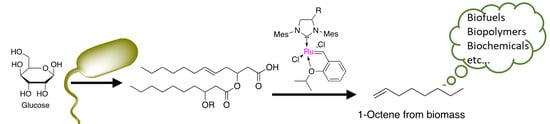
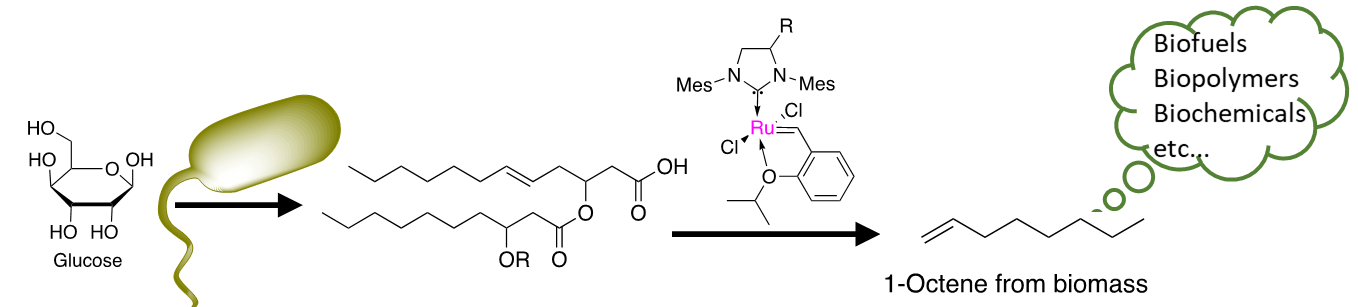
A Combined Bio-Chemical Synthesis Route for 1-Octene Sheds Light on Rhamnolipid Structure

 ,
,  ,
, 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
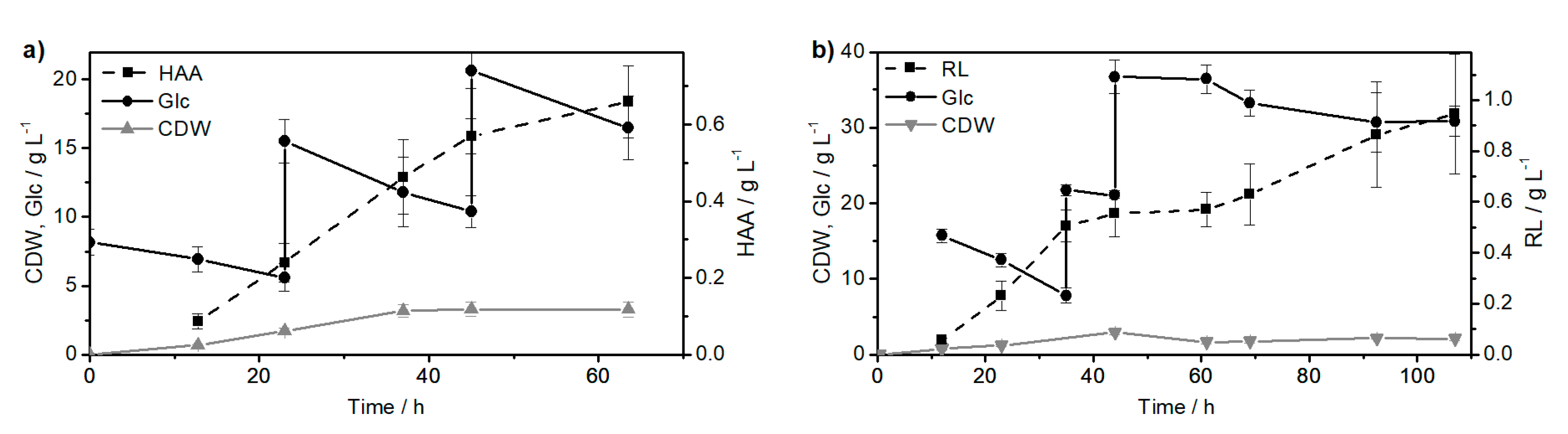
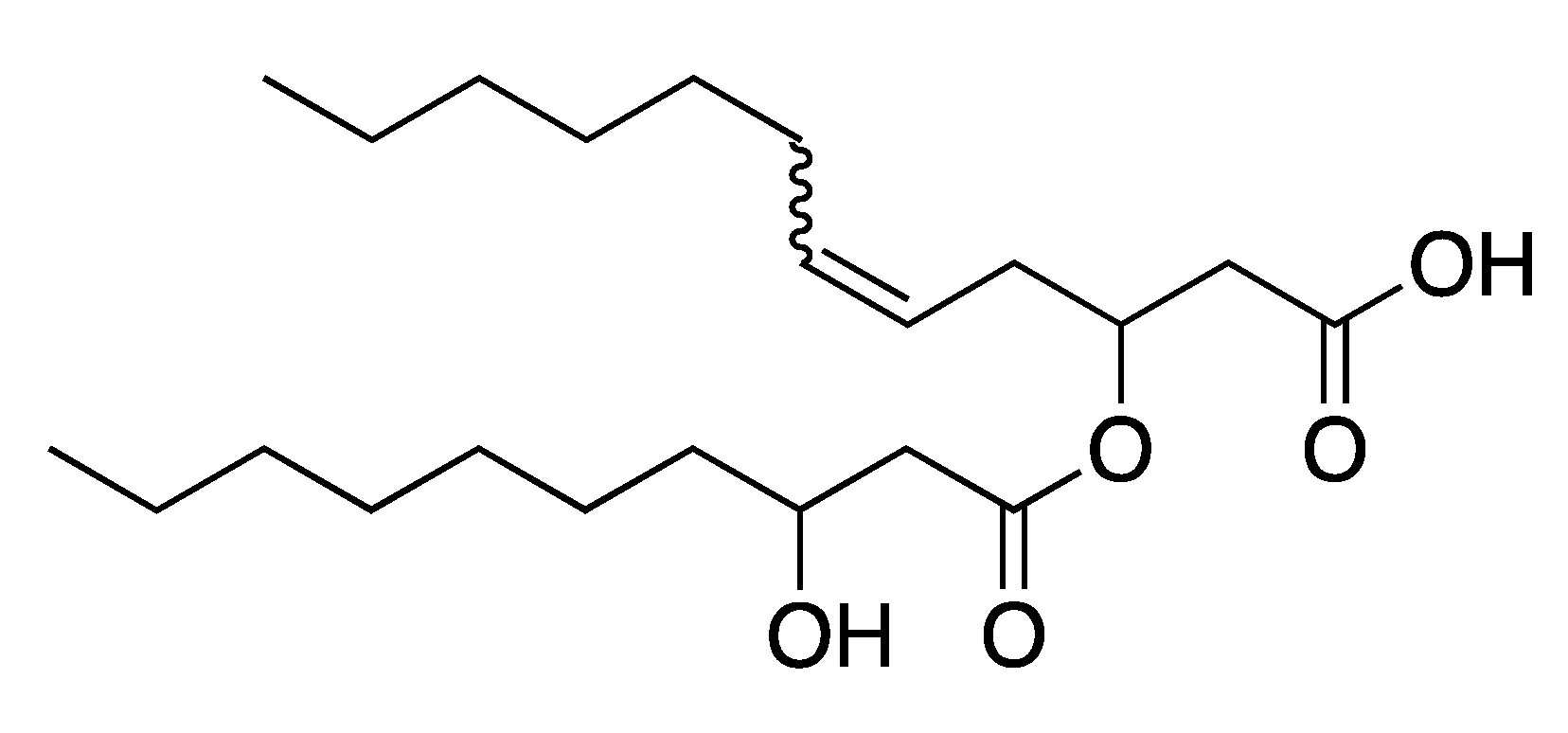
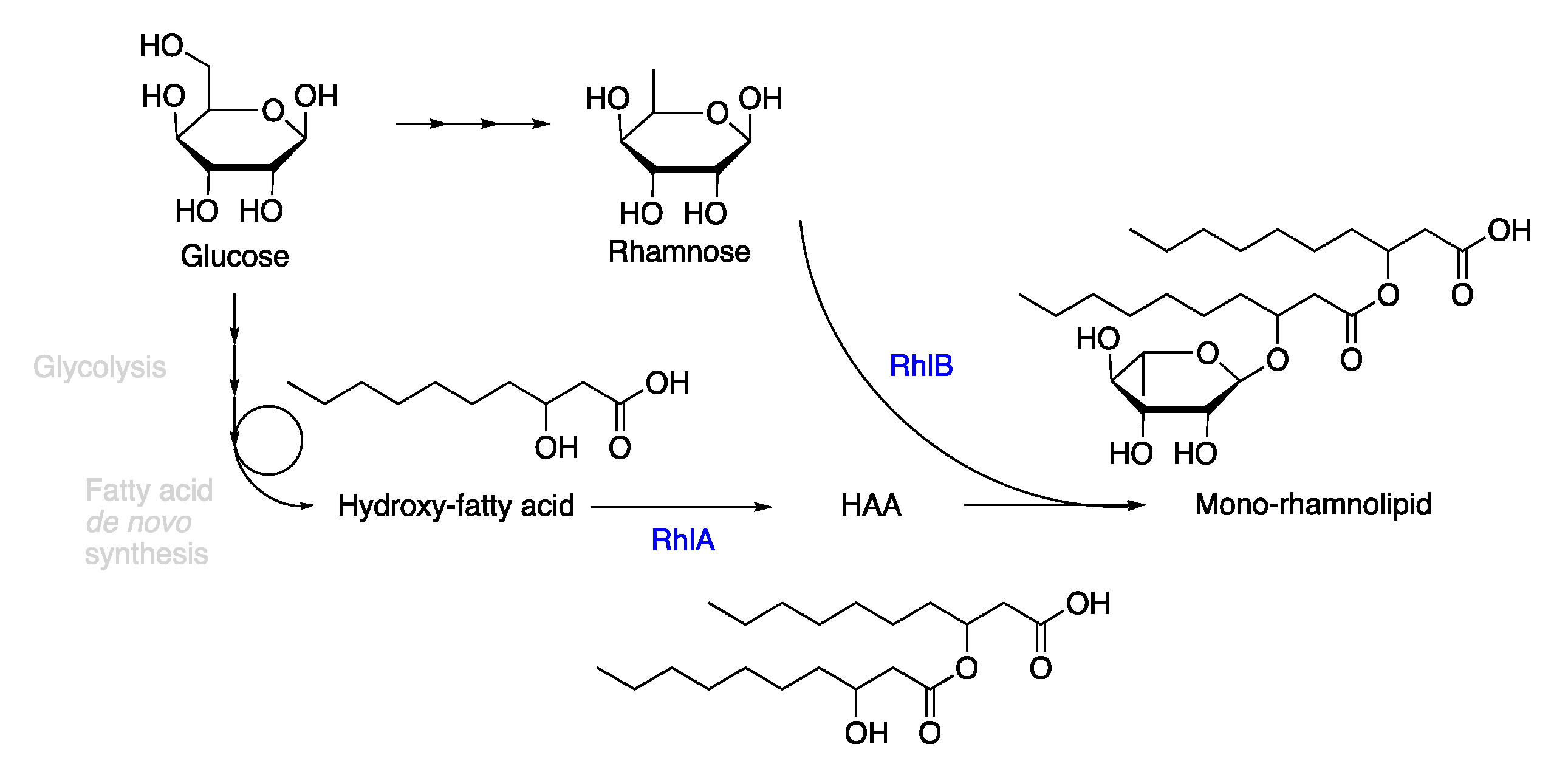
2.1. Synthesis of the Biological Intermediate
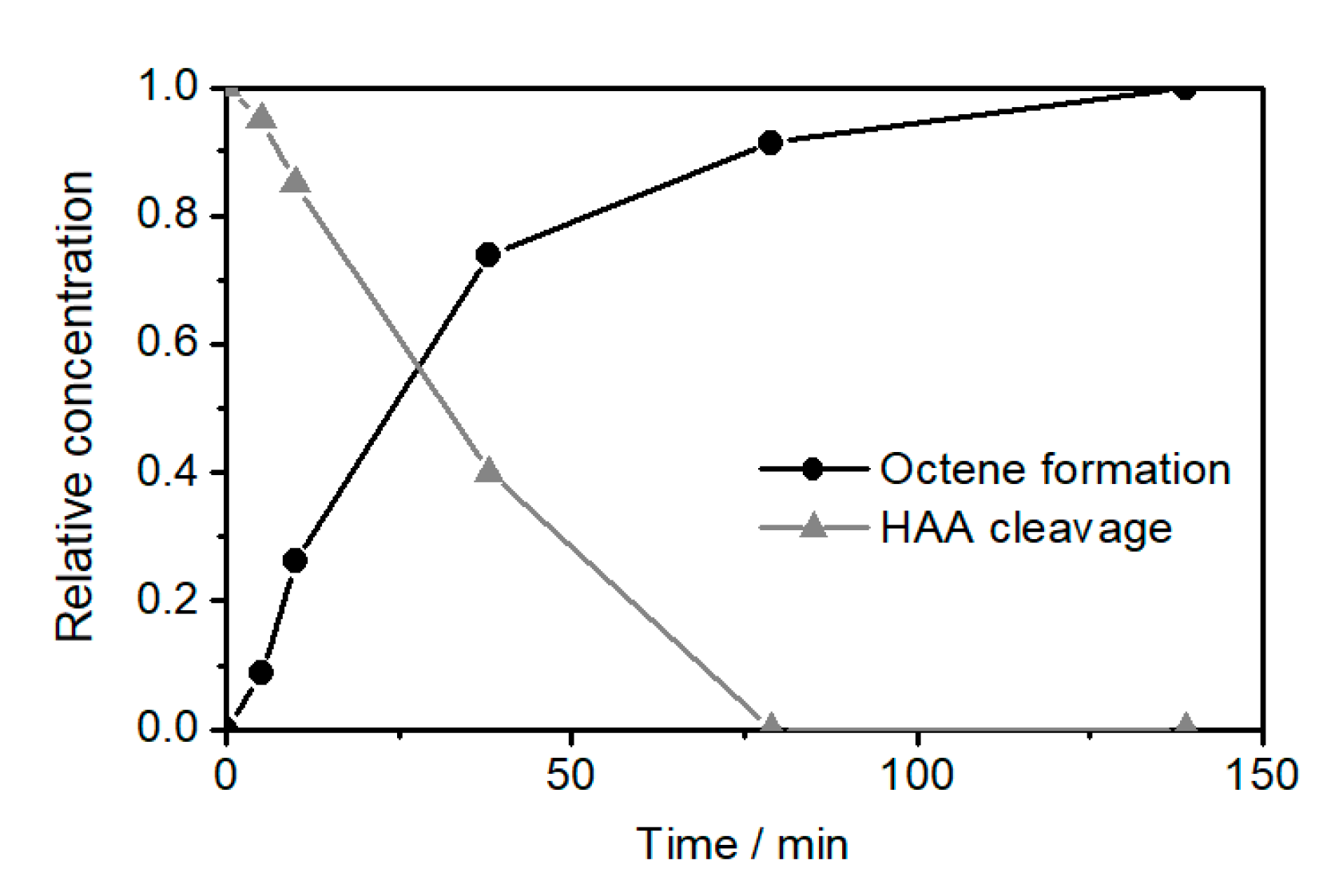
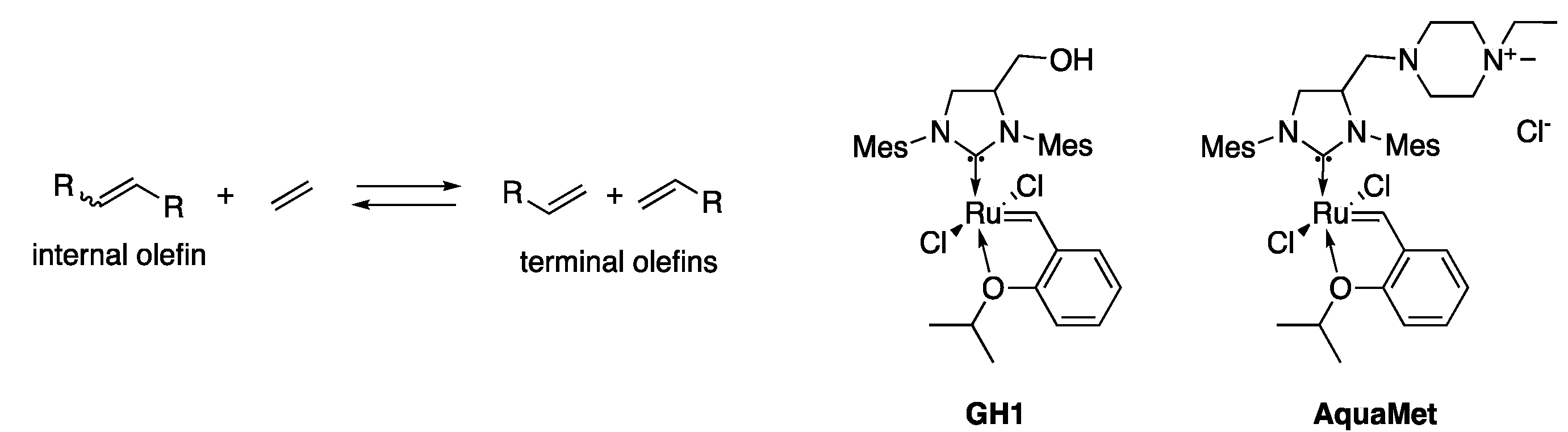
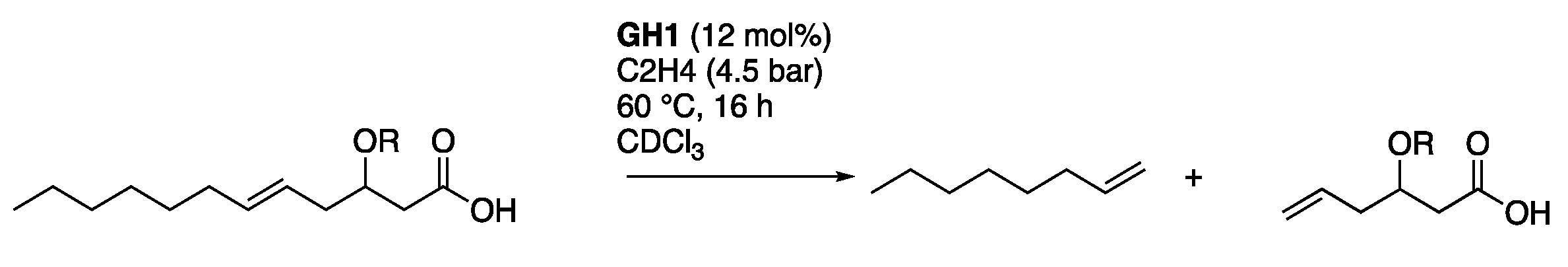
2.2. Ethenolysis of HAA/Rhamnolipids
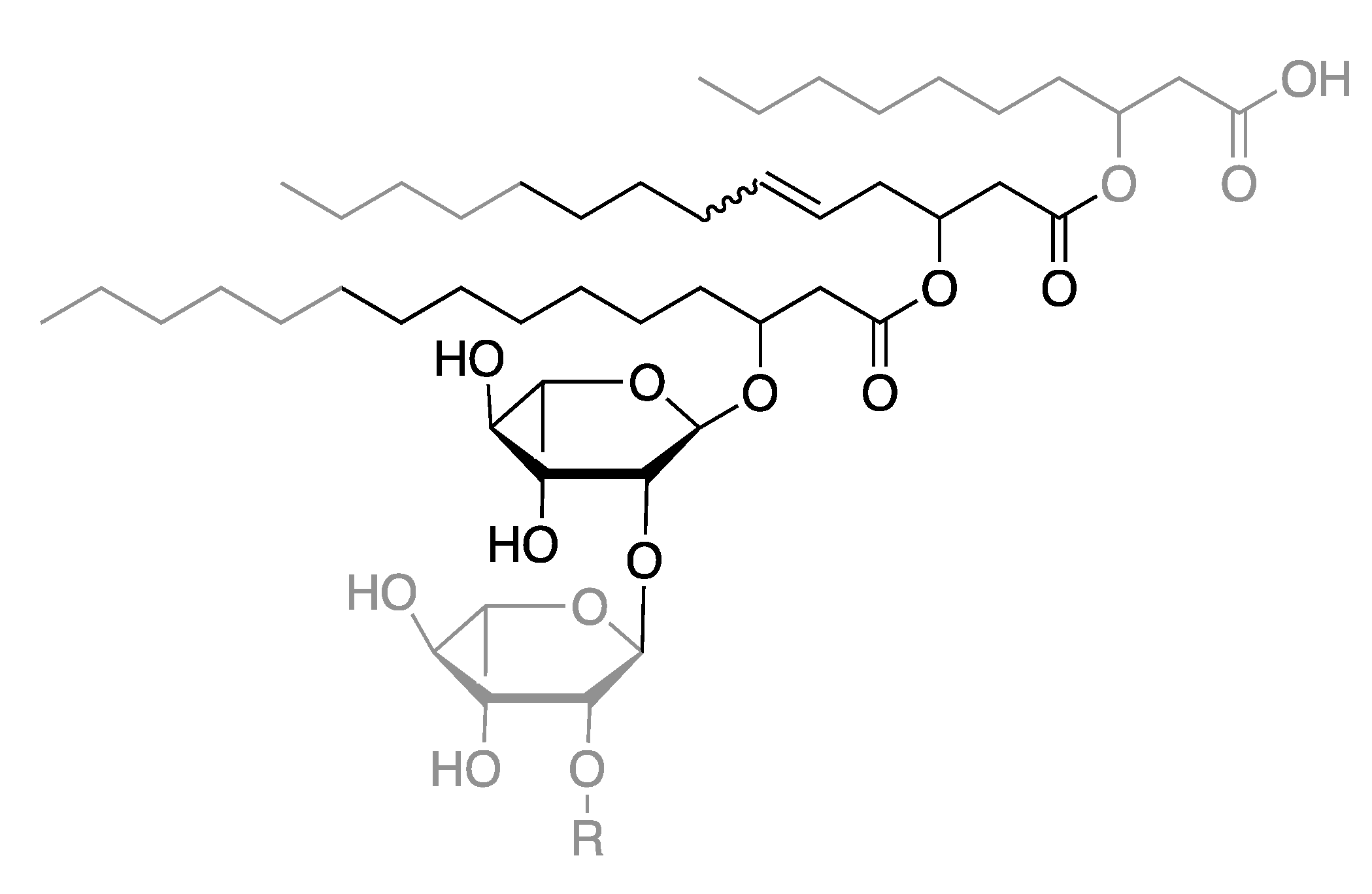
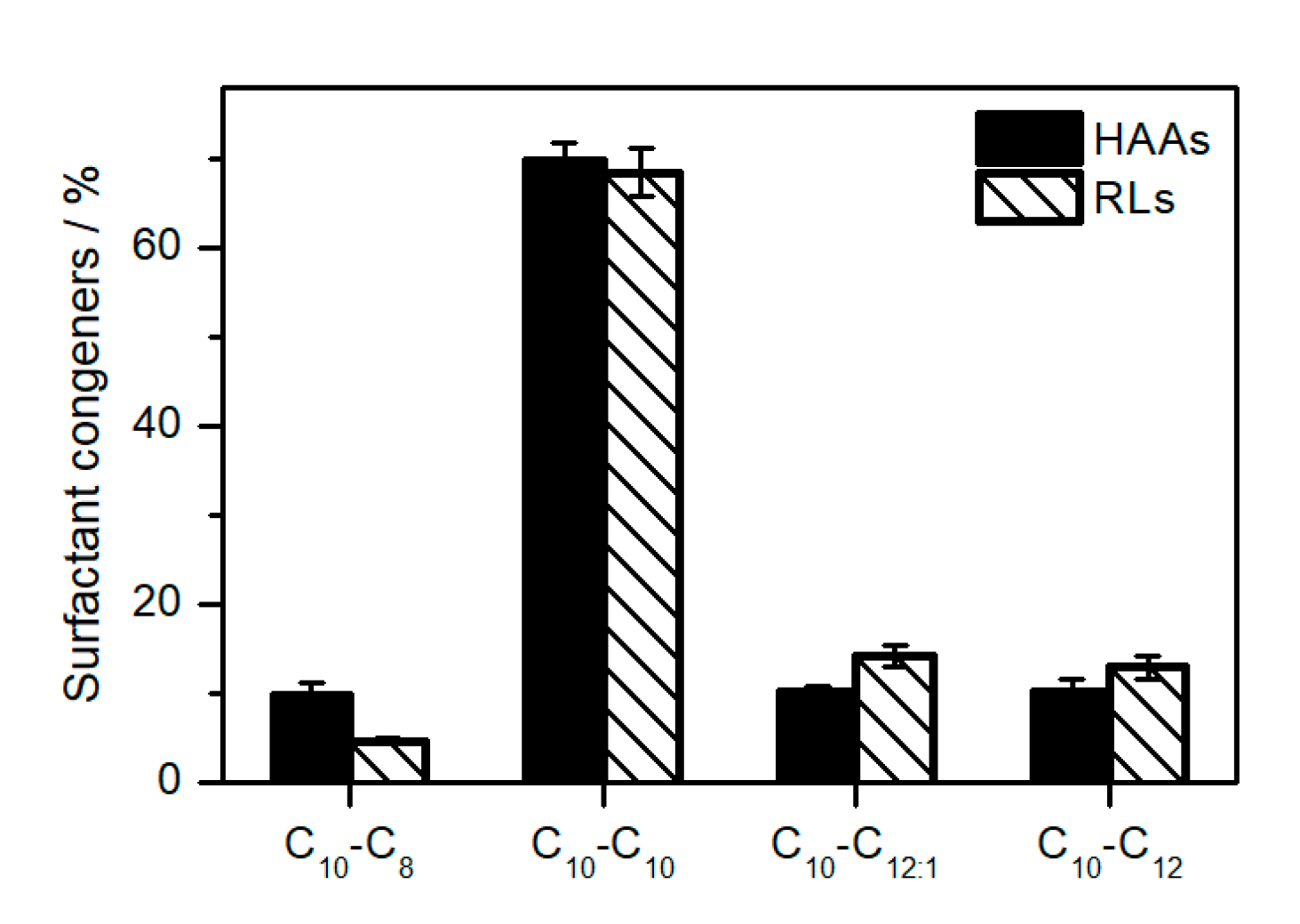
2.3. Elucidation of Rhamnolipid Structures
3. Experimental Section
3.1. Strains and Cultivation Conditions
3.2. Purification
3.3. Analytical Procedures
3.3.1. HPLC for Biosurfactant Quantification
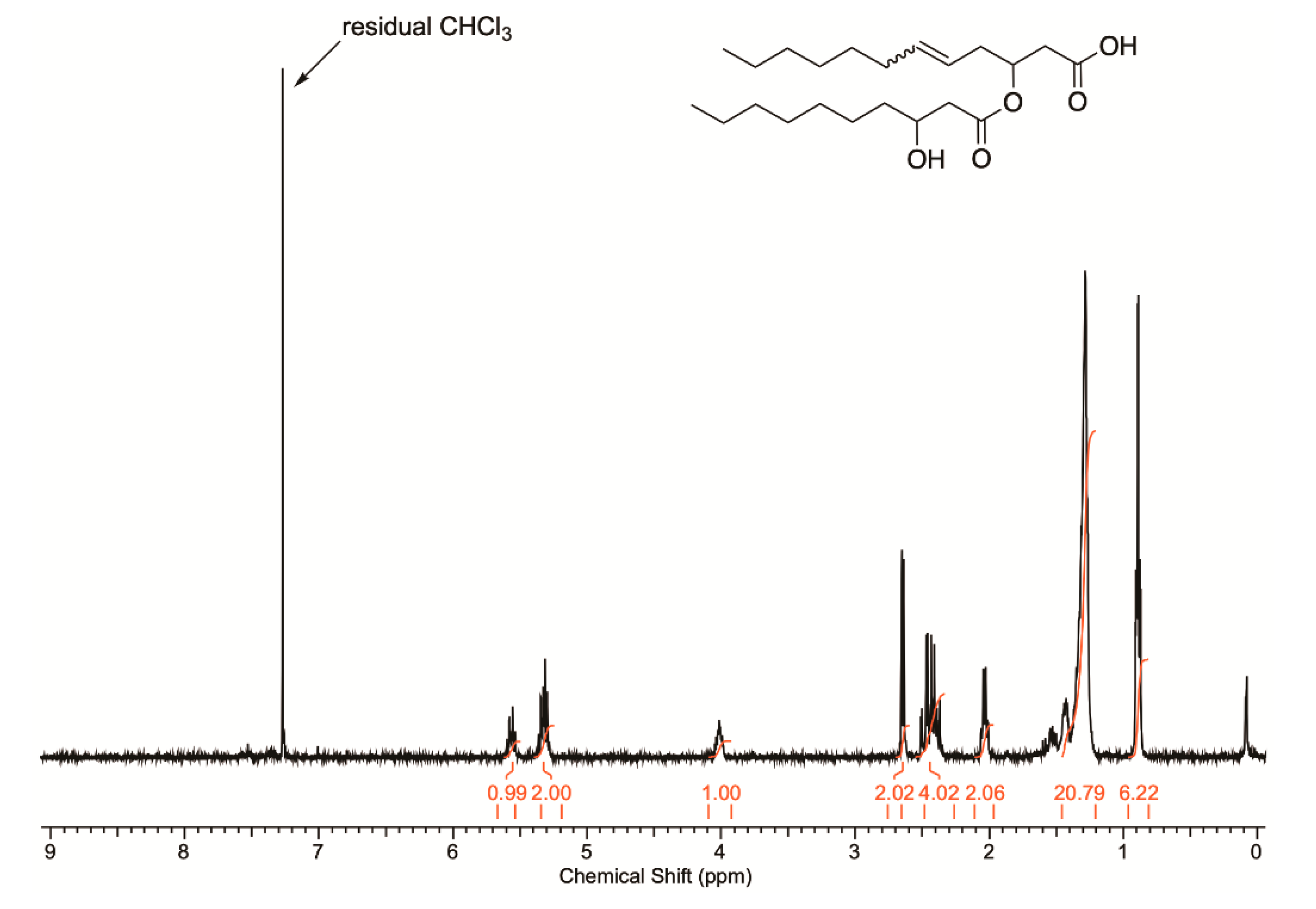
3.3.2. NMR Spectroscopy and GC MS Analysis
3.4. Ethenolysis in a Parr Autoclave
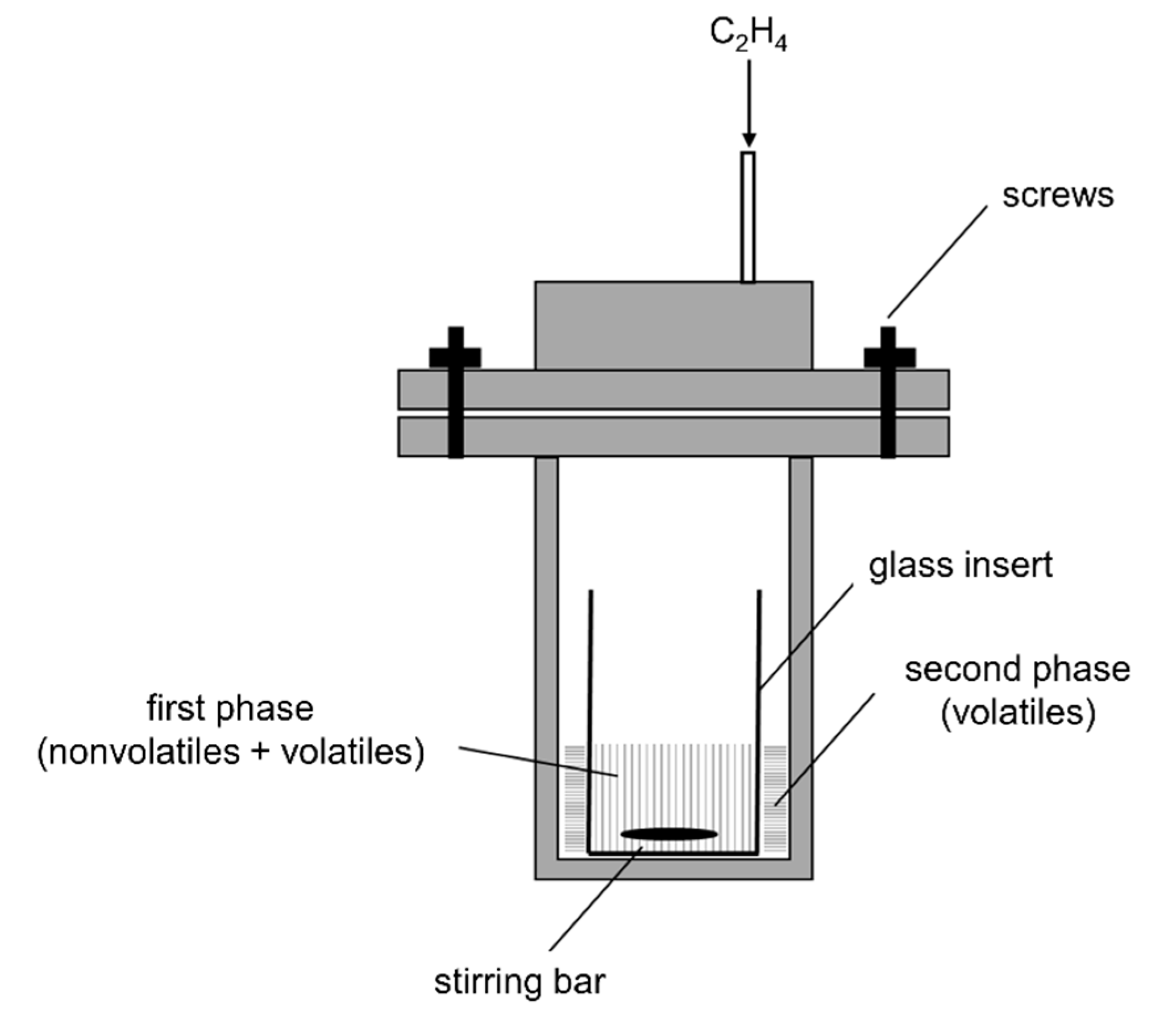
3.5. Ethenolysis in High-Pressure Norell NMR Tube
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bugge, M.M.; Hansen, T.; Klitkou, A. What Is the Bioeconomy? A Review of the Literature. Sustainability 2016, 8, 1–22. [Google Scholar] [CrossRef]
- Chen, Y.; Nielsen, J. Advances in metabolic pathway and strain engineering paving the way for sustainable production of chemical building blocks. Curr. Opin. Biotechnol. 2013, 24, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Soetaert, W.; Vandamme, E. The impact of industrial biotechnology. Biotechnol. J. 2006, 1, 756–769. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Murray, K. Metabolism of benzoate and the methylbenzoates by Pseudomonas putida (arvilla) mt-2: Evidence for the existence of a TOL plasmid. J. Bac. 1974, 120, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Bagdasarian, M.; Lurz, R.; Ruckert, B.; Franklin, F.C.H.; Bagdasarian, M.M.; Frey, J.; Timmis, K.N. Specific-purpose plasmid cloning vectors II. Broad host range, high copy number, RSF1010-derived vectors, and a host-vector system for gene cloning in Pseudomonas. Gene 1981, 16, 237–247. [Google Scholar] [CrossRef]
- Loeschcke, A.; Thies, S. Pseudomonas putida—A versatile host for the production of natural products. Appl. Microbiol. Biotechnol. 2015, 99, 6197–6214. [Google Scholar] [CrossRef]
- Wynands, B.; Lenzen, C.; Otto, M.; Koch, F.; Blank, L.M.; Wierckx, N. Metabolic engineering of Pseudomonas taiwanensis VLB120 with minimal genomic modifications for high-yield phenol production. Metab. Eng. 2018, 47, 121–133. [Google Scholar] [CrossRef]
- Tiso, T.; Sabelhaus, P.; Behrens, B.; Wittgens, A.; Rosenau, F.; Hayen, H.; Blank, L.M. Creating metabolic demand as an engineering strategy in Pseudomonas putida—Rhamnolipid synthesis as an example. Metab. Eng. Commun. 2016, 3, 234–244. [Google Scholar] [CrossRef]
- Tiso, T.; Zauter, R.; Tulke, H.; Leuchtle, B.; Li, W.-J.; Behrens, B.; Wittgens, A.; Rosenau, F.; Hayen, H.; Blank, L.M. Designer rhamnolipids by reduction of congener diversity: Production and characterization. Microb. Cell Fact. 2017, 16, 225. [Google Scholar] [CrossRef]
- Tiso, T.; Wierckx, N.J.P.; Blank, L.M. Non-pathogenic Pseudomonas as platform for industrial biocatalysis. In Industrial Biocatalysis; Grunwald, P., Ed.; Pan Stanford: Singapore, 2015; Volume 1, pp. 323–372. [Google Scholar]
- Wittgens, A.; Tiso, T.; Arndt, T.T.; Wenk, P.; Hemmerich, J.; Müller, C.; Wichmann, R.; Küpper, B.; Zwick, M.; Wilhelm, S.; et al. Growth independent rhamnolipid production from glucose using the non-pathogenic Pseudomonas putida KT2440. Microb. Cell Fact. 2011, 10, 80. [Google Scholar] [CrossRef]
- Lovaglio, R.B.; Dos Santos, F.J.; Jafelicci, M.J.; Contiero, J. Rhamnolipid emulsifying activity and emulsion stability: pH rules. Colloids. Surf. B. Biointerfaces 2011, 85, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Sarachat, T.; Pornsunthorntawee, O.; Chavadej, S.; Rujiravanit, R. Purification and concentration of a rhamnolipid biosurfactant produced by Pseudomonas aeruginosa SP4 using foam fractionation. Bioresour. Technol. 2010, 101, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.; Wullbrandt, D. Rhamnose lipids—Biosynthesis, microbial production and application potential. Appl. Microbiol. Biotechnol. 1999, 51, 22–32. [Google Scholar] [CrossRef]
- Abdel-Mawgoud, A.M.; Lépine, F.; Déziel, E. Rhamnolipids: Diversity of structures, microbial origins and roles. Appl. Microbiol. Biotechnol. 2010, 86, 1323–1336. [Google Scholar] [CrossRef] [PubMed]
- Zähringer, U.; Rettenmaier, H.; Moll, H.; Senchenkova, S.N.; Knirel, Y.A. Structure of a new 6-deoxy-alpha-D-talan from Burkholderia (Pseudomonas) plantarii strain DSM 6535, which is different from the O-chain of the lipopolysaccharide. Carbohydr. Res. 1997, 300, 143–151. [Google Scholar] [CrossRef]
- Jadhav, M.; Kalme, S.; Tamboli, D.; Govindwar, S. Rhamnolipid from Pseudomonas desmolyticum NCIM-2112 and its role in the degradation of Brown 3REL. J. Basic. Microbiol. 2011, 51, 1–12. [Google Scholar] [CrossRef]
- Rezanka, T.; Siristova, L.; Sigler, K. Rhamnolipid-producing thermophilic bacteria of species Thermus and Meiothermus. Extremophiles 2011, 15, 697–709. [Google Scholar] [CrossRef]
- Zhu, K.; Rock, C.O. RhlA converts beta-hydroxyacyl-acyl carrier protein intermediates in fatty acid synthesis to the beta-hydroxydecanoyl-beta-hydroxydecanoate component of rhamnolipids in Pseudomonas aeruginosa. J. Bac. 2008, 190, 3147–3154. [Google Scholar] [CrossRef]
- Germer, A.; Tiso, T.; Müller, C.; Behrens, B.; Vosse, C.; Scholz, K.; Froning, M.; Hayen, H.; Blank, L.M. Exploiting the natural diversity of the acyltransferase RhlA for the synthesis of the rhamnolipid precursor 3-(3-hydroxyalkanoyloxy)alkanoic acid. Appl. Environ. Microbiol. 2020, 86, e02317-02319. [Google Scholar] [CrossRef]
- Grubbs, R.H. Handbook of Metathesis; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]
- Grela, K. Olefin Metathesis: Theory and Practice; Wiley: Hoboken, NJ, USA, 2014; pp. xiii, 592. [Google Scholar]
- Mol, J.C. Application of olefin metathesis in oleochemistry: An example of green chemistry. Green Chem. 2002, 4, 5–13. [Google Scholar] [CrossRef]
- Thomas, R.M.; Keitz, B.K.; Champagne, T.M.; Grubbs, R.H. Highly selective ruthenium metathesis catalysts for ethenolysis. J. Am. Chem. Soc. 2011, 133, 7490–7496. [Google Scholar] [CrossRef] [PubMed]
- Burdett, K.A.; Harris, L.D.; Margl, P.; Maughon, B.R.; Mokhtar-Zadeh, T.; Saucier, P.C.; Wasserman, E.P. Renewable Monomer Feedstocks via Olefin Metathesis: Fundamental Mechanistic Studies of Methyl Oleate Ethenolysis with the First-Generation Grubbs Catalyst. Organometallics 2004, 23, 2027–2047. [Google Scholar] [CrossRef]
- Julis, J.; Bartlett, S.A.; Baader, S.; Beresford, N.; Routledge, E.J.; Cazin, C.S.J.; Cole-Hamilton, D.J. Selective ethenolysis and oestrogenicity of compounds from cashew nut shell liquid. Green Chem. 2014, 16, 2846–2856. [Google Scholar] [CrossRef]
- Herbert, M.B.; Marx, V.M.; Pederson, R.L.; Grubbs, R.H. Concise Syntheses of Insect Pheromones Using Z-Selective Cross Metathesis. Angew. Chem. Int. Ed. 2013, 52, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Marx, V.M.; Sullivan, A.H.; Melaimi, M.; Virgil, S.C.; Keitz, B.K.; Weinberger, D.S.; Bertrand, G.; Grubbs, R.H. Cyclic Alkyl Amino Carbene (CAAC) Ruthenium Complexes as Remarkably Active Catalysts for Ethenolysis. Angew. Chem. Int. Ed. 2015, 54, 1919–1923. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.W.; Sargeant, L.A.; Whiffin, F.M.; Santomauro, F.; Kaloudis, D.; Mozzanega, P.; Bannister, C.D.; Baena, S.; Chuck, C.J. Cross-Metathesis of Microbial Oils for the Production of Advanced Biofuels and Chemicals. ACS Sustain. Chem. Eng. 2015, 3, 1526–1535. [Google Scholar] [CrossRef]
- Marinescu, S.C.; Schrock, R.R.; Müller, P.; Hoveyda, A.H. Ethenolysis Reactions Catalyzed by Imido Alkylidene Monoaryloxide Monopyrrolide (MAP) Complexes of Molybdenum. J. Am. Chem. Soc. 2009, 131, 10840–10841. [Google Scholar] [CrossRef]
- Patel, J.; Elaridi, J.; Jackson, W.R.; Robinson, A.J.; Serelis, A.K.; Such, C. Cross-metathesis of unsaturated natural oils with 2-butene. High conversion and productive catalyst turnovers. Chem. Commun. (Cambridge, U.K.) 2005, 10, 5546–5547. [Google Scholar] [CrossRef]
- van der Klis, F.; Le Nôtre, J.; Blaauw, R.; van Haveren, J.; van Es, D.S. Renewable linear alpha olefins by selective ethenolysis of decarboxylated unsaturated fatty acids. Eur. J. Lipid Sci. Technol. 2012, 114, 911–918. [Google Scholar] [CrossRef]
- Nickel, A.; Ung, T.; Mkrtumyan, G.; Uy, J.; Lee, C.W.; Stoianova, D.; Papazian, J.; Wei, W.H.; Mallari, A.; Schrodi, Y.; et al. A Highly Efficient Olefin Metathesis Process for the Synthesis of Terminal Alkenes from Fatty Acid Esters. Top. Catal. 2012, 55, 518–523. [Google Scholar] [CrossRef]
- Zhang, J.; Song, S.; Wang, X.; Jiao, J.; Shi, M. Ruthenium-catalyzed olefin metathesis accelerated by the steric effect of the backbone substituent in cyclic (alkyl)(amino) carbenes. Chem. Commun. (Cambridge, U.K.) 2013, 49, 9491–9493. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.R.; Ung, T.; Mkrtumyan, G.; Bertrand, G.; Grubbs, R.H.; Schrodi, Y. Kinetic selectivity of olefin metathesis catalysts bearing cyclic (alkyl)(amino)carbenes. Organometallics 2008, 27, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Spekreijse, J.; Sanders, J.P.M.; Bitter, J.H.; Scott, E.L. The Future of Ethenolysis in Biobased Chemistry. ChemSusChem 2017, 10, 470–482. [Google Scholar] [CrossRef] [PubMed]
- Behr, A.; Toepell, S.; Harmuth, S. Cross-metathesis of methyl 10-undecenoate with dimethyl maleate: An efficient protocol with nearly quantitative yields. RSC Adv. 2014, 4, 16320–16326. [Google Scholar] [CrossRef]
- Fang, H.; Zhao, C.; Kong, Q.; Zou, Z.; Chen, N. Comprehensive utilization and conversion of lignocellulosic biomass for the production of long chain α,ω-dicarboxylic acids. Energy 2016, 116, 177–189. [Google Scholar] [CrossRef]
- Balcar, H.; Žilková, N.; Kubů, M.; Polášek, M.; Zedník, J. Metathesis of cardanol over ammonium tagged Hoveyda-Grubbs type catalyst supported on SBA-15. Catal. Today 2018, 304, 127–134. [Google Scholar] [CrossRef]
- Butilkov, D.; Lemcoff, N.G. Jojoba oil olefin metathesis: A valuable source for bio-renewable materials. Green Chem. 2014, 16, 4728–4733. [Google Scholar] [CrossRef]
- Ríos-Lombardía, N.; García-Álvarez, J.; González-Sabín, J. One-Pot Combination of Metal- and Bio-Catalysis in Water for the Synthesis of Chiral Molecules. Catalysts 2018, 8, 75. [Google Scholar] [CrossRef]
- Rudroff, F.; Mihovilovic, M.D.; Gröger, H.; Snajdrova, R.; Iding, H.; Bornscheuer, U.T. Opportunities and challenges for combining chemo- and biocatalysis. Nat. Catal. 2018, 1, 12–22. [Google Scholar] [CrossRef]
- Schmidt, S.; Castiglione, K.; Kourist, R. Overcoming the Incompatibility Challenge in Chemoenzymatic and Multi-Catalytic Cascade Reactions. Chem. Eur. J. 2018, 24, 1755–1768. [Google Scholar] [CrossRef]
- Gröger, H.; Hummel, W. Combining the ‘two worlds’ of chemocatalysis and biocatalysis towards multi-step one-pot processes in aqueous media. Curr. Opin. Chem. Biol. 2014, 19, 171–179. [Google Scholar] [CrossRef]
- Wilson, Y.M.; Dürrenberger, M.; Nogueira, E.S.; Ward, T.R. Neutralizing the Detrimental Effect of Glutathione on Precious Metal Catalysts. J. Am. Chem. Soc. 2014, 136, 8928–8932. [Google Scholar] [CrossRef]
- Gómez Baraibar, Á.; Reichert, D.; Mügge, C.; Seger, S.; Gröger, H.; Kourist, R. A One-Pot Cascade Reaction Combining an Encapsulated Decarboxylase with a Metathesis Catalyst for the Synthesis of Bio-Based Antioxidants. Angew. Chem. Int. Ed. 2016, 55, 14823–14827. [Google Scholar] [CrossRef] [PubMed]
- Bojarra, S.; Reichert, D.; Grote, M.; Baraibar, Á.G.; Dennig, A.; Nidetzky, B.; Mügge, C.; Kourist, R. Bio-based α,ω-Functionalized Hydrocarbons from Multi-step Reaction Sequences with Bio- and Metallo-catalysts Based on the Fatty Acid Decarboxylase OleTJE. ChemCatChem 2018, 10, 1192–1201. [Google Scholar] [CrossRef]
- Jeschek, M.; Reuter, R.; Heinisch, T.; Trindler, C.; Klehr, J.; Panke, S.; Ward, T.R. Directed evolution of artificial metalloenzymes for in vivo metathesis. Nature 2016, 537, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Sauer, D.F.; Gotzen, S.; Okuda, J. Metatheases: Artificial metalloproteins for olefin metathesis. Org. Biomol. Chem. 2016, 14, 9174–9183. [Google Scholar] [CrossRef] [PubMed]
- Sauer, D.F.; Schiffels, J.; Hayashi, T.; Schwaneberg, U.; Okuda, J. Olefin metathesis catalysts embedded in beta-barrel proteins: Creating artificial metalloproteins for olefin metathesis. Beilstein J. Org. Chem. 2018, 14, 2861–2871. [Google Scholar] [CrossRef]
- Mertens, M.A.S.; Sauer, D.F.; Markel, U.; Schiffels, J.; Okuda, J.; Schwaneberg, U. Chemoenzymatic cascade for stilbene production from cinnamic acid catalyzed by ferulic acid decarboxylase and an artificial metathease. Catal. Sci. Tech. 2019, 9, 5572–5576. [Google Scholar] [CrossRef]
- Sauer, D.F.; Qu, Y.; Mertens, M.A.S.; Schiffels, J.; Polen, T.; Schwaneberg, U.; Okuda, J. Biohybrid catalysts for sequential one-pot reactions based on an engineered transmembrane protein. Catal. Sci. Tech. 2019, 9, 942–946. [Google Scholar] [CrossRef]
- Katharina, T.; Miriam, S.; Jürgen, S.; Harald, G. Combination of Olefin Metathesis and Enzymatic Ester Hydrolysis in Aqueous Media in a One-Pot Synthesis. Adv. Synth. Catal. 2011, 353, 2363–2367. [Google Scholar] [CrossRef]
- Wu, S.; Zhou, Y.; Gerngross, D.; Jeschek, M.; Ward, T.R. Chemo-enzymatic cascades to produce cycloalkenes from bio-based resources. Nat. Commun. 2019, 10, 5060. [Google Scholar] [CrossRef] [PubMed]
- Ríos-Lombardía, N.; Rodríguez-Álvarez, M.J.; Morís, F.; Kourist, R.; Comino, N.; López-Gallego, F.; González-Sabín, J.; García-Álvarez, J. DESign of Sustainable One-Pot Chemoenzymatic Organic Transformations in Deep Eutectic Solvents for the Synthesis of 1,2-Disubstituted Aromatic Olefins. Front. Chem. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Aresta, M.; Dibenedetto, A.; Dumeignil, F. Biorefinery: From Biomass to Chemicals and Fuels; [Walter] de Gruyter: Berlin, Germany, 2012. [Google Scholar]
- Grubbs, R.H. Olefin metathesis. Tetrahedron 2004, 60, 7117–7140. [Google Scholar] [CrossRef]
- Keim, W. Oligomerization of Ethylene to α-Olefins: Discovery and Development of the Shell Higher Olefin Process (SHOP). Angew. Chem. Int. Ed. 2013, 52, 12492–12496. [Google Scholar] [CrossRef] [PubMed]
- Bollmann, A.; Blann, K.; Dixon, J.T.; Hess, F.M.; Killian, E.; Maumela, H.; McGuinness, D.S.; Morgan, D.H.; Neveling, A.; Otto, S.; et al. Ethylene Tetramerization: A New Route to Produce 1-Octene in Exceptionally High Selectivities. J. Am. Chem. Soc. 2004, 126, 14712–14713. [Google Scholar] [CrossRef]
- van Leeuwen, P.W.N.M.; Clément, N.D.; Tschan, M.J.L. New processes for the selective production of 1-octene. Coord. Chem. Rev. 2011, 255, 1499–1517. [Google Scholar] [CrossRef]
- Vasile, C. Handbook of Polyolefins, 2nd ed.; Marcel Dekker: New York, NY, USA, 2000; pp. xiii, 1014. [Google Scholar]
- Börner, A.; Franke, R. Hydroformylation: Fundamentals, Processes, and Applications in Organic Synthesis; Börner, A., Franke, R., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2016; chapter 4; pp. 285–378. [Google Scholar] [CrossRef]
- Bator, I.; Wittgens, A.; Rosenau, F.; Tiso, T.; Blank, L.M. Comparison of three xylose pathways in Pseudomonas putida KT2440 for the synthesis of valuable products. Front. Bioeng. Biotechnol. 2020, 7, 480. [Google Scholar] [CrossRef]
- Tomasek, J.; Schatz, J. Olefin metathesis in aqueous media. Green Chem. 2013, 15, 2317–2338. [Google Scholar] [CrossRef]
- Cronan, J.E.; Thomas, J. Bacterial fatty acid synthesis and its relationships with polyketide synthetic pathways. Method. Enzymol. 2009, 459, 395–433. [Google Scholar] [CrossRef]
- Behrens, B.; Engelen, J.; Tiso, T.; Blank, L.M.; Hayen, H. Characterization of rhamnolipids by liquid chromatography/mass spectrometry after solid-phase extraction. Anal. Bioanal. Chem. 2016, 408, 2505–2514. [Google Scholar] [CrossRef]
- Rendell, N.B.; Taylor, G.W.; Somerville, M.; Todd, H.; Wilson, R.; Cole, P.J. Characterisation of Pseudomonas rhamnolipids. Biochim. Biophys. Acta 1990, 1045, 189–193. [Google Scholar] [CrossRef]
- Lebron-Paler, A. Solution and Interfacial Characterization of Rhamnolipid Biosurfactant from P. aeruginosa ATCC 9027; The University of Arizona: Tucson, AZ, USA, 2008. [Google Scholar]
- Behrens, B.; Helmer, P.O.; Tiso, T.; Blank, L.M.; Hayen, H. Rhamnolipid biosurfactant analysis using online turbulent flow chromatography-liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2016, 1465, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Goldfine, H. Membrane Lipid Biogenesis. In Biogenesis of Fatty Acids, Lipids and Membranes; Geiger, O., Ed.; Springer Nature Switzerland AG: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- Nelson, K.E.; Weinel, C.; Paulsen, I.T.; Dodson, R.J.; Hilbert, H.; Martins dos Santos, V.A.P.; Fouts, D.E.; Gill, S.R.; Pop, M.; Holmes, M.; et al. Complete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440. Environ. Microbiol. 2002, 4, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Köhler, K.A.K.; Ruckert, C.; Schatschneider, S.; Vorholter, F.J.; Szczepanowski, R.; Blank, L.M.; Niehaus, K.; Goesmann, A.; Puhler, A.; Kalinowski, J.; et al. Complete genome sequence of Pseudomonas sp strain VLB120 a solvent tolerant, styrene degrading bacterium, isolated from forest soil. J. Biotechnol. 2013, 168, 729–730. [Google Scholar] [CrossRef] [PubMed]
- Panke, S.; Witholt, B.; Schmid, A.; Wubbolts, M.G. Towards a biocatalyst for (S)-styrene oxide production: Characterization of the styrene degradation pathway of Pseudomonas sp. strain VLB120. Appl. Environ. Microbiol. 1998, 64, 2032–2043. [Google Scholar] [CrossRef]
- Bertani, G. Studies on lysogenesis I.: The mode of phage liberation by lysogenic Escherichia coli. J. Bac. 1951, 62, 293–300. [Google Scholar] [CrossRef]
- Silva-Rocha, R.; Martinez-Garcia, E.; Calles, B.; Chavarria, M.; Arce-Rodriguez, A.; de las Heras, A.; Paez-Espino, A.D.; Durante-Rodriguez, G.; Kim, J.; Nikel, P.I.; et al. The Standard European Vector Architecture (SEVA): A coherent platform for the analysis and deployment of complex prokaryotic phenotypes. Nucleic Acids Res. 2013, 41, D666–D675. [Google Scholar] [CrossRef]
- Kovach, M.E.; Elzer, P.H.; Hill, D.S.; Robertson, G.T.; Farris, M.A.; Roop II, R.M.; Peterson, K.M. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 1995, 166, 175–176. [Google Scholar] [CrossRef]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- Philippart, F.; Arlt, M.; Gotzen, S.; Tenne, S.-J.; Bocola, M.; Chen, H.-H.; Zhu, L.; Schwaneberg, U.; Okuda, J. A Hybrid Ring-Opening Metathesis Polymerization Catalyst Based on an Engineered Variant of the β-Barrel Protein FhuA. Chem. Eur. J. 2013, 19, 13865–13871. [Google Scholar] [CrossRef]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tiso, T.; Sauer, D.F.; Beckerle, K.; Blesken, C.C.; Okuda, J.; Blank, L.M. A Combined Bio-Chemical Synthesis Route for 1-Octene Sheds Light on Rhamnolipid Structure. Catalysts 2020, 10, 874. https://doi.org/10.3390/catal10080874
Tiso T, Sauer DF, Beckerle K, Blesken CC, Okuda J, Blank LM. A Combined Bio-Chemical Synthesis Route for 1-Octene Sheds Light on Rhamnolipid Structure. Catalysts. 2020; 10(8):874. https://doi.org/10.3390/catal10080874
Chicago/Turabian StyleTiso, Till, Daniel F. Sauer, Klaus Beckerle, Christian C. Blesken, Jun Okuda, and Lars M. Blank. 2020. "A Combined Bio-Chemical Synthesis Route for 1-Octene Sheds Light on Rhamnolipid Structure" Catalysts 10, no. 8: 874. https://doi.org/10.3390/catal10080874
APA StyleTiso, T., Sauer, D. F., Beckerle, K., Blesken, C. C., Okuda, J., & Blank, L. M. (2020). A Combined Bio-Chemical Synthesis Route for 1-Octene Sheds Light on Rhamnolipid Structure. Catalysts, 10(8), 874. https://doi.org/10.3390/catal10080874