Discovery and Engineering of an Aldehyde Tolerant 2-deoxy-D-ribose 5-phosphate Aldolase (DERA) from Pectobacterium atrosepticum

 , ,
, ,  and
and
Abstract

1. Introduction
2. Results
2.1. Identification of PaDERA
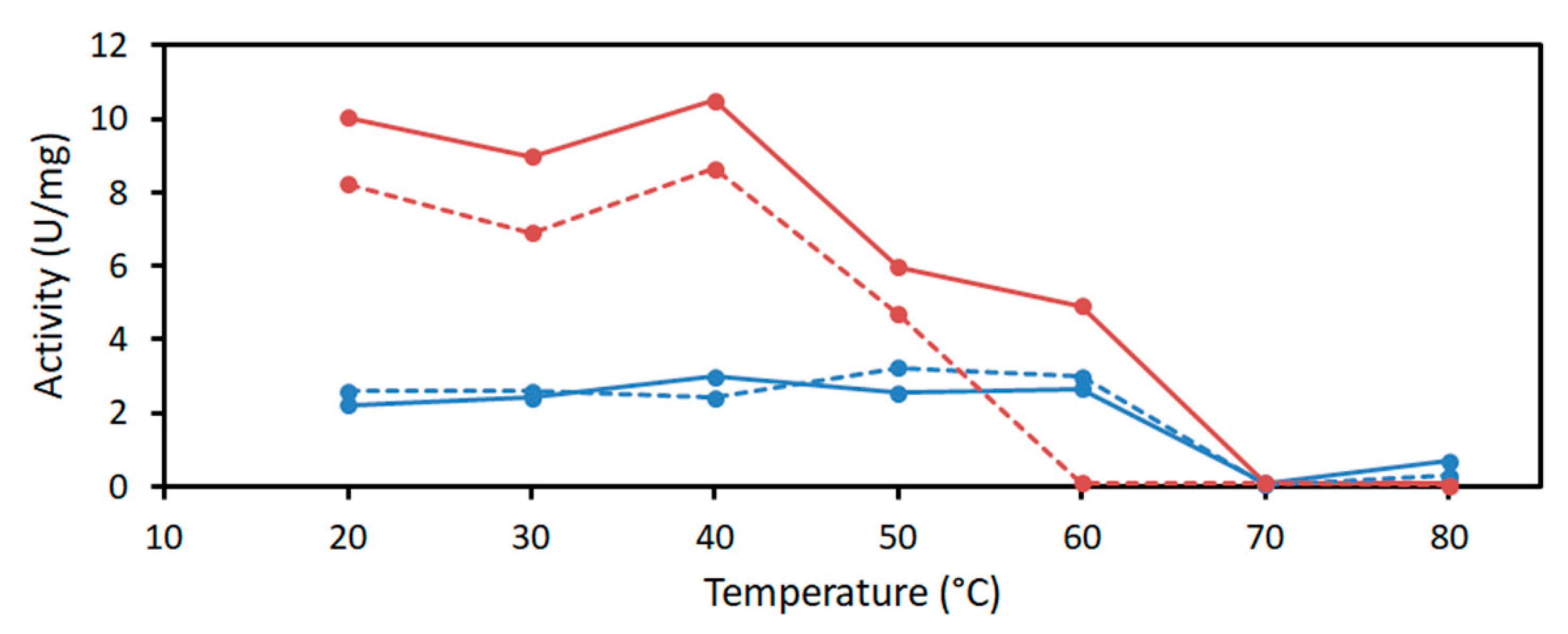
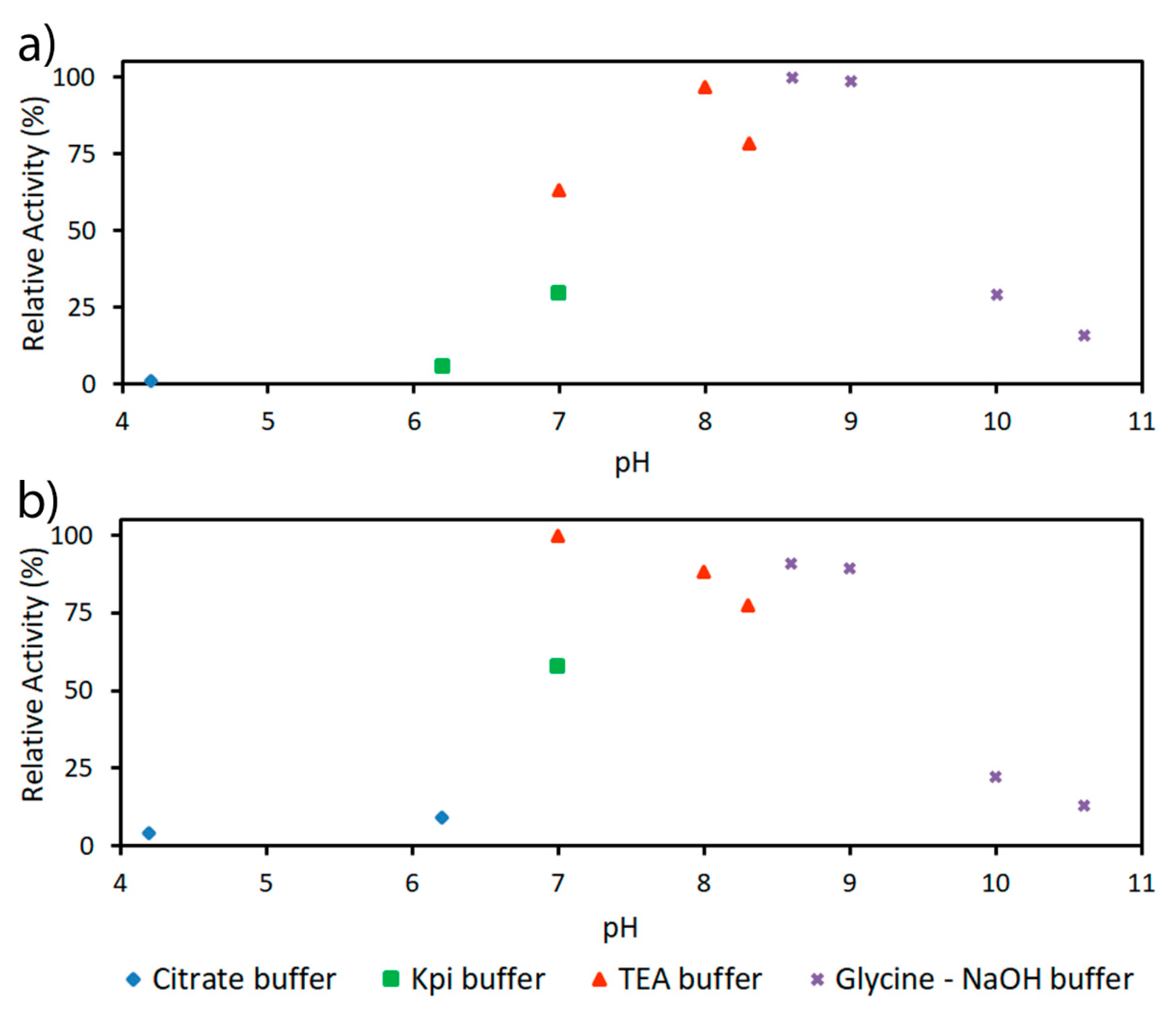
2.2. Characterization of PaDERA
2.3. Acetaldehyde Tolerance of DERA Enzymes
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Molecular Biology Methods
4.2.1. Cloning of DeoC P. atrosepticum
4.2.2. Site-Directed Mutagenesis of PaDERA
4.2.3. Protein Expression and Purification
4.3. Protein Analysis
4.3.1. Activity Assay
4.3.2. Determination of pH and Temperature Optima and Kinetic Parameters
4.3.3. Acetaldehyde Resistance of DERA
4.3.4. Sequential Aldol Reaction
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Salleron, L.; Magistrelli, G.; Mary, C.; Fischer, N.; Bairoch, A.; Lane, L. DERA is the human deoxy ribose phosphate aldolase and is involved in stress response. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 2913–2925. [Google Scholar] [CrossRef] [PubMed]
- Racker, E. Enzymatic synthesis of deoxy pentose phosphate. Nature 1951, 167, 408–409. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wong, C.-H. Aldolase-Catalyzed Asymmetric Synthesis of Novel Pyranose Synthons as a New Entry to Heterocycles and Epothilones. Angew. Chem. Int. Ed. 2002, 41, 1404–1407. [Google Scholar] [CrossRef]
- Barbas, C.F.; Wang, Y.F.; Wong, C.H. Deoxyribose-5-phosphate aldolase as a synthetic catalyst. J. Am. Chem. Soc. 1990, 112, 2013–2014. [Google Scholar] [CrossRef]
- Valino, A.L.; Iribarren, A.M.; Lewkowicz, E. New biocatalysts for one pot multistep enzymatic synthesis of pyrimidine nucleoside diphosphates from readily available reagents. J. Mol. Catal. B Enzym. 2015, 114, 58–64. [Google Scholar] [CrossRef]
- Valino, A.L.; Palazzolo, M.A.; Iribarren, A.M.; Lewkowicz, E. Selection of a new whole cell biocatalyst for the synthesis of 2-deoxyribose-5-phosphate. Appl. Biochem. Biotechnol. 2012, 166, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Gijsen, H.J.M.; Wong, C.-H. Unprecedented Asymmetric Aldol Reactions with Three Aldehyde Substrates Catalyzed by 2-Deoxyribose-5-phosphate Aldolase. J. Am. Chem. Soc. 1994, 116, 8422–8423. [Google Scholar] [CrossRef]
- Gijsen, H.J.M.; Wong, C.-H. Sequential one-pot aldol reactions catalyzed by 2-deoxyribose-5-phosphate aldolase and fructose-1,6-diphosphate aldolase. J. Am. Chem. Soc. 1995, 117, 2947–2948. [Google Scholar] [CrossRef]
- Gijsen, H.J.M.; Wong, C.-H. Sequential three- and four-substrate aldol reactions catalyzed by aldolases. J. Am. Chem. Soc. 1995, 117, 7585–7591. [Google Scholar] [CrossRef]
- Machajewski, D.T.; Wong, C.-H. The catalytic Asymmetric Aldol Reaction. Angew. Chem. Int. Ed. 2000, 39, 1352. [Google Scholar] [CrossRef]
- Xuri, W.; Jinpeng, J.; Yijun, C. Correlation between Intracellular Cofactor Concentrations and Biocatalytic Efficiency: Coexpression of Diketoreductase and Glucose Dehydrogenase for the Preparation of Chiral Diol for Statin Drugs. ACS Catal. 2011, 1, 1661. [Google Scholar]
- Haridas, M.; Abdelraheem, E.M.M.; Hanefeld, U. 2-Deoxy-D-ribose-5-phosphate aldolase (DERA): Applications and modifications. Appl. Microbiol. Biotechnol. 2018, 102, 9959–9971. [Google Scholar] [CrossRef] [PubMed]
- Gijsen, H.J.M.; Qiao, L.; Fitz, W.; Wong, C.-H. Recent Advances in the Chemoenzymatic Synthesis of Carbohydrates and Carbohydrate Mimetics. Chem. Rev. 1996, 96, 443–473. [Google Scholar] [CrossRef] [PubMed]
- Schürmann, M. An Aldolase for the synthesis of the statin side chain. In Industrial Enzymes Applications; Wiley-VCH: Weinheim, Germany, 2019; pp. 385–403. [Google Scholar]
- Huffman, M.A.; Fryszkowska, A.; Alvizo, O.; Borra-Garske, M.; Campos, K.R.; Canada, K.A.; Devine, P.A.; Duan, D.; Forstater, J.H.; Grosser, S.T.; et al. Design of an in vitro biocatalytic cascade for the manufacture of islatravir. Science 2019, 366, 1255–1259. [Google Scholar] [CrossRef] [PubMed]
- Dick, M.; Hartmann, R.; Weiergräber, O.H.; Bisterfeld, C.; Classen, T.; Schwarten, M.; Neudecker, P.; Willbold, D.; Pietruszka, J. Mechanism-based inhibition of an aldolase at high concentrations of its natural substrate acetaldehyde: Structural insights and protective strategies. Chem. Sci. 2016, 7, 4492–4502. [Google Scholar] [CrossRef] [PubMed]
- Abate, A.; Brenna, E.; Costantini, A.; Fuganti, C.; Gatti, F.G.; Malpezzi, L.; Serra, S. Enzymatic Approach to Enantiomerically Pure 5-Alken-2,4-diols and 4-Hydroxy-5-alken-2-ones: Application to the Synthesis of Chiral Synthons. J. Org. Chem. 2006, 71, 5228–5240. [Google Scholar] [CrossRef] [PubMed]
- Sakuraba, H.; Yoneda, K.; Yoshihara, K.; Satoh, K.; Kawakami, R.; Uto, Y.; Tsuge, H.; Takahashi, K.; Hori, H.; Ohshima, T. Sequential aldol condensation catalyzed by hyperthermophilic 2-deoxy-d-ribose-5-phosphate aldolase. Appl. Environ. Microbiol. 2007, 73, 7427–7434. [Google Scholar] [CrossRef] [PubMed]
- Kullartz, I.; Pietruszka, J. Cloning and characterisation of a new 2-deoxy-d-ribose-5-phosphate aldolase from Rhodococcus erythropolis. J. Biotechnol. 2012, 161, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Fei, H.; Xu, G.; Wu, J.-P.; Yang, L.-R. Improving the acetaldehyde tolerance of DERASEP by enhancing the rigidity of its protein structure. J. Mol. Catal. B Enzym. 2015, 116, 148–152. [Google Scholar] [CrossRef]
- Nicholas, P.C. Determination of fructose-1,6-diphosphate aldolase activity with glyceraldehyde-3-phosphate dehydrogenase and diformazan formation. Biochem. Soc. Trans. 1988, 16, 753–754. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | MW a (kDa) | KM (mM) | Vmax (U/mg) | kcat (s−1) | kcat/KM (mM−1s−1) |
|---|---|---|---|---|---|
| PaDERA | 28.858 | 0.22 ± 0.04 | 17.67 ± 1.01 | 8.50 ± 0.48 | 39.1 ± 9.4 |
| EcDERA | 28.556 | 0.06 ± 0.02 | 4.21 ± 0.21 | 2.00 ± 0.10 | 31.25 ± 11.7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haridas, M.; Bisterfeld, C.; Chen, L.M.; Marsden, S.R.; Tonin, F.; Médici, R.; Iribarren, A.; Lewkowicz, E.; Hagedoorn, P.-L.; Hanefeld, U.; et al. Discovery and Engineering of an Aldehyde Tolerant 2-deoxy-D-ribose 5-phosphate Aldolase (DERA) from Pectobacterium atrosepticum. Catalysts 2020, 10, 883. https://doi.org/10.3390/catal10080883
Haridas M, Bisterfeld C, Chen LM, Marsden SR, Tonin F, Médici R, Iribarren A, Lewkowicz E, Hagedoorn P-L, Hanefeld U, et al. Discovery and Engineering of an Aldehyde Tolerant 2-deoxy-D-ribose 5-phosphate Aldolase (DERA) from Pectobacterium atrosepticum. Catalysts. 2020; 10(8):883. https://doi.org/10.3390/catal10080883
Chicago/Turabian StyleHaridas, Meera, Carolin Bisterfeld, Le Min Chen, Stefan R. Marsden, Fabio Tonin, Rosario Médici, Adolfo Iribarren, Elizabeth Lewkowicz, Peter-Leon Hagedoorn, Ulf Hanefeld, and et al. 2020. "Discovery and Engineering of an Aldehyde Tolerant 2-deoxy-D-ribose 5-phosphate Aldolase (DERA) from Pectobacterium atrosepticum" Catalysts 10, no. 8: 883. https://doi.org/10.3390/catal10080883
APA StyleHaridas, M., Bisterfeld, C., Chen, L. M., Marsden, S. R., Tonin, F., Médici, R., Iribarren, A., Lewkowicz, E., Hagedoorn, P.-L., Hanefeld, U., & Abdelraheem, E. (2020). Discovery and Engineering of an Aldehyde Tolerant 2-deoxy-D-ribose 5-phosphate Aldolase (DERA) from Pectobacterium atrosepticum. Catalysts, 10(8), 883. https://doi.org/10.3390/catal10080883