CO Total and Preferential Oxidation over Stable Au/TiO2 Catalysts Derived from Preformed Au Nanoparticles

 , , ,
, , ,  and
and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Morphological and Structural Properties of the Preformed Au@NCD and Au@NCD/TiO2 Catalysts
2.1.1. TEM Characterization of Unsupported Au@NCD
2.1.2. Characterization of TiO2
2.1.3. TEM Characterization of Au/TiO2 Catalysts
2.2. In Situ XPS Studies: Thermal Stability of Au@NCD/TiO2 Catalysts and Surface Reorganization
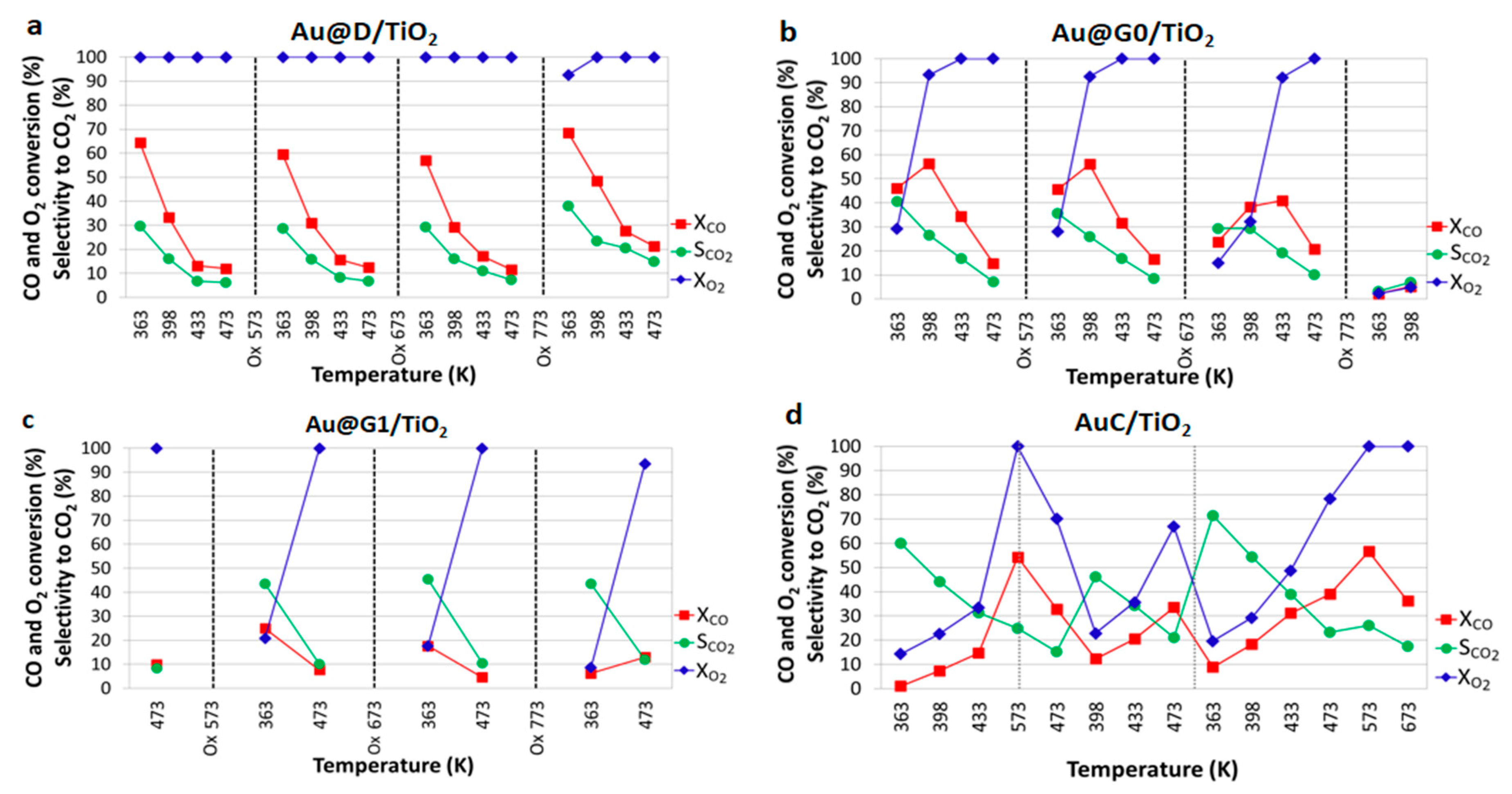
2.3. CO Oxidation and PROX Catalytic Tests
2.3.1. On Cordierite Monoliths
2.3.2. On Stainless Steel Microreactors
3. Materials and Methods
3.1. Synthesis and Characterization of Au@NCD NPs
3.2. Synthesis and Characterization of TiO2 Powder
3.3. Functionalization and Characterization of Structured Reactors
3.3.1. Functionalization of Cordierite Honeycombs with Au/TiO2 Catalysts
3.3.2. Functionalization of Stainless Steel Microreactors with Au/TiO2 Catalysts
3.4. Synthesis of Unsupported Au/TiO2 Catalysts
3.5. Characterization of Unsupported Au/TiO2 Catalysts
XPS Analyses and XPS In Situ Measurements
3.6. CO Oxidation and PROX Catalytic Tests
3.6.1. On Cordierite Monoliths
3.6.2. On Stainless Steel Microreactors
3.6.3. Long-Term Test
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gandía, L.M.; Arzamendi, G.; Diéguez, P.M. Renewable Hydrogen Energy: An Overview. In Renewable Hydrogen Technologies: Production, Purification, Storage, Applications and Safety; Elsevier B.V.: Amsterdam, The Netherlands, 2013; pp. 1–17. [Google Scholar]
- Haryanto, A.; Fernando, S.; Murali, N.; Adhikari, S. Current Status of Hydrogen Production Techniques by Steam Reforming of Ethanol: A Review. Energy Fuels 2005, 19, 2098–2106. [Google Scholar] [CrossRef]
- Choudhary, T. CO-free fuel processing for fuel cell applications. Catal. Today 2002, 77, 65–78. [Google Scholar] [CrossRef]
- Korotkikh, O.; Farrauto, R.J. Selective catalytic oxidation of CO in H2: Fuel cell applications. Catal. Today 2000, 62, 249–254. [Google Scholar] [CrossRef]
- Liu, K.; Wang, A.; Zhang, T. Recent Advances in Preferential Oxidation of CO Reaction over Platinum Group Metal Catalysts. ACS Catal. 2012, 2, 1165–1178. [Google Scholar] [CrossRef]
- Choi, Y.; Stenger, H.G. Kinetics, simulation and insights for CO selective oxidation in fuel cell applications. J. Power Sources 2004, 129, 246–254. [Google Scholar] [CrossRef]
- Pozdnyakova, O.; Teschner, D.; Wootsch, A.; Krohnert, J.; Steinhauer, B.; Sauer, H.; Tóth, L.; Jentoft, F.; Knopgericke, A.; Paal, Z. Preferential CO oxidation in hydrogen (PROX) on ceria-supported catalysts, part I: Oxidation state and surface species on Pt/CeO2 under reaction conditions. J. Catal. 2006, 237, 1–16. [Google Scholar] [CrossRef]
- Haruta, M. Size- and support-dependency in the catalysis of gold. Catal. Today 1997, 36, 153–166. [Google Scholar] [CrossRef]
- Haruta, M. Gold as a novel catalyst in the 21st century: Preparation, working mechanism and applications. Gold Bull. 2004, 37, 27–36. [Google Scholar] [CrossRef]
- Valden, M.; Lai, X.; Goodman, D.W. Onset of Catalytic Activity of Gold Clusters on Titania with the Appearance of Nonmetallic Properties. Science 1998, 281, 1647–1650. [Google Scholar] [CrossRef]
- Chen, M.; Goodman, D.W. Catalytically Active Gold: From Nanoparticles to Ultrathin Films. Accounts Chem. Res. 2006, 39, 739–746. [Google Scholar] [CrossRef]
- Lopez, N. On the origin of the catalytic activity of gold nanoparticles for low-temperature CO oxidation. J. Catal. 2004, 223, 232–235. [Google Scholar] [CrossRef]
- Beck, A.; Yang, A.-C.; Leland, A.R.; Riscoe, A.R.; Lopez, F.A.; Goodman, E.D.; Cargnello, M. Understanding the preferential oxidation of carbon monoxide (PrOx) using size-controlled Au nanocrystal catalyst. AIChE J. 2018, 64, 3159–3167. [Google Scholar] [CrossRef]
- Chen, M.; Goodman, D.W. Catalytically active gold on ordered titania supports. Chem. Soc. Rev. 2008, 37, 1860–1870. [Google Scholar] [CrossRef] [PubMed]
- Qiao, B.; Liu, J.; Wang, Y.-G.; Lin, Q.; Liu, X.; Wang, A.; Li, J.; Zhang, T.; Liu, J. Highly Efficient Catalysis of Preferential Oxidation of CO in H2-Rich Stream by Gold Single-Atom Catalysts. ACS Catal. 2015, 5, 6249–6254. [Google Scholar] [CrossRef]
- Llorca, J.; Domínguez, M.; Ledesma, C.; Chimentao, R.J.; Medina, F.; E Sueiras, J.; Angurell, I.; Seco, M.; Rossell, O. Propene epoxidation over TiO2-supported Au–Cu alloy catalysts prepared from thiol-capped nanoparticles. J. Catal. 2008, 258, 187–198. [Google Scholar] [CrossRef]
- Divins, N.J.; López, E.; Roig, M.; Trifonov, T.; Rodriguez, A.; De Rivera, F.G.; Rodríguez, L.; Seco, M.; Rossell, O.; Llorca, J. A million-channel CO-PrOx microreactor on a fingertip for fuel cell application. Chem. Eng. J. 2011, 167, 597–602. [Google Scholar] [CrossRef]
- Moreno, C.; Divins, N.J.; Gázquez, J.; Varela, M.; Angurell, I.; Llorca, J. Improved thermal stability of oxide-supported naked gold nanoparticles by ligand-assisted pinning. Nanoscale 2012, 4, 2278. [Google Scholar] [CrossRef]
- Soler, L.; Casanovas, A.; Urrich, A.; Angurell, I.; Llorca, J. CO oxidation and COPrOx over preformed Au nanoparticles supported over nanoshaped CeO2. Appl. Catal. B Environ. 2016, 197, 47–55. [Google Scholar] [CrossRef]
- Cominos, V.; Hessel, V.; Hofmann, C.; Kolb, G.; Zapf, R.; Ziogas, A.; Delsman, E.; Schouten, J. Selective oxidation of carbon monoxide in a hydrogen-rich fuel cell feed using a catalyst coated microstructured reactor. Catal. Today 2005, 110, 140–153. [Google Scholar] [CrossRef]
- Potemkin, D.; Snytnikov, P.; Belyaev, V.; Sobyanin, V. Preferential CO oxidation over Cu/CeO2−x catalyst: Internal mass transport limitation. Chem. Eng. J. 2011, 176, 165–171. [Google Scholar] [CrossRef]
- Soler, L.; Divins, N.J.; Vendrell, X.; Serrano, I.; Llorca, J. Hydrogen production in microreactors. In Current Trends and Future Developments on (Bio-) Membranes; Elsevier: Amsterdam, The Netherlands, 2020; pp. 141–182. [Google Scholar]
- Leal, G.B.; Ciotti, L.; Watacabe, B.N.; Da Silva, D.C.L.; Antoniassi, R.M.; Silva, J.C.M.; Linardi, M.; Giudicci, R.; Vaz, J.M.; Spinacé, E.V. Preparation of Au/TiO2 by a facile method at room temperature for the CO preferential oxidation reaction. Catal. Commun. 2018, 116, 38–42. [Google Scholar] [CrossRef]
- Ivanova, S.; Pitchon, V.; Petit, C.; Caps, V. Support Effects in the Gold-Catalyzed Preferential Oxidation of CO. ChemCatChem 2010, 2, 556–563. [Google Scholar] [CrossRef]
- De Rivera, F.G.; Rodríguez, L.-I.; Rossell, O.; Seco, M.; Divins, N.J.; Casanova, I.; Llorca, J. Carbosilane dendrons as stabilizing agents for the formation of gold nanoparticles. J. Organomet. Chem. 2011, 696, 2287–2293. [Google Scholar] [CrossRef]
- Wagner, C.D.; Riggs, W.M.; Davis, L.E. Handbook of X-Ray Photoelectron Spectroscopy; PerkinElmer: Waltham, MA, USA, 1979. [Google Scholar]
- Schubert, M.M.; Plzak, V.; Garche, J.; Behm, R.J. Activity, Selectivity, and Long-Term Stability of Different Metal Oxide Supported Gold Catalysts for the Preferential CO Oxidation in H2-Rich Gas. Catal. Lett. 2001, 76, 143–150. [Google Scholar] [CrossRef]
- Laguna, O.; Ngassa, E.; Oraá, S.; Alvarez, A.; Domínguez, M.; Romero-Sarria, F.; Arzamendi, G.; Gandía, L.M.; Centeno, M.A.; Odriozola, J.A.; et al. Preferential oxidation of CO (CO-PROX) over CuOx/CeO2 coated microchannel reactor. Catal. Today 2012, 180, 105–110. [Google Scholar] [CrossRef]
- Widmann, D.; Hocking, E.; Behm, R. On the origin of the selectivity in the preferential CO oxidation on Au/TiO2–Nature of the active oxygen species for H2 oxidation. J. Catal. 2014, 317, 272–276. [Google Scholar] [CrossRef]
- Green, I.X.; Tang, W.; Neurock, M.; Yates, J.T. Spectroscopic Observation of Dual Catalytic Sites During Oxidation of CO on a Au/TiO2 Catalyst. Science 2011, 333, 736–739. [Google Scholar] [CrossRef]
- Reina, T.R.; Ivanova, S.; Centeno, M.A.; Odriozola, J.A. Catalytic screening of Au/CeO2-MOx/Al2O3 catalysts (M = La, Ni, Cu, Fe, Cr, Y) in the CO-PrOx reaction. Int. J. Hydrogen Energy 2015, 40, 1782–1788. [Google Scholar] [CrossRef]
- Snytnikov, P.; Popova, M.; Men, Y.; Rebrov, E.; Kolb, G.; Hessel, V.; Schouten, J.; Sobyanin, V. Preferential CO oxidation over a copper–cerium oxide catalyst in a microchannel reactor. Appl. Catal. A Gen. 2008, 350, 53–62. [Google Scholar] [CrossRef]
- Brust, M.; Walker, M.; Bethell, D.; Schiffrin, D.J.; Whyman, R. Synthesis of thiol-derivatised gold nanoparticles in a two-phase Liquid-Liquid system. J. Chem. Soc. Chem. Commun. 1994, 801–802. [Google Scholar] [CrossRef]
- Zapf, R.; Becker-Willinger, C.; Berresheim, K.; Bolz, H.; Gnaser, H.; Hessel, V.; Kolb, G.; Löb, P.; Pannwitt, A.-K.; Ziogas, A. Detailed Characterization of Various Porous Alumina-Based Catalyst Coatings Within Microchannels and Their Testing for Methanol Steam Reforming. Chem. Eng. Res. Des. 2003, 81, 721–729. [Google Scholar] [CrossRef]
- O’Connell, M.; Kolb, G.; Zapf, R.; Men, Y.; Hessel, V. Bimetallic catalysts for the catalytic combustion of methane using microreactor technology. Catal. Today 2009, 144, 306–311. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Gold Precursor | Ligands |
|---|---|---|
| Au@D | Preformed Au@D NPs | Dodecanethiols |
| Au@G0 | Preformed Au@G0 NPs | G0 generation dendrons |
| Au@G1 | Preformed Au@G1 NPs | G1 generation dendrons |
| Au@G2 | Preformed Au@G2 NPs | G2 generation dendrons |
| AuC | HAuCl4·3H2O | None |
| Temperature (K) | Au/C (Atomic Ratio × 100) | ||
|---|---|---|---|
| Au@D/TiO2 | Au@G1/TiO2 | Au@G2/TiO2 | |
| As-synthesized | 25.6 | 6.8 | 0.6 |
| 398 | 44.8 | 7.9 | 2.6 |
| 523 | 43.5 | 9.5 | 5.1 |
| 598 | 40.0 | 12.5 | 9.1 |
| 673 | 41.9 | 17.9 | 10.4 |
| 773 | 41.0 | 13.8 | 8.1 |
| Treatment | Atomic Ratio × 100 | |||
|---|---|---|---|---|
| Au/C | Au/Ti | Si/Ti | Si/Au | |
| 1. As-synthesized | 48 | 38 | 82 | 215 |
| 2. Oxidation at 473 K | 55 | 34 | 78 | 227 |
| 3. Oxidation at 573 K | 65 | 30 | 78 | 261 |
| 4. Oxidation at 673 K | 129 | 29 | 39 | 133 |
| 5. PROX at 673 K | 296 | 29 | 44 | 154 |
| 6. Oxidation at 773 K | 239 | 27 | 58 | 220 |
| 7. Oxidation at 873 K | 289 | 26 | 363 | 1389 |
| Mixture | Concentration (vol.%) | |||
|---|---|---|---|---|
| CO | O2 | H2 | Ar | |
| A | 2 | 2 | 0 | 96 |
| B | 2 | 2 | 2 | 94 |
| C | 2 | 2 | 50 | 46 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Divins, N.J.; López, E.; Angurell, I.; Neuberg, S.; Zapf, R.; Kolb, G.; Llorca, J. CO Total and Preferential Oxidation over Stable Au/TiO2 Catalysts Derived from Preformed Au Nanoparticles. Catalysts 2020, 10, 1028. https://doi.org/10.3390/catal10091028
Divins NJ, López E, Angurell I, Neuberg S, Zapf R, Kolb G, Llorca J. CO Total and Preferential Oxidation over Stable Au/TiO2 Catalysts Derived from Preformed Au Nanoparticles. Catalysts. 2020; 10(9):1028. https://doi.org/10.3390/catal10091028
Chicago/Turabian StyleDivins, Núria J., Eduardo López, Inmaculada Angurell, Stefan Neuberg, Ralf Zapf, Gunther Kolb, and Jordi Llorca. 2020. "CO Total and Preferential Oxidation over Stable Au/TiO2 Catalysts Derived from Preformed Au Nanoparticles" Catalysts 10, no. 9: 1028. https://doi.org/10.3390/catal10091028
APA StyleDivins, N. J., López, E., Angurell, I., Neuberg, S., Zapf, R., Kolb, G., & Llorca, J. (2020). CO Total and Preferential Oxidation over Stable Au/TiO2 Catalysts Derived from Preformed Au Nanoparticles. Catalysts, 10(9), 1028. https://doi.org/10.3390/catal10091028