Reducing Immunoreactivity of Gliadins and Coeliac-Toxic Peptides Using Peptidases from L. acidophilus 5e2 and A. niger



Abstract
1. Introduction
2. Results and Discussion
2.1. Hydrolysis of Gliadins
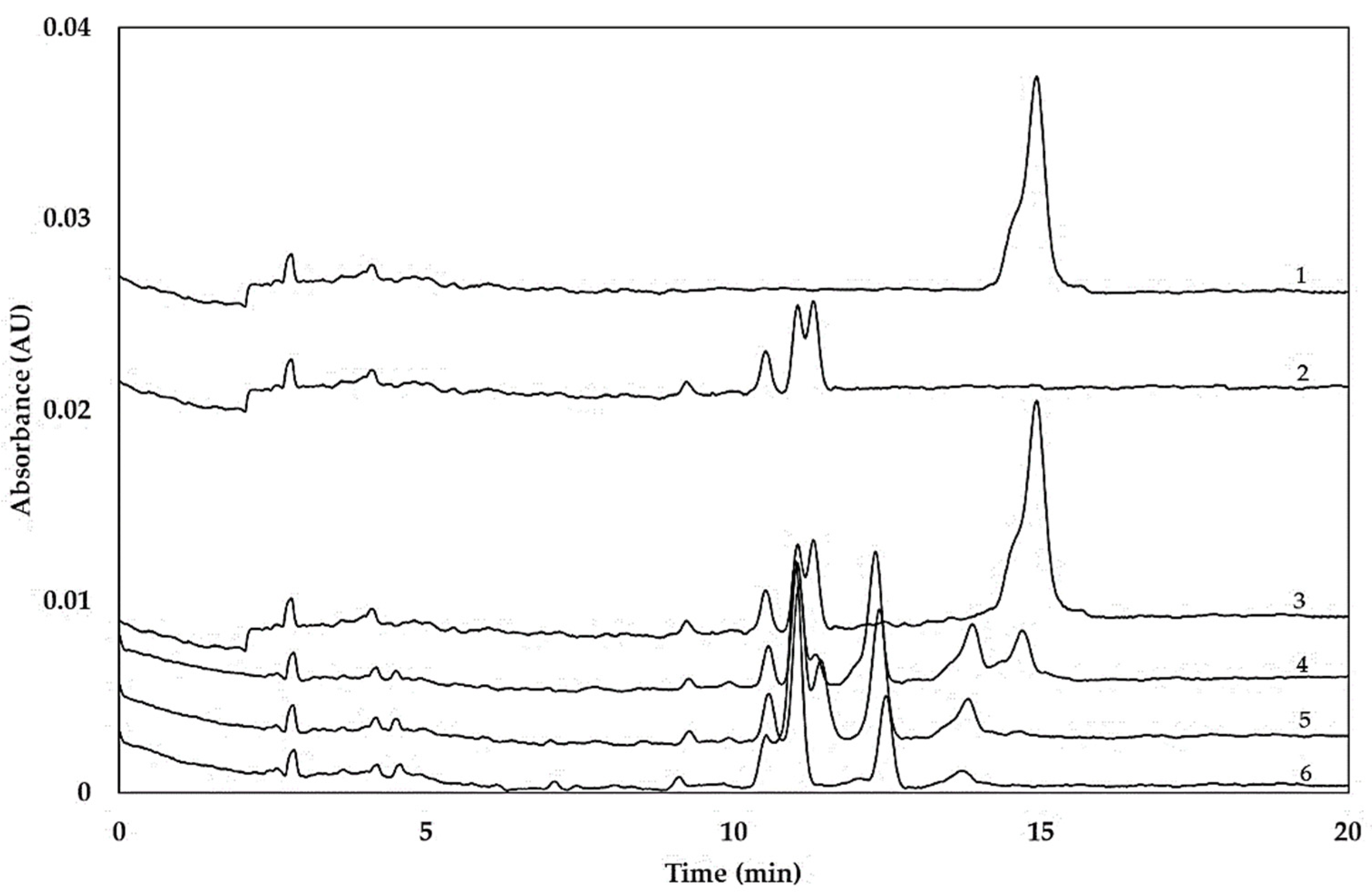
2.2. Hydrolysis of Coeliac-Toxic Peptides
3. Materials and Methods
3.1. Materials
3.2. Biosynthesis and Isolation of Peptidases
3.3. Determination of the Enzyme Preparation Activity
3.4. Gliadin Isolation
3.5. Gliadin Hydrolysis
3.6. Peptide Hydrolysis
3.7. 2D-NuPAGE Electrophoresis and Immunoblotting of Gliadins
3.7.1. Sample Preparation
3.7.2. Isoelectric Focusing of Proteins
3.7.3. Separation of Proteins in NuPAGE Gel
3.8. Immunoblotting
3.9. Degree of Protein Hydrolysis
3.10. Free Zone Capillary Electrophoresis of Peptides
3.11. Protein Immunoreactivity
3.12. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Di Sabatino, A.; Corazza, G.R. Coeliac disease. Lancet 2009, 373, 1480–1493. [Google Scholar] [CrossRef]
- Gilissen, L.J.W.J.; van der Meer, I.M.; Smulders, M.J.M. Reducing the incidence of allergy and intolerance to cereals. J. Cereal Sci. 2014, 59, 337–353. [Google Scholar]
- Wieser, H. Chemistry of gluten proteins. Food Microbiol. 2007, 24, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Wieser, H.; Koehler, P. The biochemical basis of coeliac disease. Cereal Chem. 2008, 85, 1–13. [Google Scholar]
- Brzozowski, B. Impact of food processing and simulated gastrointestinal digestion on gliadin immunoreactivity in rolls. J. Sci. Food Agric. 2018, 987, 3363–3375. [Google Scholar] [CrossRef]
- Gass, J.; Bethune, M.T.; Siegel, M.; Spencer, A.; Khosla, C. Combination enzyme therapy for gastric digestion of dietary gluten in patients with coeliac sprue. Gastroenterology 2007, 133, 472–480. [Google Scholar] [CrossRef]
- Gerez, C.L.; Dallagnol, A.; Rollán, G.; de Valdez, G.F. A combination of two lactic acid bacteria improves the hydrolysis of gliadin during wheat dough fermentation. Food Microbiol. 2012, 32, 427–430. [Google Scholar] [CrossRef]
- Rizzello, C.G.; Curiel, J.A.; Nionelli, L.; Vincentini, O.; Di Cagno, R.; Silano, M.; Gobbetti, M.; Coda, R. Use of fungal proteases and selected sourdough lactic acid bacteria for making wheat bread with an intermediate content of gluten. Food Microbiol. 2014, 37, 59–68. [Google Scholar] [CrossRef]
- Tack, G.J.; van de Water, J.M.; Bruins, M.J.; Kooy-Winkelaar, E.M.; van Bergen, J.; Bonnet, P.; Vreugdenhil, A.C.; Korponay-Szabo, I.; Edens, L.; von Blomberg, B.M.; et al. Consumption of gluten with gluten-degrading enzyme by coeliac patients: A pilot-study. World J. Gastroenterol. 2013, 19, 5837–5847. [Google Scholar] [CrossRef]
- Brzozowski, B. Immunoreactivity of wheat proteins modified by hydrolysis and polymerisation. Eur. Food Res. Technol. 2016, 242, 1025–1040. [Google Scholar] [CrossRef]
- Buddrick, O.; Cornell, H.J.; Small, D.M. Reduction of toxic gliadin content of wholegrain bread by the enzyme caricain. Food Chem. 2015, 170, 343–347. [Google Scholar] [PubMed]
- Schwalb, T.; Wieser, H.; Koehler, P. Studies on the gluten-specific peptidase activity of germinated grains from different cereal species and cultivars. Eur. Food Res. Technol. 2012, 235, 1161–1170. [Google Scholar] [CrossRef]
- Shan, L.; Marti, T.; Sollid, L.M.; Gray, G.M.; Khosla, C. Comparative biochemical analysis of three bacterial prolyl endopeptidases: Implication for coeliac sprue. Biochem. J. 2004, 383, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Tian, N.; Valery, A.C.; Zhong, Y.; Schuppan, D.; Helmerhorst, E.J. Identification of pseudolysin (lasB) as an aciduric gluten-degrading enzyme with high therapeutic potential for coeliac disease. Am. J. Gastroenterol. 2015, 110, 899–908. [Google Scholar] [CrossRef]
- Gänzle, M.; Loponen, J.; Gobbetti, M. Proteolysis in sourdough fermentations: Mechanisms and potential for improved bread quality. Trends Food Sci. Technol. 2008, 19, 513–521. [Google Scholar] [CrossRef]
- Vermeulen, N.; Pavlovic, M.; Ehrmann, M.A.; Gänzle, M.G.; Vogel, R.F. Functional characterization of the proteolytic system of Lactobacillus sanfranciscensis DSM 20451T during growth in sourdough. Appl. Environ. Microb. 2005, 71, 6260–6266. [Google Scholar]
- Thiele, C.; Gänzle, M.G.; Vogel, R.F. Fluorescence labelling of wheat proteins for determination of gluten hydrolysis and depolymerization during dough processing and sourdough fermentation. J. Agric. Food Chem. 2003, 51, 2745–2752. [Google Scholar] [CrossRef]
- De Angelis, M.; Coda, R.; Silano, M.; Minervini, F.; Rizzello, C.G.; Di Cagno, R.; Vicentini, O.; De Vincenzi, M.; Gobbetti, M. Fermentation by selected sourdough lactic acid bacteria to decrease coeliac intolerance to rye flour. J. Cereal Sci. 2006, 43, 301–314. [Google Scholar]
- Rizzello, C.G.; Nionelli, L.; Coda, R.; De Angelis, M.; Gobbetti, M. Effect of sourdough fermentation on stabilisation, and chemical and nutritional characteristics of wheat germ. Food Chem. 2010, 119, 1079–1089. [Google Scholar] [CrossRef]
- De Angelis, M.; Cassone, A.; Rizzello, C.G.; Gagliardi, F.; Minervini, F.; Calasso, M.; Di Cagno, R.; Francavilla, R.; Gobbetti, M. Mechanism of degradation of immunogenic gluten epitopes from Triticum turgidum L. var. durum by sourdough lactobacilli and fungal proteases. Appl. Environ. Microb. 2010, 76, 508–518. [Google Scholar] [CrossRef]
- Di Cagno, R.; De Angelis, M.; Auricchio, S.; Greco, L.; Clarke, C.; De Vincenzi, M.; Giovannini, C.; D’Archivio, M.; Landolfo, F.; Parrilli, G.; et al. Sourdough bread made from wheat and nontoxic flours and started with selected lactobacilli is tolerated in coeliac sprue patients. Appl. Environ. Microb. 2004, 70, 1088–1096. [Google Scholar]
- Knorr, V.; Kerpes, R.; Wieser, H.; Zarnkow, M.; Becker, T.; Koehler, P. Production and application of barley malt extract with high peptidase activity for the degradation of gluten in wort. Eur. Food Res. Technol. 2016, 242, 585–597. [Google Scholar] [CrossRef]
- Rizzello, C.G.; Angelis, M.D.; Cagno, R.D.; Camarca, A.; Silano, M.; Losito, I.; De Vincenzi, M.; De Bari, M.D.; Palmisano, F.; Maurano, F.; et al. Highly efficient gluten degradation by lactobacilli and fungal proteases during food processing: New perspectives for coeliac disease. Appl. Environ. Microb. 2007, 73, 4499–4507. [Google Scholar]
- Greco, L.; Gobbetti, M.; Auricchio, R.; Mase, R.D.; Landolfo, F.; Paparo, F.; Di Cagno, R.; De Angelis, M.; Rizzello, C.G.; Cassone, A.; et al. Safety for patients with coeliac disease of baked goods made of wheat flour hydrolysed during food processing. Clin. Gastroenterol. Hepatol. 2011, 9, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Loponen, J.; Sontag-Strohm, T.; Venäläinen, J.; Salovaara, H. Prolamin hydrolysis in wheat sourdoughs with differing proteolytic activities. J. Agric. Food Chem. 2007, 55, 978–984. [Google Scholar] [CrossRef]
- Di Cagno, R.; De Angelis, M.; Lavermicocca, P.; De Vincenzi, M.; Giovannini, C.; Faccia, M.; Gobbetti, M. Proteolysis by sourdough lactic acid bacteria: Effects on wheat flour protein fractions and gliadin peptides involved in human cereal intolerance. Appl. Environ. Microb. 2002, 68, 623–633. [Google Scholar]
- Wieser, H.; Vermeulen, N.; Gaertner, F.; Vogel, R.F. Effects of different Lactobacillus and Enterococcus strains and chemical acidification regarding degradation of gluten proteins during sourdough fermentation. Eur. Food Res. Technol. 2008, 226, 1495–1502. [Google Scholar] [CrossRef]
- Gerez, C.L.; Rollán, G.C.; de Valdez, G.F. Gluten breakdown by lactobacilli and pediococci strains isolated from sourdough. Lett. Appl. Microbiol. 2006, 42, 459–464. [Google Scholar] [CrossRef]
- Stepniak, D.; Spaenij-Dekking, L.; Mitea, C.; Moester, M.; de Ru, A.; Baak-Pablo, R.; van Veelen, P.; Edens, L.; Koning, F. Highly efficient gluten degradation with a newly identified prolyl endoprotease: Implications for coeliac disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G621–G629. [Google Scholar] [CrossRef]
- Lopponen, J. Prolamin Degradation in Sourdough. Ph.D. Dissertation, University of Helsinki, Helsinki, Finland, 2006; p. 77. [Google Scholar]
- Codex Alimentarius Commission. Codex Standard 118–1979 (Revised 2008), Foods for Special Dietary Use for Person Intolerant to Gluten; FAO: Rome, Italy, 2008. [Google Scholar]
- Kahlenberg, F.; Sanchez, D.; Lachmann, I.; Tuckova, L.; Tlaskalova, H.; Méndez, E.; Mothes, T. Monoclonal antibody R5 for detection of putatively coeliac-toxic gliadin peptides. Eur. Food Res. Technol. 2006, 222, 78–82. [Google Scholar] [CrossRef]
- Heredia-Sandoval, N.G.; de la Barca, A.C.; Carvajal-Millán, E.; Islas-Rubio, A.R. Amaranth addition to enzymatically modified wheat flour improves dough functionality, bread immunoreactivity and quality. Food Funct. 2018, 9, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Walter, T.; Wieser, H.; Koehler, P. Degradation of gluten in wheat bran and bread drink by means of a proline-specific peptidase. Nutr. Food Sci. 2014, 4, 293–299. [Google Scholar] [CrossRef]
- Mohan Kumara, B.V.; Sarabhaia, S.; Prabhasankara, P. Targeted degradation of gluten proteins in wheat flour by prolyl endoprotease and its utilization in low immunogenic pasta for gluten sensitivity population. J. Cereal Sci. 2019, 89, 59–67. [Google Scholar] [CrossRef]
- Van Eckert, R.; Bond, J.; Rawson, P.; Klein, C.L.; Stern, M.; Jordan, T.W. Reactivity of gluten detecting monoclonal antibodies to a gliadin reference material. J. Cereal Sci. 2010, 51, 198–204. [Google Scholar] [CrossRef]
- Akagawa, M.; Handoyo, T.; Ishii, T.; Kumazawa, S.; Morita, N.; Suyama, K. Proteomic analysis of wheat flour allergens. J. Agric. Food Chem. 2007, 55, 6863–8670. [Google Scholar]
- Lookhart, G.L.; Bean, S.R. Improvements in cereal protein separations by capillary electrophoresis: Resolution and reproducibility. Cereal Chem. 1996, 73, 81–87. [Google Scholar]



{kind=link}
{kind=link}
| Enzyme | Substrate | LA Activity 1 (U/mg) | PEP Activity 1 (U/mg) |
|---|---|---|---|
| Endopeptidase 2 | Acasein | 4.6 ± 0.1 | n.d. 5 |
| Carboxypeptidase 3 | Z-Phe-Ala | 1.3 ± 0.1 | n.d. |
| Aminopeptidase 4 | Leu-pNa | 452.5 ± 0.9 | n.d. |
| Prolyl endopeptidase 4 | Z-Gly-Pro-pNa | 15.4 ± 0.2 | 152.4 ± 0.8 |
| X-Prolyl-dipeptidyl-aminopeptidase 4 | Gly-Pro-pNa | 11,028.9 ± 1.2 | n.d. |
| Arg-Pro-pNa | 398.1 ± 0.6 | n.d. | |
| Proline iminopeptidase 4 | Pro-pNa | 1.6 ± 0.1 | n.d. |
| Hydrolysis Parameters | DH (%) | C (mg/kg) | EI (μg/mg) | RI (%) |
|---|---|---|---|---|
| Control | 0.0 ± 0.0 a 1 | 47,689.8 ± 221.1 a | 269,412.5 ± 4831.0 a | 100.00 ± 0.38 a |
| 30 °C; pH 4 | 66.4 ± 0.7 d | 1893.2 ± 48.1 d | 0.4 ± 0.0 c | 3.97 ± 0.08 c |
| 30 °C; pH 6 | 49.9 ± 0.8 b | 16,097.5 ± 118.1 b | 4.2 ± 0.1 b | 33.55 ± 0.20 b |
| 37 °C; pH 4 | 74.0 ± 1.2 e | 432.2 ± 4.3 e | 0.1 ± 0.0 d | 0.91 ± 0.01 d |
| 37 °C; pH 6 | 61.6 ± 1.6 c | 2452.8 ± 50.5 c | 0.5 ± 0.0 c | 5.11 ± 0.20 c |
| Hydrolysis Parameters | Molecular Weight of Fraction (kDa) | No. of Spots | No. of Immuno-Reactive Spots |
|---|---|---|---|
| Control | <28 | 0 | 0 |
| 28–65 | 35 | 18 | |
| 30 °C; pH 4 | <28 | 9 | 6 |
| 28–65 | 28 | 12 | |
| 30 °C; pH 6 | <28 | 5 | 4 |
| 28–65 | 33 | 17 | |
| 37 °C; pH 4 | <28 | 11 | 6 |
| 28–65 | 26 | 8 | |
| 37 °C; pH 6 | <28 | 8 | 5 |
| 28–65 | 30 | 14 |
| Hydrolysis Parameters | DH (%) | C (mg/kg) | RI (%) | |||
|---|---|---|---|---|---|---|
| P1 | P2 | P1 | P2 | P1 | P2 | |
| Control | 0.0 ± 0.0 a 1 | 0.0 ± 0.0 a | 403.2 ± 0.3 e | 401.8 ± 0.3 e | 100.0 ± 0.0 e | 100.0 ± 0.0 e |
| 30 °C; pH 4 | 95.7 ± 0.6 c | 93.1 ± 0.6 d | 18.2 ± 0.1 b | 23.5 ± 0.2 b | 4.5 ± 0.0 b | 5.9 ± 0.0 b |
| 30 °C; pH 6 | 93.1 ± 0.7 b | 91.3 ± 0.5 c | 22.1 ± 0.2 c | 36.1 ± 0.3 c | 5.5 ± 0.0 c | 9.0 ± 0.0 c |
| 37 °C; pH 4 | 99.8 ± 0.0 d | 97.5 ± 0.1 e | 3.2 ± 0.1 a | 12.8 ± 0.2 a | 0.8 ± 0.0 a | 3.2 ± 0.0 a |
| 37 °C; pH 6 | 94.5 ± 0.4 c | 89.4 ± 0.3 b | 35.3 ± 0.1 d | 46.2 ± 0.2 d | 8.8 ± 0.0 d | 11.5 ± 0.0 d |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brzozowski, B.; Stasiewicz, K.; Ostolski, M.; Adamczak, M. Reducing Immunoreactivity of Gliadins and Coeliac-Toxic Peptides Using Peptidases from L. acidophilus 5e2 and A. niger. Catalysts 2020, 10, 923. https://doi.org/10.3390/catal10080923
Brzozowski B, Stasiewicz K, Ostolski M, Adamczak M. Reducing Immunoreactivity of Gliadins and Coeliac-Toxic Peptides Using Peptidases from L. acidophilus 5e2 and A. niger. Catalysts. 2020; 10(8):923. https://doi.org/10.3390/catal10080923
Chicago/Turabian StyleBrzozowski, Bartosz, Katarzyna Stasiewicz, Mateusz Ostolski, and Marek Adamczak. 2020. "Reducing Immunoreactivity of Gliadins and Coeliac-Toxic Peptides Using Peptidases from L. acidophilus 5e2 and A. niger" Catalysts 10, no. 8: 923. https://doi.org/10.3390/catal10080923
APA StyleBrzozowski, B., Stasiewicz, K., Ostolski, M., & Adamczak, M. (2020). Reducing Immunoreactivity of Gliadins and Coeliac-Toxic Peptides Using Peptidases from L. acidophilus 5e2 and A. niger. Catalysts, 10(8), 923. https://doi.org/10.3390/catal10080923