
Multifunctional Electrocatalysis on Single-Site Metal Catalysts: A Computational Perspective


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experimental Insights
2.1. Different Bifunctional Single-Site Metal Catalysts
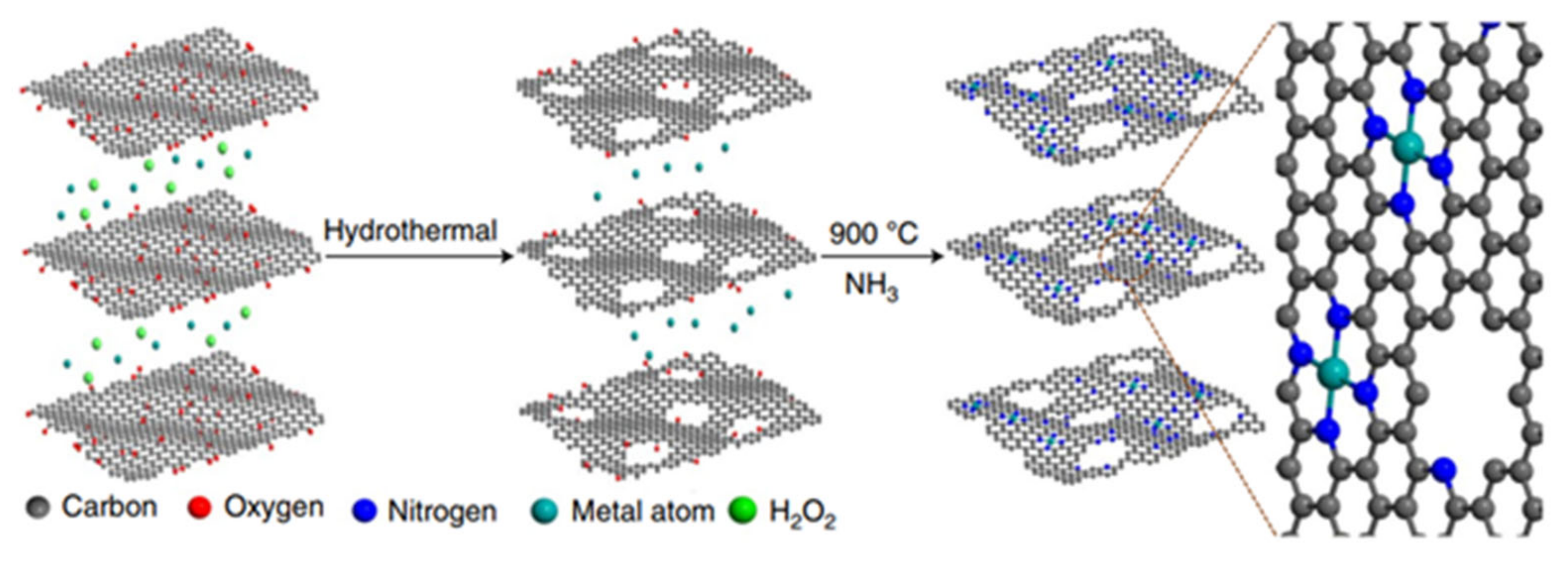
2.1.1. MNC-Derived Bifunctional Single-Site Metal Catalysts
2.1.2. Metal-Organic Framework-Derived Bifunctional Single-Site Metal Catalysts
2.2. Experimental Way of Identifying Single-Sites
3. Computational Background
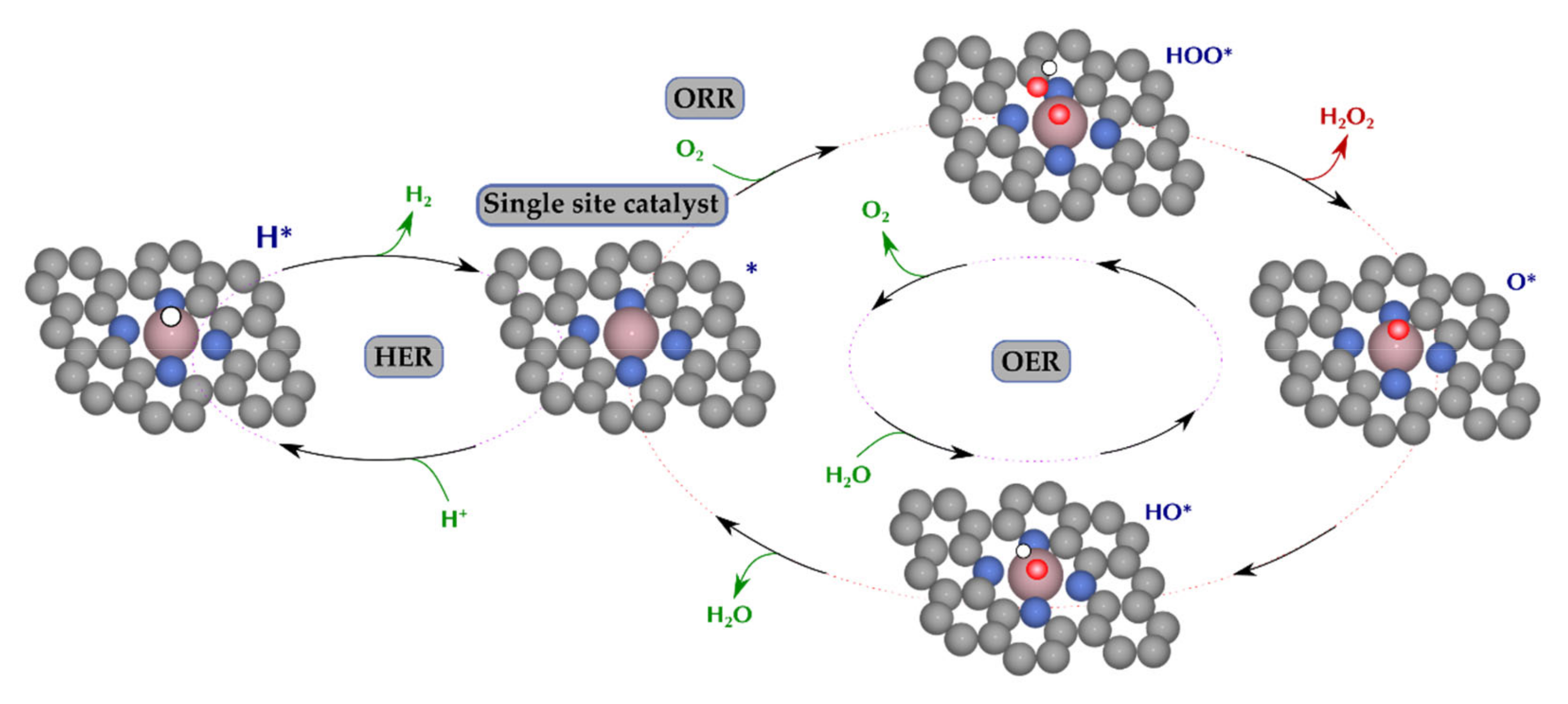
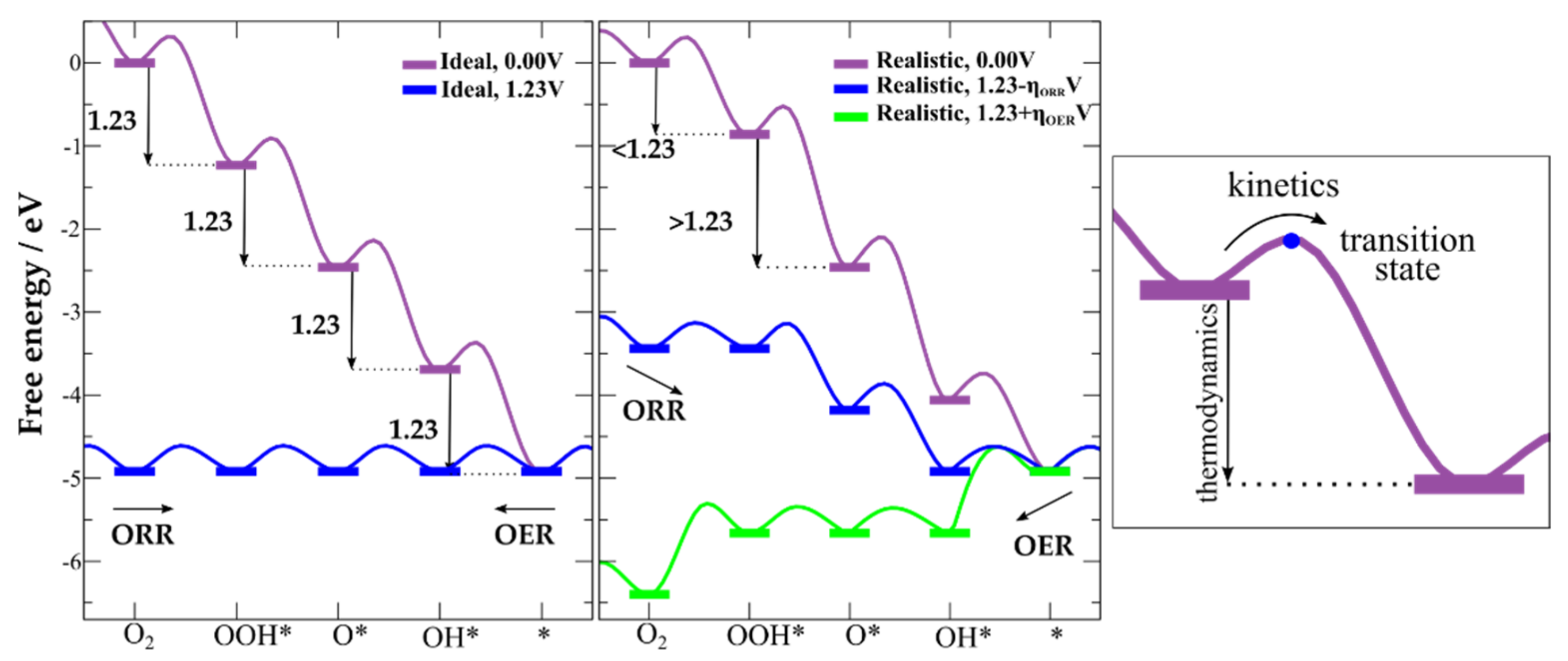
3.1. Free Energy Landscape
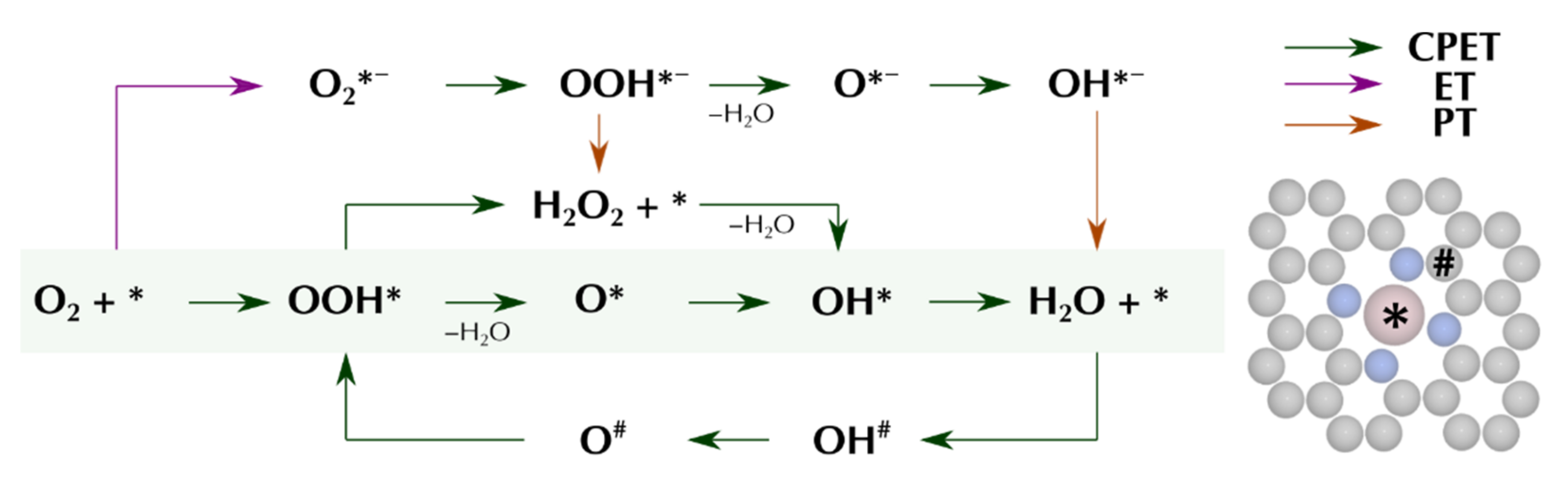
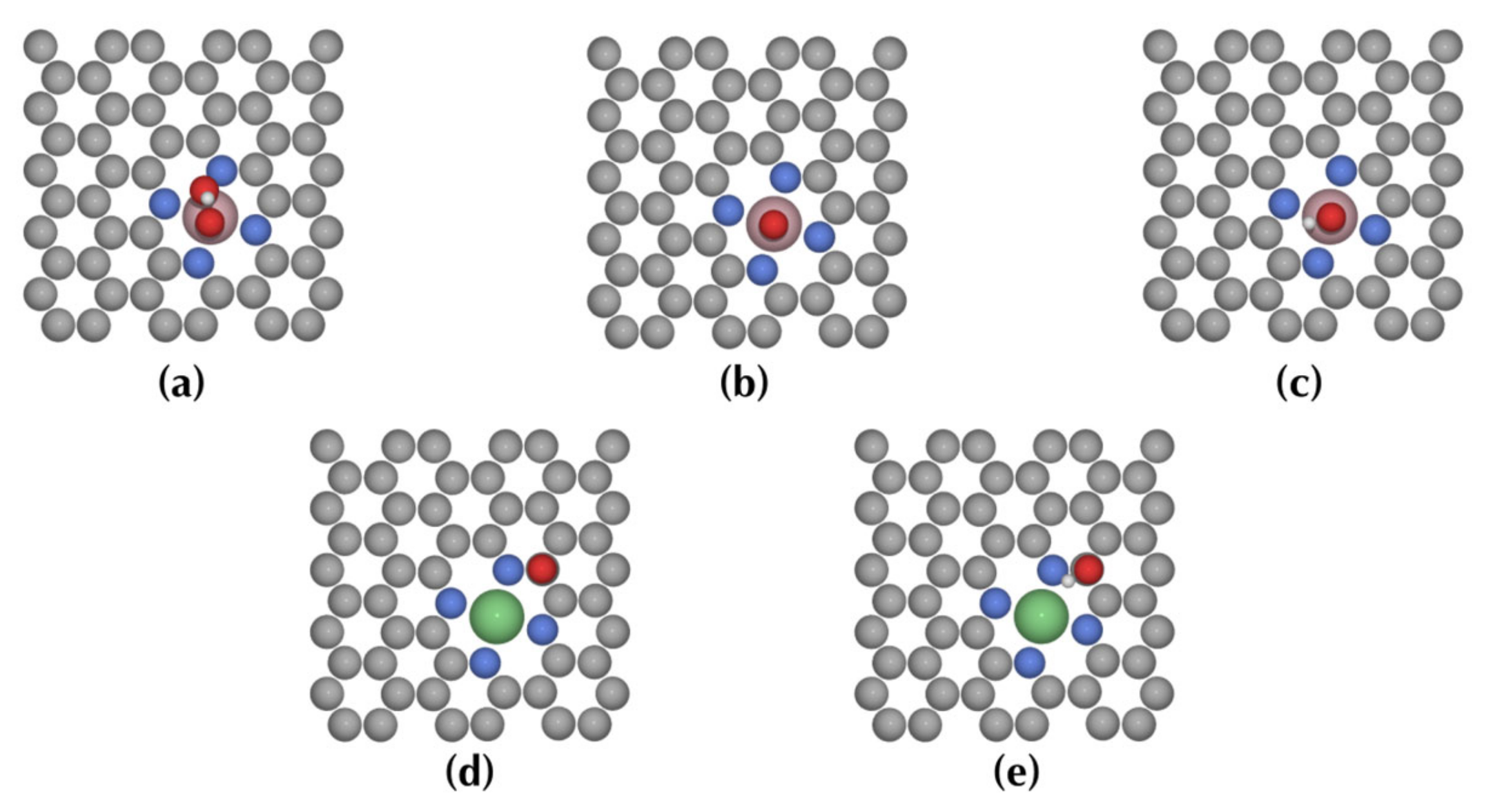
3.2. Reaction Pathways
3.3. Electrode–Electrolyte Interface Model
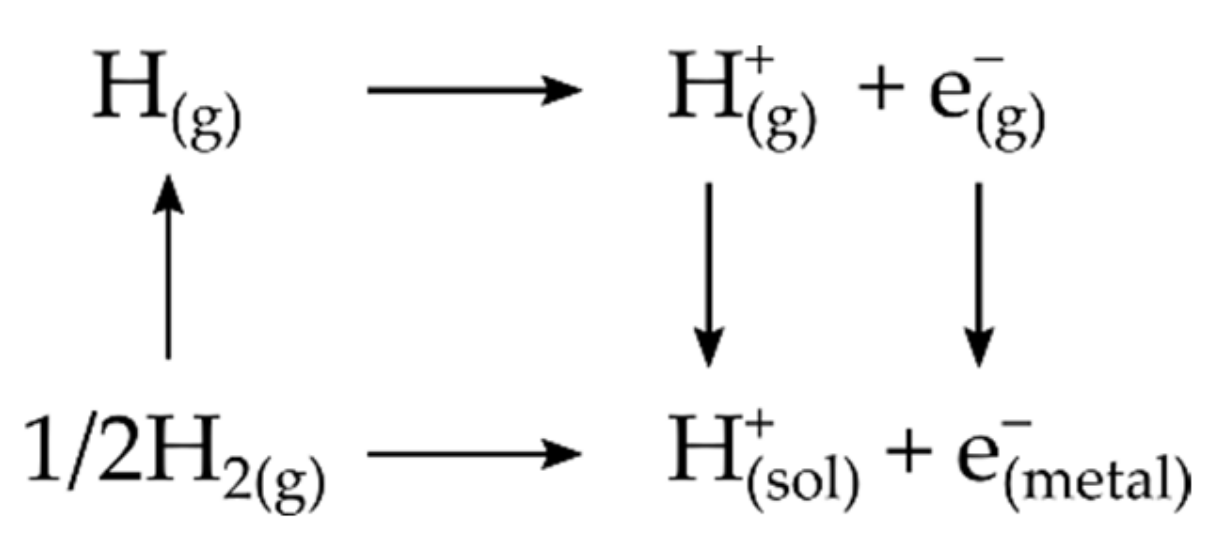
3.4. Computational Hydrogen Electrode
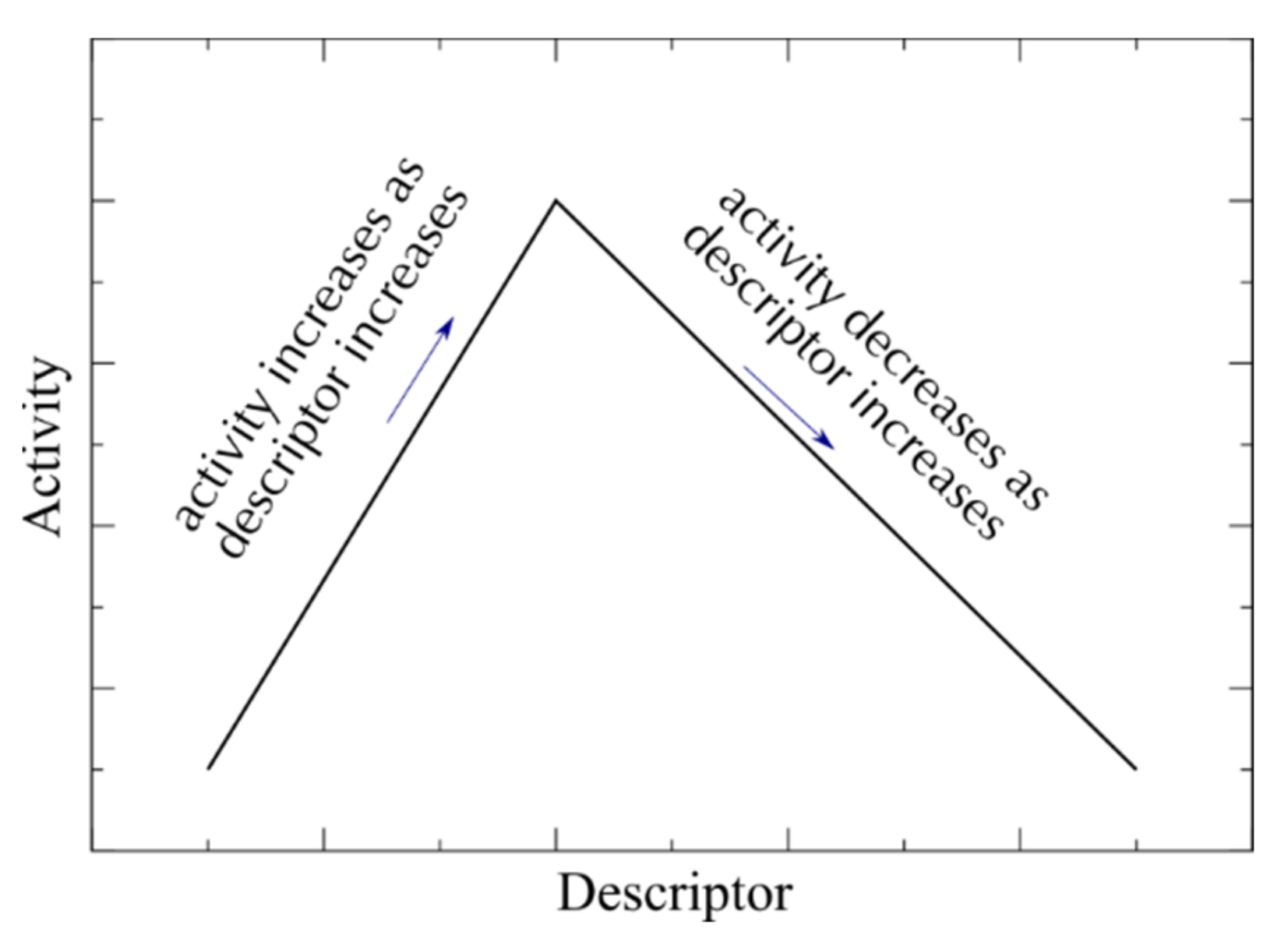
3.5. Breaking Scaling Relations
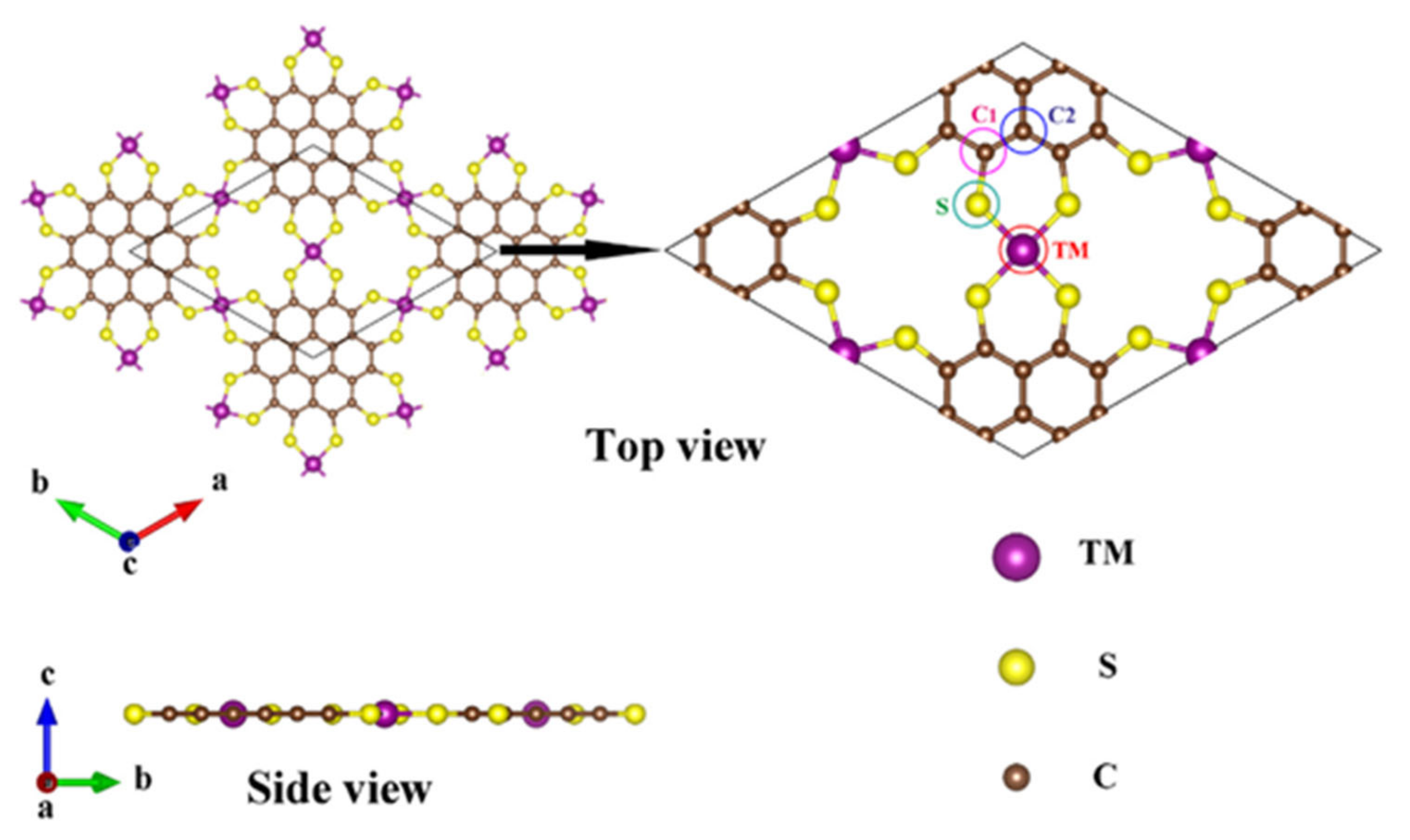
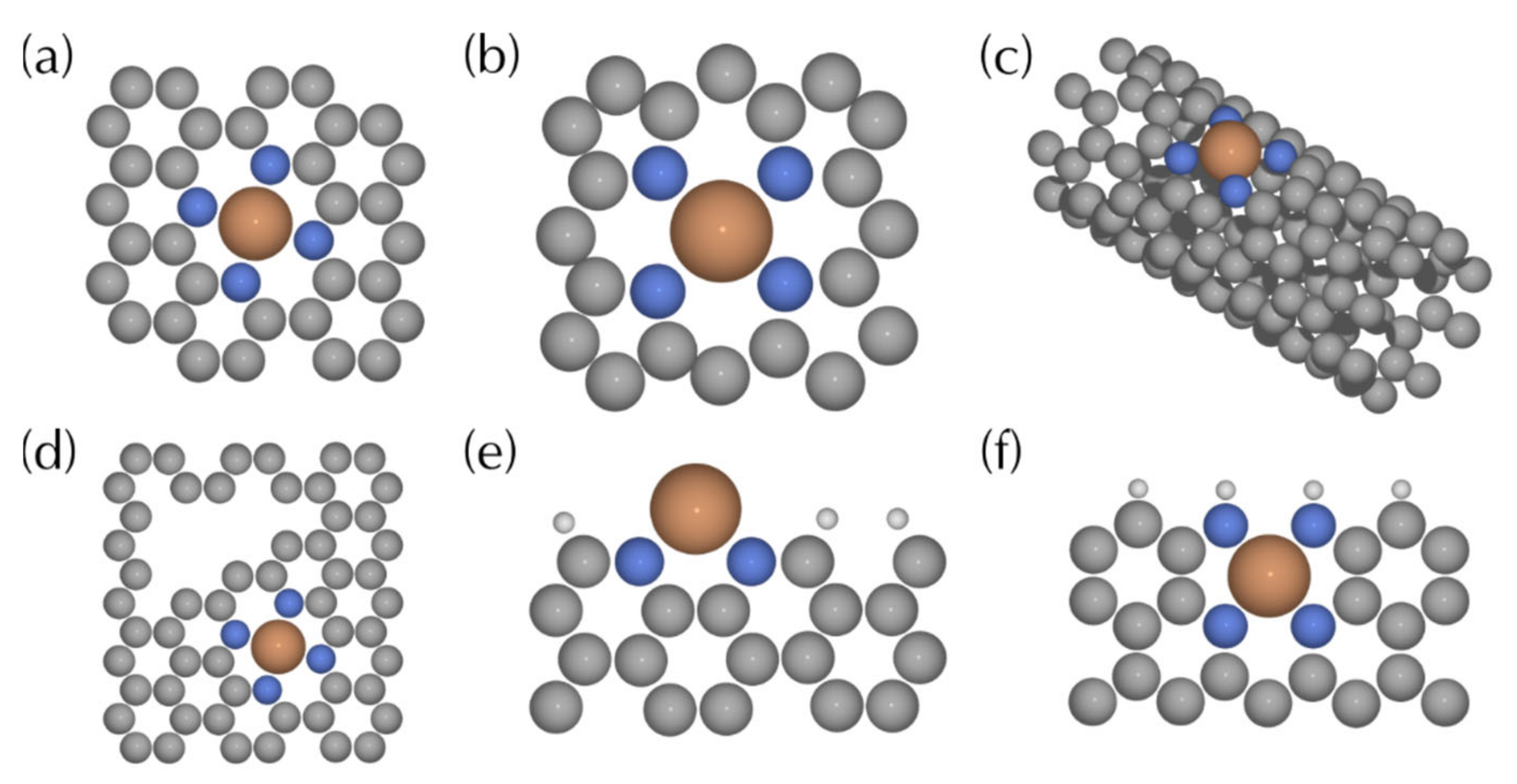
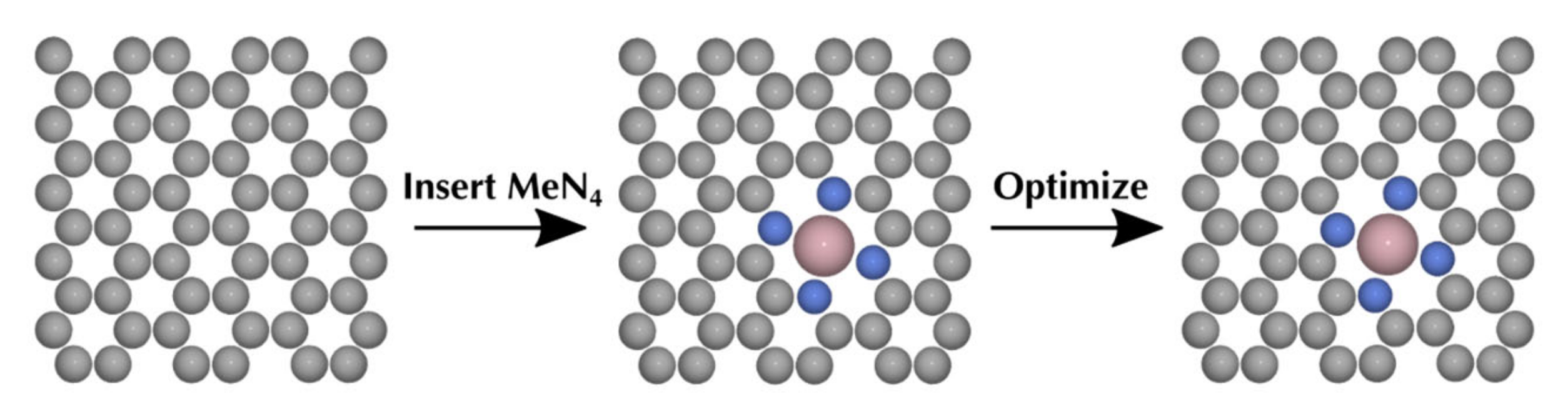
3.6. Computational Models
3.6.1. MNC Type Structures
3.6.2. Modified MNC Structures
3.6.3. MOFs
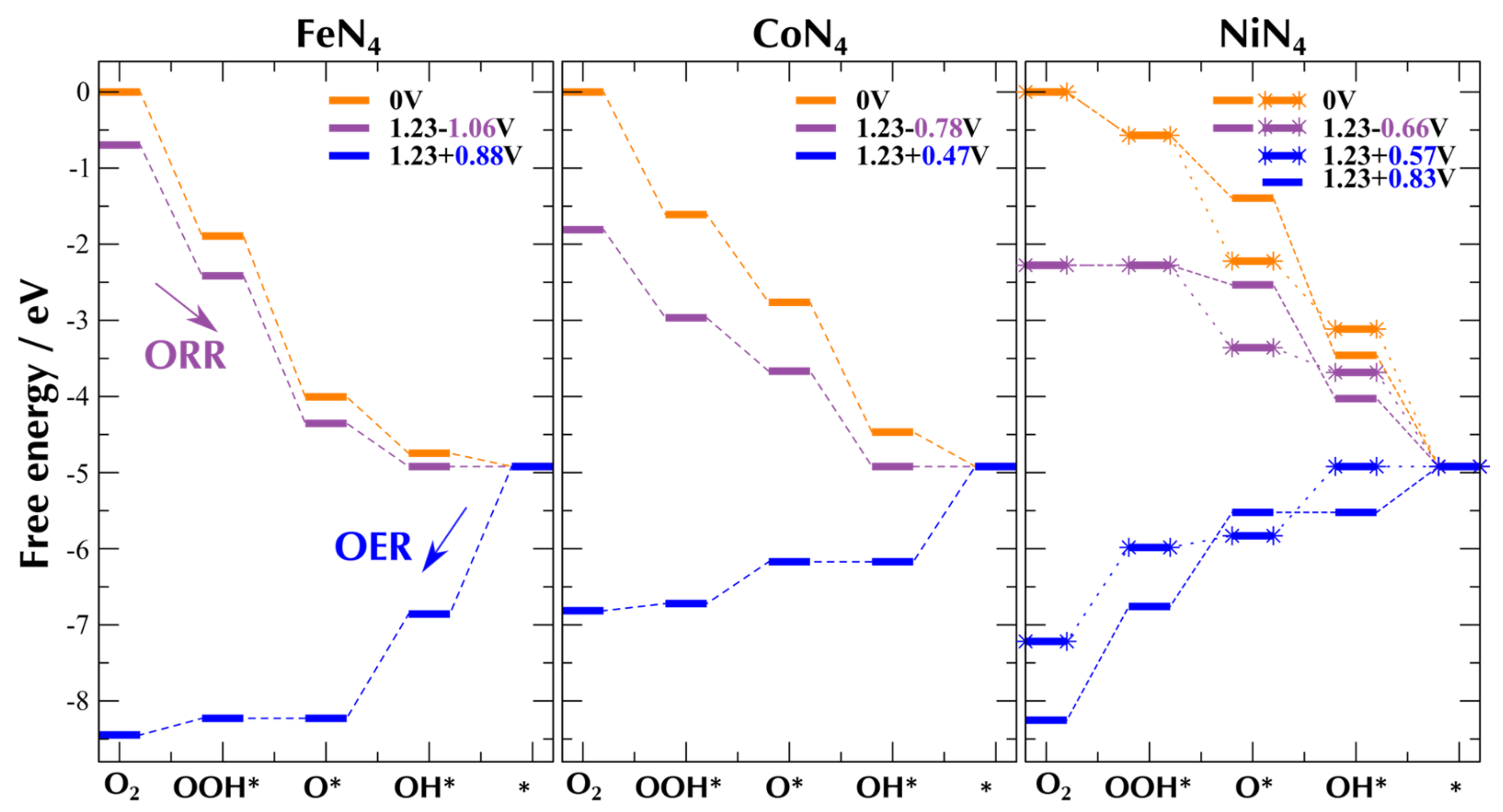
4. Computational Modeling Example
5. Perspectives
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| CHE | Computational hydrogen electrode |
| DFT | Density functional theory |
| EDL | Electrical double layer |
| ET | Electron transfer |
| PT | Proton transfer |
| CPET | Concerted proton–electron transfer |
| HER | Hydrogen evolution reaction |
| HOR | Hydrogen oxidation reaction |
| MOF | Metal-organic framework |
| MNC | Metal–nitrogen–carbon catalyst |
| COF | Covalent-organic framework |
| EXAFS | Extended X-ray absorption fine structure |
| OER | Oxygen evolution reaction |
| ORR | Oxygen reduction reaction |
| SAC | Single-atom catalyst |
| RHE | Reversible hydrogen electrode |
| ZPE | Zero-point energy |
| TS | Transition state |
| HOMO | Highest occupied molecular orbital |
| PDOS | Partial density of states |
| AIMD | Ab initio molecular dynamics |
References
- Pu, Z.; Zhang, G.; Hassanpour, A.; Zheng, D.; Wang, S.; Liao, S.; Chen, Z.; Sun, S. Regenerative fuel cells: Recent progress, challenges, perspectives and their applications for space energy system. Appl. Energy 2020, 283, 116376. [Google Scholar] [CrossRef]
- Jöerissen, L. Bifunctional Oxygen electrodes. In Encyclopedia of Electrochemical Power Sources; Elsevier: Amsterdam, The Netherlands, 2009. [Google Scholar]
- Hansen, J.N.; Prats, H.; Toudahl, K.K.; Secher, N.M.; Chan, K.; Kibsgaard, J.; Chorkendorff, I. Is there anything better than Pt for her? ACS Energy Lett. 2021, 6, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Bing, Y.; Liu, H.; Zhang, L.; Ghosh, D.; Zhang, J. Nanostructured Pt-alloy electrocatalysts for PEM fuel cell oxygen reduction reaction. Chem. Soc. Rev. 2010, 39, 2184–2202. [Google Scholar] [CrossRef]
- Ling, C.; Shi, L.; Ouyang, Y.; Zeng, X.C.; Wang, J. Nanosheet supported single-metal atom bifunctional catalyst for overall water splitting. Nano Lett. 2017, 17, 5133–5139. [Google Scholar] [CrossRef]
- Yuan, Y.; Ma, J.; Ai, H.; Kang, B.; Lee, J.Y. A simple general descriptor for rational design of graphyne-based bifunctional electrocatalysts toward hydrogen evolution and oxygen reduction reactions. J. Colloid Interface Sci. 2021, 592, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Jiao, M.; Luhua, J.; Barkholtz, H.M.; Lin, Z.; Wang, Y.; Jiang, L.; Wu, Z.; Liu, D.-J.; Zhuang, L.; et al. High performance platinum single atom electrocatalyst for oxygen reduction reaction. Nat. Commun. 2017, 8, 15938. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhou, Y.; Tana, Y.; Liua, S.; Chenga, Z.; Shenac, Z. Graphyne doped with transition-metal single atoms as effective bifunctional electrocatalysts for water splitting. Appl. Surf. Sci. 2019, 492, 8–15. [Google Scholar] [CrossRef]
- Gao, G.; Waclawik, E.; Du, A. Computational screening of two-dimensional coordination polymers as efficient catalysts for oxygen evolution and reduction reaction. J. Catal. 2017, 352, 579–585. [Google Scholar] [CrossRef]
- He, T.; Matta, S.K.; Will, G.; Du, A. Transition-metal single atoms anchored on graphdiyne as high-efficiency electrocatalysts for water splitting and oxygen reduction. Small Methods 2019, 3, 1800419. [Google Scholar] [CrossRef]
- Ji, D.; Fan, L.; Li, L.; Peng, S.; Yu, D.; Song, J.; Ramakrishna, S.; Guo, S. Atomically transition metals on self-supported porous carbon flake arrays as binder-free air cathode for wearable zinc−air batteries. Adv. Mater. 2019, 31, e1808267. [Google Scholar] [CrossRef]
- Li, Y.; Cui, M.; Yin, Z.; Chen, S.; Ma, T. Metal–organic framework based bifunctional oxygen electrocatalysts for rechargeable zinc–air batteries: Current progress and prospects. Chem. Sci. 2020, 11, 11646–11671. [Google Scholar] [CrossRef]
- Wang, S.; Qin, J.; Meng, T.; Cao, M. Metal–organic framework-induced construction of actiniae-like carbon nanotube assembly as advanced multifunctional electrocatalysts for overall water splitting and Zn-air batteries. Nano Energy 2017, 39, 626–638. [Google Scholar] [CrossRef]
- Yao, Z.-C.; Tang, T.; Hu, J.-S.; Wan, L.-J. Recent Advances on nonprecious-metal-based bifunctional oxygen electrocatalysts for zinc–air batteries. Energy Fuels 2021, 35, 6380–6401. [Google Scholar] [CrossRef]
- Chen, P.; Zhou, T.; Xing, L.; Xu, K.; Tong, Y.; Xie, H.; Zhang, L.; Yan, W.; Chu, W.; Wu, C.; et al. Atomically dispersed iron-nitrogen species as electrocatalysts for bifunctional oxygen evolution and reduction reactions. Angew. Chem. 2016, 56, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Xu, L.; Qiu, J.; Liu, X.; Zhang, S. Rational design of sustainable transition metal-based bifunctional electrocatalysts for oxygen reduction and evolution reactions. Sustain. Mater. Technol. 2020, 25, e00204. [Google Scholar] [CrossRef]
- Chen, Y.; Ji, S.; Chen, C.; Peng, Q.; Wang, D.; Li, Y. Single-atom catalysts: Synthetic strategies and electrochemical applications. Joule 2018, 2, 1242–1264. [Google Scholar] [CrossRef]
- Sun, T.; Zhao, S.; Chen, W.; Zhai, D.; Dong, J.; Wang, Y.; Zhang, S.; Han, A.; Gu, L.; Yu, R.; et al. Single-atomic cobalt sites embedded in hierarchically ordered porous nitrogen-doped carbon as a superior bifunctional electrocatalyst. Proc. Natl. Acad. Sci. USA 2018, 115, 12692–12697. [Google Scholar] [CrossRef]
- Wang, A.; Li, J.; Zhang, T. Heterogeneous single-atom catalysis. Nat. Rev. Chem. 2018, 2, 65–81. [Google Scholar] [CrossRef]
- Tavakkoli, M.; Flahaut, E.; Peljo, P.; Sainio, J.; Davodi, F.; Lobiak, E.V.; Mustonen, K.; Kauppinen, E.I. Mesoporous single-atom-doped graphene–carbon nanotube hybrid: Synthesis and tunable electrocatalytic activity for oxygen evolution and reduction reactions. ACS Catal. 2020, 10, 4647–4658. [Google Scholar] [CrossRef]
- Kaiser, S.K.; Chen, Z.; Akl, D.F.; Mitchell, S.; Pérez-Ramírez, J. Single-atom catalysts across the periodic table. Chem. Rev. 2020, 120, 11703–11809. [Google Scholar] [CrossRef]
- Li, L.; Chang, X.; Lin, X.; Zhao, Z.-J.; Gong, J. Theoretical insights into single-atom catalysts. Chem. Soc. Rev. 2020, 49, 8156–8178. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Chen, Y.; Wang, X.; Zhang, Z.; Wang, D.; Li, Y. Chemical Synthesis of single atomic site catalysts. Chem. Rev. 2020, 120, 11900–11955. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-L.; Kan, X.; Gan, L.-Y.; Fan, J.; Zhao, Y. Designing efficient single-atomic catalysts for bifunctional oxygen electrocatalysis via a general two-step strategy. Appl. Surf. Sci. 2021, 556, 149779. [Google Scholar] [CrossRef]
- Li, Z.; Wang, D.; Wu, Y.; Li, Y. Recent advances in the precise control of isolated single-site catalysts by chemical methods. Natl. Sci. Rev. 2018, 5, 673–689. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, X.; Wei, Z. Recent developments in the use of single-atom catalysts for water splitting. Chin. J. Catal. 2021, 42, 1269–1286. [Google Scholar] [CrossRef]
- Mao, X.; Zhang, L.; Kour, G.; Zhou, S.; Wang, S.; Yan, C.; Zhu, Z.; Du, A. Defective Graphene on the transition-metal surface: Formation of efficient bifunctional catalysts for oxygen evolution/reduction reactions in alkaline media. ACS Appl. Mater. Interfaces 2019, 11, 17410–17415. [Google Scholar] [CrossRef]
- Wang, P.; Zhao, D.; Yin, L.-W. Two-dimensional matrices confining metal single atoms with enhanced electrochemical reaction kinetics for energy storage applications. Energy Environ. Sci. 2020, 14, 1794–1834. [Google Scholar] [CrossRef]
- Zhang, L.; Jia, Y.; Gao, G.; Yan, X.; Chen, N.; Chen, J.; Soo, M.T.; Wood, B.; Yang, D.; Du, A.; et al. Graphene defects trap atomic ni species for hydrogen and oxygen evolution reactions. Chem 2018, 4, 285–297. [Google Scholar] [CrossRef]
- Meng, J.; Li, J.; Liu, J.; Zhang, X.; Jiang, G.; Ma, L.; Hu, Z.-Y.; Xi, S.; Zhao, Y.; Yan, M.; et al. Universal approach to fabricating graphene-supported single-atom catalysts from doped ZnO solid solutions. ACS Cent. Sci. 2020, 6, 1431–1440. [Google Scholar] [CrossRef]
- Fei, H.; Dong, J.; Feng, Y.; Allen, C.S.; Wan, C.; Volosskiy, B.; Li, M.; Zhao, Z.; Wang, Y.; Sun, H.; et al. General synthesis and definitive structural identification of MN4C4 single-atom catalysts with tunable electrocatalytic activities. Nat. Catal. 2018, 1, 63–72. [Google Scholar] [CrossRef]
- Wang, X.; Yang, Z.; Si, W.; Shen, X.; Li, X.; Li, R.; Lv, Q.; Wang, N.; Huang, C. Cobalt-nitrogen-doped graphdiyne as an efficient bifunctional catalyst for oxygen reduction and hydrogen evolution reactions. Carbon 2019, 147, 9–18. [Google Scholar] [CrossRef]
- Qin, T.; Zhao, J.; Shi, R.; Ge, C.; Li, Q. Ionic liquid derived active atomic iron sites anchored on hollow carbon nanospheres for bifunctional oxygen electrocatalysis. Chem. Eng. J. 2020, 399, 125656. [Google Scholar] [CrossRef]
- Cheng, Y.; Guo, H.; Li, X.; Wu, X.; Xu, X.; Zheng, L.; Song, R. Rational design of ultrahigh loading metal single-atoms (Co, Ni, Mo) anchored on in-situ pre-crosslinked guar gum derived N-doped carbon aerogel for efficient overall water splitting. Chem. Eng. J. 2021, 410, 128359. [Google Scholar] [CrossRef]
- Shang, H.; Sun, W.; Sui, R.; Pei, J.; Zheng, L.; Dong, J.; Jiang, Z.; Zhou, D.; Zhuang, Z.; Chen, W.; et al. Engineering Isolated Mn–N2C2 Atomic Interface Sites for Efficient Bifunctional Oxygen Reduction and Evolution Reaction. Nano Lett. 2020, 20, 5443–5450. [Google Scholar] [CrossRef]
- Du, C.; Gao, Y.; Wang, J.; Chen, W. A new strategy for engineering a hierarchical porous carbon-anchored Fe single-atom electrocatalyst and the insights into its bifunctional catalysis for flexible rechargeable Zn–air batteries. J. Mater. Chem. A 2020, 8, 9981–9990. [Google Scholar] [CrossRef]
- Wu, Y.; Xiao, Z.; Jin, Z.; Li, X.; Chen, Y. The cobalt carbide/bimetallic CoFe phosphide dispersed on carbon nanospheres as advanced bifunctional electrocatalysts for the ORR, OER, and rechargeable Zn–air batteries. J. Colloid Interface Sci. 2021, 590, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Guo, Y.; Fu, X.-Z.; Luo, J.-L.; Zhi, C. Strengthening absorption ability of Co–N–C as efficient bifunctional oxygen catalyst by modulating the d band center using MoC. Green Energy Environ. 2021. [Google Scholar] [CrossRef]
- Jose, V.; Hu, H.; Edison, E.; Manalastas, W.M., Jr.; Ren, H.; Kidkhunthod, P.; Sreejith, S.; Jayakumar, A.; Nsanzimana, J.M.V.; Srinivasan, M.; et al. Modulation of single atomic co and fe sites on hollow carbon nanospheres as oxygen electrodes for rechargeable Zn–Air batteries. Small Methods 2020, 5, 2000751. [Google Scholar] [CrossRef]
- Ban, J.; Wen, X.; Xu, H.; Wang, Z.; Liu, X.; Cao, G.; Shao, G.; Hu, J. Dual Evolution in defect and morphology of single-atom dispersed carbon based oxygen electrocatalyst. Adv. Funct. Mater. 2021, 31, 2010472. [Google Scholar] [CrossRef]
- Luo, F.; Zhu, J.; Maa, S.; Lia, M.; Xua, R.; Zhanga, Q.; Yanga, Z.; Quc, K.; Caia, W.; Chenb, Z. Regulated coordination environment of Ni single atom catalyst toward high-efficiency oxygen electrocatalysis for rechargeable Zinc-air batteries. Energy Storage Mater. 2020, 35, 723–730. [Google Scholar] [CrossRef]
- Chen, D.; Cao, W.; Liu, J.; Wang, J.; Li, X.; Jiang, L. Filling the in situ-generated vacancies with metal cations captured by C−N bonds of defect-rich 3D carbon nanosheet for bifunctional oxygen electrocatalysis. J. Energy Chem. 2020, 59, 47–54. [Google Scholar] [CrossRef]
- Jiao, L.; Jiang, H.-L. Metal-organic-framework-based single-atom catalysts for energy applications. Chem 2019, 5, 786–804. [Google Scholar] [CrossRef]
- Rogge, S.M.J.; Bavykina, A.; Hajek, J.; Garcia, H.; Olivos-Suarez, A.; Sepúlveda-Escribano, A.; Vimont, A.; Clet, G.; Bazin, P.; Kapteijn, F.; et al. Metal–organic and covalent organic frameworks as single-site catalysts. Chem. Soc. Rev. 2017, 46, 3134–3184. [Google Scholar] [CrossRef]
- Sarapuu, A.; Kibena-Põldsepp, E.; Borghei, M.; Tammeveski, K. Electrocatalysis of oxygen reduction on heteroatom-doped nanocarbons and transition metal–nitrogen–carbon catalysts for alkaline membrane fuel cells. J. Mater. Chem. A 2018, 6, 776–804. [Google Scholar] [CrossRef]
- Dilpazir, S.; He, H.; Li, Z.; Wang, M.; Lu, P.; Liu, R.; Xie, Z.; Gao, D.; Zhang, G. Cobalt Single atoms immobilized n-doped carbon nanotubes for enhanced bifunctional catalysis toward oxygen reduction and oxygen evolution reactions. ACS Appl. Energy Mater. 2018, 1, 3283–3291. [Google Scholar] [CrossRef]
- Han, X.; Ling, X.; Wang, Y.; Ma, T.; Zhong, C.; Hu, W.; Deng, Y. Generation of Nanoparticle, atomic-cluster, and single-atom cobalt catalysts from zeolitic imidazole frameworks by spatial isolation and their use in zinc–air batteries. Angew. Chem. 2019, 58, 5359–5364. [Google Scholar] [CrossRef]
- Sheng, J.; Zhu, S.; Jia, G.; Liu, X.; Li, Y. Carbon nanotube supported bifunctional electrocatalysts containing iron-nitrogen-carbon active sites for zinc-air batteries. Nano Res. 2021, 1–7. [Google Scholar] [CrossRef]
- Sun, X.; Sun, S.; Gu, S.; Liang, Z.; Zhang, J.; Yang, Y.; Deng, Z.; Wei, P.; Peng, J.; Xu, Y.; et al. High-performance single atom bifunctional oxygen catalysts derived from ZIF-67 superstructures. Nano Energy 2019, 61, 245–250. [Google Scholar] [CrossRef]
- Wei, S.; Wang, Y.; Chen, W.; Li, Z.; Cheong, W.-C.; Zhang, Q.; Gong, Y.; Gu, L.; Chen, C.; Wang, D.; et al. Atomically dispersed Fe atoms anchored on COF-derived N-doped carbon nanospheres as efficient multi-functional catalysts. Chem. Sci. 2020, 11, 786–790. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.-D.; Xu, R.; Chai, G.-L.; Zhang, T.; Zang, K.-T.; Nan, B.; Lin, H.; Liang, Y.-L.; Lv, J.; Luo, J.; et al. Cobalt single-atoms anchored on porphyrinic triazine-based frameworks as bifunctional electrocatalysts for oxygen reduction and hydrogen evolution reactions. J. Mater. Chem. A 2019, 7, 1252–1259. [Google Scholar] [CrossRef]
- Zou, L.; Wei, Y.; Hou, C.; Li, C.; Xu, Q. Single-atom catalysts derived from metal–organic frameworks for electrochemical applications. Small 2021, 17, 2004809. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Xu, L.; Wang, D.; Li, Y. Metal organic frameworks derived single atom catalysts for electrocatalytic energy conversion. Nano Res. 2019, 12, 2067–2080. [Google Scholar] [CrossRef]
- Zhu, Y.; Yue, K.; Xia, C.; Zaman, S.; Yang, H.; Wang, X.; Yan, Y.; Xia, B.Y. Recent Advances on MOF Derivatives for Non-Noble Metal Oxygen Electrocatalysts in Zinc-Air Batteries. Nanomicro Lett. 2021, 13, 1–29. [Google Scholar] [CrossRef]
- Zhang, X.; Luo, J.; Lin, H.-F.; Tang, P.; Morante, J.R.; Arbiol, J.; Wan, K.; Mao, B.-W.; Liu, L.-M.; Fransaer, J. Tailor-made metal-nitrogen-carbon bifunctional electrocatalysts for rechargeable Zn-air batteries via controllable MOF units. Energy Stor. Mater. 2019, 17, 46–61. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, P.; Yang, J.; Lu, S.; Li, K.; Liu, G.; Duan, Y.; Qiu, J. Decorating ZIF-67-derived cobalt–nitrogen doped carbon nanocapsules on 3D carbon frameworks for efficient oxygen reduction and oxygen evolution. Carbon 2021, 177, 344–356. [Google Scholar] [CrossRef]
- Chen, Z.; Ha, Y.; Jia, H.; Yan, X.; Chen, M.; Liu, M.; Wu, R. Oriented Transformation of Co-LDH into 2D/3D ZIF-67 to Achieve Co–N–C hybrids for efficient overall water splitting. Adv. Energy Mater. 2019, 9, 1803918. [Google Scholar] [CrossRef]
- Lian, Y.; Yang, W.; Zhang, C.; Sun, H.; Deng, Z.; Xu, W.; Song, L.; Ouyang, Z.; Wang, Z.; Guo, J.; et al. Unpaired 3d electrons on atomically dispersed cobalt centres in coordination polymers regulate both oxygen reduction reaction (ORR) activity and selectivity for use in Zinc–Air batteries. Angew. Chem. 2020, 59, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, J.; Luo, Y.; Chen, Y.; Luo, Y.; Zhang, C.; Xue, Y.; Liu, H.; Wang, G.; Wang, R. A defect-driven atomically dispersed Fe–N–C electrocatalyst for bifunctional oxygen electrocatalytic activity in Zn–air batteries. J. Mater. Chem. A 2021, 9, 5556–5565. [Google Scholar] [CrossRef]
- Pan, Y.; Liu, S.; Sun, K.; Chen, X.; Wang, B.; Wu, K.; Cao, X.; Cheong, W.; Shen, R.; Han, A.; et al. A bimetallic zn/fe polyphthalocyanine-derived single-atom Fe-N 4 catalytic site: A superior trifunctional catalyst for overall water splitting and Zn–Air batteries. Angew. Chem. 2018, 57, 8614–8618. [Google Scholar] [CrossRef]
- Luo, Y.; Zhang, J.; Chen, J.; Chen, Y.; Zhang, C.; Luo, Y.; Wang, G.; Wang, R. Bi-functional electrocatalysis through synergetic coupling strategy of atomically dispersed Fe and Co active sites anchored on 3D nitrogen-doped carbon sheets for Zn-air battery. J. Catal. 2021, 397, 223–232. [Google Scholar] [CrossRef]
- Zhu, X.; Zhang, D.; Chen, C.-J.; Zhang, Q.; Liu, R.-S.; Xia, Z.; Dai, L.; Amal, R.; Lu, X. Harnessing the interplay of Fe–Ni atom pairs embedded in nitrogen-doped carbon for bifunctional oxygen electrocatalysis. Nano Energy 2020, 71, 104597. [Google Scholar] [CrossRef]
- Wan, W.; Triana, C.A.; Lan, J.; Li, J.; Allen, C.S.; Zhao, Y.; Iannuzzi, M.; Patzke, G.R. Bifunctional Single Atom Electrocatalysts: Coordination–Performance Correlations and Reaction Pathways. ACS Nano 2020, 14, 13279–13293. [Google Scholar] [CrossRef]
- Chen, Y.; Ji, S.; Wang, Y.; Dong, J.; Chen, W.; Li, Z.; Shen, R.; Zheng, L.; Zhuang, Z.; Wang, D.; et al. Isolated single iron atoms anchored on n-doped porous carbon as an efficient electrocatalyst for the oxygen reduction reaction. Angew. Chem. 2017, 56, 6937–6941. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Li, J.; Zheng, J.; Chu, W.; Wang, N. Atomically dispersed metal sites stabilized on a nitrogen doped carbon carrier via N2 glow-discharge plasma. Chem. Commun. 2020, 56, 9198–9201. [Google Scholar] [CrossRef]
- Ye, C.; Zhang, N.; Wang, D.; Li, Y. Single atomic site catalysts: Synthesis, characterization, and applications. Chem. Commun. 2020, 56, 7687–7697. [Google Scholar] [CrossRef] [PubMed]
- Sakaushi, K.; Kumeda, T.; Hammes-Schiffer, S.; Melander, M.M.; Sugino, O. Advances and challenges for experiment and theory for multi-electron multi-proton transfer at electrified solid–liquid interfaces. Phys. Chem. Chem. Phys. 2020, 22, 19401–19442. [Google Scholar] [CrossRef]
- Kronberg, R.; Laasonen, K. Reconciling the experimental and computational hydrogen evolution activities of Pt (111) through DFT-based constrained MD simulations. ACS Catal. 2021, 11, 8062–8078. [Google Scholar] [CrossRef]
- Santos, E.; Quaino, P.; Schmickler, W. Theory of electrocatalysis: Hydrogen evolution and more. Phys. Chem. Chem. Phys. 2012, 14, 11224–11233. [Google Scholar] [CrossRef] [PubMed]
- Lamoureux, P.S.; Singh, A.R.; Chan, K. pH Effects on hydrogen evolution and oxidation over Pt (111): Insights from first-principles. ACS Catal. 2019, 9, 6194–6201. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Jensen, K.D.; Petersen, A.S.; Arnarson, L.; Bagger, A.; Escudero-Escribano, M. Realistic cyclic voltammograms from ab initio simulations in alkaline and acidic electrolytes. J. Phys. Chem. C 2020, 124, 20055–20065. [Google Scholar] [CrossRef]
- Tang, M.T.; Liu, X.; Ji, Y.; Norskov, J.K.; Chan, K. Modeling hydrogen evolution reaction kinetics through explicit water–metal interfaces. J. Phys. Chem. C 2020, 124, 28083–28092. [Google Scholar] [CrossRef]
- Lindgren, P.; Kastlunger, G.; Peterson, A.A. A Challenge to the G~0 interpretation of hydrogen evolution. ACS Catal. 2020, 10, 121–128. [Google Scholar] [CrossRef]
- Jerkiewicz, G. Standard and Reversible hydrogen electrodes: Theory, design, operation, and applications. ACS Catal. 2020, 10, 8409–8417. [Google Scholar] [CrossRef]
- Ooka, H.; Huang, J.; Exner, K.S. The sabatier principle in electrocatalysis: Basics, limitations, and extensions. Front. Energy Res. 2021, 9, 654460. [Google Scholar] [CrossRef]
- Quaino, P.; Juarez, F.; Santos, E.; Schmickler, W. Volcano plots in hydrogen electrocatalysis–uses and abuses. Beilstein J. Nanotechnol. 2014, 5, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Abild-Pedersen, F.; Greeley, J.; Studt, F.; Rossmeisl, J.; Munter, T.R.; Moses, P.G.; Skúlason, E.; Bligaard, T.; Nørskov, J.K. Scaling properties of adsorption energies for hydrogen-containing molecules on transition-metal surfaces. Phys. Rev. Lett. 2007, 99, 016105. [Google Scholar] [CrossRef] [PubMed]
- Abild-Pedersen, F. Computational catalyst screening: Scaling, bond-order and catalysis. Catal. Today 2016, 272, 6–13. [Google Scholar] [CrossRef]
- Montemore, M.; Medlin, J.W. Scaling relations between adsorption energies for computational screening and design of catalysts. Catal. Sci. Technol. 2014, 4, 3748–3761. [Google Scholar] [CrossRef]
- Christensen, R.; Hansen, H.A.; Dickens, C.F.; Nørskov, J.K.; Vegge, T. Functional independent scaling relation for ORR/OER catalysts. J. Phys. Chem. C 2016, 120, 24910–24916. [Google Scholar] [CrossRef]
- Divanis, S.; Kutlusoy, T.; Boye, I.M.I.; Man, I.C.; Rossmeisl, J. Oxygen evolution reaction: A perspective on a decade of atomic scale simulations. Chem. Sci. 2020, 11, 2943–2950. [Google Scholar] [CrossRef]
- Lipkowski, J.; Ross, P.N. Electrocatalysis; John Wiley & Sons: Hoboken, NJ, USA, 1998; ISBN 978-0-471-24673-2. [Google Scholar]
- Wittreich, G.R.; Alexopoulos, K.; Vlachos, D.G. Microkinetic modeling of surface catalysis. In Handbook of Materials Modeling; Andreoni, W., Yip, S., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 1377–1404. ISBN 978-3-319-44679-0. [Google Scholar]
- Dufils, T.; Jeanmairet, G.; Rotenberg, B.; Sprik, M.; Salanne, M. Simulating electrochemical systems by combining the finite field method with a constant potential electrode. Phys. Rev. Lett. 2019, 123, 195501. [Google Scholar] [CrossRef] [PubMed]
- Scalfi, L.; Limmer, D.T.; Coretti, A.; Bonella, S.; Madden, P.A.; Salanne, M.; Rotenberg, B. Charge fluctuations from molecular simulations in the constant-potential ensemble. Phys. Chem. Chem. Phys. 2020, 22, 10480–10489. [Google Scholar] [CrossRef]
- Scalfi, L.; Salanne, M.; Rotenberg, B. Molecular simulation of electrode-solution interfaces. Annu. Rev. Phys. Chem. 2021, 72, 189–212. [Google Scholar] [CrossRef] [PubMed]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.; Bligaard, T.; Jónsson, H. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Chan, K.; Ahmed, R.; Tripkovic, V.; Björketun, M.E. pH in atomic scale simulations of electrochemical interfaces. Phys. Chem. Chem. Phys. 2013, 15, 10321–10325. [Google Scholar] [CrossRef]
- Hansen, M.H.; Rossmeisl, J. pH in grand canonical statistics of an electrochemical interface. J. Phys. Chem. C 2016, 120, 29135–29143. [Google Scholar] [CrossRef]
- Koper, M.T.M. Theory of multiple proton–electron transfer reactions and its implications for electrocatalysis. Chem. Sci. 2013, 4, 2710–2723. [Google Scholar] [CrossRef]
- Kulkarni, A.; Siahrostami, S.; Patel, A.; Nørskov, J.K. Understanding catalytic activity trends in the oxygen reduction reaction. Chem. Rev. 2018, 118, 2302–2312. [Google Scholar] [CrossRef]
- Grimaud, A.; Diaz-Morales, O.; Han, B.; Hong, W.T.; Lee, Y.-L.; Giordano, L.; Stoerzinger, K.; Koper, M.T.M.; Shao-Horn, Y. Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution. Nat. Chem. 2017, 9, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Hu, P. Theory of the kinetics of chemical potentials in heterogeneous catalysis. Angew. Chem. 2011, 123, 7792–7796. [Google Scholar] [CrossRef][Green Version]
- Craig, M.J.; Coulter, G.; Dolan, E.; Soriano-López, J.; Mates-Torres, E.; Schmitt, W.; García-Melchor, M. Universal scaling relations for the rational design of molecular water oxidation catalysts with near-zero overpotential. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Huang, Z.-F.; Song, J.; Dou, S.; Li, X.; Wang, J.; Wang, X. Strategies to break the scaling relation toward enhanced oxygen electrocatalysis. Matter 2019, 1, 1494–1518. [Google Scholar] [CrossRef]
- Busch, M.; Halck, N.B.; Kramm, U.; Siahrostami, S.; Krtil, P.; Rossmeisl, J. Beyond the top of the volcano? A unified approach to electrocatalytic oxygen reduction and oxygen evolution. Nano Energy 2016, 29, 126–135. [Google Scholar] [CrossRef]
- Wan, H.; Jensen, A.W.; Escudero-Escribano, M.; Rossmeisl, J. Insights in the oxygen reduction reaction: From metallic electrocatalysts to diporphyrins. ACS Catal. 2020, 10, 5979–5989. [Google Scholar] [CrossRef]
- Wan, H.; Østergaard, T.M.; Arnarson, L.; Rossmeisl, J. Climbing the 3D volcano for the oxygen reduction reaction using porphyrin motifs. ACS Sustain. Chem. Eng. 2018, 7, 611–617. [Google Scholar] [CrossRef]
- Govindarajan, N.; Koper, M.T.M.; Meijer, E.J.; Calle-Vallejo, F. Outlining the scaling-based and scaling-free optimization of electrocatalysts. ACS Catal. 2019, 9, 4218–4225. [Google Scholar] [CrossRef]
- Calle-Vallejo, F.; Martinez, J.I.; Rossmeisl, J. Density functional studies of functionalized graphitic materials with late transition metals for oxygen reduction reactions. Phys. Chem. Chem. Phys. 2011, 13, 15639–15643. [Google Scholar] [CrossRef] [PubMed]
- Amiinu, I.S.; Liu, X.; Pu, Z.; Li, W.; Li, Q.; Zhang, J.; Tang, H.; Zhang, H.; Mu, S. From 3D ZIF Nanocrystals to Co-Nx/C nanorod array electrocatalysts for ORR, OER, and Zn-Air batteries. Adv. Funct. Mater. 2017, 28, 1704638. [Google Scholar] [CrossRef]
- Zhuang, L.; Ge, L.; Yang, Y.; Li, M.; Jia, Y.; Yao, X.; Zhu, Z. Ultrathin Iron-Cobalt Oxide Nanosheets with abundant oxygen vacancies for the oxygen evolution reaction. Adv. Mater. 2017, 29, 1606793. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, Z.; Lu, Z.; Wang, W. Bifunctional CoNx embedded graphene electrocatalysts for OER and ORR: A theoretical evaluation. Carbon 2018, 130, 112–119. [Google Scholar] [CrossRef]
- Cai, L.Q. Metal element doping graphene for the oxygen electrode: A density functional calculation. Int. J. Electrochem. Sci. 2019, 598–605. [Google Scholar] [CrossRef]
- Sun, H.; Liu, S.; Wang, M.; Qian, T.; Xiong, J.; Yan, C. Updating the intrinsic activity of a single-atom site with a P–O bond for a rechargeable Zn–Air battery. ACS Appl. Mater. Interfaces 2019, 11, 33054–33061. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhou, E.; Yu, Z.; Liu, H.; Xiong, M. Tailor-made open porous 2D CoFe/SN-carbon with slightly weakened adsorption strength of ORR/OER intermediates as remarkable electrocatalysts toward zinc-air batteries. Appl. Catal. B Environ. 2020, 269, 118771. [Google Scholar] [CrossRef]
- Lin, S.-Y.; Xia, L.-X.; Zhang, L.; Feng, J.-J.; Zhao, Y.; Wang, A.-J. Highly active Fe centered FeM-N-doped carbon (M = Co/Ni/Mn): A general strategy for efficient oxygen conversion in Zn–air battery. Chem. Eng. J. 2021, 424, 130559. [Google Scholar] [CrossRef]
- Wang, J.; Xu, R.; Sun, Y.; Liu, Q.; Xia, M.; Li, Y.; Gao, F.; Zhao, Y.; Tse, J.S. Identifying the Zn–Co binary as a robust bifunctional electrocatalyst in oxygen reduction and evolution reactions via shifting the apexes of the volcano plot. J. Energy Chem. 2020, 55, 162–168. [Google Scholar] [CrossRef]
- Mao, X.; Ling, C.; Tang, C.; Yan, C.; Zhu, Z.; Du, A. Predicting a new class of metal-organic frameworks as efficient catalyst for bi-functional oxygen evolution/reduction reactions. J. Catal. 2018, 367, 206–211. [Google Scholar] [CrossRef]
- Wang, H.-F.; Chen, L.; Pang, H.; Kaskel, S.; Xu, Q. MOF-derived electrocatalysts for oxygen reduction, oxygen evolution and hydrogen evolution reactions. Chem. Soc. Rev. 2020, 49, 1414–1448. [Google Scholar] [CrossRef]
- Larsen, A.H.; Mortensen, J.J.; Blomqvist, J.; Castelli, I.E.; Christensen, R.; Dułak, M.; Friis, J.; Groves, M.; Hammer, B.; Hargus, C.; et al. The atomic simulation environment—A Python library for working with atoms. J. Phys. Condens. Matter 2017, 29, 273002. [Google Scholar] [CrossRef]
- Enkovaara, J.; Rostgaard, C.; Mortensen, J.J.; Chen, J.; Dułak, M.; Ferrighi, L.; Gavnholt, J.; Glinsvad, C.; Haikola, V.; Hansen, H.A.; et al. Electronic structure calculations with GPAW: A real-space implementation of the projector augmented-wave method. J. Phys. Condens. Matter 2010, 22, 253202. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Tkatchenko, A.; Scheffler, M. Accurate molecular Van Der Waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef] [PubMed]
- Held, A.; Walter, M. Simplified continuum solvent model with a smooth cavity based on volumetric data. J. Chem. Phys. 2014, 141, 174108. [Google Scholar] [CrossRef] [PubMed]
- Álvarez, S.R. A cartography of the van der Waals territories. Dalton Trans. 2013, 42, 8617–8636. [Google Scholar] [CrossRef]
- Hummelshøj, J.S.; Abild-Pedersen, F.; Studt, F.; Bligaard, T.; Nørskov, J.K. CatApp: A Web application for surface chemistry and heterogeneous catalysis. Angew. Chem. 2012, 51, 272–274. [Google Scholar] [CrossRef] [PubMed]
- Computational Materials Repository, CAMd. Available online: https://cmr.fysik.dtu.dk/ (accessed on 23 September 2021).
- Chanussot, L.; Das, A.; Goyal, S.; Lavril, T.; Shuaibi, M.; Riviere, M.; Tran, K.; Heras-Domingo, J.; Ho, C.; Hu, W.; et al. Open Catalyst 2020 (OC20) Dataset and Community Challenges. ACS Catal. 2021, 11, 6059–6072. [Google Scholar] [CrossRef]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cepitis, R.; Kongi, N.; Grozovski, V.; Ivaništšev, V.; Lust, E. Multifunctional Electrocatalysis on Single-Site Metal Catalysts: A Computational Perspective. Catalysts 2021, 11, 1165. https://doi.org/10.3390/catal11101165
Cepitis R, Kongi N, Grozovski V, Ivaništšev V, Lust E. Multifunctional Electrocatalysis on Single-Site Metal Catalysts: A Computational Perspective. Catalysts. 2021; 11(10):1165. https://doi.org/10.3390/catal11101165
Chicago/Turabian StyleCepitis, Ritums, Nadezda Kongi, Vitali Grozovski, Vladislav Ivaništšev, and Enn Lust. 2021. "Multifunctional Electrocatalysis on Single-Site Metal Catalysts: A Computational Perspective" Catalysts 11, no. 10: 1165. https://doi.org/10.3390/catal11101165
APA StyleCepitis, R., Kongi, N., Grozovski, V., Ivaništšev, V., & Lust, E. (2021). Multifunctional Electrocatalysis on Single-Site Metal Catalysts: A Computational Perspective. Catalysts, 11(10), 1165. https://doi.org/10.3390/catal11101165