Review of First-Principles Studies of TiO2: Nanocluster, Bulk, and Material Interface



Abstract
1. Introduction
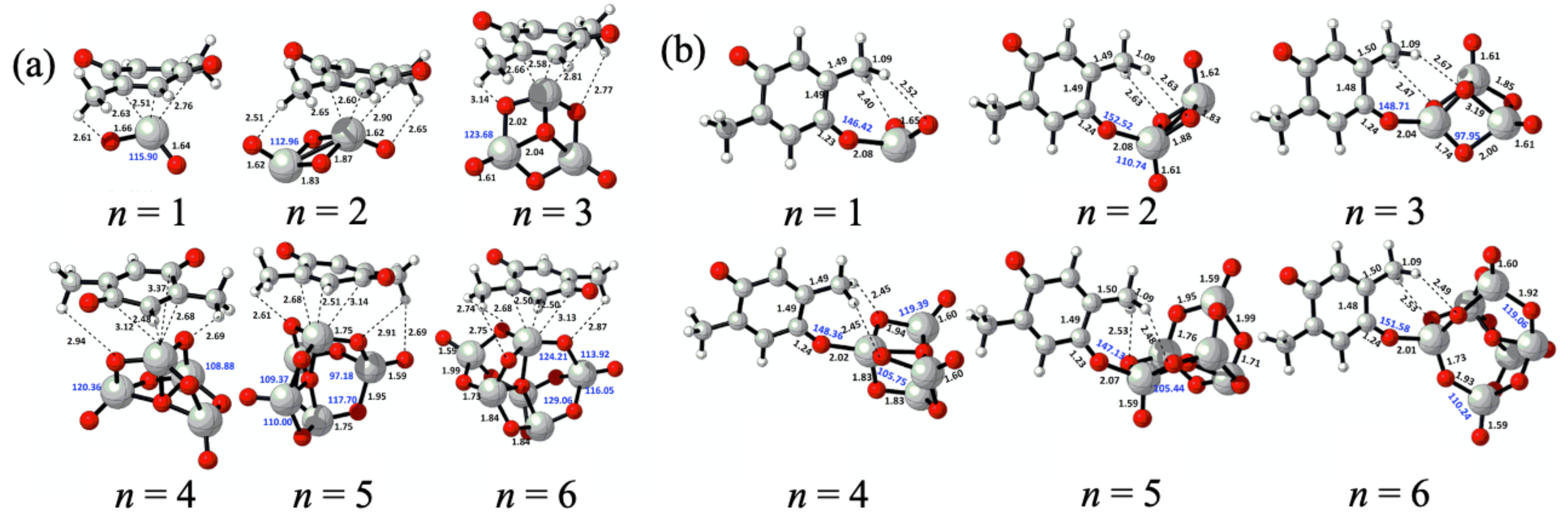
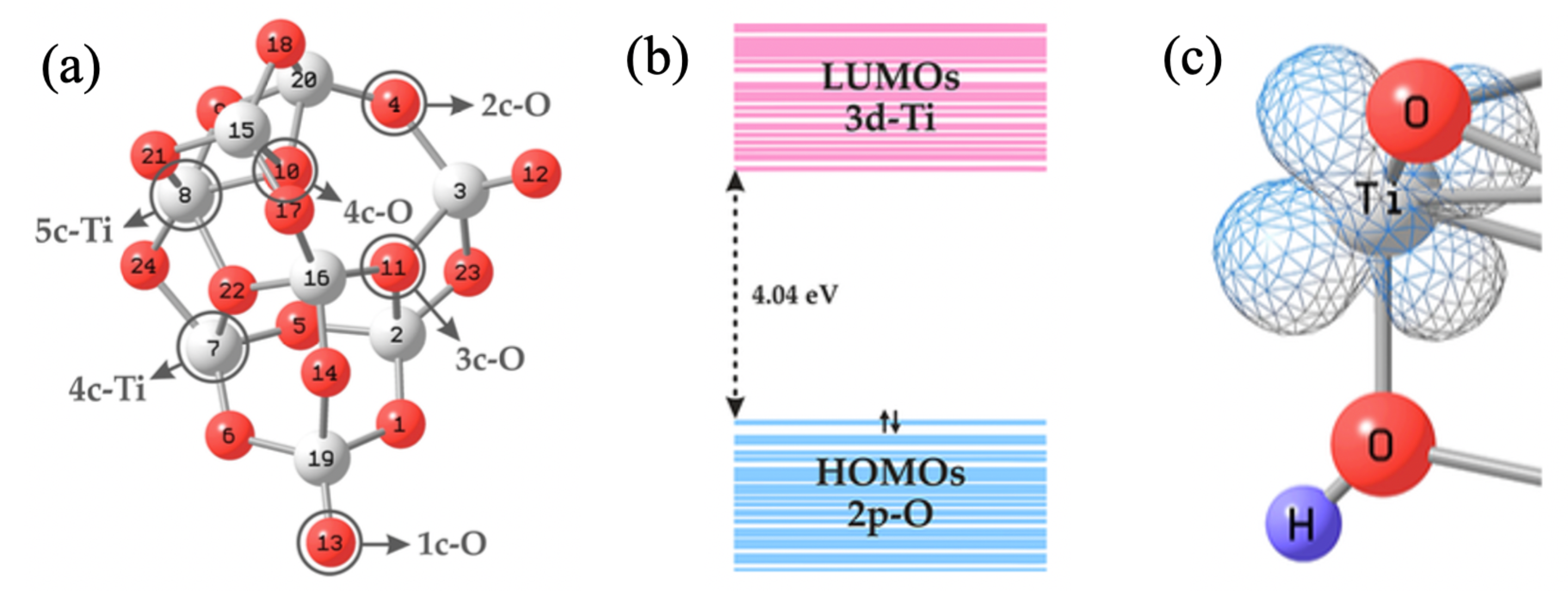
2. Nanocluster
3. Bulk
3.1. Formation Energy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dopant | Doping Site | Properties | Ref. |
|---|---|---|---|
| N | N@O | Visible light absorption due to either narrowed bandgap at a high doping level (≥~4.2 at.%) or gap states at a low doping level (≤~2.1 at.%) | [9,40,63] |
| S | S@O | Redshift of optical absorption edge due to either narrowed bandgap or gap states, depending on doping levels, as in the case of N@O doping. | [43] |
| P | P@O | Visible light absorption due to reduced optical band gap. | [43] |
| P@Ti | Unchanged band gap. | ||
| B | B@O | Redshift due to gap states. | [64] |
| B@Int | Blueshift due to Moss–Burstein shift. | ||
| C | C@O | Different optical absorption thresholds due to discrete gap states. | [65] |
| C@Ti | Visible light absorption due to narrowed band gap; forming O=C double bond. | ||
| Si | Si@Ti | Visible light optical absorption due to narrowed bandgap by 0.25 eV. | [44] |
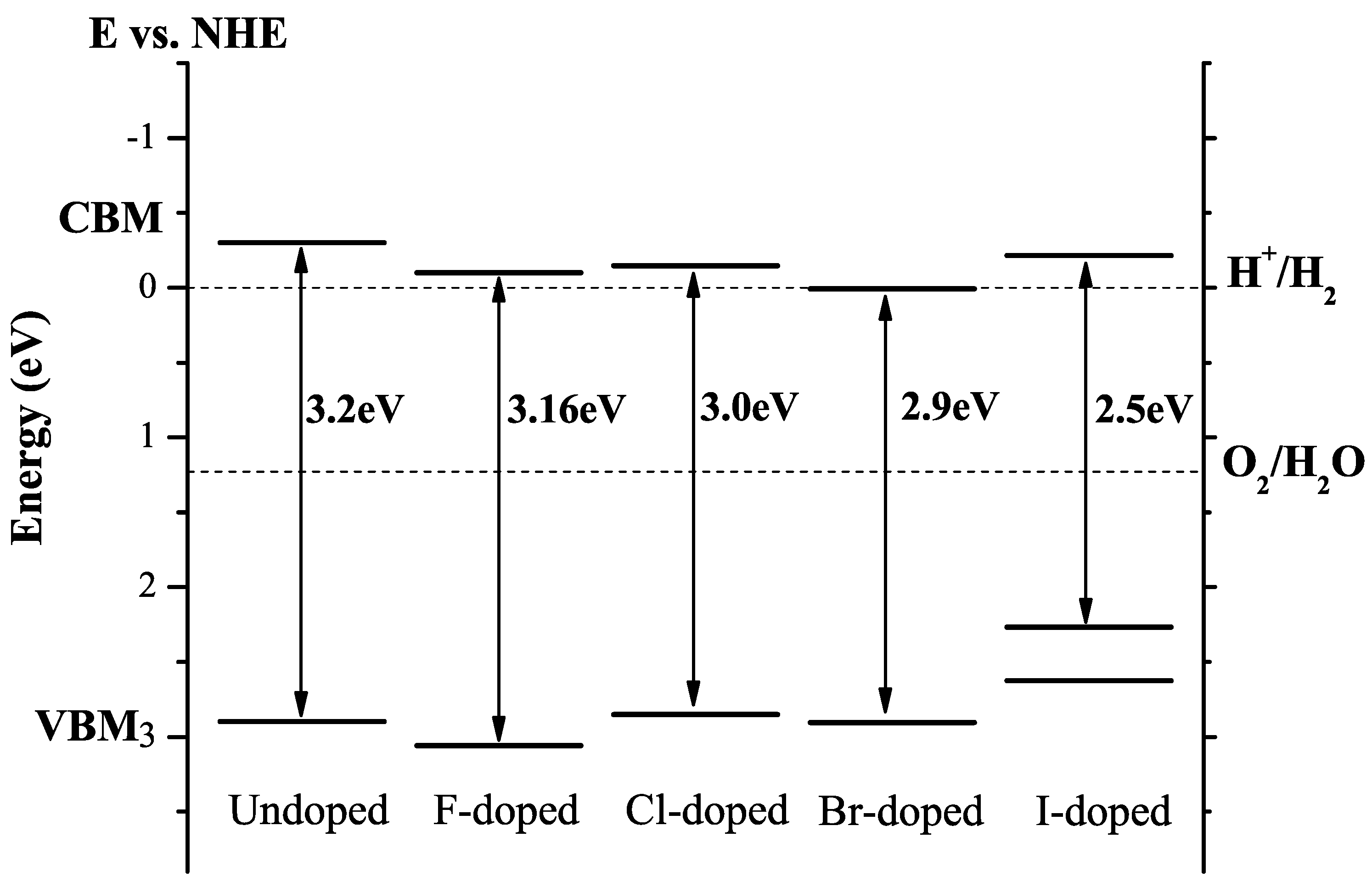
| F | F@O | Nearly unchanged band gap. | [45] |
| Cl | Cl@O | Bandgap reduces by 0.2 eV; reduced oxidation and reduction ability due to band-edge shift. | [45] |
| Br | Br@O | Bandgap reduces about 0.3 eV; CBM shifts downwards by 0.3 eV and VBM keeps unchanged. | [45] |
| I | I@Ti | n-type conductivity, visible light photocatalytic activity due to gap states. | [45] |
| H | H@Int | n-type conductivity due to interstitial H doping. | [66,67,68,69] |
3.2. Nonmetal Doping
N Doping
3.3. S Doping
3.4. P Doping
3.5. B Doping
3.6. C Doping
3.7. Si Doping
3.8. Halogen Doping
3.9. Hydrogen Impurities in TiO2
3.10. Co-Doping
3.10.1. Anion–Anion Co-Doping
3.10.2. Anion–Cation Co-Doping
3.10.3. Cation–Cation Co-Doping
4. Interface
4.1. TiO2/Perovskite Interface
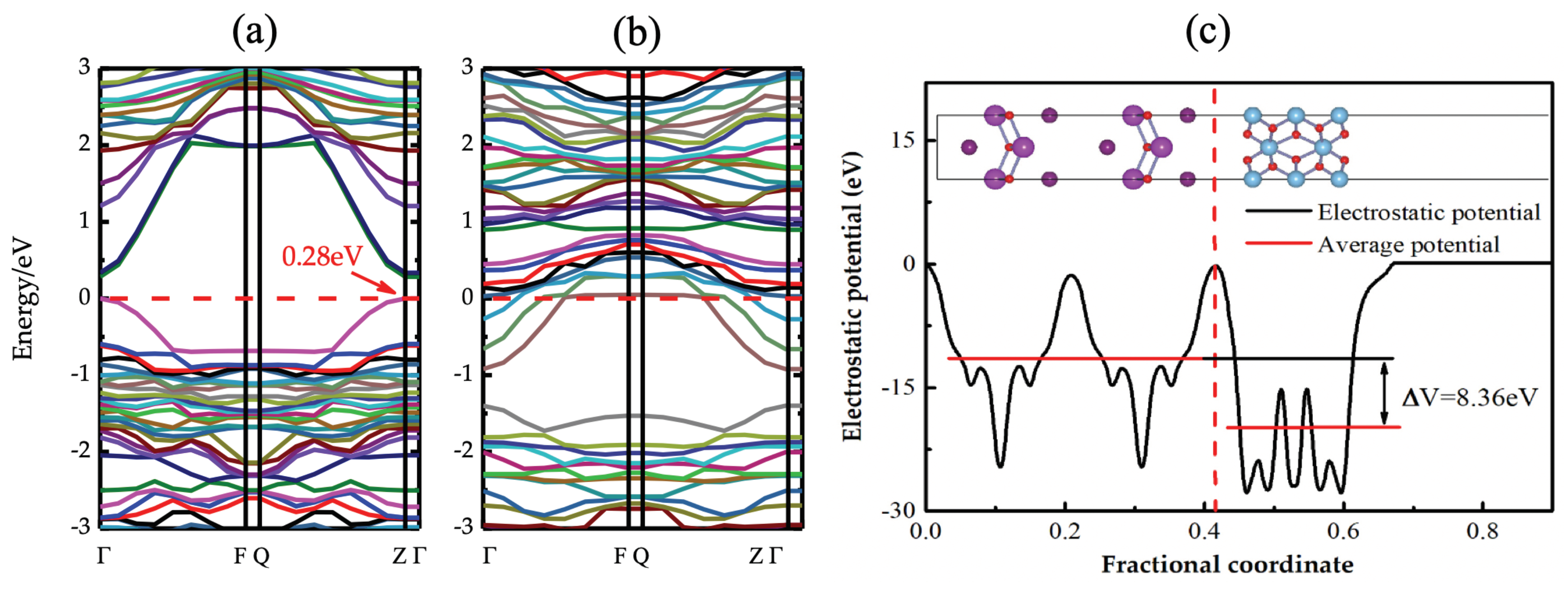
4.2. TiO2/BiOI Interface
4.3. TiO2/RuO2 Interface
5. Conclusions and Outlook
Funding
Conflicts of Interest
Abbreviations
| DFT | Density Functional Theory |
| GGA | Generalized Gradient Approximation |
| DOS | Density of States |
| VB | Valence Band |
| VBM | VB Maximum |
| CB | Conduction Band |
| CBM | CB Minimum |
| 2,5-DMBQ | Dimethylbenzoquinones |
| 4-BBA | 4-bromobenzaldehyde |
| XPS | X-ray Photoelectron Spectroscopy |
| AR-XPS | Angle-resolved XPS |
| UV | Ultraviolet |
References
- Fujishima, A.; Honda, K. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Linsebigler, A.L.; Lu, G.; Yates, J.T., Jr. Photocatalysis on TiO2 Surfaces: Principles, Mechanisms, and Selected Results. Chem. Rev. 1995, 95, 735–758. [Google Scholar] [CrossRef]
- Hoffmann, M.R.; Martin, S.T.; Wonyong, C.; Bahnemann, D.W. Environmental Applications of Semiconductor Photocatalysis. Chem. Rev. 1995, 95, 69–96. [Google Scholar] [CrossRef]
- Chen, X.; Mao, S.S. Titanium Dioxide Nanomaterials: Synthesis, Properties, Modifications, and Applications. Chem. Rev. 2007, 107, 2891–2959. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Jiang, Y.; Zhao, H.; Chen, J.; Cheng, J.; Yang, K.; Li, Y. Engineering Coexposed and 001 and 101 Facets in Oxygen-Deficient TiO2 Nanocrystals for Enhanced CO2 Photoreduction under Visible Light. ACS Catal. 2016, 6, 1097–1108. [Google Scholar] [CrossRef]
- Nie, X.; Zhuo, S.; Gloria, M.; Sohlberg, K. Doping of TiO2 Polymorphs for Altered Optical and Photocatalytic Properties. Int. J. Photoenergy 2009, 2009, 294042. [Google Scholar] [CrossRef]
- Zaleska, A. Doped-TiO2: A Review. Recent Patents Eng. 2008, 2, 157–164. [Google Scholar] [CrossRef]
- Henderson, M.A. A Surface Science Perspective on Photocatalysis. Surf. Sci. Rep. 2011, 66, 185–297. [Google Scholar] [CrossRef]
- Asahi, R.; Morikawa, T.; Ohwaki, T.; Aoki, K.; Taga, Y. Visible-Light Photocatalysis in Nitrogen-Doped Titanium Oxides. Science 2001, 293, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.U.M.; Al-Shahry, M.; William, B.I., Jr. Efficient Photochemical Water Splitting by a Chemically Modified n-TiO2. Science 2002, 297, 2243–2245. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.T.; Nicolas, Y.; Olivier, C.; Toupance, T.; Müller, M.M.; Kleebe, H.J.; Rachut, K.; Ziegler, J.; Klein, A.; Jaegermann, W. Preparation of RuO2/TiO2 Mesoporous Heterostructures and Rationalization of Their Enhanced Photocatalytic Properties by Band Alignment Investigations. J. Phys. Chem. C 2013, 117, 22098–22110. [Google Scholar] [CrossRef]
- Ismail, A.A.; Robben, L.; Bahnemann, D.W. Study of the Efficiency of UV and Visible-Light Photocatalytic Oxidation of Methanol on Mesoporous RuO2-TiO2 Nanocomposites. ChemPhysChem 2011, 12, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Kundu, S.; Vidal, A.B.; Graciani, J.; Ramirez, P.J.; Senanayake, S.D.; Stacchiola, D.; Evans, J.; Liu, P.; Sanz, J.F.; et al. Determining the Behavior of RuO(x) Nanoparticles in Mixed-Metal Oxides: Structural and Catalytic Properties of RuO2/TiO2(110) Surfaces. Angew. Chem. Int. Ed. Engl. 2011, 50, 10198–10202. [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.; Vidal, A.B.; Yang, F.; Ramà rez, P.J.; Senanayake, S.D.; Stacchiola, D.; Evans, J.; Liu, P.; Rodriguez, J.A. Special Chemical Properties of RuOx Nanowires in RuOx/TiO2(110): Dissociation of Water and Hydrogen Production. J. Phys. Chem. C 2012, 116, 4767–4773. [Google Scholar] [CrossRef]
- Xiang, G.; Shi, X.; Wu, Y.; Zhuang, J.; Wang, X. Size Effects in Atomic-Level Epitaxial Redistribution Process of RuO2 Over TiO2. Sci. Rep. 2012, 2, 801. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Xue, Y.B.; Li, X.C.; Li, C.B.; Song, H.X.; Niu, Y.S.; Liu, H.; Mai, X.M.; Zhang, J.X.; Guo, Z.H. Effects of Transition Metal Substituents on Interfacial and Electronic Structure of CH3NH3PbI3/TiO2 Interface: A First-Principles Comparative Study. Nanomaterials 2019, 9, 966. [Google Scholar] [CrossRef] [PubMed]
- Bernal, C.; Yang, K. First-Principles Hybrid Functional Study of the Organic-Inorganic Perovskites CHNH3SnBr3 and CH3NH3SnI3. J. Phys. Chem. C 2014, 118, 24383–24388. [Google Scholar] [CrossRef]
- Akbari, A.; Hashemi, J.; Mosconi, E.; De Angelis, F.; Hakala, M. First Principles Modelling of Perovskite Solar Cells Based on TiO2 and Al2O3: Stability and Interfacial Electronic Structure. J. Mater. Chem. A 2017, 5, 2339–2345. [Google Scholar] [CrossRef]
- Geng, W.; Tong, C.J.; Liu, J.; Zhu, W.J.; Lau, W.M.; Liu, L.M. Structures and Electronic Properties of Different CH3NH3PbI3/TiO2 Interface: A First-Principles Study. Sci. Rep. 2016, 6, 20131. [Google Scholar] [CrossRef] [PubMed]
- Haruyama, J.; Sodeyama, K.; Hamada, I.; Han, L.; Tateyama, Y. First-Principles Study of Electron Injection and Defects at the TiO2/CH3NH3PbI3 Interface of Perovskite Solar Cells. J. Phys. Chem. Lett. 2017, 8, 5840–5847. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, E.; Ronca, E.; De Angelis, F. First-Principles Investigation of the TiO2/Organohalide Perovskites Interface: The Role of Interfacial Chlorine. J. Phys. Chem. Lett. 2014, 5, 2619–2625. [Google Scholar] [CrossRef] [PubMed]
- Sultana, N.; Al Amin, A.; Metin, D.Z.; Gaston, N. Unveiling the Structures and Electronic Properties of CH3NH3PbI3 Interfaces with TiO2, ZnO, and SnO2: A First-Principles Study. J. Mater. Sci. 2019, 54, 13594–13608. [Google Scholar] [CrossRef]
- Xu, Z.N.; Salvador, P.; Kitchin, J.R. First-Principles Investigation of the Epitaxial Stabilization of Oxide Polymorphs: TiO2 on (Sr,Ba)TiO3. ACS Appl. Mater. Interfaces 2017, 9, 4106–4118. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.Z.; Wang, Y.X.; Liu, Y.Y. Stability and Charge Separation of Different CH3NH3SnI3/TiO2 Interface: A First-Principles Study. Appl. Sur. Sci. 2018, 441, 394–400. [Google Scholar] [CrossRef]
- Mohajeri, A.; Haghshenas, F. Global Reactivity and Site Selectivity of (TiO2) Nanoclusters (n = 5–10) Toward Hydrogen Peroxide. Mater. Chem. Phys. 2016, 183, 326–333. [Google Scholar] [CrossRef]
- Bai, F.Y.; Ni, S.; Ren, Y.; Tang, Y.Z.; Zhao, Z.; Pan, X.M. DFT Analysis on the Removal of Dimethylbenzoquinones in Atmosphere and Water Environments: Center Dot OH-Initiated Oxidation and Captured by (TiO2)n Clusters (n = 1–6). J. Hazard. Mater. 2020, 386, 121636. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.W.; Kroes, G.J. Theoretical Study of the Electronic Structure and Stability of Titanium Dioxide Clusters (TiO2)n with n = 1–9. J. Phys. Chem. B 2006, 110, 8998–9007. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.w.; Kroes, G.J. Theoretical Study of Stable, Defect-Free (TiO2)n Nanoparticles with n = 10–16. J. Phys. Chem. C 2007, 111, 16808–16817. [Google Scholar] [CrossRef]
- Qu, Z.W.; Zhu, H. Do Anionic Titanium Dioxide Nano-Clusters Reach Bulk Band Gap? A Density Functional Theory Study. J. Comput. Chem. 2010, 31, 2038–2045. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.B.; Hernandez, W.I.; Cid, A.A.P.; Garcia, J.H.C.; Villanueva, M.S. Prediction, and Physic-Chemical Properties of (TiO2)n n = 15–20 Clusters and Their Possible Catalytic Application: A DFT Study. Comput. Mater. Sci. 2019, 162, 228–235. [Google Scholar] [CrossRef]
- Arab, A.; Ziari, F.; Fazli, M. Electronic Structure and Reactivity of (TiO2)n (n = 1–10) Nano-Clusters: Global and Local Hardness Based DFT Study. Comput. Mater. Sci. 2016, 117, 90–97. [Google Scholar] [CrossRef]
- Joo, P.H.; Cheng, J.; Yang, K. Size Effects and Odd–Even Effects in MoS2 Nanosheets: First-Principles Studies. Phys. Chem. Chem. Phys. 2017, 19, 29927–29933. [Google Scholar] [CrossRef] [PubMed]
- Nollet, H.; Roels, M.; Lutgen, P.; Van der Meeren, P.; Verstraete, W. Removal of PCBs from Wastewater Using Fly Ash. Chemosphere 2003, 53, 655–665. [Google Scholar] [CrossRef]
- Gan, H.L.; Peng, L.; Gu, F.L. A DFT Study on the Mechanism of Photoselective Catalytic Reduction of 4-Bromobenzaldehyde in Different Solvents Employing an OH-Defected TiO2 Cluster Model. Phys. Chem. Chem. Phys. 2017, 19, 27755–27764. [Google Scholar] [CrossRef] [PubMed]
- Andreev, A.S.; Kuznetsov, V.N.; Chizhov, Y.V. Atomic Hydrogen Activated TiO2 Nanocluster: DFT Calculations. J. Phys. Chem. C 2012, 116, 18139–18145. [Google Scholar] [CrossRef]
- Morikawa, T.; Asahi, R.; Ohwaki, T.; Aoki, K.; Taga, Y. Band-Gap Narrowing of Titanium Dioxide by Nitrogen Doping. Jpn. J. Appl. Phys 2001, 40, L561–L563. [Google Scholar] [CrossRef]
- Van de Walle, C.G.; Neugebauer, J. First-Principles Calculations for Defects and Impurities: Applications to III-nitrides. J. Appl. Phys. 2004, 95, 3851–3879. [Google Scholar] [CrossRef]
- Di Valentin, C.; Pacchioni, G.; Selloni, A. Theory of Carbon Doping of Titanium Dioxide. Chem. Mater. 2005, 17, 6656–6665. [Google Scholar] [CrossRef]
- Livraghi, S.; Paganini, M.C.; Giamello, E.; Selloni, A.; Di Valentin, C.; Pacchioni, G. Origin of Photoactivity of Nitrogen-Doped Titanium Dioxide Under Visible Light. J. Am. Chem. Soc. 2006, 128, 15666–15671. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Dai, Y.; Huang, B.; Han, S. Theoretical Study of N-Doped TiO2 Rutile Crystals. J. Phys. Chem. B 2006, 110, 24011–24014. [Google Scholar] [CrossRef] [PubMed]
- Di Valentin, C.; Finazzi, E.; Pacchioni, G.; Selloni, A.; Livraghi, S.; Paganini, M.C.; Giamello, E. N-doped TiO2: Theory and Experiment. Chem. Phys. 2007, 339, 44–56. [Google Scholar] [CrossRef]
- Yang, K.; Dai, Y.; Huang, B. Study of Nitrogen-Concentration Influence on N-doped TiO2 Anatase from First-Principles. J. Phys. Chem. C 2007, 111, 12086–12090. [Google Scholar] [CrossRef]
- Yang, K.; Dai, Y.; Huang, B. Understanding Photocatalytic Activity of S- and P-doped TiO2 Under Visible Light from First-Principles. J. Phys. Chem. C 2007, 111, 18985–18994. [Google Scholar] [CrossRef]
- Yang, K.; Dai, Y.; Huang, B. First-Principles Calculations for Geometrical Structures and Electronic Properties of Si-doped TiO2. Chem. Phys. Lett. 2008, 456, 71–75. [Google Scholar] [CrossRef]
- Yang, K.; Dai, Y.; Huang, B.; Whangbo, M.H. Density Functional Characterization of the Band Edges, the Band Gap States, and the Preferred Doping Sites of Halogen-Doped TiO2. Chem. Mater. 2008, 20, 6528–6534. [Google Scholar] [CrossRef]
- Long, R.; Dai, Y.; Meng, G.; Huang, B. Energetic and Electronic Properties of X- (Si, Ge, Sn, Pb) Doped TiO2 from First-Principles. Phys. Chem. Chem. Phys. 2009, 11, 8165–8172. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Chen, Q.; Xu, Y.; Wu, D.; Huo, C.f. Investigation of the Silicon Concentration Effect on Si-doped Anatase TiO2 by First-Principles Calculation. J. Solid State Chem. 2011, 184, 1983–1988. [Google Scholar] [CrossRef]
- Matsushima, S.; Takehara, K.; Yamane, H.; Yamada, K.; Nakamura, H.; Arai, M.; Kobayashi, K. First-Principles Energy Band Calculation for Undoped and S-doped TiO2 with Anatase Structure. J. Phys. Chem. Solids 2007, 68, 206–210. [Google Scholar] [CrossRef]
- Zheng, J.W.; Bhattcahrayya, A.; Wu, P.; Chen, Z.; Highfield, J.; Dong, Z.; Xu, R. The Origin of Visible Light Absorption in Chalcogen Element (S, Se, and Te)-Doped Anatase TiO2 Photocatalysts. J. Phys. Chem. C 2010, 114, 7063–7069. [Google Scholar] [CrossRef]
- Yu, J.C.; Ho, W.; Yu, J.; Yip, H.; Wong, P.K.; Zhao, J. Efficient Visible-Light-Induced Photocatalytic Disinfection on Sulfur-Doped Nanocrystalline Titania. Environ. Sci. Technol. 2005, 39, 1175–1179. [Google Scholar] [CrossRef] [PubMed]
- Ohno, T.; Akiyoshi, M.; Umebayashi, T.; Asai, K.; Mitsui, T.; Matsumura, M. Preparation of S-doped TiO2 Photocatalysts and Their Photocatalytic Activities Under Visible Light. Appl. Catal. A Gen. 2004, 265, 115–121. [Google Scholar] [CrossRef]
- Ohno, T.; Mitsui, T.; Matsumura, M. Photocatalytic Activity of S-doped TiO2 Photocatalyst Under Visible Light. Chem. Lett. 2003, 32, 364. [Google Scholar] [CrossRef]
- Umebayashi, T.; Yamaki, T.; Itoh, H.; Asai, K. Band Gap Narrowing of Titanium Dioxide by Sulfur Doping. Appl. Phys. Lett. 2002, 81, 454–456. [Google Scholar] [CrossRef]
- Umebayashi, T.; Yamaki, T.; Tanaka, S.; Asai, K. Visible Light-Induced Degradation of Methylene Blue on S-doped TiO2. Chem. Lett. 2003, 32, 330. [Google Scholar] [CrossRef]
- Long, R.; Dai, Y.; Huang, B. Structural and Electronic Properties of Iodine-Doped Anatase and Rutile TiO2. Comput. Mater. Sci. 2009, 45, 223–228. [Google Scholar] [CrossRef]
- Ma, X.; Dai, Y.; Wei, W.; Huang, B.; Whangbo, M.H. Insights Into How Fluorine-Adsorption and n-Type Doping Affect the Relative Stability of the (001) and (101) Surfaces of TiO2: Enhancing the Exposure of More Active but Thermodynamically Less Stable (001). J. Phys. Chem. Lett. 2015, 6, 1876–1882. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Dai, Y.; Guo, M.; Huang, B. Relative Photooxidation and Photoreduction Activities of the 100, 101, and 001 Surfaces of Anatase TiO2. Langmuir 2013, 29, 13647–13654. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Dai, Y.; Guo, M.; Huang, B. Insights Into the Role of Surface Distortion in Promoting the Separation and Transfer of Photogenerated Carriers in Anatase TiO2. J. Phys. Chem. C 2013, 117, 24496–24502. [Google Scholar] [CrossRef]
- Reeves, K.G.; Dambournet, D.; Laberty-Robert, C.; Vuilleumier, R.; Salanne, M. A First-Principles Computational Comparison of Defect-Free and Disordered, Fluorinated Anatase TiO2 (001) Interfaces with Water. RSC Adv. 2020, 10, 8982–8988. [Google Scholar] [CrossRef]
- Di Valentin, C.; Finazzi, E.; Pacchioni, G.; Selloni, A.; Livraghi, S.; Czoska, A.M.; Paganini, M.C.; Giamello, E. Density Functional Theory and Electron Paramagnetic Resonance Study on the Effect of N-F Codoping of TiO2. Chem. Mater. 2008, 20, 3706–3714. [Google Scholar] [CrossRef]
- Geng, H.; Yin, S.; Yang, X.; Shuai, Z.; Liu, B. Geometric and Electronic Structures of the Boron-Doped Photocatalyst TiO2. J. Phys. Condens. Matter 2006, 18, 87–96. [Google Scholar] [CrossRef]
- Finazzi, E.; Di Valentin, C.; Pacchioni, G. Boron-Doped Anatase TiO2: Pure and Hybrid DFT Calculations. J. Phys. Chem. C 2008, 113, 220–228. [Google Scholar] [CrossRef]
- Lee, J.; Park, J.; Cho, J. Electronic Properties of N- and C-doped TiO2. Appl. Phys. Lett. 2005, 87, 011904. [Google Scholar] [CrossRef]
- Yang, K.; Dai, Y.; Huang, B. Origin of the Photoactivity in B-doped Anatase and Rutile TiO2 from First Principles. Phys. Rev. B 2007, 76, 195201. [Google Scholar] [CrossRef]
- Yang, K.; Dai, Y.; Huang, B.; Whangbo, M.H. Density Functional Characterization of the Visible-Light Absorption in Substitutional C-anion and C-cation Doped TiO2. J. Phys. Chem. C 2009, 113, 2624–2629. [Google Scholar] [CrossRef]
- Kilic, C.; Zunger, A. N-Type Doping of Oxides by Hydrogen. Appl. Phys. Lett. 2002, 81, 73–75. [Google Scholar] [CrossRef]
- Peacock, P.W.; Robertson, J. Behavior of Hydrogen in High Dielectric Constant Oxide Gate Insulators. Appl. Phys. Lett. 2003, 83, 2025–2027. [Google Scholar] [CrossRef]
- Robertson, J.; Peacock, P.W. Doping and Hydrogen in Wide Gap Oxides. Thin Solid Film. 2003, 445, 155–160. [Google Scholar] [CrossRef]
- Xiong, K.; Robertson, J.; Clark, S.J. Behavior of Hydrogen in Wide Band Gap Oxides. J. Appl. Phys. 2007, 102, 083710-13. [Google Scholar] [CrossRef]
- Irie, H.; Watanabe, Y.; Hashimoto, K. Nitrogen-Concentration Dependence on Photocatalytic Activity of TiO2−xNx Powders. J. Phys. Chem. B 2003, 107, 5483–5486. [Google Scholar] [CrossRef]
- Lindgren, T.; Mwabora, J.M.; Avendaño, E.; Jonsson, J.; Hoel, A.; Granqvist, C.G.; Lindquist, S.E. Photoelectrochemical and Optical Properties of Nitrogen Doped Titanium Dioxide Films Prepared by Reactive DC Magnetron Sputtering. J. Phys. Chem. B 2003, 107, 5709–5716. [Google Scholar] [CrossRef]
- Diwald, O.; Thompson, T.L.; Zubkov, T.; Goralski, E.G.; Walck, S.D.; Yates, J.T. Photochemical Activity of Nitrogen-Doped Rutile TiO2(110) in Visible Light. J. Phys. Chem. B 2004, 108, 6004–6008. [Google Scholar] [CrossRef]
- Sakthivel, S.; Janczarek, M.; Kisch, H. Visible Light Activity and Photoelectrochemical Properties of Nitrogen-Doped TiO2. J. Phys. Chem. B 2004, 108, 19384–19387. [Google Scholar] [CrossRef]
- Torres, G.R.; Lindgren, T.; Lu, J.; Granqvist, C.G.; Lindquist, S.E. Photoelectrochemical Study of Nitrogen-Doped Titanium Dioxide for Water Oxidation. J. Phys. Chem. B 2004, 108, 5995–6003. [Google Scholar] [CrossRef]
- Matsui, H.; Tabata, H.; Hasuike, N.; Harima, H.; Mizobuchi, B. Epitaxial Growth and Characteristics of N-doped Anatase TiO2 Films Grown Using a Free-Radical Nitrogen Oxide Source. J. Appl. Phys. 2005, 97, 123511. [Google Scholar] [CrossRef]
- Nakano, Y.; Morikawa, T.; Ohwaki, T.; Taga, Y. Deep-Level Optical Spectroscopy Investigation of N-doped TiO2 Films. Appl. Phys. Lett. 2005, 86, 132104. [Google Scholar] [CrossRef]
- Batzill, M.; Morales, E.H.; Diebold, U. Influence of Nitrogen Doping on the Defect Formation and Surface Properties of TiO2 Rutile and Anatase. Phys. Rev. Lett. 2006, 96, 026103. [Google Scholar] [CrossRef]
- Kitano, M.; Funatsu, K.; Matsuoka, M.; Ueshima, M.; Anpo, M. Preparation of Nitrogen-Substituted TiO2 Thin Film Photocatalysts by the Radio Frequency Magnetron Sputtering Deposition Method and Their Photocatalytic Reactivity Under Visible Light Irradiation. J. Phys. Chem. B 2006, 110, 25266–25272. [Google Scholar] [CrossRef]
- Wang, Y.; Feng, C.; Jin, Z.; Zhang, J.; Yang, J.; Zhang, S. A Novel N-doped TiO2 with High Visible Light Photocatalytic Activity. J. Mater. Chem. A 2006, 260, 1–3. [Google Scholar] [CrossRef]
- Borrás, A.; López, C.; Rico, V.; Gracia, F.; González-Elipe, A.R.; Richter, E.; Battiston, G.; Gerbasi, R.; McSporran, N.; Sauthier, G.; et al. Effect of Visible and UV Illumination on the Water Contact Angle of TiO2 Thin Films with Incorporated Nitrogen. J. Phys. Chem. C 2007, 111, 1801–1808. [Google Scholar] [CrossRef]
- Cong, Y.; Zhang, J.; Chen, F.; Anpo, M. Synthesis and Characterization of Nitrogen-Doped TiO2 Nanophotocatalyst with High Visible Light Activity. J. Phys. Chem. C 2007, 111, 6976–6982. [Google Scholar] [CrossRef]
- Livraghi, S.; Czoska, A.M.; Paganini, M.C.; Giamello, E. Preparation and Spectroscopic Characterization of Visible Light Sensitized N Doped TiO2 (rutile). J. Solid State Chem. 2009, 182, 160–164. [Google Scholar] [CrossRef]
- Ihara, T.; Miyoshi, M.; Iriyama, Y.; Matsumoto, O.; Sugihara, S. Visible-Light-Active Titanium Oxide Photocatalyst Realized by an Oxygen-Deficient Structure and by Nitrogen Doping. Appl. Catal. B Environ. 2003, 42, 403–409. [Google Scholar] [CrossRef]
- Zhao, Z.; Liu, Q. Mechanism of Higher Photocatalytic Activity of Anatase TiO2 Doped with Nitrogen Under Visible-Light Irradiation from Density Functional Theory Calculation. J. Phys. D Appl. Phys. 2008, 41, 025105. [Google Scholar] [CrossRef]
- Okato, T.; Sakano, T.; Obara, M. Suppression of Photocatalytic Efficiency in Highly N-doped Anatase Films. Phys. Rev. B 2005, 72, 115124. [Google Scholar] [CrossRef]
- Di Valentin, C.; Pacchioni, G.; Selloni, A. Origin of the Different Photoactivity of N-doped Anatase and Rutile TiO2. Phys. Rev. B 2004, 70, 085116. [Google Scholar] [CrossRef]
- Di Valentin, C.; Pacchioni, G.; Selloni, A.; Livraghi, S.; Giamello, E. Characterization of Paramagnetic Species in N-Doped TiO2 Powders by EPR Spectroscopy and DFT Calculations. J. Phys. Chem. B 2005, 109, 11414–11419. [Google Scholar] [CrossRef]
- Xu, T.H.; Song, C.L.; Liu, Y.; Han, G.R. Band Structures of TiO2 Doped with N, C and B. J. Zhejiang Univ. Sci. B 2006, 7, 299–303. [Google Scholar] [CrossRef]
- Ma, X.; Dai, Y.; Huang, B. Origin of the Increased Photocatalytic Performance of TiO2 Nanocrystal Composed of Pure Core and Heavily Nitrogen-Doped Shell: A Theoretical Study. ACS Appl. Mater. Interfaces 2014, 6, 22815–22822. [Google Scholar] [CrossRef]
- Lin, Z.; Orlov, A.; Lambert, R.M.; Payne, M.C. New Insights Into the Origin of Visible Light Photocatalytic Activity of Nitrogen-Doped and Oxygen-Deficient Anatase TiO2. J. Phys. Chem. B 2005, 109, 20948–20952. [Google Scholar] [CrossRef]
- Serpone, N. Is the Band Gap of Pristine TiO2 Narrowed by Anion- and Cation-Doping of Titanium Dioxide in Second-Generation Photocatalysts? J. Phys. Chem. B 2006, 110, 24287–24293. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, V.N.; Serpone, N. Visible Light Absorption by Various Titanium Dioxide Specimens. J. Phys. Chem. B 2006, 110, 25203–25209. [Google Scholar] [CrossRef] [PubMed]
- Graciani, J.; Ortega, Y.; Fdez Sanz, J. Carbon Doping of the TiO2 (110) Rutile Surface. A Theoretical Study Based on DFT. Chem. Mater. 2009, 21, 1431–1438. [Google Scholar] [CrossRef]
- Long, R.; English, N.J.; Dai, Y. First-Principles Study of S Doping at the Rutile TiO2 (110) Surface. J. Phys. Chem. C 2009, 113, 17464–17470. [Google Scholar] [CrossRef]
- Long, R.; English, N.J. Energetic and Electronic Properties of P Doping at the Rutile TiO2 (110) Surface from First Principles. J. Phys. Chem. C 2009, 113, 9423–9430. [Google Scholar] [CrossRef][Green Version]
- Kavan, L.; Grätzel, M.; Gilbert, S.E.; Klemenz, C.; Scheel, H.J. Electrochemical and Photoelectrochemical Investigation of Single-Crystal Anatase. J. Am. Chem. Soc. 1996, 118, 6716–6723. [Google Scholar] [CrossRef]
- Pascual, J.; Camassel, J.; Mathieu, H. Resolved Quadrupolar Transition in TiO2. Phys. Rev. Lett. 1977, 39, 1490–1493. [Google Scholar] [CrossRef]
- Diwald, O.; Thompson, T.L.; Goralski, E.G.; Walck, S.D.; Yates, J.T. The Effect of Nitrogen Ion Implantation on the Photoactivity of TiO2 Rutile Single Crystals. J. Phys. Chem. B 2004, 108, 52–57. [Google Scholar] [CrossRef]
- Umebayashi, T.; Yamaki, T.; Yamamoto, S.; Miyashita, A.; Tanaka, S. Sulfur-Doping of Rutile-Titanium Dioxide by Ion Implantation: Photocurrent Spectroscopy and First-Principles Band Calculation Studies. J. Appl. Phys. 2003, 93, 5156–5160. [Google Scholar] [CrossRef]
- Ho, W.; Yu, J.C.; Lee, S. Low-Temperature Hydrothermal Synthesis of S-doped TiO2 with Visible Light Photocatalytic Activity. J. Solid State Chem. 2006, 179, 1171–1176. [Google Scholar] [CrossRef]
- Li, H.; Zhang, X.; Huo, Y.; Zhu, J. Supercritical Preparation of a Highly Active S-Doped TiO2 Photocatalyst for Methylene Blue Mineralization. Environ. Sci. Technol. 2007, 41, 4410–4414. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Bai, Y.; Cheng, Y.; Jin, W.; Xu, N. Preparation and Characterization of Visible-Light-Driven Carbon-Sulfur-Codoped TiO2 Photocatalysts. Ind. Eng. Chem. Res. 2006, 45, 4971–4976. [Google Scholar] [CrossRef]
- Ohno, T. Preparation of Visible Light Active S-doped TiO2 Photocatalysts and Their Photocatalytic Activities. Water Sci. Technol. 2004, 49, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Yamashita, F.; Tanaka, I.; Matsubara, E.; Muramatsu, A. Electronic States of Sulfur Doped TiO2 by First Principles Calculations. Mater. Trans. 2004, 45, 1987–1990. [Google Scholar] [CrossRef][Green Version]
- Sathish, M.; Sankaran, M.; Viswanathan, B.; Viswanath, R.P. DFT Studies on Anionic Hetero Atom (N Or/and S) Substitution in TiO2. Indian J. Chem. A 2007, 46, 895–898. [Google Scholar]
- Tian, F.; Liu, C. DFT Description on Electronic Structure and Optical Absorption Properties of Anionic S-Doped Anatase TiO2. J. Phys. Chem. B 2006, 110, 17866–17871. [Google Scholar] [CrossRef]
- Shi, Q.; Yang, D.; Jiang, Z.; Li, J. Visible-Light Photocatalytic Regeneration of NADH Using P-doped TiO2 Nanoparticles. J. Mol. Catal. Enzym. 2006, 43, 44–48. [Google Scholar] [CrossRef]
- Yu, H.F. Photocatalytic Abilities of Gel-Derived P-doped TiO2. J. Phys. Chem. Solids 2007, 68, 600–607. [Google Scholar] [CrossRef]
- Lin, L.; Lin, W.; Zhu, Y.; Zhao, B.; Xie, Y. Phosphor-Doped Titania-Novel Photocatalyst Active in Visible Light. Chem. Lett. 2005, 34, 284. [Google Scholar] [CrossRef]
- Yu, J.C.; Zheng, Z.; Zhao, J. Synthesis and Characterization of Phosphated Mesoporous Titanium Dioxide with High Photocatalytic Activity. Chem. Mater. 2003, 15, 2280–2286. [Google Scholar] [CrossRef]
- Jung, K.Y.; Park, S.B.; Ihm, S.K. Local Structure and Photocatalytic Activity of B2O3-SiO2/TiO2 Ternary Mixed Oxides Prepared by Sol-Gel Method. Appl. Catal. B Environ. 2004, 51, 239–245. [Google Scholar] [CrossRef]
- Chen, D.; Yang, D.; Wang, Q.; Jiang, Z. Effects of Boron Doping on Photocatalytic Activity and Microstructure of Titanium Dioxide Nanoparticles. Ind. Eng. Chem. Res. 2006, 45, 4110–4116. [Google Scholar] [CrossRef]
- Zhao, W.; Ma, W.; Chen, C.; Zhao, J.; Shuai, Z. Efficient Degradation of Toxic Organic Pollutants with Ni2O3/TiO2−xBx Under Visible Irradiation. J. Am. Chem. Soc. 2004, 126, 4782–4783. [Google Scholar] [CrossRef] [PubMed]
- Zhukov, V.P.; Zainullina, V.M.; Chulkov, E.V. Ab Initio Approach Struct. Electron. Opt. Prop. B- C- N-Doped Anatase. Int. J. Mod. Phys. B 2010, 24, 6049–6067. [Google Scholar] [CrossRef]
- Yang, K.; Dai, Y.; Huang, B. Density Functional Study of Boron-Doped Anatase TiO2. J. Phys. Chem. C 2010, 114, 19830–19834. [Google Scholar] [CrossRef]
- Pankove, J.I. Optical Processes in Semiconductors; Dover Publications: New York, NY, USA, 1975; p. 189. [Google Scholar]
- Moss, T.S. The Interpretation of the Properties of Indium Antimonide. Proc. Phys. Soc. B 1954, 67, 775. [Google Scholar] [CrossRef]
- Burstein, E. Anomalous Optical Absorption Limit in InSb. Phys. Rev. 1954, 93, 632–633. [Google Scholar] [CrossRef]
- Jin, H.; Dai, Y.; Wei, W.; Huang, B. Density Functional Characterization of B Doping at Rutile TiO2 (110) Surface. J. Phys. D Appl. Phys. 2008, 41, 195411. [Google Scholar] [CrossRef]
- Feng, N.; Zheng, A.; Wang, Q.; Ren, P.; Gao, X.; Liu, S.B.; Shen, Z.; Chen, T.; Deng, F. Boron Environments in B-doped and (B, N)-codoped TiO2 Photocatalysts: A Combined Solid-State NMR and Theoretical Calculation Study. J. Phys. Chem. C 2011, 115, 2709–2719. [Google Scholar] [CrossRef]
- Shen, M.; Wu, Z.; Huang, H.; Du, Y.; Zou, Z.; Yang, P. Carbon-Doped Anatase TiO2 Obtained from TiC for Photocatalysis Under Visible Light Irradiation. Mater. Lett. 2006, 60, 693–697. [Google Scholar] [CrossRef]
- Hsu, S.W.; Yang, T.S.; Chen, T.K.; Wong, M.S. Ion-Assisted Electron-Beam Evaporation of Carbon-Doped Titanium Oxide Films as Visible-Light Photocatalyst. Thin Solid Film. 2007, 515, 3521–3526. [Google Scholar] [CrossRef]
- Wang, S.H.; Chen, T.K.; Rao, K.K.; Wong, M.S. Nanocolumnar Titania Thin Films Uniquely Incorporated with Carbon for Visible Light Photocatalysis. Appl. Catal. B Environ. 2007, 76, 328–334. [Google Scholar] [CrossRef]
- Wong, M.S.; Wang, S.H.; Chen, T.K.; Weng, C.W.; Rao, K.K. Co-Sputtered Carbon-Incorporated Titanium Oxide Films as Visible Light-Induced Photocatalysts. Surf. Coat. Technol. 2007, 202, 890–894. [Google Scholar] [CrossRef]
- Wu, G.; Nishikawa, T.; Ohtani, B.; Chen, A. Synthesis and Characterization of Carbon-Doped TiO2 Nanostructures with Enhanced Visible Light Response. Chem. Mater. 2007, 19, 4530–4537. [Google Scholar] [CrossRef]
- Choi, Y.; Umebayashi, T.; Yoshikawa, M. Fabrication and Characterization of C-doped Anatase TiO2 Photocatalysts. J. Mater. Sci. 2004, 39, 1837–1839. [Google Scholar] [CrossRef]
- Irie, H.; Watanabe, Y.; Hashimoto, K. Carbon-Doped Anatase TiO2 Powders as a Visible-Light Sensitive Photocatalyst. Chem. Lett. 2003, 32, 772–773. [Google Scholar] [CrossRef]
- Irie, H.; Washizuka, S.; Hashimoto, K. Hydrophilicity on Carbon-Doped TiO2 Thin Films Under Visible Light. Thin Solid Film. 2006, 510, 21–25. [Google Scholar] [CrossRef]
- Huang, Y.; Ho, W.; Lee, S.; Zhang, L.; Li, G.; Yu, J.C. Effect of Carbon Doping on the Mesoporous Structure of Nanocrystalline Titanium Dioxide and Its Solar-Light-Driven Photocatalytic Degradation of NOx. Langmuir 2008, 24, 3510–3516. [Google Scholar] [CrossRef]
- Yang, X.; Cao, C.; Hohn, K.; Erickson, L.; Maghirang, R.; Hamal, D.; Klabunde, K. Highly Visible-Light Active C- and V-doped TiO2 for Degradation of Acetaldehyde. J. Catal. 2007, 0, 1–7. [Google Scholar] [CrossRef]
- Mohapatra, S.; Misra, M.; Mahajan, V.; Raja, K. A Novel Method for the Synthesis of Titania Nanotubes Using Sonoelectrochemical Method and Its Application for Photoelectrochemical Splitting of Water. J. Catal. 2007, 246, 362–369. [Google Scholar] [CrossRef]
- Mohapatra, S.K.; Misra, M.; Mahajan, V.K.; Raja, K.S. Design of a Highly Efficient Photoelectrolytic Cell for Hydrogen Generation by Water Splitting: Application of TiO2−xCx Nanotubes as a Photoanode and Pt/TiO2 Nanotubes as a Cathode. J. Phys. Chem. C 2007, 111, 8677–8685. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, S.; Bard, A.J. Novel Carbon-Doped TiO2 Nanotube Arrays with High Aspect Ratios for Efficient Solar Water Splitting. Nano Lett. 2006, 6, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Killmeyer, R.; Gray, M.L.; Khan, S.U. Enhanced Carbon Doping of n-TiO2 Thin Films for Photoelectrochemical Water Splitting. Electrochem. Commun. 2006, 8, 1650–1654. [Google Scholar] [CrossRef]
- Kamisaka, H.; Adachi, T.; Yamashita, K. Theoretical Study of the Structure and Optical Properties of Carbon-Doped Rutile and Anatase Titanium Oxides. J. Chem. Phys. 2005, 123, 084704. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Ai, Z.; Jia, F.; Zhang, L.; Fan, X.; Zou, Z. Low Temperature Preparation and Visible Light Photocatalytic Activity of Mesoporous Carbon-Doped Crystalline TiO2. Appl. Catal. B Environ. 2007, 69, 138–144. [Google Scholar] [CrossRef]
- Tian, F.; Liu, C.; Zhang, D.; Fu, A.; Duan, Y.; Yuan, S.; Yu, J.C. On the Origin of the Visible-Light Activity of Titanium Dioxide Doped with Carbonate Species. ChemPhysChem 2010, 11, 3269–3272. [Google Scholar] [CrossRef]
- Oh, S.M.; Kim, S.S.; Lee, J.E.; Ishigaki, T.; Park, D.W. Effect of Additives on Photocatalytic Activity of Titanium Dioxide Powders Synthesized by Thermal Plasma. Thin Solid Film. 2003, 435, 252–258. [Google Scholar] [CrossRef]
- Yan, X.; He, J.; Evans, D.G.; Duan, X.; Zhu, Y. Preparation, Characterization and Photocatalytic Activity of Si-doped and Rare Earth-Doped TiO2 from Mesoporous Precursors. Appl. Catal. B Environ. 2005, 55, 243–252. [Google Scholar] [CrossRef]
- Ozaki, H.; Iwamoto, S.; Inoue, M. Enhanced Visible Light Sensitivity of Nitrogen-Doped Nanocrystalline Si-Modified Titania Prepared by the Glycothermal Method. Chem. Lett. 2005, 34, 1082–1083. [Google Scholar] [CrossRef]
- Ozaki, H.; Iwamoto, S.; Inoue, M. Effect of the Addition of a Small Amount of Vanadium on the Photocatalytic Activities of N- and Si- Co-Doped Titanias Under Visible-Light Irradiation. Catal. Lett. 2007, 113, 95–98. [Google Scholar] [CrossRef]
- Chang, J.; Jiang, Z.Y.; Zhang, Z.Y.; Lin, Y.M.; Tian, P.L.; Zhou, B.; Chen, L. Theoretical Studies of Photocatalytic Behaviors of Isoelectronic C/Si/Ge/Sn-doped TiO2: DFT+U. Appl. Surf. Sci. 2019, 484, 1304–1309. [Google Scholar] [CrossRef]
- Hattori, A.; Tada, H. High Photocatalytic Activity of F-Doped TiO2 Film on Glass. J. Sol-Gel Sci. Technol. 2001, 22, 47. [Google Scholar] [CrossRef]
- Wu, G.; Wang, J.; Thomas, D.F.; Chen, A. Synthesis of F-Doped Flower-Like TiO2 Nanostructures with High Photoelectrochemical Activity. Langmuir 2008, 24, 3503–3509. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.C.; Yu, J.; Ho, W.; Jiang, Z.; Zhang, L. Effects of F-Doping on the Photocatalytic Activity and Microstructures of Nanocrystalline TiO2 Powders. Chem. Mater. 2002, 14, 3808–3816. [Google Scholar] [CrossRef]
- Su, Y.; Zhang, X.; Han, S.; Chen, X.; Lei, L. F-B-codoping of Anodized TiO2 Nanotubes Using Chemical Vapor Deposition. Electrochem. Commun. 2007, 9, 2292–2299. [Google Scholar] [CrossRef]
- Yamaki, T.; Sumita, T.; Yamamoto, S. Formation of TiO2−xFx Compounds in Fluorine-Implanted TiO2. J. Mater. Sci. Lett. 2002, 21, 33–35. [Google Scholar] [CrossRef]
- Yamaki, T.; Umebayashi, T.; Sumita, T.; Yamamoto, S.; Maekawa, M.; Kawasuso, A.; Itoh, H. Fluorine-Doping in Titanium Dioxide by Ion Implantation Technique. Nucl. Instrum. Methods Phys. Res. Sect. B 2003, 206, 254–258. [Google Scholar] [CrossRef]
- Li, D.; Haneda, H.; Hishita, S.; Ohashi, N.; Labhsetwar, N.K. Fluorine-Doped TiO2 Powders Prepared by Spray Pyrolysis and Their Improved Hotocatalytic Activity for Decomposition of Gas-Phase Acetaldehyde. J. Fluor. Chem. 2005, 126, 69–77. [Google Scholar] [CrossRef]
- Li, D.; Haneda, H.; Hishita, S.; Ohashi, N. Visible-Light-Driven N-F-Codoped TiO2 Photocatalysts. 1. Synthesis by Spray Pyrolysis and Surface Characterization. Chem. Mater. 2005, 17, 2588–2595. [Google Scholar] [CrossRef]
- Li, D.; Haneda, H.; Hishita, S.; Ohashi, N. Visible-Light-Driven N-F-Codoped TiO2 Photocatalysts. 2. Optical Characterization, Hotocatalysis, and Potential Application to Air Purification. Chem. Mater. 2005, 17, 2596–2602. [Google Scholar] [CrossRef]
- Li, D.; Haneda, H.; Labhsetwar, N.K.; Hishita, S.; Ohashi, N. Visible-Light-Driven Photocatalysis on Fluorine-Doped TiO2 Powders by the Creation of Surface Oxygen Vacancies. Chem. Phys. Lett. 2005, 401, 579–584. [Google Scholar] [CrossRef]
- Zhou, J.K.; Lv, L.; Yu, J.; Li, H.L.; Guo, P.Z.; Sun, H.; Zhao, X.S. Synthesis of Self-Organized Polycrystalline F-doped TiO2 Hollow Microspheres and Their Photocatalytic Activity Under Visible Light. J. Phys. Chem. C 2008, 112, 5316–5321. [Google Scholar] [CrossRef]
- Huang, D.; Liao, S.; Liu, J.; Dang, Z.; Petrik, L. Preparation of Visible-Light Responsive N-F-codoped TiO2 Photocatalyst by a Sol-Gel-Solvothermal Method. J. Photochem. Photobiol. A 2006, 184, 282–288. [Google Scholar] [CrossRef]
- Huang, D.; Liao, S.; Quan, S.; Liu, L.; He, Z.; Wan, J.; Zhou, W. Preparation of Anatase F Doped TiO2 Sol and Its Performance for Photodegradation of Formaldehyde. J. Mater. Sci. 2007, 42, 8193–8202. [Google Scholar] [CrossRef]
- Tang, J.; Quan, H.; Ye, J. Photocatalytic Properties and Photoinduced Hydrophilicity of Surface-Fluorinated TiO2. Chem. Mater. 2007, 19, 116–122. [Google Scholar] [CrossRef]
- Luo, H.; Takata, T.; Lee, Y.; Zhao, J.; Domen, K.; Yan, Y. Photocatalytic Activity Enhancing for Titanium Dioxide by Co-Doping with Bromine and Chlorine. Chem. Mater. 2004, 16, 846–849. [Google Scholar] [CrossRef]
- Hong, X.; Wang, Z.; Cai, W.; Lu, F.; Zhang, J.; Yang, Y.; Ma, N.; Liu, Y. Visible-Light-Activated Nanoparticle Photocatalyst of Iodine-Doped Titanium Dioxide. Chem. Mater. 2005, 17, 1548–1552. [Google Scholar] [CrossRef]
- Liu, G.; Chen, Z.; Dong, C.; Zhao, Y.; Li, F.; Lu, G.; Cheng, H. Visible Light Photocatalyst: Iodine-Doped Mesoporous Titania with a Bicrystalline Framework. J. Phys. Chem. B 2006, 110, 20823–20828. [Google Scholar] [CrossRef]
- Guo, M.L.; Zhang, X.D.; Liang, C.T.; Jia, G.Z. Mechanism of Visible Photoactivity of F-doped TiO2. Chin. Phys. Lett. 2010, 27, 057103. [Google Scholar] [CrossRef]
- Czoska, A.M.; Livraghi, S.; Chiesa, M.; Giamello, E.; Agnoli, S.; Granozzi, G.; Finazzi, E.; Di Valentin, C.; Pacchioni, G. The Nature of Defects in Fluorine-Doped TiO2. J. Phys. Chem. C 2008, 112, 8951–8956. [Google Scholar] [CrossRef]
- Finazzi, E.; Di Valentin, C.; Pacchioni, G.; Selloni, A. Excess Electron States in Reduced Bulk Anatase TiO2: Comparison of Standard GGA, GGA+U, Hybrid DFT Calc. J. Chem. Phys. 2008, 129, 154113. [Google Scholar] [CrossRef] [PubMed]
- Long, R.; English, N.J. Electronic Properties of F/Zr Co-Doped Anatase TiO2 Photocatalysts from GGA+U Calc. Chem. Phys. Lett. 2010, 498, 338–344. [Google Scholar] [CrossRef]
- Sato, J.; Kobayashi, H.; Inoue, Y. Photocatalytic Activity for Water Decomposition of Indates with Octahedrally Coordinated D10 Configuration. II. Roles Geom. Electron. Struct. J. Phys. Chem. B 2003, 107, 7970–7975. [Google Scholar] [CrossRef]
- Long, M.; Cai, W.; Wang, Z.; Liu, G. Correlation of Electronic Structures and Crystal Structures with Photocatalytic Properties of Undoped, N-doped and I-doped TiO2. Chem. Phys. Lett. 2006, 420, 71–76. [Google Scholar] [CrossRef]
- Kuhn, W.; Wagner, H.P.; Stanzl, H.; Wolf, K.; Worle, K.; Lankes, S.; Betz, J.; Worz, M.; Lichtenberger, D.; Leiderer, H.; et al. The MOVPE Growth and Doping of ZnTe. Semicond. Sci. Technol. 1991, 6, A105–A108. [Google Scholar] [CrossRef]
- Pan, H.; Zhang, Y.W.; Shenoy, V.B.; Gao, H. Effects of H-, N-, and (H, N)-Doping on the Photocatalytic Activity of TiO2. J. Phys. Chem. C 2011, 115, 12224–12231. [Google Scholar] [CrossRef]
- Chen, W.P.; Wang, Y.; Chan, H.L.W. Hydrogen: A Metastable Donor in TiO2 Single Crystals. Appl. Phys. Lett. 2008, 92, 112907. [Google Scholar] [CrossRef]
- Herklotz, F.; Lavrov, E.V.; Weber, J. Infrared Absorption of the Hydrogen Donor in Rutile TiO2. Phys. Rev. B 2011, 83, 235202. [Google Scholar] [CrossRef]
- Yang, K.; Dai, Y.; Huang, B.; Feng, Y. Density Functional Characterization of the Antiferromagnetism in Oxygen-Deficient Anatase and Rutile TiO2. Phys. Rev. B 2010, 81, 033202. [Google Scholar] [CrossRef]
- Mi, L.; Xu, P.; Shen, H.; Wang, P.N.; Shen, W. First-Principles Calculation of N:H Codoping Effect on Energy Gap Narrowing of TiO2. Appl. Phys. Lett. 2007, 90, 171909. [Google Scholar] [CrossRef]
- Li, N.; Yao, K.L.; Li, L.; Sun, Z.Y.; Gao, G.Y.; Zhu, L. Effect of Carbon/Hydrogen Species Incorporation on Electronic Structure of anatase-TiO2. J. Appl. Phys. 2011, 110, 073513. [Google Scholar] [CrossRef]
- Yang, K.; Dai, Y.; Huang, B.; Whangbo, M.H. Density Functional Studies of the Magnetic Properties in Nitrogen Doped TiO2. Chem. Phys. Lett. 2009, 481, 99–102. [Google Scholar] [CrossRef]
- Long, R.; English, N.J. Band Gap Engineering of (N,Ta)-codoped TiO2: A First-Principles Calculation. Chem. Phys. Lett. 2009, 478, 175–179. [Google Scholar] [CrossRef]
- Yang, K.; Dai, Y.; Huang, B.; Whangbo, M.H. On the Possibility of Ferromagnetism in Carbon-Doped Anatase TiO2. Appl. Phys. Lett. 2008, 93, 132507. [Google Scholar] [CrossRef]
- Chen, X.; Liu, L.; Yu, P.Y.; Mao, S.S. Increasing Solar Absorption for Photocatalysis with Black Hydrogenated Titanium Dioxide Nanocrystals. Science 2011, 331, 746–750. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Dai, Y.; Jin, H.; Huang, B. Effective Increasing of Optical Absorption and Energy Conversion Efficiency of Anatase TiO2 Nanocrystals by Hydrogenation. Phys. Chem. Chem. Phys. 2011, 13, 18063–18068. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Irie, H.; Hashimoto, K. Conduction Band Energy Level Control of Titanium Dioxide: Toward an Efficient Visible-Light-Sensitive Photocatalyst. J. Am. Chem. Soc. 2010, 132, 6898–6899. [Google Scholar] [CrossRef]
- Gai, Y.; Li, J.; Li, S.S.; Xia, J.B.; Wei, S.H. Design of Narrow-Gap TiO2: A Passivated Codoping Approach for Enhanced Photoelectrochemical Activity. Phys. Rev. Lett. 2009, 102, 036402. [Google Scholar] [CrossRef]
- Long, R.; English, N.J. Tailoring the Electronic Structure of TiO2 by Cation Codoping from Hybrid Density Functional Theory Calculations. Phys. Rev. B 2011, 83, 155209. [Google Scholar] [CrossRef]
- Zhu, Y.; Dai, Y.; Lai, K.; Huang, B. Synergistic Modification of Electronic and Photocatalytic Properties of TiO2 Nanotubes by Implantation of Au and N Atoms. ChemPhysChem 2013, 14, 2800–2807. [Google Scholar] [CrossRef]
- Wu, G.; Wen, J.; Nigro, S.; Chen, A. One-Step Synthesis of N- and F-codoped Mesoporous TiO2 Photocatalysts with High Visible Light Activity. Nanotechnology 2010, 21, 085701. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, J.; Cui, H. Adsorption Mechanism of SF6 Decomposition Components Onto N, F-co-doped TiO2: A DFT Study. J. Fluor. Chem. 2018, 213, 18–23. [Google Scholar] [CrossRef]
- Salehi-Abar, P.; Kazempour, A. Effect of N and F Doping on the Electronic Properties of Rutile TiO2 Quantum Dot Solar Cells: A First Principle Study. Chem. Phys. Lett. 2017, 673, 56–61. [Google Scholar] [CrossRef]
- Lu, J.; Jin, H.; Dai, Y.; Yang, K.; Huang, B. Effect of Electronegativity and Charge Balance on the Visible-Light-Responsive Photocatalytic Activity of Nonmetal Doped Anatase TiO2. Int. J. Photoenergy 2012, 2012. [Google Scholar] [CrossRef]
- Obata, K.; Irie, H.; Hashimoto, K. Enhanced Photocatalytic Activities of Ta, N Co-Doped TiO2 Thin Films Under Visible Light. Chem. Phys. 2007, 339, 124–132. [Google Scholar] [CrossRef]
- Long, R.; English, N.J. Synergistic Effects on Band Gap-Narrowing in Titania by Codoping from First-Principles Calculations. Chem. Mater. 2010, 22, 1616–1623. [Google Scholar] [CrossRef]
- Liu, H.; Lu, Z.; Yue, l.; Liu, J.; Gan, Z.; Shu, C.; Zhang, T.; Shi, J.; Xiong, R. (Mo + N) Codoped TiO2 for Enhanced Visible-Light Photoactivity. Appl. Sur. Sci. 2011, 257, 9355–9361. [Google Scholar] [CrossRef]
- Wei, W.; Dai, Y.; Guo, M.; Yu, L.; Huang, B. Density Functional Characterization of the Electronic Structure and Optical Properties of N-doped, La-doped, and N/La-codoped SrTiO3. J. Phys. Chem. C 2009, 113, 15046–15050. [Google Scholar] [CrossRef]
- Wei, W.; Dai, Y.; Guo, M.; Yu, L.; Jin, H.; Han, S.; Huang, B. Codoping Synergistic Effects in N-doped SrTiO3 for Higher Energy Conversion Efficiency. Phys. Chem. Chem. Phys. 2010, 12, 7612–7619. [Google Scholar] [CrossRef]
- Sulaeman, U.; Yin, S.; Sato, T. Visible Light Photocatalytic Properties of Ta and N Codoped SrTiO3 Nanoparticles. Int. J. Opt. 2010, 2010. [Google Scholar] [CrossRef]
- Fang, Y.; Xu, T.; Zhang, Y.; Kong, X.; Liu, J.; Cui, S.; Wang, D. Structural, Electronic and Optical Properties of La, C-Codoped TiO2 Investigated by First Principle Calculations. J. Phys. Chem. Solids 2019, 132, 121–129. [Google Scholar] [CrossRef]
- Tao, J.G.; Guan, L.X.; Pan, J.S.; Huan, C.H.A.; Wang, L.; Kuo, J.L. Possible Room Temperature Ferromagnetism of Li-doped Anatase TiO2: A First-Principles Study. Phys. Lett. A 2010, 374, 4451–4454. [Google Scholar] [CrossRef]
- Colella, S.; Mosconi, E.; Pellegrino, G.; Alberti, A.; Guerra, V.L.; Masi, S.; Listorti, A.; Rizzo, A.; Condorelli, G.G.; De Angelis, F.; et al. Elusive Presence of Chloride in Mixed Halide Perovskite Solar Cells. J. Phys. Chem. Lett. 2014, 5, 3532–3538. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, F. Modeling Materials and Processes in Hybrid/Organic Photovoltaics: From Dye-Sensitized to Perovskite Solar Cells. Acc. Chem. Res. 2014, 47, 3349–3360. [Google Scholar] [CrossRef] [PubMed]
- Roiati, V.; Mosconi, E.; Listorti, A.; Colella, S.; Gigli, G.; De Angelis, F. Stark Effect in Perovskite/TiO2 Solar Cells: Evidence of Local Interfacial Order. Nano Lett. 2014, 14, 2168–2174. [Google Scholar] [CrossRef]
- Cheng, J.; Luo, J.; Yang, K. Comparison Studies of Interfacial Electronic and Energetic Properties of LaAlO3/TiO2 and TiO2/LaAlO3 Heterostructures from First-Principles Calculations. ACS Appl. Mater. Interfaces 2017, 9, 7682–7690. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lei, Y.; Li, Y.; Yu, Y.; Cai, J.; Chiu, M.H.; Rao, R.; Gu, Y.; Wang, C.; Choi, W.; et al. Strain Engineering and Epitaxial Stabilization of Halide Perovskites. Nature 2020, 577, 209–215. [Google Scholar] [CrossRef]
- Qu, Z.; Su, Y.L.; Sun, L.; Liang, F.; Zhang, G.H. Study of the Structure, Electronic and Optical Properties of BiOI/Rutile-TiO2 Heterojunction by the First-Principle Calculation. Materials 2020, 13, 323. [Google Scholar] [CrossRef]
- Wei, W.; Dai, Y.; Huang, B.; Li, X.; Nägele, F.; Over, H.; Whangbo, M.H.; Jacob, T. Density Functional Characterization of the Electronic Structures and Band Bending of Rutile RuO2/TiO2(110) Heterostructures. J. Phys. Chem. C 2015, 119, 12394–12399. [Google Scholar] [CrossRef]
- Zhou, Y.D.; Liu, Q.L.; Yang, C.; Zhao, Z.Y. Interfacial Micro-Structure and Properties of TiO2/SnO2 Heterostructures with Rutile Phase: A DFT Calculation Investigation. Appl. Surf. Sci. 2018, 451, 258–271. [Google Scholar] [CrossRef]
- Pak, Y.; Park, W.; Alaal, N.; Kumaresan, Y.; Aravindh, S.A.; Mitra, S.; Xin, B.; Min, J.W.; Kim, H.; Lim, N.; et al. Enhanced Photoresponse of WS2 Photodetectors through Interfacial Defect Engineering Using a TiO2 Interlayer. ACS Appl. Electron. Mater. 2020, 2, 838–845. [Google Scholar] [CrossRef]
- Abbasi, A.; Sardroodi, J.J. Investigation of the Adsorption of Ozone Molecules on TiO2/WSe2 Nanocomposites by DFT Computations: Applications to Gas Sensor Devices. Appl. Surf. Sci. 2018, 436, 27–41. [Google Scholar] [CrossRef]
- Zeng, X.; Xiao, X.; Zhang, W.; Wan, C.; Wang, H. Interfacial Charge Transfer and Mechanisms of Enhanced Photocatalysis of an Anatase TiO2(001)-MoS2-graphene Nanocomposite: A First-Principles Investigation. Comput. Mater. Sci. 2017, 126, 43–51. [Google Scholar] [CrossRef]
- Liberto, G.D.; Tosoni, S.; Pacchioni, G. Charge Carriers Cascade in a Ternary TiO2/TiO2/ZnS Heterojunction: A DFT Study. ChemCatChem 2020, 12, 2097–2105. [Google Scholar] [CrossRef]
- Zhao, Y.; Lin, Y.; Wang, G.; Jiang, Z.; Zhang, R.; Zhu, C. Electronic and Optical Performances of (Cu, N) Codoped TiO2/g-C3N4 Heterostructure Photocatalyst: A Spin-Polarized DFT+U Study. Sol. Energy 2018, 162, 306–316. [Google Scholar] [CrossRef]
- Wei, L.; Cui, S.; Guo, H.; Ma, X. Study on the Role of Mn Species in Low Temperature SCR on MnOx/TiO2 through Experiment and DFT Calculation. Mol. Catal. 2018, 445, 102–110. [Google Scholar] [CrossRef]
- Meng, Q.; Fan, L.; Zhu, L.; Xu, N.; Zhang, Q. Electronic and Optical Properties of α-MoO3/TiO2 Heterostructures: A DFT Study. Int. J. Quantum Chem. 2018, 118, e25681. [Google Scholar] [CrossRef]
- Long, R.; Dai, Y.; Huang, B. Fullerene Interfaced with a TiO2(110) Surface May Not Form an Efficient Photovoltaic Heterojunction: First-Principles Investigation of Electronic Structures. J. Phys. Chem. Lett. 2013, 4, 2223–2229. [Google Scholar] [CrossRef]
- Ma, X.; Dai, Y.; Yu, L.; Huang, B. New Basic Insights Into the Low Hot Electron Injection Efficiency of Gold-Nanoparticle-Photosensitized Titanium Dioxide. ACS Appl. Mater. Interfaces 2014, 6, 12388–12394. [Google Scholar] [CrossRef]
- Jin, C.; Dai, Y.; Wei, W.; Ma, X.; Li, M.; Huang, B. Effects of Single Metal Atom (Pt, Pd, Rh and Ru) Adsorption on the Photocatalytic Properties of Anatase TiO2. Appl. Surf. Sci. 2017, 426, 639–646. [Google Scholar] [CrossRef]
- Ma, X.C.; Dai, Y.; Yu, L.; Huang, B.B. Energy Transfer in Plasmonic Photocatalytic Composites. Light Sci. Appl. 2016, 5, e16017. [Google Scholar] [CrossRef] [PubMed]
- Clavero, C. Plasmon-Induced Hot-Electron Generation at Nanoparticle/metal-Oxide Interfaces for Photovoltaic and Photocatalytic Devices. Nat. Photonics 2014, 8, 95–103. [Google Scholar] [CrossRef]
- Cheng, H.; Fuku, K.; Kuwahara, Y.; Mori, K.; Yamashita, H. Harnessing Single-Active Plasmonic Nanostructures for Enhanced Photocatalysis Under Visible Light. J. Mater. Chem. A 2015, 3, 5244–5258. [Google Scholar] [CrossRef]
- Lou, Z.; Wang, Z.; Huang, B.; Dai, Y. Synthesis and Activity of Plasmonic Photocatalysts. ChemCatChem 2014, 6, 2456–2476. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, Y.L.; Liu, R.S.; Tsai, D.P. Plasmonic Photocatalysis. Rep. Prog. Phys. 2013, 76, 046401. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Huang, B.; Dai, Y.; Whangbo, M.H. Plasmonic Photocatalysts: Harvesting Visible Light with Noble Metal Nanoparticles. Phys. Chem. Chem. Phys. 2012, 14, 9813–9825. [Google Scholar] [CrossRef]
- Hou, W.; Cronin, S.B. A Review of Surface Plasmon Resonance-Enhanced Photocatalysis. Adv. Funct. Mater. 2013, 23, 1612–1619. [Google Scholar] [CrossRef]
- Jiang, R.; Li, B.; Fang, C.; Wang, J. Metal/Semiconductor Hybrid Nanostructures for Plasmon-Enhanced Applications. Adv. Mater. 2014, 26, 5274–5309. [Google Scholar] [CrossRef]
- Brongersma, M.L.; Halas, N.J.; Nordlander, P. Plasmon-Induced Hot Carrier Science and Technology. Nat. Nanotechnol. 2015, 10, 25–34. [Google Scholar] [CrossRef]
- Yang, K.; Setyawan, W.; Wang, S.; Nardelli, M.B.; Curtarolo, S. A Search Model for Topological Insulators with High-Throughput Robustness Descriptors. Nat. Mater. 2012, 11, 614–619. [Google Scholar] [CrossRef]
- Curtarolo, S.; Hart, G.L.W.; Nardelli, M.B.; Mingo, N.; Sanvito, S.; Levy, O. The High-Throughput Highway to Computational Materials Design. Nat. Mater. 2013, 12, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Yang, K. High-Throughput Design of Functional Materials Using Materials Genome Approach. Chin. Phys. B 2018, 27, 128103. [Google Scholar] [CrossRef]
- Cai, Y.; Xie, W.; Teng, Y.T.; Harikesh, P.C.; Ghosh, B.; Huck, P.; Persson, K.A.; Mathews, N.; Mhaisalkar, S.G.; Sherburne, M.; et al. High-Throughput Computational Study of Halide Double Perovskite Inorganic Compounds. Chem. Mater. 2019, 31, 5392–5401. [Google Scholar] [CrossRef]
- Yang, K.; Nazir, S.; Behtash, M.; Cheng, J. High-Throughput Design of Two-Dimensional Electron Gas Systems Based on Polar/Nonpolar Perovskite Oxide Heterostructures. Sci. Rep. 2016, 6, 34667. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Yang, K. Design of Two-Dimensional Electron Gas Systems via Polarization Discontinuity from Large-Scale First-Principles Calculations. J. Mater. Chem. C 2018, 6, 6680–6690. [Google Scholar] [CrossRef]
- Li, Y.; Yang, K. High-Throughput Computational Design of Organic-Inorganic Hybrid Halide Semiconductors Beyond Perovskites for Optoelectronics. Energy Environ. Sci. 2019, 12, 2233–2243. [Google Scholar] [CrossRef]
- Li, Y.; Maldonado-Lopez, D.; Ríos Vargas, V.; Zhang, J.; Yang, K. Stability Diagrams, Defect Tolerance, and Absorption Coefficients of Hybrid Halide Semiconductors: High-Throughput First-Principles Characterization. J. Chem. Phys. 2020, 152, 084106. [Google Scholar] [CrossRef]









| Acceptor Atom | |||
|---|---|---|---|
| Anion | N, P | 1 | [43,173] |
| C | 2 | [175] | |
| B | 3 | [115] | |
| Cation | B, Al, Ga, In, La | 1 | [115,192] |
| Be, Mg, Ca, Sr, Ba | 2 | [115] | |
| Li, Na, K, Rb, Cs | 3 | [115,193] | |
| Donor atom | |||
| Anion | F, Cl, Br, I | 1 | [45,161] |
| Cation | H, Li | 1 | [66,67,68,171] |
| I, Nb, Ta | 1 | [45,174] | |
| Mo, W | 2 | [180] | |
| Interface | Properties | Ref. |
|---|---|---|
| TiO2/MAPbI3 & TiO2/MAPbIClx | (110) perovskite surface was stabilized against (001) surface after “depositing” TiO2 for both materials interfaces; interfacial Cl atoms increase the interfacial binding energy. | [21] |
| TiO2/MAPbI3 | Higher interfacial charge transfer rate than ZnO- and SnO2-based interface. | [22] |
| TiO2/MASnI3 & TiO2/MASnI3 | TiO2/SnI2 interface is energetically most favorable among the four considered systems and has the highest electron–hole separation rate. | [24] |
| TiO2/MAPbI3 & TiO2/MAPbI3 | TiO2/PbI2 has stronger interfacial interaction but TiO2/PbI2 model is most efficient for electron–hole separation. | [19] |
| TiO2/(Ba,Sr)TiO2 | Stability of anatase TiO decreases in the order from (001) to (011) to (111) perovskite substrates. | [23] |
| TiO2/BiOX (X = Cl,Br, and I) | BiO/TiO2 interface is more stable than 1I/TiO2 interface; band gap of 1I/TiO2 interface reduced by 0.28 eV while BiO/TiO2 exhibits an n-type conductivity. | [199] |
| TiO2/RuO2 | Strong bonding interaction at the interface; oxygen vacancies at the interface changes the band bending direction. | [200] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, K.; Dai, Y.; Huang, B. Review of First-Principles Studies of TiO2: Nanocluster, Bulk, and Material Interface. Catalysts 2020, 10, 972. https://doi.org/10.3390/catal10090972
Yang K, Dai Y, Huang B. Review of First-Principles Studies of TiO2: Nanocluster, Bulk, and Material Interface. Catalysts. 2020; 10(9):972. https://doi.org/10.3390/catal10090972
Chicago/Turabian StyleYang, Kesong, Ying Dai, and Baibiao Huang. 2020. "Review of First-Principles Studies of TiO2: Nanocluster, Bulk, and Material Interface" Catalysts 10, no. 9: 972. https://doi.org/10.3390/catal10090972
APA StyleYang, K., Dai, Y., & Huang, B. (2020). Review of First-Principles Studies of TiO2: Nanocluster, Bulk, and Material Interface. Catalysts, 10(9), 972. https://doi.org/10.3390/catal10090972