Glucose–Carbon Hybrids as Pt Catalyst Supports for the Continuous Furfural Hydroconversion in Gas Phase

 ,
,  ,
, 

Abstract
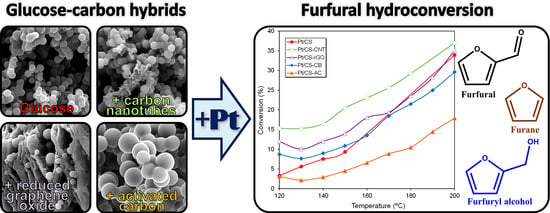
1. Introduction
2. Results and Discussion
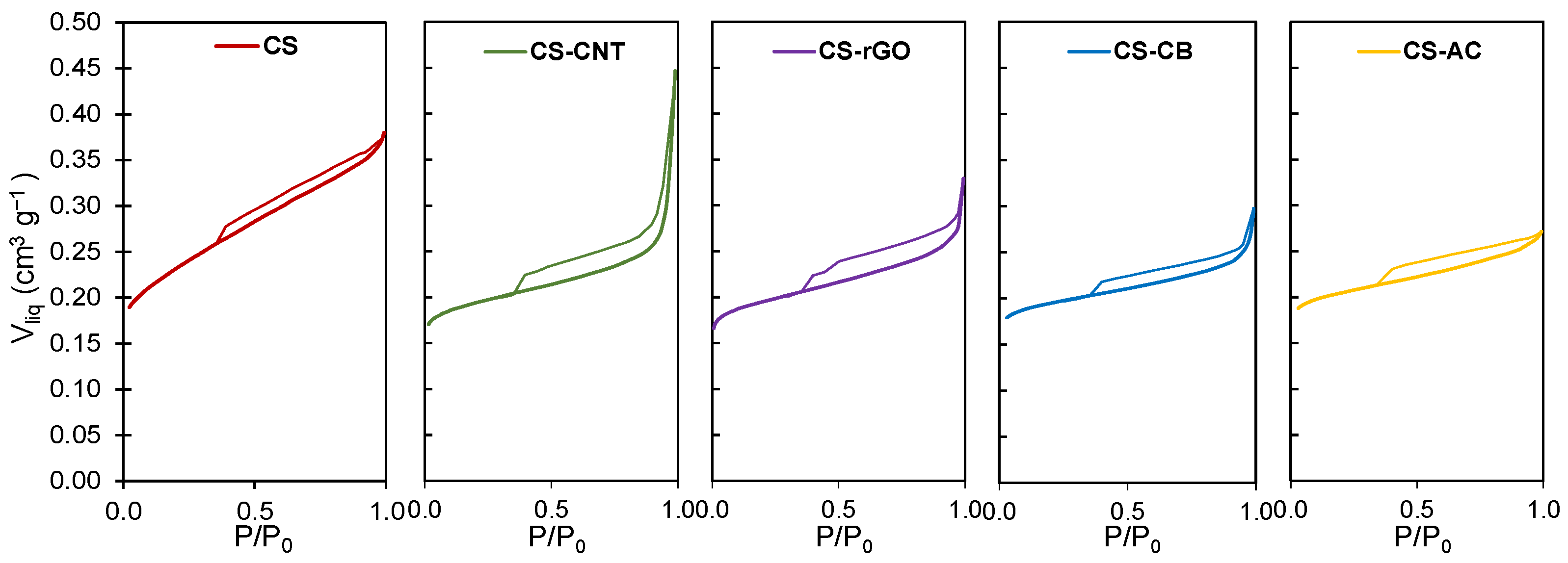
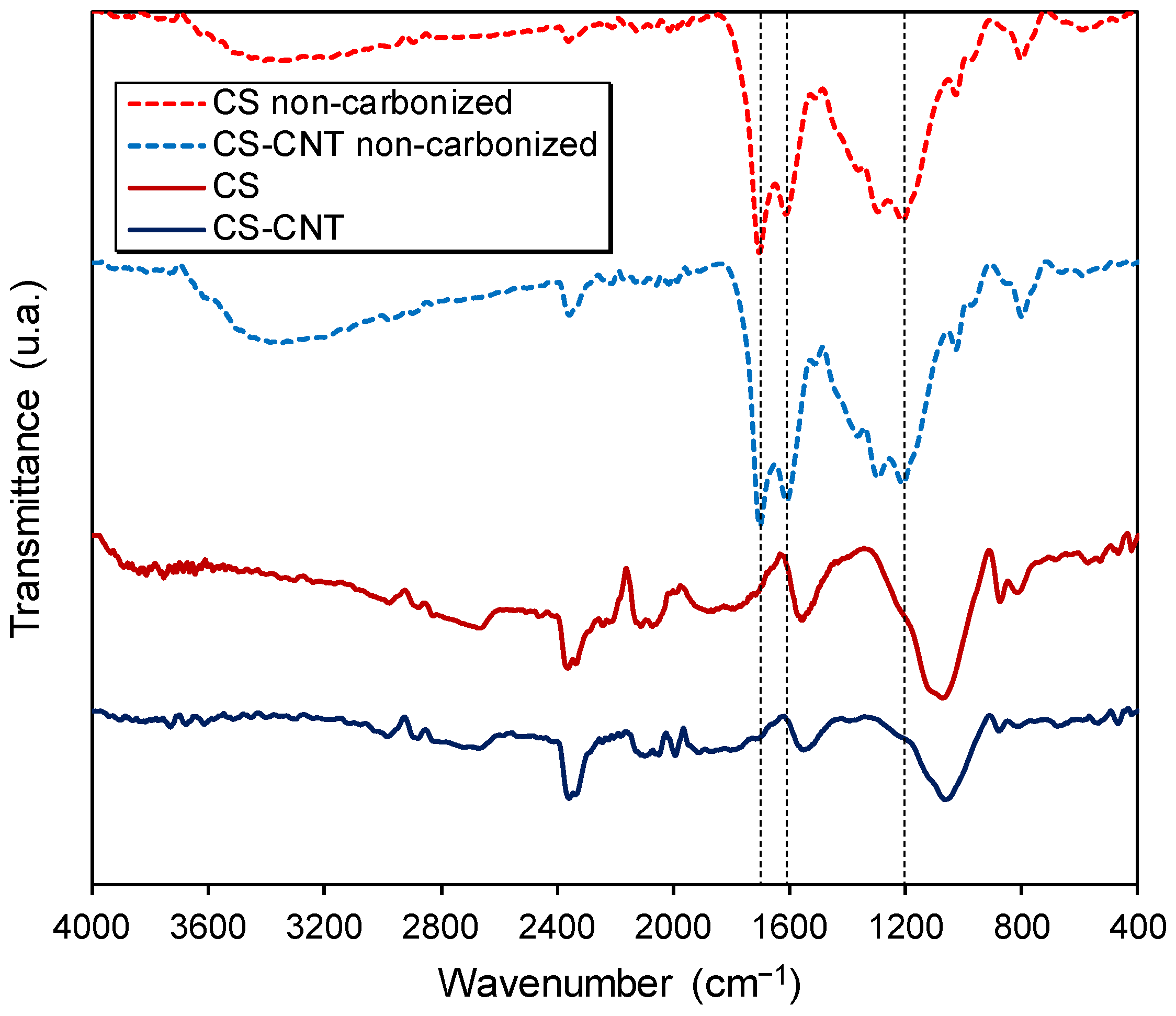
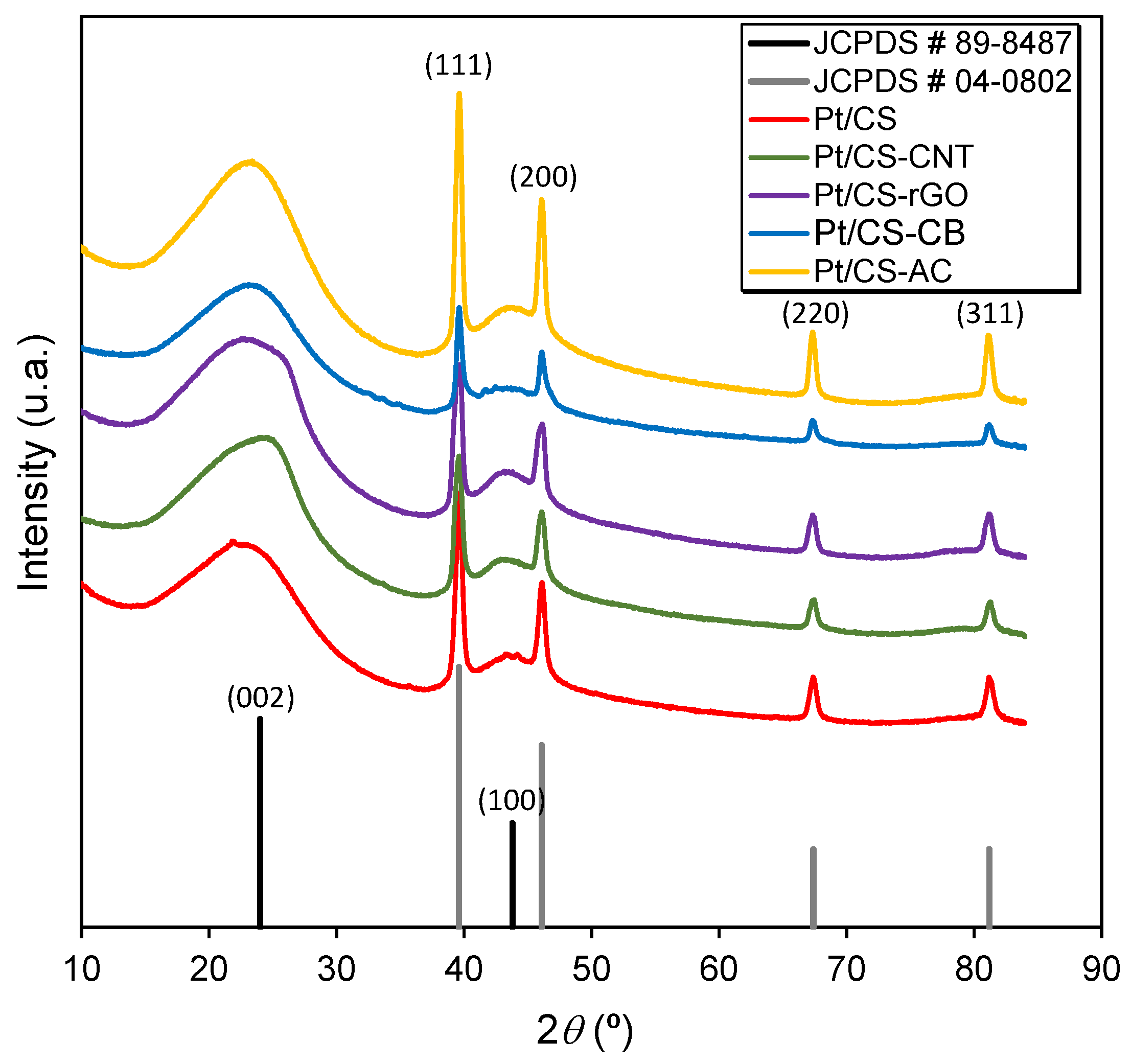
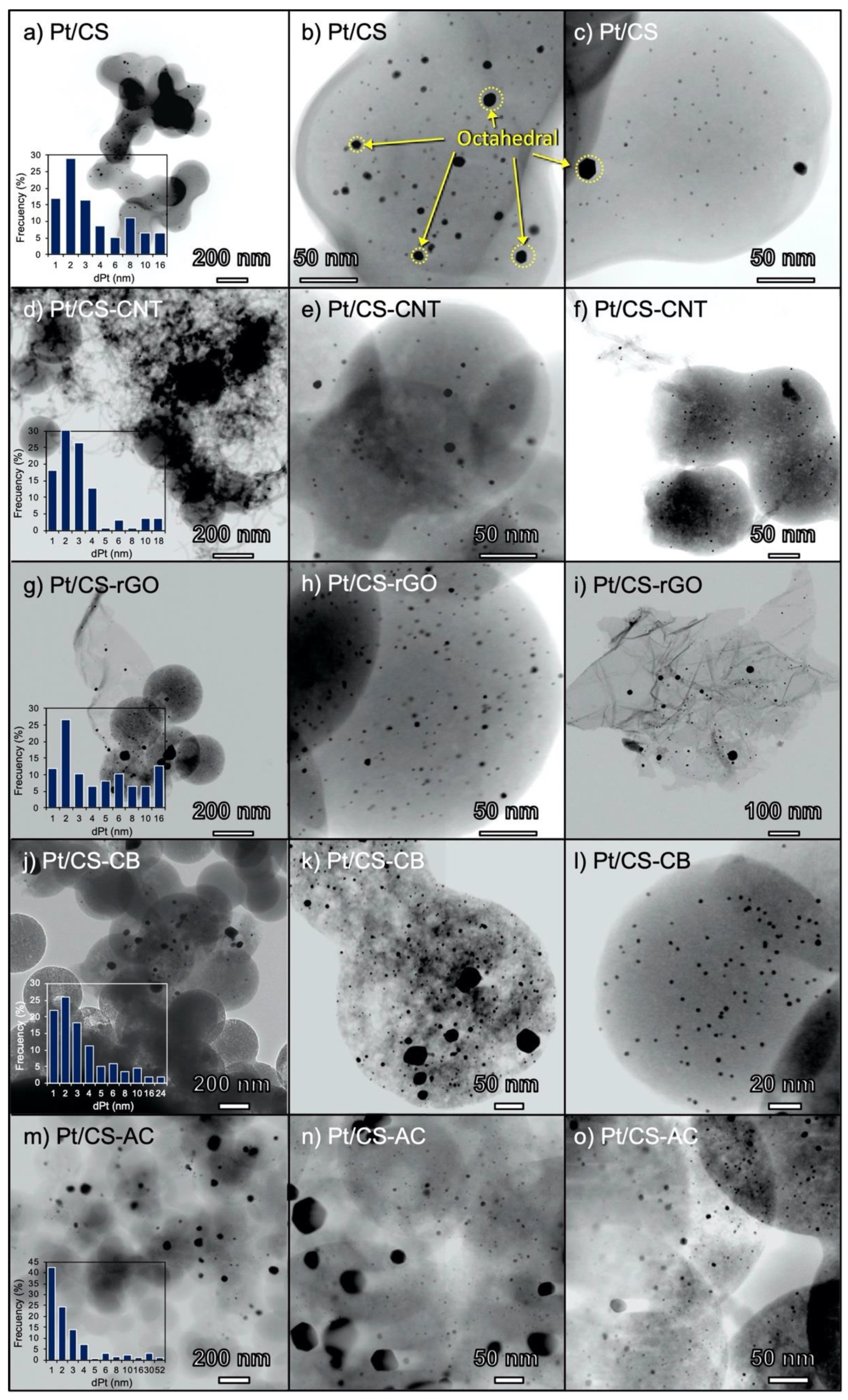
2.1. Characterization of the Carbon Supports and Pt/Carbon Catalysts
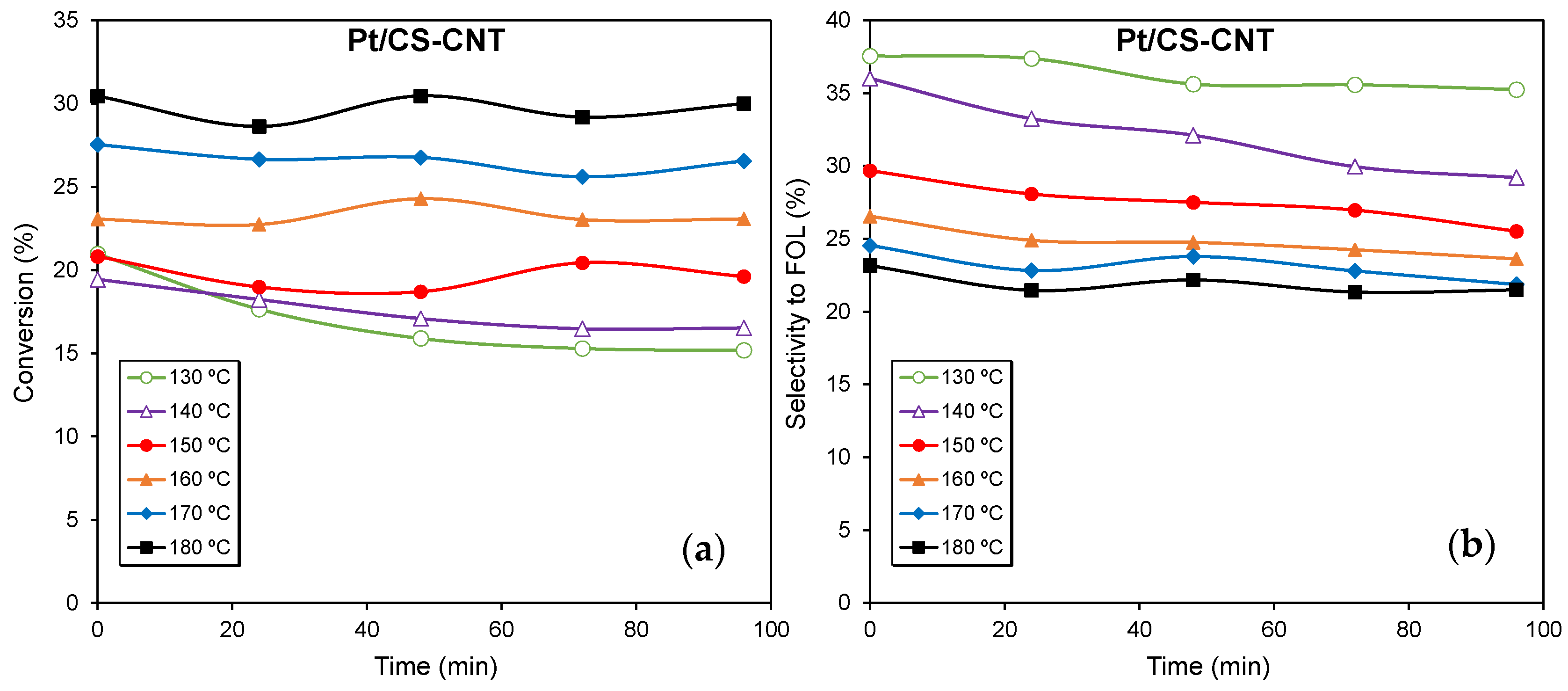
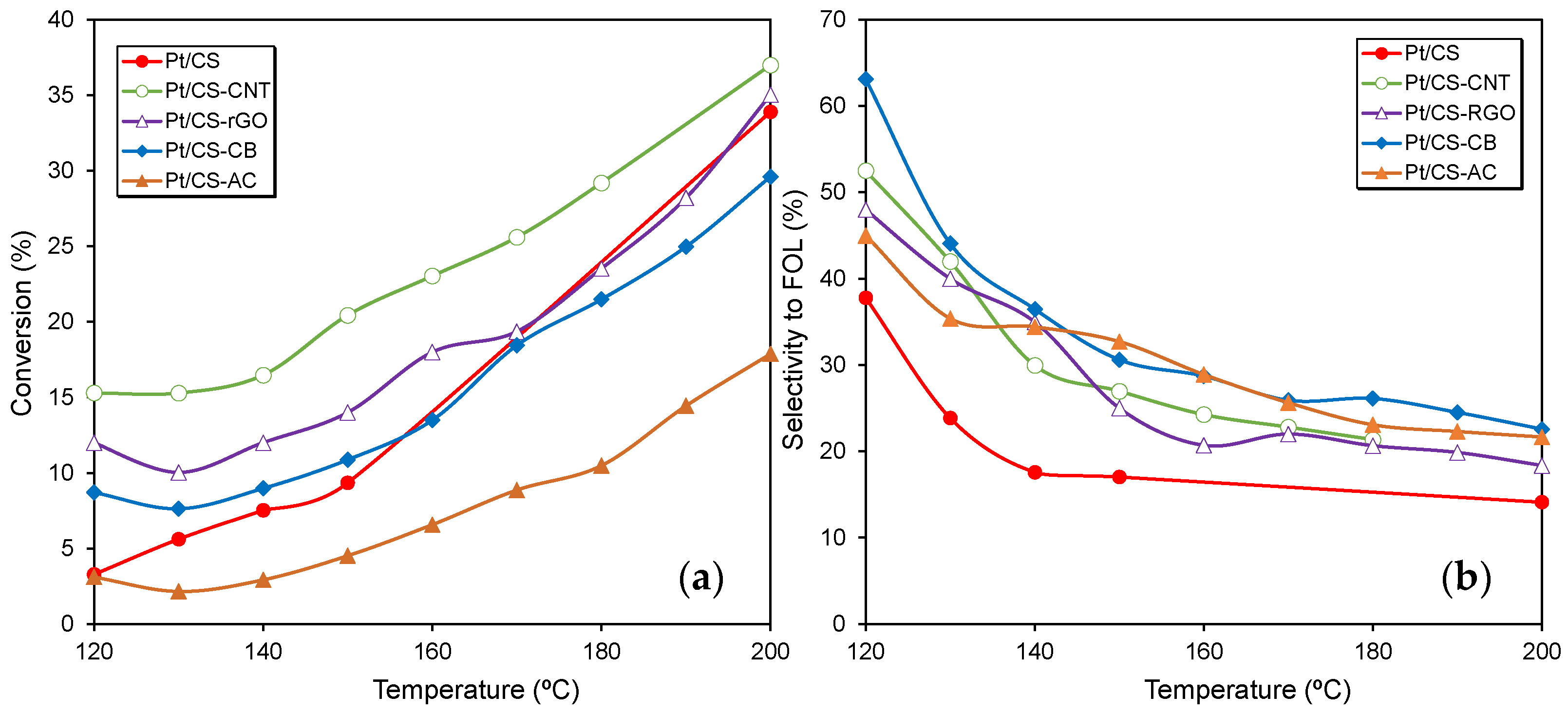
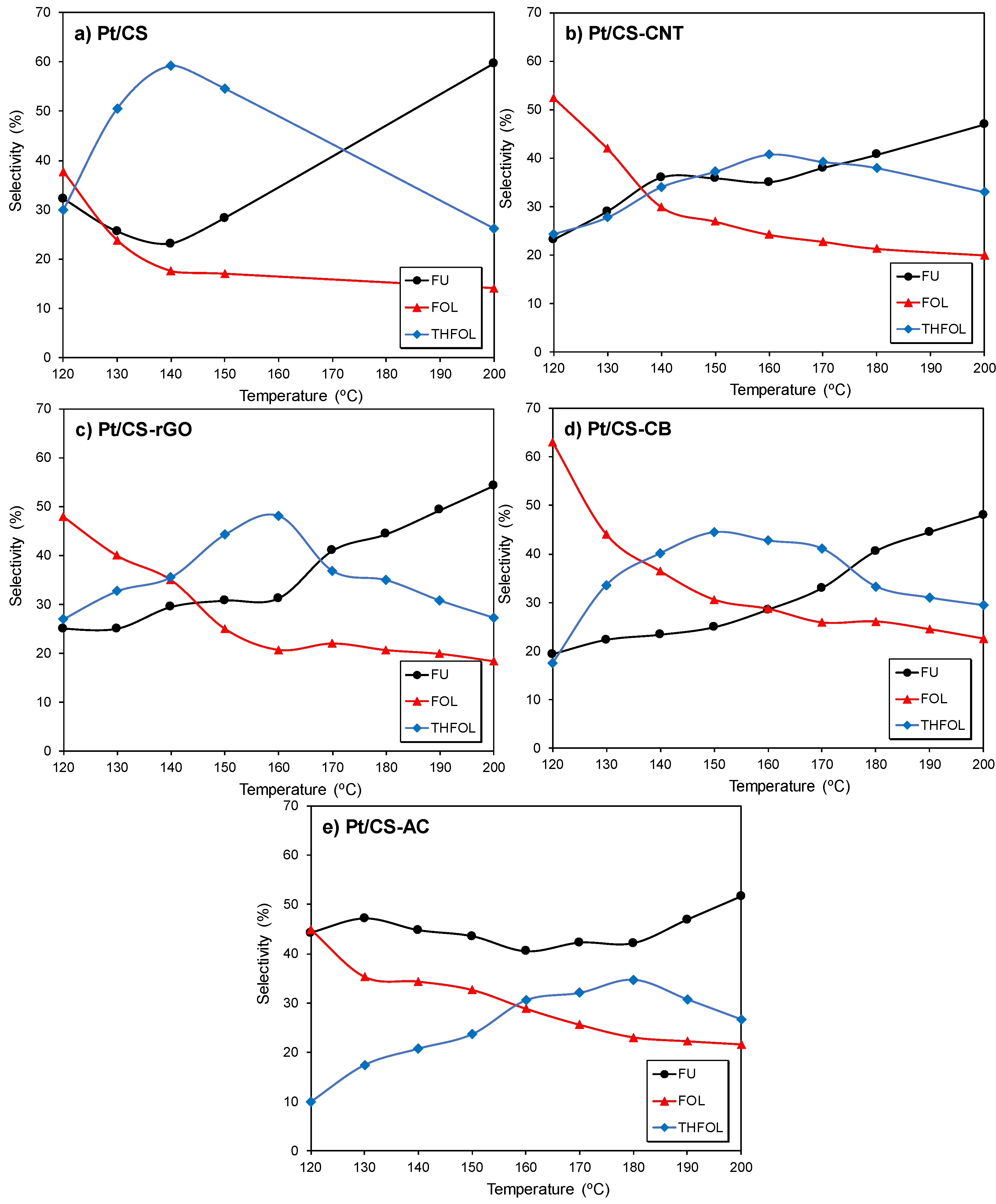
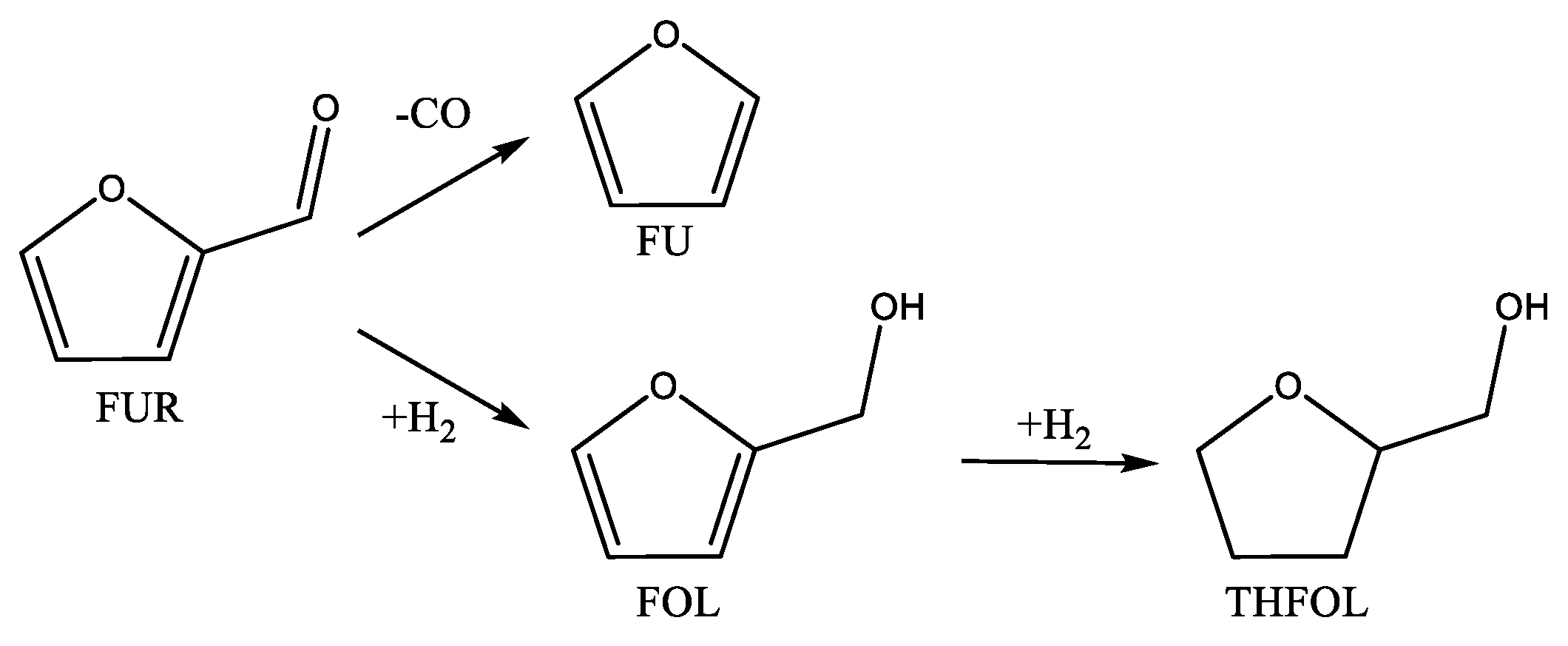
2.2. Catalytic Performance
3. Materials and Methods
3.1. Preparation of Glucose–Carbon Hybrids and Pt/Carbon Catalysts
3.2. Materials Characterisation
3.3. Catalytic Performance and Products Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Opia, A.C.; Hamid, M.K.B.A.; Syahrullail, S.; Rahim, A.B.A.; Johnson, C.A.N. Biomass as a potential source of sustainable fuel, chemical and tribological materials—Overview. Mater. Today Proc. 2020. [Google Scholar] [CrossRef]
- Perkins, G.; Batalha, N.; Kumar, A.; Bhaskar, T.; Konarova, M. Recent advances in liquefaction technologies for production of liquid hydrocarbon fuels from biomass and carbonaceous wastes. Renew. Sustain. Energy Rev. 2019, 115, 109400. [Google Scholar] [CrossRef]
- He, R.; Ye, X.P.; English, B.C.; Satrio, J.A. Influence of pyrolysis condition on switchgrass bio-oil yield and physicochemical properties. Bioresour. Technol. 2009, 100, 5305–5311. [Google Scholar] [CrossRef] [PubMed]
- Sankar, M.; Dimitratos, N.; Miedziak, P.J.; Wells, P.P.; Kiely, C.J.; Hutchings, G.J. Designing bimetallic catalysts for a green and sustainable future. Chem. Soc. Rev. 2012, 41, 8099–8139. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jia, P.; Wang, T. Furfural: A Promising Platform Compound for Sustainable Production of C4and C5Chemicals. ACS Catal. 2016, 6, 7621–7640. [Google Scholar] [CrossRef]
- Yan, K.; Wu, G.S.; Lafleur, T.; Jarvis, C. Production, properties and catalytic hydrogenation of furfural to fuel additives and value-added chemicals. Renew. Sustain. Energy Rev. 2014, 38, 663–676. [Google Scholar] [CrossRef]
- Mariscal, R.; Maireles-Torres, P.; Ojeda, M.; Sadaba, I.; Granados, M.L. Furfural: A renewable and versatile platform molecule for the synthesis of chemicals and fuels. Energy Environ. Sci. 2016, 9, 1144–1189. [Google Scholar] [CrossRef]
- Homer, A.; Ralph, C. Method of hydrogenation of furfural to furfuryl alcohol. U.S. Patent 2094975A, 5 October 1937. [Google Scholar]
- Sitthisa, S.; Sooknoi, T.; Ma, Y.G.; Balbuena, P.B.; Resasco, D.E. Kinetics and mechanism of hydrogenation of furfural on Cu/SiO2 catalysts. J. Catal. 2011, 277, 1–13. [Google Scholar] [CrossRef]
- Li, K.; Ozer, R. Vapor phase decarbonylation process. WO2010080290A1, 31 August 2010. [Google Scholar]
- Sitthisa, S.; Resasco, D.E. Hydrodeoxygenation of Furfural Over Supported Metal Catalysts: A Comparative Study of Cu, Pd and Ni. Catal. Lett. 2011, 141, 784–791. [Google Scholar] [CrossRef]
- Zhang, W.; Zhu, Y.L.; Niu, S.; Li, Y.W. A study of furfural decarbonylation on K-doped Pd/Al2O3 catalysts. J. Mol. Catal. A Chem. 2011, 335, 71–81. [Google Scholar] [CrossRef]
- Merlo, A.B.; Vetere, V.; Ruggera, J.F.; Casella, M.L. Bimetallic PtSn catalyst for the selective hydrogenation of furfural to furfuryl alcohol in liquid-phase. Catal. Commun. 2009, 10, 1665–1669. [Google Scholar] [CrossRef]
- An, K.; Musselwhite, N.; Kennedy, G.; Pushkarev, V.V.; Robert Baker, L.; Somorjai, G.A. Preparation of mesoporous oxides and their support effects on Pt nanoparticle catalysts in catalytic hydrogenation of furfural. J. Colloid Interface Sci. 2013, 392, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Baçaoui, A.; Yaacoubi, A.; Dahbi, A.; Bennouna, C.; Phan Tan Luu, R.; Maldonado-Hodar, F.J.; Rivera-Utrilla, J.; Moreno-Castilla, C. Optimization of conditions for the preparation of activated carbons from olive-waste cakes. Carbon 2001, 39, 425–432. [Google Scholar] [CrossRef]
- Morales-Torres, S.; Silva, A.M.T.; Maldonado-Hodar, F.J.; Machado, B.F.; Perez-Cadenas, A.F.; Faria, J.L.; Figueiredo, J.L.; Carrasco-Marin, F. Pt-catalysts supported on activated carbons for catalytic wet air oxidation of aniline: Activity and stability. Appl. Catal. B Environ. 2011, 105, 86–94. [Google Scholar] [CrossRef]
- Vivo-Vilches, J.F.; Bailon-Garcia, E.; Perez-Cadenas, A.F.; Carrasco-Marin, F.; Maldonado-Hodar, F.J. Tailoring activated carbons for the development of specific adsorbents of gasoline vapors. J. Hazard. Mater. 2013, 263 Pt 2, 533–540. [Google Scholar] [CrossRef]
- Esteves, B.M.; Morales-Torres, S.; Maldonado-Hodar, F.J.; Madeira, L.M. Fitting Biochars and Activated Carbons from Residues of the Olive Oil Industry as Supports of Fe- Catalysts for the Heterogeneous Fenton-Like Treatment of Simulated Olive Mill Wastewater. Nanomaterials 2020, 10, 876. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Huy, C.; Kim, J.S.; Yoon, S.; Yang, E.; Kwak, J.H.; Lee, M.S.; An, K. Supported Pd nanoparticle catalysts with high activities and selectivities in liquid-phase furfural hydrogenation. Fuel 2018, 226, 607–617. [Google Scholar] [CrossRef]
- Mironenko, R.M.; Belskaya, O.B.; Gulyaeva, T.I.; Nizovskii, A.I.; Kalinkin, A.V.; Bukhtiyarov, V.I.; Lavrenov, A.V.; Likholobov, V.A. Effect of the nature of carbon support on the formation of active sites in Pd/C and Ru/C catalysts for hydrogenation of furfural. Catal. Today 2015, 249, 145–152. [Google Scholar] [CrossRef]
- Stucchi, M.; Alijani, S.; Manzoli, M.; Villa, A.; Lahti, R.; Galloni, M.G.; Lassi, U.; Prati, L. A Pt-Mo hybrid catalyst for furfural transformation. Catal. Today 2020, 357, 122–131. [Google Scholar] [CrossRef]
- Taylor, M.J.; Beaumont, S.K.; Islam, M.J.; Tsatsos, S.; Parlett, C.A.M.; Issacs, M.A.; Kyriakou, G. Atom efficient PtCu bimetallic catalysts and ultra dilute alloys for the selective hydrogenation of furfural. Appl. Catal. B Environ. 2020, 284, 119737. [Google Scholar] [CrossRef]
- Kosydar, R.; Szewczyk, I.; Natkanski, P.; Duraczynska, D.; Gurgul, J.; Kustrowski, P.; Drelinkiewicz, A. New insight into the effect of surface oxidized groups of nanostructured carbon supported Pd catalysts on the furfural hydrogenation. Surf Interfaces 2019, 17, 100379. [Google Scholar] [CrossRef]
- Liu, L.J.; Lou, H.; Chen, M. Selective hydrogenation of furfural over Pt based and Pd based bimetallic catalysts supported on modified multiwalled carbon nanotubes (MWNT). Appl. Catal. A Gen. 2018, 550, 1–10. [Google Scholar] [CrossRef]
- Liu, L.J.; Guo, H.M.; Xue, B.; Lou, H.; Chen, M. Hydrogenation in supercritical conditions catalyzed by palladium supported on modified activated carbon. RSC Adv. 2015, 5, 66704–66710. [Google Scholar] [CrossRef]
- Deng, D.; Novoselov, K.S.; Fu, Q.; Zheng, N.; Tian, Z.; Bao, X. Catalysis with two-dimensional materials and their heterostructures. Nat. Nanotechnol. 2016, 11, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Li, Y.; Gong, Y.; Zhang, P.; Li, H.; Wang, Y. Synthesis of palladium nanoparticles supported on mesoporous N-doped carbon and their catalytic ability for biofuel upgrade. JACS 2012, 134, 16987–16990. [Google Scholar] [CrossRef]
- Gong, W.B.; Chen, C.; Zhang, H.M.; Zhang, Y.; Zhang, Y.X.; Wang, G.Z.; Zhao, H.J. Highly selective liquid-phase hydrogenation of furfural over N-doped carbon supported metallic nickel catalyst under mild conditions. Mol. Catal. 2017, 429, 51–59. [Google Scholar] [CrossRef]
- Fellah, M.F. Direct decarbonylation of furfural to furan: A density functional theory study on Pt-graphene. Appl. Surf. Sci. 2017, 405, 395–404. [Google Scholar] [CrossRef]
- Li, M.; Li, W.; Liu, S. Hydrothermal synthesis, characterization, and KOH activation of carbon spheres from glucose. Carbohydr. Res. 2011, 346, 999–1004. [Google Scholar] [CrossRef]
- Han, B.; Zhang, E.; Cheng, G.; Zhang, L.; Wang, D.; Wang, X. Hydrothermal carbon superstructures enriched with carboxyl groups for highly efficient uranium removal. Chem. Eng. J. 2018, 338, 734–744. [Google Scholar] [CrossRef]
- Shao, C.F.; Qiu, S.J.; Chu, H.L.; Zou, Y.J.; Xiang, C.L.; Xu, F.; Sun, L.X. Nitrogen-doped porous microsphere carbons derived from glucose and aminourea for high-performance supercapacitors. Catal. Today 2018, 318, 150–156. [Google Scholar] [CrossRef]
- Zhu, J.J.; Yin, H.Y.; Gong, J.Y.; Al-Furjan, M.S.H.; Nie, Q.L. Easy one pot synthesis of NiO/Nitrogen doped carbon spheres for highly sensitive enzyme free amperometric glucose sensors. Appl. Surf. Sci. 2018, 444, 56–64. [Google Scholar] [CrossRef]
- Wataniyakul, P.; Boonnoun, P.; Quitain, A.T.; Sasaki, M.; Kida, T.; Laosiripojana, N.; Shotipruk, A. Preparation of hydrothermal carbon as catalyst support for conversion of biomass to 5-hydroxymethylfurfural. Catal. Commun. 2018, 104, 41–47. [Google Scholar] [CrossRef]
- Pastrana-Martínez, L.M.; Morales-Torres, S.; Likodimos, V.; Falaras, P.; Figueiredo, J.L.; Faria, J.L.; Silva, A.M.T. Role of oxygen functionalities on the synthesis of photocatalytically active graphene–TiO2 composites. Appl. Catal. B Environ. 2014, 158–159, 329–340. [Google Scholar] [CrossRef]
- Morales-Torres, S.; Pastrana-Martínez, L.M.; Figueiredo, J.L.; Faria, J.L.; Silva, A.M.T. Graphene oxide-P25 photocatalysts for degradation of diphenhydramine pharmaceutical and methyl orange dye. Appl. Surf. Sci. 2013, 275, 361–368. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Maroto-Valiente, A.; Rodriguez-Ramos, I.; Xin, Q.; Guerrero-Ruiz, A. Synthesis and characterization of carbon black supported Pt–Ru alloy as a model catalyst for fuel cells. Catal. Today 2004, 93–95, 619–626. [Google Scholar] [CrossRef]
- Morales-Torres, S.; Maldonado-Hodar, F.J.; Perez-Cadenas, A.F.; Carrasco-Marin, F. Design of low-temperature Pt-carbon combustion catalysts for VOC’s treatments. J. Hazard. Mater. 2010, 183, 814–822. [Google Scholar] [CrossRef]
- Pushkarev, V.V.; Musselwhite, N.; An, K.; Alayoglu, S.; Somorjai, G.A. High structure sensitivity of vapor-phase furfural decarbonylation/hydrogenation reaction network as a function of size and shape of Pt nanoparticles. Nano Lett. 2012, 12, 5196–5201. [Google Scholar] [CrossRef]
- Vorotnikov, V.; Mpourmpakis, G.; Vlachos, D.G. DFT Study of Furfural Conversion to Furan, Furfuryl Alcohol, and 2-Methylfuran on Pd(111). ACS Catal. 2012, 2, 2496–2504. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Nakazawa, H.; Watanabe, H.; Tomishige, K. Total Hydrogenation of Furfural over a Silica-Supported Nickel Catalyst Prepared by the Reduction of a Nickel Nitrate Precursor. Chemcatchem 2012, 4, 1791–1797. [Google Scholar] [CrossRef]
- Tsatsos, S.; Ladas, S.; Kyriakou, G. Electronic Properties and Reactivity of Furfural on a Model Pt(111) Catalytic Surface. J. Phys. Chem. C 2020, 124, 26268–26278. [Google Scholar] [CrossRef]
- Xiong, K.; Chen, J.G. Correlating furfural reaction pathways with interactions between furfural and monometallic surfaces. Catal. Today 2020, 339, 289–295. [Google Scholar] [CrossRef]
- Wang, Y.T.; Zhao, D.Y.; Rodriguez-Padron, D.; Len, C. Recent Advances in Catalytic Hydrogenation of Furfural. Catalysts 2019, 9, 796. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, B.; Fei, B.; Chen, X.; Zhang, J.; Mu, X. Tunable and selective hydrogenation of furfural to furfuryl alcohol and cyclopentanone over Pt supported on biomass-derived porous heteroatom doped carbon. Faraday Discuss. 2017, 202, 79–98. [Google Scholar] [CrossRef]
- Wang, C.; Luo, J.; Liao, V.; Lee, J.D.; Onn, T.M.; Murray, C.B.; Gorte, R.J. A comparison of furfural hydrodeoxygenation over Pt-Co and Ni-Fe catalysts at high and low H2 pressures. Catal. Today 2018, 302, 73–79. [Google Scholar] [CrossRef]
- Wang, G.; Yao, R.; Xin, H.; Guan, Y.; Wu, P.; Li, X. At room temperature in water: Efficient hydrogenation of furfural to furfuryl alcohol with a Pt/SiC–C catalyst. RSC Adv. 2018, 8, 37243–37253. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, L.; Zhang, B.; Guo, X.; Mu, X. Highly selective hydrogenation of furfural to furfuryl alcohol over Pt nanoparticles supported on g-C3N4 nanosheets catalysts in water. Sci. Rep. 2016, 6, 28558. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, X.; Chen, Q.; Chen, L.; Liu, Q.; Wang, C.; Ma, L. One-Pot Hydrogenation of Furfural into Tetrahydrofurfuryl Alcohol under Ambient Conditions over PtNi Alloy Catalyst. Energy Fuels 2020, 34, 2178–2184. [Google Scholar] [CrossRef]
- Xu, L.; Nie, R.; Lyu, X.; Wang, J.; Lu, X. Selective hydrogenation of furfural to furfuryl alcohol without external hydrogen over N-doped carbon confined Co catalysts. Fuel Process. Technol. 2020, 197, 106205. [Google Scholar] [CrossRef]
- Hu, D.; Xu, H.; Yi, Z.; Chen, Z.; Ye, C.; Wu, Z.; Garces, H.F.; Yan, K. Green CO2-Assisted Synthesis of Mono- and Bimetallic Pd/Pt Nanoparticles on Porous Carbon Fabricated from Sorghum for Highly Selective Hydrogenation of Furfural. ACS Sustain. Chem. Eng. 2019, 7, 15339–15345. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of gases in multimolecular layers. JACS 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Stoeckli, F. Characterization of microporous carbons by adsorption and immersion techniques. In Porosity in Carbons: Characterization and Applications; Patrick, J.W., Ed.; Edward Arnold: London, UK, 1995; pp. 66–97. [Google Scholar]
- Bansal, R.C.; Donnet, J.B.; Stoeckli, F. Active Carbon; Marcel Dekker: New York, NY, USA, 1988. [Google Scholar]
- Barrett, E.P.; Joyner, L.G.; Halenda, P.P. The Determination of Pore Volume and Area Distributions in Porous Substances. I. Computations from Nitrogen Isotherms. JACS 1951, 73, 373–380. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SBET | Smicro | Vmicro | L0 | Vmeso | pHPZC |
|---|---|---|---|---|---|---|
| (m2 g−1) | (m2 g−1) | (cm3 g−1) | (nm) | (cm3 g−1) | ||
| CS | 541 | 323 | 0.22 | 1.3 | 0.14 | 7.5 |
| CS-CNT | 473 | 439 | 0.19 | 0.8 | 0.21 | 7.6 |
| CS-rGO | 478 | 504 | 0.19 | 0.7 | 0.15 | 7.3 |
| CS-CB | 473 | 433 | 0.19 | 0.9 | 0.08 | 7.0 |
| CS-AC | 496 | 442 | 0.20 | 0.9 | 0.05 | 7.1 |
| Catalyst | dPt (XRD) | dPt (TEM) |
|---|---|---|
| (nm) | (nm) | |
| Pt/CS | 14.9 | 4.5 |
| Pt/CS-CNT | 12.7 | 3.5 |
| Pt/CS-rGO | 11.7 | 5.5 |
| Pt/CS-CB | 13.6 | 4.1 |
| Pt/CS-AC | 15.9 | 3.3 |
| Catalyst | Operating Conditions | Conversion | Selectivity | Ref. |
|---|---|---|---|---|
| (%) | (%) | |||
| 1%Pt-40%Mo/AC | 150 °C, 3 bar H2, EtOH | ~80% at 2 h | ~80% FEE | [21] |
| 1%Pt/AC | ~75% at 2 h | |||
| 0.5%Pt/CNT | 130 °C, 30 bar H2, EtOH | 64.5% at 5 h | ~55% FDA | [24] |
| 5%Pt/AC | 100 °C, 10 bar H2, water | 99% at 4 h | 99% FOL | [45] |
| 10%Pt/CB | 180 °C, 1 bar H2, 1-propanol | 80.7% at 10 min g/g | 49.4% FU | [46] |
| 5%Pt/C * | 25 °C, 10 bar H2, water | ~50% at 5 h | ~95% FOL | [47] |
| 5%Pt/C3N4 | 100 °C, 10 bar H2, water | 60.9% at 5 h | >99% FOL | [48] |
| 3%Pt/AC | 35 °C, 20 bar H2, water | 99% at 12 h | 89% FOL | [49] |
| Pt/C | 150 °C, 5 bar H2, 1,4-dioxane | ~39.3% at 6 h | ~89% FOL | [50] |
| 5%Pt/C * | 140 °C, 60 bar H2, octane | ~10% at 3 h | ~55% FOL | [51] |
| 3%Pt/C | ~58% at 3 h | ~50% THFOL | ||
| 1%Pt/CS | 200 °C, 1 bar, 40 cm3 min−1 H2 | 32% | - | This work |
| 1%Pt/CS-CNT | 37% | 47% FU |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morales-Torres, S.; Pastrana-Martínez, L.M.; Pérez-García, J.A.; Maldonado-Hódar, F.J. Glucose–Carbon Hybrids as Pt Catalyst Supports for the Continuous Furfural Hydroconversion in Gas Phase. Catalysts 2021, 11, 49. https://doi.org/10.3390/catal11010049
Morales-Torres S, Pastrana-Martínez LM, Pérez-García JA, Maldonado-Hódar FJ. Glucose–Carbon Hybrids as Pt Catalyst Supports for the Continuous Furfural Hydroconversion in Gas Phase. Catalysts. 2021; 11(1):49. https://doi.org/10.3390/catal11010049
Chicago/Turabian StyleMorales-Torres, Sergio, Luisa M. Pastrana-Martínez, Juan A. Pérez-García, and Francisco J. Maldonado-Hódar. 2021. "Glucose–Carbon Hybrids as Pt Catalyst Supports for the Continuous Furfural Hydroconversion in Gas Phase" Catalysts 11, no. 1: 49. https://doi.org/10.3390/catal11010049
APA StyleMorales-Torres, S., Pastrana-Martínez, L. M., Pérez-García, J. A., & Maldonado-Hódar, F. J. (2021). Glucose–Carbon Hybrids as Pt Catalyst Supports for the Continuous Furfural Hydroconversion in Gas Phase. Catalysts, 11(1), 49. https://doi.org/10.3390/catal11010049