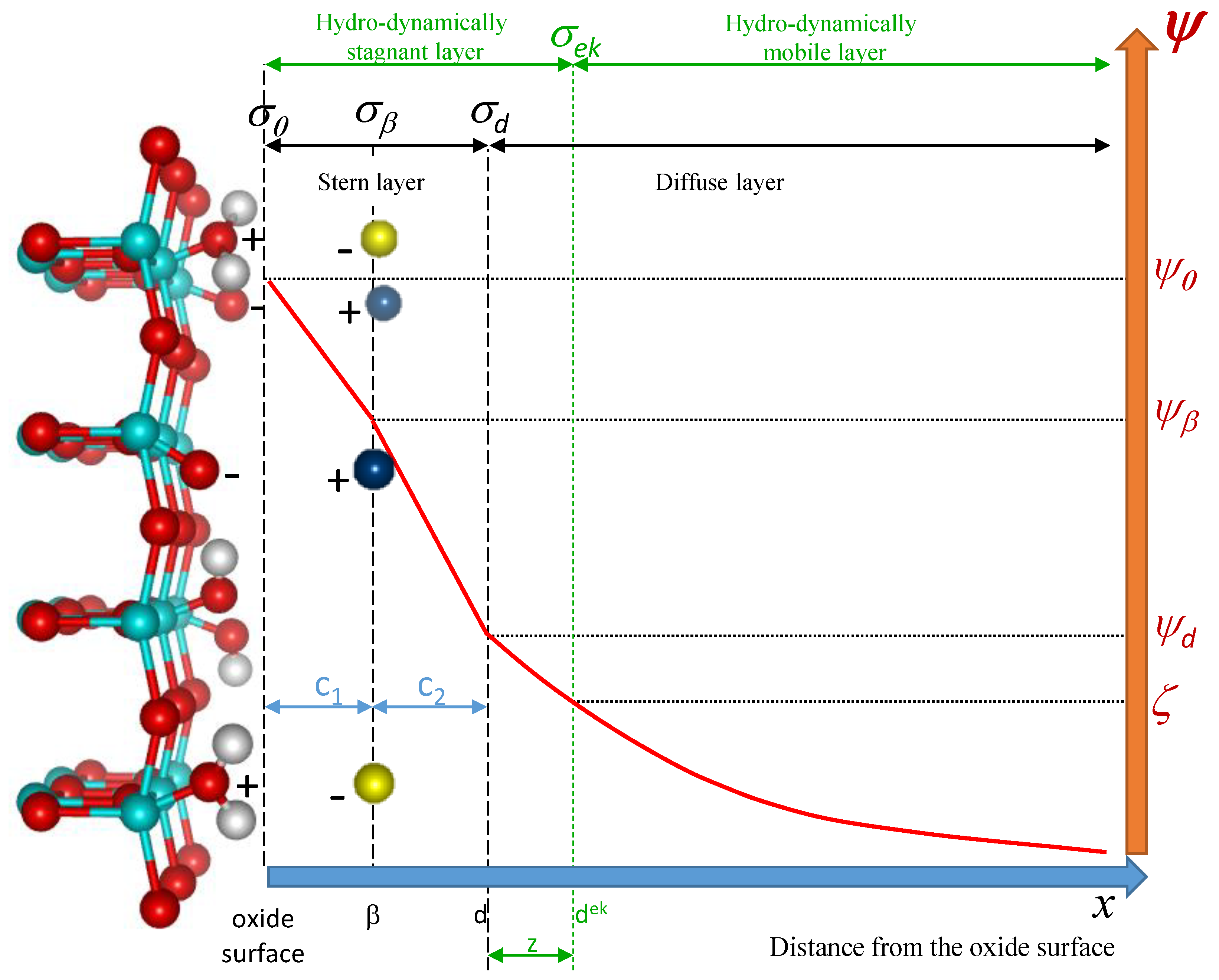
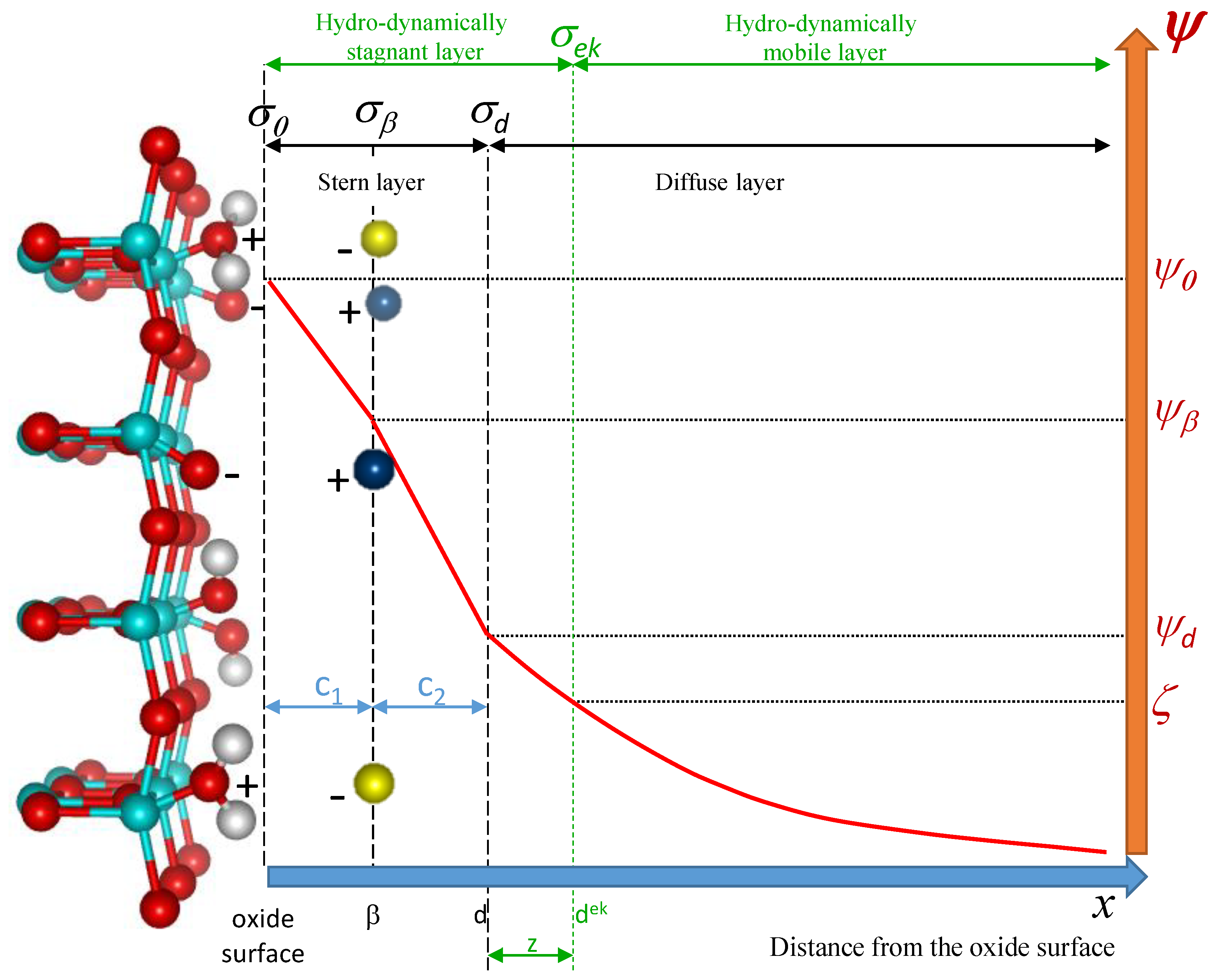
2. Theoretical Description and Simulation of the 2-pK Charging Triple-Layer Model (2-pK TLM) of the Oxide/Electrolyte Interface
2.1. Theoretical Description
2.2. Simulation of Surface Charge Density, (pH) and Surface Site Speciation
2.3. Simulation of Electrokinetic Potential, (pH)
3. Results
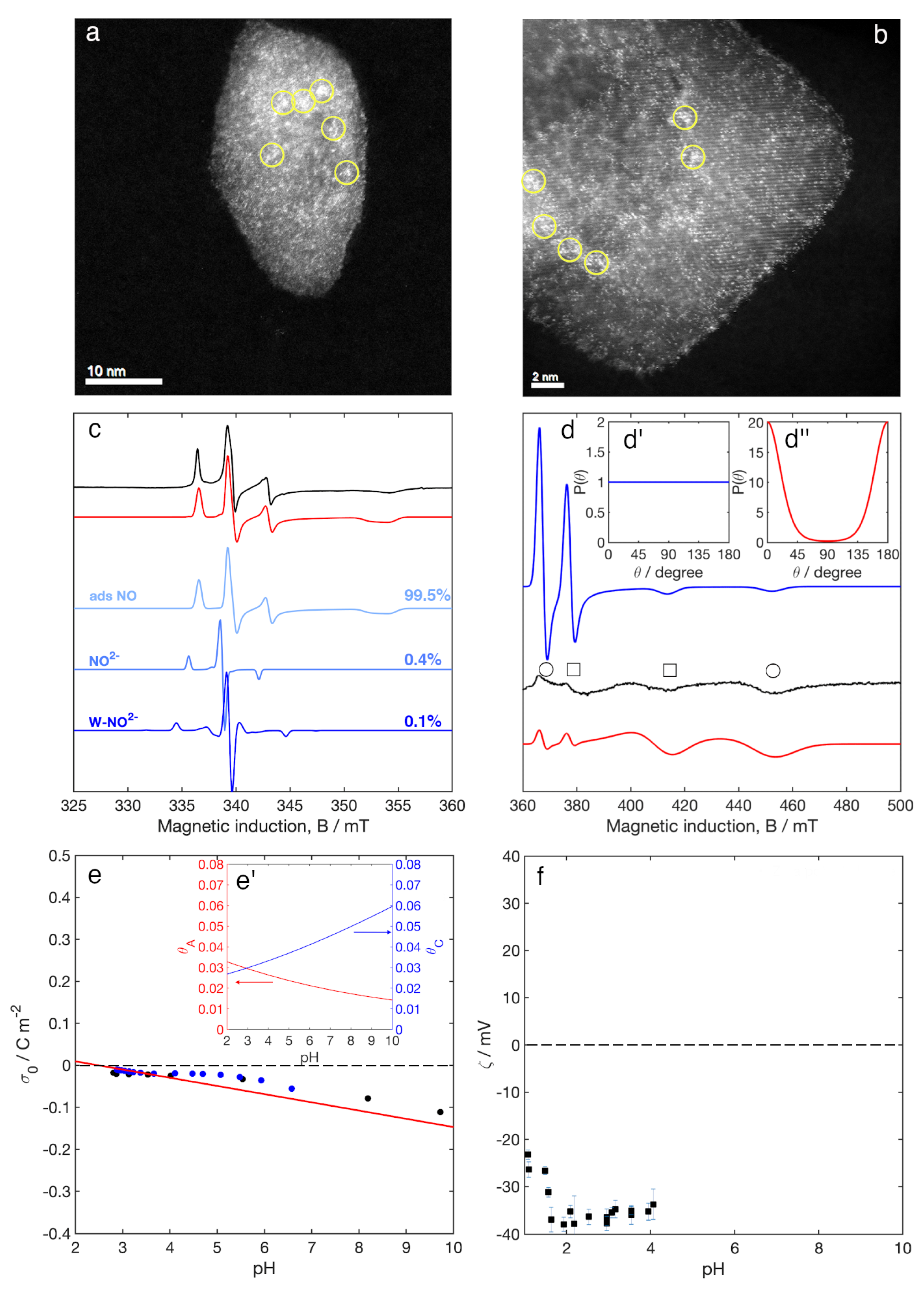
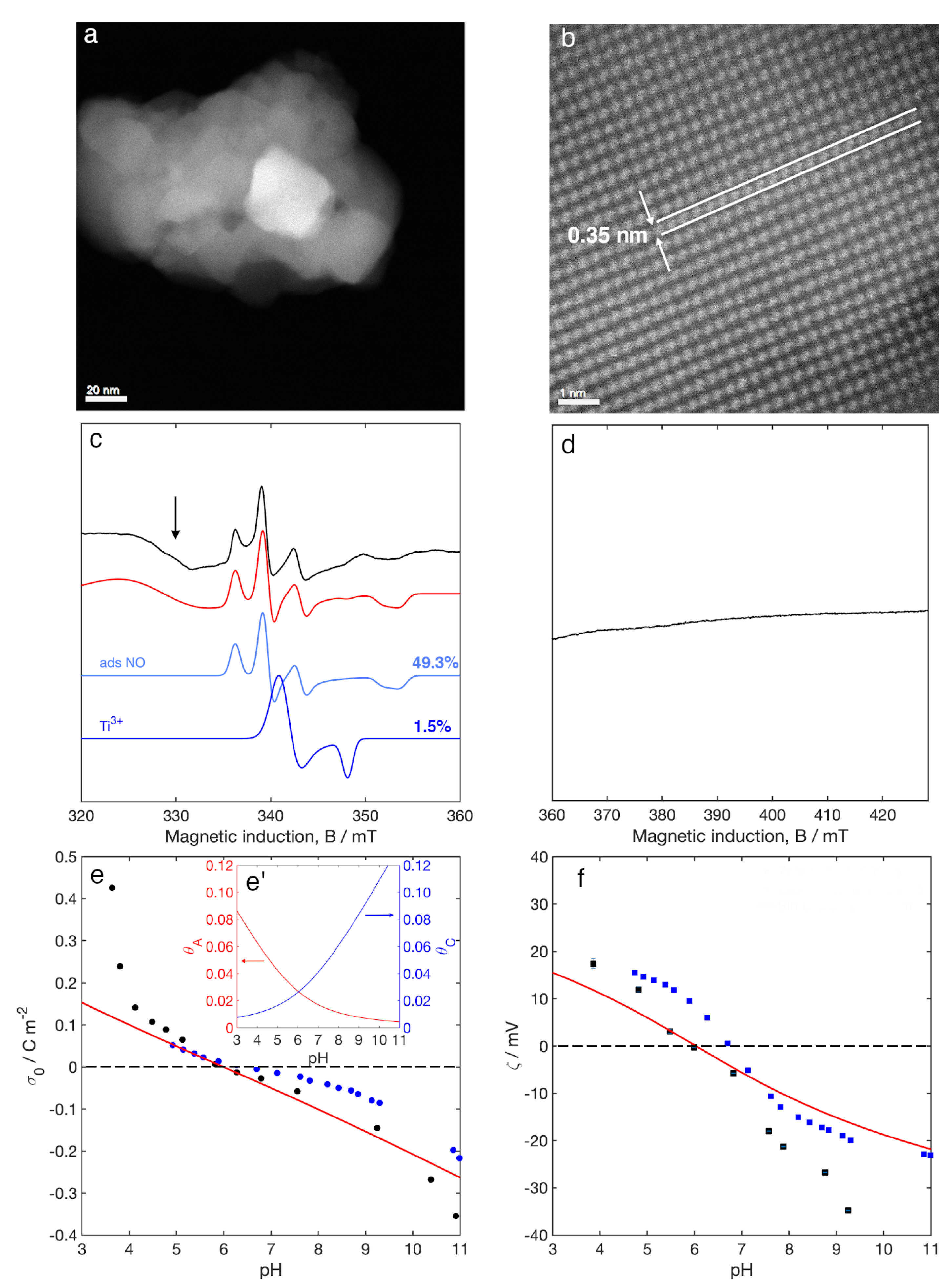
3.1. Aberration Corrected Scanning Transmission Electron Microscopy (STEM)
3.2. Electron Paramagnetic Resonance (EPR) Spectroscopy
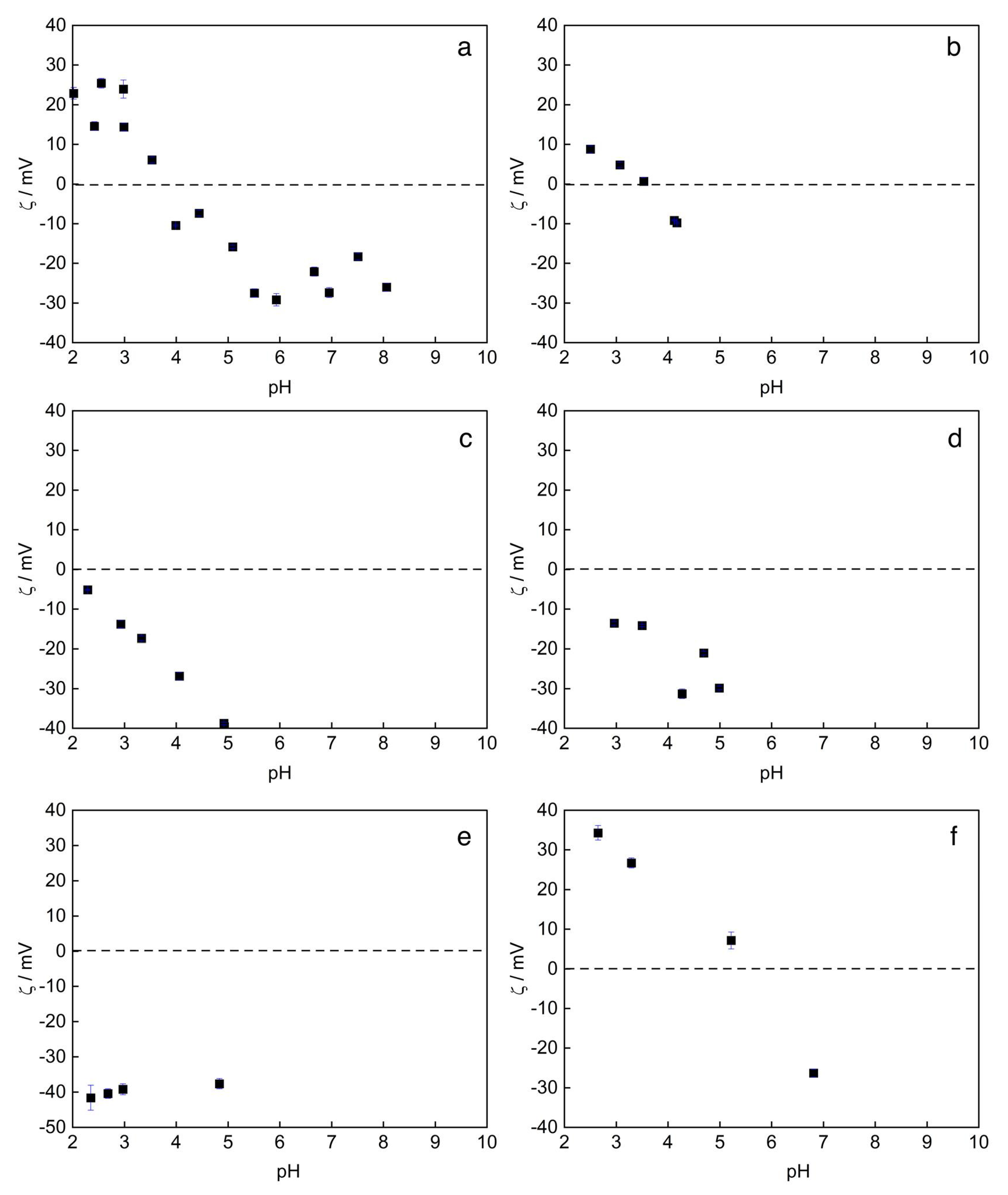
3.3. Surface Charge Density and Electrokinetic Parameters
4. Discussion
4.1. Nature of the Surface W Species
4.2. Dopant-Induced Surface Acidity
4.3. Single Contributions of W and N Dopants on Surface Acidity
4.4. Overall Applicability of the Proposed Approach
5. Materials and Methods
5.1. Synthesis
5.2. Aberration Corrected Scanning Transmission Electron Microscopy (STEM)
5.3. Electron Paramagnetic Resonance (EPR) Spectroscopy
5.4. Electrolyte and Mass Titrations
5.5. Electroacoustics and Laser Doppler Micro-Electrophoresis
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Folli, A.; Bloh, J.Z.; Strøm, M.; Pilegaard Madsen, T.; Henriksen, T.; Macphee, D.E. Efficiency of Solar-Light-Driven TiO2 Photocatalysis at Different Latitudes and Seasons. Where and When Does TiO2 Really Work? J. Phys. Chem. Lett. 2014, 5, 830–832. [Google Scholar] [CrossRef]
- Liu, L.; Chen, X. Titanium Dioxide Nanomaterials: Self-Structural Modifications. Chem. Rev. 2014, 114, 9890–9918. [Google Scholar] [CrossRef] [PubMed]
- Zuo, F.; Bozhilov, K.; Dillon, R.J.; Wang, L.; Smith, P.; Zhao, X.; Bardeen, C.; Feng, P. Active Facets on Titanium(III)-Doped TiO2: An Effective Strategy to Improve the Visible-Light Photocatalytic Activity. Angew. Chem. 2012, 124, 6327–6330. [Google Scholar] [CrossRef]
- Xing, M.; Li, X.; Zhang, J. Synergistic effect on the visible light activity of Ti3+ doped TiO2 nanorods/boron doped graphene composite. Sci. Rep. 2014, 4, 1–7. [Google Scholar] [CrossRef]
- Livraghi, S.; Paganini, M.C.; Giamello, E.; Selloni, A.; Di Valentin, C.; Pacchioni, G. Origin of Photoactivity of Nitrogen-Doped Titanium Dioxide under Visible Light. J. Am. Chem. Soc. 2006, 128, 15666–15671. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Burda, C. The Electronic Origin of the Visible-Light Absorption Properties of C-, N- and S-Doped TiO2 Nanomaterials. J. Am. Chem. Soc. 2008, 130, 5018–5019. [Google Scholar] [CrossRef] [PubMed]
- Czoska, A.M.; Livraghi, S.; Paganini, M.C.; Giamello, E.; Di Valentin, C.; Pacchioni, G. The nitrogen-boron paramagnetic center in visible light sensitized N-B co-doped TiO2. Experimental and theoretical characterization. Phys. Chem. Chem. Phys. 2011, 13, 136–143. [Google Scholar] [CrossRef]
- Barolo, G.; Livraghi, S.; Chiesa, M.; Paganini, M.C.; Giamello, E. Mechanism of the Photoactivity under Visible Light of N-Doped Titanium Dioxide. Charge Carriers Migration in Irradiated N-TiO2 Investigated by Electron Paramagnetic Resonance. J. Phys. Chem. C 2012, 116, 20887–20894. [Google Scholar] [CrossRef]
- Xu, J.; Wang, F.; Liu, W.; Cao, W. Nanocrystalline N-Doped TiO2 Powders: Mild Hydrothermal Synthesis and Photocatalytic Degradation of Phenol under Visible Light Irradiation. Int. J. Photoenergy 2013, 2013, 1–7. [Google Scholar]
- Yuan, W.; Li, J.; Wang, L.; Chen, P.; Xie, A.; Shen, Y. Nanocomposite of N-Doped TiO2 Nanorods and Graphene as an Effective Electrocatalyst for the Oxygen Reduction Reaction. ACS Appl. Mater. Interfaces 2014, 6, 21978–21985. [Google Scholar] [CrossRef]
- Bauer, D.; Roberts, A.J.; Matsumi, N.; Darr, J.A. Nano-sized Mo- and Nb-doped TiO2 as anode materials for high energy and high power hybrid Li-ion capacitors. Nanotechnology 2017, 28, 195403. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.T. Paramagnetic Resonance Spectrum of W5+ in Rutile (TiO2). Phys. Rev. 1966, 147, 264–267. [Google Scholar] [CrossRef]
- De Trizio, L.; Buonsanti, R.; Schimpf, A.M.; Llordes, A.; Gamelin, D.R.; Simonutti, R.; Milliron, D.J. Nb-Doped Colloidal TiO2 Nanocrystals with Tunable Infrared Absorption. Chem. Mater. 2013, 25, 3383–3390. [Google Scholar] [CrossRef]
- Nosaka, Y.; Takahashi, S.; Sakamoto, H.; Nosaka, A.Y. Reaction Mechanism of Cu(II)-Grafted Visible-Light Responsive TiO2 and WO3 Photocatalysts Studied by Means of ESR Spectroscopy and Chemiluminescence Photometry. J. Phys. Chem. C 2011, 115, 21283–21290. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Li, X.; Wang, C. Electrospun mesoporous W6+-doped TiO2 thin films for efficient visible-light photocatalysis. Mater. Lett. 2009, 63, 331–333. [Google Scholar] [CrossRef]
- Zimmermann, P.H. Temperature Dependence of the EPR Spectra of Niobium-Doped TiO2. Phys. Rev. B 1973, 8, 3917–3927. [Google Scholar] [CrossRef]
- Yue, J.; Suchomski, C.; Voepel, P.; Ellinghaus, R.; Rohnke, M.; Leichtweiss, T.; Elm, M.T.; Smarsly, B.M. Mesoporous niobium-doped titanium dioxide films from the assembly of crystalline nanoparticles: Study on the relationship between the band structure, conductivity and charge storage mechanism. J. Mater. Chem. A 2017, 5, 1978–1988. [Google Scholar] [CrossRef] [Green Version]
- Atashbar, M.Z.; Sun, H.T.; Gong, B.; Wlodarski, W.; Lamb, R. XPS study of Nb-doped oxygen sensing TiO2 thin films prepared by sol-gel method. Thin Solid Films 1998, 326, 238–244. [Google Scholar] [CrossRef]
- Tobaldi, D.M.; Pullar, R.C.; Gualtieri, A.F.; Seabra, M.P.; Labrincha, J.A. Sol-gel synthesis, characterisation and photocatalytic activity of pure, W-, Ag- and W/Ag co-doped TiO2 nanopowders. Chem. Eng. J. 2013, 214, 364–375. [Google Scholar] [CrossRef]
- Tasaki, C.; Oka, N.; Yagi, T.; Taketoshi, N.; Baba, T.; Kamiyama, T.; Nakamura, S.-I.; Shigesato, Y. Thermophysical Properties of Transparent Conductive Nb-Doped TiO2 Films. Jpn. J. App. Phys. 2012, 51, 035802. [Google Scholar]
- Lee, S.; Noh, J.H.; Han, H.S.; Yim, D.K.; Kim, D.H.; Lee, J.-K.; Kim, J.Y.; Jung, H.S.; Hong, K.S. Nb-Doped TiO2: A New Compact Layer Material for TiO2 Dye-Sensitized Solar Cells. J. Phys. Chem. C 2009, 113, 6878–6882. [Google Scholar] [CrossRef]
- Lee, H.Y.; Robertson, J. Doping and compensation in Nb-doped anatase and rutile TiO2. J. App. Phys. 2013, 113, 213706. [Google Scholar] [CrossRef]
- Couselo, N.; GarciaEinschlag, F.; Candal, R.; Jobbagy, M. Tungsten-Doped TiO2 vs. Pure TiO2 Photocatalysts: Effects on Photobleaching Kinetics and Mechanism. J. Phys. Chem. C 2008, 112, 1094–1100. [Google Scholar] [CrossRef]
- Tung, W.S.; Daoud, W.A. New Approach Toward Nanosized Ferrous Ferric Oxide and Fe3O4-doped Titanium Dioxide Photocatalysts. ACS Appl. Mater. Interfaces 2009, 1, 2453–2461. [Google Scholar] [CrossRef]
- Thind, S.S.; Wu, G.; Chen, A. Synthesis of mesoporous nitrogen–tungsten co-doped TiO2 photocatalysts with high visible light activity. Appl. Catal. B Environ. 2012, 111–112, 38–45. [Google Scholar] [CrossRef]
- Çelik, V.; Mete, E. Range-separated hybrid exchange-correlation functional analyses of anatase TiO2 doped with W, N, S, W/N, or W/S. Phys. Rev. B 2012, 86, 205112. [Google Scholar] [CrossRef] [Green Version]
- Biedrzycki, J.; Livraghi, S.; Giamello, E.; Agnoli, S.; Granozzi, G. Fluorine- and Niobium-Doped TiO2: Chemical and Spectroscopic Properties of Polycrystalline n-Type-Doped Anatase. J. Phys. Chem. C 2014, 118, 8462–8473. [Google Scholar] [CrossRef]
- Kubacka, A.; Colón, G.; Fernández-García, M. N- and/or W-(co)doped TiO2-anatase catalysts: Effect of the calcination treatment on photoactivity. Appl. Catal. B 2010, 95, 238–244. [Google Scholar] [CrossRef]
- Sajjad, A.K.L.; Shamaila, S.; Zhang, J. Study of new states in visible light active W, N co-doped TiO2 photo catalyst. Mater. Res. Bull. 2012, 47, 3083–3089. [Google Scholar] [CrossRef]
- Folli, A.; Bloh, J.Z.; Beukes, E.P.; Howe, R.F.; Macphee, D.E. Photogenerated Charge Carriers and Paramagnetic Species in (W,N)- Codoped TiO2 Photocatalysts under Visible-Light Irradiation: An EPR Study. J. Phys. Chem. C 2013, 117, 22149–22155. [Google Scholar] [CrossRef]
- Folli, A.; Bloh, J.Z.; Armstrong, K.; Richards, E.; Murphy, D.M.; Lu, L.; Kiely, C.J.; Morgan, D.J.; Smith, R.I.; Mclaughlin, A.C.; et al. Improving the Selectivity of Photocatalytic NOx Abatement through Improved O2 Reduction Pathways Using Ti0.909W0.091O2Nx Semiconductor Nanoparticles: From Characterization to Photocatalytic Performanc. ACS Catal. 2018, 8, 6927–6938. [Google Scholar] [CrossRef]
- Bloh, J.Z.; Folli, A.; Macphee, D.E. Adjusting Nitrogen Doping Level in Titanium Dioxide by Codoping with Tungsten: Properties and Band Structure of the Resulting Materials. J. Phys. Chem. C 2014, 118, 21281–21292. [Google Scholar] [CrossRef]
- Folli, A.; Bloh, J.Z.; Walker, R.; Lecaplain, A.; Macphee, D.E. Properties and Photochemistry of Valence-Induced-Ti3+ Enriched (Nb,N)-Codoped Anatase TiO2 Semiconductors. Phys. Chem. Chem. Phys. 2015, 17, 4849–4853. [Google Scholar] [CrossRef]
- Borlaf, M.; Colomer, M.T.; De Andrés, A.; Cabello, F.; Serna, R.; Moreno, R. TiO2/Eu3+ thin films with high photoluminescence emission prepared by electrophoretic deposition from nanoparticulate sols. Eur. J. Inorg. Chem. 2014, 2014, 5152–5159. [Google Scholar] [CrossRef]
- us Saqib, N.; Adnan, R.; Shah, I. A mini-review on rare earth metal-doped TiO2 for photocatalytic remediation of wastewater. Environ. Sci. Pollut. Res. 2016, 23, 15941–15951. [Google Scholar] [CrossRef]
- D’ Arienzo, M.; Siedl, N.; Sternig, A.; Scotti, R.; Morazzoni, F.; Bernardi, J.; Diwald, O. Solar light and dopant-induced recombination effects: Photoactive nitrogen in TiO2 as a case study. J. Phys. Chem. C 2010, 114, 18067–18072. [Google Scholar] [CrossRef]
- Blesa, M.A.; Kallay, N. The metal oxide - electrolyte solution interface revisited. Adv. Colloid Interf. Sci. 1988, 28, 111–134. [Google Scholar] [CrossRef]
- Piasecki, W.; Rudzinski, W.; Charmas, R. 1-p K and 2-p K Protonation Models in the Theoretical Description of Simple Ion Adsorption at the Oxide/Electrolyte Interface: A Comparative Study of the Behavior of the Surface Charge, the Individual Isotherms of Ions, and the Accompanying Electrokinetic. J. Phys. Chem. B 2001, 105, 9755–9771. [Google Scholar] [CrossRef]
- Tejedor-Tejedor, M.I.; Anderson, M.A. In situ attenuated total reflection fourier transform infrared studies of the Goethite (alpha-FeOOH)-acqueous solution interface. Langmuir 1986, 2, 203–210. [Google Scholar] [CrossRef]
- Folli, A.; Pochard, I.; Nonat, A.; Jakobsen, U.; Shepherd, A.; Macphee, D. Engineering photocatalytic Cements: Understanding TiO2 surface chemistry to control and modulate photocatalytic performances. J. Amer. Ceram. Soc. 2010, 93, 3360–3369. [Google Scholar] [CrossRef]
- Di Valentin, C.; Finazzi, E.; Pacchioni, G.; Selloni, A.; Livraghi, S.; Paganini, M.C.; Giamello, E. N-doped TiO2: Theory and Experiment. Chem. Phys. 2007, 339, 44–56. [Google Scholar] [CrossRef]
- Livraghi, S.; Chierotti, M.R.; Giamello, E.; Magnacca, G.; Paganini, M.C.; Cappelletti, G.; Bianchi, C.L. Nitrogen-Doped Titanium Dioxide Active in Photocatalytic Reactions with Visible Light: A Multi-Technique Characterization of Differently Prepared Materials. J. Phys. Chem. C 2008, 112, 17244–17252. [Google Scholar] [CrossRef]
- Messai, Y.; Vileno, B.; Martel, D.; Turek, P.; Mekki, D.E. Milling effect on the photo-activated properties of TiO2 nanoparticles: Electronic and structural investigations. Bull. Mater. Sci. 2018, 41, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kuba, S.; Heydorn, P.; Grasselli, R.K.; Gates, B.C.; Chec, M.; Kno, H. Redox Properties of Tungstated Zirconia Catalysts: Relevance to the Activation of n-Alkanes. Phys. Chem. Chem. Phys. 2001, 3, 146–154. [Google Scholar] [CrossRef]
- Occhiuzzi, M.; Cordischi, D.; De Rossi, S.; Ferraris, G.; Gazzoli, D.; Valigi, M. Pd-Promoted WOx/ZrO2 Catalysts: Characterization and Catalytic Activity for n-Butane Isomerization. Appl. Catal. A 2008, 351, 29–35. [Google Scholar] [CrossRef]
- Occhiuzzi, M.; Cordischi, D.; Gazzoli, D.; Valigi, M.; Heydorn, P.C. WOx/ZrO2 Catalysts Part 4. Redox Properties as Investigated by Redox Cycles, XPS and EPR. Appl. Catal. A 2004, 269, 169–177. [Google Scholar] [CrossRef]
- Weil, J.A.; Bolton, J.R.; Wertz, J.E. Electron Paramagnetic Resonance: Elementary Theory and Practical Applications, 1st ed.; John Wiley & Sons: New York, NY, USA, 1994; p. 568. [Google Scholar]
- Castner, T.; Newell, G.S.; Holton, W.C.; Slighter, C.P. Note on the paramagnetic resonance of iron in glass. J. Chem. Phys. 1960, 32, 668–673. [Google Scholar] [CrossRef]
- Howe, R.F.; Gratzel, M. EPR observation of trapped electrons in colloidal titanium dioxide. J. Phys. Chem. 1985, 89, 4495–4499. [Google Scholar] [CrossRef]
- Micic, O.I.; Zhang, Y.; Cromack, K.R.; Trifunac, A.D.; Thurnauer, M.C. Photoinduced Hole Transfer from TiO2 to Methanol Molecules in Aqueous Solution Studied by Electron Paramagnetic Resonance. J. Phys. Chem. 1993, 97, 13284–13288. [Google Scholar] [CrossRef]
- Micic, O.I.; Zhang, Y.; Cromack, K.; Trifunac, A.D.; Thurnauer, M.C. Trapped holes on TiO2 colloids studied by electron paramagnetic resonance. J. Phys. Chem. 1993, 97, 7277–7283. [Google Scholar] [CrossRef]
- Jenkins, C.A.; Murphy, D.M. Thermal and photoreactivity of TiO2 at the gas-solid interface with aliphatic and aromatic aldehydes. J. Phys. Chem. B 1999, 103, 1019–1026. [Google Scholar] [CrossRef]
- Preocanin, T.; Kallay, N. Point of zero charge and surface charge density of TiO2 in aqueous electrolyte solution as obtained by potentiometric mass titration. Croat. Chem. Acta 2006, 79, 95–106. [Google Scholar]
- Zalac, S.; Kallay, N. Application of Mass Titration to the Point of Zero Charge Determination. J. Colloid Interf. Sci. 1992, 149, 233–240. [Google Scholar] [CrossRef]
- Fernandez-Garcia, M.; Martínez-Arias, A.; Fuerte, A.; Conesa, J.C. Nanostructured Ti-W Mixed-Metal Oxides: Structural and Electronic Properties. J. Phys. Chem. B 2005, 109, 6075–6083. [Google Scholar] [CrossRef]
- Spencer, J.; Folli, A.; Richards, E.; Murphy, D.M. Applications of electron paramagnetic resonance spectroscopy for interrogating catalytic systems. In Electron Paramagnetic Resonance; Chechik, V., Murphy, D.M., Eds.; Royal Society of Chemistry: Cambridge, UK, 2019; Volume 26, pp. 130–170. [Google Scholar]
- Li, X.Z.; Li, F.B.; Yang, C.L.; Ge, W.K. Photocatalytic activity of WOx-TiO2 under visible light irradiation. J. Photochem. Photobiol. A 2001, 141, 209–217. [Google Scholar] [CrossRef]
- Park, H.; Yang, D.-J.; Yoo, J.-S.; Mun, K.-S.; Kim, W.-R.; Kim, H.-G.; Choi, W.-Y. Surface passivation of highly ordered TiO2 nanotube arrays and application to dye-sensitized solar cells using the concept of isoelectric point. J. Ceram. Soc. Japan 2009, 117, 596–599. [Google Scholar] [CrossRef] [Green Version]
- Andersson, K.M.; Bergström, L. DLVO interactions of tungsten oxide and cobalt oxide surfaces measured with the colloidal probe technique. J. Colloid Interf. Sci. 2002, 246, 309–315. [Google Scholar] [CrossRef]
- Miyauchi, M.; Ikezawa, A.; Tobimatsu, H.; Irie, H.; Hashimoto, K. Zeta potential and photocatalytic activity of nitrogen doped TiO2 thin films. Phys. Chem. Chem. Phys. 2004, 6, 865–870. [Google Scholar] [CrossRef]
- Folli, A.; Campbell, S.B.; Anderson, J.A.; Macphee, D.E. Role of TiO2 Surface Hydration on NO Oxidation Photo-Activity. J. Photochem. Photobiol. A 2011, 220, 85–93. [Google Scholar] [CrossRef]
- Chen, F.; Zhao, J.; Hidaka, H. Highly selective deethylation of rhodamine B: Adsorption and photooxidation pathways of the dye on the TiO2/SiO2 composite photocatalyst. Intern. J. Photoenergy 2003, 5, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Chen, C.; Zhao, D.; Ma, W.; Zhao, J. Change of Adsorption Modes of Dyes on Fluorinated TiO2 and Its Effect on Photocatalytic Degradation of Dyes under Visible Irradiation. Langmuir 2008, 24, 7338–7345. [Google Scholar] [CrossRef]
- Macphee, D.E.; Bloh, J.Z.; Folli, A.; Greenhalgh, D. A method of photocatalytically oxidising nitrogen oxides. Patent WO/2016/005760A1, 14 January 2016. [Google Scholar]
- Stoll, S.; Schweiger, A. EasySpin, a Comprehensive Software Package for Spectral Simulation and Analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Reymond, J.P.; Kolenda, F. Estimation of the point of zero charge of simple and mixed oxides by mass titration. Powder Tech. 1999, 103, 30–36. [Google Scholar] [CrossRef]
- Sonnefeld, J. On the influence of background electrolyte concentration on the position of the isoelectric point and the point of zero charge. Colloids Surfaces Physicochem. Eng. Asp. 2001, 190, 179–183. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Temp./K | /MHz | /MHz | /MHz | /MHz | /MHz | /MHz | |||
|---|---|---|---|---|---|---|---|---|---|---|
| ads NO | 50 | 2.001 | 1.998 | 1.921 | <2.8 | 89.7 | 26.9 | |||
| 50 | 2.005 | 2.004 | 2.003 | 6.5 | 15.7 | 89.7 | ||||
| W- | 50 | 2.001 | 2.000 | 1.999 | 42.0 | 22.4 | 140.1 | 44.8 | <2.8 | 154.1 |
| Species | Temp. | ||
|---|---|---|---|
| / | |||
| WxOy | 50 | 1.85 e | 1.50 e |
| 50 | 1.80 e | 1.64 e | |
| 50 | 1.988 | 1.950 |
| Sample | |||||||
|---|---|---|---|---|---|---|---|
| /Fm−2 | /Fm−2 | /mol m−2 | |||||
| (anatase) | 3.20 | 8.80 | 5.55 | 6.45 | 1.05 | inf | 1.99 × 10−5 |
| −1.50 | 4.50 | 2.00 | 3.00 | 0.35 | inf | 3.00 × 10−5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorman, S.; Rickaby, K.; Lu, L.; Kiely, C.J.; Macphee, D.E.; Folli, A. A Combination of EPR, Microscopy, Electrophoresis and Theory to Elucidate the Chemistry of W- and N-Doped TiO2 Nanoparticle/Water Interfaces. Catalysts 2021, 11, 1305. https://doi.org/10.3390/catal11111305
Gorman S, Rickaby K, Lu L, Kiely CJ, Macphee DE, Folli A. A Combination of EPR, Microscopy, Electrophoresis and Theory to Elucidate the Chemistry of W- and N-Doped TiO2 Nanoparticle/Water Interfaces. Catalysts. 2021; 11(11):1305. https://doi.org/10.3390/catal11111305
Chicago/Turabian StyleGorman, Sam, Kirstie Rickaby, Li Lu, Christopher J. Kiely, Donald E. Macphee, and Andrea Folli. 2021. "A Combination of EPR, Microscopy, Electrophoresis and Theory to Elucidate the Chemistry of W- and N-Doped TiO2 Nanoparticle/Water Interfaces" Catalysts 11, no. 11: 1305. https://doi.org/10.3390/catal11111305
APA StyleGorman, S., Rickaby, K., Lu, L., Kiely, C. J., Macphee, D. E., & Folli, A. (2021). A Combination of EPR, Microscopy, Electrophoresis and Theory to Elucidate the Chemistry of W- and N-Doped TiO2 Nanoparticle/Water Interfaces. Catalysts, 11(11), 1305. https://doi.org/10.3390/catal11111305