Alkaline-Based Catalysts for Glycerol Polymerization Reaction: A Review




Abstract
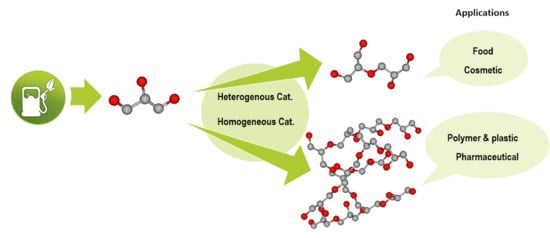
1. Introduction
2. Polyglycerols Applications
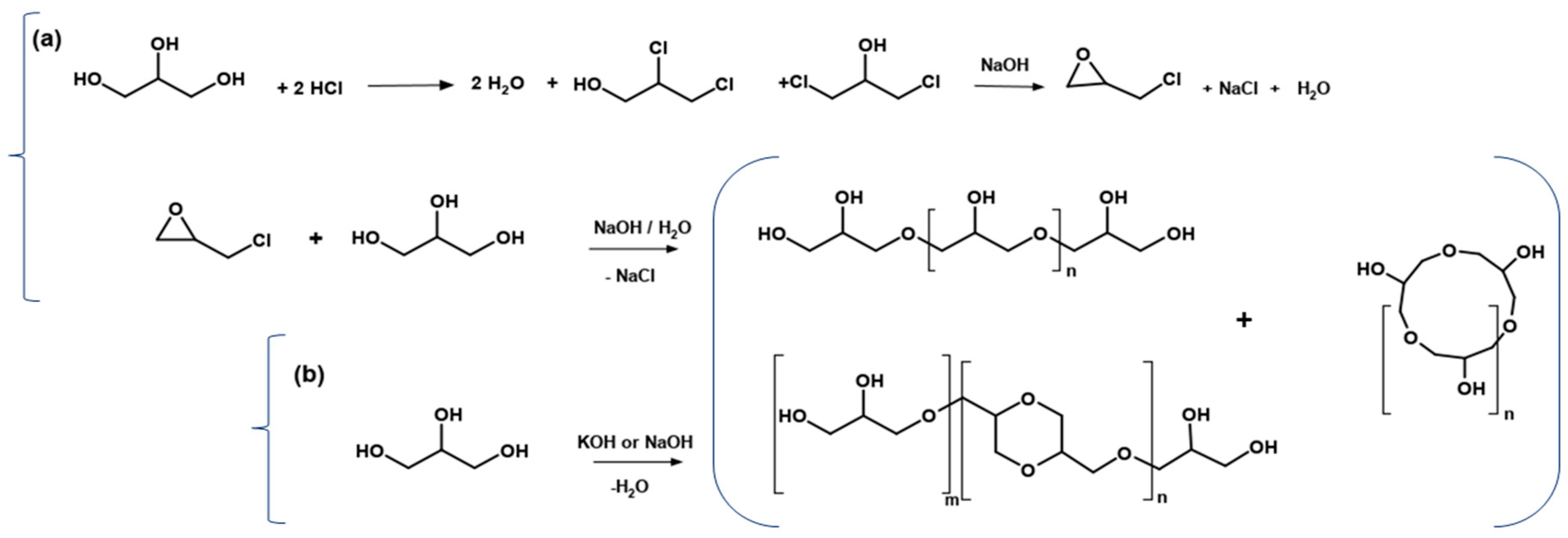
3. Industrial Routes for Polyglycerols Production
- (a)
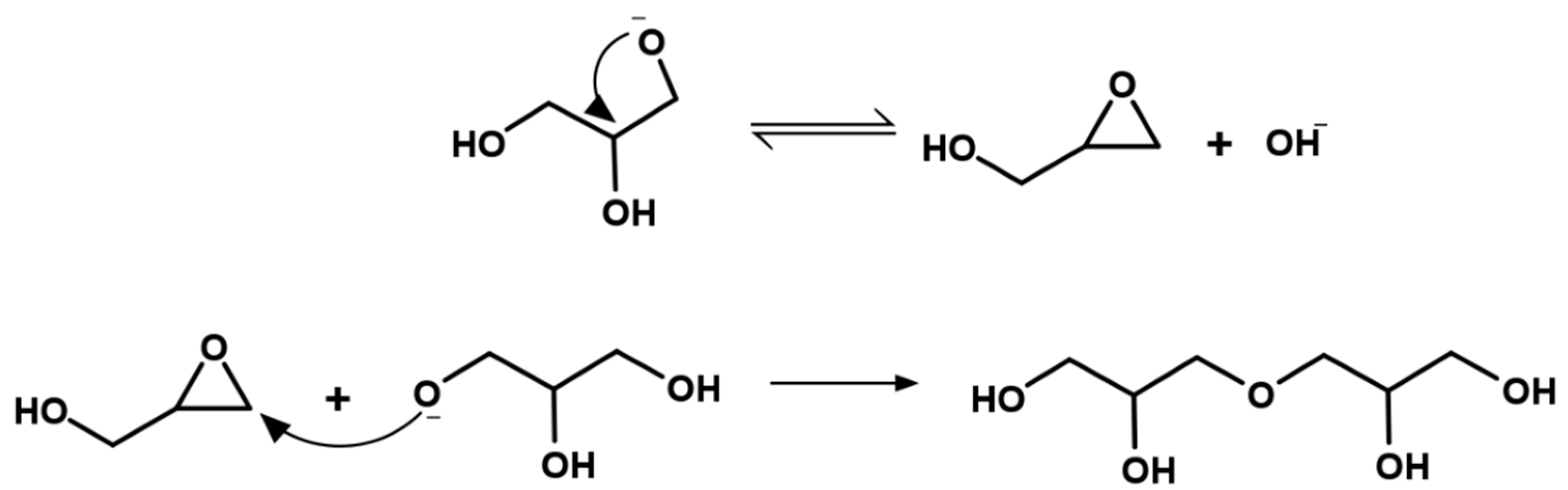
- Basic hydrolysis of epichlorohydrin and glycerol
- (b)
- Direct polymerization of glycerol in the presence of a strong homogeneous base
4. Catalytic Systems
4.1. Homogenous Catalysis
4.2. Heterogeneous Catalysts
4.3. Partial Dissolution of Heterogeneous Catalysts
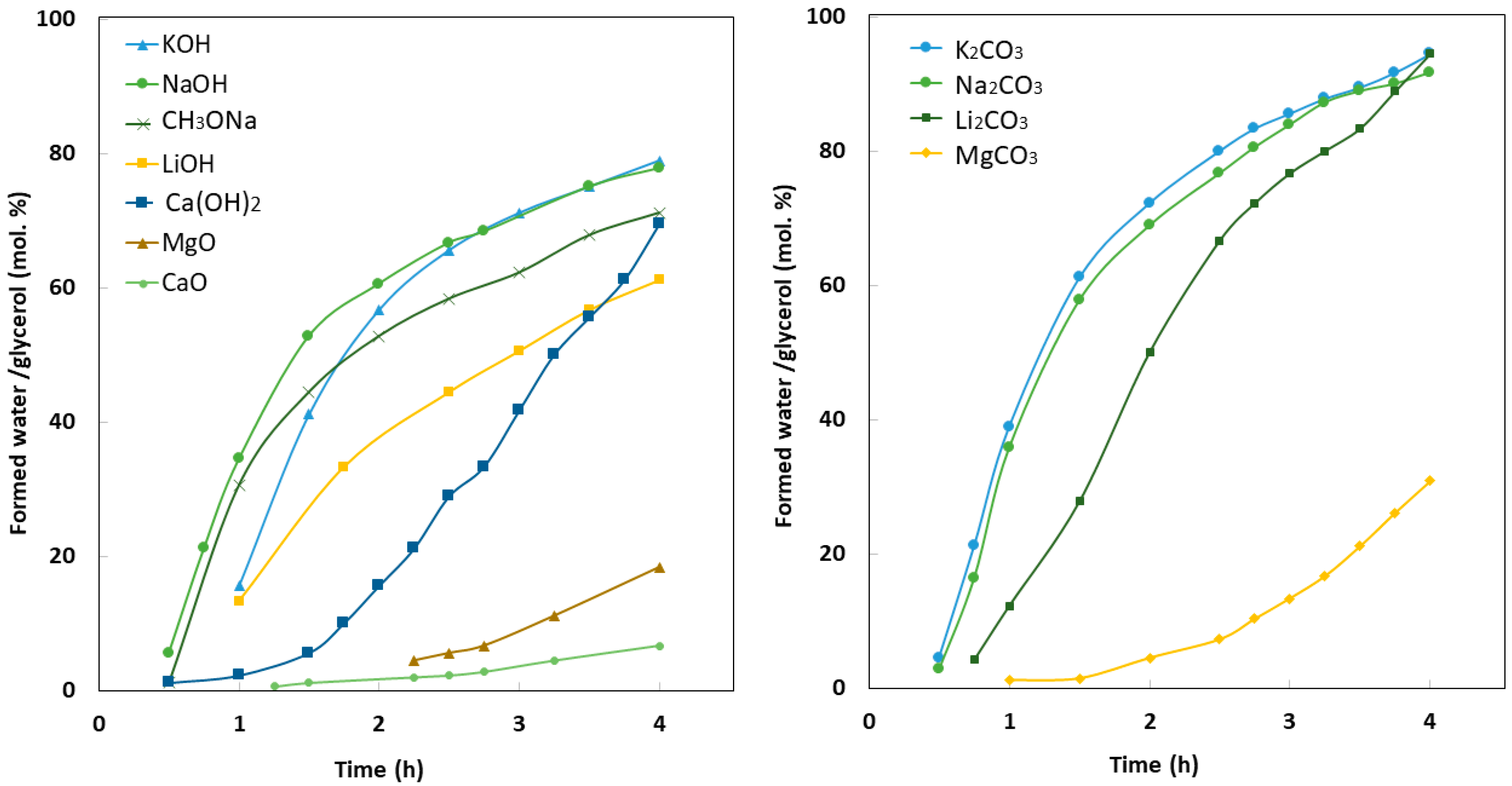
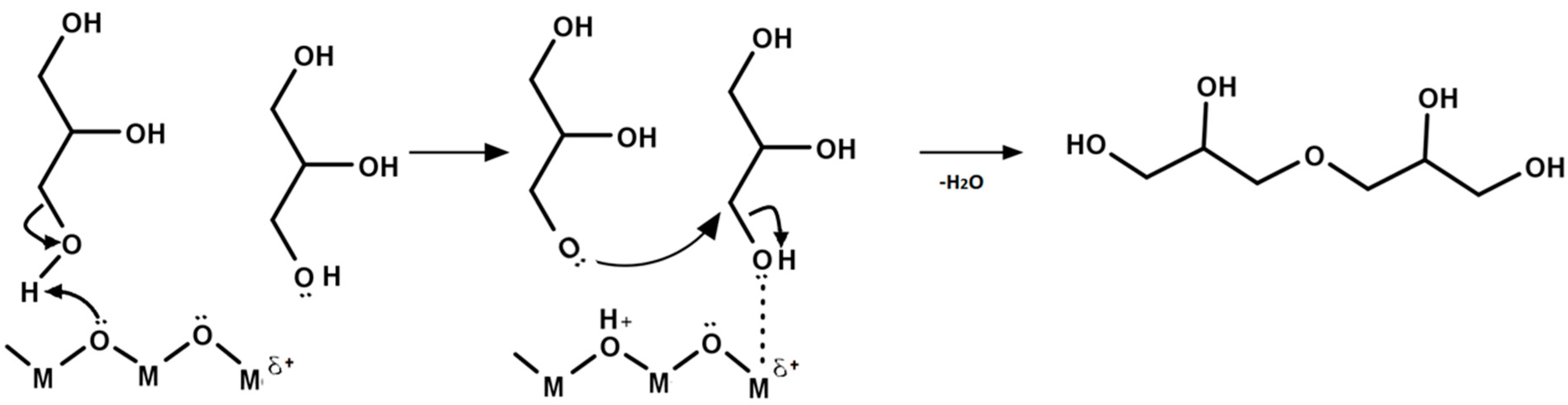
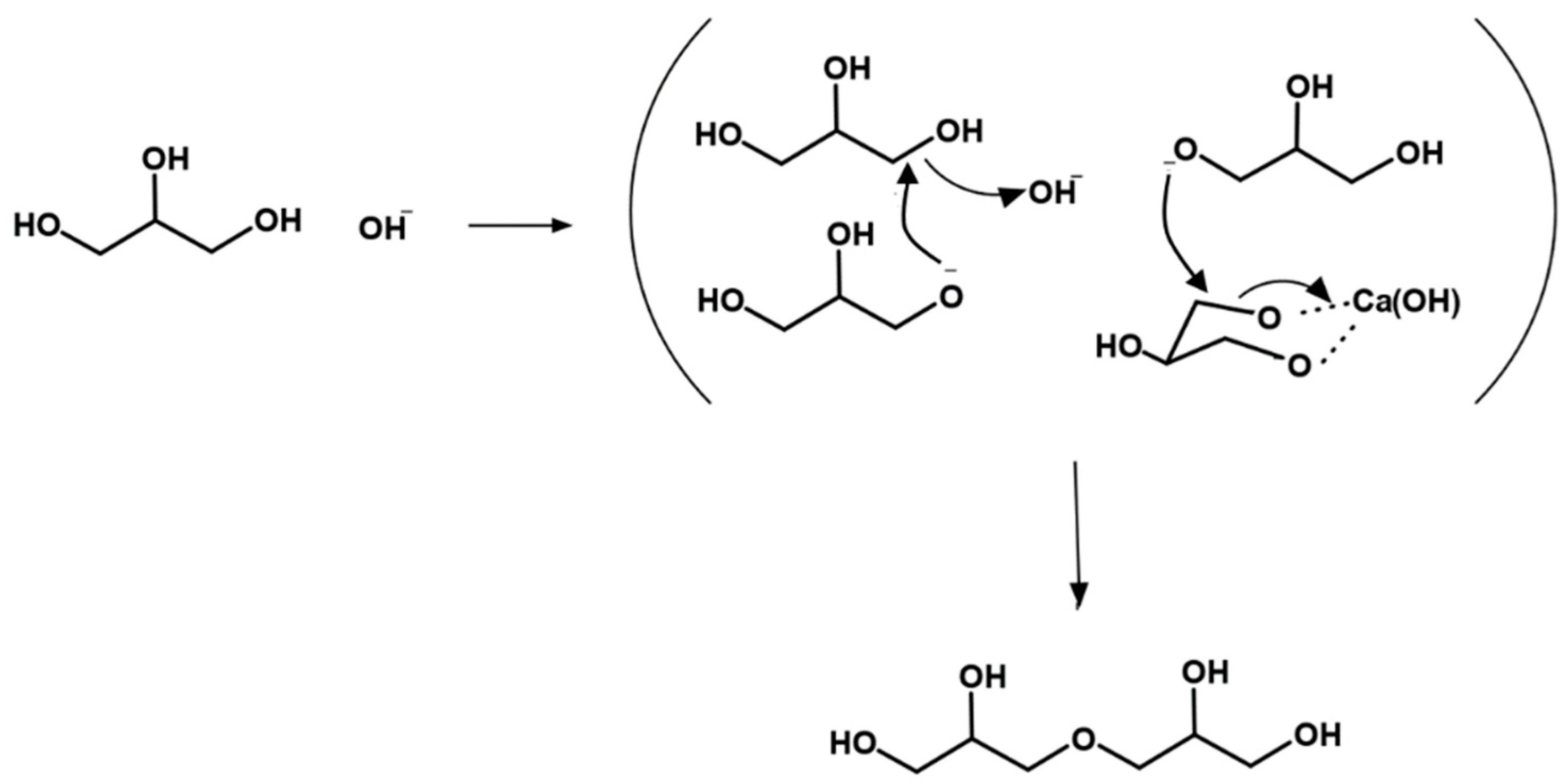
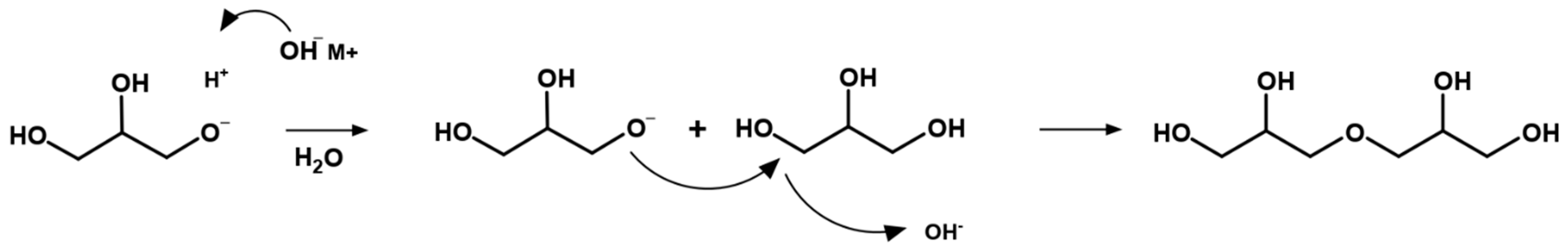
5. Mechanism of Glycerol Polymerization over Alkaline Catalysts
6. Reaction Conditions
6.1. Temperature
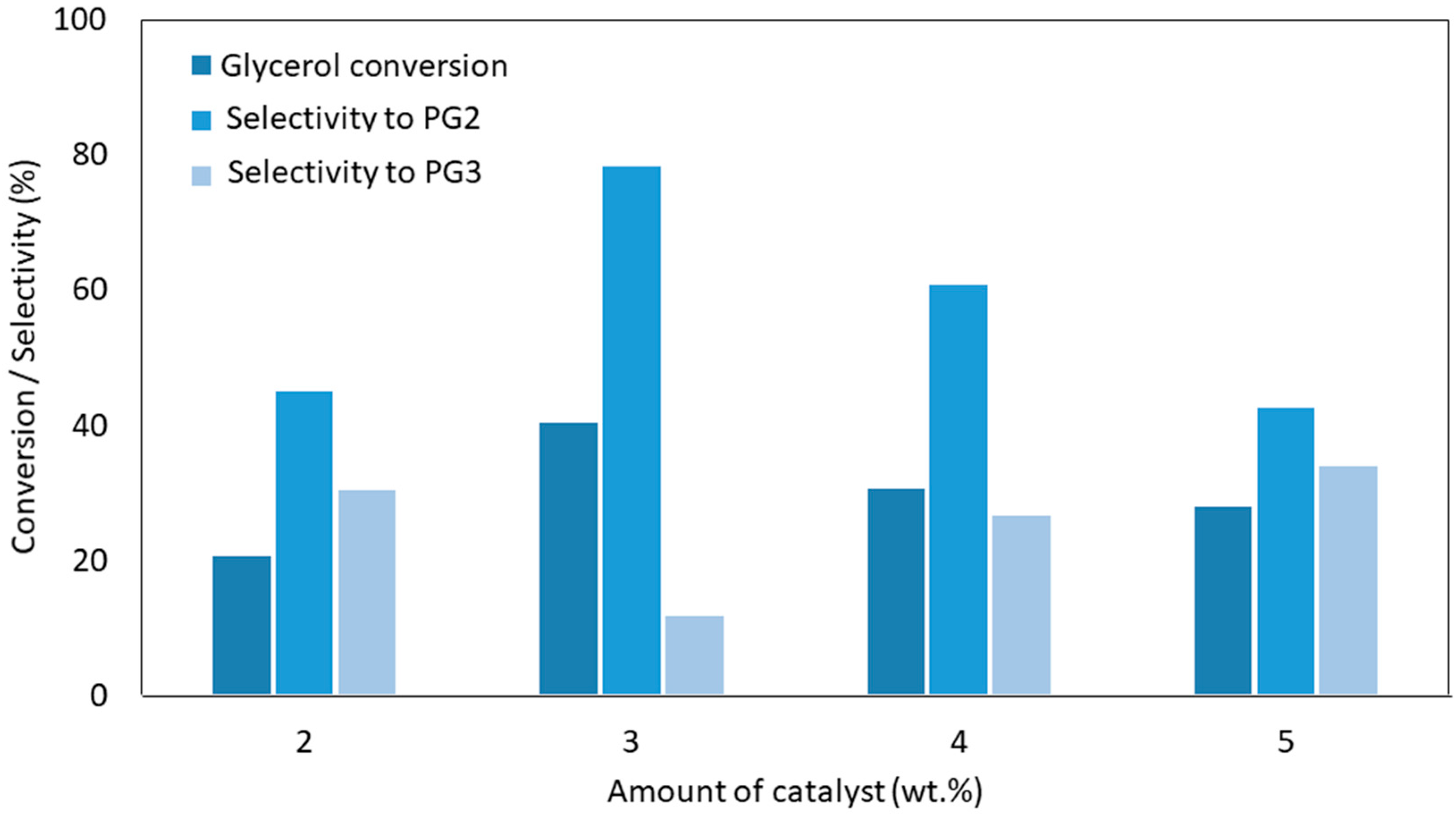
6.2. Effect of Catalyst Loading
6.3. Pressure
6.4. Atmosphere
6.5. Reaction Time
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Polyglycerol Market Size. Available online: https://www.grandviewresearch.com/industry-analysis/polyglycerol-market (accessed on 5 May 2020).
- Ciriminna, R.; Katryniok, B.; Paul, S.; Dumeignil, F.; Pagliaro, M. Glycerol-Derived Renewable Polyglycerols: A Class of Versatile Chemicals of Wide Potential Application. Org. Process Res. Dev. 2015, 19, 748–754. [Google Scholar] [CrossRef]
- Barros, F.J.S.; Moreno-Tost, R.; Cecilia, J.A.; Ledesma-Muñoz, A.L.; de Oliveira, L.C.C.; Luna, F.M.T.; Vieira, R.S. Glycerol Oligomers Production by Etherification Using Calcined Eggshell as Catalyst. Mol. Catal. 2017, 433, 282–290. [Google Scholar] [CrossRef]
- García-Sancho, C.; Moreno-Tost, R.; Mérida-Robles, J.M.; Santamaría-González, J.; Jiménez-López, A.; Torres, P.M. Etherification of Glycerol to Polyglycerols over MgAl Mixed Oxides. Catal. Today 2011, 167, 84–90. [Google Scholar] [CrossRef]
- Ruppert, A.M.; Meeldijk, J.D.; Kuipers, B.W.M.; Erné, B.H.; Weckhuysen, B.M. Glycerol Etherification over Highly Active CaO-Based Materials: New Mechanistic Aspects and Related Colloidal Particle Formation. Chem. Eur. J. 2008, 14, 2016–2024. [Google Scholar] [CrossRef] [PubMed]
- Nieuwelink, A.-E. CaO/CNF for the Oligomerization of Glycerol. Master’s Thesis, Department of Chemistry, Utrecht University, Utrecht, The Netherlands, 2015. [Google Scholar]
- Barros, F.J.S.; Cecilia, J.A.; Moreno-Tost, R.; de Oliveira, M.F.; Rodríguez-Castellón, E.; Luna, F.M.T.; Vieira, R.S. Glycerol Oligomerization Using Low Cost Dolomite Catalyst. Waste Biomass Valorization 2018, 11, 1499–1512. [Google Scholar] [CrossRef]
- Ionescu, M.; Petrović, Z.S. On the Mechanism of Base-Catalyzed Glycerol Polymerization and Copolymerization. Eur. J. Lipid Sci. Technol. 2018, 120, 1800004. [Google Scholar] [CrossRef]
- Gholami, Z.; Abdullah, A.Z.; Lee, K.-T. Dealing with the Surplus of Glycerol Production from Biodiesel Industry through Catalytic Upgrading to Polyglycerols and Other Value-Added Products. Renew. Sustain. Energy Rev. 2014, 39, 327–341. [Google Scholar] [CrossRef]
- Chong, C.C.; Aqsha, A.; Ayoub, M.; Sajid, M.; Abdullah, A.Z.; Yusup, S.; Abdullah, B. A Review over the Role of Catalysts for Selective Short-Chain Polyglycerol Production from Biodiesel Derived Waste Glycerol. Environ. Technol. Innov. 2020, 19, 100859. [Google Scholar] [CrossRef]
- Jungermann, E.; Sonntag, N. (Eds.) Glycerine: A Key Cosmetic Ingredient; Cosmetic Science and Technology Series; Dekker: New York, NY, USA, 1991. [Google Scholar]
- Martin, A.; Richter, M. Oligomerization of Glycerol—A Critical Review. Eur. J. Lipid Sci. Technol. 2010, 113, 100–117. [Google Scholar] [CrossRef]
- Hejna, A.; Kosmela, P.; Formela, K.; Piszczyk, Ł.; Haponiuk, J.T. Potential Applications of Crude Glycerol in Polymer Technology–Current State and Perspectives. Renew. Sustain. Energy Rev. 2016, 66, 449–475. [Google Scholar] [CrossRef]
- Cassel, S.; Debaig, C.; Benvegnu, T.; Chaimbault, P.; Lafosse, M.; Plusquellec, D.; Rollin, P. Original Synthesis of Linear, Branched and Cyclic Oligoglycerol Standards. Eur. J. Org. Chem. 2001, 2001, 875–896. [Google Scholar] [CrossRef]
- Sunder, A.; Mülhaupt, R.; Frey, H. Hyperbranched Polyether−Polyols Based on Polyglycerol: Polarity Design by Block Copolymerization with Propylene Oxide. Macromolecules 2000, 33, 309–314. [Google Scholar] [CrossRef]
- Kainthan, R.K.; Muliawan, E.B.; Hatzikiriakos, S.G.; Brooks, D.E. Synthesis, Characterization, and Viscoelastic Properties of High Molecular Weight Hyperbranched Polyglycerols. Macromolecules 2006, 39, 7708–7717. [Google Scholar] [CrossRef]
- Nieberle, J. Hyperbranched Polyglycerols as Building Blocks for Complex Amphiphilic Structures: Synthesis, Characterization and Applications. Ph.D. Thesis, Johannes Gutenberg University, Mainz, Germany, 2008. [Google Scholar]
- Kainthan, R.K.; Hester, S.R.; Levin, E.; Devine, D.V.; Brooks, D.E. In Vitro Biological Evaluation of High Molecular Weight Hyperbranched Polyglycerols. Biomaterials 2007, 28, 4581–4590. [Google Scholar] [CrossRef] [PubMed]
- Pagliaro, M.; Rossi, M. The Future of Glycerol; RSC: Philadelphia, PA, USA, 2008. [Google Scholar] [CrossRef]
- Babayan, V.K.; Lehman, H. Process for Preparation and Purification of Polyglycerols and Esters Thereof. U.S. Patent 3,637,774, 25 January 1972. [Google Scholar]
- Lemke, D.W. Processes for Preparing Linear Polyglycerols and Polyglycerol Esters. U.S. Patent 6,620,904, 16 September 2003. [Google Scholar]
- Hasenhuettl, G.L. Synthesis and Commercial Preparation of Food Emulsifiers. In Food Emulsifiers and Their Applications; Hasenhuettl, G.L., Hartel, R.W., Eds.; Springer: New York, NY, USA, 2008; pp. 11–37. [Google Scholar] [CrossRef]
- Żołek-Tryznowska, Z.; Tryznowski, M.; Królikowska, J. Hyperbranched Polyglycerol as an Additive for Water-Based Printing Ink. J. Coat. Technol. Res. 2015, 12, 385–392. [Google Scholar] [CrossRef]
- Meyer, J.; Friedrich, A.; Foetsch, H.; Springer, O.; von Hof, J.M.; Wenk, H.H. Polyglycerol Esters with a Particular Oligomer Distribution of the Polyglycerol. U.S. Patent 9,427,385, 30 August 2016. [Google Scholar]
- Thomas, A.; Müller, S.S.; Frey, H. Beyond Poly(Ethylene Glycol): Linear Polyglycerol as a Multifunctional Polyether for Biomedical and Pharmaceutical Applications. Biomacromolecules 2014, 15, 1935–1954. [Google Scholar] [CrossRef]
- Daly, S.; Maitra, P.; Setiawan, B. Sunscreen Compositions Containing a Uv-Absorbing Polyglycerol and a Non-Uv-Absorbing Polyglycerol. U.S. Patent 14/674,536, 12 November 2015. [Google Scholar]
- Zhang, H.; Grinstaff, M.W. Recent Advances in Glycerol Polymers: Chemistry and Biomedical Applications. Macromol. Rapid Commun. 2014, 35, 1906–1924. [Google Scholar] [CrossRef]
- Ayoub, M.; Khayoon, M.S.; Abdullah, A.Z. Synthesis of Oxygenated Fuel Additives via the Solventless Etherification of Glycerol. Bioresour. Technol. 2012, 112, 308–312. [Google Scholar] [CrossRef]
- Morlino, N.M.; Sweeny, P.G.; Curham, B.D. Polyglycerol Antifoam Agents in Paper Processing. U.S. Patent 5,429,718, 4 July 1995. [Google Scholar]
- Nilewski, M.; Favresse, P.; Scharf, A.; Gehrmann, P.; Mettin, T.; Schwan, M.; Springer, O.; Wied, T. Use of Polyglycerol Partial Esters as Defoamers. U.S. Patent 9,738,797, 22 August 2017. [Google Scholar]
- Lu, Y. 10-20 Polyglycerol and Production Method Thereof. Patent CN104650340A, 27 May 2015. [Google Scholar]
- Pinto, B.P.; De Araujo Mota, C.J. Developments in Glycerol Byproduct-Based Biorefineries. In Advances in Biorefineries; Woodhead Publishing: Cambridge, UK, 2014; pp. 364–385. [Google Scholar] [CrossRef]
- Milewski, A.; Dydo, P.; Jakóbik-Kolon, A.; Czechowicz, D.; Babilas, D.; Burek, M.; Waśkiewicz, S.; Byczek-Wyrostek, A.; Krawczyk, T.; Kasprzycka, A. Preparation of Triglycerol from Glycerol and Epichlorohydrin at Room Temperature: Synthesis Optimization and Toxicity Studies. ACS Sustain. Chem. Eng. 2018, 6, 13208–13216. [Google Scholar] [CrossRef]
- Endo, T.; Omori, H. Process for Producing Polyglycerol. Patent EP1568677B1, 8 January 2014. [Google Scholar]
- Cespi, D.; Cucciniello, R.; Ricciardi, M.; Capacchione, C.; Vassura, I.; Passarini, F.; Proto, A. A Simplified Early Stage Assessment of Process Intensification: Glycidol as a Value-Added Product from Epichlorohydrin Industry Wastes. Green Chem. 2016, 18, 4559–4570. [Google Scholar] [CrossRef]
- Mukbaniani, O.V.; Abadie, M.J.M.; Tatrishvili, T.N. (Eds.) Chemical Engineering of Polymers: Production of Functional and Flexible Materials, 1st ed.; Apple Academic Press: Palm Bay, FL, USA, 2017. [Google Scholar] [CrossRef]
- Lemke, D.W.; Nivens, S. Process for Prepapring Polycerol and Mixed Ethers. U.S. Patent 12/221,608, 11 December 2008. [Google Scholar]
- Plasman, V.; Caulier, T.; Boulos, N. Polyglycerol Esters Demonstrate Superior Antifogging Properties for Films. Plast. Addit. Compd. 2005, 7, 30–33. [Google Scholar] [CrossRef]
- Sayoud, N.; De Oliveira Vigier, K.; Cucu, T.; De Meulenaer, B.; Fan, Z.; Lai, J.; Clacens, J.-M.; Liebens, A.; Jérôme, F. Homogeneously-Acid Catalyzed Oligomerization of Glycerol. Green Chem. 2015, 17, 4307–4314. [Google Scholar] [CrossRef]
- Salehpour, S.; Dubé, M.A. Towards the Sustainable Production of Higher-Molecular-Weight Polyglycerol. Macromol. Chem. Phys. 2011, 212, 1284–1293. [Google Scholar] [CrossRef]
- Medeiros, M.A.; Araujo, M.H.; Augusti, R.; de Oliveira, L.C.A.; Lago, R.M. Acid-Catalyzed Oligomerization of Glycerol Investigated by Electrospray Ionization Mass Spectrometry. J. Braz. Chem. Soc. 2009, 20, 1667–1673. [Google Scholar] [CrossRef]
- Krisnandi, Y.K.; Eckelt, R.; Schneider, M.; Martin, A.; Richter, M. Glycerol Upgrading over Zeolites by Batch-Reactor Liquid-Phase Oligomerization: Heterogeneous versus Homogeneous Reaction. ChemSusChem 2008, 1, 835–844. [Google Scholar] [CrossRef]
- Harris, E.G.; Hees, U.; Bunte, R.; Hachgenei, J.W.; Kuhm, P. Process for the Production of Oligoglycerol Mixtures of Increased Diglycerol Content. U.S. Patent 5,349,094, 20 September 1994. [Google Scholar]
- Kirby, F.; Nieuwelink, A.-E.; Kuipers, B.W.M.; Kaiser, A.; Bruijnincx, P.C.A.; Weckhuysen, B.M. CaO as Drop-In Colloidal Catalysts for the Synthesis of Higher Polyglycerols. Chem. Eur. J. 2015, 21, 5101–5109. [Google Scholar] [CrossRef]
- Richter, M.; Krisnandi, Y.; Eckelt, R.; Martin, A. Homogeneously Catalyzed Batch Reactor Glycerol Etherification by CsHCO3. Catal. Commun. 2008, 9, 2112–2116. [Google Scholar] [CrossRef]
- Garti, N.; Aserin, A.; Zaidman, B. Polyglycerol Esters: Optimization and Techno-Economic Evaluation. J. Am. Oil Chem. Soc. 1981, 58, 878–883. [Google Scholar] [CrossRef]
- Clacens, J.-M.; Pouilloux, Y.; Barrault, J. Selective Etherification of Glycerol to Polyglycerols over Impregnated Basic MCM-41 Type Mesoporous Catalysts. Appl. Catal. Gen. 2002, 227, 181–190. [Google Scholar] [CrossRef]
- Harris, B. Polyglycerol Esters of Aliphatic Acids of Relatively High Molecular Weight. U.S. Patent 2,023,388, 3 December 1935. [Google Scholar]
- Charles, G.; Clacens, J.-M.; Pouilloux, Y.; Barrault, J. Préparation de Diglycérol et Triglycérol Par Polymérisation Directe Du Glycérol En Présence de Catalyseurs Mésoporeux Basiques. Ol. Corps Gras Lipides 2003, 10, 74–82. [Google Scholar] [CrossRef]
- Nosal, H.; Nowicki, J.; Warzała, M.; Nowakowska-Bogdan, E.; Zarębska, M. Synthesis and Characterization of Alkyd Resins Based on Camelina Sativa Oil and Polyglycerol. Prog. Org. Coat. 2015, 86, 59–70. [Google Scholar] [CrossRef]
- Ayoub, M.; Sufian, S.; Hailegiorgis, S.M.; Ullah, S.; Uemura, Y. Conversion of Glycerol to Polyglycerol over Waste Duck-Bones as a Catalyst in Solvent Free Etherification Process. IOP Conf. Ser. Mater. Sci. Eng. 2017, 226, 012073. [Google Scholar] [CrossRef]
- Chen, X.; Peng, M. Method for Preparing Polyglycerine with High Degree of Polymerization. Patent CN102532515A, 4 July 2012. [Google Scholar]
- Sivaiah, M.V.; Robles-Manuel, S.; Valange, S.; Barrault, J. Recent Developments in Acid and Base-Catalyzed Etherification of Glycerol to Polyglycerols. Catal. Today 2012, 198, 305–313. [Google Scholar] [CrossRef]
- Ayoub, M.; Abdullah, A.Z.; Ahmad, M.; Sultana, S. Performance of Lithium Modified Zeolite Y Catalyst in Solvent-Free Conversion of Glycerol to Polyglycerols. J. Taibah Univ. Sci. 2014, 8, 231–235. [Google Scholar] [CrossRef]
- Gholami, Z.; Abdullah, A.Z.; Lee, K.-T. Selective Etherification of Glycerol over Heterogeneous Mixed Oxide Catalyst: Optimization of Reaction Parameters. Chem. Eng. Sci. 2013, 1, 79–86. [Google Scholar] [CrossRef]
- Gholami, Z.; Abdullah, A.Z.; Lee, K.-T. Glycerol Etherification to Polyglycerols Using Ca1+xAl1−xLaxO3 Composite Catalysts in a Solventless Medium. J. Taiwan Inst. Chem. Eng. 2013, 44, 117–122. [Google Scholar] [CrossRef]
- Gholami, Z.; Abdullah, A.Z.; Lee, K.T. Heterogeneously Catalyzed Etherification of Glycerol to Diglycerol over Calcium–Lanthanum Oxide Supported on MCM-41: A Heterogeneous Basic Catalyst. Appl. Catal. Gen. 2014, 479, 76–86. [Google Scholar] [CrossRef]
- Guerrero-Urbaneja, P.; García-Sancho, C.; Moreno-Tost, R.; Mérida-Robles, J.; Santamaría-González, J.; Jiménez-López, A.; Maireles-Torres, P. Glycerol Valorization by Etherification to Polyglycerols by Using Metal Oxides Derived from MgFe Hydrotalcites. Appl. Catal. Gen. 2014, 470, 199–207. [Google Scholar] [CrossRef]
- Pérez-Barrado, E.; Pujol, M.C.; Aguiló, M.; Llorca, J.; Cesteros, Y.; Díaz, F.; Pallarès, J.; Marsal, L.F.; Salagre, P. Influence of Acid–Base Properties of Calcined MgAl and CaAl Layered Double Hydroxides on the Catalytic Glycerol Etherification to Short-Chain Polyglycerols. Chem. Eng. J. 2015, 264, 547–556. [Google Scholar] [CrossRef]
- Sangkhum, P.; Yanamphorn, J.; Wangriya, A.; Ngamcharussrivichai, C. Ca–Mg–Al Ternary Mixed Oxides Derived from Layered Double Hydroxide for Selective Etherification of Glycerol to Short-Chain Polyglycerols. Appl. Clay Sci. 2019, 173, 79–87. [Google Scholar] [CrossRef]
- Kaiser, A.; Weckhuysen, B.M.; Leinweber, D.; Kirby, F.; Scherl, F.X.; Metz, H.J.; Bruijnincx, P.C.A. Preparation of Polyglycerols. Patent WO2015122770A1, 20 August 2015. [Google Scholar]
- Han, Y.H.; Kim, H.R.; Han, I.S.; Choi, H.O.; Kim, H.S.; An, S.Y.; Youn, Y.H. Metal Oxide Catalysts for Etherification, Method for Its Preparation Thereof, and Method for the Production of Linear Polyglycerol Using the Same. Patent WO2010044531A2, 22 April 2010. [Google Scholar]
- Barrault, J.; Clacens, J.-M.; Pouilloux, Y. Methods for Etherifying Glycerol, and Catalysts for Implementing Said Methods. Patent WO2001098243A1, 27 December 2001. [Google Scholar]
- Barriau, E. Hyperbranched Polyether Polyols as Building Blocks for Complex Macromolecular Architectures. Ph.D. Thesis, Johannes Gutenberg University, Mainz, Germany, 2005. [Google Scholar]
- Cammenga, H.K.; Schulze, F.W.; Theuerl, W. Vapor Pressure and Evaporation Coefficient of Glycerol. J. Chem. Eng. Data 1977, 22, 131–134. [Google Scholar] [CrossRef]
- Wilson, R.; van Schie, B.J.; Howes, D. Overview of the Preparation, Use and Biological Studies on Polyglycerol Polyricinoleate (PGPR). Food Chem. Toxicol. 1998, 36, 711–718. [Google Scholar] [CrossRef]
- Nalawade, S.P.; Picchioni, F.; Janssen, L.P.B.M. Supercritical Carbon Dioxide as a Green Solvent for Processing Polymer Melts: Processing Aspects and Applications. Prog. Polym. Sci. 2006, 31, 19–43. [Google Scholar] [CrossRef]
- de Caro, P.; Bandres, M.; Urrutigoïty, M.; Cecutti, C.; Thiebaud-Roux, S. Recent Progress in Synthesis of Glycerol Carbonate and Evaluation of Its Plasticizing Properties. Front. Chem. 2019, 7. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
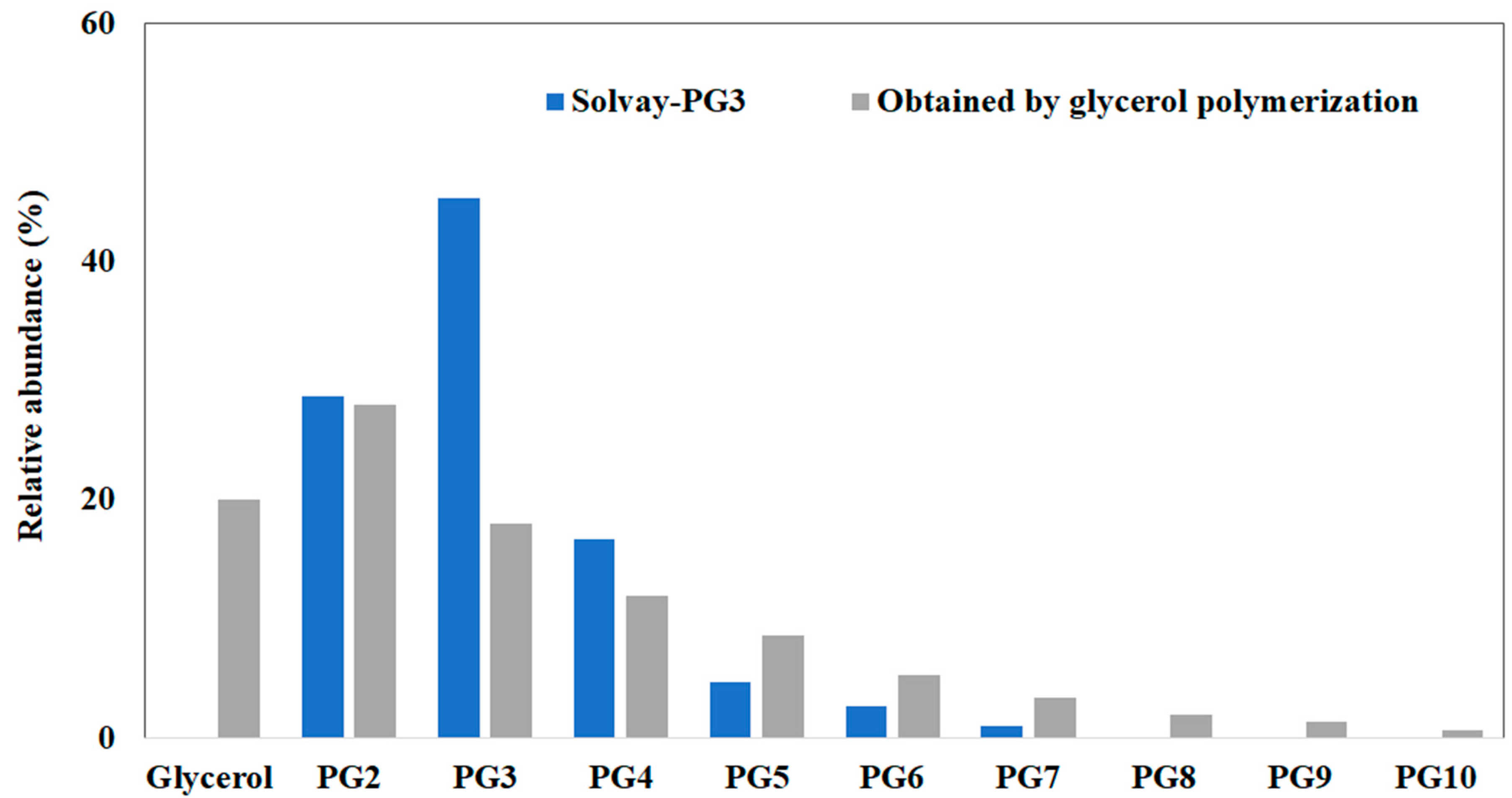
| Name (Abbreviation) | Molecular Weight (g mol−1) | OH Groups | Viscosity (CTKS 1 @ 50 °C) | Density (g/cm3) | Hydroxyl Value (mg KOH/g) 2 |
|---|---|---|---|---|---|
| Glycerol (G) | 92 | 3 | 45 | 1.256 | 1830 |
| Diglycerol (PG2) | 166 | 4 | 287 | 1.279 | 1352 |
| Triglycerol (PG3) | 240 | 5 | 647 | 1.2646 (40 °C) | 1169 |
| Tetraglycerol (PG4) | 314 | 6 | 1067 | 1.2687 (40 °C) | 1071 |
| Pentaglycerol (PG5) | 388 | 7 | 1408 | - | 1012 |
| Hexaglycerol (PG6) | 462 | 8 | 1671 | - | 970 |
| Heptaglycerol (PG7) | 536 | 9 | 2053 | - | 941 |
| Octaglycerol (PG8) | 610 | 10 | 2292 | - | 920 |
| Nonaglycerol (PG9) | 684 | 11 | 2817 | - | 903 |
| Decaglycerol (PG10) | 758 | 12 | 3199 | - | 880 |
| Pentadecaglycerol (PG15) | 1128 | 17 | 4893 | - | 846 |
| Catalyst | Temp (°C) | Cat (wt.%) | Time (h) | Reaction Conditions | Glycerol Conversion (%) | Selectivity in PGx | Ref |
|---|---|---|---|---|---|---|---|
| LiOH | 240 | 2 | 6 | Continuous N2 flow using Dean–Stark system to condensed water | 100 | SPG2: 20% | [51] |
| LiOH | 230 260 | 0.1 | 7 | Low N2 flow using Dean–Stark system to condensed water | 24 | SPG2-3: 100% SPG2-3: 68% | [50] |
| 80 | |||||||
| KOH NaOH | 260–280 | 2–4 | 1–4 | Continuous N2 flow to remove formed water | 90–95 | PG10-25 | [52] |
| CsHCO3 | 260 | 0.4 | 8 | Under atmospheric pressure | 64 | SPG2: 23% SPG2: 39% SPG2: 32% | [45] |
| Cs2CO3 | 0.7 | 71 | |||||
| CsOH | 0.3 | 75 | |||||
| Ca(OH)2 | 230 | 0.1 | - | Under vacuum (200 mmHg) | 57 | SPG2-3: 87% | [21] |
| Na2CO3 NaOH | 240 | 2 | 9 | - | 76 | SPG2-3: 93% SPG2-3: 99% | [49] |
| 63 | |||||||
| CsOH | 260 | 2 | 4 | Under N2 atmosphere using Dean–Stark system to condensed water | 90 | SPG2-3: 63% | [47] |
| KOH NaOH | 260 | - | - | Continuous N2 flow to remove formed water | 50–100 | - | [20] |
| Catalyst | Temp (°C) | Cat (wt.%) | Time (h) | Reaction Conditions | Glycerol Conversion (%) | Selectivity to PGx | Ref |
|---|---|---|---|---|---|---|---|
| Ca-MgAl LDH | 220 | 3 | 24 | Under N2 flow using Dean–Stark system to collect water | 40.4 | SPG2: 78.3% | [60] |
| Dolomite (mixed oxide CaO-MgO) | 245 | 2 | 24 | Under N2 atmosphere using Dean–Stark system to condense water | 90 | SPG2: 23% SPG3: 22% | [7] |
| Calcined eggshell | 220 245 | 2 | 24 | Under N2 atmosphere | 85 | SPG2: 35% SPG3: 15% | [3] |
| 100 | |||||||
| Duck bones | 240 | 2 | 12 | Under N2 atmosphere using Dean–Stark system to condense water | 99 | - | [51] |
| 14wt.% CaO/CNF | 220 | 0.46 * | 24 | Under Ar gas flow using Dean–Stark system to condense water | 76 | SPG2: 40% SPG3: 15% | [44] |
| MgAl-LDHs–CaAl-LDHs: cHT1 | 235 | 2 | 24 | Under N2 atmosphere using Dean–Stark system to condense water | 24 | SPG2-3: 100% | [59] |
| MgAl-LDHs–CaAl-LDHs: cHT2 | 235 | 2 | 24 | Under N2 atmosphere using Dean–Stark system to condensed water | 96 | SPG2-3: 12% Sacrolein: 88% | [59] |
| MgFeO4 | 220 | - | 24 | Under N2 atmosphere using Dean–Stark system to condensed water | 41 | SPG2: 90% SPG3: 10% | [58] |
| Ca1.6La0.4Al0.6O3 | 250 | 2 | 8 | Under N2 atmosphere using Dean–Stark system to condensed water | 96.3 | SPG2-3: 86% | [55] |
| MgAl-Na | 220 | 2 | 24 | Under N2 atmosphere using Dean–Stark system to condensed water | 50 | SPG2: 85% SPG3: 15% | [4] |
| Ca(OH)2 | 140 | 2.4 | ~6 | Under reduced pressure using Dean–Stark system to condensed water | 12 | - | [40] |
| CaCO3 | 11 | ||||||
| CaO/Ca12Al14O33 | 200–250 | - | 24 | Under N2 atmosphere using Dean–Stark system to condensed water | - | Linear PGs | [62] |
| Zeolite NaX | 260 | 2 | 24 | Under Ar atmosphere | 100 | SPG2: 15% | [42] |
| Zeolite NaY | 80 | SPG2: 58% | |||||
| Zeolite Na Beta | 50 | SPG2: 90% | |||||
| MgO | 220 | 2 | 20 | Under Ar atmosphere using Dean–Stark system to condensed water and Ice trap for acrolein | 10 | SPG2-3: 90% | [5] |
| BaO-SrO | 80 | ||||||
| CaO | > 80 | ||||||
| Cs25Al(Si/Al:20) | 260 | 2 | 20 | Under N2 atmosphere using Dean–Stark system to condensed water | 80 | SPG2: 62% SPG3: 33% | [47,63] |
| Zeolite NaA Zeolite NaZ | 200–260 | 22 | Under N2 with a reflux condenser and water separator | 84.6 | SPG2-3: 62% SPG2-3: 52% | [43] | |
| 90.5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ebadipour, N.; Paul, S.; Katryniok, B.; Dumeignil, F. Alkaline-Based Catalysts for Glycerol Polymerization Reaction: A Review. Catalysts 2020, 10, 1021. https://doi.org/10.3390/catal10091021
Ebadipour N, Paul S, Katryniok B, Dumeignil F. Alkaline-Based Catalysts for Glycerol Polymerization Reaction: A Review. Catalysts. 2020; 10(9):1021. https://doi.org/10.3390/catal10091021
Chicago/Turabian StyleEbadipour, Negisa, Sébastien Paul, Benjamin Katryniok, and Franck Dumeignil. 2020. "Alkaline-Based Catalysts for Glycerol Polymerization Reaction: A Review" Catalysts 10, no. 9: 1021. https://doi.org/10.3390/catal10091021
APA StyleEbadipour, N., Paul, S., Katryniok, B., & Dumeignil, F. (2020). Alkaline-Based Catalysts for Glycerol Polymerization Reaction: A Review. Catalysts, 10(9), 1021. https://doi.org/10.3390/catal10091021