Recent Advances in Metal Catalyst Design for CO2 Hydroboration to C1 Derivatives




Abstract
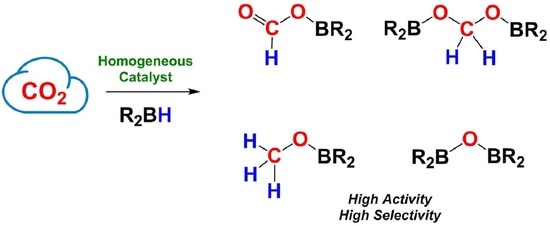
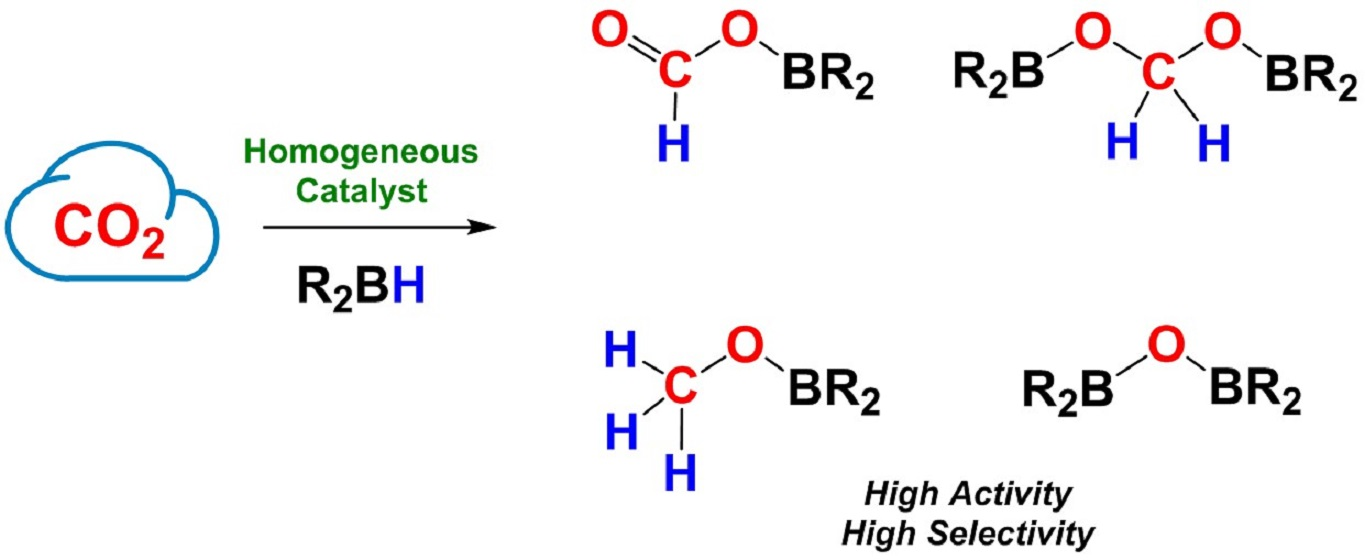
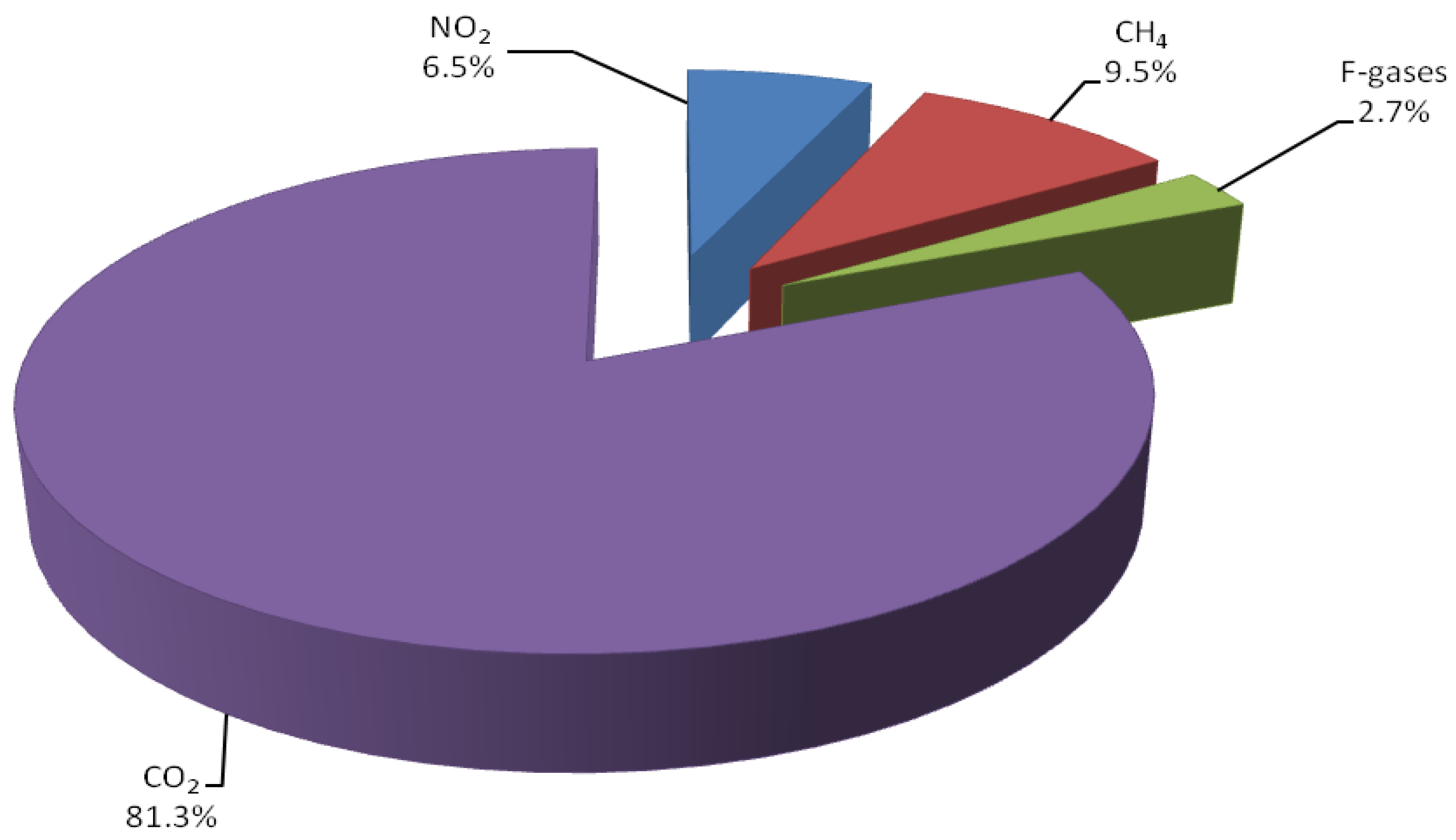
1. Introduction
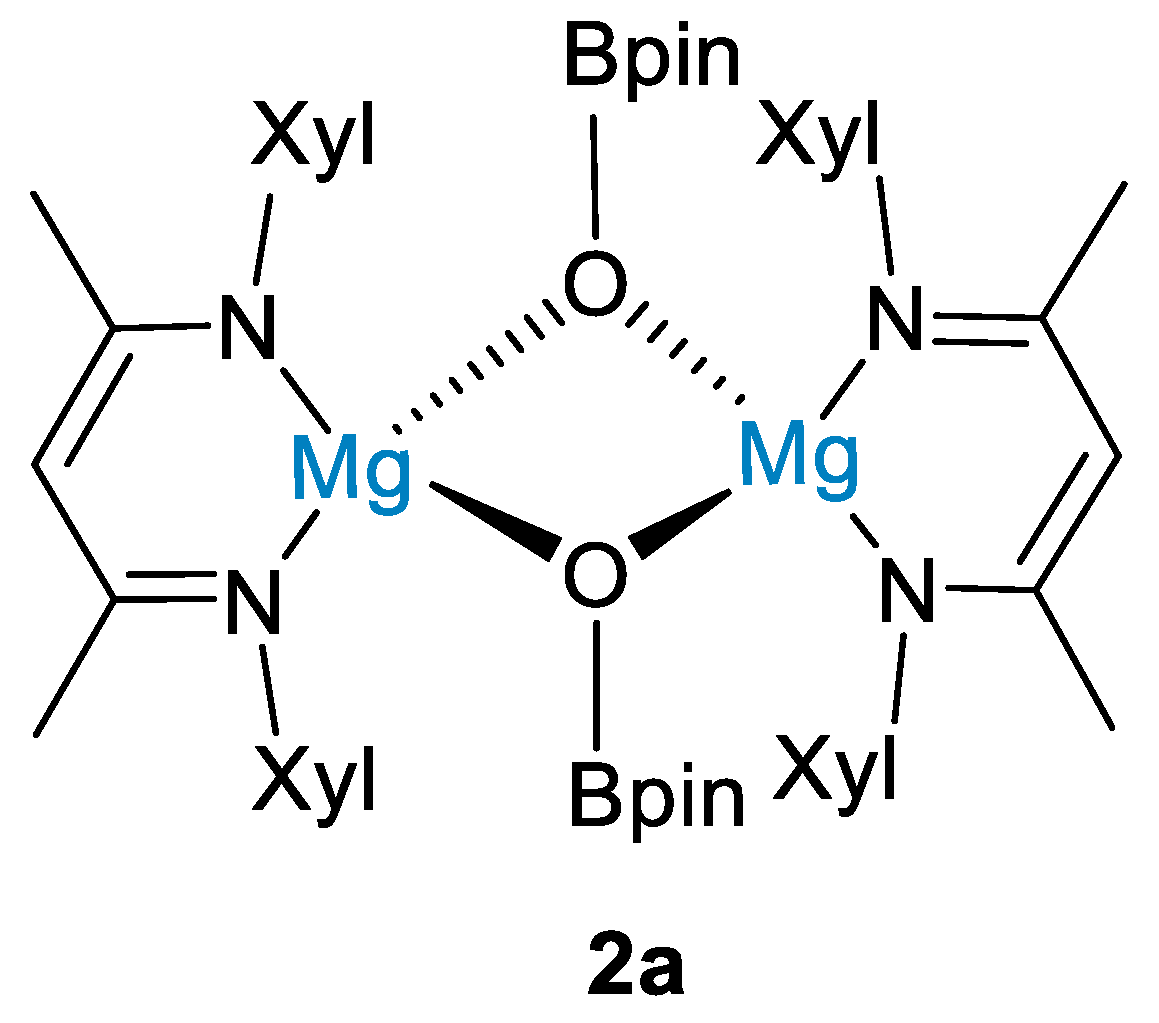
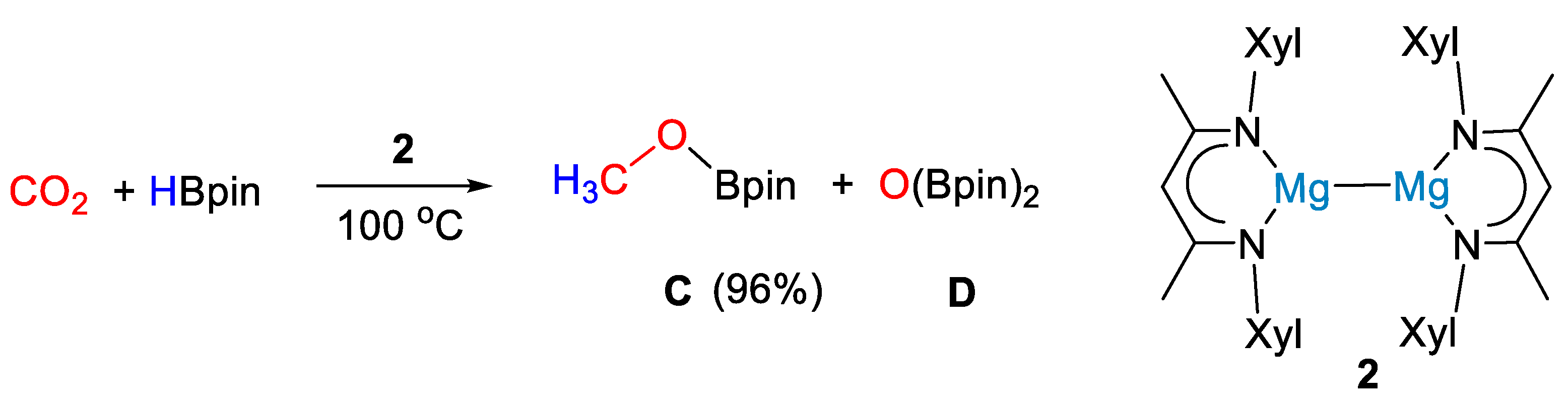
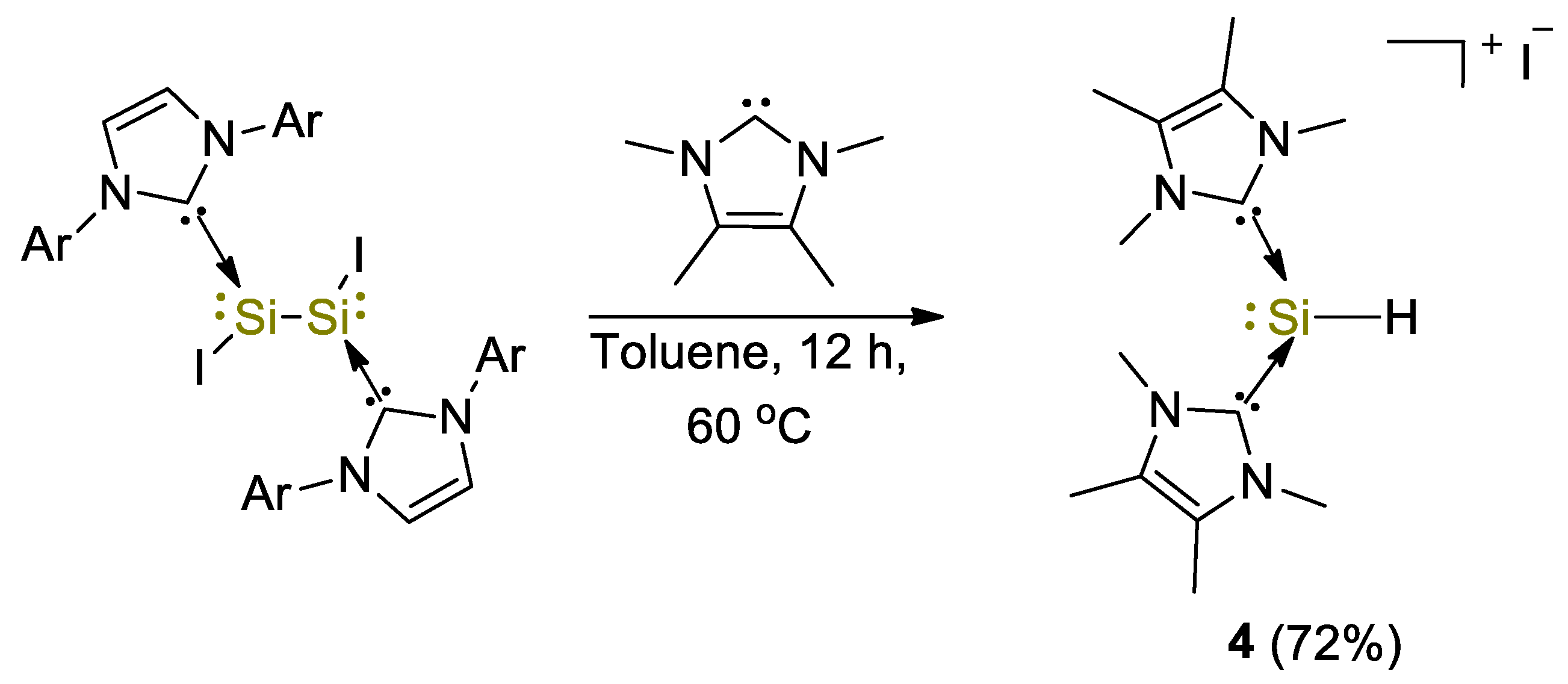
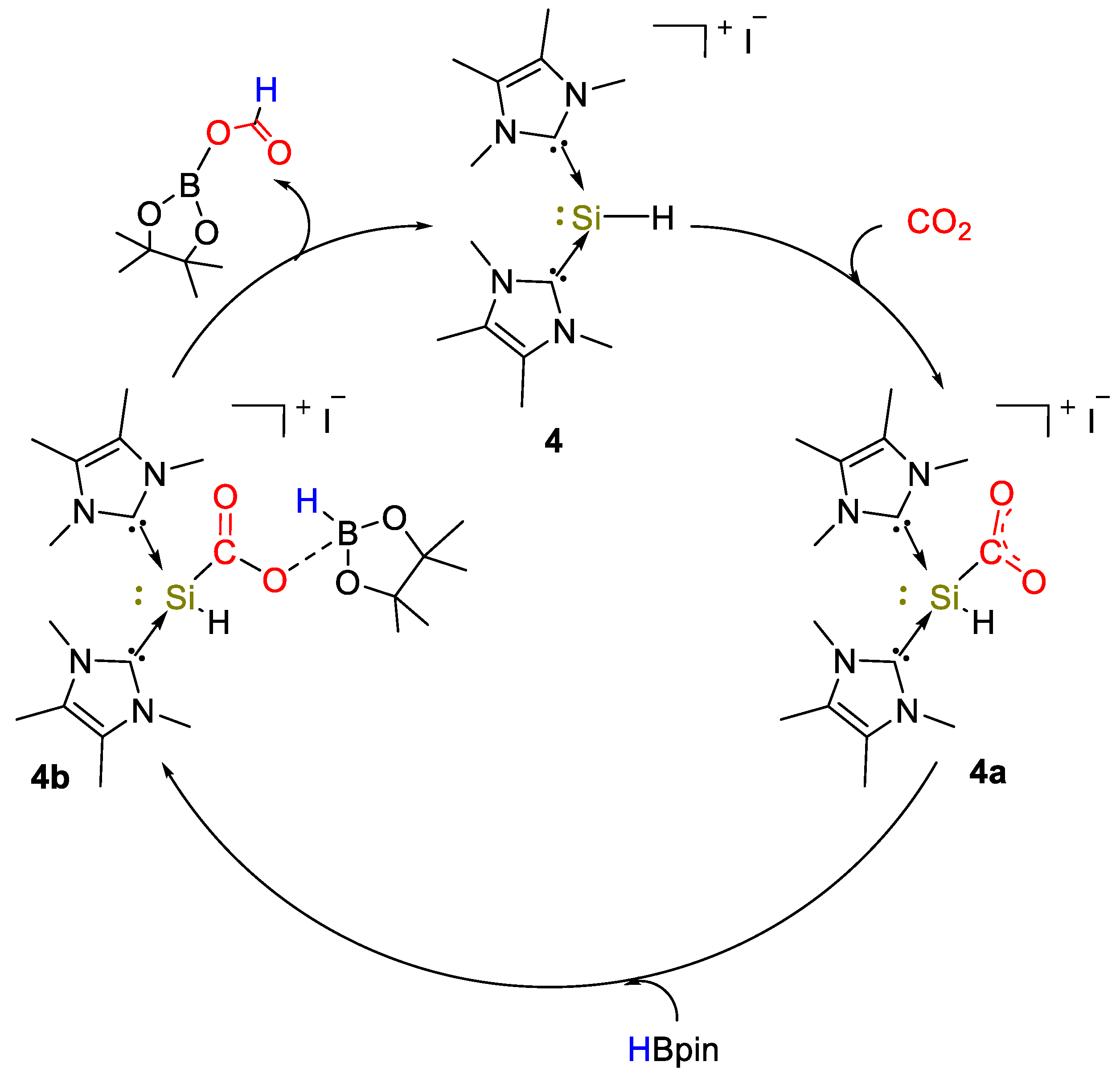
2. Main Group Metal Catalysts for CO2 Hydroboration
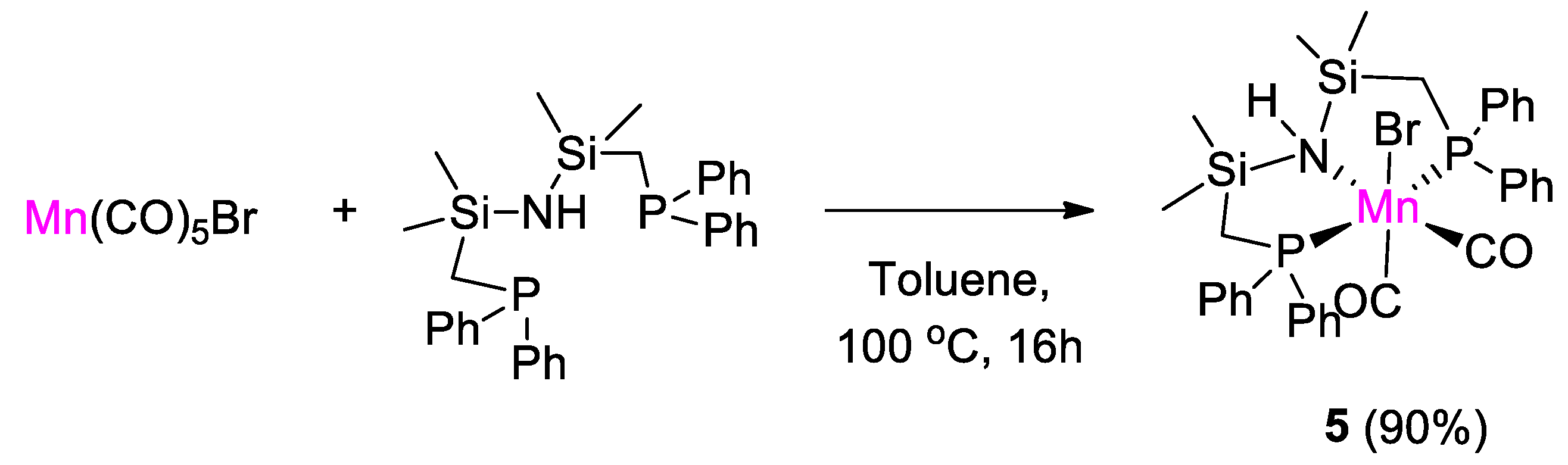
3. First-Row Transition Metal Catalysts for CO2 Hydroboration
4. Second-Row Transition Metal Catalysts for CO2 Hydroboration
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Data from United States Environmental Protection Agency (EPA). 2018. Available online: https://www.epa.gov/ghgemissions/overview-greenhouse-gases (accessed on 23 November 2020).
- Data from International Energy Agency Editions. Available online: https://www.iea.org (accessed on 23 November 2020).
- Mleczko, L.; Wolf, A.; Lolli, G. New Feedstocks and Chemistry for Lower CO2-Footprint: Today, Tomorrow, and in the Future. ChemBioEng. Rev. 2016, 3, 204–218. [Google Scholar] [CrossRef]
- Zhang, Y.-Y.; Wu, G.-P.; Darensbourg, D.J. CO2-Based Block Copolymers: Present and Future Designs. Trends Chem. 2020, 2, 750–763. [Google Scholar] [CrossRef]
- Jiang, X.; Nie, X.; Guo, X.; Song, C.; Chen, J.G. Recent Advances in Carbon Dioxide Hydrogenation to Methanol via Heterogeneous Catalysis. Chem. Rev. 2020, 120, 7984–8034. [Google Scholar] [CrossRef] [PubMed]
- Onishi, N.; Laurenczy, G.; Beller, M.; Himeda, Y. Recent progress for reversible homogeneous catalytic hydrogen storage in formic acid and in methanol. Coord. Chem. Rev. 2018, 373, 317–332. [Google Scholar] [CrossRef]
- Yaashikaa, P.R.; Senthil Kumar, P.; Varjani, S.J.; Saravanan, A. A review on photochemical, biochemical and electrochemical transformation of CO2 into value-added products. J. CO2 Util. 2019, 33, 131–147. [Google Scholar] [CrossRef]
- Daiyan, R.; Saputera, W.H.; Masood, H.; Leverett, J.; Lu, X.; Amal, R. A Disquisition on the Active Sites of Heterogeneous Catalysts for Electrochemical Reduction of CO2 to Value-Added Chemicals and Fuel. Adv. Energy Mater. 2020, 10, 1902106. [Google Scholar] [CrossRef]
- Modak, A.; Bhanja, P.; Dutta, S.; Chowdhury, B.; Bhaumik, A. Catalytic reduction of CO2 into fuels and fine chemicals. Green Chem. 2020, 22, 4002–4033. [Google Scholar] [CrossRef]
- Singh, A.K.; Singh, S.; Kumar, A. Hydrogen energy future with formic acid: A renewable chemical hydrogen storage system. Catal. Sci. Technol. 2016, 6, 12–40. [Google Scholar] [CrossRef]
- Sordakis, K.; Tang, C.; Vogt, L.K.; Junge, H.; Dyson, P.J.; Beller, M.; Laurenczy, G. Homogeneous Catalysis for Sustainable Hydrogen Storage in Formic Acid and Alcohols. Chem. Rev. 2018, 118, 372–433. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, T.; Das, S. Catalytic transformation of CO2 into C1 chemicals using hydrosilanes as a reducing agent. Green Chem. 2020, 22, 1800–1820. [Google Scholar] [CrossRef]
- Fernández-Alvarez, L.A.; Oro, L.A. Homogeneous Catalytic Reduction of CO2 with Silicon-Hydrides, State of the Art. ChemmatChem 2018, 10, 4783–4796. [Google Scholar] [CrossRef]
- Fernández-Alvarez, F.J.; Aitani, A.M.; Oro, L.A. Homogeneous catalytic reduction of CO2 with hydrosilanes. Catal. Sci. Technol. 2014, 4, 611–624. [Google Scholar] [CrossRef]
- Tlili, A.; Blondiaux, E.; Frogneux, X.; Cantat, T. Reductive functionalization of CO2 with amines: An entry to formamide, formamidine and methylamine derivatives. Green Chem. 2015, 17, 157–168. [Google Scholar] [CrossRef]
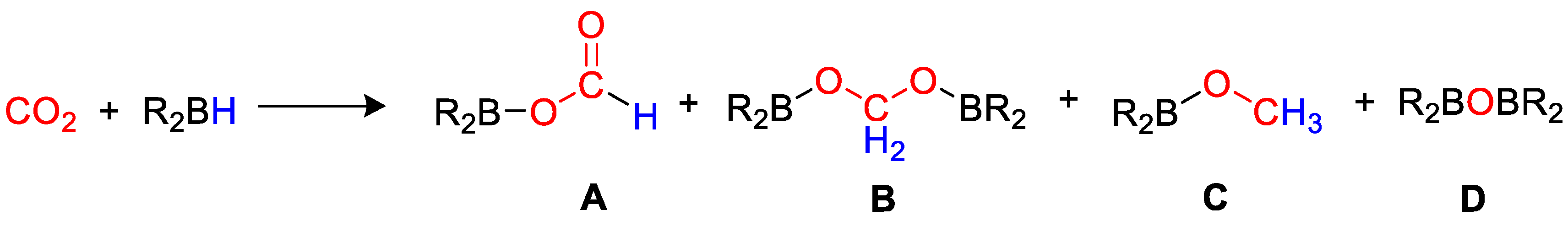
- Bontemps, S. Boron-mediated activation of carbon dioxide. Coord. Chem. Rev. 2016, 308, 117–130. [Google Scholar] [CrossRef]
- Geier, S.J.; Vogels, C.M.; Westcott, S.A. Boron Reagents in Synthesis; ACS Symposium Series; Coca, A., Ed.; American Chemical Society: Washington, DC, USA, 2016; Chapter 6; pp. 209–225. ISBN 139780841231832. [Google Scholar]
- Wu, X.-F.; Beller, M. (Eds.) Chemical Transformations of Carbon Dioxide. In Topics in Current Chemistry Collections, 1st ed.; Springer International Publishing: New York, NY, USA, 2018. [Google Scholar]
- Aresta, M.; Dibenedetto, A.; Angelini, A. Catalysis for the Valorization of Exhaust Carbon: From CO2 to Chemicals, Materials, and Fuels. Technological Use of CO2. Chem. Rev. 2014, 114, 1709–1742. [Google Scholar] [CrossRef]
- Peters, M.; Koehler, B.; Kuckshinrichs, W.; Leitner, W.; Markewitz, P.; Mueller, T.E. Chemical Technologies for Exploiting and Recycling Carbon Dioxide into the Value Chain. ChemSusChem 2011, 4, 1216–1240. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A. Utilisation of CO2 as a Chemical Feedstock: Opportunities and Challenges. Dalton Trans. 2007, 2975–2992. [Google Scholar] [CrossRef]
- Pinaka, A.; Vougioukalakis, G.C. Using Sustainable Metals to Carry out “Green” Transformations: Fe- and Cu-Catalyzed CO2 Monetization. Coord. Chem. Rev. 2015, 288, 69–97. [Google Scholar] [CrossRef]
- Grice, K.A. Carbon dioxide reduction with homogenous early transition metal complexes: Opportunities and challenges for developing CO2 catalysis. Coord. Chem. Rev. 2017, 336, 78–95. [Google Scholar] [CrossRef]
- Dagorne, S.; Wehmschulte, W. Recent Developments on the Use of Group 13 Metal Complexes in Catalysis. ChemCatChem 2018, 10, 2509–2520. [Google Scholar] [CrossRef]
- Wang, X.; Xia, C.; Wu, L. Homogeneous carbon dioxide reduction with p-block element-containing reductants. Green Chem. 2018, 20, 5415–5426. [Google Scholar] [CrossRef]
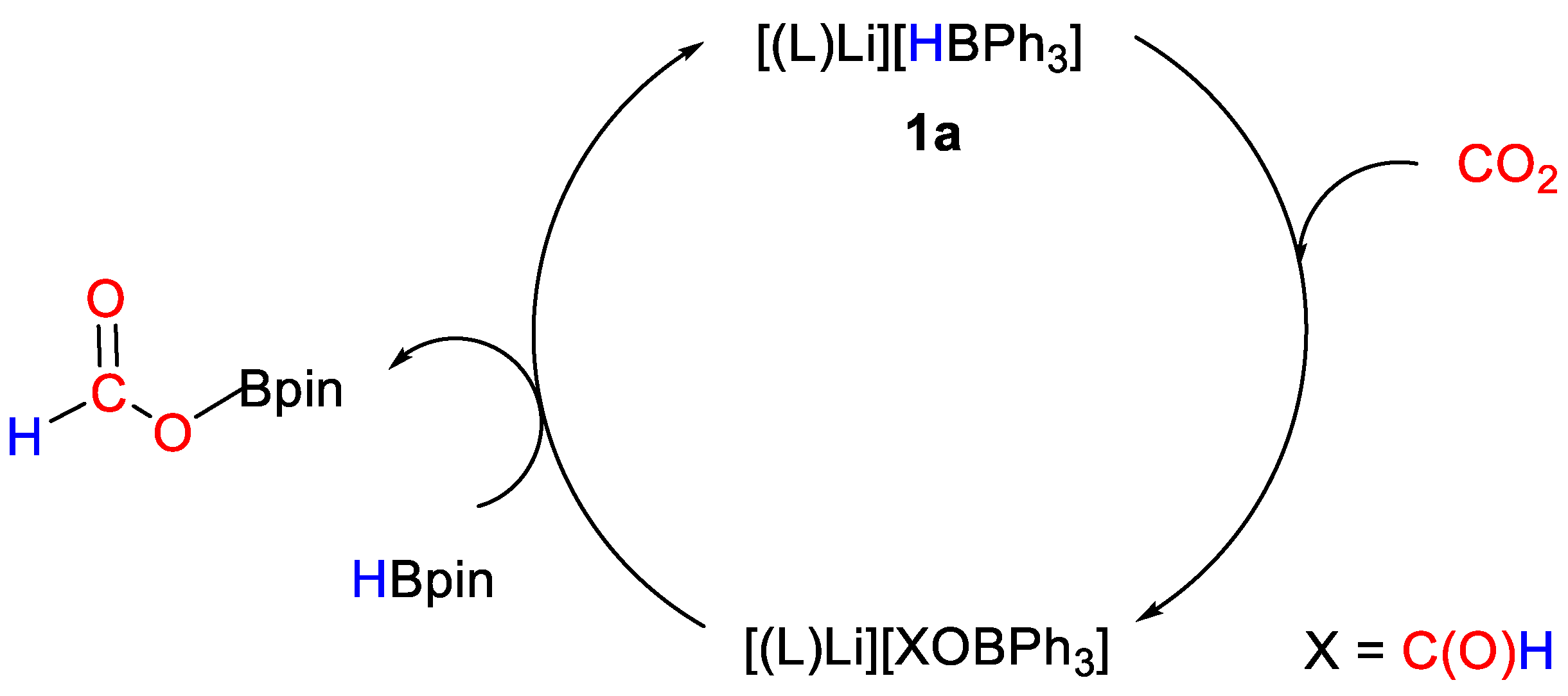
- Mukherjee, D.; Osseili, H.; Spaniol, T.P.; Okuda, J. Alkali Metal Hydridotriphenylborates [(L)M][HBPh3] (M = Li, Na, K): Chemoselective Catalysts for Carbonyl and CO2 Hydroboration. J. Am. Chem. Soc. 2016, 138, 10790–10793. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Wang, W.; Lu, K.; Yao, W.; Xue, F.; Ma, M. Magnesium-catalyzed hydroboration of organic carbonates, carbon dioxide and esters. Dalton Trans. 2020, 49, 2776–2780. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, D.; Shirase, S.; Spaniol, T.P.; Mashima, K.; Okuda, J. Magnesium hydridotriphenylborate [Mg(thf)6][HBPh3]2: A versatile hydroboration catalyst. Chem. Commun. 2016, 52, 13155–13158. [Google Scholar] [CrossRef]
- Leong, B.X.; Lee, J.; Li, Y.; Yang, M.-C.; Siu, C.-K.A.; Su, M.-D.; So, C.-W. A Versatile NHC-Parent Silyliumylidene Cation for Catalytic Chemo- and Regioselective Hydroboration. J. Am. Chem. Soc. 2019, 141, 17629–17636. [Google Scholar] [CrossRef]
- Li, Y.; Chan, Y.-C.; Leong, B.-X.; Li, Y.; Richards, E.; Indu, P.; De, S.; Parameswaran, P.; So, C.-W. Trapping a Silicon(I) Radical with Carbenes: A Cationic CAAC-Silicon(I) Radical and an NHC-Parent-Silyliumylidene Cation. Angew. Chem. Int. Ed. 2017, 56, 7573–7578. [Google Scholar] [CrossRef]
- Erken, C.; Kaithal, A.; Sen, S.; Weyhermüller, T.; Hölscher, M.; Werlé, C.; Leitner, W. Manganese-catalyzed hydroboration of carbon dioxide and other challenging carbonyl groups. Nat. Commun. 2018, 9, 4521. [Google Scholar] [CrossRef]
- Kostera, S.; Peruzzini, M.; Kirchner, K.; Gonsalvi, L. Mild and Selective Carbon Dioxide Hydroboration to Methoxyboranes Catalyzed by Mn(I) PNP Pincer Complexes. ChemCatChem 2020, 12, 4625–4631. [Google Scholar] [CrossRef]
- Mastalir, M.; Glatz, M.; Gorgas, N.; Stöger, B.; Pittenauer, E.; Allmaier, G.; Veiros, L.F.; Kirchner, K. Divergent Coupling of Alcohols and Amines Catalyzed by Isoelectronic Hydride MnI and FeII PNP Pincer Complexes. Chem. Eur. J. 2016, 22, 12316–12320. [Google Scholar] [CrossRef]
- Desmons, S.; Zhang, D.; Fajardo, A.M.; Bontemps, S. Versatile CO2 Transformations into Complex Products: A One-pot Two-step Strategy. J. Vis. Exp. 2019, 153, 60348. [Google Scholar]
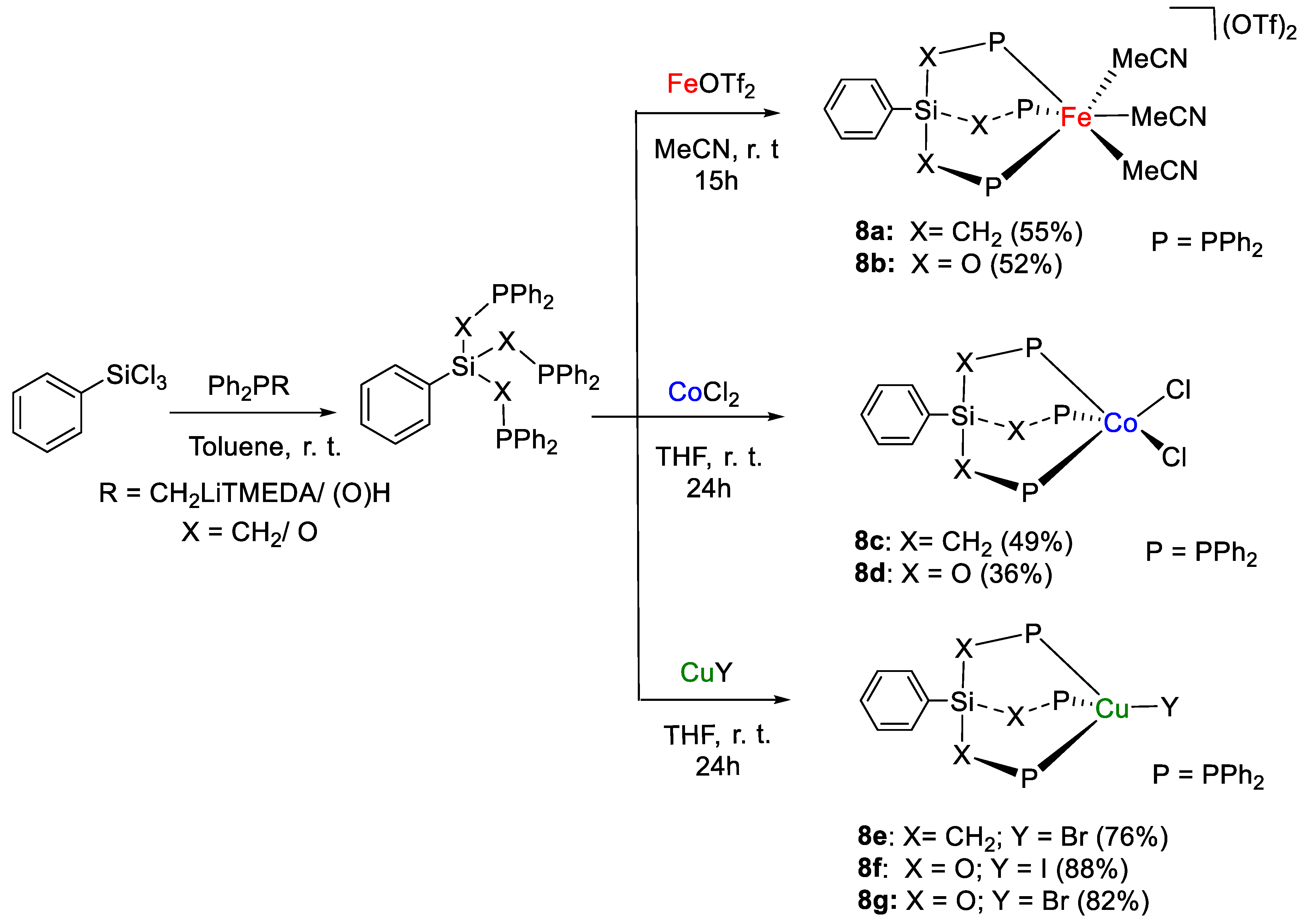
- Aloisi, A.; Berthet, J.-C.; Genre, C.; Thuéry, P.; Cantat, T. Complexes of the tripodal phosphine ligands PhSi(XPPh2)3 (X = CH2, O): Synthesis, structure and catalytic activity in the hydroboration of CO2. Dalton Trans. 2016, 45, 14774–14788. [Google Scholar] [CrossRef] [PubMed]
- Tamang, S.R.; Findlater, M. Cobalt catalysed reduction of CO2 via hydroboration. Dalton Trans. 2018, 47, 8199–8203. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chang, J.; Liu, T.; Cao, B.; Ding, Y.; Chen, X. Application of POCOP Pincer Nickel Complexes to the Catalytic Hydroboration of Carbon Dioxide. Catalysts 2018, 8, 508. [Google Scholar] [CrossRef]
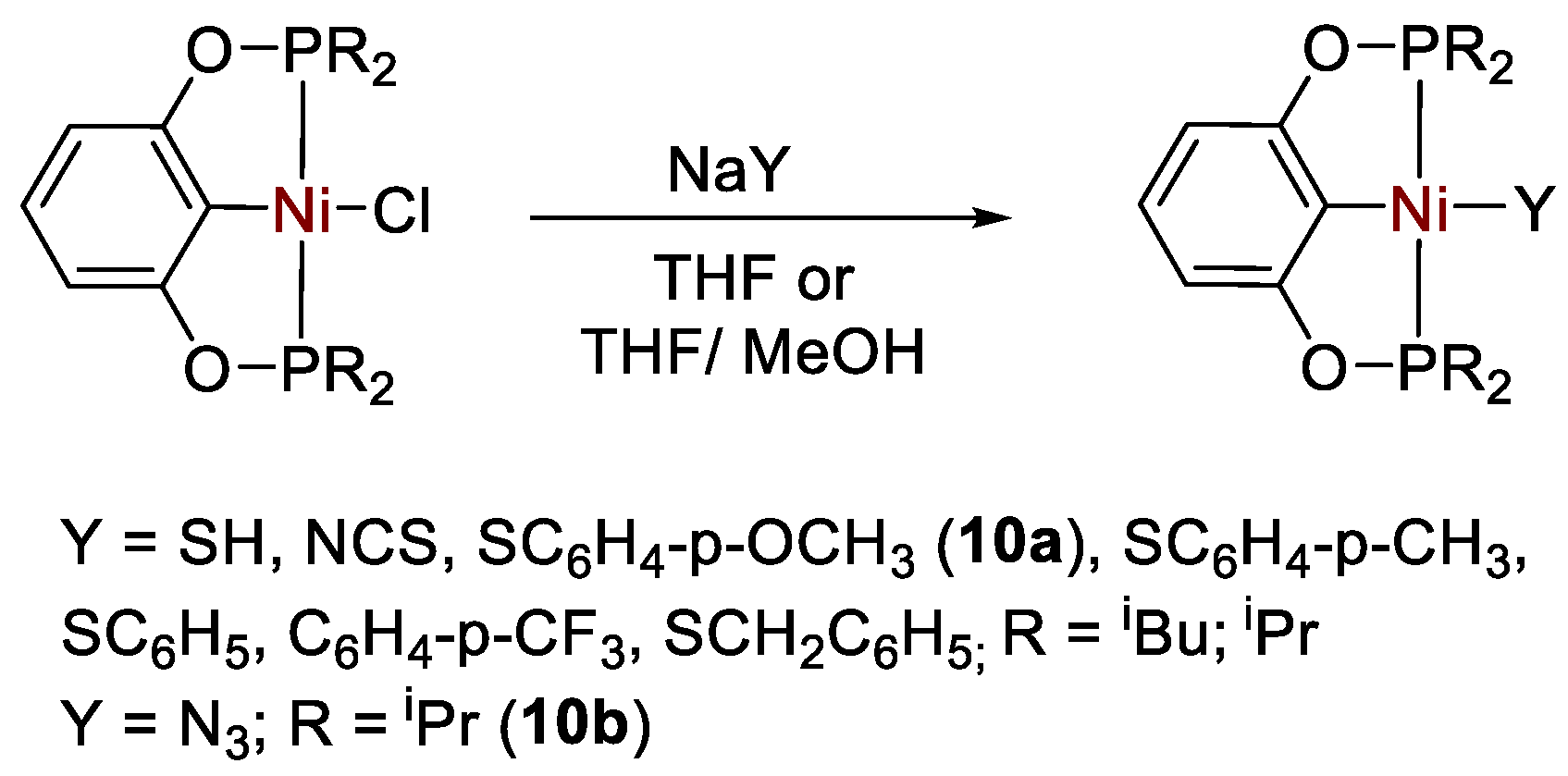
- Liu, T.; Meng, W.; Ma, Q.-Q.; Zhang, J.; Li, H.; Li, S.; Zhao, Q.; Chen, X. Hydroboration of CO2 catalyzed by bis(phosphinite) pincer ligated nickel thiolate complexes. Dalton Trans. 2017, 46, 4504–4509. [Google Scholar] [CrossRef] [PubMed]
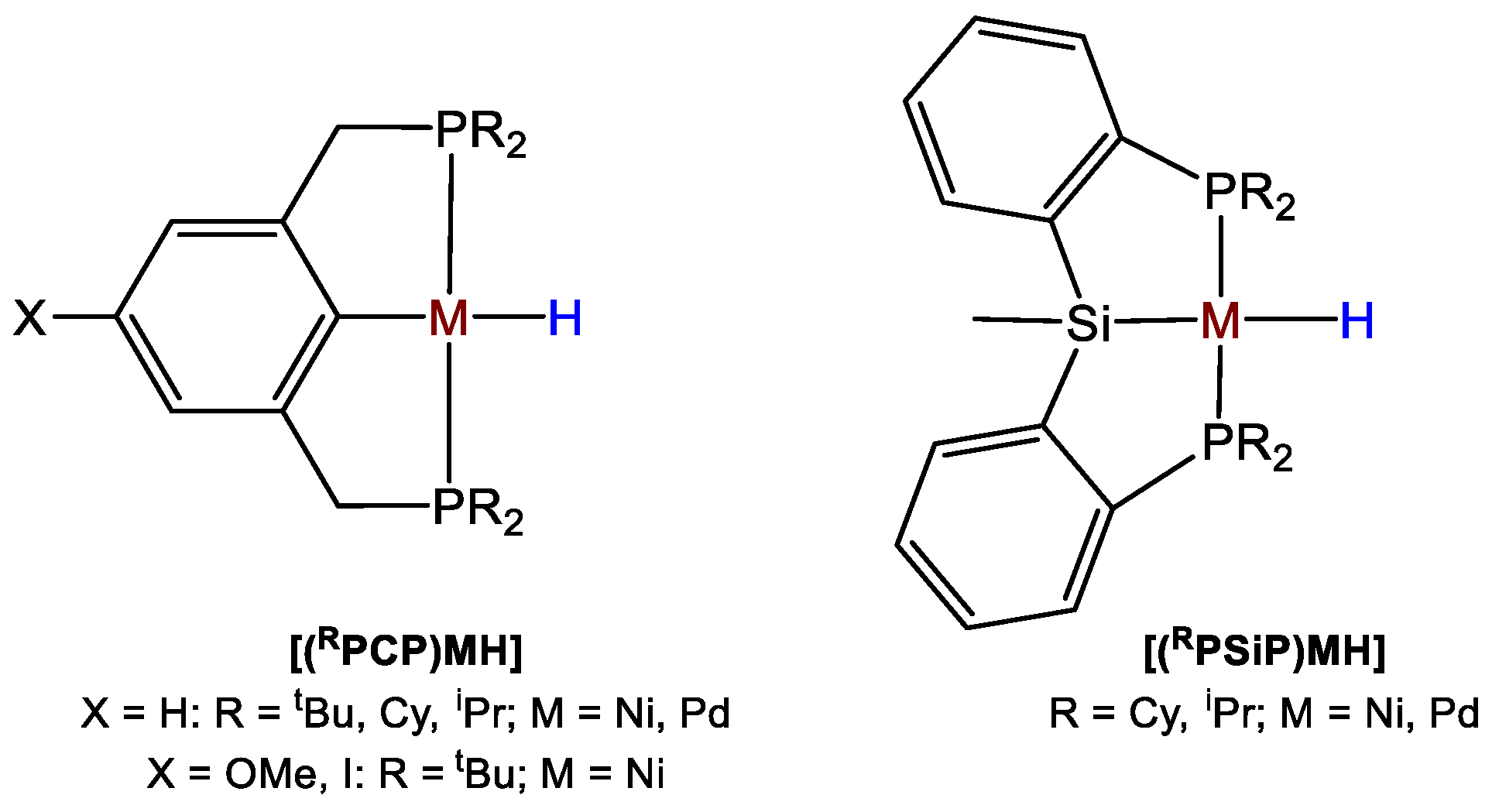
- Murphy, L.J.; Hollenhorst, H.; McDonald, R.; Ferguson, M.; Lumsden, M.D.; Turculet, L. Selective Ni-Catalyzed Hydroboration of CO2 to the Formaldehyde Level Enabled by New PSiP Ligation. Organometallics 2017, 36, 3709–3720. [Google Scholar] [CrossRef]
- Wellala, N.P.N.; Dong, H.T.; Krause, J.A.; Guan, H. Janus POCOP Pincer Complexes of Nickel. Organometallics 2018, 37, 4031–4039. [Google Scholar] [CrossRef]
- Smirnova, E.S.; Acuña-Parés, F.; Escudero-Adán, E.C.; Jelsch, C.; Lloret-Fillol, J. Synthesis and Reactivity of Copper(I) Complexes Based on C3-Symmetric Tripodal HTIM(PR2)3 Ligands. Eur. J. Inorg. Chem. 2018, 2612–2620. [Google Scholar] [CrossRef]
- Mukherjee, D.; Wiegand, A.-K.; Spaniol, T.P.; Okuda, J. Zinc hydridotriphenylborates supported by a neutral macrocyclic polyamine. Dalton Trans. 2017, 46, 6183–6186. [Google Scholar] [CrossRef]
- Janes, T.; Osten, K.M.; Pantaleo, A.; Yan, E.; Yang, Y.; Song, D. Insertion of CO2 into the carbon–boron bond of a boronic ester ligand. Chem. Commun. 2016, 52, 4148–4151. [Google Scholar] [CrossRef]
- Wang, X.; Chang, K.; Xu, X. Hydroboration of carbon dioxide enabled by molecular zinc dihydrides. Dalton Trans. 2020, 49, 7324–7327. [Google Scholar] [CrossRef]
- Li, L.; Zhu, H.; Liu, L.; Song, D.; Lei, M. A Hydride-Shuttle Mechanism for the Catalytic Hydroboration of CO2. Inorg. Chem. 2018, 57, 3054–3060. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.K.; Wu, J.; Hor, T.S.A.; Luo, H.-K. A binary catalyst system of a cationic Ru–CNC pincer complex with an alkali metal salt for selective hydroboration of carbon dioxide. Chem. Commun. 2016, 52, 11842–11845. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.-Q.; Liu, T.; Li, S.; Zhang, J.; Chen, X.; Guan, H. Highly efficient reduction of carbon dioxide with a borane catalyzed by bis(phosphinite) pincer ligated palladium thiolate complexes. Chem. Commun. 2016, 52, 14262–14265. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, M.R.; Charboneau, D.J.; de Oliveira, A.G.; Hazari, N. Controlling Selectivity in the Hydroboration of Carbon Dioxide to the Formic Acid, Formaldehyde, and Methanol Oxidation Levels. ACS Catal. 2019, 9, 301–314. [Google Scholar] [CrossRef]
- Sánchez, P.; Hernández-Juárez, M.; Rendón, N.; López-Serrano, J.; Álvarez, E.; Paneque, M.; Suárez, A. Hydroboration of carbon dioxide with catechol- and pinacolborane using an Ir–CNP* pincer complex. Water influence on the catalytic activity. Dalton Trans. 2018, 47, 16766–16776. [Google Scholar] [CrossRef]
- Field, L.D.; Shaw, W.J.; Turner, P. Addition of Nitrogen-Containing Heteroallenes to Iron(II)-Hydrides. Organometallics 2001, 20, 3491–3499. [Google Scholar] [CrossRef]
- Li, H.; Meng, W.; Adhikary, A.; Li, S.; Ma, N.; Zhao, Q.; Yang, Q.; Eberhardt, N.A.; Leahy, K.M.; Krause, J.A.; et al. Metathesis reactivity of bis(phosphinite) pincer ligated nickel chloride, isothiocyanate and azidecomplexes. J. Organomet. Chem. 2016, 804, 132–141. [Google Scholar] [CrossRef]
- Boro, B.J.; Duesler, E.N.; Goldberg, K.I.; Kemp, R.A. Synthesis, Characterization, and Reactivity of Nickel Hydride Complexes Containing 2,6-C6H3(CH2PR2)2 (R = tBu, cHex, and iPr) Pincer Ligands. Inorg. Chem. 2009, 48, 5081–5087. [Google Scholar] [CrossRef]
- Suh, H.-W.; Schmeier, T.J.; Hazari, N.; Kemp, R.A.; Takase, M.K. Experimental and Computational Studies of the Reaction of Carbon Dioxide with Pincer-Supported Nickel and Palladium Hydride. Organometallics 2012, 31, 8225–8236. [Google Scholar] [CrossRef]
- Moulton, C.J.; Shaw, B.L. Transition Metal-Carbon Bonds. Part XLII. Complexes of Nickel, Palladium, Platinum, Rhodium and Iridium with the Tridentate Ligand 2,6-Bis[(di-t-butylphosphino)methyl]phenyl. J. Chem. Soc. Dalton Trans 1976, 1020–1024. [Google Scholar] [CrossRef]
- Martínez-Prieto, L.M.; Melero, C.; del Río, D.; Palma, P.; Cámpora, J.; Álvarez, E. Synthesis and Reactivity of Nickel and Palladium Fluoride Complexes with PCP Pincer Ligands. NMR-Based Assessment of Electron-Donating Properties of Fluoride and Other Monoanionic Ligands. Organometallics 2012, 31, 1425–1438. [Google Scholar] [CrossRef]
- Suh, H.-W.; Balcells, D.; Edwards, A.J.; Guard, L.M.; Hazari, N.; Mader, E.A.; Mercado, B.Q.; Repisky, M. Understanding the Solution and Solid-State Structures of Pd and Pt PSiP Pincer-Supported Hydrides. Inorg. Chem. 2015, 54, 11411–11422. [Google Scholar] [CrossRef] [PubMed]











































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
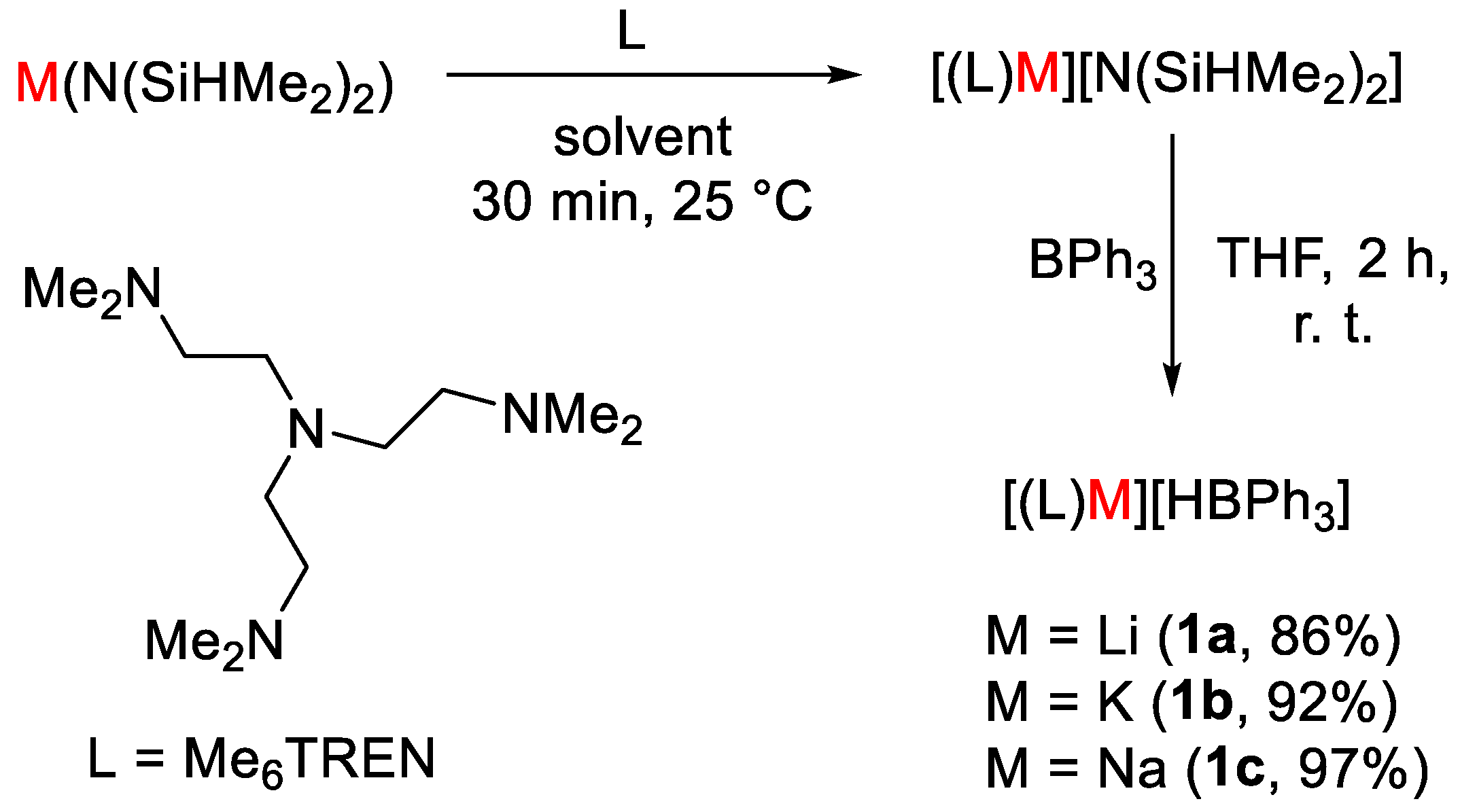{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
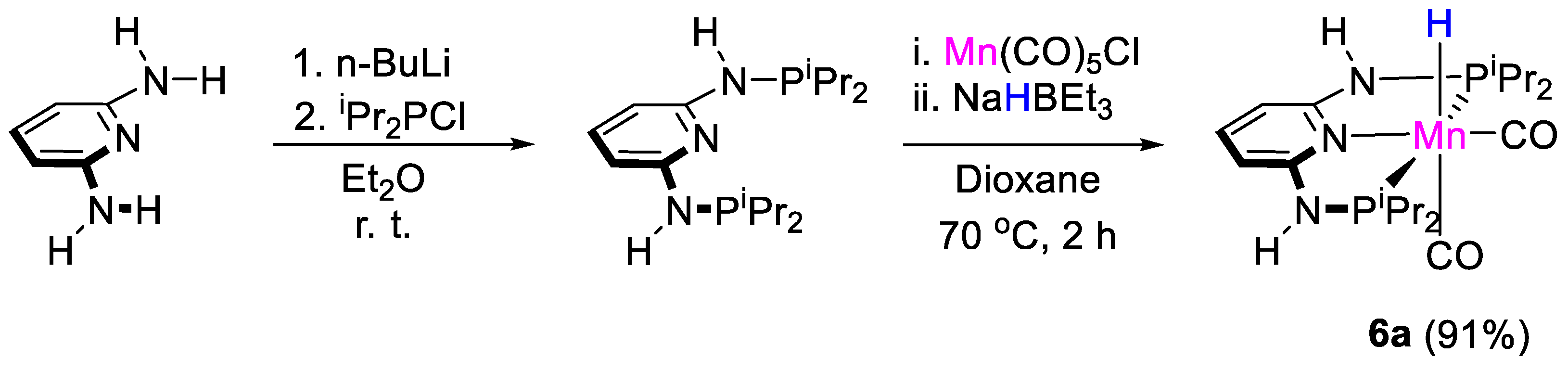{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
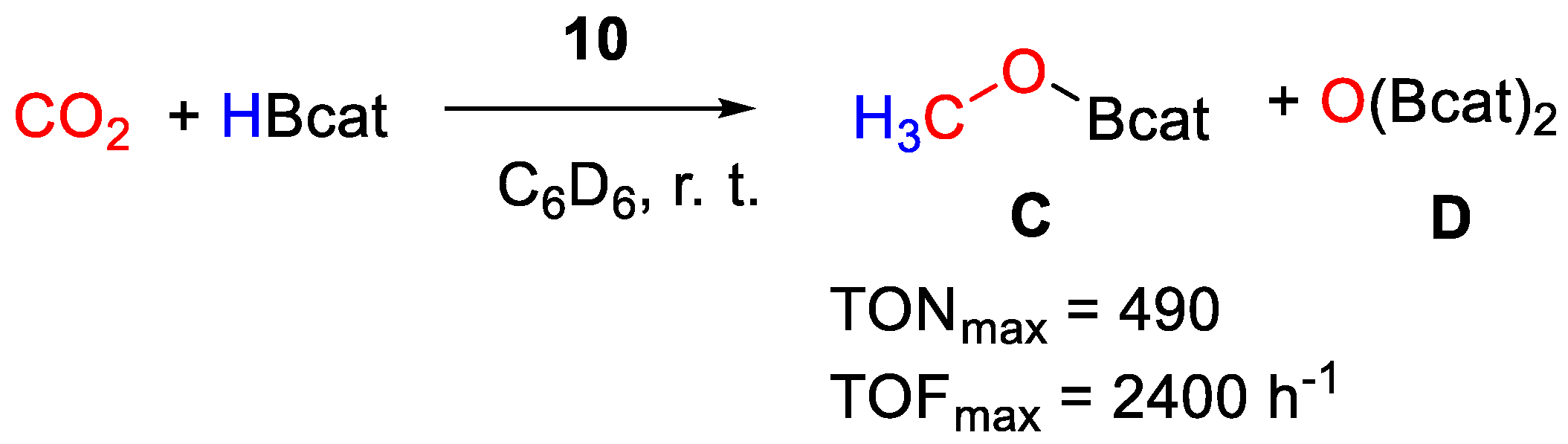{kind=link}
{kind=link}
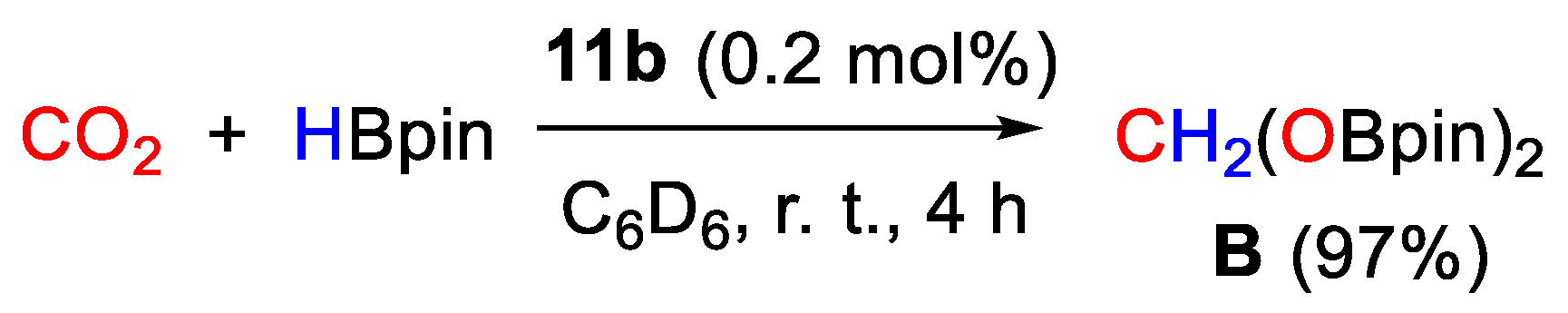{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
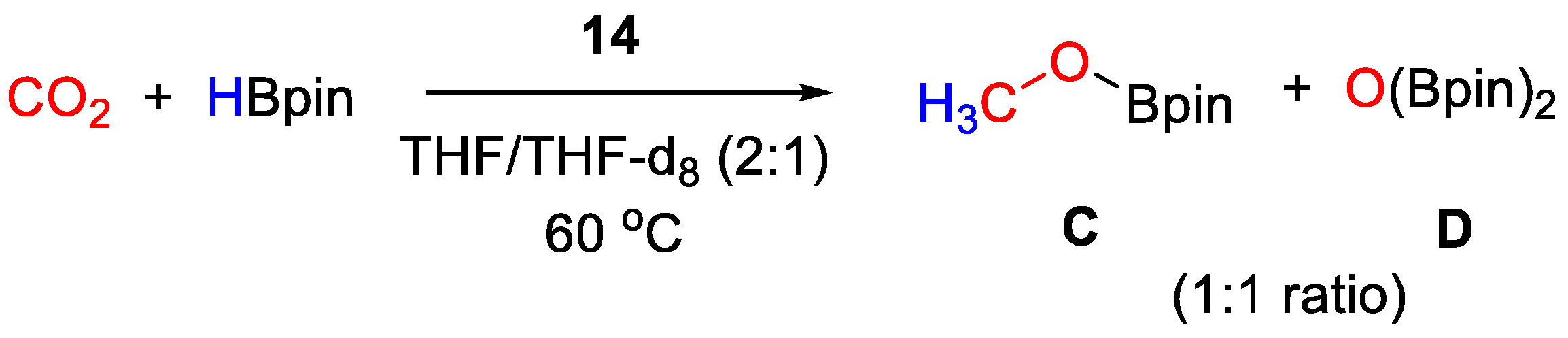{kind=link}
{kind=link}
{kind=link}
{kind=link}
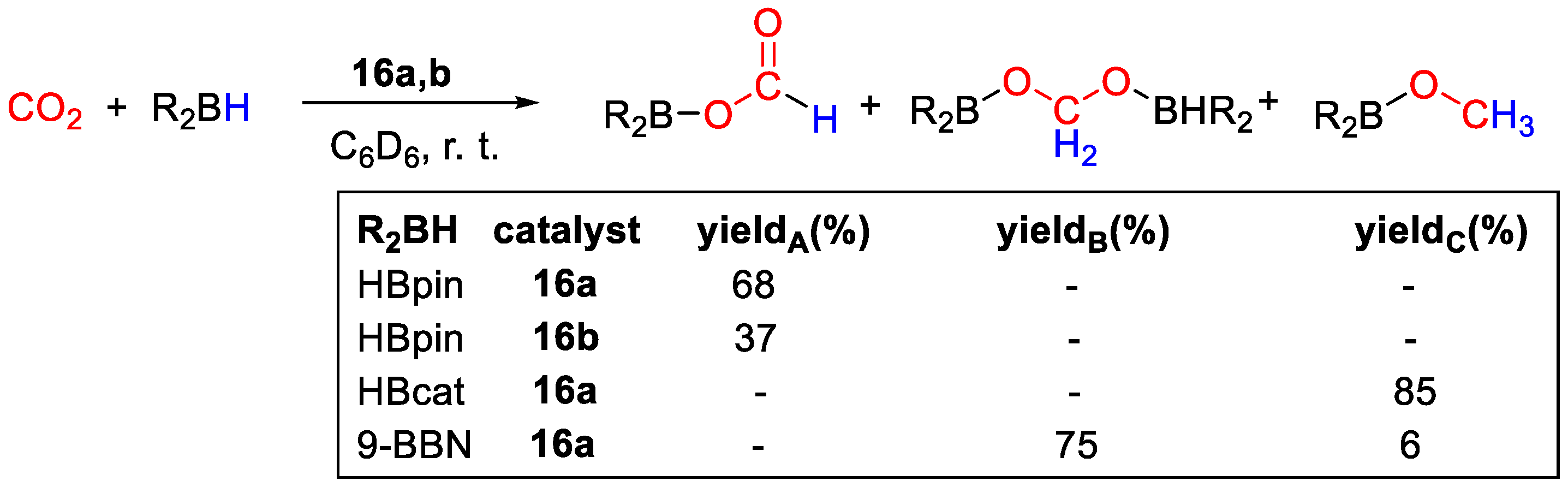{kind=link}
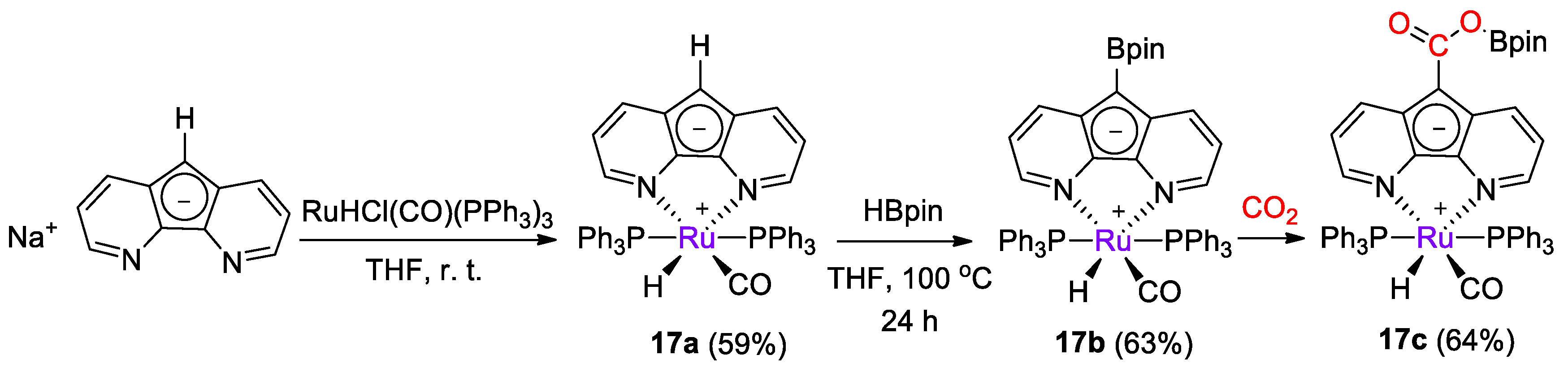{kind=link}
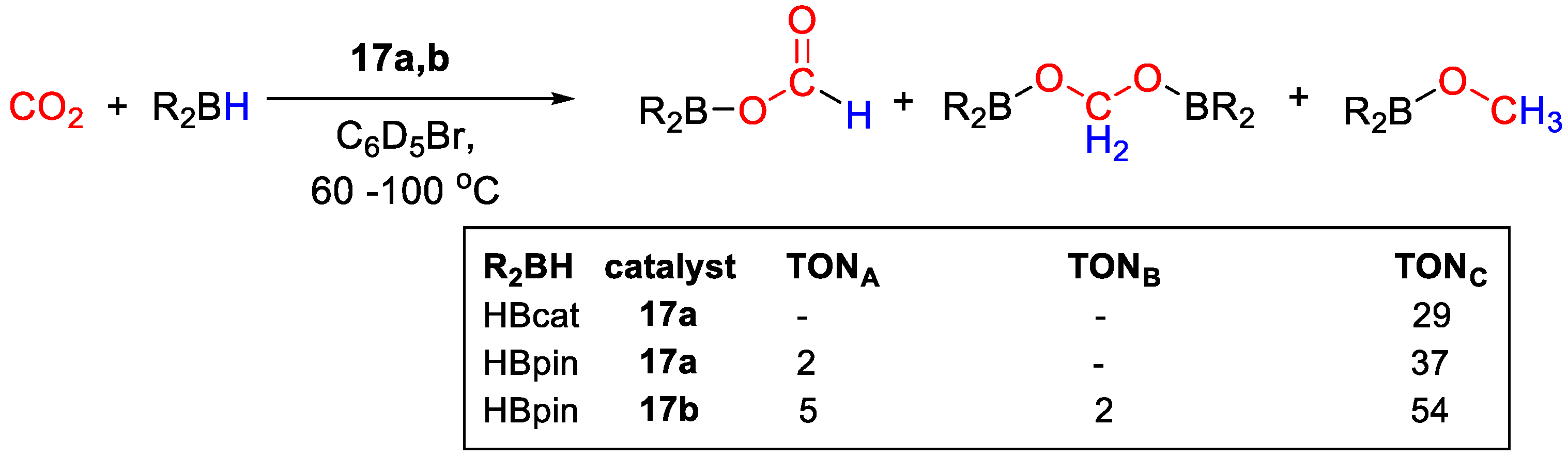{kind=link}
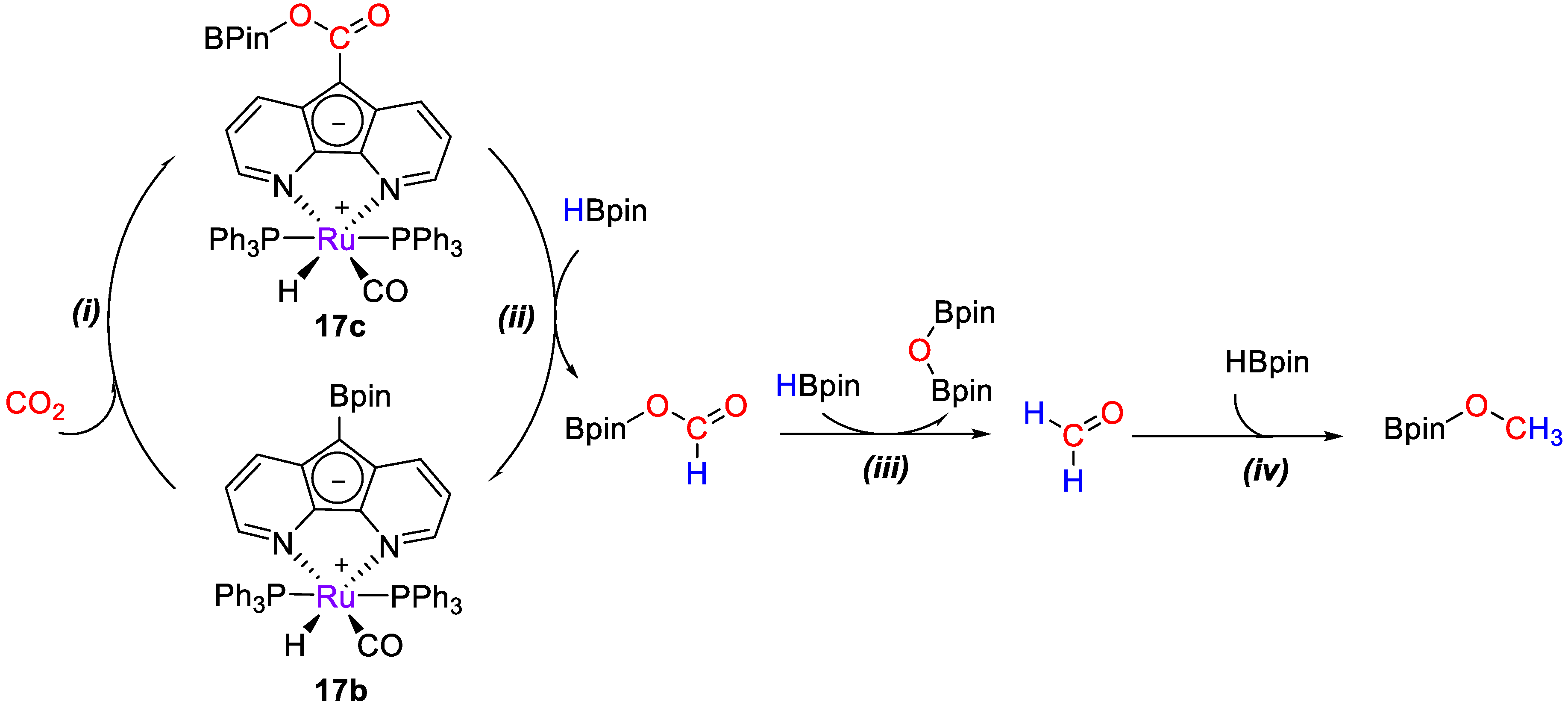{kind=link}
{kind=link}
{kind=link}
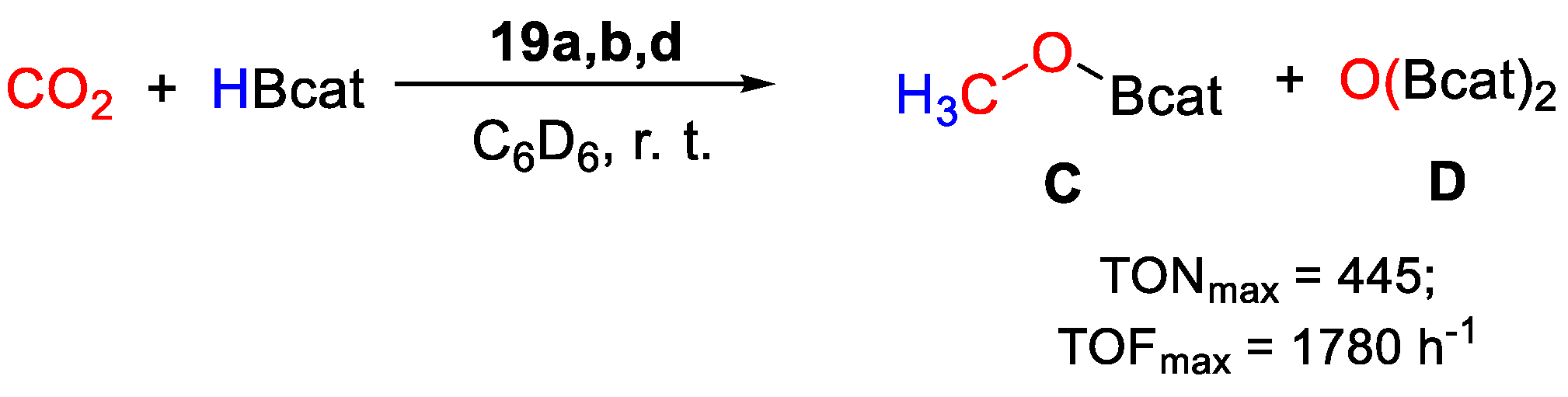{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Metal | Amount of Cat. (mol%) | Solvent | Temp. (°C) | Time | Additives | Type of Product(s) a (type of HBR2) | Best TON/TOF (h−1) | Ref |
|---|---|---|---|---|---|---|---|---|---|
| 1a | Li | 1 | THF | 25 | 10 h | - | A (HBpin) | -/10 | [26] |
| 1b | K | 1 | THF | 25 | 16 h | - | A (HBpin) | -/6.25 | [26] |
| 1c | Na | 1 | THF | 25 | 16 h | - | A (HBpin) | -/6.25 | [26] |
| 2 | Mg | 5 | - | 100 | 15 h | - | C, D (HBpin) | -/- | [27] |
| 3 | Mg | 10 | THF-d8 | 25 | 3 h | - | C, D (HBpin) | -/- | [28] |
| 4 | Si | 10 | C6D6 | 90 | 10 min | - | A, D (HBpin) | -/60 | [29] |
| 4 | Si | 10 | C6D6 | 90 | 30 min | - | C, E b (BH3·S(Me)2) | -/19.8 | [29] |
| 4 | Si | 10 | C6D6 | 90 | 24 h | - | C, D (HBcat) | -/0.34 | [29] |
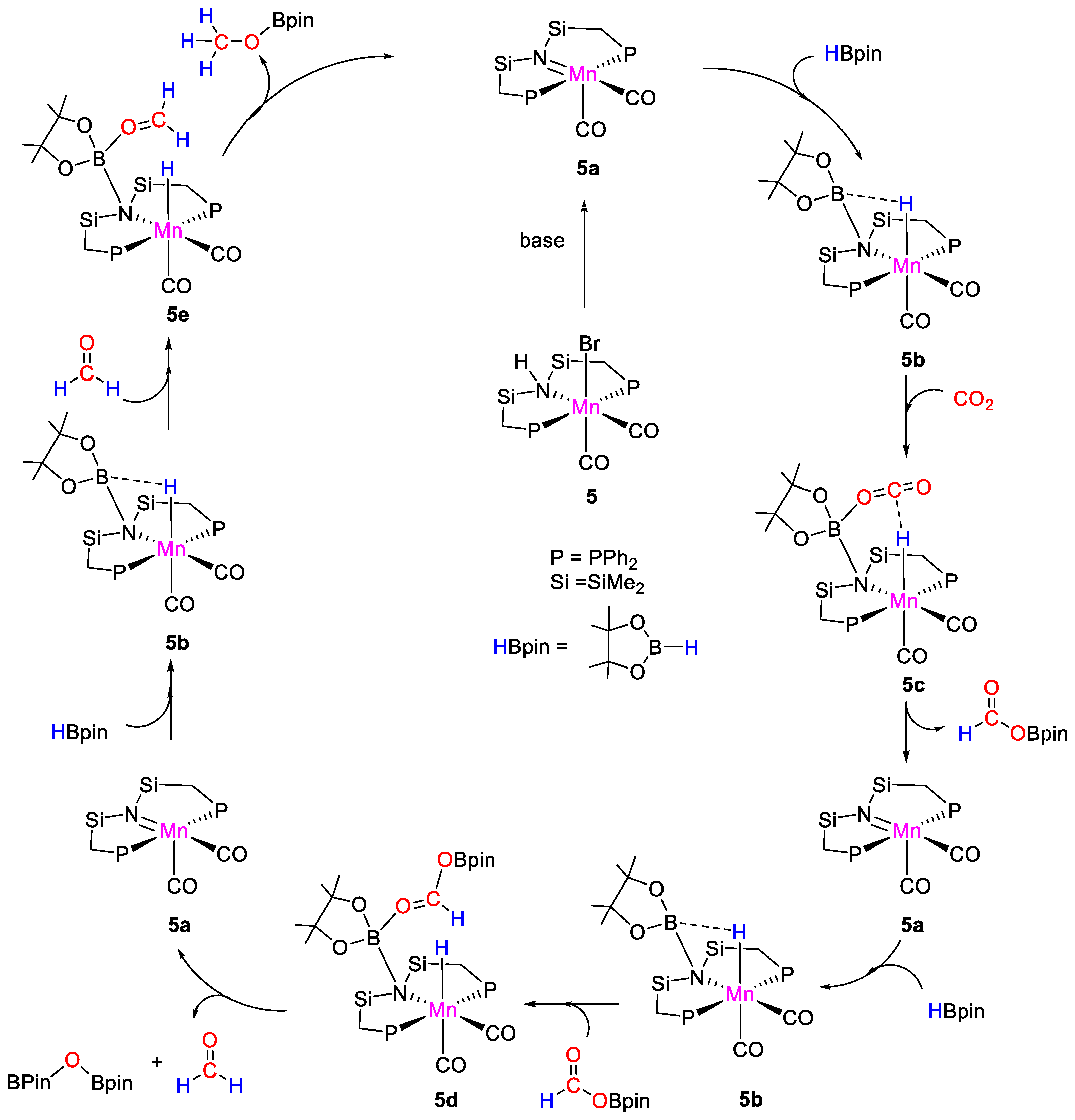
| 5 | Mn | 0.072 | - | 100 | 14 h | NaOtBu c | C, D (HBpin) | 883/- | [31] |
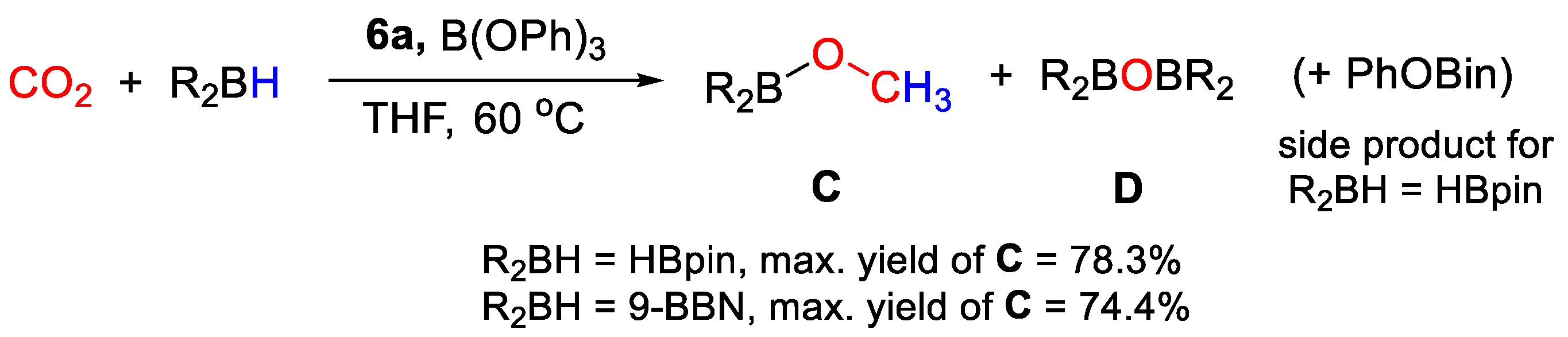
| 6a | Mn | 1 | THF-d8 | 60 | 24 h | B(OPh)3 d | C, D (HBpin) | -/- | [33] |
| 6a | Mn | 1 | THF-d8 | 60 | 24 h | B(OPh)3 d | C, D (9-BBN) | -/- | [33] |
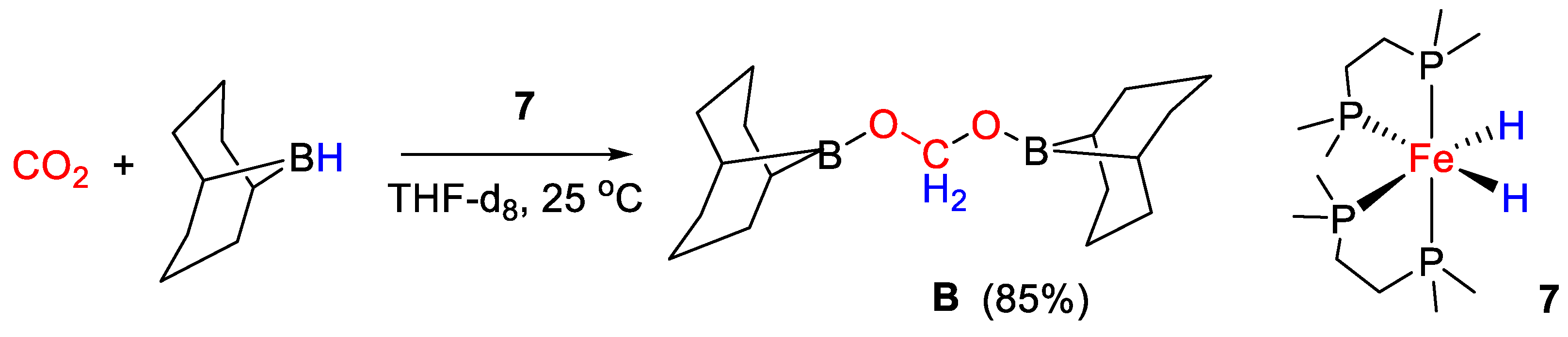
| 7 | Fe | 1 | CH3CN | 25 | 45 min | - | B (9-BBN) | -/- | [34] |
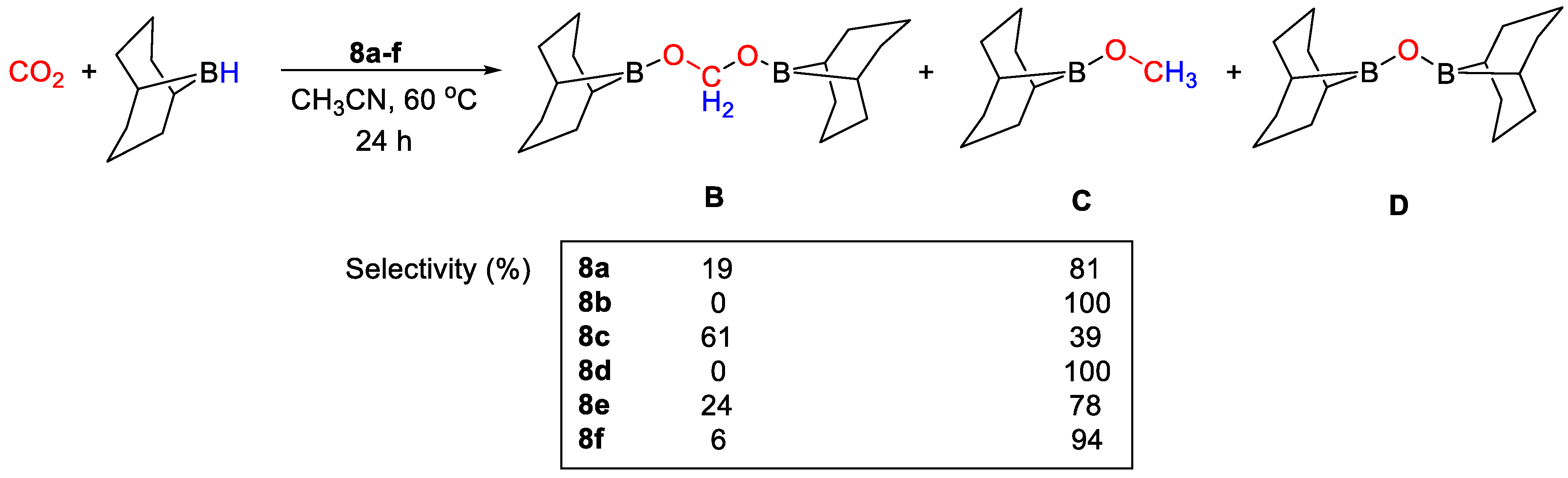
| 8a | Fe | 1.5 | CH3CN | 60 | 24–40 h | - | B, C (9-BBN) | 66/2.1 | [35] |
| 8c | Co | 1.5 | CH3CN | 60 | 24 h | - | B, C (9-BBN) | 66/2.8 | [35] |
| 8f | Cu | 1.5 | THF | 60 | 4–24 h | - | B, C (9-BBN) | 59/14.8 | [35] |
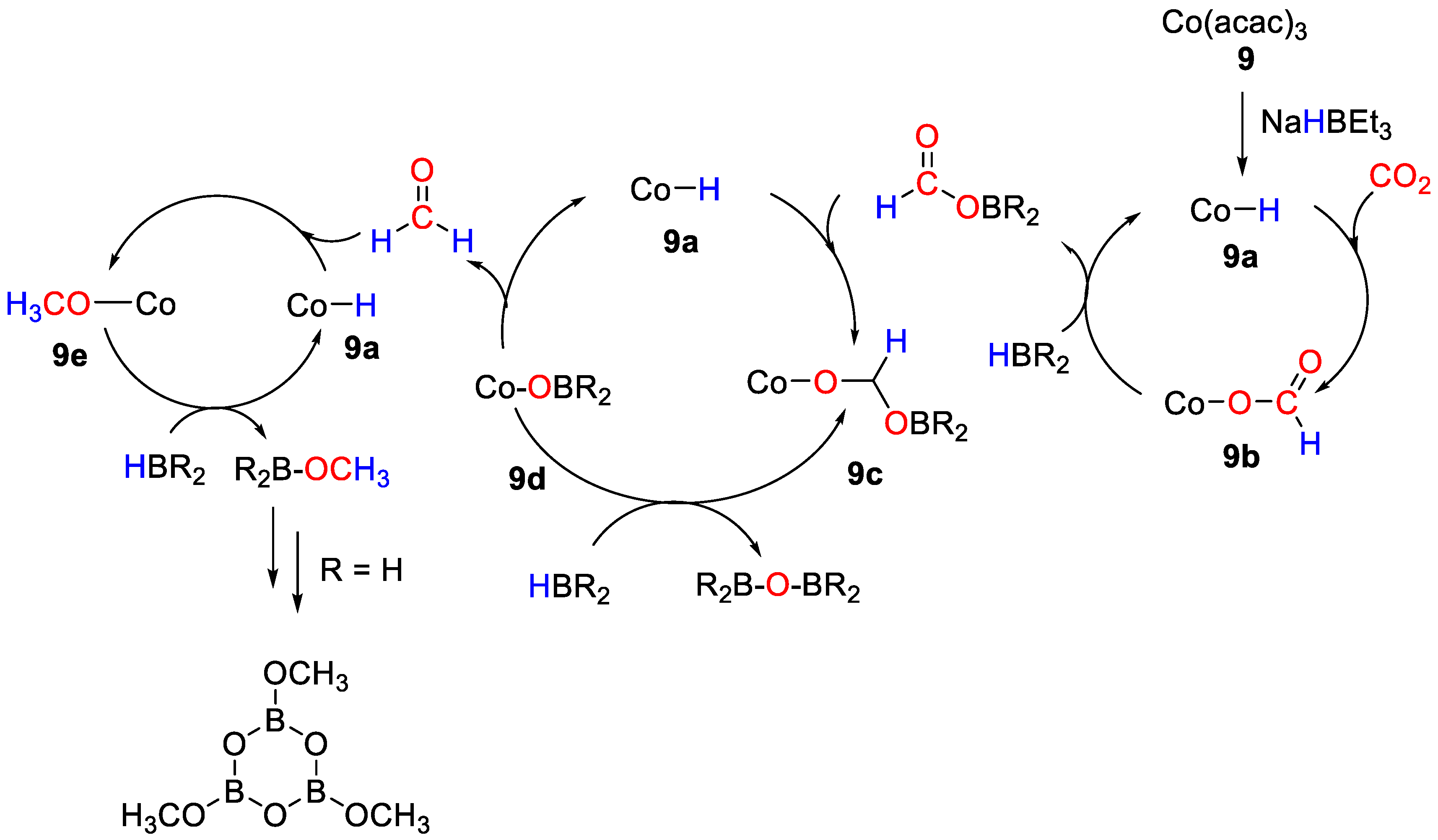
| 9 | Co | 1 | THF | 50 | 16 h | NaHBEt3 e | A, C, D (HBpin) | -/- | [36] |
| 9 | Co | 1 | THF | 50 | 20 h | NaHBEt3 e | E b (BH3·S(Me)2) | 300/15 | [36] |
| 9 | Co | 1 | C6D6 | 50 | 72 h | NaHBEt3 e | C, D (HBcat) | -/- | [36] |
| 10a | Ni | 0.2 | C6D6 | 25 | 15 min | - | C, D (HBcat) | 490/2400 | [38] |
| 10b | Ni | 0.2 | C6D6 | 25 | 15 min | - | C, D (HBcat) | 477/ 1908 | [37] |
| 11b | Ni | 0.2 | C6D6 | 25 | 4 h | - | B (HBpin) | 487/- | [39] |
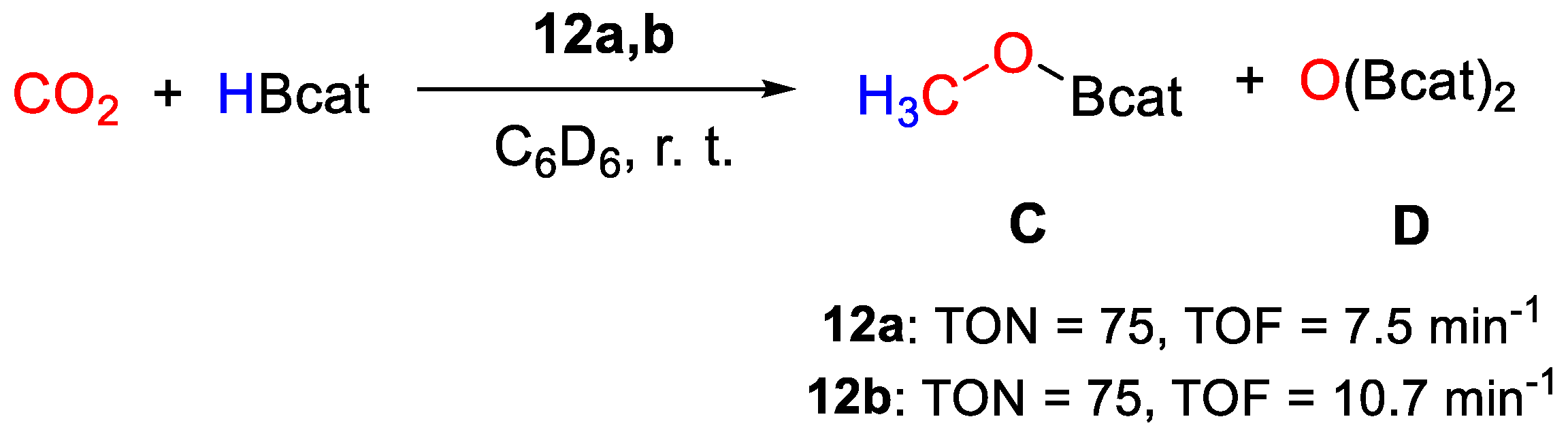
| 12a | Ni | 0.5 | C6D6 | 25 | 10 min | - | C, D (HBcat) | 75/7.5 | [40] |
| 12b | Ni | 0.25 | THF-d8 | 25 | 7 min | - | C, D (HBcat) | 75/10.7 | [40] |
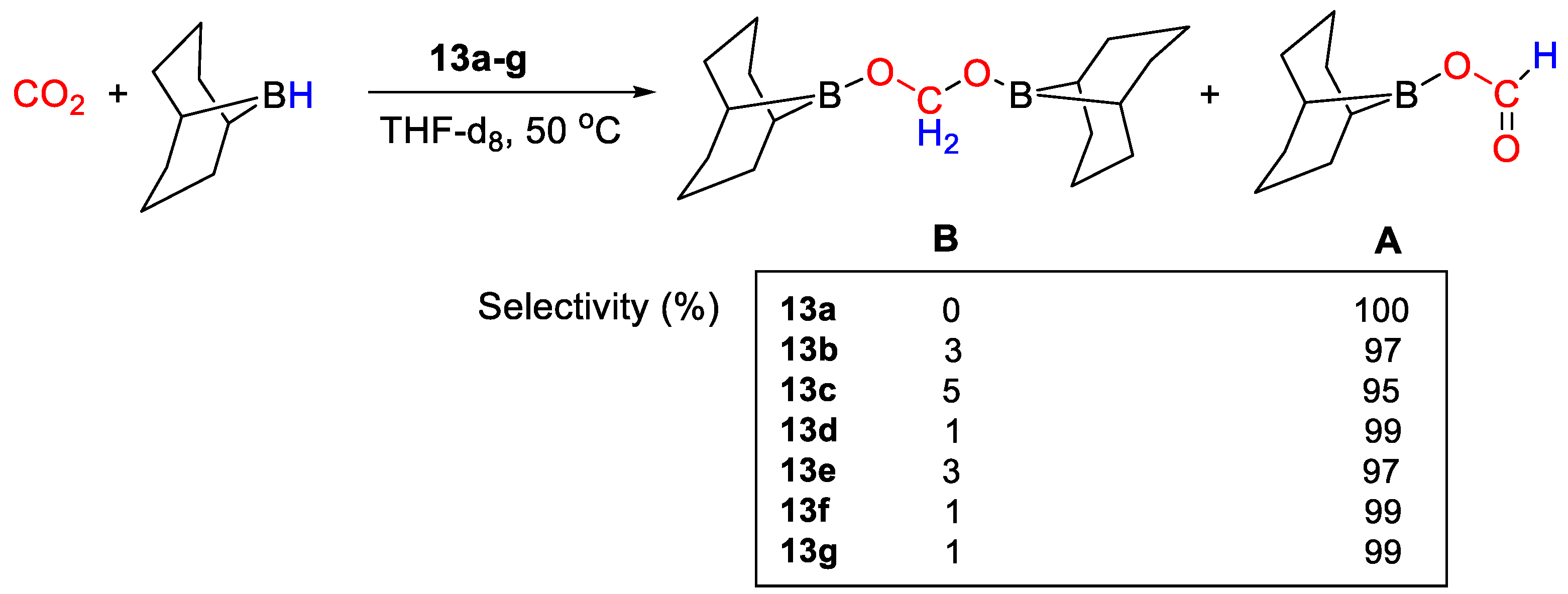
| 13g | Cu | 2 | THF-d8 | 50 | 18 h | - | A, B (9-BBN) | 31/- | [41] |
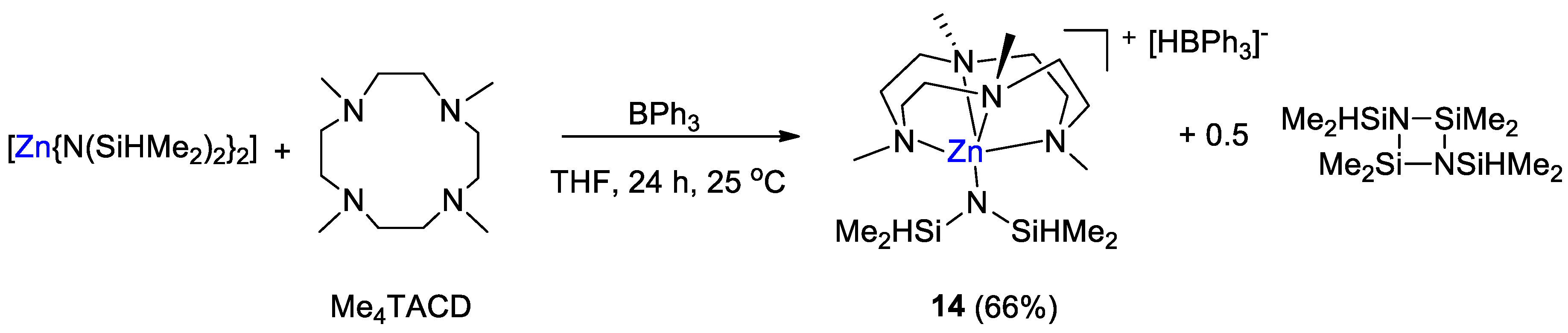
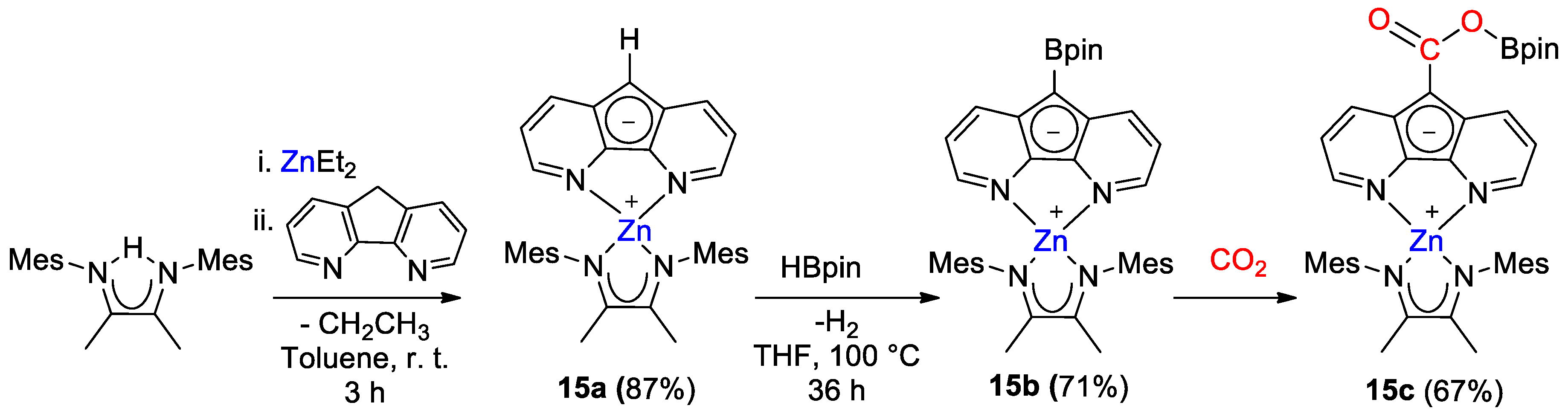
| 14 | Zn | 10 | THF/THF-d8 | 60 | 16 h | - | C, D (HBpin) | -/- | [42] |
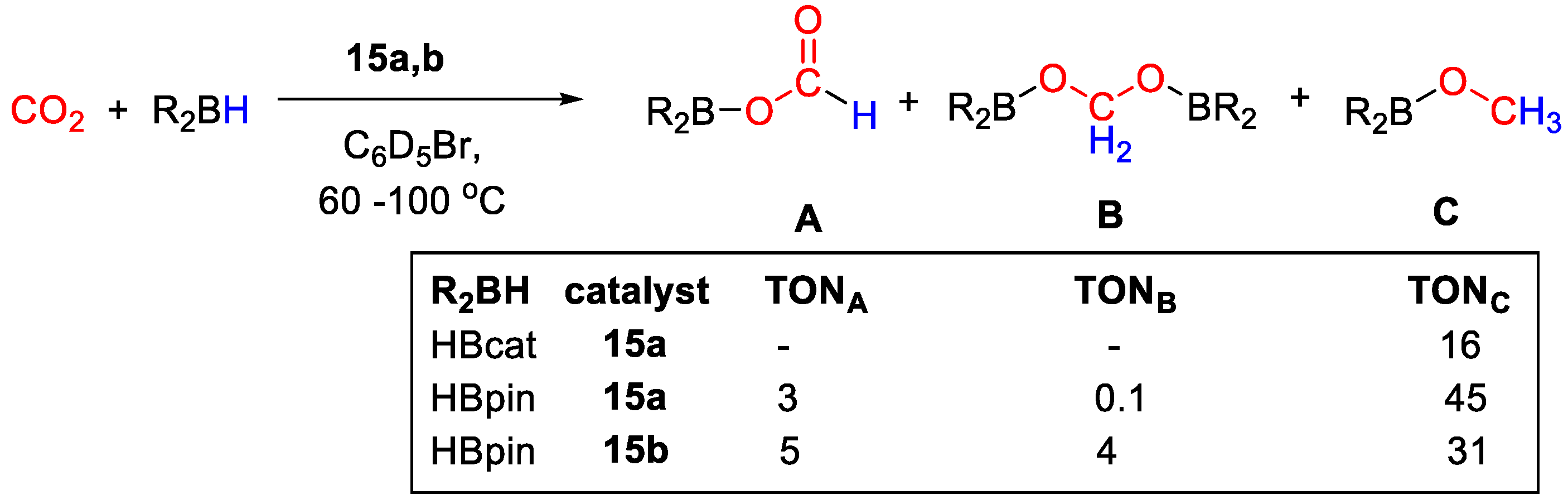
| 15a | Zn | 10 | C6D5Br | 90 | 20 h | - | A, B, C (HBpin) | 45/- | [43] |
| 15b | Zn | 10 | C6D5Br | 60 | 20 h | - | C (HBcat) | 16/- | [43] |
| 16a | Zn | 5 | C6D6 | 25 | 12 h | - | A (HBpin) | -/- | [44] |
| 16a | Zn | 5 | C6D6 | 25 | 4 h | - | B, C (9-BBN) | -/- | [44] |
| 16a | Zn | 5 | C6D6 | 25 | 12 h | - | C (HBcat) | -/- | [44] |
| 17a | Ru | 10 | C6D5Br | 90 | 45 h | - | C (HBcat) | 29/- | [43] |
| 17a | Ru | 10 | C6D5Br | 90 | 45 h | - | A, C (HBpin) | 39/- | [43] |
| 17b | Ru | 10 | C6D5Br | 100 | 45 h | - | A, B, C (HBpin) | 60/- | [43] |
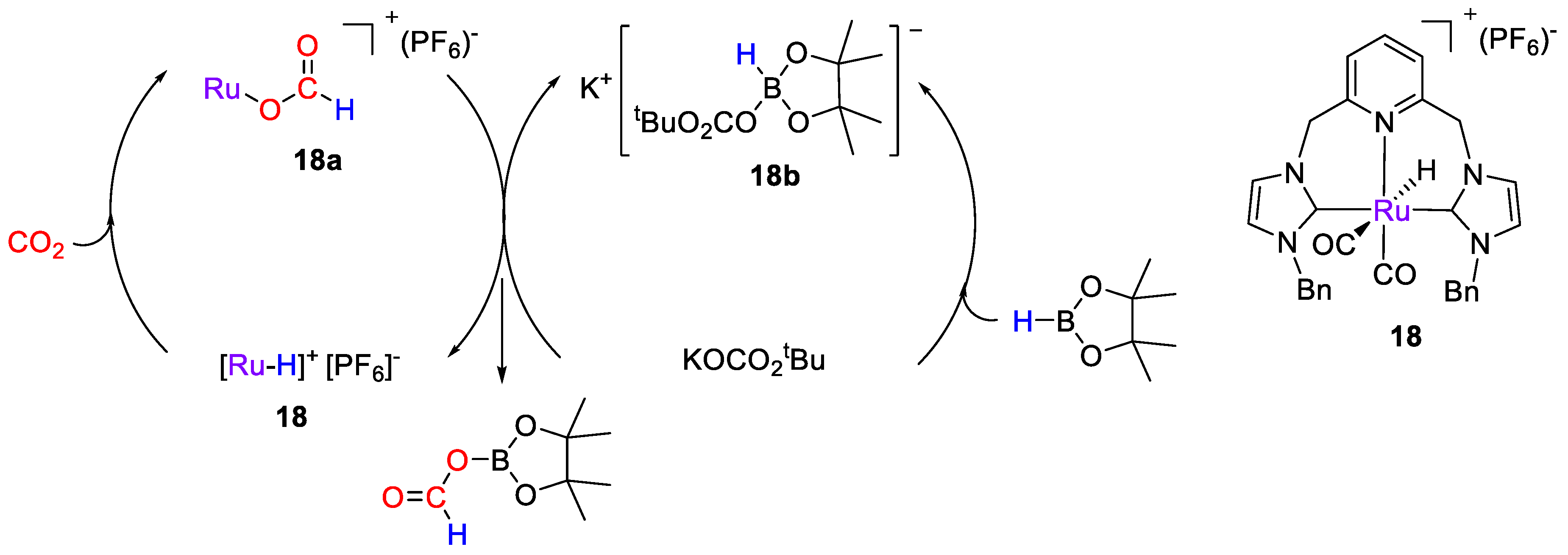
| 18 | Ru | 1 | CD2Cl2 | 25 | 30 min | KOCO2tBu f | A, D (HBpin) | -/- | [46] |
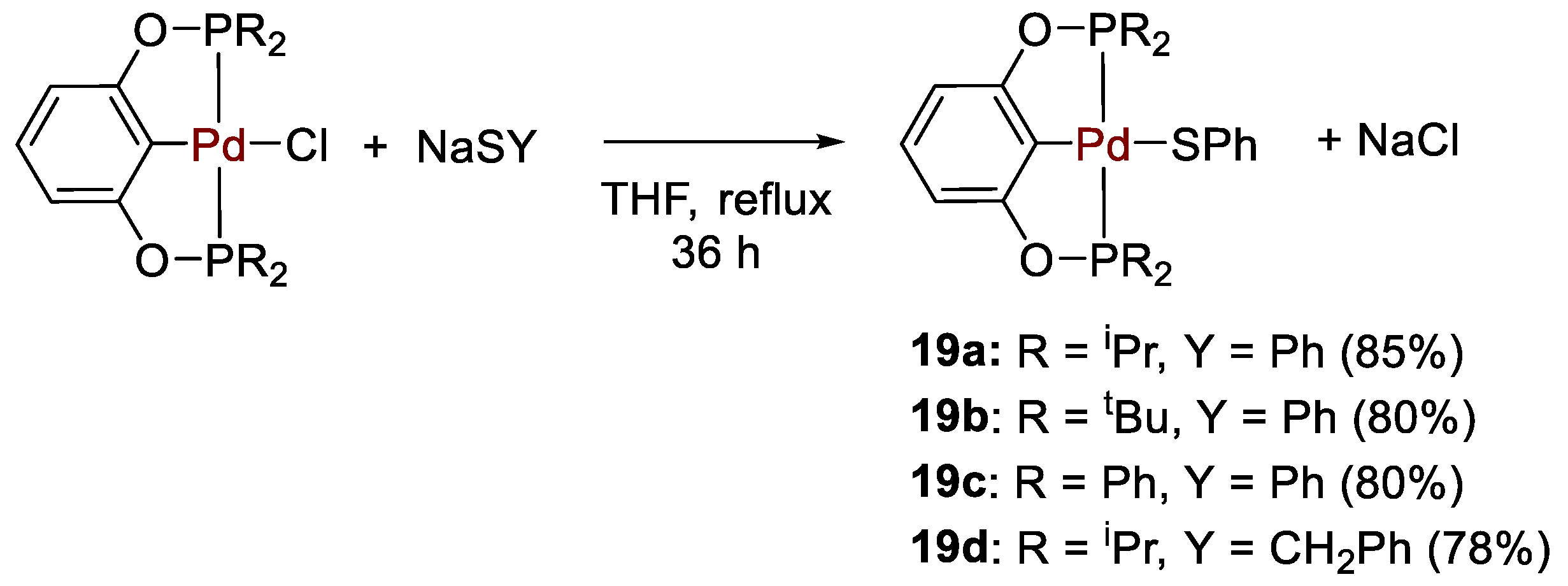
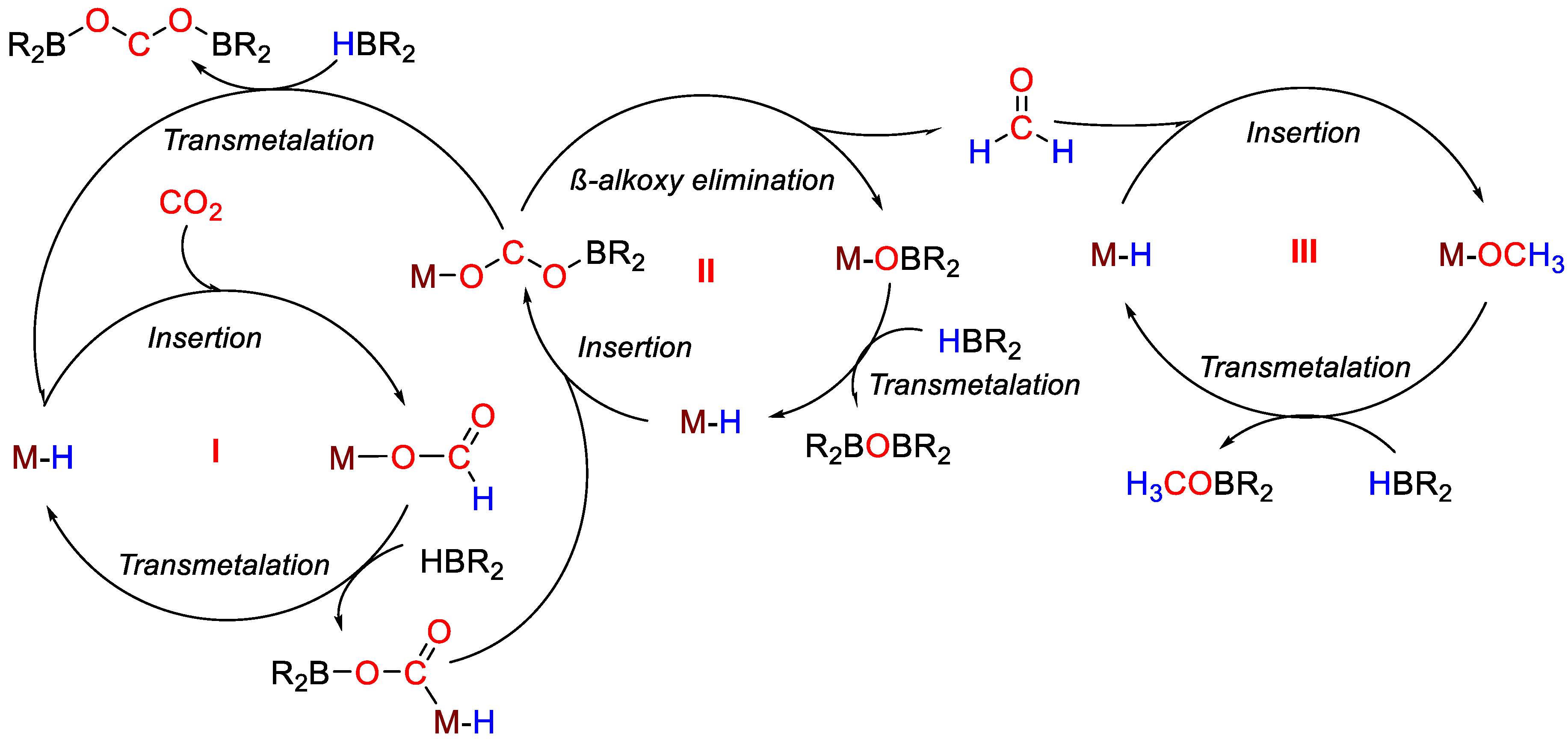
| 19 | Pd | 0.2 | C6D6 | 25 | 15 min | - | C, D (HBcat) | 445/1780 | [47] |
| 20a–j | Pd, Ni | 1 | C6D6 | 25 | 10 min–10 days | B(OPh)3 d | A, B, C (HBpin, HBcat, 9-BBN) | -/- | [48] |
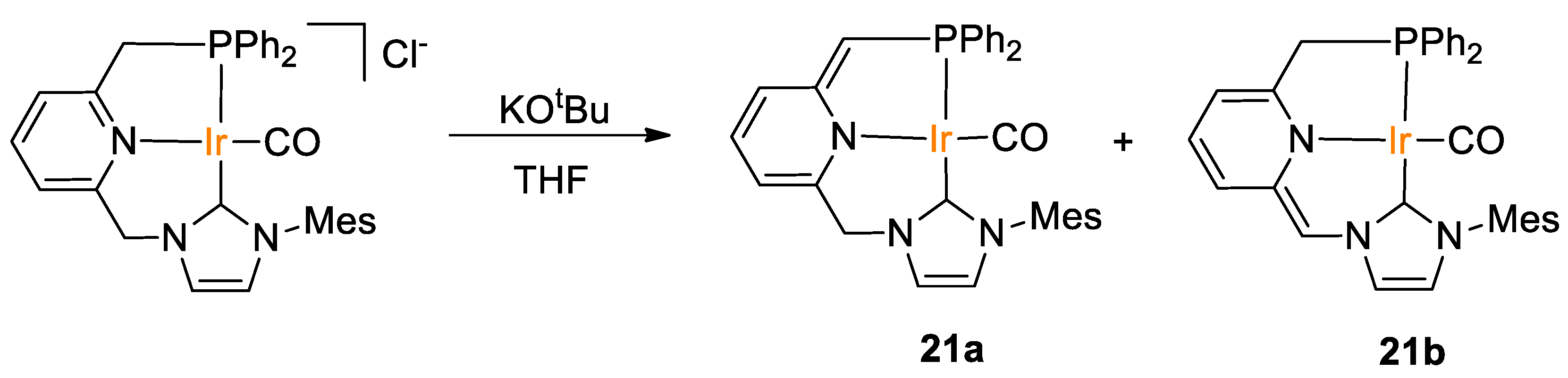
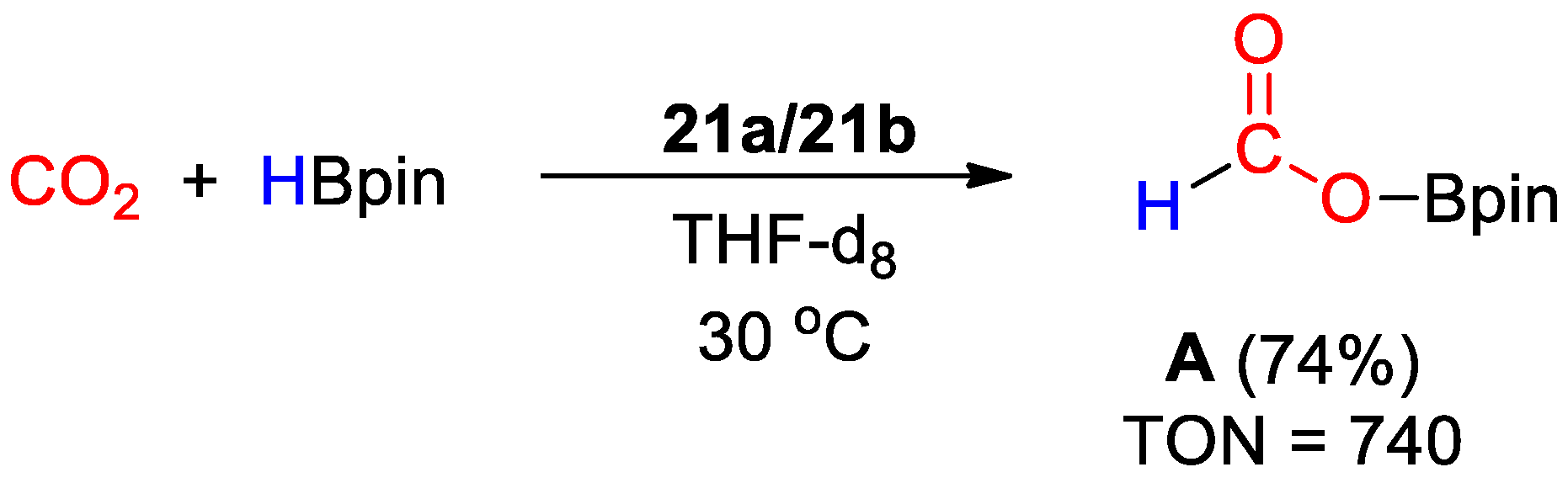
| 21a,b | Ir | 0.2 | THF-d8 | 30 | 20 min | H2O g | A (HBpin) | 740/1245 | [49] |
| 21a,b | Ir | 0.2 | THF-d8 | 30 | 20 min | H2O g | C (HBcat) | 78/56 | [49] |
| Catalyst | Solvent | 1H NMR δ (ppm) | 31P NMR δ (ppm) | 11B NMR δ (ppm) | 29Si NMR δ (ppm) | Ref. |
|---|---|---|---|---|---|---|
| 1a | THF-d8 | - | - | −8.2 (d, JBH= 79 Hz) | - | [26] |
| 1b | THF-d8 | - | - | −8.0 (d, JBH = 76 Hz) | - | [26] |
| 1c | THF-d8 | - | - | −8.2 (d, JBH = 76Hz) | - | [26] |
| 3 | DMSO-d6 | - | - | −8.3 (d, JBH = 80 Hz) | - | [28] |
| 4 | Pyridine-d5 | 9.73 (s, SiH) | - | - | −77.9 (d, 1JSiH = 283 Hz) | [30] |
| 5 | THF-d8 | - | 48.5 | - | - | [31] |
| 6a | C6D6 | −5.72 (t, JHP = 51.4 Hz, MnH) | 164.8 | - | - | [32] |
| 7 | C6D6 | −14.33 | 83.7, 74.1 | - | - | [50] |
| 8a | CD3CN | - | 30.2 | - | - | [35] |
| 8b | CD3CN | - | 147.9 | - | - | [35] |
| 8e | CD2Cl2 | - | −33 (m, JPCu∼840 Hz); | - | - | [35] |
| 8f | CD2Cl2 | - | 80 (m, JPCu∼860 Hz) | - | - | [35] |
| 10b | CDCl3 | - | 185.2 | - | - | [51] |
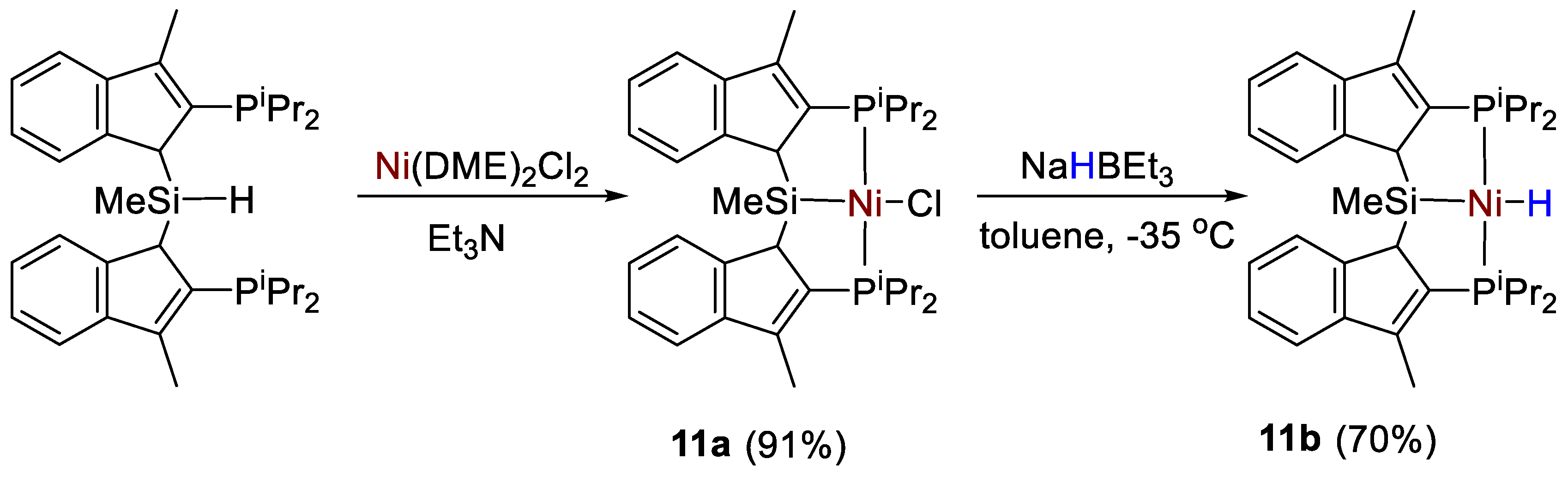
| 11a | C6D6 | - | 36.0 | - | 43.8 | [39] |
| 11b | C6D6 | −4.79 (brs, NiH) | 65.6 | - | 59.8 | [39] |
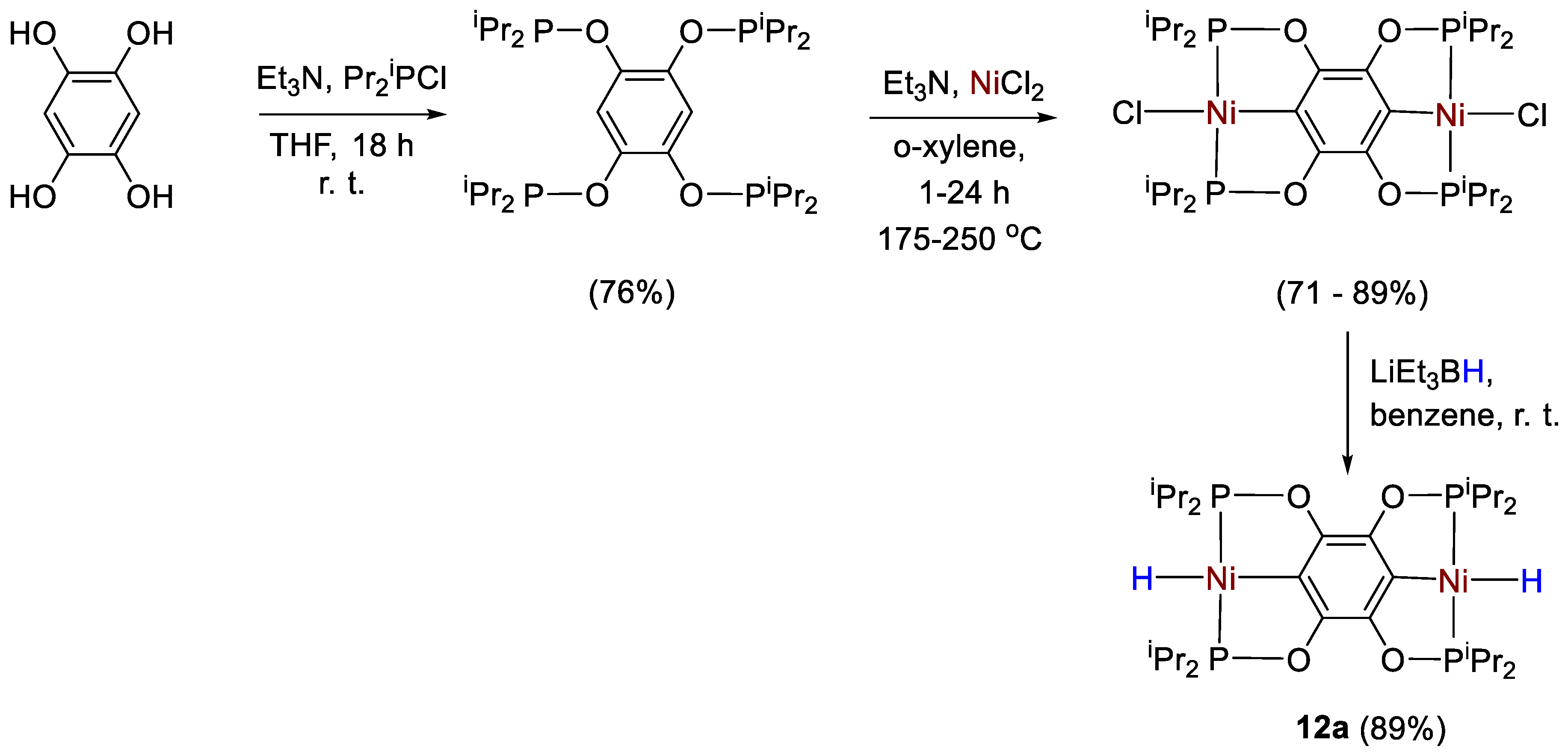
| 12a | C6D6 | −8.25 (t, JHP = 55.6 Hz, Ni-H) | 205.7 | - | - | [40] |
| 12b | C7D8 | −7.90 (t, JHP = 55.2 Hz, Ni-H) | 206.6 | - | - | [40] |
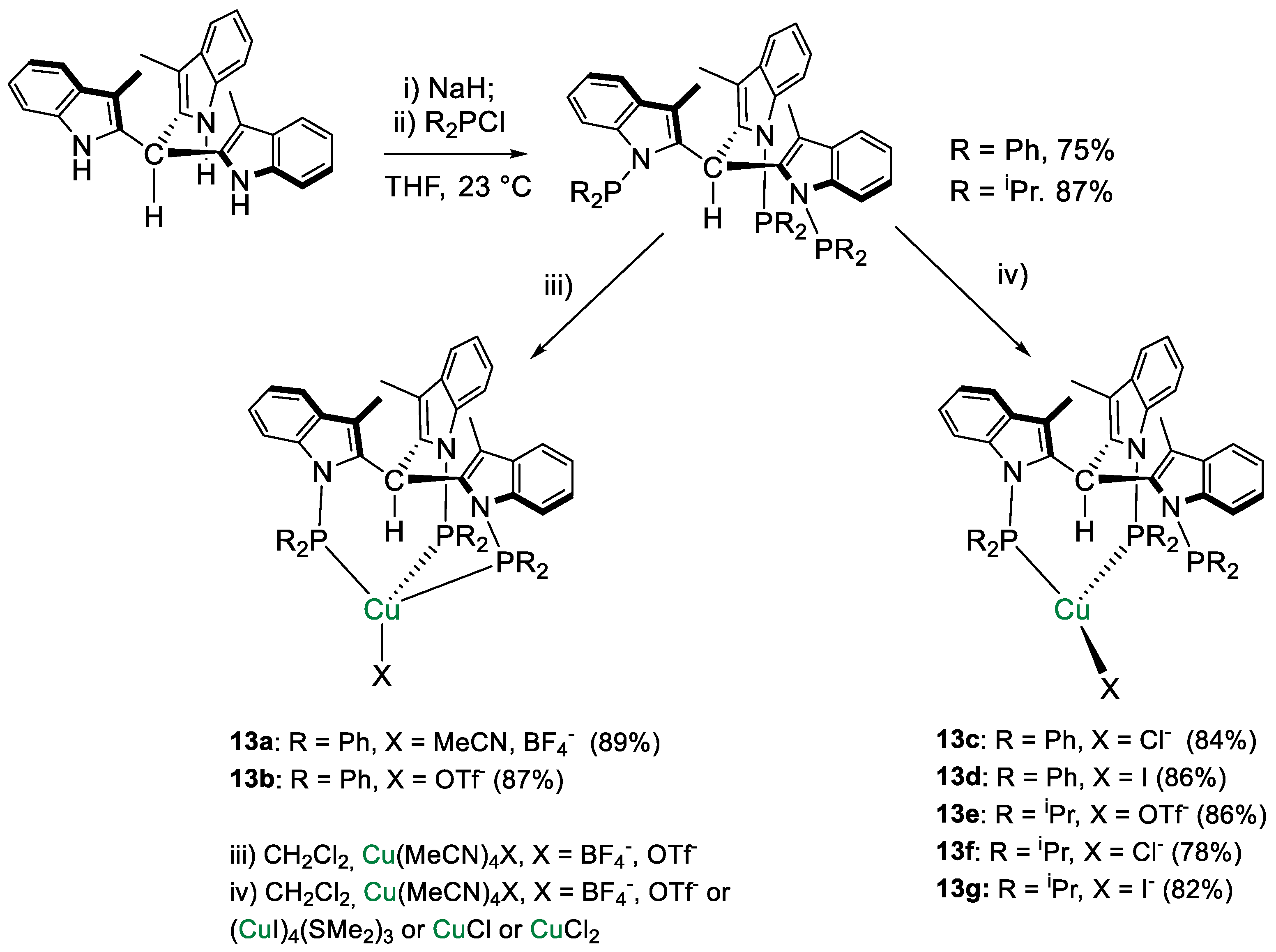
| 13a | CD2Cl2 | - | 31.4 | - | - | [41] |
| 13b | CD2Cl2 | - | 31.8 | - | - | [41] |
| 13c | CD2Cl2 | - | 29.7 | - | - | [41] |
| 13d | CD2Cl2 | - | 25.9 | - | - | [41] |
| 13e | CD2Cl2 | - | 64.1 | - | - | [41] |
| 13f | CD2Cl2 | - | 62.8 | - | - | [41] |
| 13g | CD2Cl2 | - | 61.9 | - | - | [41] |
| 14 | THF-d8 | - | - | −7.9 (d, 1JBH = 79 Hz) | −15.1 | [42] |
| 15b | C6D6 | - | - | 31.40 | - | [43] |
| 16a | C7D8 | 4.15 (s), 3.50 (s) | −20.0 | - | - | [44] |
| 16b | C7D8 | 4.32 (s), 3.46 (s) | −19.5 | - | - | [44] |
| 17a | C6D6 | - | 48.6 | - | - | [43] |
| 17b | C6D6 | - | 48.5 | 31.30 | - | [43] |
| 18 | DMSO-d6 | −4.99 (s, Ru-H); | - | - | - | [46] |
| 19a | C6D6 | - | 189.7 | - | - | [47] |
| 19b | C6D6 | - | 192.4 | - | - | [47] |
| 19c | C6D6 | - | 147.8 | - | - | [47] |
| 19d | C6D6 | - | 191.4 | - | - | [47] |
| 20a | C6D6 | −9.9 (t, 2JHP = 55.5 Hz, Ni-H); | 66.9 | - | - | [52] |
| 20b | C6D6 | −10.0 (t, 2JHP = 52.8 Hz, NiH) | 99.8 | - | - | [52] |
| 20c | C6D6 | −9.9 (t, 2JHP = 55.6 Hz, Ni-H) | 78.2 | - | - | [52] |
| 20d | C6D6 | −3.50 (t, 2JHP = 46 Hz, Ni−H) | 78.5 | - | - | [53] |
| 20g | C6D6 | −3.86 (t, 2JHP = 13.5 Hz, Pd-H) | - | - | - | [54] |
| 20h | CDCl3 | −3.77 (t, 2JHP = 17.0 Hz, Pd-H) | 71.7 | - | - | [55] |
| 20i | C6D6 | 1.28 | 81.0 | - | 62.5 (t, 3JSiH = 47 Hz) | [53] |
| 20j | C6D6 | 1.01 (t, 2JHP = 22.6 Hz, Pd-H) | 88.3 | - | 60.8 (t, 3JSiH = 6.7 Hz) | [56] |
| 21a | THF-d8 | - | 28.3 | - | - | [49] |
| 21b | THF-d8 | - | 33.3 | - | - | [49] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kostera, S.; Peruzzini, M.; Gonsalvi, L. Recent Advances in Metal Catalyst Design for CO2 Hydroboration to C1 Derivatives. Catalysts 2021, 11, 58. https://doi.org/10.3390/catal11010058
Kostera S, Peruzzini M, Gonsalvi L. Recent Advances in Metal Catalyst Design for CO2 Hydroboration to C1 Derivatives. Catalysts. 2021; 11(1):58. https://doi.org/10.3390/catal11010058
Chicago/Turabian StyleKostera, Sylwia, Maurizio Peruzzini, and Luca Gonsalvi. 2021. "Recent Advances in Metal Catalyst Design for CO2 Hydroboration to C1 Derivatives" Catalysts 11, no. 1: 58. https://doi.org/10.3390/catal11010058
APA StyleKostera, S., Peruzzini, M., & Gonsalvi, L. (2021). Recent Advances in Metal Catalyst Design for CO2 Hydroboration to C1 Derivatives. Catalysts, 11(1), 58. https://doi.org/10.3390/catal11010058