Green Catalysts: Applied and Synthetic Photosynthesis †

 , and
, and
Abstract
1. Introduction
2. Applied Photocurrent
2.1. Light Absorption, Electronic Considerations, and Optical Cross Sections of Reaction Centers
2.2. Novel Semiconductor Materials, Architectures, and Electrode Surface Modifications
2.2.1. Semiconductor Materials, Architectures, and Modifications
2.2.2. Electrode Materials and Modifications
2.3. Redox Mediators
2.4. Biofilms, Microbial Solar Cells, and Thylakoid Membrane-Based Solar Cells
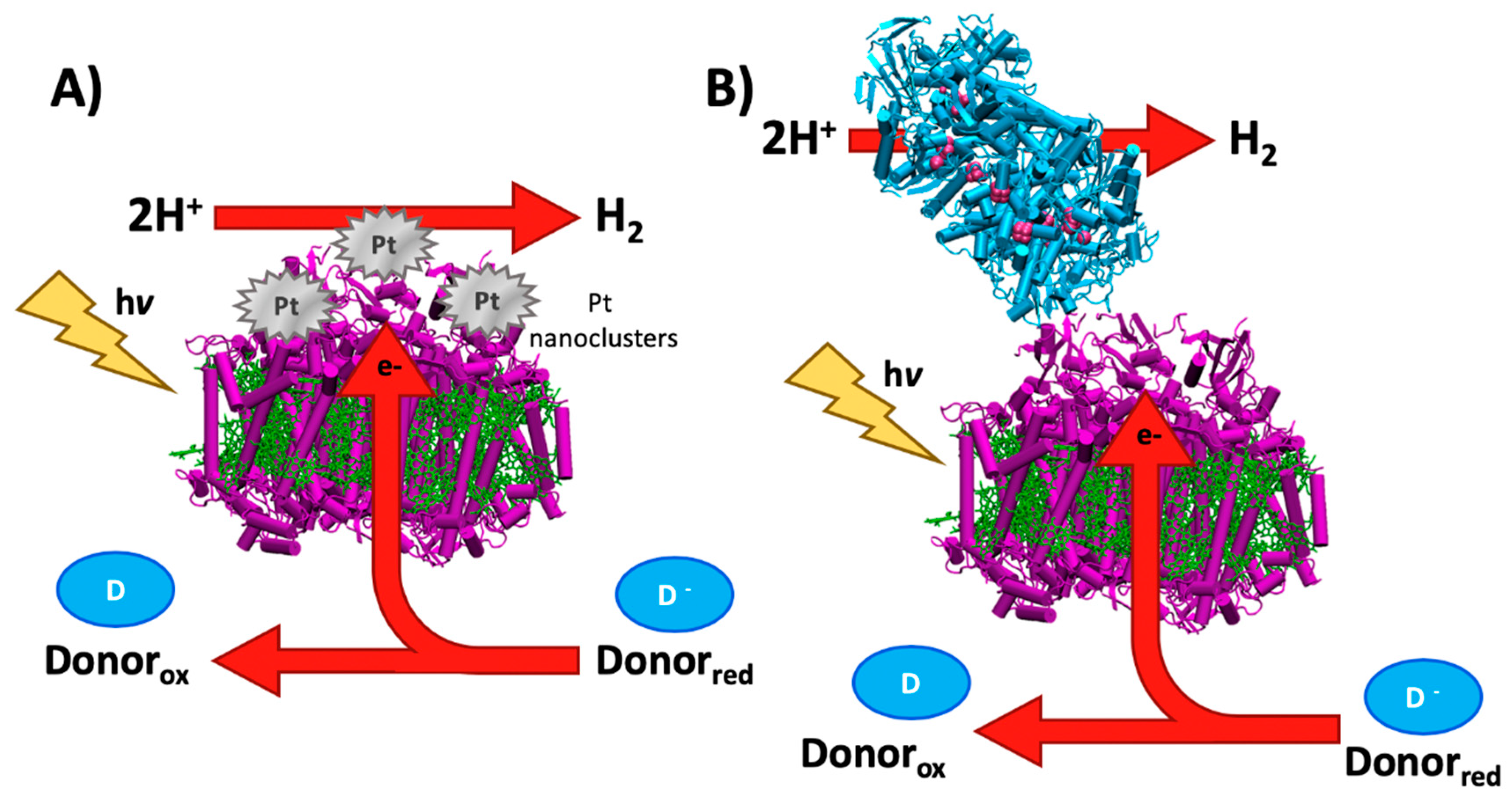
3. Hydrogen Evolution Photocatalyzed by Biological Reaction Centers
3.1. Early History of Photosynthetic Hydrogen Production
3.2. Bioengineered In Vivo Hydrogen Production
3.3. Overcoming Kinetic Limitations of Hydrogen Evolution
3.4. In Vitro Strategies for Hydrogen Evolution
3.5. Emerging Technologies for Bioengineering Hydrogen Evolution
4. Biohybrid Photosynthesis with Artificial Reaction Centers
4.1. Light-Driven ATP Synthesis
4.2. Bacteriorhodopsin as a Catalyst to Improve Device Performances
4.3. Light-Driven Enzymatic Catalysis by Inorganic Photocatalysts
4.4. Light-Driven Enzyme Systems for CO2 Reduction
4.5. Light-Activated Nitrogenase
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kothe, T.; Poller, S.; Zhao, F.; Fortgang, P.; Rogner, M.; Schuhmann, W.; Plumere, N. Engineered electron-transfer chain in photosystem 1 based photocathodes outperforms electron-transfer rates in natural photosynthesis. Chemistry 2014, 20, 11029–11034. [Google Scholar] [CrossRef] [PubMed]
- Kazemzadeh, S.; Riazi, G.; Ajeian, R. Novel approach of biophotovoltaic solid state solar cells based on a multilayer of PS1 complexes as an active layer. ACS Sustain. Chem. Eng. 2017, 5, 9836–9840. [Google Scholar] [CrossRef]
- Teodor, A.H.; Bruce, B.D. Putting photosystem I to work: Truly green energy. Trends Biotechnol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bennett, T.; Niroomand, H.; Pamu, R.; Ivanov, I.; Mukherjee, D.; Khomami, B. Elucidating the role of methyl viologen as a scavenger of photoactivated electrons from photosystem I under aerobic and anaerobic conditions. Phys. Chem. Chem. Phys. 2016, 18, 8512–8521. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, G.; Gizzie, E.; Yang, S.; Cliffel, D.E.; Jennings, G.K. Photosystem I protein films at electrode surfaces for solar energy conversion. Langmuir 2014, 30, 10990–11001. [Google Scholar] [CrossRef]
- Nurnberg, D.J.; Morton, J.; Santabarbara, S.; Telfer, A.; Joliot, P.; Antonaru, L.A.; Ruban, A.V.; Cardona, T.; Krausz, E.; Boussac, A.; et al. Photochemistry beyond the red limit in chlorophyll f-containing photosystems. Science 2018, 360, 1210–1213. [Google Scholar] [CrossRef]
- Baker, D.R.; Simmerman, R.F.; Sumner, J.J.; Bruce, B.D.; Lundgren, C.A. Photoelectrochemistry of photosystem I bound in nafion. Langmuir 2014, 30, 13650–13655. [Google Scholar] [CrossRef]
- Gordiichuk, P.I.; Rimmerman, D.; Paul, A.; Gautier, D.A.; Gruszka, A.; Saller, M.; de Vries, J.W.; Wetzelaer, G.J.; Manca, M.; Gomulya, W.; et al. Filling the green gap of a megadalton photosystem I complex by conjugation of organic dyes. Bioconjug. Chem. 2016, 27, 36–41. [Google Scholar] [CrossRef]
- Balaban, T.S. Tailoring porphyrins and chlorins for self-assembly in biomimetic artificial antenna systems. Acc. Chem. Res. 2005, 38, 612–623. [Google Scholar] [CrossRef]
- Linke-Schaetzel, M.; Bhise, A.D.; Gliemann, H.; Koch, T.; Schimmel, T.; Balaban, T.S. Self-assembled chromophores for hybrid solar cells. Thin Solid Films 2004, 451, 16–21. [Google Scholar] [CrossRef]
- Gundlach, K.; Werwie, M.; Wiegand, S.; Paulsen, H. Filling the “green gap” of the major light-harvesting chlorophyll a/b complex by covalent attachment of Rhodamine Red. BBA Bioenerg. 2009, 1787, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.K.; Lin, S.; Loskutov, A.; Levenberg, S.; Jun, D.; Saer, R.; Beatty, J.T.; Liu, Y.; Yan, H.; Woodbury, N.W. Reengineering the optical absorption cross-section of photosynthetic reaction centers. J. Am. Chem. Soc. 2014, 136, 4599–4604. [Google Scholar] [CrossRef] [PubMed]
- Nagakawa, H.; Takeuchi, A.; Takekuma, Y.; Noji, T.; Kawakami, K.; Kamiya, N.; Nango, M.; Furukawa, R.; Nagata, M. Efficient hydrogen production using photosystem I enhanced by artificial light harvesting dye. Photochem. Photobiol. Sci. 2019, 18, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Carmeli, I.; Lieberman, I.; Kraversky, L.; Fan, Z.; Govorov, A.O.; Markovich, G.; Richter, S. Broad band enhancement of light absorption in photosystem I by metal nanoparticle antennas. Nano Lett. 2010, 10, 2069–2074. [Google Scholar] [CrossRef] [PubMed]
- Dervishogullari, D.; Gizzie, E.A.; Jennings, G.K.; Cliffel, D.E. Polyviologen as electron transport material in photosystem I-based biophotovoltaic cells. Langmuir 2018, 34, 15658–15664. [Google Scholar] [CrossRef]
- Mackowski, S.; Wormke, S.; Maier, A.J.; Brotosudarmo, T.H.; Harutyunyan, H.; Hartschuh, A.; Govorov, A.O.; Scheer, H.; Brauchle, C. Metal-enhanced fluorescence of chlorophylls in single light-harvesting complexes. Nano Lett. 2008, 8, 558–564. [Google Scholar] [CrossRef]
- Kim, I.; Bender, S.L.; Hranisavljevic, J.; Utschig, L.M.; Huang, L.; Wiederrecht, G.P.; Tiede, D.M. Metal nanoparticle plasmon-enhanced light-harvesting in a photosystem I thin film. Nano Lett. 2011, 11, 3091–3098. [Google Scholar] [CrossRef]
- Torres, R.; Diz, V.E.; Lagorio, M.G. Effects of gold nanoparticles on the photophysical and photosynthetic parameters of leaves and chloroplasts. Photochem. Photobiol. Sci. 2018, 17, 505–516. [Google Scholar] [CrossRef]
- Niroomand, H.; Pamu, R.; Mukherjee, D.; Khomami, B. Tuning the photocurrent generations from photosystem I assembled in tailored biotic-abiotic interfaces. MRS Commun. 2018, 8, 823–829. [Google Scholar]
- Antonacci, A.; Scognamiglio, V. Photosynthesis-based hybrid nanostructures: Electrochemical sensors and photovoltaic cells as case studies. TrAC Trend Anal. Chem. 2019, 115, 100–109. [Google Scholar] [CrossRef]
- Murray, C.B.; Kagan, C.R.; Bawendi, M.G. Synthesis and characterization of monodisperse nanocrystals and close-packed nanocrystal assemblies. Annu. Rev. Mater. Sci. 2000, 30, 545–610. [Google Scholar] [CrossRef]
- Jung, H.; Gulis, G.; Gupta, S.; Redding, K.; Gosztola, D.J.; Wiederrecht, G.P.; Stroscio, M.A.; Dutta, M. Optical and electrical measurement of energy transfer between nanocrystalline quantum dots and photosystem I. J. Phys. Chem. B 2010, 114, 14544–14549. [Google Scholar] [CrossRef] [PubMed]
- Nabiev, I.; Rakovich, A.; Sukhanova, A.; Lukashev, E.; Zagidullin, V.; Pachenko, V.; Rakovich, Y.P.; Donegan, J.F.; Rubin, A.B.; Govorov, A.O. Fluorescent quantum dots as artificial antennas for enhanced light harvesting and energy transfer to photosynthetic reaction centers. Angew. Chem. Int. Ed. 2010, 49, 7217–7221. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.T.; Mantell, J.; Di Bartolo, N.; Jones, M.R. Mechanisms of self-assembly and energy harvesting in tuneable conjugates of quantum dots and engineered photovoltaic proteins. Small 2019, 15, 1804267. [Google Scholar] [CrossRef]
- Yu, D.; Wang, M.; Zhu, G.; Ge, B.; Liu, S.; Huang, F. Enhanced photocurrent production by bio-dyes of photosynthetic macromolecules on designed TiO2 film. Sci. Rep. 2015, 5, 9375. [Google Scholar] [CrossRef]
- Hagfeldt, A.; Boschloo, G.; Sun, L.; Kloo, L.; Pettersson, H. Dye-sensitized solar cells. Chem. Rev. 2010, 110, 6595–6663. [Google Scholar] [CrossRef]
- Kondo, M.; Amano, M.; Joke, T.; Ishigure, S.; Noji, T.; Dewa, T.; Amao, Y.; Nango, M. Immobilization of photosystem I or II complexes on electrodes for preparation of photoenergy-conversion devices. Res. Chem. Intermediat. 2014, 40, 3287–3293. [Google Scholar] [CrossRef]
- Ciornii, D.; Riedel, M.; Stieger, K.R.; Feifel, S.C.; Hejazi, M.; Lokstein, H.; Zouni, A.; Lisdat, F. Bioelectronic circuit on a 3D electrode architecture: Enzymatic catalysis interconnected with photosystem I. J. Am. Chem. Soc. 2017, 139, 16478–16481. [Google Scholar] [CrossRef]
- Riedel, M.; Parak, W.J.; Ruff, A.; Schuhmann, W.; Lisdat, F. Light as trigger for biocatalysis: Photonic Wiring of flavin adenine dinucleotide-dependent glucose dehydrogenase to quantum dot-sensitized inverse opal TiO2 architectures via redox polymers. ACS Catal. 2018, 8, 5212–5220. [Google Scholar] [CrossRef]
- Simmerman, R.F.; Zhu, T.; Baker, D.R.; Wang, L.; Mishra, S.R.; Lundgren, C.A.; Bruce, B.D. Engineering photosystem I complexes with metal oxide binding peptides for bioelectronic applications. Bioconjug. Chem. 2015, 26, 2097–2105. [Google Scholar] [CrossRef]
- Ocakoglu, K.; Krupnik, T.; van den Bosch, B.; Harputlu, E.; Gullo, M.P.; Olmos, J.D.J.; Yildirimcan, S.; Gupta, R.K.; Yakuphanoglu, F.; Barbieri, A.; et al. Photosystem I-based biophotovoltaics on nanostructured hematite. Adv. Funct. Mater. 2014, 24, 7467–7477. [Google Scholar] [CrossRef]
- Gizzie, E.A.; LeBlanc, G.; Jennings, G.K.; Cliffel, D.E. Electrochemical preparation of Photosystem I-polyaniline composite films for biohybrid solar energy conversion. ACS Appl. Mater. Interfaces 2015, 7, 9328–9335. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.T.; Simons, C.E.; Cliffel, D.E.; Jennings, G.K. Photocatalytic photosystem I/PEDOT composite films prepared by vapor-phase polymerization. Nanoscale 2017, 9, 6158–6166. [Google Scholar] [CrossRef] [PubMed]
- Feifel, S.C.; Stieger, K.R.; Lokstein, H.; Lux, H.; Lisdat, F. High photocurrent generation by photosystem I on artificial interfaces composed of pi-system-modified graphene. J. Mater. Chem. A 2015, 3, 12188–12196. [Google Scholar] [CrossRef]
- Ciornii, D.; Feifel, S.C.; Hejazi, M.; Kolsch, A.; Lokstein, H.; Zouni, A.; Lisdat, F. Construction of photobiocathodes using multi-walled carbon nanotubes and photosystem I. Phys. Status Solidi A 2017, 214, 1700017. [Google Scholar] [CrossRef]
- Feifel, S.C.; Lokstein, H.; Hejazi, M.; Zouni, A.; Lisdat, F. Unidirectional photocurrent of photosystem I on pi-system-modified graphene electrodes: Nanobionic approaches for the construction of photobiohybrid systems. Langmuir 2015, 31, 10590–10598. [Google Scholar] [CrossRef]
- Darby, E.; LeBlanc, G.; Gizzie, E.A.; Winter, K.M.; Jennings, G.K.; Cliffel, D.E. Photoactive films of photosystem I on transparent reduced graphene oxide electrodes. Langmuir 2014, 30, 8990–8994. [Google Scholar] [CrossRef]
- LeBlanc, G.; Chen, G.; Gizzie, E.A.; Jennings, G.K.; Cliffel, D.E. Enhanced photocurrents of photosystem I films on p-doped silicon. Adv. Mater. 2012, 24, 5959–5962. [Google Scholar] [CrossRef]
- Beam, J.C.; LeBlanc, G.; Gizzie, E.A.; Ivanov, B.L.; Needell, D.R.; Shearer, M.J.; Jennings, G.K.; Lukehart, C.M.; Cliffel, D.E. Construction of a semiconductor-biological interface for solar energy conversion: P-doped silicon/photosystem I/Zinc oxide. Langmuir 2015, 31, 10002–10007. [Google Scholar] [CrossRef]
- Stieger, K.R.; Feifel, S.C.; Lokstein, H.; Lisdat, F. Advanced unidirectional photocurrent generation via cytochrome c as reaction partner for directed assembly of photosystem I. Phys. Chem. Chem. Phys. 2014, 16, 15667–15674. [Google Scholar] [CrossRef]
- Olmos, J.D.J.; Becquet, P.; Gront, D.; Sar, J.; Dabrowski, A.; Gawlik, G.; Teodorczyk, M.; Pawlak, D.; Kargul, J. Biofunctionalisation of p-doped silicon with cytochrome c(553) minimises charge recombination and enhances photovoltaic performance of the all-solid-state photosystem I-based biophotoelectrode. RSC Adv. 2017, 7, 47854–47866. [Google Scholar] [CrossRef]
- Ciesielski, P.N.; Scott, A.M.; Faulkner, C.J.; Berron, B.J.; Cliffel, D.E.; Jennings, G.K. Functionalized nanoporous gold leaf electrode films for the immobilization of photosystem I. ACS Nano 2008, 2, 2465–2472. [Google Scholar] [CrossRef]
- Yamanoi, Y.; Terasaki, N.; Miyachi, M.; Inoue, Y.; Nishihara, H. Enhanced photocurrent production by photosystem I with modi fi ed viologen derivatives. Thin Solid Films 2012, 520, 5123–5127. [Google Scholar] [CrossRef]
- Yang, S.Y.; Robinson, M.T.; Mwambutsa, F.; Cliffel, D.E.; Jennings, G.K. Effect of cross-linking on the performance and stability of photocatalytic photosystem I films. Electrochim. Acta 2016, 222, 926–932. [Google Scholar] [CrossRef]
- Pang, H.; Zhao, G.; Liu, G.; Zhang, H.; Hai, X.; Wang, S.; Song, H.; Ye, J. Interfacing photosynthetic membrane protein with mesoporous WO3 photoelectrode for solar water oxidation. Small 2018, 14, e1800104. [Google Scholar] [CrossRef]
- Ooi, E.J.; Medina, J.; Teodor, A.; Brady, N.; Vaughn, M.; Bruce, B.; Bergkamp, J. Cobalt redox mediators for integration in photosystem I biohybrid solar cells. In Proceedings of the American Chemical Society Meeting, New Orleans, LA, USA, 18–22 March 2018. [Google Scholar]
- Zhao, F.; Ruff, A.; Rogner, M.; Schuhmann, W.; Conzuelo, F. Extended operational lifetime of a photosystem-based bioelectrode. J. Am. Chem. Soc. 2019, 141, 5102–5106. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.T.; Cliffel, D.E.; Jennings, G.K. An electrochemical reaction-diffusion model of the photocatalytic effect of photosystem I multilayer films. J. Phys. Chem. B 2018, 122, 117–125. [Google Scholar] [CrossRef]
- Zhao, F.; Plumere, N.; Nowaczyk, M.M.; Ruff, A.; Schuhmann, W.; Conzuelo, F. Interrogation of a PS1-based photocathode by means of scanning photoelectrochemical microscopy. Small 2017, 13, 1604093. [Google Scholar] [CrossRef]
- Faulkner, C.J.; Lees, S.; Ciesielski, P.N.; Cliffel, D.E.; Jennings, G.K. Rapid assembly of photosystem I monolayers on gold electrodes. Langmuir 2008, 24, 8409–8412. [Google Scholar] [CrossRef]
- Ciesielski, P.N.; Hijazi, F.M.; Scott, A.M.; Faulkner, C.J.; Beard, L.; Emmett, K.; Rosenthal, S.J.; Cliffel, D.; Jennings, G.K. Photosystem I—Based biohybrid photoelectrochemical cells. Bioresour. Technol. 2010, 101, 3047–3053. [Google Scholar] [CrossRef]
- Gizzie, E.A.; Niezgoda, J.S.; Robinson, M.T.; Harris, A.G.; Jennings, G.K.; Rosenthal, S.J.; Cliffel, D.E. Photosystem I-polyaniline/TiO2 solid-state solar cells: Simple devices for biohybrid solar energy conversion. Energy Environ. Sci. 2015, 8, 3572–3576. [Google Scholar] [CrossRef]
- Shah, V.B.; Henson, W.R.; Chadha, T.S.; Lakin, G.; Liu, H.J.; Blankenship, R.E.; Biswas, P. Linker-free deposition and adhesion of photosystem i onto nanostructured TiO2 for biohybrid photoelectrochemical cells. Langmuir 2015, 31, 1675–1682. [Google Scholar] [CrossRef] [PubMed]
- Stieger, K.R.; Feifel, S.C.; Lokstein, H.; Hejazi, M.; Zouni, A.; Lisdat, F. Biohybrid architectures for efficient light-to-current conversion based on photosystem I within scalable 3D mesoporous electrodes. J. Mater. Chem. A 2016, 4, 17009–17017. [Google Scholar] [CrossRef]
- Cadirci, B.H. An electricity production study by Rhodobacter sphaeroides. Int. J. Hydrog. Energy 2018, 43, 18001–18006. [Google Scholar] [CrossRef]
- Csiki, R.; Drieschner, S.; Lyuleeva, A.; Cattani-Scholz, A.; Stutzmann, M.; Garrido, J.A. Photocurrent generation of biohybrid systems based on bacterial reaction centers and graphene electrodes. Diam. Relat. Mater. 2018, 89, 286–292. [Google Scholar] [CrossRef]
- Wenzel, T.; Hartter, D.; Bombelli, P.; Howe, C.J.; Steiner, U. Porous translucent electrodes enhance current generation from photosynthetic biofilms. Nat. Commun. 2018, 9, 1299. [Google Scholar] [CrossRef]
- Wey, L.T.; Bombelli, P.; Chen, X.; Lawrence, J.M.; Rabideau, C.M.; Rowden, S.J.L.; Zhang, J.Z.; Howe, C.J. The development of biophotovoltaic systems for power generation and biological analysis. ChemElectroChem 2019, 6, 5375–5386. [Google Scholar] [CrossRef]
- Kavadiya, S.; Chadha, T.S.; Liu, H.; Shah, V.B.; Blankenship, R.E.; Biswas, P. Directed assembly of the thylakoid membrane on nanostructured TiO2 for a photo-electrochemical cell. Nanoscale 2016, 8, 1868–1872. [Google Scholar] [CrossRef]
- Zeynali, A.; Ghiasi, T.S.; Riazi, G.; Ajeian, R. Organic solar cell based on photosystem I pigment-protein complex, fabrication and optimization. Org. Electron. 2017, 51, 341–348. [Google Scholar] [CrossRef]
- Gaffron, H. Reduction of carbon dioxide with molecular hydrogen in green algae. Nature 1939, 143, 204–205. [Google Scholar] [CrossRef]
- Stuart, T.S.; Gaffron, H. Mechanism of hydrogen photoproduction by several algae: II. Contribution of Photosystem-Ii. Planta 1972, 106, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, E. Photosynthetic unit of hydrogen evolution. Science 1977, 196, 879–880. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, E. Simultaneous photoproduction of hydrogen and oxygen by photosynthesis. Biotechnol. Bioeng. 1980, 22, 1–13. [Google Scholar]
- Maione, T.E.; Gibbs, M. Hydrogenase-mediated activities in isolated chloroplasts of chlamydomonas reinhardii. Plant Physiol. 1986, 80, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Wunschiers, R.; Stangier, K.; Senger, H.; Schulz, R. Molecular evidence for a Fe-hydrogenase in the green alga Scenedesmus obliquus. Curr. Microbiol. 2001, 42, 353–360. [Google Scholar] [CrossRef]
- Winkler, M.; Heil, B.; Happe, T. Isolation and molecular characterization of the [Fe]-hydrogenase from the unicellular green alga Chlorella fusca. Biochim. Biophys. Acta 2002, 1576, 330–334. [Google Scholar] [CrossRef]
- Florin, L.; Tsokoglou, A.; Happe, T. A novel type of iron hydrogenase in the green alga Scenedesmus obliquus is linked to the photosynthetic electron transport chain. J. Biol. Chem. 2001, 276, 6125–6132. [Google Scholar] [CrossRef]
- Ludwig, M.; Schulz-Friedrich, R.; Appel, J. Occurrence of hydrogenases in cyanobacteria and anoxygenic photosynthetic bacteria: Implications for the phylogenetic origin of cyanobacterial and algal hydrogenases. J. Mol. Evol. 2006, 63, 758–768. [Google Scholar] [CrossRef]
- Ghirardi, M.L.; Posewitz, M.C.; Maness, P.C.; Dubini, A.; Yu, J.; Seibert, M. Hydrogenases and hydrogen photoproduction in oxygenic photosynthetic organisms. Annu. Rev. Plant Biol. 2007, 58, 71–91. [Google Scholar] [CrossRef]
- Peters, J.W.; Schut, G.J.; Boyd, E.S.; Mulder, D.W.; Shepard, E.M.; Broderick, J.B.; King, P.W.; Adams, M.W. [FeFe]- and [NiFe]-hydrogenase diversity, mechanism, and maturation. Biochim. Biophys. Acta 2015, 1853, 1350–1369. [Google Scholar] [CrossRef]
- Morra, S.; Arizzi, M.; Valetti, F.; Gilardi, G. Oxygen stability in the new [FeFe]-hydrogenase from clostridium beijerinckii SM10 (CbA5H). Biochemistry 2016, 55, 5897–5900. [Google Scholar] [CrossRef] [PubMed]
- Iwuchukwu, I.J.; Vaughn, M.; Myers, N.; O’Neill, H.; Frymier, P.; Bruce, B.D. Self-organized photosynthetic nanoparticle for cell-free hydrogen production. Nat. Nanotechnol. 2010, 5, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Khanna, N.; Lindblad, P. Cyanobacterial hydrogenases and hydrogen metabolism revisited: Recent progress and future prospects. Int. J. Mol. Sci. 2015, 16, 10537–10561. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, E. Photosynthetic hydrogen and oxygen production: Kinetic studies. Science 1982, 215, 291–293. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, E.; Guillard, R.R.L.; Sunda, W.G. Hydrogen and oxygen photoproduction by marine-algae. Photochem. Photobiol. 1983, 37, 649–655. [Google Scholar] [CrossRef]
- Greenbaum, E. Energetic efficiency of hydrogen photoevolution by algal water splitting. Biophys. J. 1988, 54, 365–368. [Google Scholar] [CrossRef]
- Stuart, T.S.; Gaffron, H. Kinetics of hydrogen photoproduction by adapted scenedesmus. Planta 1971, 100, 228–243. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.S.; Gaffron, H. Kinetics of H2 photoproduction by scenedesmus. Plant Physiol. 1971, 47, 32. [Google Scholar]
- Stuart, T.S.; Gaffron, H. Mechanism of hydrogen photoproduction by several algae: I. Effect of inhibitors of photophosphorylation. Planta 1972, 106, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Benemann, J.R.; Berenson, J.A.; Kaplan, N.O.; Kamen, M.D. Hydrogen evolution by a chloroplast-ferredoxin-hydrogenase system. Proc. Natl. Acad. Sci. USA 1973, 70, 2317–2320. [Google Scholar] [CrossRef] [PubMed]
- Melis, A.; Zhang, L.; Forestier, M.; Ghirardi, M.L.; Seibert, M. Sustained photobiological hydrogen gas production upon reversible inactivation of oxygen evolution in the green alga Chlamydomonas reinhardtii. Plant Physiol. 2000, 122, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Melis, A. Photosynthetic H2 metabolism in Chlamydomonas reinhardtii (unicellular green algae). Planta 2007, 226, 1075–1086. [Google Scholar] [CrossRef] [PubMed]
- Liran, O.; Semyatich, R.; Milrad, Y.; Eilenberg, H.; Weiner, I.; Yacoby, I. Microoxic niches within the thylakoid stroma of air-grown chlamydomonas reinhardtii protect [FeFe]-hydrogenase and support hydrogen production under fully aerobic environment. Plant Physiol. 2016, 172, 264–271. [Google Scholar] [CrossRef]
- Saper, G.; Kallmann, D.; Conzuelo, F.; Zhao, F.; Toth, T.N.; Liveanu, V.; Meir, S.; Szymanski, J.; Aharoni, A.; Schuhmann, W.; et al. Live cyanobacteria produce photocurrent and hydrogen using both the respiratory and photosynthetic systems. Nat. Commun. 2018, 9, 2168. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Kalathil, S.; Reisner, E. Semi-biological approaches to solar-to-chemical conversion. Chem. Soc. Rev. 2020, 49, 4926–4952. [Google Scholar] [CrossRef]
- Evans, B.R.; O’Neill, H.M.; Hutchens, S.A.; Bruce, B.D.; Greenbaum, E. Enhanced photocatalytic hydrogen evolution by covalent attachment of plastocyanin to photosystem I. Nano Lett. 2004, 4, 1815–1819. [Google Scholar] [CrossRef]
- Lubner, C.E.; Applegate, A.M.; Knorzer, P.; Ganago, A.; Bryant, D.A.; Happe, T.; Golbeck, J.H. Solar hydrogen-producing bionanodevice outperforms natural photosynthesis. Proc. Natl. Acad. Sci. USA 2011, 108, 20988–20991. [Google Scholar] [CrossRef]
- Hohner, R.; Pribil, M.; Herbstova, M.; Lopez, L.S.; Kunz, H.H.; Li, M.; Wood, M.; Svoboda, V.; Puthiyaveetil, S.; Leister, D.; et al. Plastocyanin is the long-range electron carrier between photosystem II and photosystem I in plants. Proc. Natl. Acad. Sci. USA 2020, 117, 15354–15362. [Google Scholar] [CrossRef]
- Kovalenko, I.B.; Knyazeva, O.S.; Antal, T.K.; Ponomarev, V.Y.; Riznichenko, G.Y.; Rubin, A.B. Multiparticle Brownian dynamics simulation of experimental kinetics of cytochrome bf oxidation and photosystem I reduction by plastocyanin. Physiol. Plant. 2017, 161, 88–96. [Google Scholar] [CrossRef]
- Ihara, M.; Nishihara, H.; Yoon, K.S.; Lenz, O.; Friedrich, B.; Nakamoto, H.; Kojima, K.; Honma, D.; Kamachi, T.; Okura, I. Light-driven hydrogen production by a hybrid complex of a [NiFe]-hydrogenase and the cyanobacterial photosystem I. Photochem. Photobiol. 2006, 82, 676–682. [Google Scholar] [CrossRef]
- Appel, J.; Hueren, V.; Boehm, M.; Gutekunst, K. Cyanobacterial in vivo solar hydrogen production using a photosystem I–hydrogenase (PsaD-HoxYH) fusion complex. Nat. Energy 2020, 5, 458–467. [Google Scholar] [CrossRef]
- Eilenberg, H.; Weiner, I.; Ben-Zvi, O.; Pundak, C.; Marmari, A.; Liran, O.; Wecker, M.S.; Milrad, Y.; Yacoby, I. The dual effect of a ferredoxin-hydrogenase fusion protein in vivo: Successful divergence of the photosynthetic electron flux towards hydrogen production and elevated oxygen tolerance. Biotechnol. Biofuels 2016, 9, 182. [Google Scholar] [CrossRef] [PubMed]
- Kanygin, A.; Milrad, Y.; Thummala, C.; Reifschneider, K.; Baker, P.; Marco, P.; Yacoby, I.; Redding, K.E. Rewiring photosynthesis: A photosystem I-hydrogenase chimera that makes H2 in vivo. Energy Environ. Sci. 2020. [Google Scholar] [CrossRef]
- Utschig, L.M.; Soltau, S.R.; Tiede, D.M. Light-driven hydrogen production from Photosystem I-catalyst hybrids. Curr. Opin. Chem. Biol. 2015, 25, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Lee, J.W.; Stubna, A.; Greenbaum, E. Measurement of electrostatic potentials above oriented single photosynthetic reaction centers. J. Phys. Chem. B 2000, 104, 2439–2443. [Google Scholar] [CrossRef]
- Brettel, K. Electron transfer and arrangement of the redox cofactors in photosystem I. Biochim. Biophys. Acta Bioenerg. 1997, 1318, 322–373. [Google Scholar] [CrossRef]
- Zankel, K.L.; Reed, D.W.; Clayton, R.K. Fluorescence and photochemical quenching in photosynthetic reaction centers. Proc. Natl. Acad. Sci. USA 1968, 61, 1243. [Google Scholar] [CrossRef]
- Hiyama, T. Quantum yield and requirement for the photooxidation of P-700. Physiol. Veg. 1985, 23, 605–610. [Google Scholar]
- Millsaps, J.F.; Bruce, B.D.; Lee, J.W.; Greenbaum, E. Nanoscale photosynthesis: Photocatalytic production of hydrogen by platinized photosystem I reaction centers. Photochem. Photobiol. 2001, 73, 630–635. [Google Scholar] [CrossRef]
- Ihara, M.; Nakamoto, H.; Kamachi, T.; Okura, I.; Maeda, M. Photoinduced hydrogen production by direct electron transfer from photosystem I cross-linked with cytochrome c3 to [NiFe]-hydrogenase. Photochem. Photobiol. 2006, 82, 1677–1685. [Google Scholar] [CrossRef]
- Greenbaum, E. Platinized chloroplasts: A novel photocatalytic material. Science 1985, 230, 1373–1375. [Google Scholar] [CrossRef] [PubMed]
- Grimme, R.A.; Lubner, C.E.; Bryant, D.A.; Golbeck, J.H. Photosystem I/molecular wire/metal nanoparticle bioconjugates for the photocatalytic production of H-2. J. Am. Chem. Soc. 2008, 130, 6308–6309. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Conzuelo, F.; Hartmann, V.; Li, H.; Nowaczyk, M.M.; Plumere, N.; Rogner, M.; Schuhmann, W. Light Induced H2 evolution from a biophotocathode based on photosystem 1—Pt nanoparticles complexes integrated in solvated redox polymers films. J. Phys. Chem. B 2015, 119, 13726–13731. [Google Scholar] [CrossRef] [PubMed]
- Applegate, A.M.; Lubner, C.E.; Knörzer, P.; Happe, T.; Golbeck, J.H. Quantum yield measurements of light-induced H2 generation in a photosystem I-[FeFe]-H2ase nanoconstruct. Photosynth. Res. 2016, 127, 5–11. [Google Scholar] [CrossRef]
- Ban, S.; Lin, W.; Luo, Z.; Luo, J. Improving hydrogen production of Chlamydomonas reinhardtii by reducing chlorophyll content via atmospheric and room temperature plasma. Bioresour. Technol. 2019, 275, 425–429. [Google Scholar] [CrossRef]
- Kiley, P.; Zhao, X.; Vaughn, M.; Baldo, M.A.; Bruce, B.D.; Zhang, S. Self-assembling peptide detergents stabilize isolated photosystem I on a dry surface for an extended time. PLoS Biol. 2005, 3, e230. [Google Scholar] [CrossRef]
- Prince, R.C.; Kheshgi, H.S. The photobiological production of hydrogen: Potential efficiency and effectiveness as a renewable fuel. Crit. Rev. Microbiol. 2005, 31, 19–31. [Google Scholar] [CrossRef]
- Lam, E.; Malkin, R. Reconstruction of the chloroplast noncyclic electron transport pathway from water to NADP with three integral protein complexes. Proc. Natl. Acad. Sci. USA 1982, 79, 5494–5498. [Google Scholar] [CrossRef]
- Pernil, R.; Schleiff, E. Metalloproteins in the biology of heterocysts. Life 2019, 9, 32. [Google Scholar] [CrossRef]
- Queiroz, M.I.; Vieira, J.G.; Maroneze, M.M. Morphophysiological, structural, and metabolic aspects of microalgae. In Handbook of Microalgae-Based Processes and Products; Elsevier: Amsterdam, The Netherlands, 2020; pp. 25–48. [Google Scholar] [CrossRef]
- Swainsbury, D.J.; Scheidelaar, S.; van Grondelle, R.; Killian, J.A.; Jones, M.R. Bacterial reaction centers purified with styrene maleic acid copolymer retain native membrane functional properties and display enhanced stability. Angew. Chem. Int. Ed. Engl. 2014, 53, 11803–11807. [Google Scholar] [CrossRef]
- Brady, N.G.; Li, M.; Ma, Y.; Gumbart, J.C.; Bruce, B.D. Non-detergent isolation of a cyanobacterial photosystem I using styrene maleic acid alternating copolymers. RSC Adv. 2019, 9, 31781–31796. [Google Scholar] [CrossRef]
- Sun, C.; Benlekbir, S.; Venkatakrishnan, P.; Wang, Y.; Hong, S.; Hosler, J.; Tajkhorshid, E.; Rubinstein, J.L.; Gennis, R.B. Structure of the alternative complex III in a supercomplex with cytochrome oxidase. Nature 2018, 557, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, V.; Sturgis, J.N. Modifying styrene-maleic acid co-polymer for studying lipid nanodiscs. Biochim. Biophys. Acta Biomembr. 2018, 1860, 777–783. [Google Scholar] [CrossRef]
- Wheatley, M.; Charlton, J.; Jamshad, M.; Routledge, S.J.; Bailey, S.; La-Borde, P.J.; Azam, M.T.; Logan, R.T.; Bill, R.M.; Dafforn, T.R.; et al. GPCR-styrene maleic acid lipid particles (GPCR-SMALPs): Their nature and potential. Biochem. Soc. Trans. 2016, 44, 619–623. [Google Scholar] [CrossRef]
- Gulamhussein, A.A.; Meah, D.; Soja, D.D.; Fenner, S.; Saidani, Z.; Akram, A.; Lallie, S.; Mathews, A.; Painter, C.; Liddar, M.K.; et al. Examining the stability of membrane proteins within SMALPs. Eur. Polym. J. 2019, 112, 120–125. [Google Scholar] [CrossRef]
- Cherepanov, D.A.; Brady, N.G.; Shelaev, I.V.; Nguyen, J.; Gostev, F.E.; Mamedov, M.D.; Nadtochenko, V.A.; Bruce, B.D. PSI-SMALP, a detergent-free cyanobacterial photosystem I, reveals faster femtosecond photochemistry. Biophys. J. 2020, 118, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Kruger, A.; Schafers, C.; Schroder, C.; Antranikian, G. Towards a sustainable biobased industry—Highlighting the impact of extremophiles. N. Biotechnol. 2018, 40, 144–153. [Google Scholar] [CrossRef]
- Spieck, E.; Spohn, M.; Wendt, K.; Bock, E.; Shively, J.; Frank, J.; Indenbirken, D.; Alawi, M.; Lucker, S.; Hupeden, J. Extremophilic nitrite-oxidizing Chloroflexi from Yellowstone hot springs. ISME J. 2020, 14, 364–379. [Google Scholar] [CrossRef]
- Shrestha, N.; Chilkoor, G.; Vemuri, B.; Rathinam, N.; Sani, R.K.; Gadhamshetty, V. Extremophiles for microbial-electrochemistry applications: A critical review. Bioresour. Technol. 2018, 255, 318–330. [Google Scholar] [CrossRef]
- Macia-Agullo, J.A.; Corma, A.; Garcia, H. Photobiocatalysis: The power of combining photocatalysis and enzymes. Chemistry 2015, 21, 10940–10959. [Google Scholar] [CrossRef]
- Fukuzumi, S.; Lee, Y.M.; Nam, W. Artificial photosynthesis for production of ATP, NAD(P)H, and hydrogen peroxide. ChemPhotoChem 2017, 2, 121–135. [Google Scholar] [CrossRef]
- Lee, S.H.; Choi, D.S.; Kuk, S.K.; Park, C.B. Photobiocatalysis: Activating redox enzymes by direct or indirect transfer of photoinduced electrons. Angew. Chem. Int. Ed. Engl. 2018, 57, 7958–7985. [Google Scholar] [CrossRef] [PubMed]
- Gust, D.; Moore, T.A.; Moore, A.L. Solar fuels via artificial photosynthesis. Acc. Chem. Res. 2009, 42, 1890–1898. [Google Scholar] [CrossRef]
- Sherman, B.D.; Vaughn, M.D.; Bergkamp, J.J.; Gust, D.; Moore, A.L.; Moore, T.A. Evolution of reaction center mimics to systems capable of generating solar fuel. Photosynth. Res. 2014, 120, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Steinberg-Yfrach, G.; Rigaud, J.L.; Durantini, E.N.; Moore, A.L.; Gust, D.; Moore, T.A. Light-driven production of ATP catalysed by F0F1-ATP synthase in an artificial photosynthetic membrane. Nature 1998, 392, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Numata, T.; Murakami, T.; Kawashima, F.; Morone, N.; Heuser, J.E.; Takano, Y.; Ohkubo, K.; Fukuzumi, S.; Mori, Y.; Imahori, H. Utilization of photoinduced charge-separated state of donor-acceptor-linked molecules for regulation of cell membrane potential and ion transport. J. Am. Chem. Soc. 2012, 134, 6092–6095. [Google Scholar] [CrossRef]
- Bhosale, S.; Sisson, A.L.; Talukdar, P.; Furstenberg, A.; Banerji, N.; Vauthey, E.; Bollot, G.; Mareda, J.; Roger, C.; Wurthner, F.; et al. Photoproduction of proton gradients with pi-stacked fluorophore scaffolds in lipid bilayers. Science 2006, 313, 84–86. [Google Scholar] [CrossRef]
- Hvasanov, D.; Peterson, J.R.; Thordarson, P. Self-assembled light-driven photosynthetic-respiratory electron transport chain hybrid proton pump. Chem. Sci. 2013, 4, 3833–3838. [Google Scholar] [CrossRef]
- Ernst, O.P.; Lodowski, D.T.; Elstner, M.; Hegemann, P.; Brown, L.S.; Kandori, H. Microbial and animal rhodopsins: Structures, functions, and molecular mechanisms. Chem. Rev. 2014, 114, 126–163. [Google Scholar] [CrossRef]
- Schulten, K.; Tavan, P. A mechanism for the light-driven proton pump of Halobacterium halobium. Nature 1978, 272, 85–86. [Google Scholar] [CrossRef]
- Mahyad, B.; Janfaza, S.; Hosseini, E.S. Bio-nano hybrid materials based on bacteriorhodopsin: Potential applications and future strategies. Adv. Colloid Interface Sci. 2015, 225, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.T.; Tian, Y.; Tian, H.; Tu, T.; Gou, G.Y.; Wang, Q.; Qiao, Y.C.; Yang, Y.; Ren, T.L. A review on bacteriorhodopsin-based bioelectronic devices. Sensors 2018, 18, 1368. [Google Scholar] [CrossRef] [PubMed]
- Thavasi, V.; Lazarova, T.; Filipek, S.; Kolinski, M.; Querol, E.; Kumar, A.; Ramakrishna, S.; Padros, E.; Renugopalakrishnan, V. Study on the feasibility of bacteriorhodopsin as bio-photosensitizer in excitonic solar cell: A first report. J. Nanosci. Nanotechnol. 2009, 9, 1679–1687. [Google Scholar] [CrossRef] [PubMed]
- Renugopalakrishnan, V.; Barbiellini, B.; King, C.; Molinari, M.; Mochalov, K.; Sukhanova, A.; Nabiev, I.; Fojan, P.; Tuller, H.L.; Chin, M.; et al. Engineering a robust photovoltaic device with quantum dots and bacteriorhodopsin. J. Phys. Chem. C Nanomater. Interfaces 2014, 118, 16710–16717. [Google Scholar] [CrossRef]
- Mohammadpour, R.; Janfaza, S. Efficient nanostructured biophotovoltaic cell based on bacteriorhodopsin as biophotosensitizer. ACS Sustain. Chem. Eng. 2015, 3, 809–813. [Google Scholar] [CrossRef]
- Molaeirad, A.; Rezaeian, N. Oriented assembly of bacteriorhodopsin on ZnO nanostructured electrode for enhanced photocurrent generation. Biotechnol. Appl. Biochem. 2015, 62, 489–493. [Google Scholar] [CrossRef]
- Das, S.; Wu, C.; Song, Z.; Hou, Y.; Koch, R.; Somasundaran, P.; Priya, S.; Barbiellini, B.; Venkatesan, R. Bacteriorhodopsin enhances efficiency of perovskite solar cells. ACS Appl. Mater. Interfaces 2019, 11, 30728–30734. [Google Scholar] [CrossRef]
- Chu, L.K.; Yen, C.W.; El-Sayed, M.A. Bacteriorhodopsin-based photo-electrochemical cell. Biosens. Bioelectron. 2010, 26, 620–626. [Google Scholar] [CrossRef]
- Yen, C.W.; Hayden, S.C.; Dreaden, E.C.; Szymanski, P.; El-Sayed, M.A. Tailoring plasmonic and electrostatic field effects to maximize solar energy conversion by bacteriorhodopsin, the other natural photosynthetic system. Nano Lett. 2011, 11, 3821–3826. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Wang, P.; Schaller, R.D.; Rajh, T.; Rozhkova, E.A. High-performance bioassisted nanophotocatalyst for hydrogen production. Nano Lett. 2013, 13, 3365–3371. [Google Scholar] [CrossRef]
- Johnson, K.E.; Gakhar, S.; Risbud, S.H.; Longo, M.L. Development and characterization of titanium dioxide gel with encapsulated bacteriorhodopsin for hydrogen production. Langmuir 2018, 34, 7488–7496. [Google Scholar] [CrossRef]
- Allam, N.K.; Yen, C.-W.; Near, R.D.; El-Sayed, M.A. Bacteriorhodopsin/TiO2 nanotube arrays hybrid system for enhanced photoelectrochemical water splitting. Energy Environ. Sci. 2011, 4, 2909–2914. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, P.; Xu, X.; Sheves, M.; Jin, Y. Bacteriorhodopsin/Ag nanoparticle-based hybrid nano-bio electrocatalyst for efficient and robust H2 evolution from water. J. Am. Chem. Soc. 2015, 137, 2840–2843. [Google Scholar] [CrossRef] [PubMed]
- Skubi, K.L.; Blum, T.R.; Yoon, T.P. Dual catalysis strategies in photochemical synthesis. Chem. Rev. 2016, 116, 10035–10074. [Google Scholar] [CrossRef] [PubMed]
- Mandler, D.; Willner, I. Solar light induced formation of chiral 2-butanol in an enzyme-catalyzed chemical system. J. Am. Chem. Soc. 1984, 106, 5352–5353. [Google Scholar] [CrossRef]
- Mandler, D.; Willner, I. Photoinduced enzyme-catalysed synthesis of amino acids by visible light. J. Chem. Soc. Chem. Commun. 1986, 851–853. [Google Scholar] [CrossRef]
- Ruppert, R.; Steckhan, E. Efficient photoelectrochemical in-situ regeneration of NAD(P)+ coupled to enzymatic oxidation of alcohols. J. Chem. Soc. Perkin Trans. 2 1989, 811–814. [Google Scholar] [CrossRef]
- Gandomkar, S.; Dennig, A.; Dordic, A.; Hammerer, L.; Pickl, M.; Haas, T.; Hall, M.; Faber, K. Biocatalytic oxidative cascade for the conversion of fatty acids into alpha-ketoacids via internal H2 O2 recycling. Angew. Chem. Int. Ed. Engl. 2018, 57, 427–430. [Google Scholar] [CrossRef]
- Zhang, W.; Fernandez-Fueyo, E.; Ni, Y.; van Schie, M.; Gacs, J.; Renirie, R.; Wever, R.; Mutti, F.G.; Rother, D.; Alcalde, M.; et al. Selective aerobic oxidation reactions using a combination of photocatalytic water oxidation and enzymatic oxyfunctionalisations. Nat. Catal. 2018, 1, 55–62. [Google Scholar] [CrossRef]
- Choi, D.S.; Lee, H.; Tieves, F.; Lee, Y.W.; Son, E.J.; Zhang, W.; Shin, B.; Hollmann, F.; Park, C.B. Bias-free in situ H2O2 generation in a photovoltaic-photoelectrochemical tandem cell for biocatalytic oxyfunctionalization. ACS Catal. 2019, 9, 10562–10566. [Google Scholar] [CrossRef]
- Zachos, I.; Gassmeyer, S.K.; Bauer, D.; Sieber, V.; Hollmann, F.; Kourist, R. Photobiocatalytic decarboxylation for olefin synthesis. Chem. Commun. 2015, 51, 1918–1921. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.; Baeg, J.O.; Park, N.J.; Yadav, R.K. A photocatalyst/enzyme couple that uses solar energy in the asymmetric reduction of acetophenones. Angew. Chem. Int. Ed. Engl. 2012, 51, 11624–11628. [Google Scholar] [CrossRef] [PubMed]
- Hutton, G.A.; Reuillard, B.; Martindale, B.C.; Caputo, C.A.; Lockwood, C.W.; Butt, J.N.; Reisner, E. Carbon dots as versatile photosensitizers for solar-driven catalysis with redox enzymes. J. Am. Chem. Soc. 2016, 138, 16722–16730. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Dong, C.L.; Guan, Z.; He, Y.H. Concurrent asymmetric reactions combining photocatalysis and enzyme catalysis: Direct enantioselective synthesis of 2,2-disubstituted indol-3-ones from 2-arylindoles. Angew. Chem. Int. Ed. Engl. 2019, 58, 118–124. [Google Scholar] [CrossRef]
- Osman, A.I.; Skillen, N.C.; Robertson, P.K.J.; Rooney, D.W.; Morgan, K. Exploring the photocatalytic hydrogen production potential of titania doped with alumina derived from foil waste. Int. J. Hydrog. Energy 2020. [Google Scholar] [CrossRef]
- Bard, A.J.; Fox, M.A. Artificial photosynthesis: Solar splitting of water to hydrogen and oxygen. Acc. Chem. Res. 1995, 28, 141–145. [Google Scholar] [CrossRef]
- Fujishima, A.; Honda, K. Electrochemical photolysis of water at a semiconductor electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef]
- Khaselev, O. A Monolithic photovoltaic-photoelectrochemical device for hydrogen production via water splitting. Science 1998, 280, 425–427. [Google Scholar] [CrossRef]
- Ashford, D.L.; Gish, M.K.; Vannucci, A.K.; Brennaman, M.K.; Templeton, J.L.; Papanikolas, J.M.; Meyer, T.J. Molecular chromophore-catalyst assemblies for solar fuel applications. Chem. Rev. 2015, 115, 13006–13049. [Google Scholar] [CrossRef]
- Xu, P.; McCool, N.S.; Mallouk, T.E. Water splitting dye-sensitized solar cells. Nano Today 2017, 14, 42–58. [Google Scholar] [CrossRef]
- Hambourger, M.; Moore, G.F.; Kramer, D.M.; Gust, D.; Moore, A.L.; Moore, T.A. Biology and technology for photochemical fuel production. Chem. Soc. Rev. 2009, 38, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Willner, I.; Mandler, D.; Riklin, A. Photoinduced carbon dioxide fixation forming malic and isocitric acid. J. Chem. Soc. Chem. Commun. 1986, 1022–1024. [Google Scholar] [CrossRef]
- Hamby, H.; Li, B.; Shinopoulos, K.E.; Keller, H.R.; Elliott, S.J.; Dukovic, G. Light-driven carbon-carbon bond formation via CO2 reduction catalyzed by complexes of CdS nanorods and a 2-oxoacid oxidoreductase. Proc. Natl. Acad. Sci. USA 2020, 117, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Woolerton, T.W.; Sheard, S.; Pierce, E.; Ragsdale, S.W.; Armstrong, F.A. CO2 photoreduction at enzyme-modified metal oxide nanoparticles. Energy Environ. Sci. 2011, 4, 2393–2399. [Google Scholar] [CrossRef]
- Miller, M.; Robinson, W.E.; Oliveira, A.R.; Heidary, N.; Kornienko, N.; Warnan, J.; Pereira, I.A.C.; Reisner, E. Interfacing formate dehydrogenase with metal oxides for the reversible electrocatalysis and solar-driven reduction of carbon dioxide. Angew. Chem. Int. Ed. Engl. 2019, 58, 4601–4605. [Google Scholar] [CrossRef]
- Yadav, R.K.; Baeg, J.O.; Oh, G.H.; Park, N.J.; Kong, K.J.; Kim, J.; Hwang, D.W.; Biswas, S.K. A photocatalyst-enzyme coupled artificial photosynthesis system for solar energy in production of formic acid from CO2. J. Am. Chem. Soc. 2012, 134, 11455–11461. [Google Scholar] [CrossRef]
- Parkinson, B.A.; Weaver, P.F. Photoelectrochemical pumping of enzymatic CO2 reduction. Nature 1984, 309, 148–149. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, S.H.; Lee, J.S.; Lee, M.; Park, C.B. Zn-containing porphyrin as a biomimetic light-harvesting molecule for biocatalyzed artificial photosynthesis. Chem. Commun. 2011, 47, 10227–10229. [Google Scholar] [CrossRef]
- Ji, X.; Su, Z.; Wang, P.; Ma, G.; Zhang, S. Integration of artificial photosynthesis system for enhanced electronic energy-transfer efficacy: A case study for solar-energy driven bioconversion of carbon dioxide to methanol. Small 2016, 12, 4753–4762. [Google Scholar] [CrossRef]
- Hofler, G.T.; Fernandez-Fueyo, E.; Pesic, M.; Younes, S.H.; Choi, E.G.; Kim, Y.H.; Urlacher, V.B.; Arends, I.; Hollmann, F. A photoenzymatic NADH regeneration system. ChemBioChem 2018, 19, 2344–2347. [Google Scholar] [CrossRef]
- Yadav, R.K.; Kumar, A.; Park, N.-J.; Kong, K.-J.; Baeg, J.-O. A highly efficient covalent organic framework film photocatalyst for selective solar fuel production from CO2. J. Mater. Chem. A 2016, 4, 9413–9418. [Google Scholar] [CrossRef]
- Park, C.B.; Lee, S.H.; Subramanian, E.; Kale, B.B.; Lee, S.M.; Baeg, J.O. Solar energy in production of L-glutamate through visible light active photocatalyst—Redox enzyme coupled bioreactor. Chem. Commun. 2008, 5423–5425. [Google Scholar] [CrossRef] [PubMed]
- Nam, D.H.; Lee, S.H.; Park, C.B. CdTe, CdSe, and CdS nanocrystals for highly efficient regeneration of nicotinamide cofactor under visible light. Small 2010, 6, 922–926. [Google Scholar] [CrossRef] [PubMed]
- Dibenedetto, A.; Stufano, P.; Macyk, W.; Baran, T.; Fragale, C.; Costa, M.; Aresta, M. Hybrid technologies for an enhanced carbon recycling based on the enzymatic reduction of CO2 to methanol in water: Chemical and photochemical NADH regeneration. ChemSusChem 2012, 5, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Liu, C.; Wang, J.; Su, Z.; Ma, G.; Zhang, S. Integration of functionalized two-dimensional TaS2 nanosheets and an electron mediator for more efficient biocatalyzed artificial photosynthesis. J. Mater. Chem. A 2017, 5, 5511–5522. [Google Scholar] [CrossRef]
- Son, E.J.; Lee, Y.W.; Ko, J.W.; Park, C.B. Amorphous carbon nitride as a robust photocatalyst for biocatalytic solar-to-chemical conversion. ACS Sustain. Chem. Eng. 2018, 7, 2545–2552. [Google Scholar] [CrossRef]
- Ryu, J.; Nam, D.H.; Lee, S.H.; Park, C.B. Biocatalytic photosynthesis with water as an electron donor. Chemistry 2014, 20, 12020–12025. [Google Scholar] [CrossRef]
- Ganesan, V.; Sivanesan, D.; Yoon, S. Correlation between the structure and catalytic activity of [Cp*Rh(Substituted Bipyridine)] complexes for NADH regeneration. Inorg. Chem. 2017, 56, 1366–1374. [Google Scholar] [CrossRef]
- Nam, D.H.; Kuk, S.K.; Choe, H.; Lee, S.; Ko, J.W.; Son, E.J.; Choi, E.-G.; Kim, Y.H.; Park, C.B. Enzymatic photosynthesis of formate from carbon dioxide coupled with highly efficient photoelectrochemical regeneration of nicotinamide cofactors. Green Chem. 2016, 18, 5989–5993. [Google Scholar] [CrossRef]
- Lee, S.Y.; Lim, S.Y.; Seo, D.; Lee, J.-Y.; Chung, T.D. Light-driven highly selective conversion of CO2 to formate by electrosynthesized enzyme/cofactor thin film electrode. Adv. Energy Mater. 2016, 6, 1502207. [Google Scholar] [CrossRef]
- Lee, Y.W.; Boonmongkolras, P.; Son, E.J.; Kim, J.; Lee, S.H.; Kuk, S.K.; Ko, J.W.; Shin, B.; Park, C.B. Unbiased biocatalytic solar-to-chemical conversion by FeOOH/BiVO4/perovskite tandem structure. Nat. Commun. 2018, 9, 4208. [Google Scholar] [CrossRef] [PubMed]
- Kuk, S.K.; Ham, Y.; Gopinath, K.; Boonmongkolras, P.; Lee, Y.; Lee, Y.W.; Kondaveeti, S.; Ahn, C.; Shin, B.; Lee, J.K.; et al. Continuous 3D titanium nitride nanoshell structure for solar-driven unbiased biocatalytic CO2 reduction. Adv. Energy Mater. 2019, 9, 1900029. [Google Scholar] [CrossRef]
- Yadav, R.K.; Oh, G.H.; Park, N.J.; Kumar, A.; Kong, K.J.; Baeg, J.O. Highly selective solar-driven methanol from CO2 by a photocatalyst/biocatalyst integrated system. J. Am. Chem. Soc. 2014, 136, 16728–16731. [Google Scholar] [CrossRef] [PubMed]
- Kuk, S.K.; Singh, R.K.; Nam, D.H.; Singh, R.; Lee, J.K.; Park, C.B. Photoelectrochemical reduction of carbon dioxide to methanol through a highly efficient enzyme cascade. Angew. Chem. Int. Ed. Engl. 2017, 56, 3827–3832. [Google Scholar] [CrossRef] [PubMed]
- Roth, L.E.; Nguyen, J.C.; Tezcan, F.A. ATP- and iron-protein-independent activation of nitrogenase catalysis by light. J. Am. Chem. Soc. 2010, 132, 13672–13674. [Google Scholar] [CrossRef]
- Roth, L.E.; Tezcan, F.A. ATP-uncoupled, six-electron photoreduction of hydrogen cyanide to methane by the molybdenum-iron protein. J. Am. Chem. Soc. 2012, 134, 8416–8419. [Google Scholar] [CrossRef]
- Brown, K.A.; Harris, D.F.; Wilker, M.B.; Rasmussen, A.; Khadka, N.; Hamby, H.; Keable, S.; Dukovic, G.; Peters, J.W.; Seefeldt, L.C.; et al. Light-driven dinitrogen reduction catalyzed by a CdS: Nitrogenase MoFe protein biohybrid. Science 2016, 352, 448–450. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrode Surface/Immobilization | Redox Mediators | Photocurrent (µA cm−2) | Current Density (µA cm−2 mW−1) | Ref. |
|---|---|---|---|---|
| Terephthalic-dialdehyde SAM on nanoporous gold | Sodium ascorbate; 2,6-dichloroindophenol | 0.3 | 0.08 | [42] |
| Bare gold | Sodium ascorbate; 2,6-dichlorophenolindophenol | N/A | 0.1 | [50] |
| PSI-based biohybrid cells | Sodium ascorbate; 2,6-dichloroindophenol | 2 | 0.138 | [51] |
| PSI films/p-doped silicon | Methyl viologen | 875 | 4.6 | [38] |
| PSI multilayer/reduced graphene oxide | Ferrocyanide; methylene blue; sodium ascorbate; methyl viologen; 2,6-dichlorophenolindophenol; ruthenium(II) hexamine | 1.2 | N/A | [37] |
| PSI multilayer film on gold | Ferricyanide | 0.9 | N/A | [5] |
| Polyaniline–PSI film on gold surface | Sodium ascorbate | 5.7 | N/A | [32] |
| PSI–polyaniline/titanium dioxide | Methyl viologen | 72 | N/A | [52] |
| Solid-state unetched p-doped silicon/PSI | Methyl viologen | Non-etched: 21 Etched: 127 | N/A | [39] |
| PSI multilayer film/SAM on gold-coated silicon | Osmium-based redox hydrogel | 2-iminothiolane-cross-linked: ~8 | N/A | [44] a |
| PSI–poly(3,4-ethylene-dioxythiophene): poly-styrenesulfonate/fluorine-doped tin oxide | N/A | 960 | N/A | [2] |
| PSI multilayer film/gold/SAM/aminoethanethiol | [Fe(CN)6]4−; [Fe(CN)6]3− | 0–0.84 for PSI film thickness 0–1.3 µm | N/A | [48] |
| PSI–p-doped silicon | Methyl viologen; polyviologen | N/A | N/A | [15] |
| PSI Source | Electrode Surface/Immobilization | Redox Mediators | Photocurrent (µA cm−2) | Current Density (µA cm−2 mW−1) | Ref. |
|---|---|---|---|---|---|
| T. elongatus | PSI/SAM on gold surface | Sodium ascorbate | 0.088 | N/A | [43] a |
| T. elongatus | PSI/Nafion film | Osmium bis(2,2′-bipyridine)chloride; methyl viologen | 4 | N/A | [7] |
| T. elongatus | PSI/osmium complex-modified polymer | Methyl viologen; osmium-based redox hydrogel | N/A | 322 | [1] a |
| T. elongatus | PSI/thiol-modified gold | Ascorbate-reduced 2,6-dichloroindophenol; methyl viologen | 1 | 0.97 | [40] a |
| T. elongatus | PSI/pi system-modified graphene | Methyl viologen | 135 | N/A | [34] b |
| HT3 cells | PSI/1D nanostructured titanium dioxide thin films | Sodium ascorbate; 2,6-dichlorophenolindophenol | 4150 | N/A | [53] |
| T. elongatus | ZOBiP–PSI- or TOBiP-Fd–PSI-based biohybrid dye-sensitized solar cells | Cyt c6 | N/A | N/A | [30] a |
| T. elongatus | PSI/transparent mesoporous indium tin oxide | N/A | 150 | N/A | [54] |
| T. elongatus | PSI on C9 alkanethiolate SAM/Au | Methyl viologen | 0.006 | N/A | [4] |
| T. elongatus | PSI/pi system-modified graphene | Methyl viologen | 4.5 | N/A | [36] |
| T. elongatus | PSI/carboxylated pyrene derivative multiwalled carbon nanotubes | Sodium ascorbate; methyl viologen | No cyt c: 0.8 Cyt c present: 18 | N/A | [35] |
| R. sphaeroides | PSI-based biohybrid cells | N/A | 405,630 | N/A | [55] |
| R. sphaeroides | PSI/graphene | Aminomethylferrocene; coenzyme Q0 | 1.2, 0.4 | N/A | [56] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teodor, A.H.; Sherman, B.D.; Ison, Z.Y.; Ooi, E.-J.; Bergkamp, J.J.; Bruce, B.D. Green Catalysts: Applied and Synthetic Photosynthesis. Catalysts 2020, 10, 1016. https://doi.org/10.3390/catal10091016
Teodor AH, Sherman BD, Ison ZY, Ooi E-J, Bergkamp JJ, Bruce BD. Green Catalysts: Applied and Synthetic Photosynthesis. Catalysts. 2020; 10(9):1016. https://doi.org/10.3390/catal10091016
Chicago/Turabian StyleTeodor, Alexandra H., Benjamin D. Sherman, Zeah Yvette Ison, Eu-Jee Ooi, Jesse J. Bergkamp, and Barry D. Bruce. 2020. "Green Catalysts: Applied and Synthetic Photosynthesis" Catalysts 10, no. 9: 1016. https://doi.org/10.3390/catal10091016
APA StyleTeodor, A. H., Sherman, B. D., Ison, Z. Y., Ooi, E.-J., Bergkamp, J. J., & Bruce, B. D. (2020). Green Catalysts: Applied and Synthetic Photosynthesis. Catalysts, 10(9), 1016. https://doi.org/10.3390/catal10091016