From Alkynes to Heterocycles through Metal-Promoted Silylformylation and Silylcarbocyclization Reactions



Abstract

{kind=link}
{kind=link}
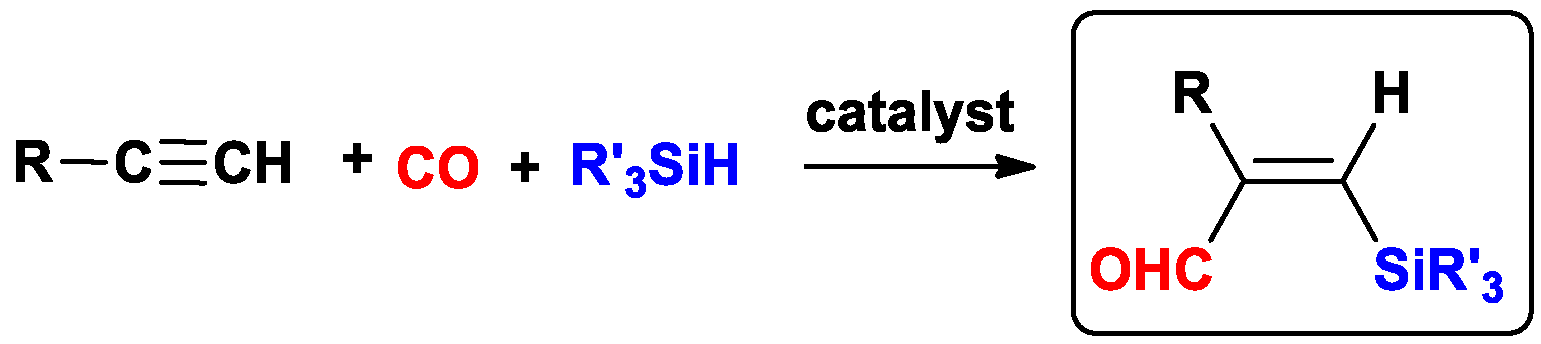{kind=link}
{kind=link}
{kind=link}
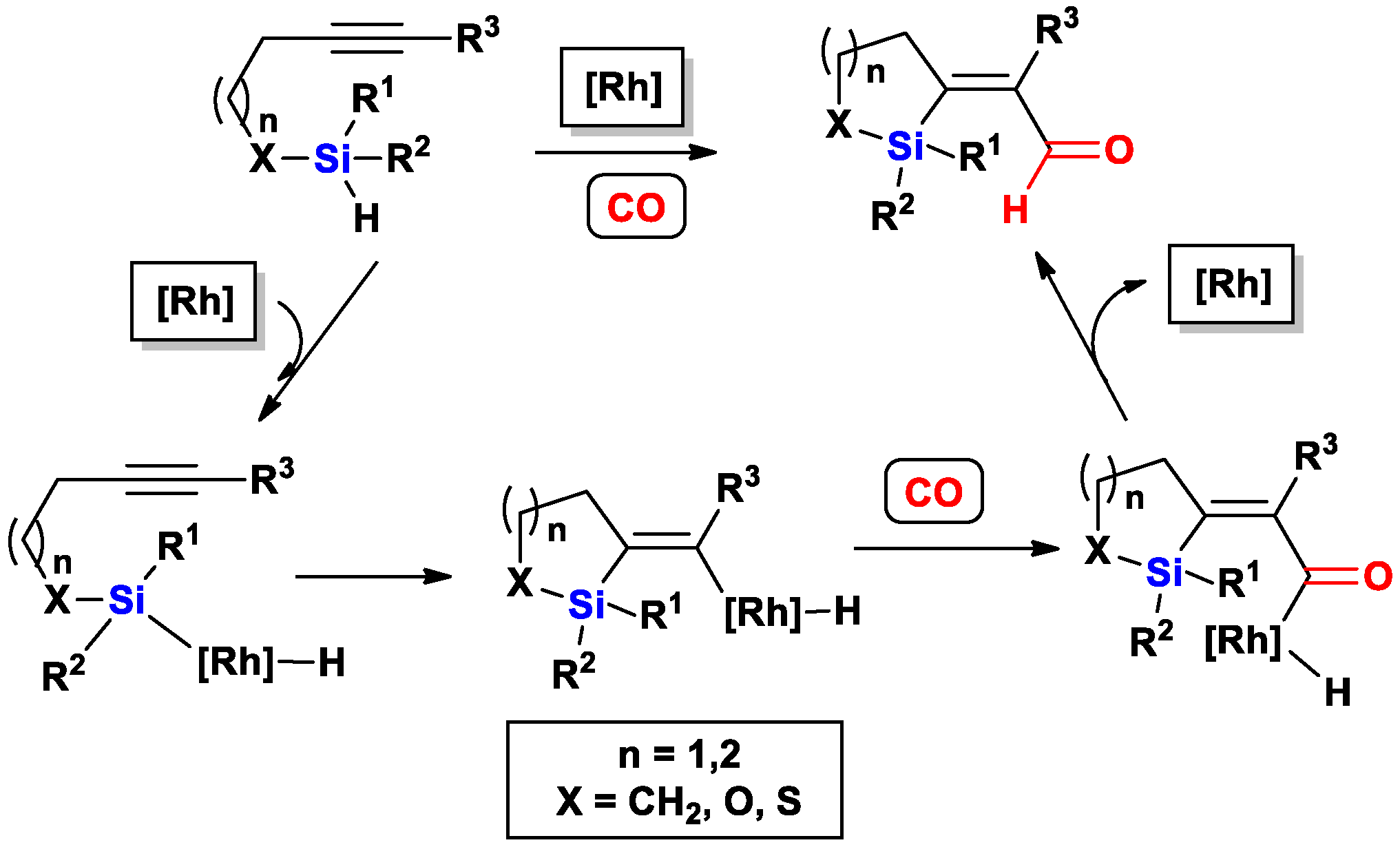{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
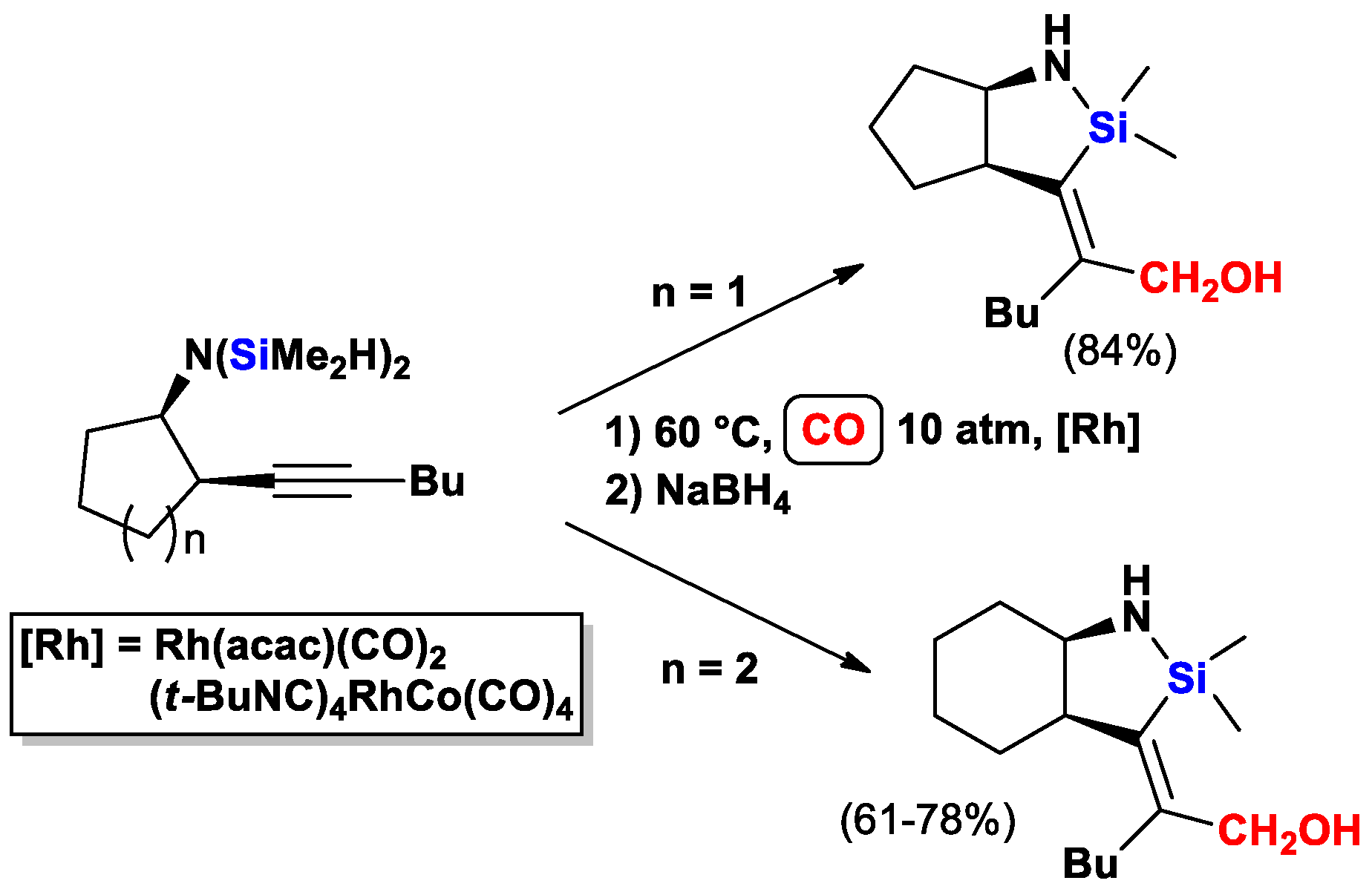{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
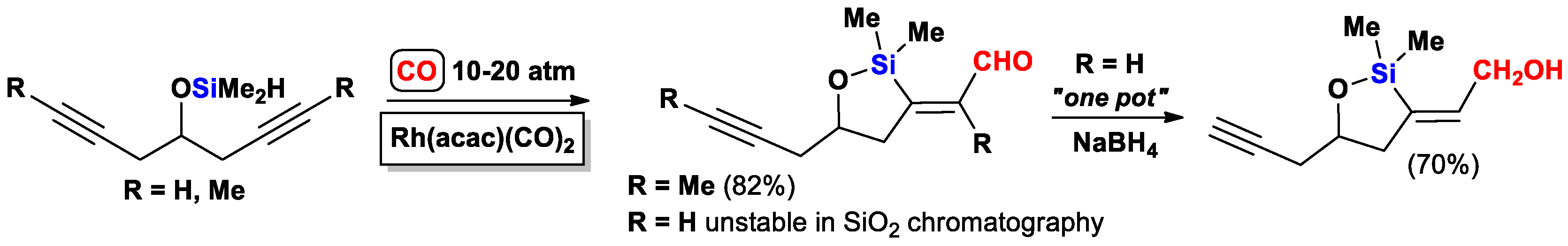{kind=link}
{kind=link}
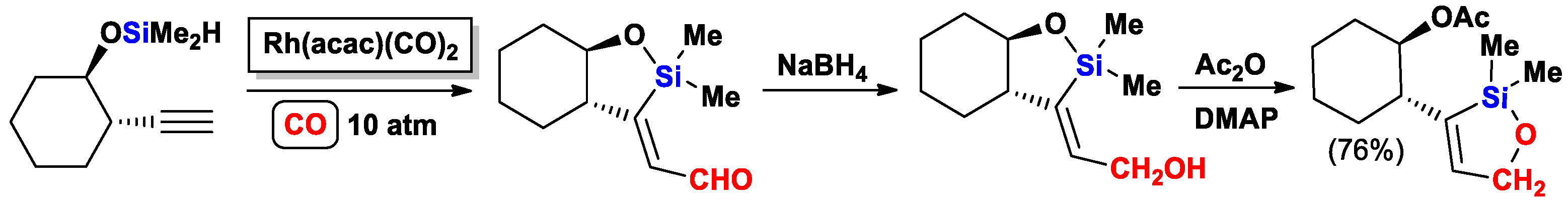{kind=link}
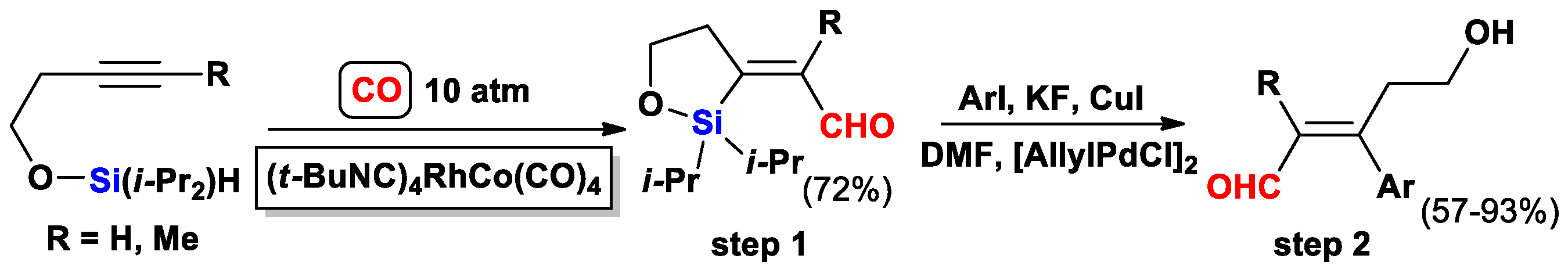{kind=link}
{kind=link}
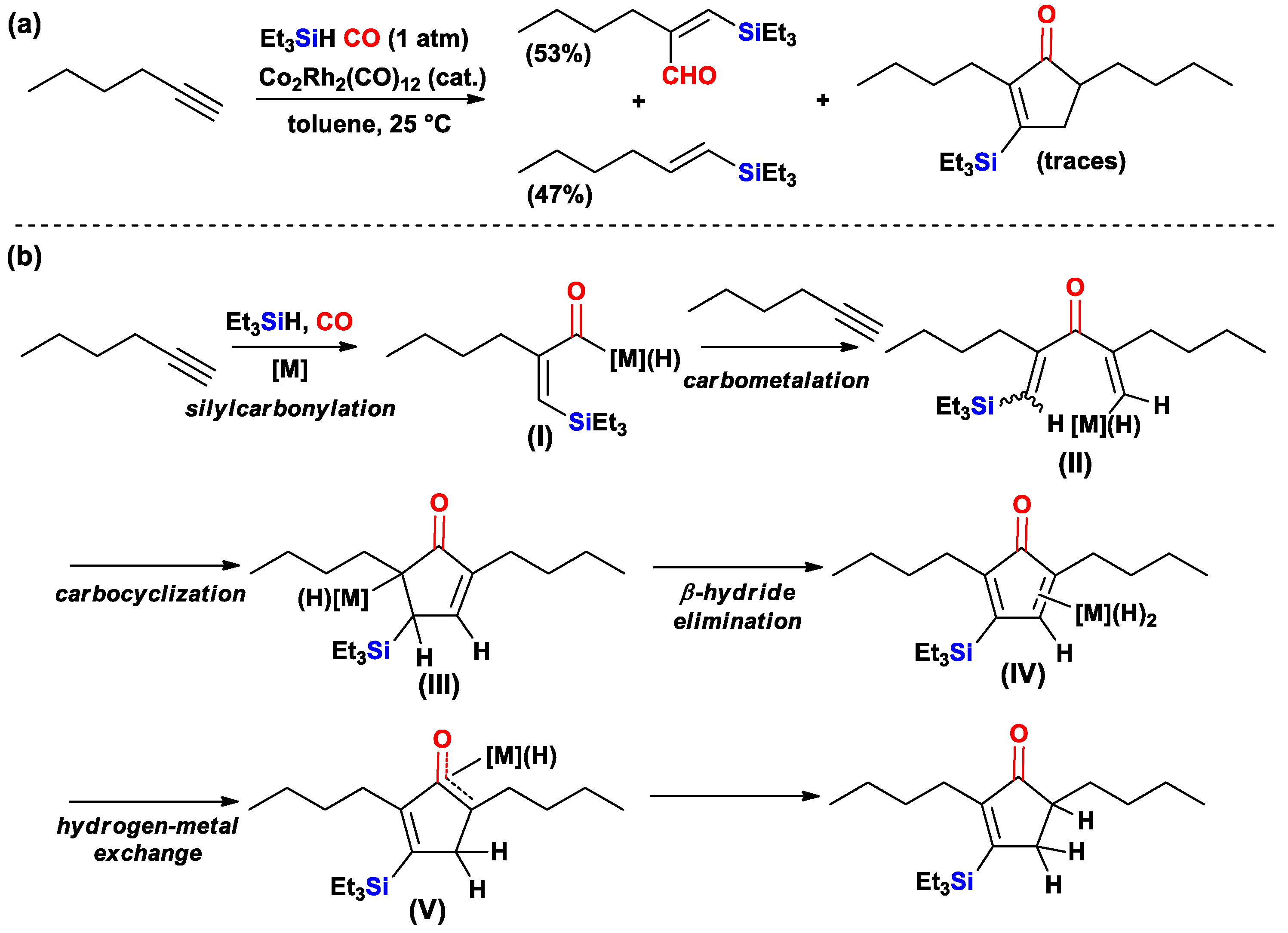{kind=link}
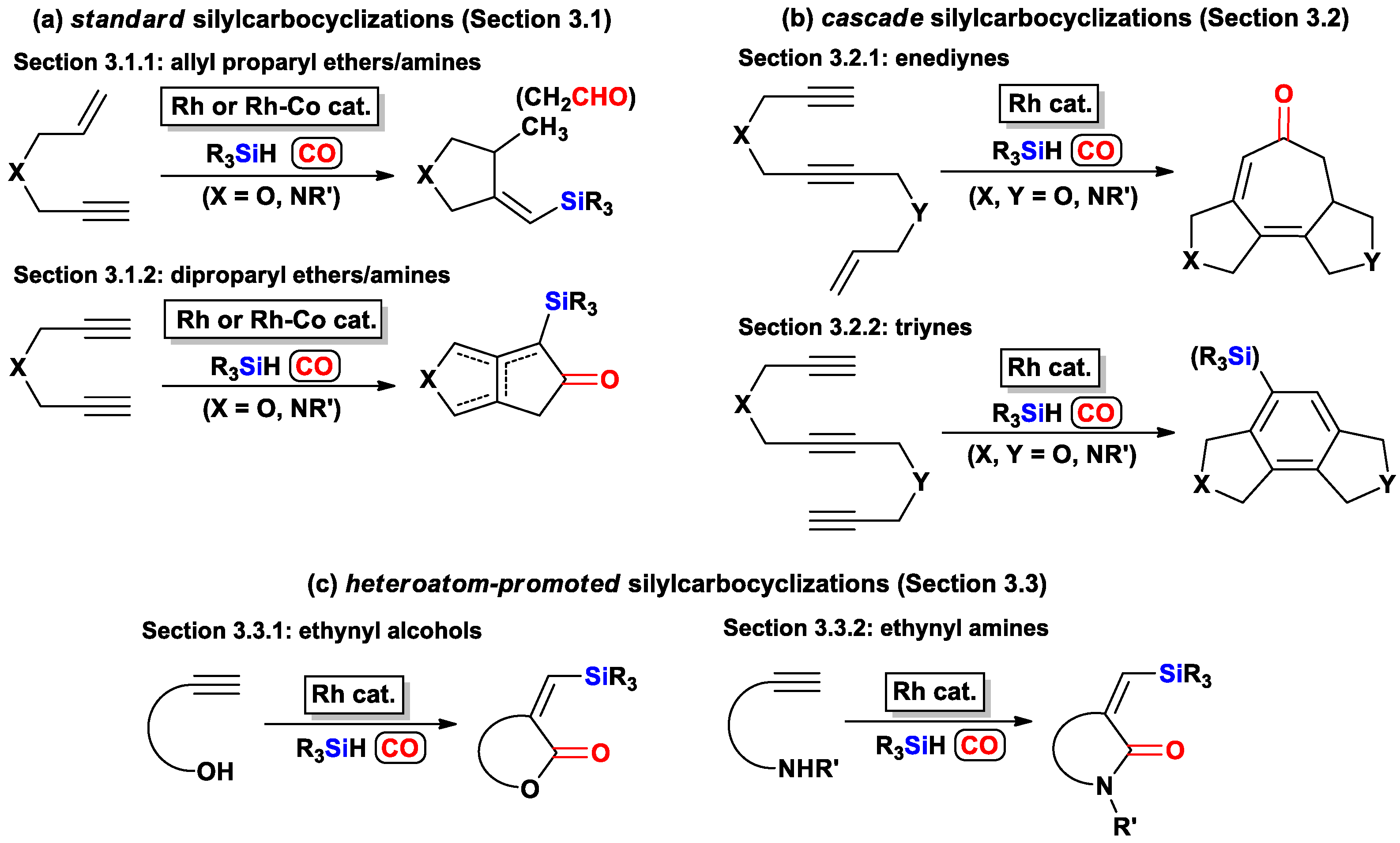{kind=link}
{kind=link}
{kind=link}
{kind=link}
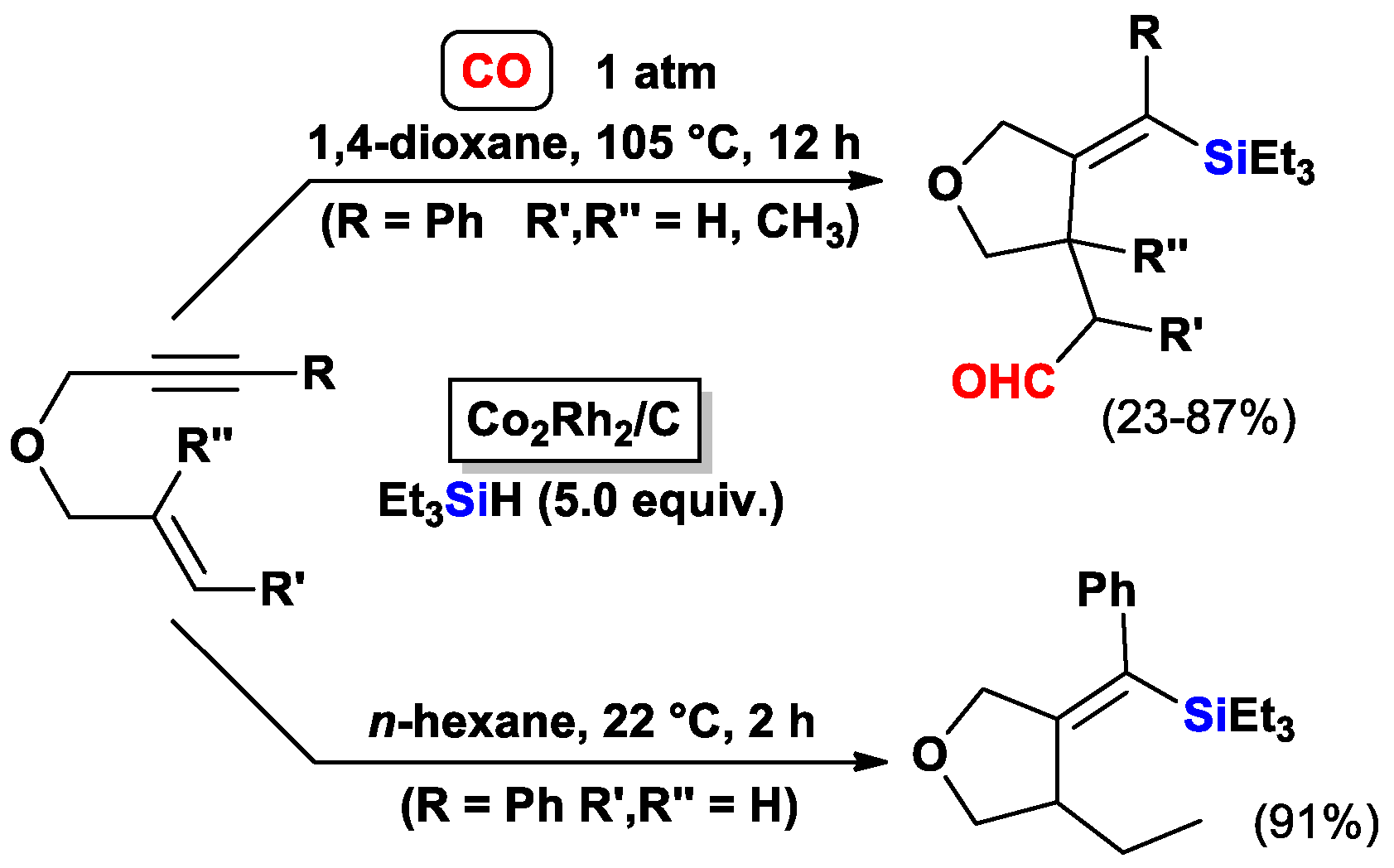{kind=link}
{kind=link}
{kind=link}
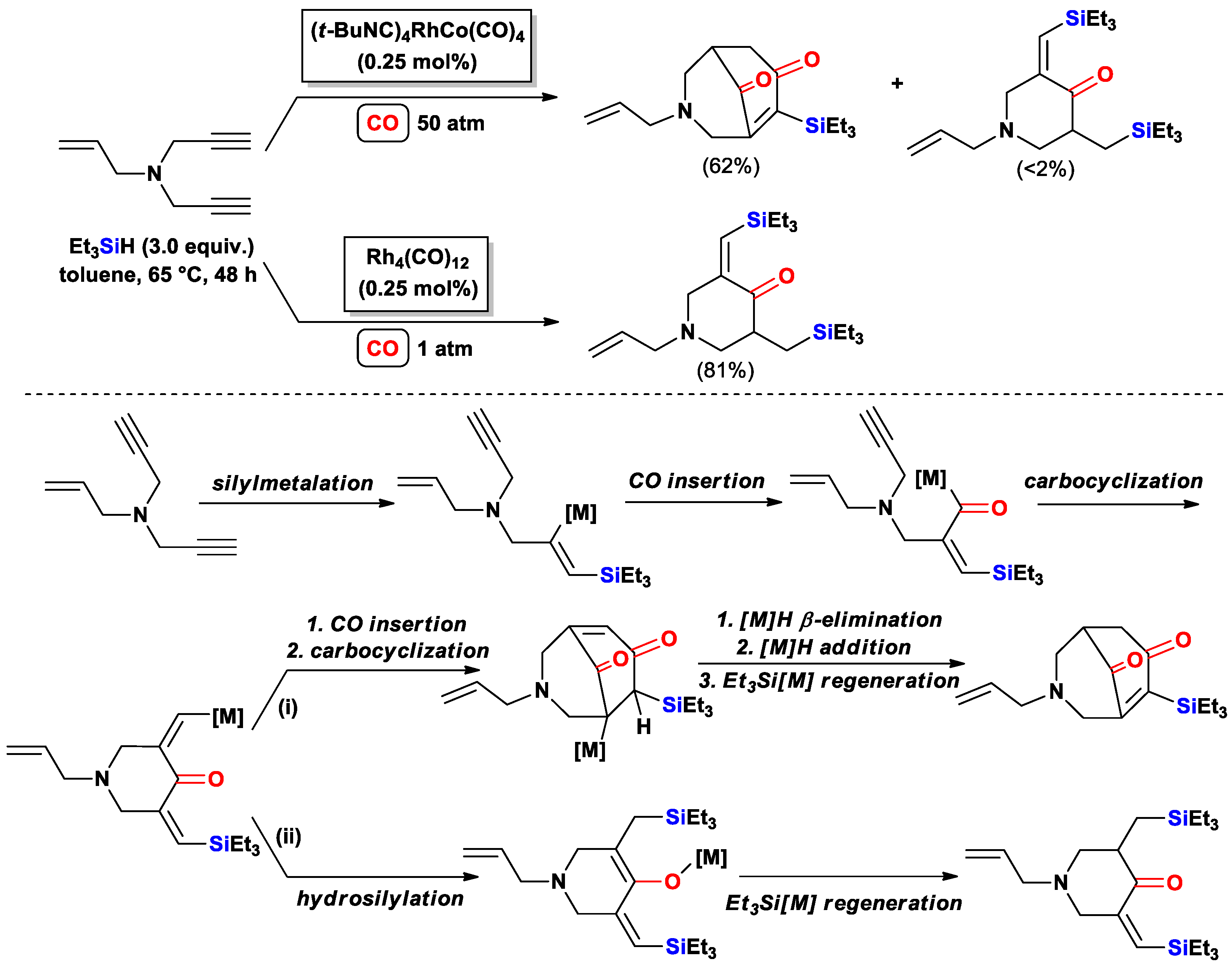{kind=link}
{kind=link}
{kind=link}
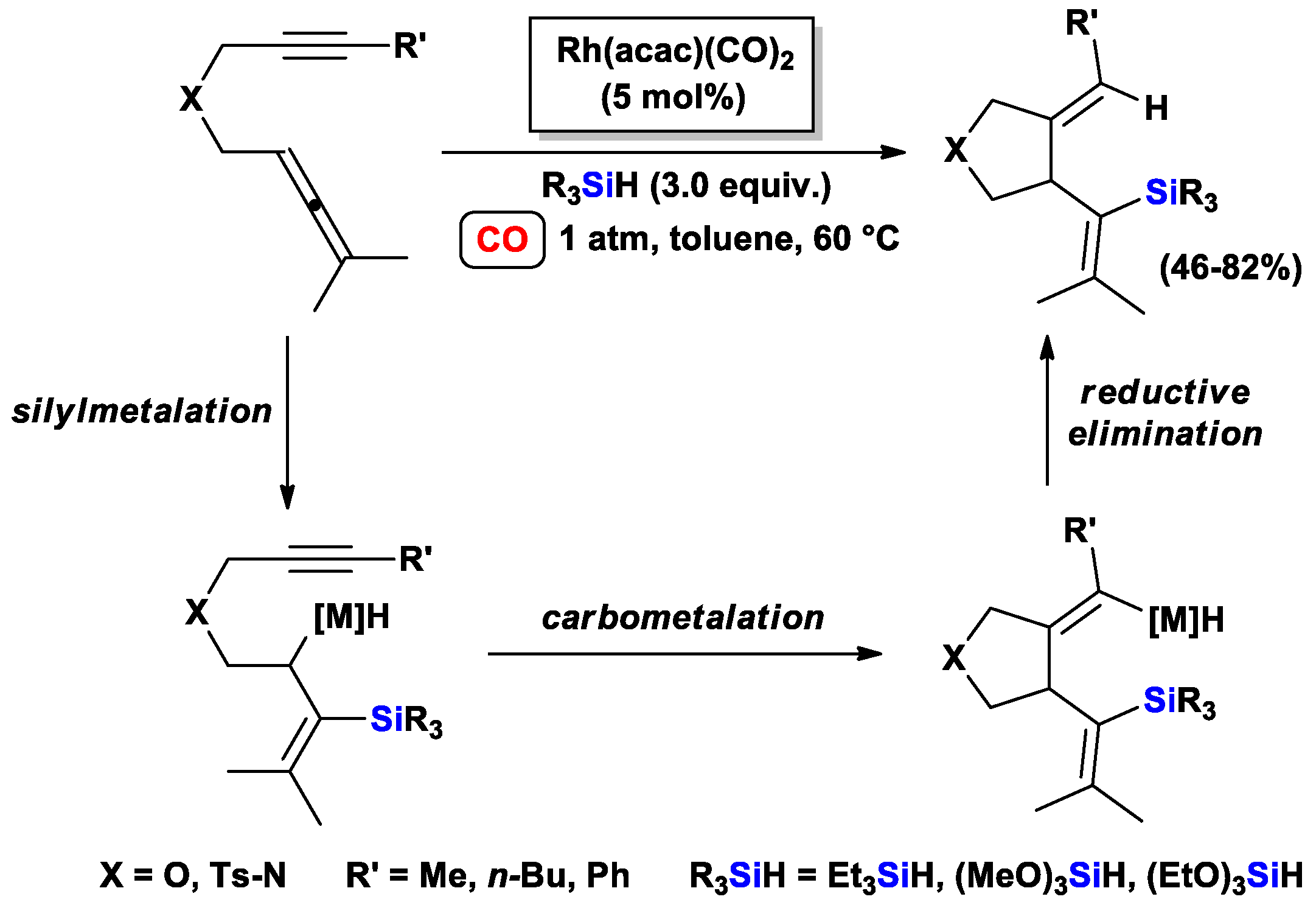{kind=link}
{kind=link}
{kind=link}
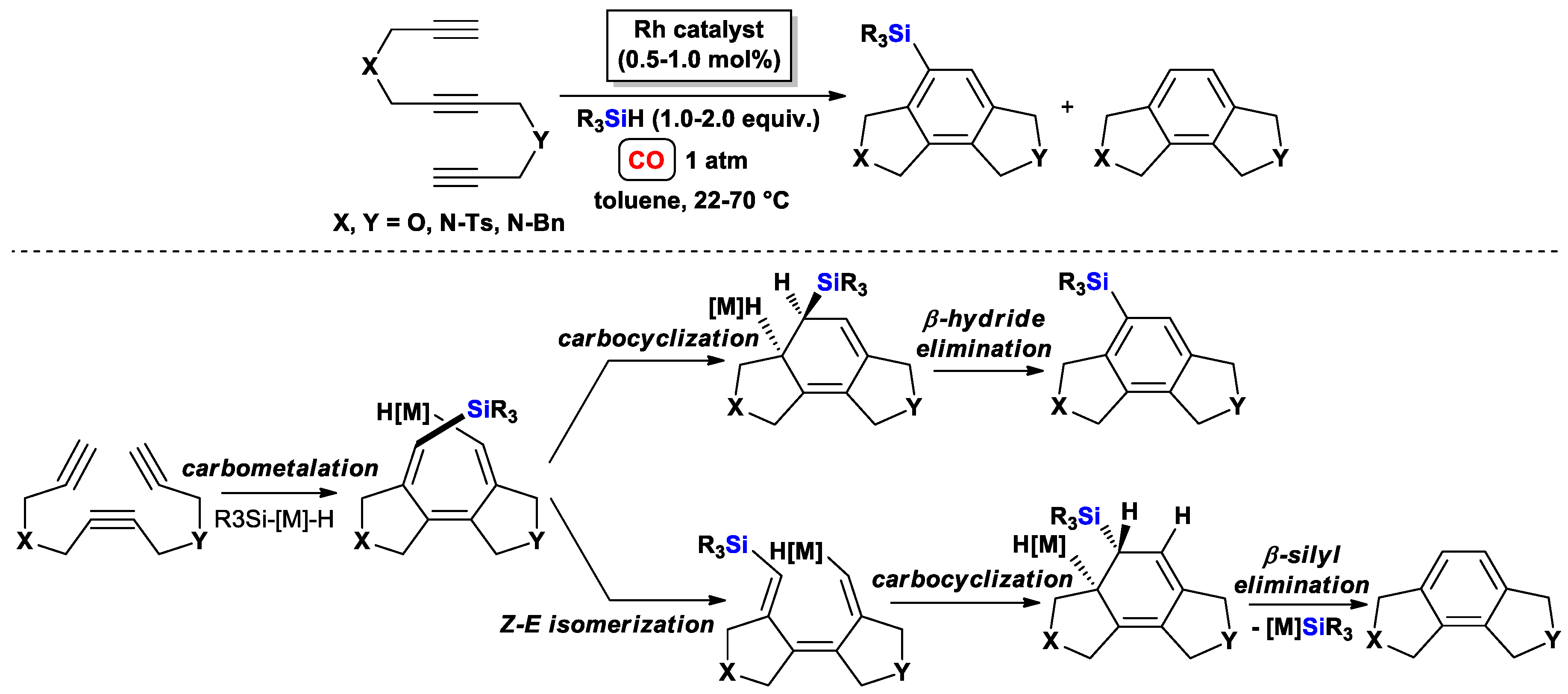{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
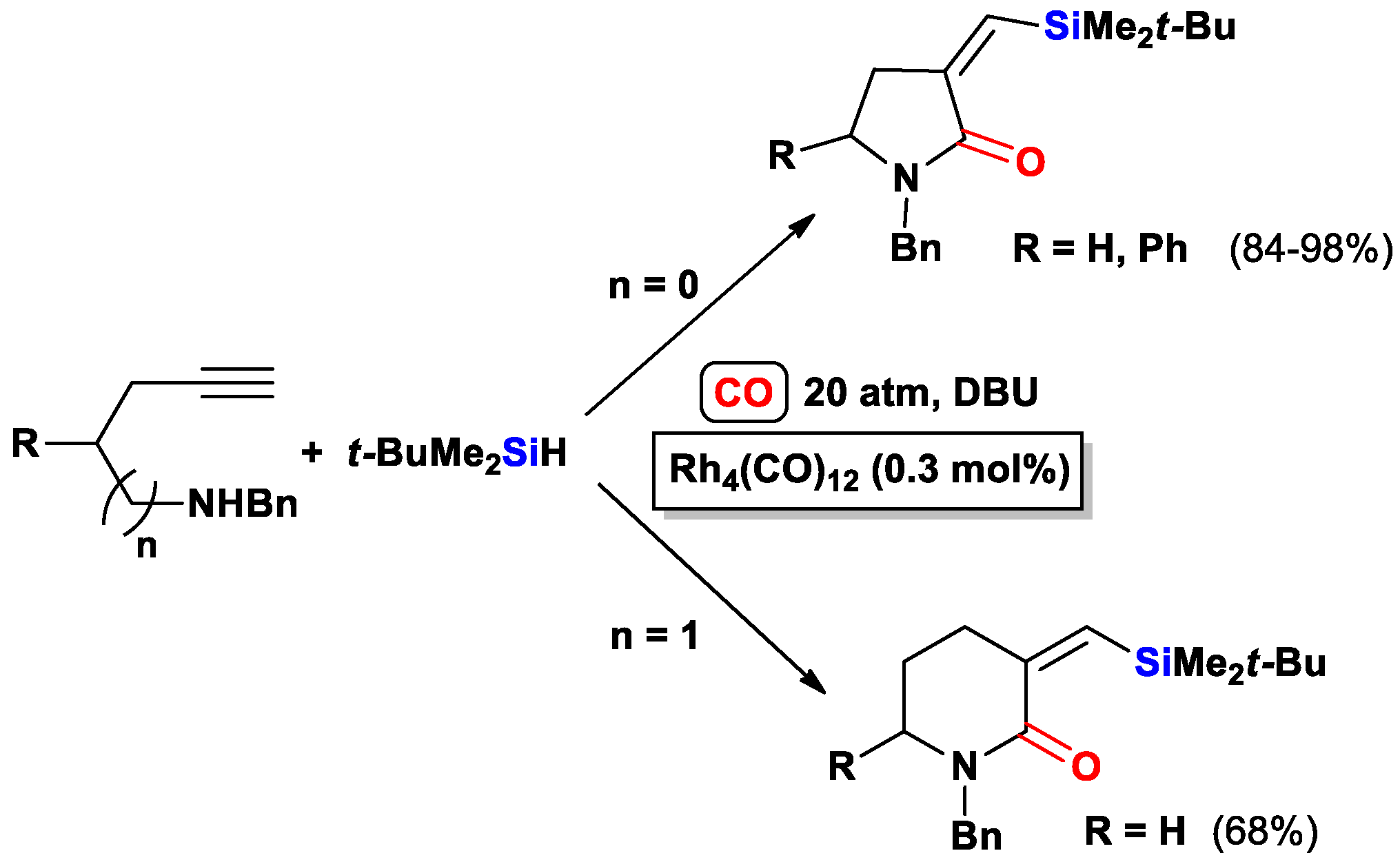{kind=link}
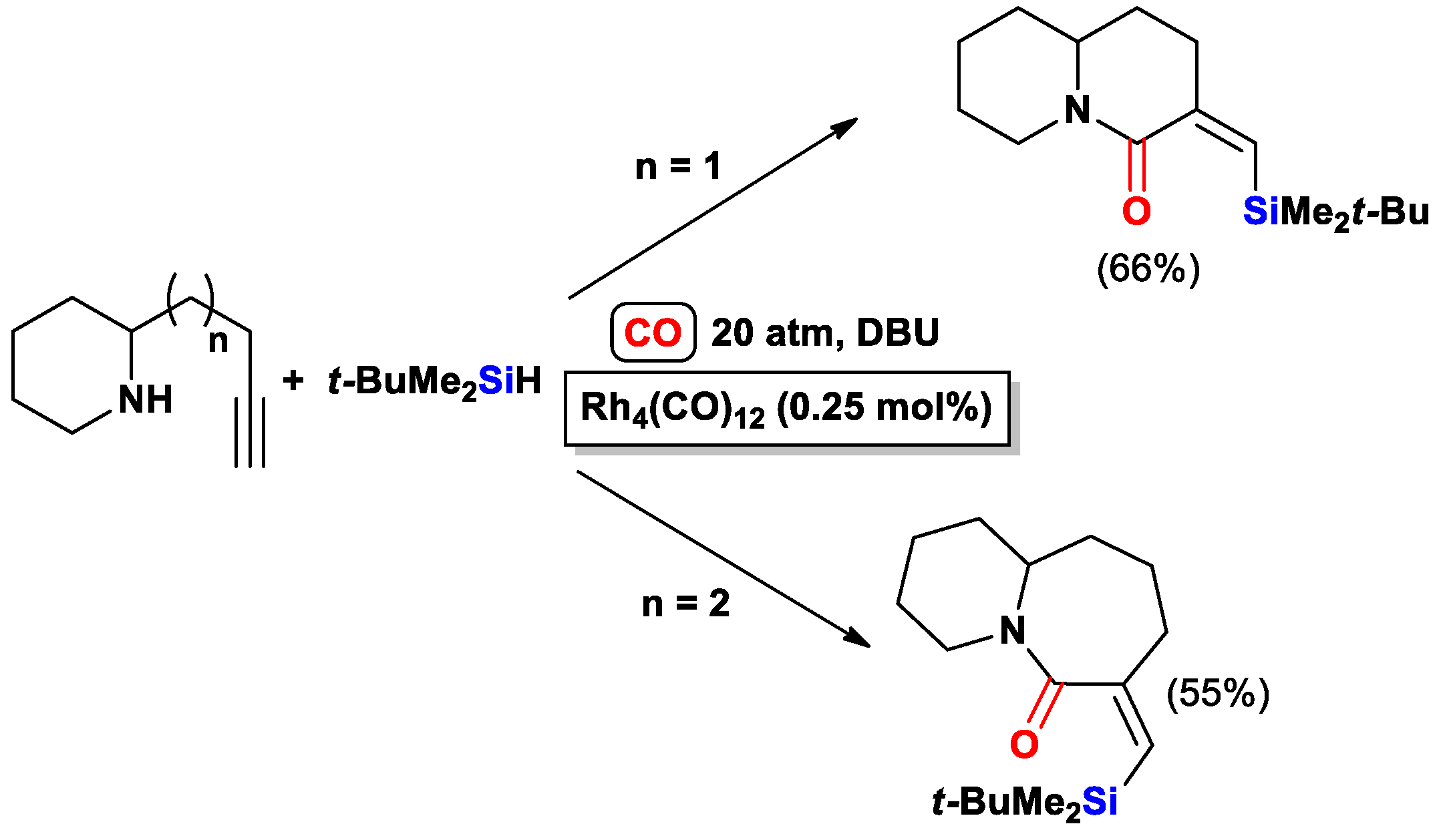{kind=link}
{kind=link}
{kind=link}
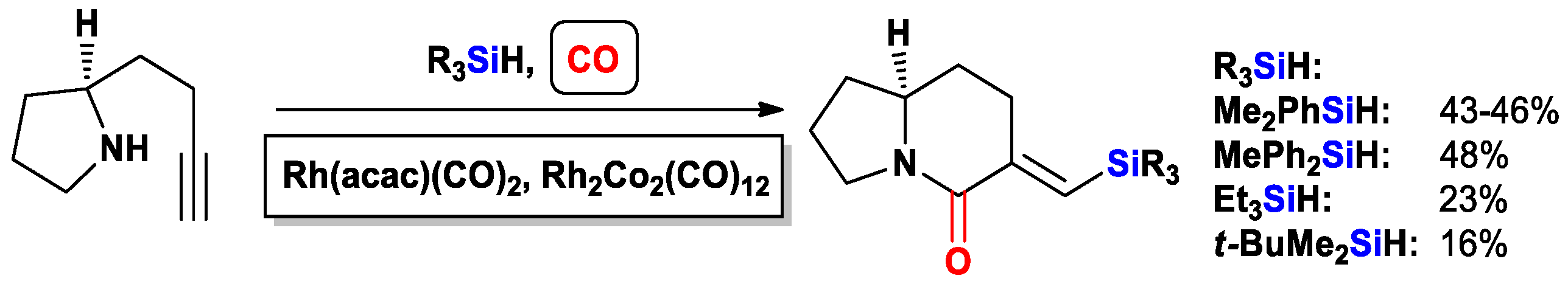{kind=link}
{kind=link}
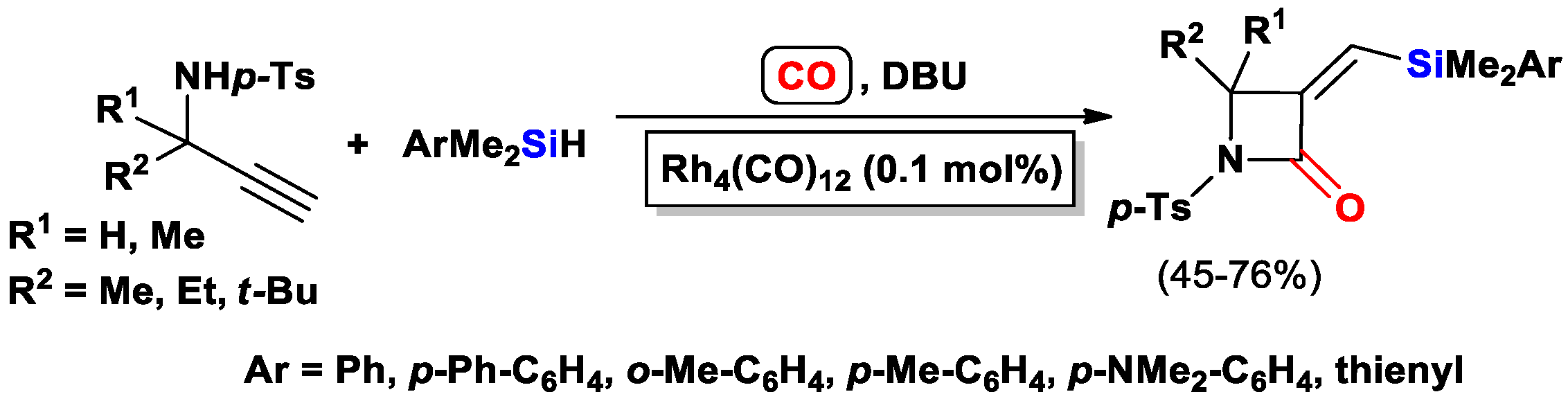{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Heterocycles Synthesis via Metal-Catalyzed Intramolecular Silylformylation of Alkynes
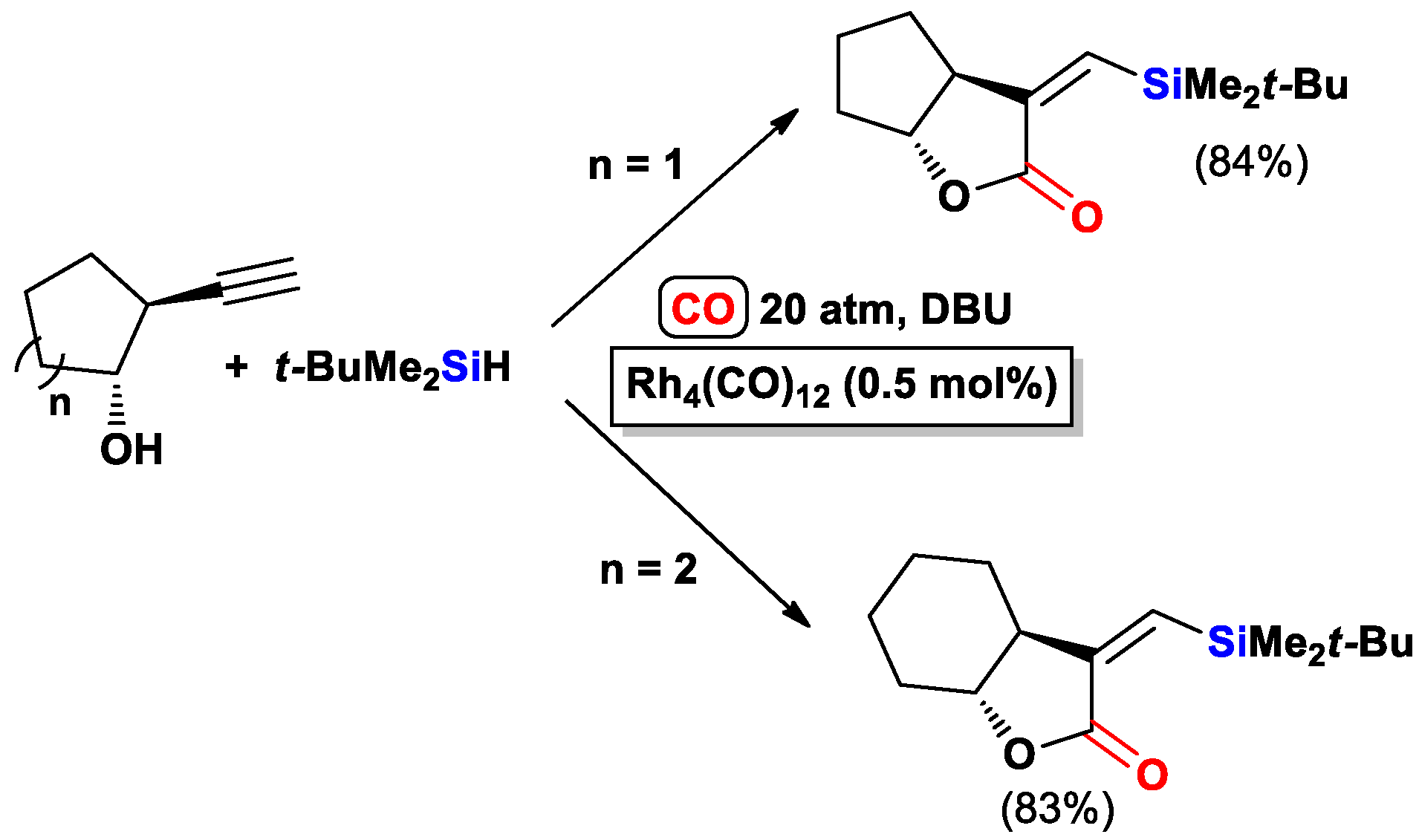
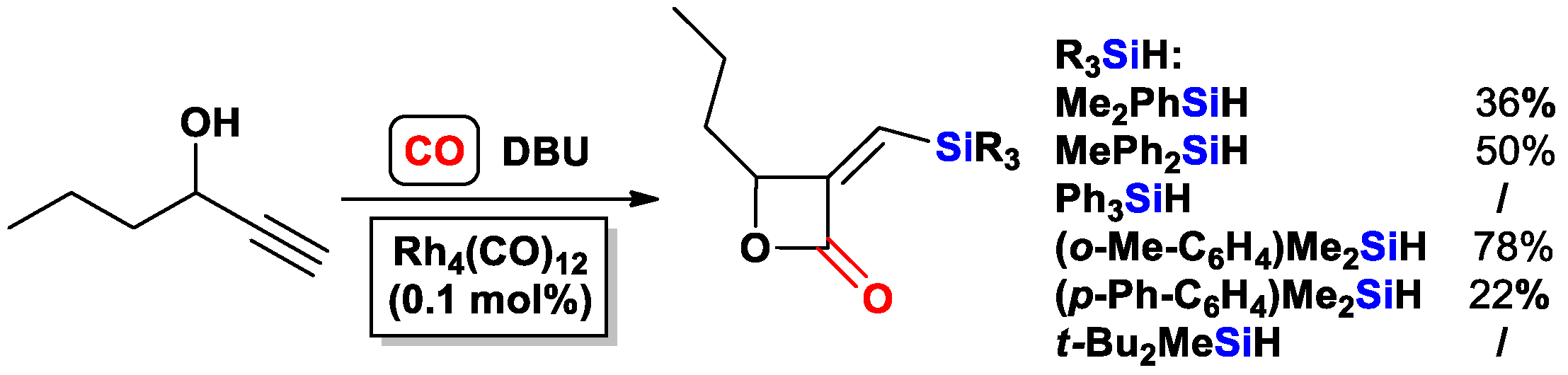
2.1. The Intramolecular Silylformylation of ω-Silylalkynes: Synthesis of Silacyclanes
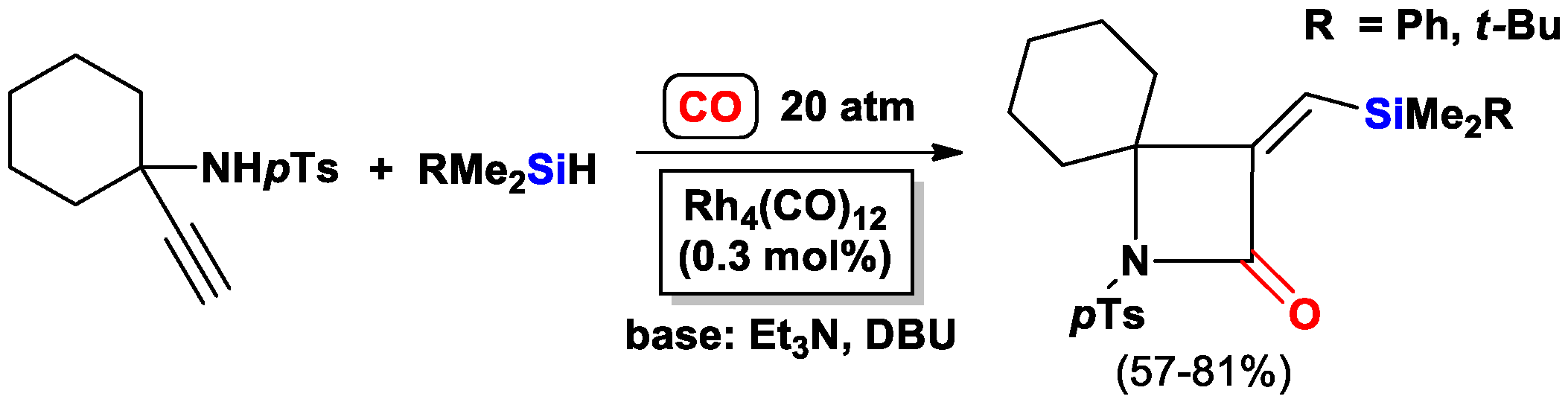
2.2. The Intramolecular Silylformylation of ω-Bis(Dimethylsilylamino)Alkynes: Synthesis of Azasilacyclanes
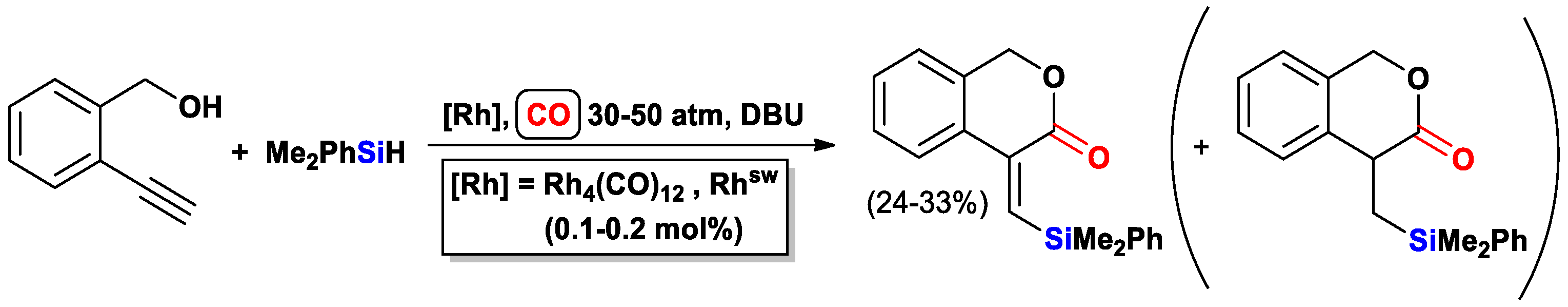
2.3. The Intramolecular Silylformylation of ω-Silyloxyalkynes: Synthesis of Oxasilacyclanes
3. Heterocycles Synthesis via Metal-Catalyzed Silylcarbocyclization of Alkynes
3.1. Synthesis of Heterocycles via Metal-Catalyzed Standard Silylcarbocyclizations of Alkynes
3.1.1. Standard Silylcarbocyclizations of Allyl Propargyl Ethers/Amines
3.1.2. Standard Silylcarbocyclizations of Dipropargyl Ethers/Amines
3.2. Synthesis of Heterocycles via Metal-Catalyzed Cascade Silylcarbocyclizations of Alkynes
3.2.1. Cascade Silylcarbocyclizations of Enediynes
3.2.2. Cascade Silylcarbocyclizations of Triynes
3.3. Synthesis of Heterocycles Via Metal-Catalyzed Heteroatom-Promoted Silylcarbocyclizations of Alkynes
3.3.1. Heteroatom-Promoted Silylcarbocyclizations of Ethynyl Alcohols
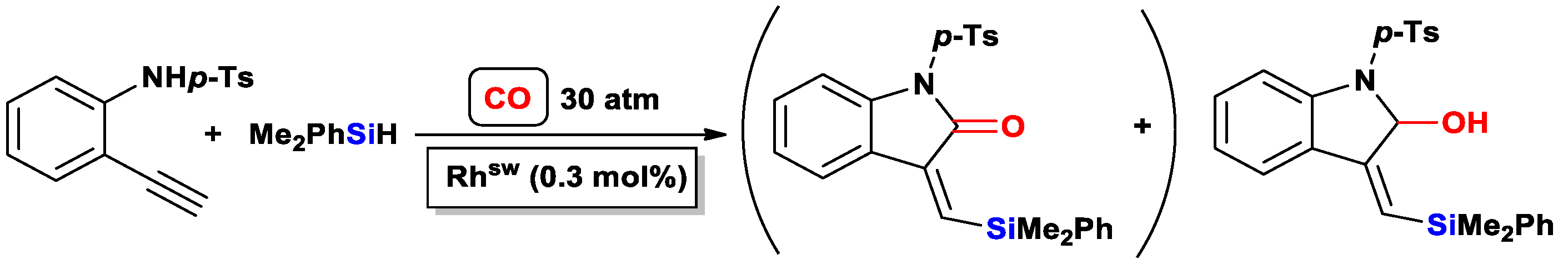
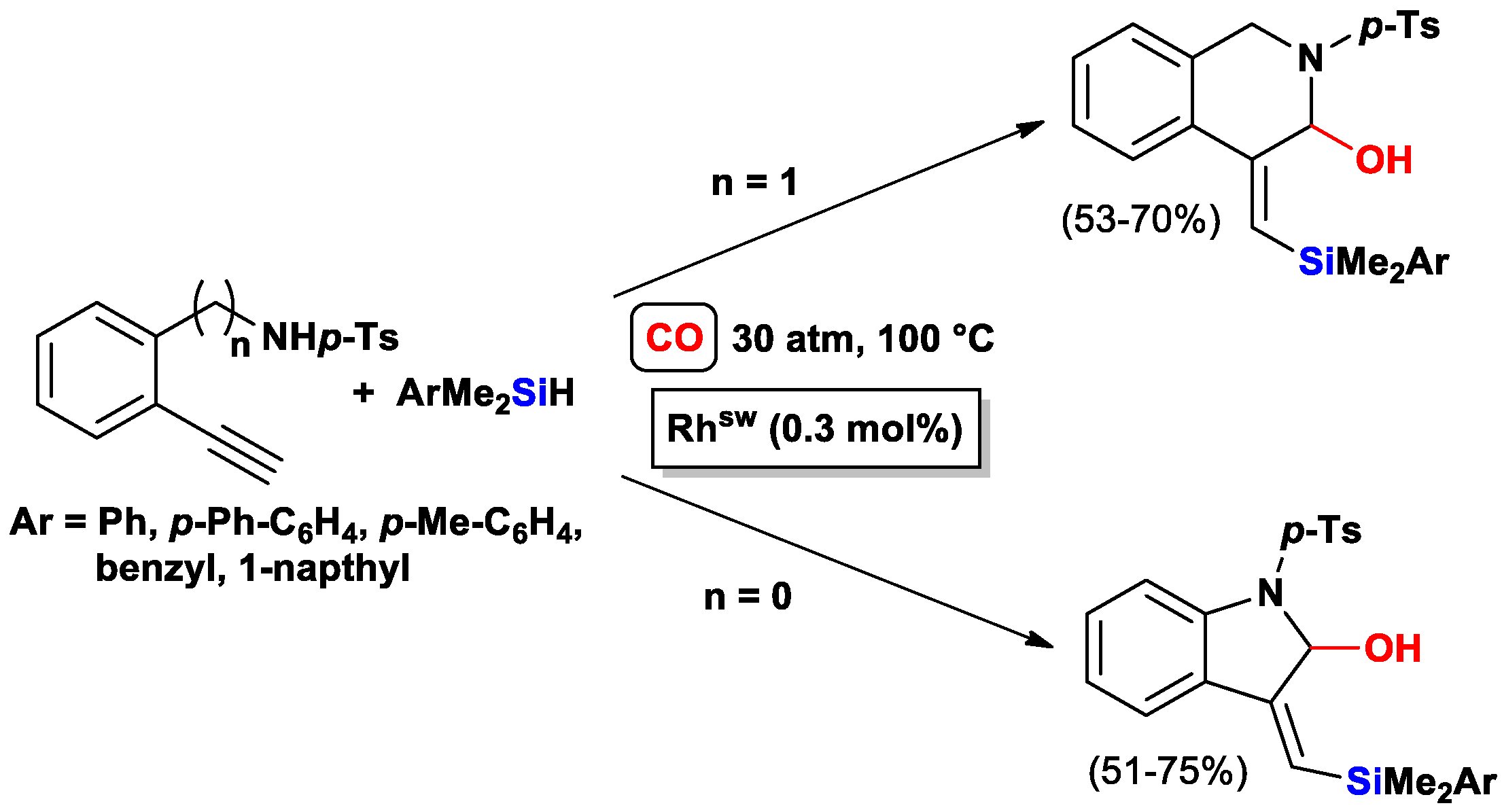
3.3.2. Heteroatom-Promoted Silylcarbocyclizations of Ethynyl Amines
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chatani, N.; Murai, S. HSiR3/CO as the potent reactant combination in developing new transition-metal-catalyzed reactions. Synlett 1996, 1996, 414–424. [Google Scholar] [CrossRef]
- Matsuda, I.; Fukuta, Y.; Tsuchihashi, T.; Nagashima, H.; Itoh, K. Rhodium-catalyzed silylformylation of Acetylenic bonds: Its scope and mechanistic considerations. Organometallics 1997, 16, 4327–4345. [Google Scholar] [CrossRef]
- Leighton, J.L. Stereoselective rhodium(I)-catalyzed hydroformylation and silylformylation reactions and their application to organic synthesis. In Modern Rhodium-Catalyzed Organic Reactions; Evans, P.A., Ed.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005; pp. 93–110. [Google Scholar]
- Matsuda, I. Silylformylation. In Comprehensive Organometallic Chemistry III; Mingos, D.M.P., Crabtree, R.H., Eds.; Elsevier: Amsterdam, The Netherlands, 2007; pp. 473–510. [Google Scholar]
- Aronica, L.A.; Caporusso, A.M.; Salvadori, P. Synthesis and reactivity of silylformylation products derived from alkynes. Eur. J. Org. Chem. 2008, 2008, 3039–3060. [Google Scholar] [CrossRef]
- Beller, M.; Cornils, B.; Frohning, C.D.; Kohlpaintner, C.W. Progress in hydroformylation and carbonylation. J. Mol. Catal. A Chem. 1995, 104, 17–85. [Google Scholar] [CrossRef]
- Reiser, O. Metal-catalyzed hydroformylations. In Organic Synthesis Highlights IV; Schmalz, H.-G., Ed.; WILEY-VCH Verlag GmbH: Weinheim, Germany, 2000; pp. 97–103. [Google Scholar]
- Ojima, I.; Tsai, C.-Y.; Tzamarioudaki, M.; Bonafoux, D. The hydroformylation reaction. In Organic Reactions; Overman, L.E., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2000; Volume 56. [Google Scholar]
- Breit, B.; Seiche, W. Recent advances on chemo-, regio-and stereoselective hydroformylation. Synthesis 2001, 2001, 0001–0036. [Google Scholar] [CrossRef]
- Wiese, K.-D.; Obst, D. Hydroformylation. Top. Organomet. Chem. 2006, 18, 1–33. [Google Scholar]
- Franke, R.; Selent, D.; Börner, A. Applied hydroformylation. Chem. Rev. 2012, 112, 5675–5732. [Google Scholar] [CrossRef]
- Pospech, J.; Fleischer, I.; Franke, R.; Buchholz, S.; Beller, M. Alternative metals for homogeneous catalyzed hydroformylation reactions. Angew. Chem. Int. Ed. 2013, 52, 2852–2872. [Google Scholar] [CrossRef]
- Breit, B.; Diab, L. Hydroformylation and related carbonylation reactions of alkenes, alkynes, and allenes. In Comprehensive Organic Synthesis, 2nd ed.; Knochel, P., Molander, G.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 4, pp. 995–1053. [Google Scholar]
- Wu, X.-F.; Fang, X.; Wu, L.; Jackstell, R.; Neumann, H.; Beller, M. Transition-metal-catalyzed carbonylation reactions of olefins and alkynes: A personal account. Acc. Chem. Res. 2014, 47, 1041–1053. [Google Scholar] [CrossRef]
- Hanf, S.; Alvarado Rupflin, L.; Gläser, R.; Schunk, S.A. Current state of the art of the solid rh-based catalyzed hydroformylation of short-chain olefins. Catalysts 2020, 10, 510. [Google Scholar] [CrossRef]
- Matsuda, I.; Ogiso, A.; Sato, S.; Izumi, Y. An efficient silylformylation of alkynes catalyzed by tetrarhodium dodecacarbonyl. J. Am. Chem. Soc. 1989, 111, 2332–2333. [Google Scholar] [CrossRef]
- Aronica, L.A.; Raffa, P.; Valentini, G.; Caporusso, A.M.; Salvadori, P. Silylformylation—Fluoride-assisted aryl migration of acetylenic derivatives in a versatile approach to the synthesis of polyfunctionalised compounds. Eur. J. Org. Chem. 2006, 2006, 1845–1851. [Google Scholar] [CrossRef]
- Aronica, L.A.; Raffa, P.; Caporusso, A.M.; Salvadori, P. Fluoride-promoted rearrangement of organo silicon compounds: A new synthesis of 2-(arylmethyl)aldehydes from 1-alkynes. J. Org. Chem. 2003, 68, 9292–9298. [Google Scholar] [CrossRef] [PubMed]
- Aronica, L.A.; Raffa, P.; Valentini, G.; Caporusso, A.M.; Salvadori, P. Silylformylation–desilylation of propargyl amides: Synthesis of α,β-unsaturated aldehydes. Tetrahedron Lett. 2006, 47, 527–530. [Google Scholar] [CrossRef]
- Donskaya, N.A.; Yur’eva, N.M.; Sigeev, A.S.; Voevodskaya, T.I.; Beletskaya, I.P.; Tretyakov, V.F. Silylformylation of alkynes catalysed by Di-μ-chlorotetrakis(η2-methylene-cyclopropane)dirhodium. Mendeleev Commun. 1995, 5, 220–221. [Google Scholar] [CrossRef]
- Alonso, M.A.; Casares, J.A.; Espinet, P.; Vallés, E.; Soulantica, K. Efficient silylformylation of alkynes catalyzed by rhodium complexes with P,N donor ligands. Tetrahedron Lett. 2001, 42, 5697–5700. [Google Scholar] [CrossRef]
- Basato, M.; Biffis, A.; Martinati, G.; Zecca, M.; Ganis, P.; Benetollo, F.; Aronica, L.A.; Caporusso, A.M. Cationic carboxylato complexes of dirhodium(II) with oxo thioethers: Promising catalysts with unusual coordination modes. Organometallics 2004, 23, 1947–1952. [Google Scholar] [CrossRef]
- Basato, M.; Biffis, A.; Martinati, G.; Tubaro, C.; Graiff, C.; Tiripicchio, A.; Aronica, L.A.; Caporusso, A.M. Cationic complexes of dirhodium(II) with 1,8-naphthyridine: Catalysis of reactions involving silanes. J. Organomet. Chem. 2006, 691, 3464–3471. [Google Scholar] [CrossRef]
- Biffis, A.; Conte, L.; Tubaro, C.; Basato, M.; Aronica, L.A.; Cuzzola, A.; Caporusso, A.M. Highly selective silylformylation of internal and functionalised alkynes with a cationic dirhodium(II) complex catalyst. J. Organomet. Chem. 2010, 695, 792–798. [Google Scholar] [CrossRef]
- Ojima, I.; Ingallina, P.; Donovan, R.J.; Clos, N. Silylformylation of 1-hexyne catalyzed by rhodium-cobalt mixed-metal carbonyl clusters. Organometallics 1991, 10, 38–41. [Google Scholar] [CrossRef]
- Ojima, I.; Donovan, R.J.; Eguchi, M.; Shay, W.R.; Ingallina, P.; Korda, A.; Zeng, Q. Silylformylation catalyzed by Rh and Rh-Co mixed metal complexes and its application to the synthesis of pyrrolizidine alkaloids. Tetrahedron 1993, 49, 5431–5444. [Google Scholar] [CrossRef]
- Ojima, I.; Donovan, R.J.; Ingallina, P.; Clos, N.; Shay, W.R.; Eguchi, M.; Zeng, Q.; Korda, A. Organometallic chemistry and homogeneous catalysis of Rh and Rh-Co mixed metal carbonyl clusters. J. Cluster Sci. 1992, 3, 423–438. [Google Scholar] [CrossRef]
- Ojima, I.; Zhaoyang, L.; Donovan, R.J.; Ingallina, P. On the mechanism of silylformylation catalyzed by Rh-Co mixed metal complexes. Inorg. Chim. Acta 1998, 270, 279–284. [Google Scholar] [CrossRef]
- Yoshikai, N.; Yamanaka, M.; Ojima, I.; Morokuma, K.; Nakamura, E. Bimetallic synergism in alkyne silylformylation catalyzed by a cobalt−rhodium mixed-metal cluster. Organometallics 2006, 25, 3867–3875. [Google Scholar] [CrossRef]
- Doyle, M.P.; Shanklin, M.S. Highly Regioselective and stereoselective silylformylation of alkynes under mild conditions promoted by dirhodium(II) perfluorobutyrate. Organometallics 1994, 13, 1081–1088. [Google Scholar] [CrossRef]
- Doyle, M.P.; Shanklin, M.S. Highly efficient regioselective silylcarbonylation of alkynes catalyzed by dirhodium(II) perfluorobutyrate. Organometallics 1993, 12, 11–12. [Google Scholar] [CrossRef]
- Zhou, Z.; Facey, G.; James, B.R.; Alper, H. Interconversion between zwitterionic and cationic rhodium(I) complexes of demonstrated value as catalysts in hydroformylation, silylformylation, and hydrogenation reactions. dynamic 31P{1H} NMR studies of (η6-PhBPh3)-Rh+(DPPB) and [Rh(DPPB)2]+BPh4-in solution. Organometallics 1996, 15, 2496–2503. [Google Scholar]
- Zhou, J.-Q.; Alper, H. Zwitterionic rhodium(I) complex catalyzed net silylhydroformylation of terminal alkynes. Organometallics 1994, 13, 1586–1591. [Google Scholar] [CrossRef]
- Okazaki, H.; Kawanami, Y.; Yamamoto, K. The Silylformylation of simple 1-alkynes catalyzed by [Rh(cod)][BPh4] in an Ionic liquid, [Bmim][PF6], under biphasic conditions: An efficiently reusable Catalyst system. Chem. Lett. 2001, 30, 650–651. [Google Scholar] [CrossRef]
- Aronica, L.A.; Terreni, S.; Caporusso, A.M.; Salvadori, P. Silylformylation of chiral 1-alkynes, catalysed by solvated rhodium atoms. Eur. J. Org. Chem. 2001, 2001, 4321–4329. [Google Scholar] [CrossRef]
- Aronica, L.A.; Valentini, G.; Caporusso, A.M.; Salvadori, P. Silylation–desilylation of propargyl amides: Rapid synthesis of functionalised aldehydes and β-lactams. Tetrahedron 2007, 63, 6843–6854. [Google Scholar] [CrossRef]
- Ojima, I.; Tzamarioudaki, M.; Tsai, C.-Y. Extremely chemoselective silylformylation and silylcarbocyclization of alkynals. J. Am. Chem. Soc. 1994, 116, 3643–3644. [Google Scholar] [CrossRef]
- Carter, M.J.; Fleming, I. Allyl silanes in organic synthesis: Some reactions of 3-trimethylsilylcyclohex-4-ene-1,2-dicarboxylic acid and its derivatives. J. Chem. Soc. Chem. Commun. 1976, 679–680. [Google Scholar] [CrossRef]
- Fleming, I.; Perry, D.A. The synthesis of αβ-unsaturated ketones from β-silylenones and β-silylynones. Tetrahedron 1981, 37, 4027–4034. [Google Scholar] [CrossRef]
- Jain, N.F.; Cirillo, P.F.; Schaus, J.V.; Panek, J.S. An efficient procedure for the preparation of chiral β-substituted (E)-crotylsilanes: Application of a rhodium(II) catalyzed silylformylation of terminal alkynes. Tetrahedron Lett. 1995, 36, 8723–8726. [Google Scholar] [CrossRef]
- Eilbracht, P.; Hollmann, C.; Schmidt, A.M.; Bärfacker, L. Tandem silylformylation/Wittig olefination of terminal alkynes: Stereoselective synthesis of 2,4-dienoic esters. Eur. J. Org. Chem. 2000, 2000, 1131–1135. [Google Scholar] [CrossRef]
- Bärfacker, L.; Hollmann, C.; Eilbracht, P. Rhodium(I)-catalysed hydrocarbonylation and silylcarbonylation reactions of alkynes in the presence of primary amines leading to 2-pyrrolidinones and 4-silylated 1-aza-1,3-butadienes. Tetrahedron 1998, 54, 4493–4506. [Google Scholar] [CrossRef]
- Jung, M.E.; Gaede, B. Synthesis and diels-alder reactions of e-1-trimethylsilylbuta-1,3-diene. Tetrahedron 1979, 35, 621–625. [Google Scholar] [CrossRef]
- Jones, T.K.; Denmark, S.E. Silicon-directed nazarov reactions II. Preparation and cyclization of β-silyl-substituted divinyl ketones. Helv. Chim. Acta 1983, 66, 2377–2396. [Google Scholar] [CrossRef]
- Denmark, S.E.; Jones, T.K. Silicon-directed Nazarov cyclization. J. Am. Chem. Soc. 1982, 104, 2642–2645. [Google Scholar] [CrossRef]
- Aronica, L.A.; Morini, F.; Caporusso, A.M.; Salvadori, P. New synthesis of α-benzylaldehydes from 2-(dimethylphenylsilylmethylene)alkanals by fluoride promoted phenyl migration. Tetrahedron Lett. 2002, 43, 5813–5815. [Google Scholar] [CrossRef]
- Monteil, F.; Matsuda, I.; Alper, H. Rhodium-catalyzed intramolecular silylformylation of acetylenes: A vehicle for complete regio-and stereoselectivity in the formylation of acetylenic bonds. J. Am. Chem. Soc. 1995, 117, 4419–4420. [Google Scholar] [CrossRef]
- Baldwin, J.E. Rules for ring closure. J. Chem. Soc. Chem. Commun. 1976, 734–736. [Google Scholar] [CrossRef]
- Gilmore, K.; Alabugin, I.V. Cyclizations of alkynes: Revisiting baldwin’s rules for ring closure. Chem. Rev. 2011, 111, 6513–6556. [Google Scholar] [CrossRef] [PubMed]
- Aronica, L.A.; Caporusso, A.M.; Salvadori, P.; Alper, H. Diastereoselective intramolecular silylformylation of ω-silylacetylenes. J. Org. Chem. 1999, 64, 9711–9714. [Google Scholar] [CrossRef]
- Aronica, L.A.; Caporusso, A.M.; Tuci, G.; Evangelisti, C.; Manzoli, M.; Botavina, M.; Martra, G. Palladium nanoparticles supported on Smopex® metal scavengers as catalyst for carbonylative Sonogashira reactions: Synthesis of α,β-alkynyl ketones. Appl. Catal. A Gen. 2014, 480, 1–9. [Google Scholar] [CrossRef]
- Albano, G.; Evangelisti, C.; Aronica, L.A. Hydrogenolysis of benzyl protected phenols and aniline promoted by supported palladium nanoparticles. ChemistrySelect 2017, 2, 384–388. [Google Scholar] [CrossRef]
- Albano, G.; Interlandi, S.; Evangelisti, C.; Aronica, L.A. Polyvinylpyridine-supported palladium nanoparticles: A valuable catalyst for the synthesis of alkynyl ketones via acyl sonogashira reactions. Catal. Lett. 2020, 150, 652–659. [Google Scholar] [CrossRef]
- Lukevics, E.; Abele, E.; Ignatovich, L. Biologically active silacyclanes. In Advances in Heterocyclic Chemistry; Katritzky, A.R., Ed.; Academic Press, Elsevier: Amsterdam, The Netherlands, 2010; Volume 99, pp. 107–141. [Google Scholar]
- Ojima, I.; Vidal, E.S. Extremely regioselective intramolecular silylformylation of bis(silylamino)alkynes. Organometallics 1999, 18, 5103–5107. [Google Scholar] [CrossRef]
- Ojima, I.; Vidal, E.; Tzamarioudaki, M.; Matsuda, I. Extremely regioselective intramolecular silylformylation of alkynes. J. Am. Chem. Soc. 1995, 117, 6797–6798. [Google Scholar] [CrossRef]
- Aronica, L.A. Hydrosilylation and Silylformylation of Aceytlenic Compounds. Ph.D. Thesis, University of Pisa, Pisa, Italy, 1999. [Google Scholar]
- Bonafoux, D.; Ojima, I. Desymmetrization of 4-dimethylsiloxy-1,6-heptadiynes through sequential double silylformylation. Org. Lett. 2001, 3, 1303–1305. [Google Scholar] [CrossRef]
- Bonafoux, D.; Ojima, I. Novel DMAP-Catalyzed Skeletal Rearrangement of 5-exo-(2-Hydroxy- ethylene)oxasilacyclopentanes. Org. Lett. 2001, 3, 2333–2335. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Kobayashi, T. Tandem Intramolecular silylformylation and silicon-assisted cross-coupling reactions. Synthesis of geometrically defined α,β-unsaturated aldehydes. J. Org. Chem. 2003, 68, 5153–5159. [Google Scholar] [CrossRef] [PubMed]
- Zacuto, M.J.; O’Malle, S.J.; Leighton, J.L. Tandem intramolecular silylformylation−crotylsilylation: Highly efficient synthesis of polyketide fragments. J. Am. Chem. Soc. 2002, 124, 7890–7891. [Google Scholar] [CrossRef]
- Zacuto, M.J.; O’Malley, S.J.; Leighton, J.L. Tandem silylformylation–allyl(crotyl)silylation: A new approach to polyketide synthesis. Tetrahedron 2003, 59, 8889–8900. [Google Scholar] [CrossRef]
- Spletstoser, J.T.; Zacuto, M.J.; Leighton, J.L. Tandem silylformylation−crotylsilylation/tamao oxidation of internal alkynes: A remarkable example of generating complexity from simplicity. Org. Lett. 2008, 10, 5593–5596. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Ho, S.; Leighton, J.L. A More comprehensive and highly practical solution to enantioselective aldehyde crotylation. Org. Lett. 2008, 10, 5593–5596. [Google Scholar] [CrossRef] [PubMed]
- Harrison, T.J.; Rabbat, P.M.A.; Leighton, J.L. An “aprotic” tamao oxidation/syn-selective tautomerization reaction for the efficient synthesis of the C(1)–C(9) fragment of fludelone. Org. Lett. 2012, 14, 4890–4893. [Google Scholar] [CrossRef]
- Foley, C.N.; Leighton, J.L. Beyond the roche ester: A new approach to polypropionate stereotriad synthesis. Org. Lett. 2014, 16, 1180–1183. [Google Scholar] [CrossRef]
- Foley, C.N.; Leighton, J.L. A Highly stereoselective, efficient, and scalable synthesis of the C(1)–C(9) Fragment of the epothilones. Org. Lett. 2015, 17, 5858–5861. [Google Scholar] [CrossRef]
- Ojima, I. New cyclization reactions in organic syntheses. Pure Appl. Chem. 2002, 74, 159. [Google Scholar] [CrossRef][Green Version]
- Ojima, I.; Moralee, A.C.; Vassar, V.C. Recent advances in rhodium-catalyzed cyclization reactions. Top. Catal. 2002, 19, 89–99. [Google Scholar] [CrossRef]
- Varchi, G.; Ojima, I. Synthesis of heterocycles through hydrosilylation, silylformylation, silylcarbocyclization and cyclohydrocarbonylation reactions. Curr. Org. Chem. 2006, 10, 1341–1362. [Google Scholar] [CrossRef]
- Ojima, I.; Donovan, R.J.; Shay, W.R. Silylcarbocyclization reactions catalyzed by rhodium and rhodium-cobalt complexes. J. Am. Chem. Soc. 1992, 114, 6580–6582. [Google Scholar] [CrossRef]
- Ojima, I.; McCullagh, J.V.; Shay, W.R. New cascade silylcarbocyclization (SiCaC) of enediynes. J. Organomet. Chem. 1996, 521, 421–423. [Google Scholar] [CrossRef]
- Ojima, I.; Vu, A.T.; Lee, S.-Y.; McCullagh, J.V.; Moralee, A.C.; Fujiwara, M.; Hoang, T.H. Rhodium-catalyzed silylcarbocyclization (SiCaC) and carbonylative silylcarbocyclization (CO−SiCaC) reactions of enynes. J. Am. Chem. Soc. 2002, 124, 9164–9174. [Google Scholar] [CrossRef] [PubMed]
- Fukuta, Y.; Matsuda, I.; Itoh, K. Rhodium-catalyzed domino silylformylation of enynes involving carbocyclization. Tetrahedron Lett. 1999, 40, 4703–4706. [Google Scholar] [CrossRef]
- Park, K.H.; Jung, I.G.; Kim, S.Y.; Chung, Y.K. Immobilized cobalt/rhodium heterobimetallic nanoparticle-catalyzed silylcarbocylization and carbonylative silylcarbocyclization of 1,6-enynes. Org. Lett. 2003, 5, 4967–4970. [Google Scholar] [CrossRef] [PubMed]
- Murai, T.; Toshio, R.; Mutoh, Y. Sequential addition reaction of lithium acetylides and Grignard reagents to thioiminium salts from thiolactams leading to 2,2-disubstituted cyclic amines. Tetrahedron 2006, 62, 6312–6320. [Google Scholar] [CrossRef]
- Denmark, S.E.; Liu, J.H.-C. Sequential silylcarbocyclization/silicon-based cross-coupling reactions. J. Am. Chem. Soc. 2007, 129, 3737–3744. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Liu, J.H.-C.; Muhuhi, J.M. Total syntheses of isodomoic acids G and H. J. Am. Chem. Soc. 2009, 131, 14188–14189. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Liu, J.H.-C.; Muhuhi, J.M. Stereocontrolled total syntheses of isodomoic acids G and H via a unified strategy. J. Org. Chem. 2011, 76, 201–215. [Google Scholar] [CrossRef][Green Version]
- Ojima, I.; Fracchiolla, D.A.; Donovan, R.J.; Banerji, P. Silylcarbobicyclization of 1,6-diynes: A novel Catalytic route to bicyclo[3.3.0]octenones. J. Org. Chem. 1994, 59, 7594–7595. [Google Scholar] [CrossRef]
- Ojima, I.; Kass, D.F.; Zhu, J. New and efficient catalytic route to bicyclo[3.3.0]octa-1,5-dien-3-ones. Organometallics 1996, 15, 5191–5195. [Google Scholar] [CrossRef]
- Ojima, I.; Zhu, J.; Vidal, E.S.; Kass, D.F. Silylcarbocyclizations of 1,6-diynes. J. Am. Chem. Soc. 1998, 120, 6690–6697. [Google Scholar] [CrossRef]
- Matsuda, I.; Ishibashi, H.; Ii, N. Silylative cyclocarbonylation of acetylenic bonds catalyzed by Rh4(CO)12: An easy access to bicyclo[3.3.0]octenone skeletons. Tetrahedron Lett. 1995, 36, 241–244. [Google Scholar] [CrossRef]
- Shibata, T.; Kadowaki, S.; Takagi, K. Chemo-and regioselective intramolecular hydrosilylative carbocyclization of allenynes. Organometallics 2004, 23, 4116–4120. [Google Scholar] [CrossRef]
- Ojima, I.; Lee, S.-Y. Rhodium-catalyzed novel carbonylative carbotricyclization of enediynes. J. Am. Chem. Soc. 2000, 122, 2385–2386. [Google Scholar] [CrossRef]
- Bennacer, B.; Fujiwara, M.; Ojima, I. Novel [2 + 2 + 2 + 1] Cycloaddition of enediynes catalyzed by rhodium complexes. Org. Lett. 2004, 6, 3589–3591. [Google Scholar] [CrossRef]
- Bennacer, B.; Fujiwara, M.; Lee, S.-Y.; Ojima, I. Silicon-initiated carbonylative carbotricyclization and [2+2+2+1] cycloaddition of enediynes catalyzed by rhodium complexes. J. Am. Chem. Soc. 2005, 127, 17756–17767. [Google Scholar] [CrossRef]
- Ojima, I.; Vu, A.T.; McCullagh, J.V.; Kinoshita, A. Rhodium-catalyzed intramolecular silylcarbotricyclization (SiCaT) of triynes. J. Am. Chem. Soc. 1999, 121, 3230–3231. [Google Scholar] [CrossRef]
- Matsuda, I.; Ogiso, A.; Sato, S. Ready access of .alpha.-(triorganosilyl)methylene .beta.-lactones by means of rhodium-catalyzed cyclocarbonylation of substituted propargyl alcohols. J. Am. Chem. Soc. 1990, 112, 6120–6121. [Google Scholar] [CrossRef]
- Matsuda, I.; Sakakibara, J.; Nagashima, H. A novel approach to α-silylmethylene-β-lactams via Rh-catalyzed silylcarbonylation of propargylamine derivatives. Tetrahedron Lett. 1991, 32, 7431–7434. [Google Scholar] [CrossRef]
- Fukuta, Y.; Matsuda, I.; Itoh, K. Synthesis of 3-silyl-2(5H)-furanone by rhodium-catalyzed cyclocarbonylation. Tetrahedron Lett. 2001, 42, 1301–1304. [Google Scholar] [CrossRef]
- Aronica, L.A.; Mazzoni, C.; Caporusso, A.M. Synthesis of functionalised β-lactones via silylcarbocyclisation/desilylation reactions of propargyl alcohols. Tetrahedron 2010, 66, 265–273. [Google Scholar] [CrossRef]
- Aronica, L.A.; Caporusso, A.M.; Evangelisti, C.; Botavina, M.; Alberto, G.; Martra, G. Metal vapour derived supported rhodium nanoparticles in the synthesis of β-lactams and β-lactones derivatives. J. Organomet. Chem. 2012, 700, 20–28. [Google Scholar] [CrossRef]
- Albano, G.; Morelli, M.; Aronica, L.A. Synthesis of functionalised 3-isochromanones by silylcarbocyclisation/desilylation reactions. Eur. J. Org. Chem. 2017, 2017, 3473–3480. [Google Scholar] [CrossRef]
- Kong, K.-F.; Schneper, L.; Mathee, K. Beta-lactam antibiotics: From antibiosis to resistance and bacteriology. APMIS 2010, 118, 1–36. [Google Scholar] [CrossRef]
- Shahid, M.; Sobia, F.; Singh, A.; Malik, A.; Khan, H.M.; Jonas, D.; Hawkey, P.M. Beta-lactams and Beta-lactamase-inhibitors in current-or potential-clinical practice: A comprehensive update. Crit. Rev. Microbiol. 2009, 35, 81–108. [Google Scholar] [CrossRef]
- Romano, A.; Gaeta, F.; Poves, M.F.A.; Valluzzi, R.L. Cross-reactivity among beta-lactams. Curr. Allergy Asthma Rep. 2016, 16, 24. [Google Scholar] [CrossRef]
- Broccolo, F.; Cainelli, G.; Caltabiano, G.; Cocuzza, C.E.A.; Fortuna, C.G.; Galletti, P.; Giacomini, D.; Musumarra, G.; Musumeci, R.; Quintavalla, A. Design, synthesis, and biological evaluation of 4-alkyliden-beta lactams: New products with promising antibiotic activity against resistant bacteria. J. Med. Chem. 2006, 49, 2804–2811. [Google Scholar] [CrossRef] [PubMed]
- Bouthillier, G.; Mastalerz, H.; Menard, M.; Fung-Tomc, J.; Gradelski, E. The synthesis, antibacterial, and beta-lactamase inhibitory activity of a novel asparenomycin analog. J. Antibiot. 1992, 45, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Sinner, E.K.; Lichstrahl, M.S.; Li, R.; Marous, D.R.; Townsend, C.A. Methylations in complex carbapenem biosynthesis are catalyzed by a single cobalamin-dependent radical S-adenosylmethionine enzyme. Chem. Commun. 2019, 55, 14934–14937. [Google Scholar] [CrossRef] [PubMed]
- Rolinson, G.N.; Geddes, A.M. The 50th anniversary of the discovery of 6-aminopenicillanic acid (6-APA). Int. J. Antimicrob. Agents 2007, 29, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Golemi, D.; Maveyraud, L.; Ishiwata, A.; Tranier, S.; Miyashita, K.; Nagase, T.; Massova, I.; Mourey, L.; Samama, J.P.; Mobashery, S. 6-(hydroxyalkyl)penicillanates as probes for mechanisms of beta-lactamases. J. Antibiot. 2000, 53, 1022–1027. [Google Scholar] [CrossRef]
- Liang, Y.; Raju, R.; Le, T.; Taylor, C.D.; Howell, A.R. Cross-metathesis of α-methylene-β-lactams: The first tetrasubstituted alkenes by CM. Tetrahedron Lett. 2009, 50, 1020–1022. [Google Scholar] [CrossRef]
- Zhu, L.; Xiong, Y.; Li, C. Synthesis of α-methylene-β-lactams via PPh3-catalyzed umpolung cyclization of propiolamides. J. Org. Chem. 2015, 80, 628–633. [Google Scholar] [CrossRef]
- Hussein, M.; Nasr El Dine, A.; Farès, F.; Dorcet, V.; Hachem, A.; Grée, R. A new direct synthesis of α-methylene- and α-alkylidene-β-lactams. Tetrahedron Lett. 2016, 57, 1990–1993. [Google Scholar] [CrossRef]
- Zhang, L.; Ma, L.; Zhou, H.; Yao, J.; Li, X.; Qiu, G. Synthesis of α-methylene-β-lactams enabled by base-promoted intramolecular 1,2-addition of N-propiolamide and C–C bond migrating cleavage of aziridine. Org. Lett. 2018, 20, 2407–2411. [Google Scholar] [CrossRef]
- Ojima, I.; Machnik, D.; Donovan, R.J.; Mneimne, O. Silylcyclocarbonylation (SiCCa) of alkynylamines catalyzed by rhodium complexes. Inorg. Chim. Acta 1996, 251, 299–307. [Google Scholar] [CrossRef]
- Ojima, I.; Clos, N.; Donovan, R.J.; Ingallina, P. Hydrosilylation of 1-hexyne catalyzed by rhodium and cobalt-rhodium mixed-metal complexes. Mechanism of apparent trans addition. Organometallics 1990, 9, 3127–3133. [Google Scholar] [CrossRef]
- Albano, G.; Morelli, M.; Lissia, M.; Aronica, L.A. Synthesis of functionalised indoline and isoquinoline derivatives through a silylcarbocyclisation/desilylation sequence. ChemistrySelect 2019, 4, 2505–2511. [Google Scholar] [CrossRef]
- Xu, X.; Doyle, M.P. The [3 + 3]-cycloaddition alternative for heterocycle syntheses: Catalytically generated metalloenolcarbenes as dipolar adducts. Acc. Chem. Res. 2014, 47, 1396–1405. [Google Scholar] [CrossRef] [PubMed]
- Albano, G.; Aronica, L.A. Cyclization reactions for the synthesis of phthalans and isoindolines. Synthesis 2018, 50, 1209–1227. [Google Scholar]
- Pucci, A.; Albano, G.; Pollastrini, M.; Lucci, A.; Colalillo, M.; Oliva, F.; Evangelisti, C.; Marelli, M.; Santalucia, D.; Mandoli, A. Supported tris-triazole ligands for batch and continuous-flow copper-catalyzed huisgen 1,3-dipolar cycloaddition reactions. Catalysts 2020, 10, 434. [Google Scholar] [CrossRef]
- Albano, G.; Aronica, L.A. Potentiality and synthesis of o-and n-heterocycles: Pd-catalyzed cyclocarbonylative sonogashira coupling as a valuable route to phthalans, isochromans, and isoindolines. Eur. J. Org. Chem. 2017, 2017, 7204–7221. [Google Scholar] [CrossRef]
- Aronica, L.A.; Albano, G.; Giannotti, L.; Meucci, E. Synthesis of n-heteroaromatic compounds through cyclocarbonylative sonogashira reactions. Eur. J. Org. Chem. 2017, 2017, 955–963. [Google Scholar] [CrossRef]
- Albano, G.; Aronica, L.A. Acyl sonogashira cross-coupling: State of the art and application to the synthesis of heterocyclic compounds. Catalysts 2020, 10, 25. [Google Scholar] [CrossRef]
- Albano, G.; Giuntini, S.; Aronica, L.A. Synthesis of 3-Alkylideneisoindolin-1-ones via sonogashira cyclocarbonylative reactions of 2-ethynylbenzamides. J. Org. Chem. 2020, 85, 10022–10034. [Google Scholar] [CrossRef]
- Konishi, H.; Manabe, K. Formic acid derivatives as practical carbon monoxide surrogates for metal-catalyzed carbonylation reactions. Synlett 2014, 25, 1971–1986. [Google Scholar] [CrossRef]
- Wu, L.; Liu, Q.; Jackstell, R.; Beller, M. Carbonylations of alkenes with CO surrogates. Angew. Chem. Int. Ed. 2014, 53, 6310–6320. [Google Scholar] [CrossRef] [PubMed]
- Gautam, P.; Bhanage, B.M. Recent advances in the transition metal catalyzed carbonylation of alkynes, arenes and aryl halides using CO surrogates. Catal. Sci. Technol. 2015, 5, 4663–4702. [Google Scholar] [CrossRef]


























































© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albano, G.; Aronica, L.A. From Alkynes to Heterocycles through Metal-Promoted Silylformylation and Silylcarbocyclization Reactions. Catalysts 2020, 10, 1012. https://doi.org/10.3390/catal10091012
Albano G, Aronica LA. From Alkynes to Heterocycles through Metal-Promoted Silylformylation and Silylcarbocyclization Reactions. Catalysts. 2020; 10(9):1012. https://doi.org/10.3390/catal10091012
Chicago/Turabian StyleAlbano, Gianluigi, and Laura Antonella Aronica. 2020. "From Alkynes to Heterocycles through Metal-Promoted Silylformylation and Silylcarbocyclization Reactions" Catalysts 10, no. 9: 1012. https://doi.org/10.3390/catal10091012
APA StyleAlbano, G., & Aronica, L. A. (2020). From Alkynes to Heterocycles through Metal-Promoted Silylformylation and Silylcarbocyclization Reactions. Catalysts, 10(9), 1012. https://doi.org/10.3390/catal10091012