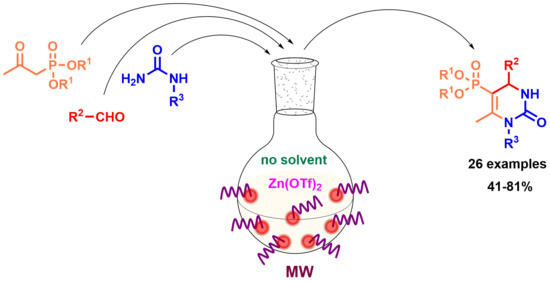
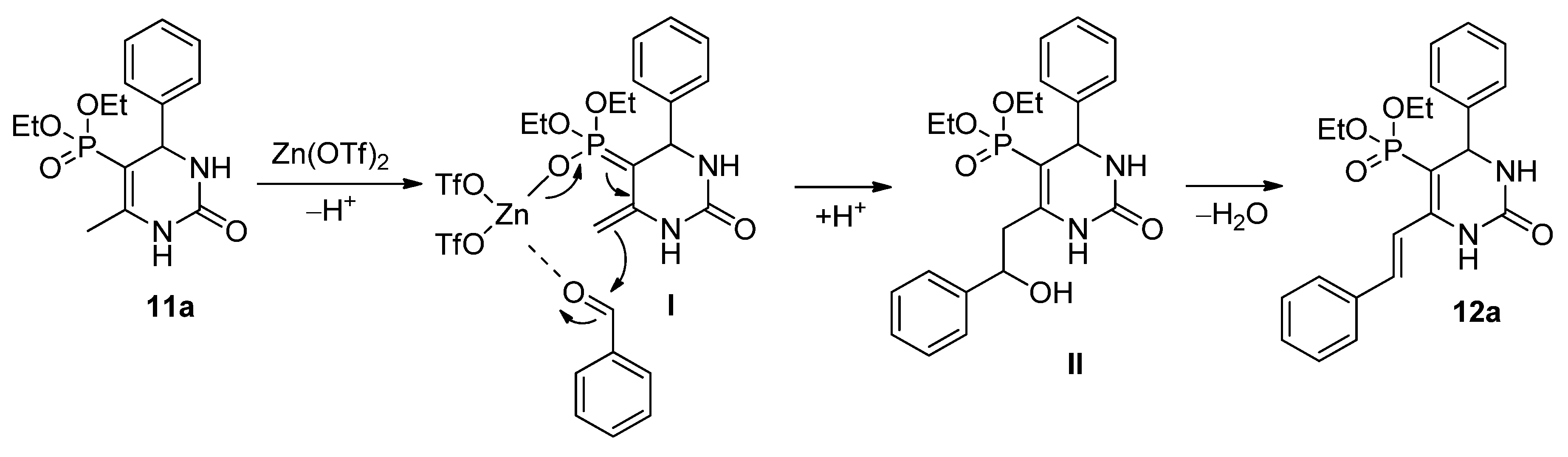
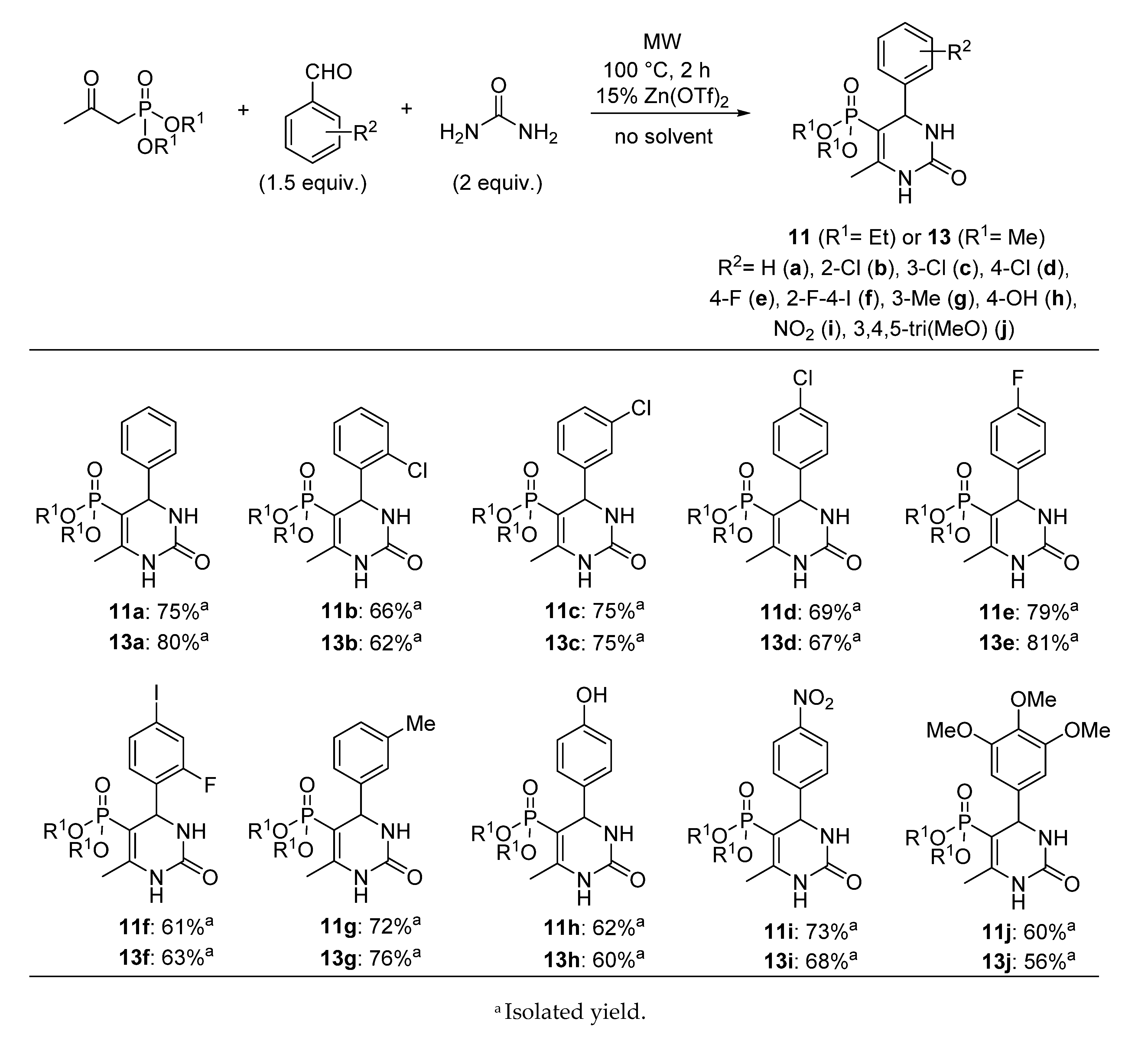
3.4. General Procedure for the Synthesis of 5-Phosphonato-3,4-dihydropyrimidin-2(1H)-one (11, 13–16) Derivatives
A mixture of 1.0 mmol β-ketophosphonate (diethyl (2-oxopropyl)phosphonate (0.19 mL) or 0.14 mL dimethyl (2-oxopropyl)phosphonate), 1.5 mmol aldehyde (0.15 mL benzaldehyde, 0.17 mL 2-chlorobenzaldehyde, 0.17 mL 3-chlorobenzaldehyde, 0.17 mL 4-chlorobenzaldehyde, 0.16 mL 4-fluorobenzaldehde, 0.38 g 2-fluoro-4-iodobenzaldehyde, 0.18 mL 3-methylbenzaldehyde, 0.18 g 4-hydroxybenzaldehyde, 0.23 g 4-nitrobenzaldehyde, 0.29 g 3,4,5-trimethoxybenzaldehyde, 0.14 mL butyraldehyde or 0.16 mL isovaleraldehyde), 2.0 mmol urea derivative (0.12 g urea or 0.14 g N-methylurea) and 0.15 mmol (0.05 g) zinc triflate was irradiated in a sealed tube at 100 °C for 2 h in a CEM Microwave reactor equipped with a pressure controller. The volatile components were removed in vacuum, and the residue was analyzed by 31P NMR and HPLC-MS. The 5-phosphonato-3,4-dihydropyrimidin-2(1H)-ones (4, 6–8) were obtained by column chromatography using silica gel as the absorbent and dichloromethane:methanol (9:1) as the eluent, or by reversed-phase column chromatography using C18-reversed phase silica gel and the composition of eluent A was 0.1% (NH4)(HCOO) in water; eluent B was 0.1% (NH4)(HCOO) and 8% water in acetonitrile. The following products were thus prepared:
5-Diethoxyphosphoryl-6-methyl-4-phenyl-3,4-dihydropyrimidin-2(1H)-one (
11a): Yield: 75% (0.24 g), white solid; Mp: 168–169 °C; Mp [
27]: 169–170 °C;
31P (DMSO-d
6) δ 19.8;
31P (DMSO-d
6) δ [
27] 19.1; [M + H]
+found = 325.1314, [M + H]
+calculated = 325.1317.
5-Diethoxyphosphoryl-6-methyl-4-(2-chlorophenyl)-3,4-dihydropyrimidin-2(1H)-one (11b): Yield: 66% (0.24 g), white solid; Mp: 230–231 °C; 31P (DMSO-d6) δ 19.0; 13C NMR (DMSO-d6) δ 16.1 (d, 3JCP = 7.0, CH3CH2OI), 16.6 (d, 3JCP = 6.4, CH3CH2OII), 17.7 (d, 3JCP = 3.4, CCH3), 52.3 (d, 2JCP = 15.1, CHNH), 60.8 (d, 2JCP = 5.0, CH3CH2OI), 61.1 (d, 2JCP = 5.2, CH3CH2OII), 93.1 (d, 1JCP = 207.0, PC=C), 128.1 (C6), 129.5 (C4), 129.7 (C5), 129.8 (C3), 132.1 (C2), 141.6 (C1), 149.8 (d, 2JCP = 21.8, PC=C), 152.4 (C=O); 1H NMR (DMSO-d6) δ 0.85 (t, JHH = 7.0, 3H, CH3CH2OI), 1.18 (t, JHH = 7.1, 3H, CH3CH2OII), 2.20 (d, JHP = 2.0, 3H, CCH3), 3.36–3.47 (m, 1H, CHA, CH3CH2OI), 3.59–3.76 (m, 1H, CHB, CH3CH2OI), 3.76–3.92 (m, 2H, CH3CH2OII), 5.36 (dd, 3JHP = 8.5, JHH = 3.3, 1H, CHNH), 7.20–7.49 (m, 4H, ArH), 7.64 (br s, 1H, NH), 9.18 (d, JHH = 4.6, 1H, NH); [M + H]+found = 359.0924, [M + H]+calculated = 359.0927.
5-Diethoxyphosphoryl-6-methyl-4-(3-chlorophenyl)-3,4-dihydropyrimidin-2(1H)-one (
11c): Yield: 75% (0.27 g), light yellow solid; Mp: 170–171 °C, Mp [
28]: 171–172 °C;
31P (DMSOd
6) δ 19.5;
13C NMR (DMSO-d
6) δ 16.3 (d,
3JCP = 6.7,
CH
3CH
2O
I), 16.5 (d,
3JCP = 6.4,
CH
3CH
2O
II), 17.9 (d,
3JCP = 3.4, C
CH
3), 55.1 (d,
2JCP = 15.2, CHNH), 61.1 (d,
2JCP = 5.1, CH
3CH
2O
I), 61.2 (d,
2JCP = 5.2, CH
3CH
2O
II), 93.8 (d,
1JCP = 206.3, P
C=C), 125.6 (C
6), 126.9 (C
2), 127.9 (C
4), 130.9 (C
5), 133.4 (C
3), 147.4 (C
1), 149.3 (d,
2JCP = 20.7, PC=
C), 152.9 (C=O);
1H NMR (DMSO-d
6) δ 1.00 (t,
JHH = 6.3, 3H, C
H3CH
2O
I), 1.14 (t,
JHH = 7.0, 3H, C
H3CH
2O
II), 2.11 (d,
JHP = 2.2, 3H, CCH
3), 3.55–3.68 (m, 1H, CH
A, CH
3C
H2O
I), 3.72–3.87 (m, 3H, CH
B, CH
3C
H2O
I, CH
3C
H2O
II), 4.88 (dd,
3JHP = 8.8,
JHH = 3.6, 1H, C
HNH), 7.15–7.45 (m, 4H, ArH), 7.70 (br s, 1H, NH), 9.19 (d,
JHH = 4.6, 1H, NH);
1H NMR (CDCl
3 + DMSO-d
6) δ [
28] 1.10 (t,
JHH = 6.9, 3H, C
H3CH
2O
I), 1.22 (t,
JHH = 6.9, 3H, C
H3CH
2O
II), 2.17 (s, 3H, CCH
3), 3.80–3.91 (m, 4H, CH
3C
H2O), 4.96–4.99 (m, 1H, C
HNH), 7.24–7.34 (m, 3H, ArH), 7.63 (s, 1H, ArH), 8.01 (s, 1H, NH), 9.12 (s, 1H, NH); [M + H]
+found = 359.0916, [M + H]
+calculated = 359.0927.
5-Diethoxyphosphoryl-6-methyl-4-(4-chlorophenyl)-3,4-dihydropyrimidin-2(1H)-one (
11d): Yield: 69% (0.25 g), white solid; Mp: 120–121 °C; Mp [
27]: 120–122°C;
31P (DMSO-d
6) δ 19.6;
31P (DMSO-d
6) δ [
27] 19.8; [M + H]
+found = 359.0923, [M + H]
+calculated = 359.0927.
5-Diethoxyphosphoryl-6-methyl-4-(4-fluorophenyl)-3,4-dihydropyrimidin-2(1H)-one (
11e): Yield: 79% (0.27 g), white solid; Mp: 109–110 °C; Mp [
28]: 110–112 °C;
31P NMR (DMSO-d
6) δ 19.7;
13C NMR (DMSO-d
6) δ 16.3 (d,
3JCP = 6.7,
CH
3CH
2O
I), 16.5 (d,
3JCP = 6.2,
CH
3CH
2O
II), 17.8 (d,
3JCP = 3.4, C
CH
3), 54.9 (d,
2JCP = 15.0, CHNH), 61.0 (d,
2JCP = 5.0, CH
3CH
2O
I), 61.1 (d,
2JCP = 5.1, CH
3CH
2O
II), 94.2 (d,
1JCP = 205.5, P
C=C), 115.5 (d,
2JCF = 21.4, C
3), 129.0 (d,
3JCF = 8.3, C
2), 141.3 (d,
JCF = 2.5, C
1), 148.8 (d,
2JCP = 20.7, PC=
C), 152.9 (C=O), 161.9 (d,
1JCF = 243.0, C
4);
1H NMR (DMSO-d
6) δ 0.98 (t,
JHH = 7.0, 3H, C
H3CH
2O
I), 1.13 (t,
JHH = 7.0, 3H, C
H3CH
2O
II), 2.10 (d,
JHP = 2.5, 3H, CCH
3), 3.49–3.65 (m, 1H, CH
A, CH
3C
H2O
I), 3.68–3.85 (m, 3H, CH
B, CH
3C
H2O
I, CH
3C
H2O
II), 4.86 (dd,
3JHP = 8.6,
JHH = 3.5, 1H, C
HNH), 7.10–7.22 (m, 2H, ArH), 7.24–7.37 (m, 2H, ArH), 7.65 (br s, 1H, NH), 9.15 (d,
JHH = 4.7, 1H, NH);
1H NMR (CDCl
3 + DMSO-d
6) δ [
28] 1.04 (t,
JHH = 7.0, 3H, C
H3CH
2O
I), 1.19 (t,
JHH = 7.0, 3H, C
H3CH
2O
II), 2.13 (s, 3H, CCH
3), 3.70–3.85 (m, 4H, CH
3C
H2O), 4.92 (dd,
3JHP = 8.5,
JHH = 3.2, 1H, C
HNH), 7.04–7.34 (m, 4H, ArH), 7.60 (s, 1H, NH), 9.12 (d,
JHH = 3.6, 1H, NH); [M + H]
+found = 343.1221, [M + H]
+calculated = 343.1222.
5-Diethoxyphosphoryl-6-methyl-4-(2-fluoro-4-iodophenyl)-3,4-dihydropyrimidin-2(1H)-one (11f): Yield: 61% (0.29 g), white solid; Mp: 103–104 °C; 31P (DMSO-d6) δ 25.9; 13C NMR (DMSO-d6) δ 20.1 (d, 3JCP = 7.0, CH3CH2OI), 20.5 (d, 3JCP = 6.2, CH3CH2OII), 21.8 (d, 3JCP = 3.5, CCH3), 53.3 (dd, 2JCP = 24.4, 3JCF = 2.6, CHNH), 64.9 (d, 2JCP = 5.1, CH3CH2OI), 65.1 (d, 2JCP = 5.2, CH3CH2OII), 96.0 (d, 1JCP = 207.0, PC=C), 97.8 (d, 3JCF = 8.7, C6), 128.7 (d, 2JCF = 24.8, C3), 135.3 (d, 3JCF = 4.0, C5), 135.7 (d, 2JCF = 13.8, C1), 138.0 (d, 3JCF = 3.4, C4), 153.6 (d, 2JCP = 21.4, PC=C), 156.3 (C=O), 163.5 (d, 1JCF = 252.0, C2); 1H NMR (DMSO-d6) δ 0.93 (t, JHH = 7.0, 3H, CH3CH2OI), 1.14 (t, JHH = 7.1, 3H, CH3CH2OII), 2.13 (d, JHP = 2.4, 3H, CCH3), 3.43–3.58 (m, 1H, CHA, CH3CH2OI), 3.64–3.88, (m, 3H, CHB, CH3CH2OI, CH3CH2OII), 5.14 (dd, 3JHP = 8.0, JHH = 3.2, 1H, CHNH), 7.07 (t, JHH = 8.0, 1H, ArH), 7.47–7.78 (m, 3H, ArH, NH), 9.16 (d, JHH = 3.0, 1H, NH); [M + H]+found = 469.0184, [M + H]+calculated = 469.0189.
5-Diethoxyphosphoryl-6-methyl-4-(3-methylphenyl)-3,4-dihydropyrimidin-2(1H)-one (11g): Yield: 72% (0.24 g), white solid; Mp: 192–193 °C; 31P (DMSO-d6) δ 19.9; 13C NMR (DMSO-d6) δ 16.2 (d, 3JCP = 6.8, CH3CH2OI), 16.5 (d, 3JCP = 6.3, CH3CH2OII), 17.9 (d, 3JCP = 3.5, CCH3), 21.6 (s, C3CH3), 55.5 (d, 2JCP = 15.1, CHNH), 60.9 (d, 2JCP = 4.9, CH3CH2OI), 61.0 (d, 2JCP = 5.1, CH3CH2OII), 94.2 (d, 1JCP = 205.3, PC=C), 124.1 (C6), 127.6 (C2), 128.4 (C4), 128.7 (C5), 137.7 (C3), 144.9 (C1), 148.7 (d, 2JCP = 20.7, PC=C), 153.0 (C=O); 1H NMR (DMSO-d6) δ 0.98 (t, JHH = 7.1, 3H, CH3CH2OI), 1.14 (t, JHH = 7.1, 3H, CH3CH2OII), 2.11 (d, JHP = 2.3, 3H, CCH3), 2.28 (s, 3H, C3CH3), 3.51–3.60 (m, 1H, CHA, CH3CH2OI), 3.72–3.82 (m, 3H, CHB, CH3CH2OI, CH3CH2OII), 4.82 (dd, 3JHP = 8.7, JHH = 3.4, 1H, CHNH), 7.07–7.13 (m, 3H, ArH), 7.19–7.25 (m, 1H, ArH), 7.60 (br s, 1H, NH), 9.11 (d, JHH = 3.1, 1H, NH); [M + H]+found = 339.1473, [M + H]+calculated = 339.1473.
5-Diethoxyphosphoryl-6-methyl-4-(4-hydroxyphenyl)-3,4-dihydropyrimidin-2(1H)-one (11h): Yield: 62% (0.21 g), yellow solid; Mp: 186–188 °C; 31P (DMSO-d6) δ 20.1; 13C NMR (DMSO-d6) δ 16.3 (d, 3JCP = 7.0, CH3CH2OI), 16.6 (d, 3JCP = 6.3, CH3CH2OII), 17.8 (d, 3JCP = 3.7, CCH3), 55.0 (d, 2JCP = 14.8, CHNH), 60.9 (d, 2JCP = 4.5, CH3CH2OI), 61.0 (d, 2JCP = 4.7, CH3CH2OII), 94.7 (d, 1JCP = 204.6, PC=C), 115.4 (C3), 127.7 (C2), 135.6 (C1), 148.2 (d, 2JCP = 21.0, PC=C), 153.1 (C=O), 157.1 (C4); 1H NMR (DMSO-d6) δ 0.92 (t, JHH = 7.0, 3H, CH3CH2OI), 1.07 (t, JHH = 7.0, 3H, CH3CH2OII), 2.03 (d, JHP = 2.1, 3H, CCH3), 3.41–3.52 (m, 1H, CHA, CH3CH2OI), 3.61–3.77 (m, 3H, CHB, CH3CH2OI, CH3CH2OII), 4.67 (dd, 3JHP = 9.1, JHH = 3.3, 1H, CHNH), 6.53–6.72 (m, 2H, ArH), 6.93–7.09 (m, 2H, ArH), 7.45 (br s, 1H, NH), 8.98 (br s, 1H, NH), 9.28 (s, 1H, OH); [M + H]+found = 341.1262, [M + H]+calculated = 341.1266.
5-Diethoxyphosphoryl-6-methyl-4-(4-nitrophenyl)-3,4-dihydropyrimidin-2(1H)-one (
11i): Yield: 73% (0.27 g), yellow solid; Mp: 218–219 °C; Mp [
27]: 218–220 °C;
31P (DMSO-d
6) δ 19.1;
31P (DMSO-d
6) δ [
27] 19.2; [M + H]
+found = 370.1166, [M + H]
+calculated = 370.1167.
5-Diethoxyphosphoryl-6-methyl-4-(3,4,5-trimethoxyphenyl)-3,4-dihydropyrimidin-2(1H)-one (11j): Yield: 60% (0.25 g), white solid; Mp: 175–177 °C; 31P (DMSO-d6) δ 20.0; 13C NMR (DMSO-d6) δ 16.3 (d, 3JCP = 6.6, CH3CH2OI), 16.6 (d, 3JCP = 6.3, CH3CH2OII), 17.9 (d, 3JCP = 3.5, CCH3), 55.3 (d, 2JCP = 15.3, CHNH), 56.3 (C3OCH3), 60.5 (C4OCH3), 61.10 (d, 2JCP = 5.2, CH3CH2OI), 61.13 (d, 2JCP = 5.6, CH3CH2OII), 94.0 (d, 1JCP = 206.7, PC=C), 104.4 (C2), 137.4 (C3), 140.4 (C4), 149.0 (d, 2JCP = 20.7, PC=C), 152.7 (C=O), 153.2 (C1); 1H NMR (DMSO-d6) δ 1.03 (t, JHH = 7.0, 3H, CH3CH2OI), 1.16 (t, JHH = 7.1, 3H, CH3CH2OII), 2.11 (d, JHP = 2.5, 3H, CCH3), 3.57–3.90 [3.63 (s, C4OCH3), 3.73 (s, C3OCH3) overlapped by the multiplet of CH3CH2O total int. 13 H], 4.83 (dd, 3JHP = 8.9, JHH = 3.5, 1H, CHNH), 6.61 (s, 2H, ArH), 7.62 (br s, 1H, NH), 9.11 (d, JHH = 3.0, 1H, NH); [M + H]+found = 415.1631, [M + H]+calculated = 415.1634.
5-Dimethoxyphosphoryl-6-methyl-4-phenyl-3,4-dihydropyrimidin-2(1H)-one (
13a): Yield: 80% (0.24 g), white solid; Mp: 197–199 °C; Mp [
27]: 198–200 °C;
31P (DMSO-d
6) δ 23.0;
31P (DMSO-d
6) δ [
27] 22.8; [M + H]
+found = 297.1001, [M + H]
+calculated = 297.1004.
5-Dimethoxyphosphoryl-6-methyl-4-(2-chlorophenyl)-3,4-dihydropyrimidin-2(1H)-one (13b) Yield: 62% (0.20 g), white solid; Mp: 230–231 °C; 31P (DMSO-d6) δ 22.1; 13C NMR (DMSO-d6) δ 17.8 (d, 3JCP = 3.4, CCH3), 51.7 (d, 2JCP = 5.1, CH3OI), 52.0 (d, 2JCP = 5.4, CH3OII), 52.3 (d, 2JCP = 15.0, CHNH), 91.9 (d, 1JCP = 207.4, PC=C), 128.1 (C6), 129.5 (C4), 129.7 (C5), 129.9 (C3), 132.0 (C2), 141.5 (C1), 150.4 (d, 2JCP = 21.7, PC=C), 152.3 (C=O); 1H NMR (DMSO-d6) δ 2.18 (s, 3H, CCH3), 3.18 (d, 3JHP = 11.1, 3H, CH3OI), 3.48 (d, 3JHP = 11.2, 3H, CH3OII), 5.34 (dd, 3JHP = 8.4, JHH = 3.4, 1H, CHNH), 7.21–7.50 (m, 4H, ArH), 7.68 (br s, 1H, NH), 9.24 (d, JHH = 6.1, 1H, NH); [M + H]+found = 331.0612, [M + H]+calculated = 331.0614.
5-Dimethoxyphosphoryl-6-methyl-4-(3-chlorophenyl)-3,4-dihydropyrimidin-2(1H)-one (13c): Yield: 75% (0.25 g), white solid; Mp: 126–127 °C; 31P (DMSO-d6) δ 22.6; 13C NMR (DMSO-d6) δ 17.9 (d, 3JCP = 3.6, CCH3), 51.9 (d, 2JCP = 5.0, CH3OI), 52.1 (d, 2JCP = 5.3, CH3OII), 55.0 (d, 2JCP = 15.2, CHNH), 92.5 (d, 1JCP = 206.7, PC=C), 125.6 (C6), 126.9 (C2), 127.9 (C4), 131.0 (C5), 133.4 (C3), 147.3 (C1), 149.9 (d, 2JCP = 20.5, PC=C), 152.8 (C=O); 1H NMR (DMSO-d6) δ 2.09 (d, JHP = 2.4, 3H, CCH3), 3.36 (d, 3JHP = 11.4, 3H, CH3OI), 3.44 (d, 3JHP = 11.3, 3H, CH3OII), 4.87 (dd, 3JHP = 8.6, JHH = 3.5, 1H, CHNH), 7.23–7.31 (m, 2H, ArH), 7.32–7.42 (m, 2H, ArH), 7.74 (d, JHH = 2.0, 1H, NH), 9.27 (d, JHH = 2.7, 1H, NH); [M + H]+found = 331.0617, [M + H]+calculated = 331.0614.
5-Dimethoxyphosphoryl-6-methyl-4-(4-chlorophenyl)-3,4-dihydropyrimidin-2(1H)-one (
13d): Yield: 67% (0.22 g), white solid; Mp: 226–227 °C; Mp [
27]: 226–227 °C;
31P (DMSO-d
6) δ 22.8;
31P (DMSO-d
6) δ [
27] 22.6; [M + H]
+found= 331.0613, [M + H]
+calculated = 331.0614.
5-Dimethoxyphosphoryl-6-methyl-4-(4-fluorophenyl)-3,4-dihydropyrimidin-2(1H)-one (
13e): Yield: 81% (0.25 g), white solid; Mp: 234–235 °C; Mp [
28]: 232–233 °C;
31P (DMSO-d
6) δ 22.7;
13C NMR (DMSO-d
6) δ 17.8 (d,
3JCP = 3.6, C
CH
3), 51.9 (d,
2JCP = 5.0, CH
3O
I), 52.0 (d,
2JCP = 5.3, CH
3O
II), 54.8 (d,
2JCP = 15.3, CHNH), 93.0 (d,
1JCP = 206.2, P
C=C), 115.6 (d,
2JCF = 21.3, C
3), 129.0 (d,
3JCF = 8.0, C
2), 141.3 (d,
JCF = 3.0, C
1), 149.4 (d,
2JCP = 20.6, PC=
C), 152.8 (C=O), 161.9 (d,
1JCF = 243.2, C
4);
1H NMR (DMSO-d
6) δ 2.09 (d,
JHP = 2.4, 3H, CCH
3), 3.32 (d,
3JHP = 11.4, 3H, CH
3O
I), 3.42 (d,
3JHP = 11.4, 3H, CH
3O
II), 4.85 (dd,
3JHP = 8.6,
JHH = 3.4, 1H, C
HNH), 7.12–7.20 (m, 2H, ArH), 7.27–7.33 (m, 2H, ArH), 7.68 (br s, 1H, NH), 9.21 (d,
JHH = 2.7, 1H, NH);
1H NMR (CDCl
3 + DMSO-d
6) δ [
28] 2.11 (s, 3H, CCH
3), 3.34 (d,
3JHP = 11.2, 3H, CH
3O
I), 3.39 (d,
3JHP = 11.3, 3H, CH
3O
II), 4.90 (dd,
3JHP = 8.5,
JHH = 3.2, 1H, C
HNH), 7.02–7.09 (m, 2H, ArH), 7.31–7.35 (m, 2H, ArH), 7.63 (s, 1H, NH), 9.17 (d,
JHH = 3.4, 1H, NH); [M + H]
+found= 315.0910, [M + H]
+calculated = 315.0909.
5-Dimethoxyphosphoryl-6-methyl-4-(2-fluoro-4-iodophenyl)-3,4-dihydropyrimidin-2(1H)-one (13f): Yield: 63% (0.28 g), white solid; Mp: 250–251 °C; 31P (DMSO-d6) δ 22.2; 13C NMR (DMSO-d6) δ 17.8 (d, 3JCP = 3.5, CCH3), 49.5 (dd, 2JCP = 14.9, 3JCF = 2.9, CHNH), 51.8 (d, 2JCP = 5.1, CH3OI), 52.0 (d, 2JCP = 5.3, CH3OII), 90.7 (d, 1JCP = 207.5, PC=C), 94.0 (d, 3JCF = 8.2, C6), 124.7 (d, 2JCF = 24.7, C3), 131.1 (d, 3JCF = 4.1, C5), 131.6 (d, 2JCF = 13.9, C1), 134.0 (d, 3JCF = 3.1, C4), 150.2 (d, 2JCP = 21.1, PC=C), 152.3 (C=O), 159.4 (d, 1JCF = 252.6, C2); 1H NMR (DMSO-d6) δ 2.11 (d, JHP = 2.4, 3H, CCH3), 3.27 (d, 3JHP = 11.2, 3H, CH3OI), 3.44 (d, 3JHP = 11.3, 3H, CH3OII), 5.11 (dd, 3JHP = 8.0, JHH = 3.2, 1H, CHNH), 7.03 (t, JHH = 8.0, 1H, ArH), 7.48–7.77 [7.67 (br s, NH) overlapped by the multiplet of ArH total int. 3H], 9.26 (d, JHH = 2.8, 1H, NH); [M + H]+found = 440.9870, [M + H]+calculated = 440.9876.
5-Dimethoxyphosphoryl-6-methyl-4-(3-methylphenyl)-3,4-dihydropyrimidin-2(1H)-one (13g) Yield: 76% (0.24 g), white solid; Mp: 205–206 °C; 31P (DMSO-d6) δ 22.9; 13C NMR (DMSO-d6) δ 17.9 (d, 3JCP = 3.5, CCH3), 21.7 (C3CH3), 51.8 (d, 2JCP = 5.0, CH3OI), 52.0 (d, 2JCP = 5.3, CH3OII), 55.4 (d, 2JCP = 15.1, CHNH), 93.0 (d, 1JCP = 205.8, PC=C), 124.1 (C6), 127.6 (C2), 128.6 (C4), 128.8 (C5), 137.9 (C3), 144.9 (C1), 149.3 (d, 2JCP = 20.8, PC=C), 153.0 (C=O); 1H NMR (DMSO-d6) δ 2.09 (d, JHP = 2.5, 3H, CCH3), 2.29 (s, 3H, C3CH3), 3.30 (d, 3JHP = 11.2, 3H, CH3OI), 3.42 (d, 3JHP = 11.5, 3H, CH3OII), 4.80 (dd, 3JHP = 8.8, JHH = 3.5, 1H, CHNH), 7.04–7.11 (m, 3H, ArH), 7.18–7.25 (m, 1H, ArH), 7.64 (d, 1H, JHH = 2.2, NH), 9.16 (d, JHH = 2.7, 1H, NH); [M + H]+found = 311.1160, [M + H]+calculated = 311.1160.
5-Dimethoxyphosphoryl-6-methyl-4-(4-hydroxyphenyl)-3,4-dihydropyrimidin-2(1H)-one (13h): Yield: 60% (0.19 g), white solid; Mp: 230–231 °C; 31P (DMSO-d6) δ 23.2; 13C NMR (DMSO-d6) δ 17.8 (d, 3JCP = 3.6, CCH3), 51.8 (d, 2JCP = 5.0, CH3OI), 51.9 (d, 2JCP = 5.2, CH3OII), 54.9 (d, 2JCP = 15.3, CHNH), 93.5 (d, 1JCP = 205.3, PC=C), 115.5 (C3), 128.2 (C2), 135.6 (C1), 148.8 (d, 2JCP = 20.8, PC=C), 153.0 (C=O), 157.2 (C4); 1H NMR (DMSO-d6) δ 2.08 (d, JHP = 2.5, 3H, CCH3), 3.28 (d, 3JHP = 11.2, 3H, CH3OI), 3.40 (d, 3JHP = 11.3, 3H, CH3OII), 4.72 (dd, 3JHP = 8.7, JHH = 3.4, 1H, CHNH), 6.61–6.81 (m, 2H, ArH), 6.99–7.12 (m, 2H, ArH), 7.54 (br s, 1H, NH), 9.10 (d, JHH = 2.9, 1H, NH), 9.34 (s, 1H, OH); [M+H]+found = 313.0956, [M + H]+calculated = 313.0953.
5-Dimethoxyphosphoryl-6-methyl-4-(4-nitrophenyl)-3,4-dihydropyrimidin-2(1H)-one (
13i): Yield: 68% (0.23 g), yellow solid; Mp: 235–236 °C; Mp [
27]: 235–237 °C;
31P (DMSO-d
6) δ 22.3;
31P (DMSO-d
6) δ [
27] 22.3; [M + H]
+found = 342.0856, [M + H]
+calculated = 342.0854.
5-Dimethoxyphosphoryl-6-methyl-4-(3,4,5-trimethoxyphenyl)-3,4-dihydropyrimidin-2(1H)-one (13j): Yield: 56% (0.22 g), white solid; Mp: 219–220 °C; 31P (DMSO-d6) δ 23.0; 13C NMR (DMSO-d6) δ 17.8 (d, 3JCP = 3.6, CCH3), 52.0 (d, 2JCP = 4.7, CH3OI), 52.1 (d, 2JCP = 5.5, CH3OII), 55.1 (d, 2JCP = 15.1, CHNH), 56.3 (C3OCH3), 60.5 (C4OCH3), 96.8 (d, 1JCP = 206.6, PC=C), 104.4 (C2), 137.4 (C3), 140.3 (C4), 149.6 (d, 2JCP = 20.6, PC=C), 153.0 (C=O), 153.2 (C1); 1H NMR (DMSO-d6) δ 2.06 (d, JHP = 2.4, 3H, CCH3), 3.37 (d, 3JHP = 11.2, 3H, CH3OI), 3.46 (d, 3JPH = 11.3, 3H, CH3OII), 3.61 (s, 3H, C4OCH3), 3.71 (s, 6H, C3OCH3), 4.80 (dd, 3JHP = 9.0, JHH = 3.4, 1H, CHNH), 6.58 (s, 2H, ArH), 7.64 (br s, 1H, NH), 9.16 (d, JHH = 5.5, 1H, NH); [M + H]+found = 387.1322, [M + H]+calculated = 387.1321.
5-Diethoxyphosphoryl-1,6-dimethyl-4-phenyl-3,4-dihydropyrimidin-2(1H)-one (14a): Yield: 56% (0.19 g), light yellow oil; 31P (DMSO-d6) δ 19.9, 20.9; 13C NMR (DMSO-d6) δ 16.3 (d, 3JCP = 6.8, CH3CH2OI), 16.5 (d, 3JCP = 6.5, CH3CH2OII), 17.6 (d, 3JCP = 3.7, CCH3), 29.2 (NCH3), 53.7 (d, 2JCP = 14.1, CHNH), 61.16 (d, 3JCP = 4.1, CH3CH2OI), 61.23 (d, 3JCP = 4.9, CH3CH2OII), 97.3 (d, 1JCP = 203.4, PC=C), 126.9 (C3), 127.9 (C4), 128.8 (C2), 144.2 (d, 3JCP = 1.5, C1), 151.1 (d, 2JCP = 21.5, PC=C), 153.9 (d, JCP = 1.5, C=O); 1H NMR (DMSO-d6) δ 1.00 (t, JHH = 7.1, 3H, CH3CH2OI), 1.15 (t, JHH = 7.0, 3H, CH3CH2OII), 2.32 (d, JHP = 2.5, 3H, CCH3), 3.08 (s, 3H, NCH3), 3.51–3.68 (m, 1H, CHA, CH3CH2OI), 3.71–3.91 (m, 3H, CHB, CH3CH2OI, CH3CH2OII), 4.84 (dd, 3JHP = 10.1, JHH = 3.9, 1H, CHNH), 7.17–7.42, (m, 5H, ArH) 7.85 (br s, 1H, NH); [M + H]+found = 339.1473, [M + H]+calculated = 339.1474.
5-Dimethoxyphosphoryl-1,6-dimethyl-4-phenyl-3,4-dihydropyrimidin-2(1H)-one (14b): Yield: 53% (0.16 g), white solid; Mp: 127–128 °C; 31P (DMSO-d6) δ 23.1, 23.9; 13C NMR (DMSO-d6) δ 17.6 (d, 3JCP = 3.7, CCH3), 30.0 (NCH3), 52.0 (d, 2JCP = 5.0, CH3OI), 52.2 (d, 2JCP = 5.4, CH3OII), 53.6 (d, 2JCP = 14.3, CHNH), 96.0 (d, 1JCP = 204.4, PC=C), 126.9 (C3), 127.9 (C4), 128.89 (C2), 144.2 (d, 3JCP = 1.4, C1), 151.7 (d, 2JCP = 21.4, PC=C), 153.9 (d, JCP = 1.5, C=O); 1H NMR (DMSO-d6) δ 2.31 (d, JHP = 2.6, 3H, CCH3), 3.08 (s, 3H, NCH3), 3.36 (d, 3JHP = 11.2, 3H, CH3OI), 3.46 (d, 3JHP = 11.4, 3H, CH3OII), 4.82 (dd, 3JHP = 10.1, JHH = 3.9, 1H, CHNH), 7.21–7.41, (m, 5H, ArH) 7.89 (t, JHH = 3.9, 1H, NH); [M+H]+found = 311.1159, [M + H]+calculated = 311.1161.
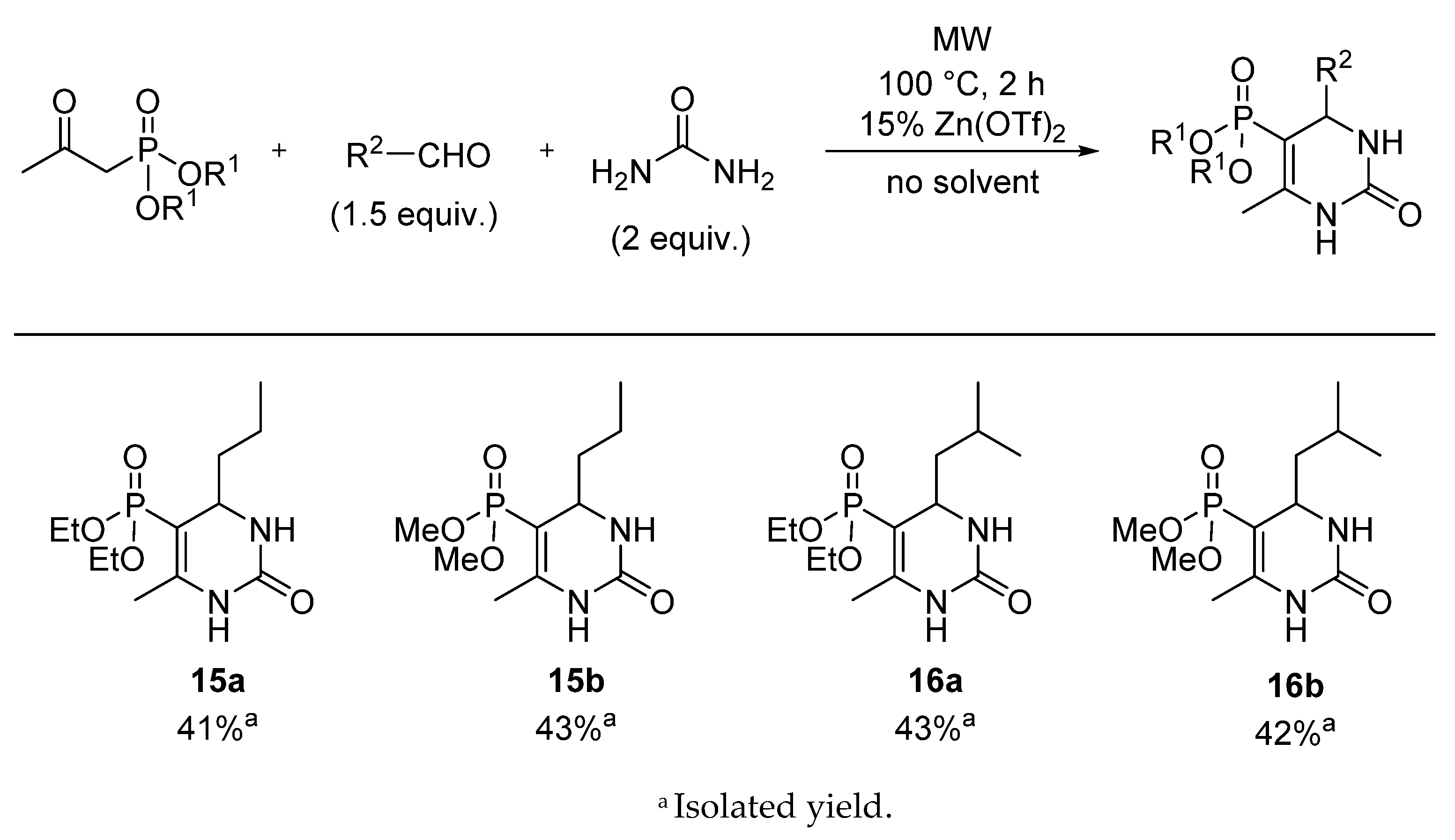
5-Diethoxyphosphoryl-6-methyl-4-propyl-3,4-dihydropyrimidin-2(1H)-one (15a): Yield: 41% (0.12 g), yellow oil; 31P (DMSO-d6) δ 20.5; 13C NMR (DMSO-d6) δ 14.2 (CH2CH2CH3), 16.60 (d, 3JCP = 6.6, CH3CH2OI), 16.61 (d, 3JCP = 6.6, CH3CH2OII), 17.3 (CH2CH2CH3), 17.7 (d, 3JCP = 3.7, CCH3), 39.7 (CH2CH2CH3), 51.4 (d, 2JCP = 16.4, CHNH), 61.0 (d, 2JCP = 5.3, CH3CH2OI), 61.1 (d, 2JCP = 5.3, CH3CH2OII), 94.3 (d, 1JCP = 204.2, PC=C), 148.5 (d, 2JCP = 21.2, PC=C), 153.6 (d, JCP = 1.3, C=O); 1H NMR (DMSO-d6) δ 0.86 (t, JHH = 6.7, 3H, CH2CH2CH3), 1.12–1.53 [1.22 (t, JHH = 7.0, CH3CH2O) overlapped by the multiplet of CH2CH2CH3, CH2CH2CH3 total int. 10H], 2.00 (d, JHP = 2.6, 3H, CCH3), 3.71–3.82 (m, 1H, CHA, CH3CH2OI), 3.84–4.00 (m, 4H, CHB, CH3CH2OI, CH3CH2OII, CHNH), 7.25 (br s, 1H, NH), 8.85 (d, JHH = 3.2, 1H, NH); [M + H]+found = 291.1472, [M + H]+calculated = 291.1473.
5-Dimethoxyphosphoryl-6-methyl-4-propyl-3,4-dihydropyrimidin-2(1H)-one (15b): Yield: 43% (0.11 g), yellow oil; 31P (DMSO-d6) δ 23.6; 13C NMR (DMSO-d6) δ 14.2 (CH2CH2CH3), 17.3 (CH2CH2CH3), 17.7 (d, 3JCP = 3.7, CCH3), 39.7 (CH2CH2CH3), 51.4 (d, 2JCP = 14.7, CHNH), 52.0 (d, 2JCP = 5.2, CH3OI), 52.1 (d, 2JCP = 5.4, CH3OII), 93.0 (d, 1JCP = 204.6, PC=C), 149.2 (d, 2JCP = 21.2, PC=C), 153.6 (d, JCP = 1.3, C=O); 1H NMR (DMSO-d6) δ 0.85 (t, JHH = 7.0, 3H, CH2CH2CH3), 1.16–1.26 (m, 1H, CH2CH2CH3, CHA), 1.30–1.46 (m, 3H, CH2CH2CH3, CHB, CH2CH2CH3), 2.00 (d, JHP = 2.4, 3H, CCH3), 3.71–3.82 (m, 1H, CHA, CH3CH2OI), 3.57 (d, 3JHP = 11.3, 3H, CH3OI), 3.57 (d, 3JHP = 11.2, 3H, CH3OII), 43.71–3.79 (m, 1H, CHNH), 7.26–7.30 (m, 1H, NH), 8.92 (d, JHH = 2.8, 1H, NH); [M + H]+found = 263.1156, [M + H]+calculated = 263.1160.
5-Diethoxyphosphoryl-6-methyl-4-isobutyl-3,4-dihydropyrimidin-2(1H)-one (16a): Yield: 43% (0.13 g), yellow oil; 31P (DMSO-d6) δ 20.2; 13C NMR (DMSO-d6) δ 16.2 (d, 3JCP = 6.4, CH3CH2O), 17.3 (d, 3JCP = 3.5, CCH3), 21.1 (CH2CHCH3I), 22.7 (CH2CHCH3II), 23.9 (CH2CH(CH3)2), 45.6 (CH2CH(CH3)2), 49.1 (d, 2JCP = 14.1, CHNH), 60.5 (d, 2JCP = 5.3, CH3CH2OI), 60.7 (d, 2JCP = 5.4, CH3CH2OII), 95.0 (d, 1JCP = 204.5, PC=C), 147.9 (d, 2JCP = 21.7, PC=C), 153.2 (C=O); 1H NMR (DMSO-d6) δ 0.84 (d, JHH = 6.5, 3H, (CH2CHCH3I), 0.86 (d, JHH = 6.7, 3H, (CH2CHCH3II), 1.08–1.15 (m, 1H, CH2CH(CH3)2, CHA), 1.22 (t, JHH = 7.1, 6H, CH3CH2O), 1.39–1.46 (m, 1H, CH2CH(CH3)2, CHB), 1.68–1.77 (m, 1H, CH2CH(CH3)2), 2.01 (d, JHP = 2.4, 3H, CCH3), 3.72–3.80 (m, 1H, CHNH), 3.87–3.98 (m, 4H, CH3CH2O), 7.37–7.41 (m, 1H, NH), 8.88 (d, JHH = 4.8, 1H, NH); [M + H]+found = 305.1632, [M + H]+calculated = 305.1630.
5-Dimethoxyphosphoryl-6-methyl-4-isobutyl-3,4-dihydropyrimidin-2(1H)-one (16b): Yield: 42% (0.12 g), yellow oil; 31P (DMSO-d6) δ 20.2; 13C NMR (DMSO-d6) δ 17.3 (d, 3JCP = 3.6, CCH3), 21.6 (CH2CHCH3I), 23.2 (CH2CHCH3II), 24.3 (CH2CH(CH3)2), 46.2 (CH2CH(CH3)2), 49.5 (d, 2JCP = 14.3, CHNH), 52.0 (d, 2JCP = 10.2, CH3OI), 52.1 (d, 2JCP = 10.1, CH3OII), 94.2 (d, 1JCP = 204.8, PC=C), 149.0 (d, 2JCP = 21.6, PC=C), 153.5 (d, JCP = 1.2, C=O); 1H NMR (DMSO-d6) δ 0.83 (d, JHH = 6.5, 3H, (CH2CHCH3I), 0.86 (d, JHH = 6.8, 3H, CH2CHCH3II), 1.02–1.18 (m, 1H, CH2CH(CH3)2, CHA), 1.37–1.49 (m, 1H, CH2CH(CH3)2, CHB), 1.64–1.80 (m, 1H, CH2CH(CH3)2), 2.00 (d, JHP = 2.6, 3H, CCH3), 3.57 (d, 3JHP = 11.3, 3H, CH3OI), 3.57 (d, 3JPH = 11.2, 3H, CH3OII), 3.68–3.80 (m, 1H, CHNH), 7.33–7.45 (m, 1H, NH), 8.93 (d, JHH = 3.4, 1H, NH); [M + H]+found = 277.1317, [M + H]+calculated = 277.1317.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
